
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 12 September 2024
Sec. Infectious Agents and Disease
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1452564
This article is part of the Research TopicRecent advances in Campylobacter researchView all 10 articles
 Chian Teng Ong1
Chian Teng Ong1 Patrick. J. Blackall2
Patrick. J. Blackall2 Gry B. Boe-Hansen3
Gry B. Boe-Hansen3 Sharon deWet4
Sharon deWet4 Ben J. Hayes1Lea Indjein3
Ben J. Hayes1Lea Indjein3 Victoria Korolik5
Victoria Korolik5 Catherine Minchin6
Catherine Minchin6 Loan To Nguyen1Yusralimuna Nordin1
Loan To Nguyen1Yusralimuna Nordin1 Hannah Siddle1
Hannah Siddle1 Conny Turni2
Conny Turni2 Bronwyn Venus1Mark E. Westman7Zhetao Zhang1
Bronwyn Venus1Mark E. Westman7Zhetao Zhang1 Ala E. Tabor1,8*
Ala E. Tabor1,8*Introduction: Bovine Genital Campylobacteriosis (BGC), caused by Campylobacter fetus subsp. venerealis, is a sexually transmitted bacterium that significantly impacts cattle reproductive performance. However, current detection methods lack consistency and reliability due to the close genetic similarity between C. fetus subsp. venerealis and C. fetus subsp. fetus. Therefore, this study aimed to utilize complete genome analysis to distinguish genetic features between C. fetus subsp. venerealis and other subspecies, thereby enhancing BGC detection for routine screening and epidemiological studies.
Methods and results: This study reported the complete genomes of four C. fetus subsp. fetus and five C. fetus subsp. venerealis, sequenced using long-read sequencing technologies. Comparative whole-genome analyses (n = 25) were conducted, incorporating an additional 16 complete C. fetus genomes from the NCBI database, to investigate the genomic differences between these two closely related C. fetus subspecies. Pan-genomic analyses revealed a core genome consisting of 1,561 genes and an accessory pangenome of 1,064 genes between the two C. fetus subspecies. However, no unique predicted genes were identified in either subspecies. Nonetheless, whole-genome single nucleotide polymorphisms (SNPs) analysis identified 289 SNPs unique to one or the C. fetus subspecies. After the removal of SNPs located on putative genomic islands, recombination sites, and those causing synonymous amino acid changes, the remaining 184 SNPs were functionally annotated. Candidate SNPs that were annotated with the KEGG “Peptidoglycan Biosynthesis” pathway were recruited for further analysis due to their potential association with the glycine intolerance characteristic of C. fetus subsp. venerealis and its biovar variant. Verification with 58 annotated C. fetus genomes, both complete and incomplete, from RefSeq, successfully classified these seven SNPs into two groups, aligning with their phenotypic identification as CFF (Campylobacter fetus subsp. fetus) or CFV/CFVi (Campylobacter fetus subsp. venerealis and its biovar variant). Furthermore, we demonstrated the application of mraY SNPs for detecting C. fetus subspecies using a quantitative PCR assay.
Discussion: Our results highlighted the high genetic stability of C. fetus subspecies. Nevertheless, Campylobacter fetus subsp. venerealis and its biovar variants encoded common SNPs in genes related to glycine intolerance, which differentiates them from C. fetus subsp. fetus. This discovery highlights the potential of employing a multiple-SNP assay for the precise differentiation of C. fetus subspecies.
Campylobacter spp. are Gram-negative, microaerophilic bacteria that are generally curved-shaped rods (Sebald and Veron, 1963; Smibert, 1981). Campylobacter fetus was first described as Vibrio fetus in 1919 (Smith and Taylor, 1919), and it was identified as a pathogenic species that can cause disease in humans and a number of other hosts (Smibert, 1981). There are three subspecies of C. fetus—C. fetus subsp. fetus, C. fetus subsp. testudinum, and C. fetus subsp. venerealis (Tu et al., 2004; Smibert, 1978). These C. fetus subspecies form a distinct host dichotomy (Gilbert et al., 2016; Tu et al., 2001). Specifically, the host for C. fetus subsp. testudinum is primarily reptiles (Fitzgerald et al., 2014), while C. fetus subsp. fetus and C. fetus subsp. venerealis are primarily associated with mammals (Marsh and Firehammer, 1953).
Campylobacter fetus subsp. fetus has been isolated from a broader range of hosts than C. fetus subsp. venerealis, including cattle, sheep, and humans, mainly from the gastrointestinal tract and occasionally from aborted fetuses (Smibert, 1978). The epidemiology of C. fetus subsp. fetus infection features persistent but mild infection and sporadic abortion only. In contrast, the colonization of C. fetus subsp. venerealis is highly host- and niche-specific, as this organism is confined to the bovine genital tract (Penner, 1988; Hoffer, 1981; Hum, 1996; OIE, 2019). Campylobacter fetus subsp. venerealis is recognized as the etiologic agent of Bovine Genital Campylobacteriosis (BGC), which is a venereal disease associated with low herd fertility and high economic loss across multiple geographical locations (Sprenger et al., 2012). Transmission of C. fetus subsp. venerealis is through natural mating with asymptomatic bulls or insemination with contaminated semen or equipment. The persistent infection of this subspecies in the female reproductive tract results in BGC, which is often manifested as infertility, embryonic loss, and abortions in the first part of pregnancy (Mshelia et al., 2010).
The expression of Surface Array Proteins (SAP) correlates with serovars of the C. fetus subsp. fetus (serovars A, B, and AB), C. fetus subsp. venerealis (serovar A), and C. fetus subsp. testudinum (serovars A, B and AB). However, it is not distinctive at the subspecies level (Moran et al., 1994; Perez-Perez et al., 1986; Dworkin et al., 1995). Nonetheless, C. fetus subsp. testudinum has been demonstrated to be genetically distinct from its two closely related subspecies (Fitzgerald et al., 2014; Dingle et al., 2010). A phylogenetic reconstruction, which involved 61 C. fetus genomes, revealed a barrier to lateral gene transfer between C. fetus subsp. testudinum and the other C. fetus subspecies (Gilbert et al., 2016). Additionally, several genetic features segregating the reptile-associated C. fetus subsp. testudinum from mammal-associated C. fetus were reported, including the exclusive presence of a putative locus encoding for tricarballylate catabolism pathway in C. fetus subsp. testudinum (Gilbert et al., 2016). Biomarkers based on proteotyping have also been identified for the differentiation of C. fetus subsp. testudinum from the other two C. fetus subspecies (Emele et al., 2019).
In comparison, despite their differences in niche specificity, biochemical properties, and pathogenicity, there is no universally recognized method for the differentiation between C. fetus subsp. fetus and C. fetus subsp. venerealis (Nadin-Davis et al., 2021; van der Graaf-van Bloois et al., 2014). The two biochemical tests described in the OIE Terrestrial Manual (OIE, 2019) for differentiating the C. fetus subspecies are the 1% glycine tolerance test and H2S production from cysteine-rich medium, with C. fetus subsp. The fetus was positive for both tests, while C. fetus subsp. venerealis is negative in both tests. Genome analysis has also confirmed the partial deletion of a putative cysteine transporter in C. fetus subsp. venerealis strains (van der Graaf-van Bloois et al., 2016a). However, the reliability of the biochemical tests for subspecies differentiation is complicated by the presence of the biovar C. fetus subsp. venerealis bv. intermedius, which possesses phenotypic characteristics from both C. fetus subsp. fetus and C. fetus subsp. venerealis (Sprenger et al., 2012; Iraola et al., 2013). Campylobacter fetus subsp. venerealis bv. intermedius has been isolated from both bovine intestinal and genital tracts and is glycine intolerant, which is typical of C. fetus subsp. venerealis but H2S positive as is typical of C. fetus subsp. fetus (OIE, 2019).
Amplified fragment length polymorphism (AFLP) and pulsed-field gel electrophoresis (PFGE) were shown to be effective in correlating the phenotypic and genotypic characteristics of C. fetus subsp. fetus and C. fetus subsp. venerealis; the high labor costs and the difficulty in isolating pure cultures of C. fetus from clinical samples required for these methods render them not ideal for routine testing by diagnostic laboratories (On and Harrington, 2001; van Bergen et al., 2005a; Wagenaar et al., 2001). Other discriminatory methods, for example, serotyping (Moran et al., 1994; Perez-Perez et al., 1986), DNA hybridization tests (Harvey and Greenwood, 1983; Basden et al., 1968), and protein banding patterns (Vandamme et al., 1990) were not able to accurately distinguish C. fetus subsp. fetus from C. fetus subsp. venerealis. Genomic studies suggested that these two C. fetus subspecies share a high level of genome synteny, with C. fetus subsp. venerealis possessing increased genome length and plasticity compared to C. fetus subsp. fetus (van der Graaf-van Bloois et al., 2014; Ali et al., 2012; Kienesberger et al., 2014). The C. fetus subsp. venerealis adaptation was attributed to the presence of hypervariable regions, pathogenicity islands and the acquisition of transposable elements, including prophages, transposons, and plasmids encoding for virulence factors (Nordin, 2013). Subspecies discrimination between C. fetus subsp. fetus and C. fetus subsp. venerealis using molecular methods has been attempted. For example, a multiple locus sequence typing (MLST) scheme based on the seven housekeeping genes categorized sequence type 4 (ST-4) exclusively with C. fetus subsp. venerealis (van Bergen et al., 2005b). However, ST-4 was later found to also be present in C. fetus subsp. fetus strain H1-UY (Iraola et al., 2015).
Several polymerase chain reaction (PCR) targets, including the plasmid partition protein A (parA; Hum et al., 1997) and the putative VirB6 protein truncated by the insertion element (ISCfe1; Abril et al., 2007), have been developed for the subspecies identification after investigating the different pathogenicity of C. fetus subspecies. The early research proposed that C. fetus subsp. venerealis displays a higher level of pathogenicity because of the genomic island in its genome (Iraola et al., 2012; Moolhuijzen et al., 2009). Several molecular targets were developed to target the genomic island, which encodes a type IV secretion system (T4SS). However, these assays with transfer-associated genes lacked specificity later when they were tested against multiple strains from both subspecies and related Campylobacter strains (Hum et al., 1997; Abril et al., 2007). For instance, the parA gene was not detected in 10 C. fetus subsp. venerealis in the previous study (Silva et al., 2021). Moreover, the previous PCR tests using various molecular targets on T4SS, including VirB6, also did not consistently align the C. fetus strains with their phenotypic identification (Nadin-Davis et al., 2021; Abril et al., 2007; van der Graaf-van Bloois et al., 2016b). There was no strong evidence of subspecies misidentification with ISCfe1, except its absence in C. fetus subsp. venerealis CCUG 34111 (Abril et al., 2007). Since T4SS-encoding regions are not exclusive to C. fetus subsp. venerealis and were found present in C. fetus subsp. fetus (Kienesberger et al., 2014; van der Graaf-van Bloois et al., 2016b) as well as other related Campylobacter bacteria, including C. jejuni, C. lari, and C. coli (Moolhuijzen et al., 2009), the reliability of ISCfe1, which is the insertion element truncated the T4SS VirB6 protein, for C. fetus subsp. venerealis, detection should be tested with a large number of Campylobacter strains from multiple continents. Another successful diagnostic test was developed using the L-Cys transporter-deletion polymorphism as the potential marker for H2S-positive C. fetus strains (van der Graaf-van Bloois et al., 2016a; Farace et al., 2019). However, this PCR assay was not able to capture the intermedius biovar variant, which is positive for H2S production (Farace et al., 2019, 2021).
The results from the previous study have suggested that the full genome sequence of the C. fetus subsp. venerealis and its biovars from different geographical continents will benefit the C. fetus subsp. venerealis detection research (Moolhuijzen et al., 2009). The genome completeness was demonstrated to be beneficial for whole-genome comparisons, particularly genomic regions with low coverage, high GC content, and/or high repetitiveness (Malmberg et al., 2019; Goldstein et al., 2019). Using complete genomes can therefore prevent the identification of false positives that arise from analyzing incomplete genomes, as missing data can lead to incorrect conclusions (Ribeiro et al., 2015; Ceres et al., 2022).
Therefore, in this study, we aimed to generate closed and complete genomes for nine C. fetus strains, including four C. fetus subsp. fetus, three C. fetus subsp. venerealis, and two C. fetus subsp. venerealis bv. intermedius, using Oxford Nanopore Technologies (ONT) long-read sequencing. Using these closed genomes in addition to the complete C. fetus genomes available on NCBI RefSeq, we conducted a whole-genome comparison to investigate the phylogenetic relationship between C. fetus subsp. fetus and C. fetus subsp. venerealis by examining their genome identity, differentially expressed gene orthologs, and single nucleotide polymorphisms (SNPs).
The bacterial strains sequenced in this study were from three different culture collections, the details of which are summarized in Table 1. Briefly, M20-08756/1A and M20-04752/1B were kindly gifted by the Department of Primary Industries in New South Wales, Australia, while isolates BT268/06 and BT376/03 were kindly gifted by the Institute for Glycomics at Griffith University in Queensland, Australia. The other isolates, including A8, 957, 76223, 924, and 926, were in-house isolates at the Queensland Alliance for Agriculture and Food Innovation at the University of Queensland, Australia, which were previously isolated from a local abattoir (Indjein, 2013). In total, four C. fetus subsp. fetus (M20-08756/1A, M20-04752/1B, BT268/06, and BT376/03), three C. fetus subsp. venerealis (A8, 957, and 76223), and two C. fetus subsp. venerealis bv. intermedius (924 and 926) strains were used in this study (Table 1). These strains were previously phenotyped using the standard OIE biochemical assays, and their subspecies identity was confirmed by cpn60 gene sequencing (Nordin, 2013; Indjein, 2013; Koya, 2016). The type strains for C. fetus subsp. fetus (ATCC 27374T) and C. fetus subsp. venerealis (ATCC 19438T), which had their complete genome published on the National Center for Biotechnology Information (NCBI) database, were also sequenced in this study to serve as internal controls. The cultures were stored at −80°C and were revived by culturing on the tryptone soya agar supplemented with 5% defibrinated sheep blood (Thermo Scientific, Delaware, USA) under micro-aerophilic conditions at 37°C for 48 h. Colonies of each bacterial strain were resuspended in sterile phosphate-buffered saline to reach an optical density measured at a wavelength of 600 nm (OD600) to yield ~1 x 109 colony-forming units per mL (cfu/mL). Genomic DNA extraction of the pure bacterial culture was conducted using the Genomic-tip extraction kit (QIAGEN, Hilden, Germany). The quantity and quality of extracted gDNA were assessed using a QubitTM 4 fluorometer (Thermo Scientific, Delaware, USA) and pulsed-field gel electrophoresis (Pippin Pulse, Sage Science, Massachusetts, USA).
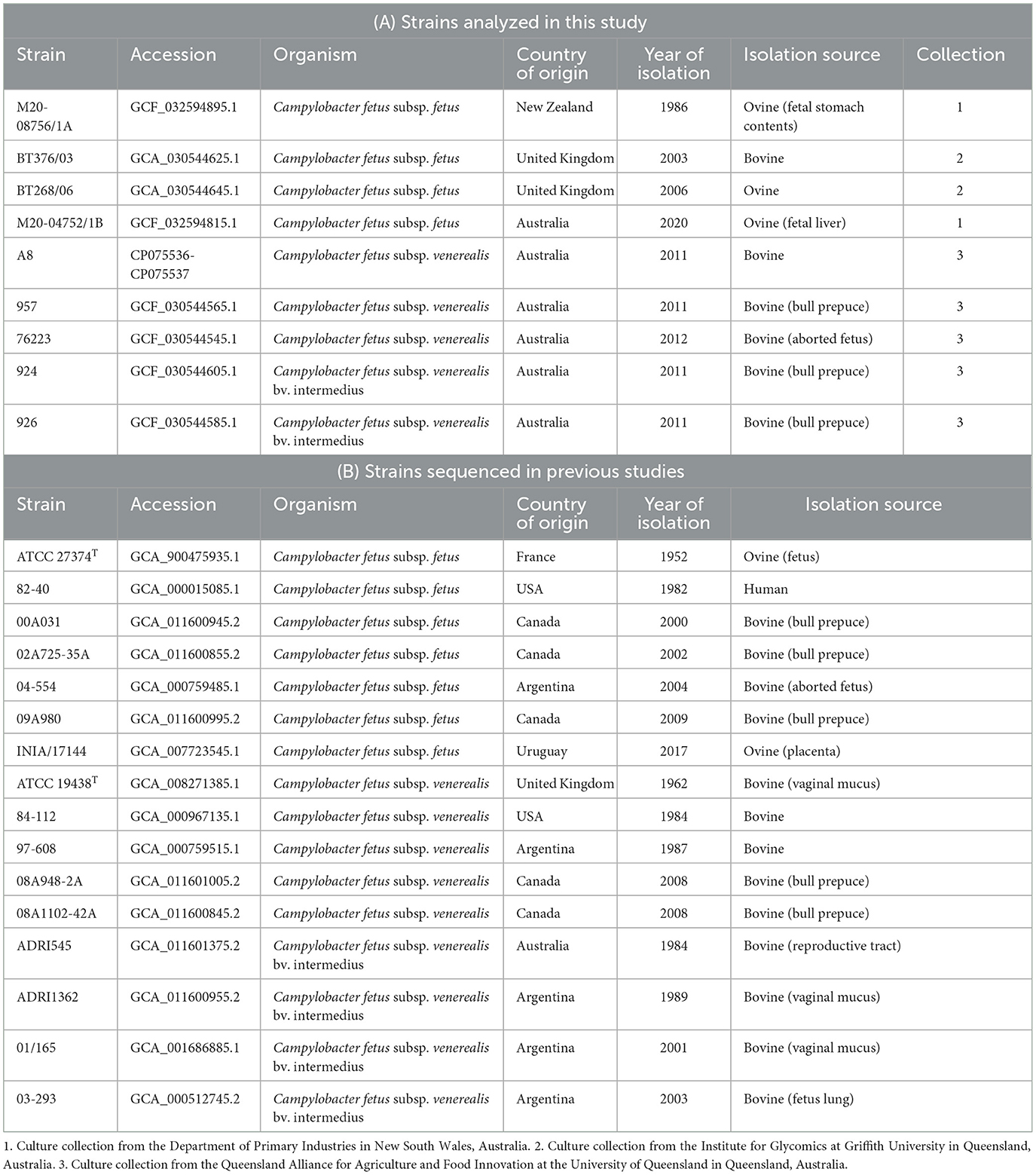
Table 1. List of Campylobacter fetus strains sequenced and analyzed in this study.
The Ligation Sequencing Kit SQK-LSK-109 (Oxford Nanopore Technologies, Cambridge, UK) was used to prepare sequencing libraries from the double-stranded high molecular weight genomic DNA. The sequencing libraries were loaded onto MinION (Oxford Nanopore Technologies, Cambridge, UK) for long-read sequencing with MinKNOW software (Oxford Nanopore Technologies, Cambridge, UK). Approximately 1 Gbp of data were generated for each isolate, and modified base-calling from raw signal data with minimum quality score filtering of 8 was performed using Guppy 5.0.7.
Extracted DNA was sent to the Ramaciotti Center for Genomics (University of New South Wales, Sydney, Australia) for short-read sequencing to generate 4 million read pairs or 1 Gbp of data. The libraries were prepared using the Nextera DNA Flex library prep kit (Illumina, California, USA), and the paired-end sequencing was executed on an iSeq 100 i1 sequencer with >80% bases higher than Q30 at 2 × 150 bp. The quality of the reads was assessed using FastQC 0.11.4 (Andrew, 2010) and was trimmed with Trimmomatic 0.39.1 using the paired-end mode (Bolger et al., 2014).
Porechop 0.2.4 (Wick et al., 2017a) was utilized to first remove the adapters, while NanoFilt 2.7.0 (De Coster et al., 2018) was implemented to select for reads that were >8,000 bp in length and >10 in quality score. The quality of the Nanopore long-read sequencing data was assessed and visualized using FastQC 0.11.4 (Andrew, 2010) and NanoPlot 1.3.0 (De Coster et al., 2018).
Quality long reads were assembled into contigs using Trycycler 0.5.0 (Wick et al., 2017b). Briefly, the read files were divided into 12 subsets, with three subsets of each assembled using Flye 2.9 (Kolmogorov et al., 2019), Miniasm+Minipolish v0.1.3 (Vaser et al., 2017), Raven v1.5.1 (Vaser and Šikić, 2021), and Redbean v2.5 (Ruan and Li, 2020). The resulting long-read assemblies were grouped into per-replicon clusters. The cluster containing contigs with a genome size closest to the reference genome was manually selected for the reconciliation step, aiming to circularize the replicons.
Trycycler then conducted multiple sequence alignments of the contigs within each cluster and generated a consensus sequence for the final assembly. The expected genome size for each bacterial strain was determined based on their respective published reference genome available in the NCBI database (NCBI Resource Coordinators, 2018).
To polish the complete genomes derived from Nanopore long reads, Medaka 1.4.2 with model r941_min_high_g303 and Nanopolish 0.13.2 (Loman et al., 2015) were used. Genome polishing was accelerated using GNU Parallel (Tange, 2011). The draft assemblies were further refined by polishing with their corresponding Illumina short-read data using Pilon 1.24 (Walker et al., 2014).
The quality of the complete genomes was evaluated with Samtools 1.10 (Li et al., 2009) and QUAST 5.0.2 (Gurevich et al., 2013) using both long and short reads. The quality assessments generated for each polished assembly were combined and presented using MultiQC 1.10.1 (Ewels et al., 2016). The polished genomes were then visualized and validated using Artemis (Carver et al., 2011). The identities of the complete genomes were confirmed using BLAST (Altschul et al., 1990). Finally, the polished assemblies were submitted to the NCBI and annotated using the Prokaryotic Genome Annotation Pipeline (PGAP; Tatusova et al., 2016).
For a more comprehensive whole-genome comparison, other complete genomes of C. fetus subsp. fetus (n = 7) and C. fetus subsp. venerealis (n = 9) were downloaded from the NCBI Genome database (NCBI Resource Coordinators, 2018) (Table 1). The inconsistencies of different annotation tools employed in previous studies were taken into consideration. Therefore, the complete genome sequence fasta files of the 16 C. fetus strains sequenced in previous studies were reannotated using the same parameters as the nine C. fetus strains sequenced in this study. The whole-genome average nucleotide identity (ANI) of the 25 C. fetus subspecies genomes was computed using FastANI 1.33 using the all-against-all mode (Jain et al., 2018). The correlation of the 25 C. fetus subspecies' complete whole genomes based on their ANI was computed in R (R Core Team, 2018) using the “corrplot” package (Wei, 2021).
Prokka 1.14.6 (Seemann, 2014) was utilized to annotate the complete genome sequences (n = 25). Briefly, Prokka 1.14.6 (Seemann, 2014) identified the protein-coding regions using Prodigal 2.6.3 (Hyatt et al., 2010), followed by the functional annotation of the encoded protein by similarity search against protein databases. Campylobacter fetus subsp. fetus 04/554 (GCA_000759485.1) and C. fetus subsp. venerealis ATCC 19438T (GCA_008271385.1) were provided as reference genomes for Prokka annotations to minimize the biases in annotation files for downstream analysis. The putative genomic islands (GIs) were predicted using IslandViewer 4 (Bertelli et al., 2017). The annotated assemblies were submitted to Roary 3.13.0 (Page et al., 2015) for pan-genome calculation. A heatmap was computed in R (R Core Team, 2018) using the “pheatmap” package (Kolde, 2019) to visualize the relationship of the 25 C. fetus subspecies based on the Roary results. The gene content and differences between the two closely related subspecies were also computed with GenAPI 1.0 (Gabrielaite and Marvig, 2019). The virulence factors known to be associated with Campylobacter were downloaded from the Virulence Factor Database (VFDB; Chen et al., 2005) for building a Campylobacter-specific VFDB database using ABRicate 1.0.1 (Seemann, 2016). The virulence factor encoding regions in each of the C. fetus subspecies were identified using the ABRicate 1.0.1 (Seemann, 2016) program.
SNPs were identified using Parsnp 1.5.6 (Treangen et al., 2014) from the whole-genome alignment generated with the complete genomes of the 25 C. fetus subspecies. A phylogenetic tree based on the SNPs identified in the whole-genome alignment of the 25 C. fetus strains was generated in R (R Core Team, 2018) using the “ape” package (Paradis and Schliep, 2018). The putative recombination regions with high SNP density were detected using Gubbins 3.0.0 (Croucher et al., 2014). The recombination-filtered SNPs were analyzed and annotated with SnpEff 4.3t (Cingolani et al., 2012) to filter out SNPs that potentially induce synonymous amino acid changes. The amino acid change was verified by examining the translated protein sequences of the SNP-coding coding sequences (CDS). The recombination-filtered synonymous SNPs that were different between C. fetus subsp. fetus and C. fetus subsp. venerealis, which are not present in putative GIs, were labeled as “candidate SNPs.” Additionally, SNPs that were different between C. fetus subsp. venerealis and its biovar intermedius variant were labeled as “biovar SNPs.” Any CDS that encoded for candidate SNPs were functionally annotated using eggNOG-mapper 2.1.6 (Cantalapiedra et al., 2021).
The annotated proteins of the SNP-coding CDS were retrieved from Prokka 1.14.6 (Seemann, 2014), and the interactions between the proteins were computed using STRING v11 (Szklarczyk et al., 2019) with C. fetus subsp. venerealis set as the organism of interest. The CDS, which were annotated with the “Peptidoglycan Biosynthesis” KEGG pathway, and their neighboring CDS with ≥8-degree functional association were taken for further investigation due to their potential association to the differential glycine tolerance among the subspecies. These SNPs were labeled as “Peptidoglycan SNPs.” To further verify the potential of the peptidoglycan SNPs for differentiation assay, an additional 33 curated and contamination-free RefSeq assemblies (13 C. fetus subsp. fetus and 20 C. fetus subsp. venerealis and its biovar) were downloaded from the NCBI Genome database (https://www.ncbi.nlm.nih.gov/datasets/genome) on 24th May 2024. The search terms “Campylobacter fetus subsp. fetus” and “Campylobacter fetus subsp. venerealis” were used, with the filter “Annotated by NCBI RefSeq” applied. The base change and amino acid change resulting from the peptidoglycan SNPs were verified across 58 C. fetus assemblies. The subspecies identification of each strain was compared with the phenotypic and molecular tests reported in previous studies.
Whole-genome alignment of the 25 C. fetus complete genomes was computed and visualized using the Blast Ring Image Generator (BRIG) 0.95 (Alikhan et al., 2011), which incorporated BLAST+ 2.10.1 (Camacho et al., 2009) for genome alignment. Additionally, the genes that were used in the published C. fetus subspecies identification PCR assays, including sodium/hydrogen exchanger family protein (nahE; Abril et al., 2007), ISCfe1 (Abril et al., 2007), peptide transporter carbon starvation (cstA; Hum et al., 1997) and parA (Hum et al., 1997; McMillen et al., 2006) are downloaded from the NCBI nucleotide database (NCBI Resource Coordinators, 2018). The location of putative GIs, candidate SNPs, and existing PCR targets for C. fetus subspecies identification was identified and labeled on the alignment image generated using BRIG (Alikhan et al., 2011).
One of the above-identified C. fetus subspecies SNPs (mraY gene SNP) was further exploited as a subspecies differentiating qPCR assay due to its potential association with the different glycine tolerance among C.fetus subspecies. Custom TaqMan MGB probes were designed using the Thermo Fisher Scientific online tool (Custom TaqMan™ SNP Genotyping Assay) targeting the C. fetus subsp. venerealis and C. fetus subsp. fetus mraY genes and labeled with VIC™ and FAM™ reporter dyes, respectively. mraY Forward primer: 5′ AAAATGATGATGAATTGGCGCCATT 3′; mraY Reverse primer: 5′ TGTGATGGAAACCTTATCTGTTATATTGCA 3′; C. fetus subsp. fetus mraY Probe: 5′FAM- CGTTTTTTGCGTATTTT-3′MGBNFQ; C. fetus subsp. venerealis mraY Probe: 5′VIC- CCGTTTTTTGTGTATTTT 3′MGBNFQ. The two probes and the forward and reverse primers were pre-mixed by Applied Biosystems and provided as a 20x mix for use in custom assays (Thermo Fisher Scientific, Australia). A 10 μL qPCR reaction was set up in duplicate using the AgPath-ID™ One-Step RT-PCR Reagent (Thermo Fisher Scientific) without the reverse transcriptase step consisting of 1x buffer, 900 nM of forward and reverse primers, 200 nM of the C. fetus subsp. fetus FAM probe, 200 nM of the C. fetus subsp. venerealis VIC probe, and 0.4 μl of the 25X RT-PCR Enzyme Mix (AmpliTaq Gold™ DNA Polymerase at 0.025 units per reaction). For positive controls, 2 ng of C. fetus subsp. fetus DNA (ATCC 27374) or 2 ng of C. fetus subsp. venerealis DNA (ATCC 19438) was added. A No Template Control (NTC, negative control) was included in every run. The assays were cycled in the Bio-Rad CFX96 Touch™ Real-Time PCR Detection System under the following conditions: activation at 95°C 10 min, followed by 45 cycles of 95°C 15 s, 69°C 1 min, and a final extension at 69°C for 7 min. Raw amplification data [quantification cycle (Cq) values and Relative Fluorescence Units (RFU)] were exported for analysis in Excel and R Studio (R Core Team, 2018) using the ggplot2 package (Wickham, 2016). The assay was screened against the following isolates: C. fetus subsp. venerealis ATCC19438 (positive control), A8, 957, 76223, 924, and 926; C. fetus subsp. fetus ATCC27374 (positive control) and BT376/03 (Supplementary Table 1). Other closely related bacterial species were also included as further controls: Campylobacter hyointestinalis strain 337, Arcobacter cryaerophilus strain 312, Campylobacter ureolyticus strain 412, and Campylobacter sputorum strain 530, which were isolated in a previous study screening abattoir bull penises and have been shown to cross-react in previous C. fetus venerealis molecular assays (McMillen et al., 2006; Spence et al., 2011).
The average read length, average N50, and average read quality of the nine strains sequenced with ONT were 33.52 kb, 38.43 kb, and 12.61, respectively (Supplementary Table 2). Illumina short-read sequencing yielded a total mean of 416,870 paired-end raw reads for each sample, which provided a mean coverage of 64X for each bacterial isolate (Supplementary Table 3).
All C. fetus strains were assembled into complete circular genomes. The average assembly size of C. fetus. fetus and C. fetus subsp. venerealis were 1,818,690 and 2,136,077 bp, respectively (Table 2). The QUAST quality assessment of each assembly showed that the long reads and short reads provided an average of 745.73X and 64.56X, respectively, to the assemblies (Supplementary Tables 4, 5). The average percentage of mapped long reads and mapped short reads were 98.56 and 97.50%, respectively. The complete genomes generated in this study and published genomes of C. fetus subsp. fetus and C. fetus subsp. venerealis were annotated with an average of 1,901 and 2,177 genes, respectively (Supplementary Table 6).
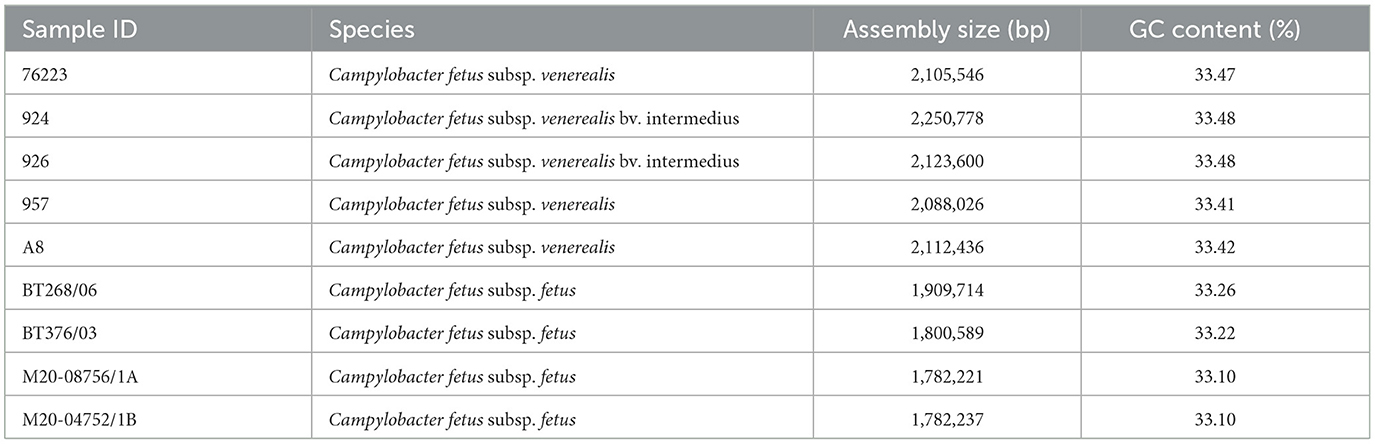
Table 2. Details of Campylobacter fetus complete genomes assembled in this study.
Pangenome analysis revealed that 1,561 core genes and 1,064 accessory genes were shared among the 25 C. fetus strains. The C. fetus subsp. fetus and C. fetus subsp. venerealis strains were separated in the hierarchical tree generated based on the presence and absence of gene orthologs (Figure 1). The pangenome analysis demonstrated the gene ortholog, which encoded for a peptidase S24 LexA-like protein, was exclusively encoded in all the C. fetus subsp. venerealis genomes but not in any of the C. fetus subsp. fetus genomes. However, three paralogs also encoded for peptidase S24 LexA-like proteins and were predicted in some of the C. fetus subsp. fetus and C. fetus subsp. venerealis strains (Supplementary Table 7).
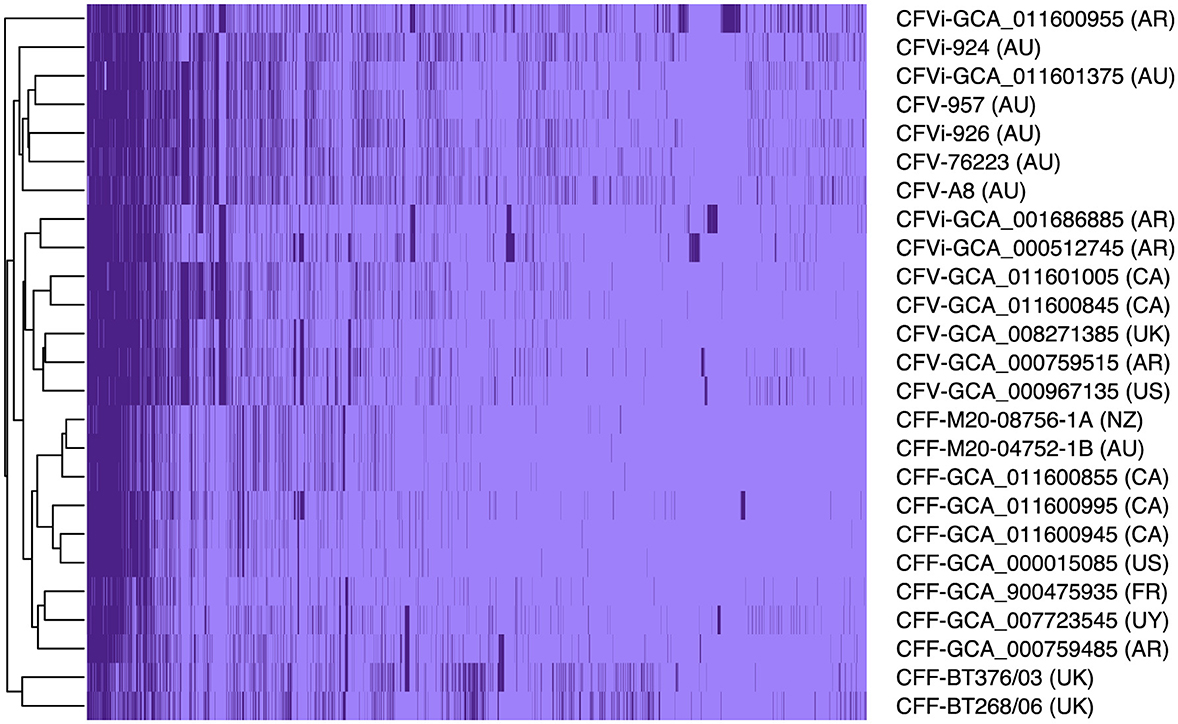
Figure 1. The pangenome of the 25 Campylobacter fetus strains. Dark purple indicates the presence of the gene ortholog, while light purple represents the absence of the gene ortholog. International Organization for Standardization (ISO) country code: Canada (CA), United States (US), France (FR), Uruguay (UY), Argentina (AR), United Kingdom (UK), Australia (AU), and New Zealand (NZ).
In contrast, C. fetus subsp. venerealis and the bv. intermedius variant did not fall into distinct branches because none of the gene orthologs was able to differentiate C. fetus subsp. venerealis and C. fetus subsp. venerealis bv. intermedius. Interestingly, the six Australian C. fetus subsp. venerealis strains (GCA_011601375, A8, 924, 926, 957, and 76223) formed a separated clade from the other non-Australian C. fetus subsp. venerealis. A total of 14 gene orthologs were identified in the Australian C. fetus subsp. venerealis strains but not in the non-Australian strains, of which one was present in all C. fetus subsp. fetus, and nine were absent in all the C. fetus subsp. fetus strains. On the other hand, 37 gene orthologs were present in the non-Australian C. fetus subsp. venerealis and not the Australian strains, none of which were identified in all C. fetus subsp. fetus, and 34 were absent in all the C. fetus subsp. fetus strains.
A closer look at the genomic regions encoding the Campylobacter-specific virulence factors demonstrated that the 25 C. fetus subspecies commonly expressed genomic regions encoding 88 virulence factors (Figure 2). Nine virulence factors were not expressed in some of the C. fetus subspecies but not explicitly in either C. fetus subsp. fetus or C. fetus subsp. venerealis. The highest variation was observed within the S-layer proteins, which are categorized under the class of “colonization and immune evasion” in VFDB. Nonetheless, the variations among the predicted proteins were not consistent within either of the subspecies or the biovar.
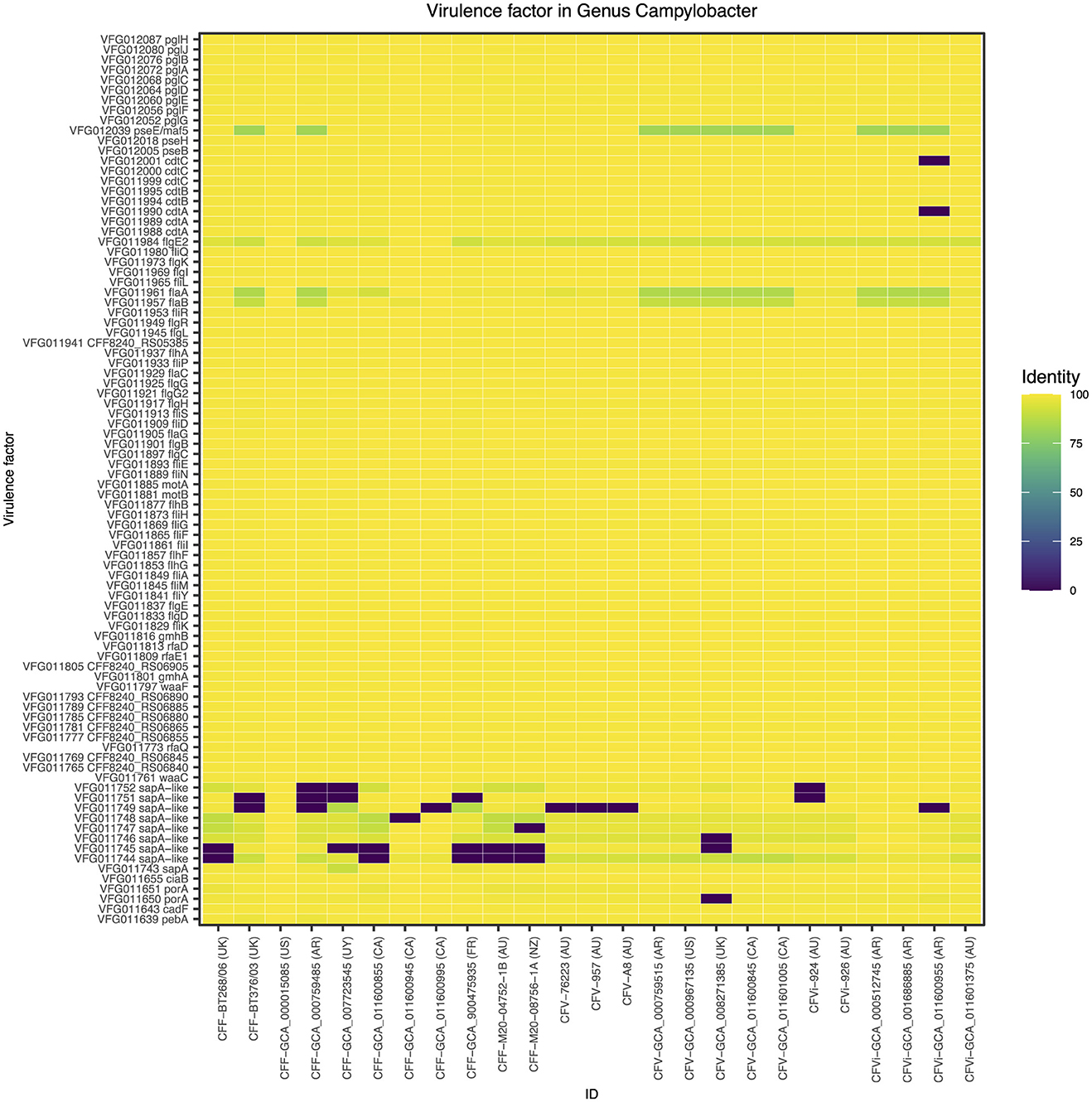
Figure 2. The Campylobacter-specific virulence factors among the 25 Campylobacter fetus strains. The color key indicates the percentage of identity between the genomic regions identified in each isolate and the Campylobacter-specific virulence factors, in which yellow represents 100% identity and purple represents 0% identity. International Organization for Standardization (ISO) country code: Canada (CA), United States (US), France (FR), Uruguay (UY), Argentina (AR), United Kingdom (UK), Australia (AU), and New Zealand (NZ).
The average nucleotide identity between 25 C. fetus strains was more than 95%. The correlation tree based on the ANI showed that eight of the C. fetus subsp. fetus strains, including GCA_011600855, BT268/06, M20-04752/1B, M20-08756/1A, GCA_900475935T, GCA_007723545, BT376/03, and GCA_00759485, formed a separate branch from the C. fetus subsp. venerealis strains (Figure 3). The other three C. fetus subsp. fetus strains (GCA_000015085, GCA_011600995, and GCA_011600945) shared the same ancestor with three C. fetus subsp. venerealis bv. intermedius strains (GCA_011600955, GCA_001686885, and GCA_000512745) from Argentina. The remaining C. fetus subsp. venerealis strains (n = 11) formed a separate clade, in which the C. fetus subsp. venerealis, including both biovars from Australia (GCA_011601375, A8, 924, 926, 957, and 76223) clustered separately to all other C. fetus subsp. venerealis from around the world.
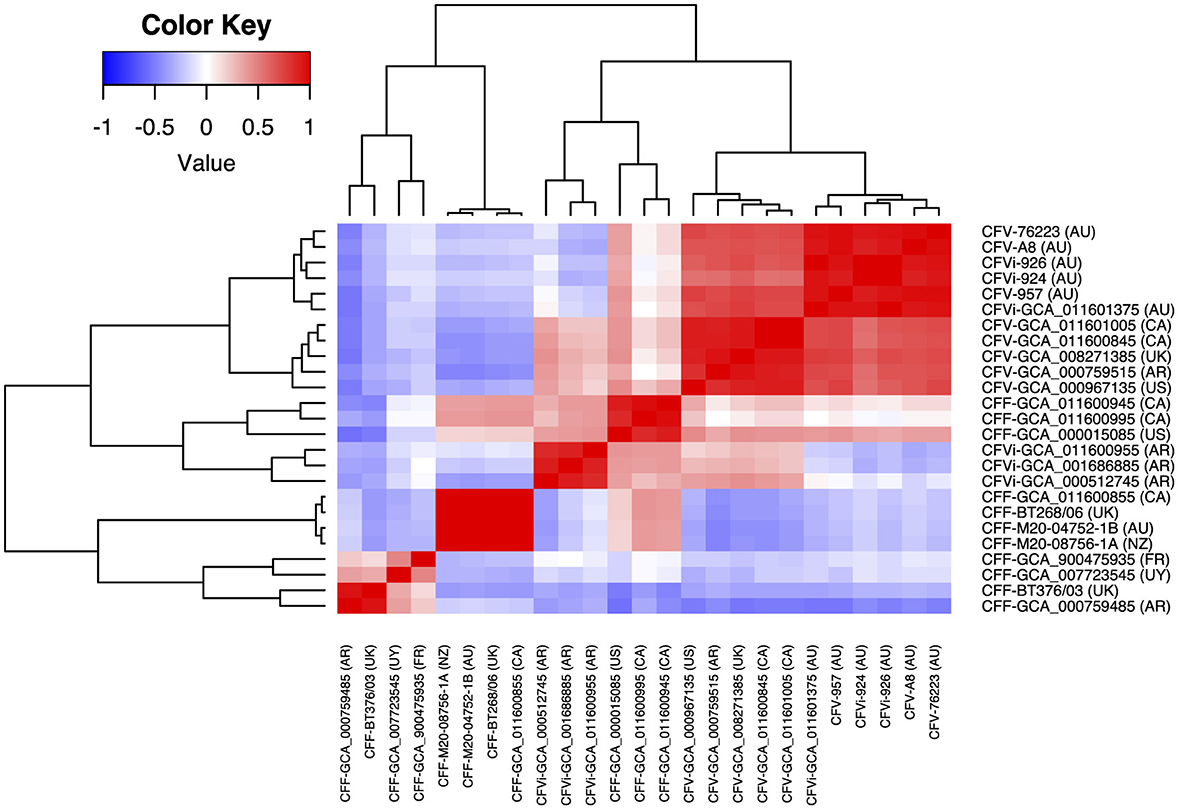
Figure 3. Correlation between the 25 Campylobacter fetus strains based on their average nucleotide identity. The color key indicates the degree of correlation between the strains, in which red represents the highest correlation while blue represents the lowest correlation. The dendrogram demonstrates the hierarchical relationship between the 25 Campylobacter fetus strains based on their average nucleotide identity. International Organization for Standardization (ISO) country code: Canada (CA), United States (US), France (FR), Uruguay (UY), Argentina (AR), United Kingdom (UK), Australia (AU), and New Zealand (NZ).
Comparative genome alignment of the C. fetus strains (n = 25) illustrated the high level of genome synteny between the C. fetus subspecies (Figure 4). Missing genomic regions were inconsistently observed among the 25 C. fetus strains where putative GIs were predicted. PCR target genes, including sapB2 and parA, were located on putative GIs.
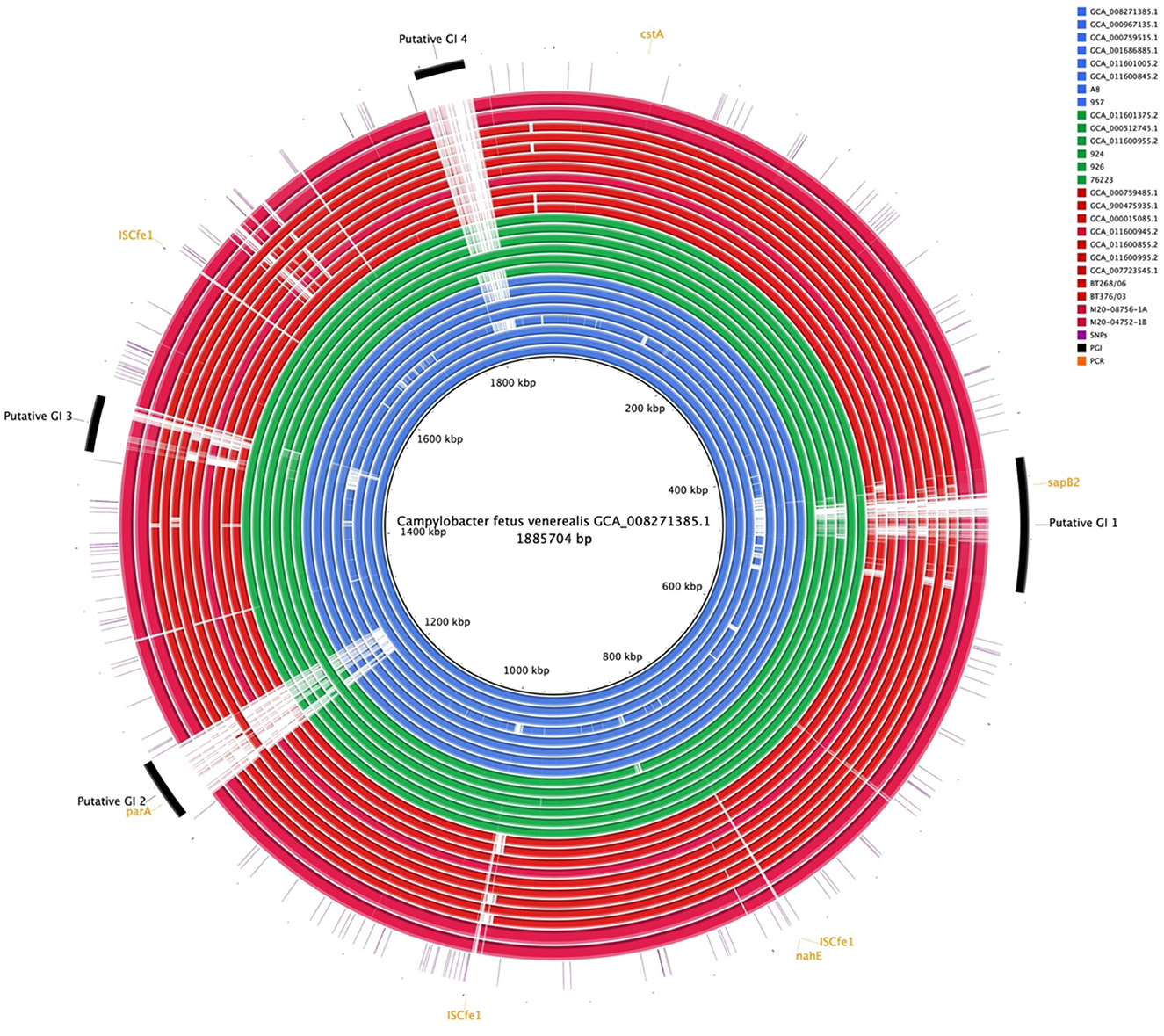
Figure 4. Whole-genome alignment of 25 Campylobacter fetus subspecies. Reference: published C. fetus subsp. venerealis ATCC 19438T [GCA_008271385.1]. Query were complete genome sequences of C. fetus subsp. Fetus (n = 11) and C. fetus subsp. venerealis (n = 14) subspecies. Strains labeled in red are C. fetus subsp. fetus, isolated and labeled in blue, are C. fetus subsp. venerealis and strains labeled in green are C. fetus subsp. venerealis bv. intermedius. Black arcs represent the putative genomic islands. Orange arcs represent the genes that were used in PCR C. fetus subspecies identification. Purple lines represent candidate SNPs identified in the 25 C. fetus subspecies.
The phylogenetic tree that resulted from the whole-genome alignment clustered C. fetus subsp. venerealis into a separate branch from the C. fetus subsp. fetus (Figure 5). Moreover, the phylogenetic tree also clustered the Australian C. fetus subsp. venerealis in a separate branch from the C. fetus subsp. venerealis identified in the United Kingdom, United States, Canada, and Argentina (Figure 5). Both within the Australian and non-Australian C. fetus subsp. venerealis clades, the C. fetus subsp. venerealis bv. intermedius were clustered separately from the normal venerealis variant.
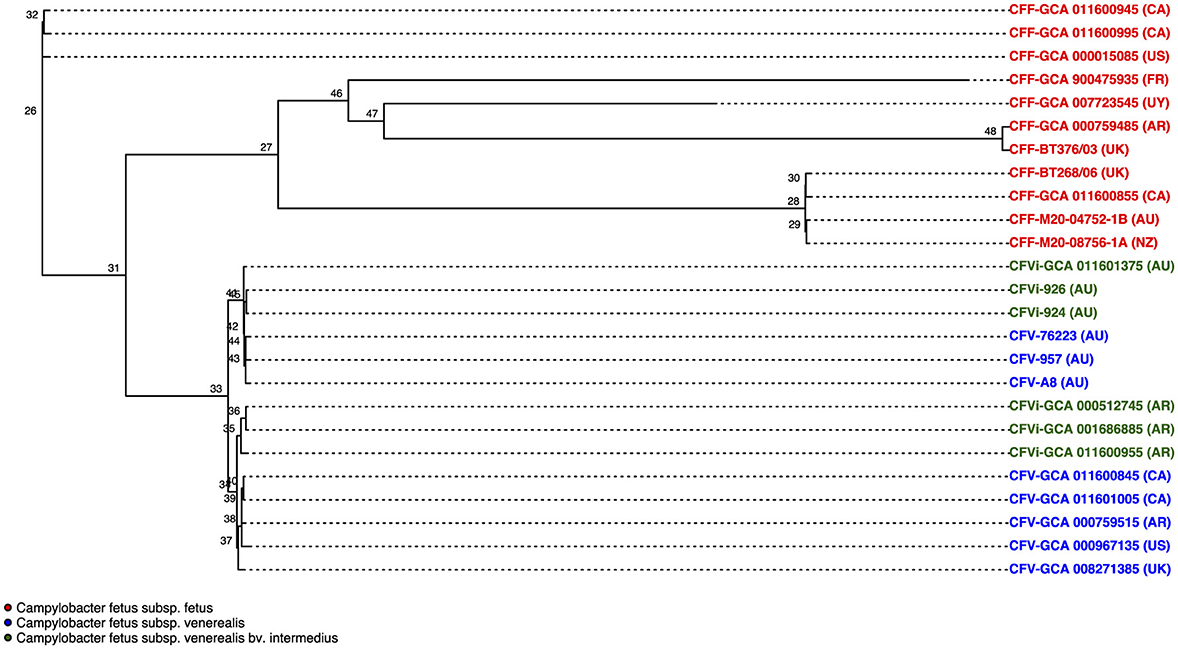
Figure 5. Phylogenetic tree based on single nucleotide polymorphisms (SNPs) between the 25 Campylobacter fetus strains. Strains labeled in red are C. fetus subsp. fetus, strains labeled in blue are C. fetus subsp. venerealis, and strains labeled in green are C. fetus subsp. venerealis bv. intermedius. All C. fetus subsp. fetus isolates clustered into a separate branch from all C. fetus subsp. venerealis and C. fetus subsp. venerealis bv. intermedius isolates, indicating that there were distinctive SNPs. C. fetus subsp. venerealis and C. fetus subsp. venerealis bv. intermedius isolates from Australia clustered separately from non-Australian isolates. International Organization for Standardization (ISO) country code: Canada (CA), United States (US), France (FR), Uruguay (UY), Argentina (AR), United Kingdom (UK), Australia (AU), and New Zealand (NZ).
Nine thousand and 44 SNPs were identified from the core genome, of which only 269 SNPs were different between all C. fetus subsp. fetus and all C. fetus subsp. venerealis strains (Figure 6A). The SNPs that contributed to putative synonymous amino acid change, located on putative GIs, and involved in recombination events were filtered out from downstream analysis. The remaining 184 SNPs were labeled as “candidate SNPs,” of which 17 were located in non-coding regions, and 167 were located in CDS (Supplementary Table 8). A total of 16 of the CDSs were encoded for two candidate SNPs, with one CDS encoded for three candidate SNPs and the other CDS encoded for five candidate SNPs. In total, the 167 candidate SNPs were located on 145 CDS, of which 15 of the CDS were encoding for hypothetical proteins. The majority of the SNPs-encoding CDS were responsible for “cellular processes and signaling” (n = 45), followed by “metabolism” (n = 44) and “information storage and processing” (n = 21; Figure 6B, Supplementary Table 9).
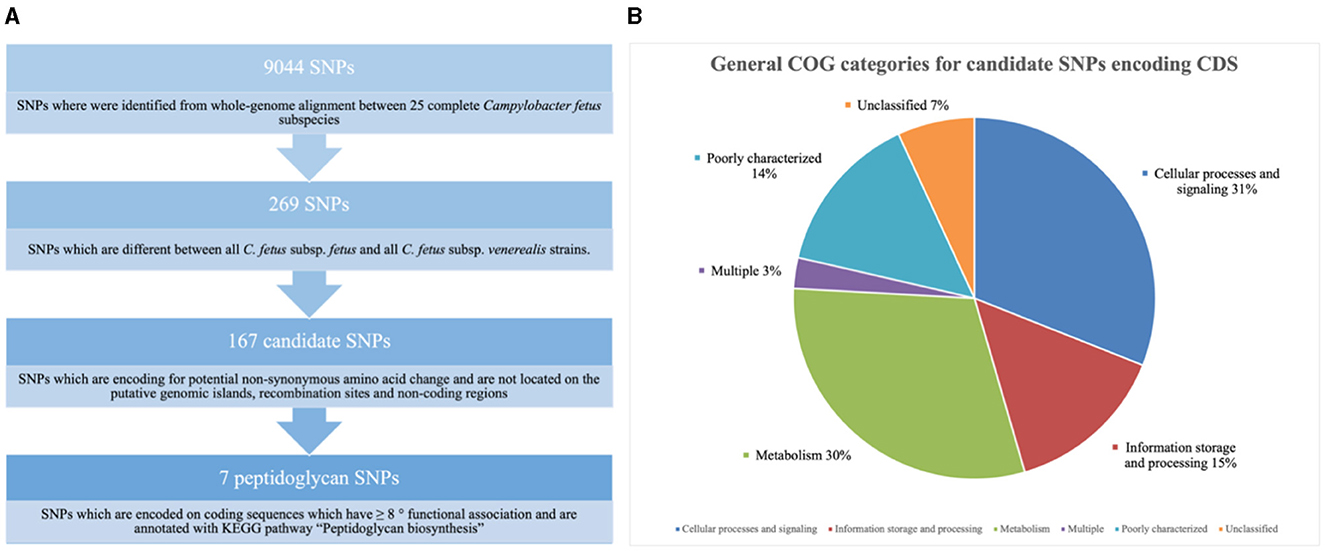
Figure 6. (A) Screening for single nucleotide polymorphisms (SNPs) suitable for reliable differentiation between Campylobacter fetus subsp. fetus and Campylobacter fetus subsp. venerealis. (B) Candidate single nucleotide polymorphisms (SNPs) are categorized into Clusters of Orthologous Groups (COGs). The blue section represents coding sequences (CDS) responsible for “cellular processes and signaling” (n = 45). The orange section represents the CDS responsible for “information storage and processing” (n = 21). The gray section represents CDS responsible for “metabolism” (n = 44). The yellow section represents CDS belonging to multiple groups of COGs (n = 4). The light blue section represents CDS, which was poorly categorized (n = 21). The green section represents CDS with no match return from querying the COG database (n = 10).
The potential functional association between the SNP-coding CDS was determined, and 41 candidate SNPs were identified on the SNP-coding CDS, which posed more than eight degrees of association. A subset of the 41 SNPs that were annotated with the KEGG pathway “Peptidoglycan biosynthesis” (Supplementary Figure 1) was chosen for further analysis due to their potential association with the differential glycine production in the C. fetus subspecies. The subset included murC, ftsI, uppP, and mraY, and their first neighbors (Figure 7). Among them, the SNP-coding CDS encoded for the candidate SNPs associated with significant amino acid change, including rpoC, cysS, rpoB, flgG, mfd, mraY, and mutS2, were manually verified (Table 3). The candidate SNPs in these SNP-coding CDS were referred to as peptidoglycan SNPs (Figure 6A).
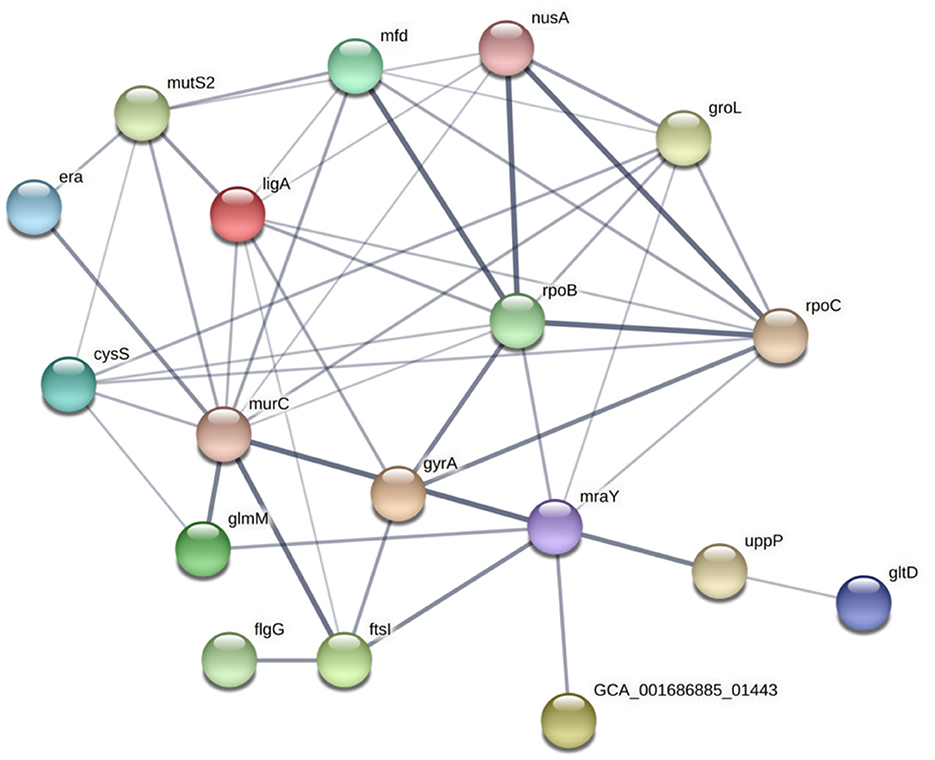
Figure 7. STRING network of SNP-coding CDS, which had more than eight degrees of association. The thickness of the network edges indicates the confidence level of the functional interactions.
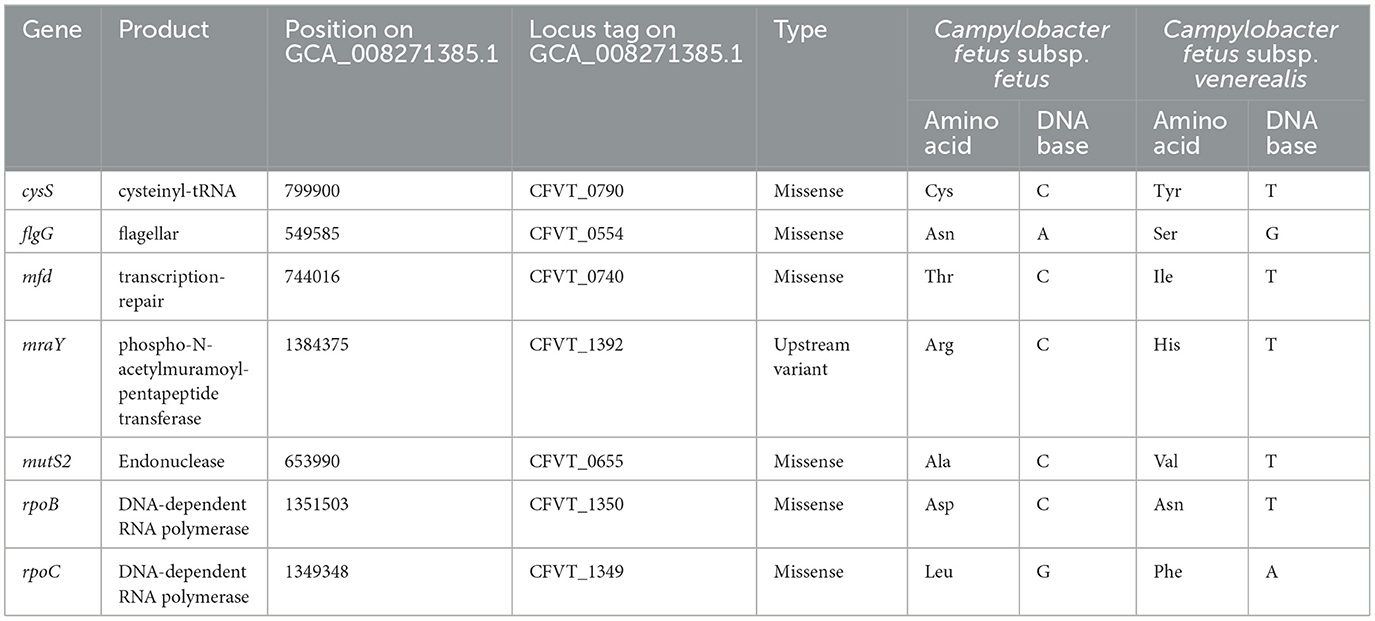
Table 3. Amino acid and DNA base changes on the candidate SNP-coding CDS encoded on C. fetus subsp. fetus and C. fetus subsp. venerealis.
Interestingly, there were no common SNPs that could absolutely separate all the C. fetus subsp. venerealis and its biovar intermedius variant regardless of the country of origin. Therefore, this study only tested the potential of the seven peptidoglycan SNPs in differentiating the C. fetus subsp. fetus (CFF) from C. fetus subsp. venerealis and its biovar (CFV/CFVi). On top of the 25 complete genomes, an additional 33 curated RefSeq assemblies (13 C. fetus subsp. fetus and 20 C. fetus subsp. venerealis and its biovar) were recruited in the evaluation (Table 4). All seven peptidoglycan SNPs performed consistently and reliably divided the assemblies into two groups (Supplementary Table 10). The 24 C. fetus subsp. fetus assemblies, except NWU_ED24 (GCF_013406925.1), 13/344 (GCF_008527615.1), and 08/421 (GCF_008526335.1), posed SNP pattern conformed to the CFF subset. Interestingly, the three assemblies (NWU_ED24, 13/344, and 08/421) posed an SNP pattern that conformed to the CFV/CFVi subset. On the other hand, the 34 C. fetus subsp. venerealis assemblies, except P4531 (GCF_016406645.1), posed an SNP pattern conformed to the CFV/CFVi subset. The P4531 strain posed SNP pattern conformed to the CFF subset instead. Therefore, in this study, NWU_ED24, 13/344, and 08/421 were classified as C. fetus subsp. venerealis and its biovar, while the P4531 strain was classified as C. fetus subsp. fetus.
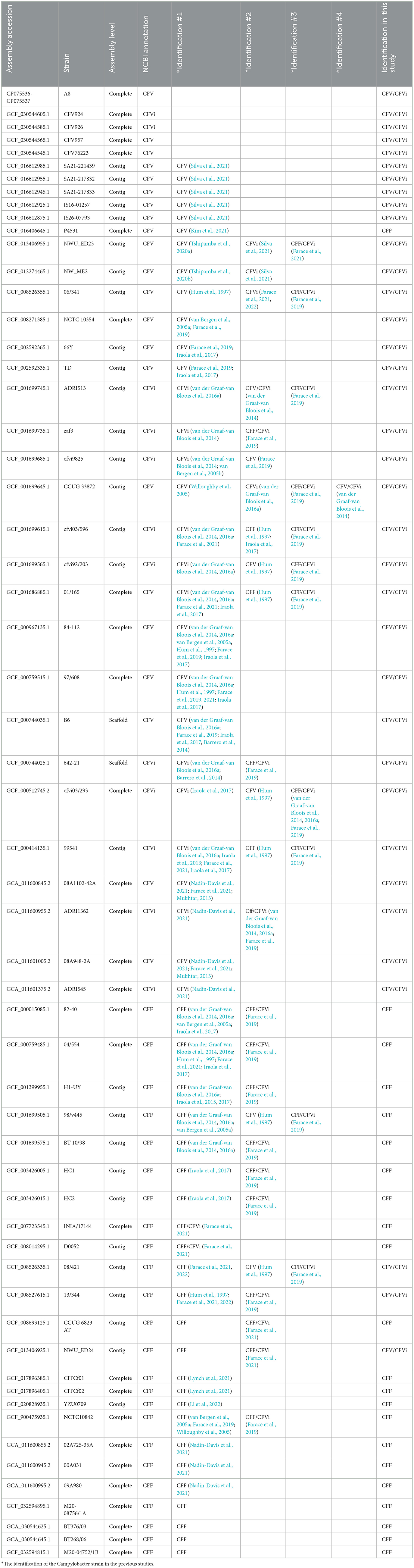
Table 4. Characterization of 58 Campylobacter fetus subspecies according to previous studies and the current study.
One of the SNPs differentiating the C. fetus subspecies located in the mraY gene (Table 3) was further exploited in a TaqMan SNP qPCR assay where the FAM channel detected the C. fetus subsp. fetus and the VIC channel C. fetus subsp. venerealis. The sensitivity of the assay was 1 pg when using pure bacterial DNA, and following testing of mixed Campylobacter-like species, a positive cut-off was determined at Cq 33 cycles (Figure 8A). The mraY assay was able to distinguish between C. fetus subsp. venerealis and C. fetus subsp. fetus controls, as well as other C. fetus isolates listed in Table 1 and other closely related Campylobacter species (C. sputorum, C. ureolyticus, C. hyointestinalis, and A. cryoaerophilus; Figure 8B).
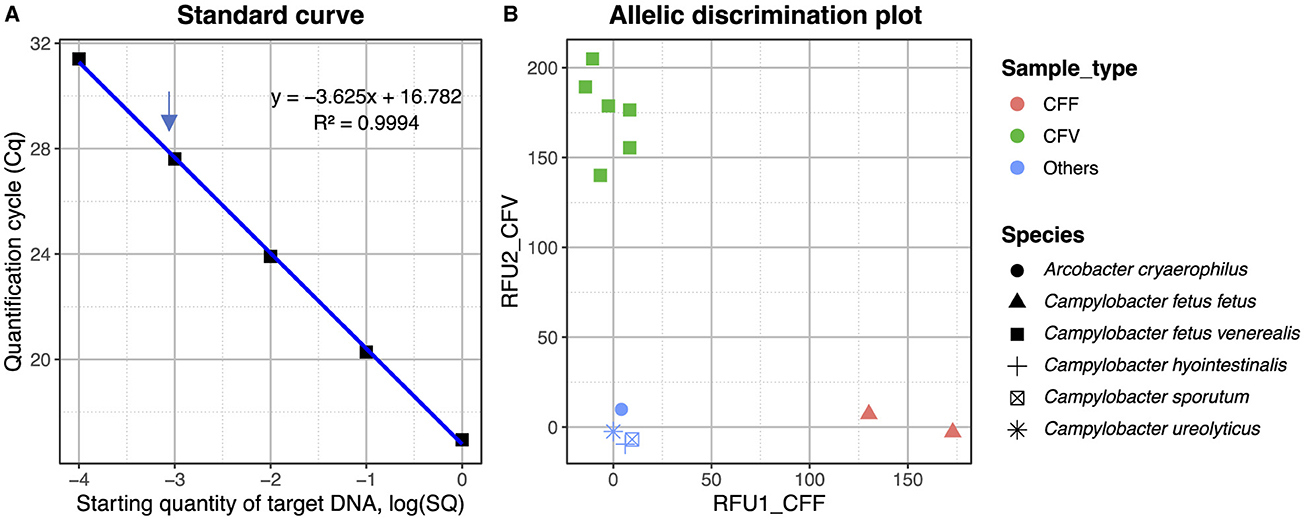
Figure 8. (A) Standard curve for the mraY SNP assay plotting the quantification cycle (Cq) value against the starting quantity of C. fetus venerealis DNA. Data points are from 1 ng to 0.1 pg, with 1 pg indicated by an arrow. The R2 of the standard curve is 0.994. (B) Allelic discrimination plot for mraY Taqman SNP assay using Relative Fluorescent Units (RFU) for VIC and FAM. Red—CFF control (ATCC 27374), brown—CFF isolate (strain BT376/03), green—CFV control (ATCC 19438), blue—CFV isolates (strains A8, 957, 76223, 924, and 926), and light blue—other Campylobacter species (C. sputorum, C. ureolyticus, C. hyointestinalis, and A. cryoaerophilus).
Differentiating C. fetus subspecies is crucial for the routine screening of ruminants for import and export and epidemiological investigation. Subspecies differentiation has been investigated using multiple methods, including biochemical analysis (OIE, 2019), AFLP (Wagenaar et al., 2001), PFGE (On and Harrington, 2001), MLST (van Bergen et al., 2005b; Iraola et al., 2015), PCR (Hum et al., 1997; Abril et al., 2007; Wang et al., 2002), and genome comparison methods (van der Graaf-van Bloois et al., 2014; Kienesberger et al., 2014). The results of this study demonstrated an improved and less laborious genome analysis method that successfully separated C. fetus subsp. fetus and C. fetus subsp. venerealis into two distinct clusters. According to their phenotypes, similar to previous AFLP (Wagenaar et al., 2001) and PFGE analyses (On and Harrington, 2001), but with a more efficient procedure.
In this study, the Australian C. fetus subsp. venerealis were separated from the non-Australian C. fetus subsp. venerealis, which was different from the indistinguishable geographical genotypes described in the previous core genome analysis (van der Graaf-van Bloois et al., 2014) and PFGE analysis (On and Harrington, 2001). Unique microbial lineages due to geographical separation are not novel. For example, most Australian Glaesserella (Haemophilus) parasuis strains were found to belong to unique sequence types not previously recorded in the relevant MLST database (Turni et al., 2018). In the phylogenetic tree generated using whole-genome alignment, the C. fetus subsp. venerealis clade shared the same ancestor with eight other C. fetus subsp. fetus strains, potentially indicating that C. fetus subsp. venerealis was derived from C. fetus subsp. fetus. The identification of a small number of SNPs (n = 184), which are differentially expressed in the C. fetus subsp. fetus and C. fetus subsp. venerealis strains, which were supported by the observations from the MLST analyses, indicating a limited genetic variation between C. fetus subspecies was driven mainly by the slow accumulation of point mutations (van Bergen et al., 2005b). Additionally, this study did not identify any SNPs that are unique to the C. fetus subsp. venerealis bv. intermedius regardless of geographical origins, suggesting that clonal evolution occurred separately within the geographically different C. fetus subsp. venerealis clades. It appears that C. fetus subsp. venerealis and its biovar-acquired point mutations, which have been vertically transmitted and enabled the development of their highly niche-specific and pathogenicity-specific characteristics.
For the comparative whole-genome analysis, only complete C. fetus genomes were used. This approach ensured a comprehensive comparison and prevented false positives that could result from aligning the complete genome against the gaps of an incomplete genome. The previous investigation, compared one complete genome of C. fetus subsp. fetus (GCA_000015085) against one incomplete genome of C. fetus subsp. venerealis (GCA_000222425) reported that 88 and 428 gene families were unique to C. fetus subsp. fetus and C. fetus subsp. venerealis, respectively (Ali et al., 2012). The current study only identified one gene ortholog, which was different between the two complete genomes of the C. fetus subspecies. Our results aligned with the previous suggestion, based on methods other than genome sequencing, that there is a lack of genetic diversity between the C. fetus subspecies because the C. fetus strains are at an early evolutionary stage (van Bergen et al., 2005a; Wagenaar et al., 2001). The only unique gene ortholog encoded for a peptidase S24 LexA-like protein was only identified in C. fetus subsp. venerealis and not in the C. fetus subsp. fetus strains. However, three other paralogs coding for peptidase S24 LexA-like protein were identified in both C. fetus subspecies in this study. While LexA is a global transcription factor responsible for regulating host SOS response, several studies have also suggested that mobile genetic elements utilize the LexA activity of the hosts for their induction (Fornelos et al., 2016; Quinones et al., 2005; Kimsey and Waldor, 2009). The previous comparison of 14 C. fetus strains observed that the lexA gene, which served as a prophage regulator, was mostly identified at the boundary of a prophage element (Nadin-Davis et al., 2021).
Additionally, we found no gene ortholog that was unique to either C. fetus subsp. venerealis or the C. fetus subsp. venerealis biovar variant. This is in contrast to the previous pangenome analysis of 31 C. fetus subsp. venerealis strains using a mixture of complete and incomplete genomes, which identified inconsistent expressions of parA and T4SS encoding genes (virB2-virB11 and virD4) in the C. fetus strains (Silva et al., 2021). Additionally, the virulence gene investigation in this study demonstrated variations in the sap genes among the 25 C. fetus strains. This result corresponded to the previous studies, which categorized C. fetus subspecies to serovars based on the variable expression of genes in the sap locus that encode for surface layer protein (SLP; Tu et al., 2001; Dworkin et al., 1995). Our results reinforced that the serovar classification is not unique at the C. fetus subspecies level.
The biochemical tests that are currently recommended by OIE (2019) for identifying C. fetus subspecies include glycine tolerance test and H2S production. C. fetus subsp. fetus is positive for both tests, while C. fetus subsp. venerealis is negative for both. Previous studies have suggested the ATP-binding cassette-type L-cysteine transporter as a potential marker for H2S-positive C. fetus strains, leading to the development of an accurate diagnostic test based on the L-Cys transporter-deletion polymorphism (van der Graaf-van Bloois et al., 2016a; Farace et al., 2019).
However, unlike C. fetus subsp. venerealis, the intermedius biovar variant is positive for H2S production. As a result, previous tests using molecular techniques to detect the L-Cys transporter-deletion polymorphism were unable to distinguish C. fetus subsp. venerealis biovar intermedius strains from C. fetus (Farace et al., 2019, 2021). This study provided 167 SNPs as new candidates for C. fetus subspecies genotyping, which may be valuable for precise and efficient routine screening on farms, international trade, and for epidemiological investigations and diagnostics.
In this study, SNP-coding CDS was investigated for a correlation with the differential tolerance to glycine among the C. fetus subspecies, which separates the C. fetus subsp. fetus from the C. fetus subsp. venerealis and its biovar intermedius. Glycine has been suggested to inhibit bacterial cell wall biosynthesis, particularly the peptidoglycan component, and thus can show an antibacterial effect (Hishinuma et al., 1969). Therefore, candidate CDS annotated with the “peptidoglycan biosynthesis” KEGG pathway and posed potential functional association with one another, including cysS, flgG, mfd, mraY, mutS2, rpoB, and rpoC, were recruited for further analysis. Gene mraY was part of the gene set found in peptidoglycan-intermediate obligate intracellular bacteria (Otten et al., 2018). Phospho-N-acetylmuramoyl-pentapeptide-transferase (mraY) is a catalytic enzyme that initiates the lipid cycle reactions during bacterial peptidoglycan synthesis (Struve et al., 1966). Under normal circumstances, mraY has high specificity to L-alanine and D-alanine (Hammes and Neuhaus, 1974). However, the non-synonymous amino acid change introduced by the peptidoglycan SNP identified in our study potentially modified mraY and thus allowed glycine substitution. As a result, bacteriolysis or morphological aberrations of the bacterial cells could be induced and lead to differential glycine tolerance between the C. fetus subspecies. The presence of this SNP was further exploited in a novel C. fetus subspecies TaqMan SNP qPCR, which demonstrated specific detection of each subspecies using different fluorophores and no detection of other closely related species such as C. hyointestinalis as reported for other diagnostic targets present on mobile elements (Spence et al., 2011).
Other peptidoglycan SNPs were previously reported to be associated with variations in niche adaption and virulence between closely related bacterial strains. The rpoB and rpoC genes code for the β- and β'-like subunits of the DNA-dependent RNA polymerase, which regulates gene expression in bacteria and archaea (Zakharova et al., 1998). Mutations in rpoB and rpoC genes were reported to be responsible for the different degrees of antibiotic resistance observed in Staphylococcus aureus (Matsuo et al., 2015), Mycobacterium tuberculosis (Ma et al., 2021) and Clostridium difficile (Kuehne et al., 2018). Similar observations on the impact of these mutations have been demonstrated for the virulence of C. difficile strains (Kuehne et al., 2018), as well as the differences in the ability of E. coli (Conrad et al., 2010) and Helicobacter pylori (Zakharova et al., 1998) to colonize different niches. SNPs in both rpoB and cysS were suggested to contribute to the niche adaptation of the multi-host pathogen S. aureus (Bacigalupe et al., 2019). The interaction between the “mutation frequency decline” (mfd) gene and RNA polymerase, for example, rpoB, plays a role in the development of antimicrobial resistance in highly divergent bacterial species (Ragheb et al., 2019). Additionally, mfd is recognized as the “pro-evolutionary factor,” as studies suggested that mfd plays a significant role in prokaryotic virulence and survival (Lindsey-Boltz and Sancar, 2021; Strick and Portman, 2019). Mutations on the outer membrane proteins, including flgG (Palau et al., 2021) and genes of the hop family (Linz et al., 2013), facilitated the niche adaptations of different strains of Helicobacter pylori and allowed the colonization of the new host. Non-synonymous mutations in the mutS2, a gene coding for endonuclease responsible for suppressing homologous recombination, were linked to increased mutation rates during niche adaptation in Streptococcus pneumoniae (Green et al., 2021) and species-specific variation among the Aquilegia species (Wang et al., 2022).
Each of the seven peptidoglycan SNPs consistently divided the 58 C. fetus strains into two distinct groups, CFF and CFV/CFVi, suggesting the potential of developing a reliable assay for the subspecies differentiation. The CFF or CFV/CFVi identification in this study is consistent with the reported identity of the C. fetus strains, except for P4531 (GCF_016406645.1). P4531 was announced as a C. fetus subsp. venerealis but was typed as a C. fetus subsp. fetus using the seven peptidoglycan SNPs in this study. Nonetheless, P4531 was previously identified as a C. fetus subsp. venerealis using a microbial identification system and 16S rRNA sequence analysis (Kim et al., 2021), which are not sufficient to accurately differentiate between the subspecies (van der Graaf-van Bloois et al., 2016a). In some cases where there was inconsistency between the reported phenotypic and genomic identification of the C. fetus subspecies, including cfvi03/596, 01/165, 99541, 98/v445, and 08/421, the identification using the seven peptidoglycan SNPs in this study demonstrated consistent alignment with the reported phenotypic identification, which was typed with the biochemical tests, glycine tolerance and H2S production, recommended by OIE.
Moreover, the identification of subspecies reported in this study was consistent with biochemical and other molecular tests. For example, the C. fetus subsp. fetus reference strain NCTC10842 (GCF_900475935.1) was reported as a CFF/CFVi (Farace et al., 2019) by the L-Cys transporter PCR assay. However, several phenotypic and genomic tests have confirmed the identity of NCTC10842 as a C. fetus subsp. fetus. A similar observation was observed at multiple strains, including NWU_ED23, 06/341, ADRI513, zaf3, 82-40, 04/554, H1-UY, and so on. A reliable and accurate identification of C. fetus subsp. venerealis biovars, which are commonly identified in the bovine genital tract, has been an objective goal in BGC diagnostics research. Hence, we propose the seven peptidoglycan SNPs described in this study could be potential tools for BGC diagnostics and differentiation from C. fetus subsp. fetus.
One limitation of this study was the number of C. fetus strains (n = 25) recruited for SNP calling. However, we carefully conducted the whole-genome comparison with only the complete genomes to avoid the potential of false positive results arising from incomplete genomes. Additionally, the peptidoglycan SNPs were further verified using all the annotated and contamination-free C. fetus genomes (n = 58) available on the NCBI RefSeq database, which is a curated database with a non-redundant, high-quality set of sequences with detailed annotations. Ideally, more C. fetus strains from multiple geographical regions and sources should be included in the whole-genome comparison to provide a more comprehensive view of the evolutionary events of C. fetus subspecies as well as the branching of C. fetus subsp. venerealis bv. intermedius at different geographical regions. To obtain a detailed evolutionary history of the genus Campylobacter, future investigations should also include the complete genomes of the other subspecies, such as Campylobacter fetus subsp. testudinum, as well as other closely related Campylobacter species. Additionally, the impacts of amino acid change on SNP-coding CDSs should be further investigated and validated with phenotypic assays to examine the effect on glycine tolerance.
Our results have reinforced the high genetic stability of C. fetus subspecies, suggesting that they are at the early stage of their evolutionary history and their genomic diversity is at the nucleotide base level. Clonal evolution was found to have occurred separately within the non-Australian and Australian strains. Regardless of geographical regions, C. fetus subsp. venerealis and the biovar variants have potentially acquired common point mutations from the vertical transmission that have enabled their niche-specificity and pathogenicity that separates them from C. fetus subsp. fetus. The C. fetus subsp. venerealis and the biovar intermedius variant also acquired different SNPs. The peptidoglycan SNPs identified and verified in this study are key candidates for the development of more accurate multi-SNP genotyping assays because of their association with the differential glycine tolerance between C. fetus subsp. fetus and C. fetus subsp. venerealis.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA675960.
The manuscript presents research on animals that do not require ethical approval for their study.
CO: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. PB: Supervision, Writing – review & editing. GB-H: Supervision, Writing – review & editing. Sd: Investigation, Methodology, Writing – review & editing. BH: Supervision, Writing – review & editing. LI: Formal analysis, Methodology, Writing – review & editing. VK: Resources, Writing – review & editing. CM: Formal analysis, Methodology, Writing – review & editing. LN: Methodology, Supervision, Writing – review & editing. YN: Formal analysis, Methodology, Writing – review & editing. HS: Formal analysis, Investigation, Methodology, Writing – review & editing. CT: Methodology, Supervision, Validation, Writing – review & editing. BV: Methodology, Writing – review & editing. MW: Resources, Validation, Writing – review & editing. ZZ: Formal analysis, Investigation, Methodology, Writing – review & editing. AT: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Meat and Livestock Australia Donor Company (grant number: P.PSH.0799) and the Queensland Department of Agriculture and Fisheries.
The authors thank Meat and Livestock Australia Donor Company Project P.PSH.0799 for funding this research. The authors thank Diane Newell for sharing the bacterial strains BT268/06 and BT376/03 and Ingrid Flemming of IFM Quality Services for sharing the bacterial isolate M20-08756/1A.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1452564/full#supplementary-material
SNP, Single Nucleotide Polymorphisms; BGC, Bovine Genital Campylobacteriosis; SAP, Surface Array Proteins; MLST, Multiple Locus Sequence Typing; ST, Sequence Type; PCR, Polymerase Chain Reaction; AFLP, Amplified Fragment Length Polymorphism; PFGE, Pulsed-Field Gel Electrophoresis; ONT, Oxford Nanopore Technologies; PGAP, Prokaryotic Genome Annotation Pipeline; ANI, Average Nucleotide Identity; CDS, Coding Sequences; NTC, No Template Control; Cq, Quantification Cycle; RFU, Relative Fluorescence Units; CFF, Campylobacter fetus subsp. fetus; CFV, Campylobacter fetus subsp. venerealis; CFVi, Campylobacter fetus subsp. venerealis biovar intermedius.
Abril, C., Vilei, E. M., Brodard, I., Burnens, A., Frey, J., Miserez, R., et al. (2007). Discovery of insertion element ISCfe 1: a new tool for Campylobacter fetus subspecies differentiation. Clin. Microbiol. Infect. 13, 993–1000. doi: 10.1111/j.1469-0691.2007.01787.x
Ali, A., Soares, S. C., Santos, A. R., Guimarães, L. C., Barbosa, E., Almeida, S. S., et al. (2012). Campylobacter fetus subspecies: comparative genomics and prediction of potential virulence targets. Gene 508, 145–156. doi: 10.1016/j.gene.2012.07.070
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genom. 12:402. doi: 10.1186/1471-2164-12-402
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrew, S. (2010). FastQC: a Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed January 15, 2020).
Bacigalupe, R., Tormo-Mas, M. Á., Penadés, J. R., and Fitzgerald, J. R. (2019). A multihost bacterial pathogen overcomes continuous population bottlenecks to adapt to new host species. Sci. Adv. 5:eaax0063. doi: 10.1126/sciadv.aax0063
Barrero, R. A., Moolhuijzen, P., Indjein, L., Venus, B., Keeble-Gagnère, G., Power, J., et al. (2014). Draft genome sequences of Campylobacter fetus subsp. venerealis bv. venerealis Strain B6 and bv. intermedius Strain 642-21. Genome Announc. 2:e00943–14. doi: 10.1128/genomeA.00943-14
Basden, E. H. 2nd., Tourtellotte, M. E., Plastridge, W. N., and Tucker, J. S. (1968). Genetic relationship among bacteria classified as Vibrios. J. Bacteriol. 95, 439–443. doi: 10.1128/jb.95.2.439-443.1968
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., and Hoad, G. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucl. Acids Res. 45, W30-W35. doi: 10.1093/nar/gkx343
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021:msab293. doi: 10.1093/molbev/msab293
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2011). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Ceres, K. M., Stanhope, M. J., and Gröhn, Y. T. (2022). A critical evaluation of Mycobacterium bovis pangenomics, with reference to its utility in outbreak investigation. Microb. Genom. 8:839. doi: 10.1099/mgen.0.000839
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucl. Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118) iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Conrad, T. M., Frazier, M., Joyce, A. R., Cho, B. K., Knight, E. M., Lewis, N. E., et al. (2010). RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc. Natl. Acad. Sci. U. S. A. 107, 20500–20505. doi: 10.1073/pnas.0911253107
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2014). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucl. Acids Res. 43:e15. doi: 10.1093/nar/gku1196
De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Dingle, K. E., Blaser, M. J., Tu, Z. C., Pruckler, J., Fitzgerald, C., van Bergen, M. A. P., et al. (2010). Genetic relationships among reptilian and mammalian Campylobacter fetus strains determined by multilocus sequence typing. J. Clin. Microbiol. 48, 977–980. doi: 10.1128/JCM.01439-09
Dworkin, J., Tummuru, M. K. R., and Blaser, M. J. (1995). Segmental conservation of sapA sequences in type B Campylobacter fetus cells. J. Biol. Chem. 270, 15093–15101. doi: 10.1074/jbc.270.25.15093
Emele, M. F., Karg, M., Hotzel, H., Bloois, L. G. V., Groß, U., Bader, O., et al. (2019). Differentiation of Campylobacter fetus subspecies by proteotyping. Eur. J. Microbiol. Immunol. 9, 62–71. doi: 10.1556/1886.2019.00006
Ewels, P., Magnusson, M., Lundin, S., and Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. doi: 10.1093/bioinformatics/btw354
Farace, P., Cravero, S., Taibo, C., Diodati, J., Morsella, C., Paolicchi, F., et al. (2022). Campylobacter fetus releases S-layered and immunoreactive outer membrane vesicles. Rev. Argent Microbiol. 54, 74–80. doi: 10.1016/j.ram.2021.06.001
Farace, P. D., Irazoqui, J. M., Morsella, C. G., García, J. A., Méndez, M. A., Paolicchi, F. A., et al. (2021). Phylogenomic analysis for Campylobacter fetus ocurring in Argentina. Vet. World 14, 1165–1179. doi: 10.14202/vetworld.2021.1165-1179
Farace, P. D., Morsella, C. G., Cravero, S. L., Sioya, B. A., Amadio, A. F., Paolicchi, F. A., et al. (2019). L-cysteine transporter-PCR to detect hydrogen sulfide-producing Campylobacter fetus. PeerJ 7:e7820. doi: 10.7717/peerj.7820
Fitzgerald, C., Tu, Z., Patrick, M., Stiles, T., Lawson, A. J., Santovenia, M., et al. (2014). Campylobacter fetus subsp. testudinum subsp. nov., isolated from humans and reptiles. Int. J. Syst. Evol. Microbiol. 64, 2944–2948. doi: 10.1099/ijs.0.057778-0
Fornelos, N., Browning, D. F., and Butala, M. (2016). The use and abuse of LexA by mobile genetic elements. Trends Microbiol. 24, 391–401. doi: 10.1016/j.tim.2016.02.009
Gabrielaite, M., and Marvig, R. L. (2019). GenAPI: a tool for gene absence-presence identification in fragmented bacterial genome sequences. bioRxiv. 2019:658476. doi: 10.1101/658476
Gilbert, M. J., Miller, W. G., Yee, E., Zomer, A. L., van der Graaf-van Bloois, L., Fitzgerald, C., et al. (2016). Comparative genomics of Campylobacter fetus from reptiles and mammals reveals divergent evolution in host-associated lineages. Genome Biol. Evol. 8, 2006–2019. doi: 10.1093/gbe/evw146
Goldstein, S., Beka, L., Graf, J., and Klassen, J. L. (2019). Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genom. 20:23. doi: 10.1186/s12864-018-5381-7
Green, A. E., Howarth, D., Chaguza, C., Echlin, H., Langendonk, R. F., Munro, C., et al. (2021). Pneumococcal colonization and virulence factors identified via experimental evolution in infection models. Mol. Biol. Evol. 38, 2209–2226. doi: 10.1093/molbev/msab018
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hammes, W. P., and Neuhaus, F. C. (1974). On the specificity of phospho-N-acetylmuramyl-pentapeptide translocase. J. Biol. Chem. 249, 3140–3150. doi: 10.1016/S0021-9258(19)42649-5
Harvey, S. M., and Greenwood, J. R. (1983). Relationships among catalase-positive Campylobacters determined by deoxyribonucleic acid-deoxyribonucleic acid hybridization. Int. J. Syst. Evol. Microbiol. 33, 275–284. doi: 10.1099/00207713-33-2-275
Hishinuma, F., Izaki, K., and Takahashi, H. (1969). Effects of glycine and D-amino acids on growth of various microorganisms. Agric. Biol. Chem. 33, 1577–1586. doi: 10.1080/00021369.1969.10859511
Hum, S. (1996). “Bovine venereal campylobacteriosis,” in Campylobacters, Helicobacters, and Related Organisms, eds. D. G. Newell, J. M. Ketley, and R. A. Feldman (Boston, MA: Springer US), 355–358.
Hum, S., Quinn, K., Brunner, J., and On, S. (1997). Evaluation of a PCR assay for identification and differentiation of Campylobacter fetus subspecies. Aust. Vet. J. 75, 827–831. doi: 10.1111/j.1751-0813.1997.tb15665.x
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., Hauser, L. J., et al. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Indjein, L. (2013). Molecular Diagnostic Protocols for Bovine genital campylobacteriosis Using Comparative Genomics and Virulence Studies (Ph.D. Thesis). The University of Queensland, St Lucia, QLD, Australia.
Iraola, G., Betancor, L., Calleros, L., Gadea, P., Algorta, G., Galeano, S., et al. (2015). A rural worker infected with a bovine-prevalent genotype of Campylobacter fetus subsp. fetus supports zoonotic transmission and inconsistency of MLST and whole-genome typing. Eur. J. Clin. Microbiol. Infect. Dis. 34, 1593–1596. doi: 10.1007/s10096-015-2393-y
Iraola, G., Forster, S. C., Kumar, N., Lehours, P., Bekal, S., Garcia-Pena, F. J., et al. (2017). Distinct Campylobacter fetus lineages adapted as livestock pathogens and human pathobionts in the intestinal microbiota. Nat. Commun. 8:1367. doi: 10.1038/s41467-017-01449-9
Iraola, G., Hernández, M., Calleros, L., Paolicchi, F., Silveyra, S., Velilla, A., et al. (2012). Application of a multiplex PCR assay for Campylobacter fetus detection and subspecies differentiation in uncultured samples of aborted bovine fetuses. J. Vet. Sci. 13, 371–376. doi: 10.4142/jvs.2012.13.4.371
Iraola, G., Perez, R., Naya, H., Paolicchi, F., Harris, D., Lawley, T. D., et al. (2013). Complete genome sequence of Campylobacter fetus subsp. venerealis biovar intermedius, isolated from the prepuce of a bull. Genome Announc. 1, e00526–e00513. doi: 10.1128/genomeA.00526-13
Jain, C., Rodriguez, R. L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Kienesberger, S., Sprenger, H., Wolfgruber, S., Halwachs, B., Thallinger, G. G., Perez-Perez, G. I., et al. (2014). Comparative genome analysis of Campylobacter fetus subspecies revealed horizontally acquired genetic elements important for virulence and niche specificity. PLoS ONE 9:e85491. doi: 10.1371/journal.pone.0085491
Kim, S. G., Summage-West, C. V., Sims, L. M., and Foley, S. L. (2021). Complete genome sequence of Campylobacter fetus subsp. venerealis P4531 from a Rhesus Monkey. Microbiol. Resour. Announc. 10:e0073921. doi: 10.1128/MRA.00739-21
Kimsey, H. H., and Waldor, M. K. (2009). Vibrio cholerae LexA coordinates CTX prophage gene expression. J. Bacteriol. 191, 6788–6795. doi: 10.1128/JB.00682-09
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Koya, A. (2016). Bovine genital campylobacteriosis: Isolation, Identification and Virulence Profiling of Campylobacter fetus subsp. venerealis in a small animal model (Ph.D. Thesis). The University of Queensland, St Lucia, QLD, Australia.
Kuehne, S. A., Dempster, A. W., Collery, M. M., Joshi, N., Jowett, J., Kelly, M. L., et al. (2018). Characterization of the impact of rpoB mutations on the in vitro and in vivo competitive fitness of Clostridium difficile and susceptibility to fidaxomicin. J. Antimicrob. Chemother. 73, 973–980. doi: 10.1093/jac/dkx486
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, X., Tang, H., Xu, Z., Tang, H., Fan, Z., Jiao, X., et al. (2022). Prevalence and characteristics of Campylobacter from the genital tract of primates and ruminants in Eastern China. Transbound. Emerg. Dis. 69, e1892–e8. doi: 10.1111/tbed.14524
Lindsey-Boltz, L. A., and Sancar, A. (2021). The transcription-repair coupling factor Mfd prevents and promotes mutagenesis in a context-dependent manner. Front. Mol. Biosci. 8:668290. doi: 10.3389/fmolb.2021.668290
Linz, B., Windsor, H. M., Gajewski, J. P., Hake, C. M., Drautz, D. I., Schuster, S. C., et al. (2013). Helicobacter pylori genomic microevolution during naturally occurring transmission between adults. PLoS ONE 8:e82187. doi: 10.1371/journal.pone.0082187
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
Lynch, C. T., Buttimer, C., Epping, L., O'Connor, J., Walsh, N., McCarthy, C., et al. (2021). Phenotypic and genetic analyses of two Campylobacter fetus isolates from a patient with relapsed prosthetic valve endocarditis. Pathog Dis. 79:ftab055. doi: 10.1093/femspd/ftab055
Ma, P., Luo, T., Ge, L., Chen, Z., Wang, X., Zhao, R., et al. (2021). Compensatory effects of M. tuberculosis rpoB mutations outside the rifampicin resistance-determining region. Emerg. Microbes Infect. 10, 743–752. doi: 10.1080/22221751.2021.1908096
Malmberg, M. M., Spangenberg, G. C., Daetwyler, H. D., and Cogan, N. O. I. (2019). Assessment of low-coverage nanopore long read sequencing for SNP genotyping in doubled haploid canola (Brassica napus L.). Sci. Rep. 9:8688. doi: 10.1038/s41598-019-45131-0
Marsh, H., and Firehammer, B. D. (1953). Serological relationships of twenty three ovine and three bovine strains of Vibrio fetus. Am. J. Vet. Res. 14, 396–398.
Matsuo, M., Hishinuma, T., Katayama, Y., and Hiramatsu, K. (2015). A mutation of RNA polymerase β' subunit (RpoC) converts heterogeneously vancomycin-intermediate Staphylococcus aureus (hVISA) into “slow VISA”. Antimicrob. Agents Chemother. 59, 4215–4225. doi: 10.1128/AAC.00135-15
McMillen, L., Fordyce, G., Doogan, V. J., and Lew, A. E. (2006). Comparison of culture and a novel 5′ Taq nuclease assay for direct detection of Campylobacter fetus subsp. venerealis in clinical specimens from cattle. J. Clin. Microbiol. 44, 938–945. doi: 10.1128/JCM.44.3.938-945.2006
Moolhuijzen, P. M., Lew-Tabor, A. E., Wlodek, B. M., Aguero, F. G., Comerci, D. J., Ugalde, R. A., et al. (2009). Genomic analysis of Campylobacter fetus subspecies: identification of candidate virulence determinants and diagnostic assay targets. BMC Microbiol. 9:86. doi: 10.1186/1471-2180-9-86
Moran, A. P., O'Malley, D. T., Kosunen, T. U., and Helander, I. M. (1994). Biochemical characterization of Campylobacter fetus lipopolysaccharides. Infect. Immun. 62, 3922–3929. doi: 10.1128/iai.62.9.3922-3929.1994
Mshelia, G. D., Amin, J. D., Woldehiwet, Z., Murray, R. D., and Egwu, G. O. (2010). Epidemiology of Bovine venereal campylobacteriosis: geographic distribution and recent advances in molecular diagnostic techniques. Reprod. Domest. Anim. 45, e221–e230. doi: 10.1111/j.1439-0531.2009.01546.x
Mukhtar, L. (2013). Evaluation of the Genetic Differences Between Two Subtypes of Campylobacter fetus (fetus and venerealis) in Canada (Ph.D. Thesis). University of Ottawa, Ottawa, ON, Canada.
Nadin-Davis, S. A., Chmara, J., Carrillo, C. D., Amoako, K., Goji, N., Duceppe, M., et al. (2021). A comparison of fourteen fully characterized mammalian-associated Campylobacter fetus isolates suggests that loss of defense mechanisms contribute to high genomic plasticity and subspecies evolution. PeerJ. 9:e10586. doi: 10.7717/peerj.10586
NCBI Resource Coordinators (2018). Database resources of the National Center for Biotechnology Information. Nucl. Acids Res. 46, D8–D13. doi: 10.1093/nar/gkx1095
Nordin, Y. (2013). Campylobacter fetus subspecies Molecular Typing, Polymerase Chain Reaction (PCR) and High- Resolution Melt (HRM) Analyses for Bovine campylobacteriosis diagnosis (Honours Thesis). Univerisity of Queensland, St Lucia, QLD, Australia.
OIE (2019). “Bovine genital campylobacteriosis,” in Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2019. World Organisation For Animal Health OIE, 1031–1044. Available at: https://www.oie.int/en/what-we-do/standards/codes-and-manuals/terrestrial-manual-online-access/ (accessed December 19, 2019).
On, S. L. W., and Harrington, C. S. (2001). Evaluation of numerical analysis of PFGE-DNA profiles for differentiating Campylobacter fetus subspecies by comparison with phenotypic, PCR and 16S rDNA sequencing methods. J. Appl. Microbiol. 90, 285–293. doi: 10.1046/j.1365-2672.2001.01247.x
Otten, C., Brilli, M., Vollmer, W., Viollier, P. H., and Salje, J. (2018). Peptidoglycan in obligate intracellular bacteria. Mol. Microbiol. 107, 142–163. doi: 10.1111/mmi.13880
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palau, M., Piqué, N., Ramírez-Lázaro, M. J., Lario, S., Calvet, X., Miñana-Galbis, D., et al. (2021). Whole-genome sequencing and comparative genomics of three Helicobacter pylori strains isolated from the stomach of a patient with Adenocarcinoma. Pathogens 10:331. doi: 10.3390/pathogens10030331
Paradis, E., and Schliep, K. (2018). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Penner, J. L. (1988). The genus Campylobacter: a decade of progress. Clin. Microbiol. Rev. 1, 157–172. doi: 10.1128/CMR.1.2.157
Perez-Perez, G. I., Blaser, M. J., and Bryner, J. H. (1986). Lipopolysaccharide structures of Campylobacter fetus are related to heat-stable serogroups. Infect. Immun. 51, 209–212. doi: 10.1128/iai.51.1.209-212.1986
Quinones, M., Kimsey, H. H., and Waldor, M. K. (2005). LexA cleavage is required for CTX prophage induction. Mol. Cell 17, 291–300. doi: 10.1016/j.molcel.2004.11.046
R Core Team (2018). R: a Language and Environment for Statistical Computing. Available at: https://www.R-project.org/ (accessed June 7, 2020).
Ragheb, M. N., Thomason, M. K., Hsu, C., Nugent, P., Gage, J., Samadpour, A. N., et al. (2019). Inhibiting the evolution of antibiotic resistance. Mol. Cell 73, 157–165.e5. doi: 10.1016/j.molcel.2018.10.015
Ribeiro, A., Golicz, A., Hackett, C. A., Milne, I., Stephen, G., Marshall, D., et al. (2015). An investigation of causes of false positive single nucleotide polymorphisms using simulated reads from a small eukaryote genome. BMC Bioinform. 16:382. doi: 10.1186/s12859-015-0801-z
Ruan, J., and Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158. doi: 10.1038/s41592-019-0669-3
Sebald, M., and Veron, M. (1963). Base DNA content and classification of Vibrios. Ann. Inst. Pasteur. 105, 897–910.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seemann, T. (2016). ABRicate: Mass Screening of Contigs for Antibiotic Resistance Genes. Available at: https://github.com/tseemann/abricate (accessed August 31, 2020).
Silva, M. F., Pereira, A. L., Fraqueza, M. J., Pereira, G., Mateus, L., Lopes-da-Costa, L., et al. (2021). Genomic and phenotypic characterization of Campylobacter fetus subsp. venerealis strains. Microorganisms 9:340. doi: 10.3390/microorganisms9020340
Smibert, R. M. (1978). The genus Campylobacter. Annu. Rev. Microbiol. 32, 673–709. doi: 10.1146/annurev.mi.32.100178.003325
Smibert, R. M. (1981). “The genus Campylobacter,” in The Prokaryotes: A Handbook on Habitats, Isolation, and Identification of Bacteria, eds. M. P. Starr, H. Stolp, H. G. Trüper, A. Balows, and H. G. Schlegel (Berlin; Heidelberg: Springer Berlin Heidelberg), 609–617.
Smith, T., and Taylor, M. S. (1919). Some morphological and biological characters of the spirilla (Vibrio fetus, n. sp.) associated with disease of the fetal membranes in cattle. J. Exp. Med. 30, 299–311. doi: 10.1084/jem.30.4.299
Spence, R. P., Bruce, I. R., McFadden, A. M. J., Hill, F. I., Tisdall, D., Humphrey, S., et al. (2011). Cross-reaction of a Campylobacter fetus subspecies venerealis real-time PCR. Vet. Rec. 168:131. doi: 10.1136/vr.c5264
Sprenger, H., Zechner, E. L., and Gorkiewicz, G. (2012). So close and yet so far—molecular microbiology of Campylobacter fetus subspecies. Eur. J. Microbiol. Immunol. 2, 66–75. doi: 10.1556/EuJMI.2.2012.1.10
Strick, T. R., and Portman, J. R. (2019). Transcription-coupled repair: from cells to single molecules and back again. J. Mol. Biol. 431, 4093–4102. doi: 10.1016/j.jmb.2019.05.040
Struve, W. G., Sinha, R. K., Neuhaus, F. C., and Prime, M. S. (1966). On the initial stage in peptidoglycan synthesis. Phospho-N-acetylmuramyl-pentapeptide translocase (Uridine monophosphate). Biochemistry 5, 82–93. doi: 10.1021/bi00865a012
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Tange, O. (2011). GNU parallel—the command-line power tool. USENIX Mag. 36, 42–47. Available at: https://www.usenix.org/publications/login/february-2011-volume-36-number-1/gnu-parallel-command-line-power-tool
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucl. Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest Suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Tshipamba, M. E., Akinola, S. A., Ngoma, L., and Mwanza, M. (2020a). Genome sequence of Campylobacter fetus subsp. venerealis NW_ED23, isolated from bovine sheath wash. Microbiol. Resour. Announc. 9:20. doi: 10.1128/MRA.00854-20
Tshipamba, M. E., Lubanza, N., and Mwanza, M. (2020b). Genome analysis of antimicrobial resistance genes and virulence factors in multidrug-resistant Campylobacter fetus subspecies isolated from sheath wash. World's Vet. J. 10, 465–480. doi: 10.54203/scil.2020.wvj57
Tu, Z. C., Dewhirst, F. E., and Blaser, M. J. (2001). Evidence that the Campylobacter fetus sap locus is an ancient genomic constituent with origins before mammals and reptiles diverged. Infect. Immun. 69, 2237–2244. doi: 10.1128/IAI.69.4.2237-2244.2001
Tu, Z. C., Zeitlin, G., Gagner, J. P., Keo, T., Hanna, B. A., Blaser, M. J., et al. (2004). Campylobacter fetus of reptile origin as a human pathogen. J. Clin. Microbiol. 42, 4405–4407. doi: 10.1128/JCM.42.9.4405-4407.2004
Turni, C., Singh, R., and Blackall, P. J. (2018). Virulence-associated gene profiling, DNA fingerprinting and multilocus sequence typing of Haemophilus parasuis isolates in Australia. Aust. Vet. J. 96, 196–202. doi: 10.1111/avj.12705
van Bergen, M. A., Dingle, K. E., Maiden, M. C., Newell, D. G., van der Graaf-Van Bloois, L., van Putten, J. P., et al. (2005b). Clonal nature of Campylobacter fetus as defined by multilocus sequence typing. J. Clin. Microbiol. 43, 5888–5898. doi: 10.1128/JCM.43.12.5888-5898.2005
van Bergen, M. A., Simons, G., Graaf-van Bloois, L., van Putten, J. P., Rombout, J., Wesley, I., et al. (2005a). Amplified fragment length polymorphism based identification of genetic markers and novel PCR assay for differentiation of Campylobacter fetus subspecies. J. Med. Microbiol. 54, 1217–1224. doi: 10.1099/jmm.0.46186-0
van der Graaf-van Bloois, L., Duim, B., Miller, W. G., Forbes, K. J., Wagenaar, J. A., and Zomer, A. (2016a). Whole genome sequence analysis indicates recent diversification of mammal-associated Campylobacter fetus and implicates a genetic factor associated with H2S production. BMC Genom. 17:713. doi: 10.1186/s12864-016-3058-7
van der Graaf-van Bloois, L., Miller, W. G., Yee, E., Gorkiewicz, G., Forbes, K. J., Zomer, A. L., et al. (2016b). Campylobacter fetus subspecies contain conserved type IV secretion systems on multiple genomic islands and plasmids. PLoS ONE 11:e0152832. doi: 10.1371/journal.pone.0152832
van der Graaf-van Bloois, L., Miller, W. G., Yee, E., Rijnsburger, M., Wagenaar, J. A., and Duim, B. (2014). Inconsistency of phenotypic and genomic characteristics of Campylobacter fetus subspecies requires reevaluation of current diagnostics. J. Clin. Microbiol. 52, 4183–4188. doi: 10.1128/JCM.01837-14
Vandamme, P., Pot, B., Falsen, E., Kersters, K., and De Ley, J. (1990). Intra- and interspecific relationships of veterinary Campylobacters revealed by numerical analysis of electrophoretic protein profiles and DNA:DNA hybridizations. Syst. Appl. Microbiol. 13, 295–303. doi: 10.1016/S0723-2020(11)80201-8
Vaser, R., and Šikić, M. (2021). Raven: a de novo genome assembler for long reads. bioRxiv. 2021:242461. doi: 10.1101/2020.08.07.242461
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Wagenaar, J. A., van Bergen, M. A. P., Newell, D. G., Grogono-Thomas, R., and Duim, B. (2001). Comparative study using amplified fragment length polymorphism fingerprinting, PCR genotyping, and phenotyping to differentiate Campylobacter fetus strains isolated from animals. J. Clin. Microbiol. 39:2283. doi: 10.1128/JCM.39.6.2283-2286.2001
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, G., Clark, C. G., Taylor, T. M., Pucknell, C., Barton, C., Price, L., et al. (2002). Colony multiplex PCR assay for identification and differentiation of Campylobacter jejuni, C. coli, C. lari, C. upsaliensis, and C. fetus subsp. fetus. J. Clin. Microbiol. 40, 4744–4747. doi: 10.1128/JCM.40.12.4744-4747.2002
Wang, Z., Lu, T., Li, M., Ding, N., Lan, L., Gao, X., et al. (2022). Genetic and epigenetic signatures associated with the divergence of Aquilegia species. Genes 13:793. doi: 10.3390/genes13050793
Wei, T. V. S. (2021). R Package “corrplot”: Visualization of a Correlation Matrix.0.91. Available at: https://github.com/taiyun/corrplot (accessed July 22, 2022).
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017a). Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 3:e000132. doi: 10.1099/mgen.0.000132
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017b). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. Available at: https://ggplot2.tidyverse.org (accessed June 30, 2020).
Willoughby, K., Nettleton, P. F., Quirie, M., Maley, M. A., Foster, G., Toszeghy, M., et al. (2005). A multiplex polymerase chain reaction to detect and differentiate Campylobacter fetus subspecies fetus and Campylobacter fetus subspecies venerealis: use on UK isolates of C. fetus and other Campylobacter spp. J. Appl. Microbiol. 99, 758–766. doi: 10.1111/j.1365-2672.2005.02680.x
Keywords: Campylobacter fetus, complete genomes, subspecies, SNPs, genome comparison, glycine, veterinary science
Citation: Ong CT, Blackall PJ, Boe-Hansen GB, deWet S, Hayes BJ, Indjein L, Korolik V, Minchin C, Nguyen LT, Nordin Y, Siddle H, Turni C, Venus B, Westman ME, Zhang Z and Tabor AE (2024) Whole-genome comparison using complete genomes from Campylobacter fetus strains revealed single nucleotide polymorphisms on non-genomic islands for subspecies differentiation. Front. Microbiol. 15:1452564. doi: 10.3389/fmicb.2024.1452564
Received: 21 June 2024; Accepted: 27 August 2024;
Published: 12 September 2024.
Edited by:
Ozan Gundogdu, University of London, United KingdomReviewed by:
Maria Jorge Campos, Polytechnic Institute of Leiria, PortugalCopyright © 2024 Ong, Blackall, Boe-Hansen, deWet, Hayes, Indjein, Korolik, Minchin, Nguyen, Nordin, Siddle, Turni, Venus, Westman, Zhang and Tabor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ala E. Tabor, YS50YWJvckB1cS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.