 Xiaohong Chen1†
Xiaohong Chen1† Mengke Dou
Mengke Dou Xiong Huang
Xiong Huang- 1College of Forestry, Sichuan Agricultural University, Chengdu, China
- 2Ganzi Prefecture Forestry Research Institute, Kangding, China
Plant tissues harbor abundant endophytes, which are crucial for plant growth. Endophytes present in Alsophila spinulosa, which is enriched with medicinal components, have not been isolated and characterized yet. Here we employed meta-amplicon sequencing to identify endophytic species and examined their diversity in the leaves, petioles, roots and stems of A. spinulosa. Our findings revealed 1,247 operational taxonomic units (OTUs) for endophytic bacteria across 210 species and 476 OTUs for endophytic fungi across 222 species. Alpha diversity analysis showed the highest endophytic bacterial diversity in A. spinulosa roots, whereas fungal diversity was similar across the leaf, petiole and root tissues. Fungal diversity in the leaves and petioles was markedly higher than that in the stems. Furthermore, beta diversity analysis revealed similarities in the endophytic bacterial and fungal compositions between the leaves and petioles, whereas the compositions in roots and stems considerably differed from those in the leaves and petioles. At the genus level, the predominant endophytic bacteria were Methylobacterium-Methylorubrum and Pseudomonas, whereas the predominant endophytic fungi were Cutaneotrichosporon and Pseudofabraea. Linear discriminant analysis effect size revealed characteristic endophytic bacterial genera specific to each tissue type and characteristic endophytic fungal genera specifically in the leaves, petioles and roots. The co-occurrence network analysis indicated that the complexity of endophyte networks was the highest in the leaves and the lowest in the stems of A. spinulosa. Overall, this study elucidates the distribution patterns of endophytes in A. spinulosa across various tissues, offering valuable microbial resources for the development of natural products for medicinal application.
Introduction
Endophytes are microorganisms that inhabit the cells and interstitial spaces of various plant organs, including roots, stems, leaves, flowers, fruits, and seeds (Yan et al., 2022). These microorganisms can be present in plants at any stage of their lifecycle without causing visible disease symptoms (Liang et al., 2023). Endophytes are present either as symbiotic microbes that engage in beneficial or neutral interactions or as latent pathogens that reside in the host without causing immediate harm (Omomowo and Babalola, 2019). Fungi and bacteria are the predominant microbial types functioning as endophytes (Asad et al., 2023). Endophytes have synergistically evolved with their host plants, forming dynamic and balanced symbiotic relationships, which are beneficial to plant growth, resilience, and resistance to pests and diseases; in particular, endophytes promote nitrogen fixation, secrete growth hormones, and produce active metabolites to support plant growth (Kushwaha et al., 2020). Endophytes can be isolated from surface-sterilized plant tissues or directly detected within the tissues by using microbial DNA (macrogenomic) extraction techniques. Recently, numerous endophytes have been isolated and characterized from the roots, stems, and leaves of various species, including citrus trees, tea trees, grapevines, and Taxus. Endophytic species are the most abundant in the roots of forest trees, followed by stems and leaves (Abdalla and Matasyoh, 2014; Lin et al., 2022).
Medicinal plants are used to prevent and treat diseases (Aye et al., 2019). Either the whole plants or their specific parts serve as medicinal agents or raw materials in the pharmaceutical industry, providing diverse medicinal and economic benefits (Tang et al., 2023). Despite rapid advancements in modern medicine, including the methods for extracting metabolites from plants and other natural sources, synthetic chemistry, combinatorial chemistry, and molecular modeling for drug discovery (Ley and Baxendale, 2002; Mario Geysen et al., 2003; Lombardino and Lowe, 2004), medicinal plants, with their distinct advantages, continue to play a valuable role in disease treatment. They are not only culturally accepted and highly compatible with the human body but also more effective than synthetic compounds. Thus, natural products extracted from medicinal plants are still used in the synthesis of many clinical drugs (Tang et al., 2023). Endophytes have been isolated from various medicinal plants (Zhou et al., 2022; Zotchev, 2024). Many endophytes can secrete phytohormones, growth factors, and other compounds that support the growth and development of plants (Rani et al., 2022). In addition, endophytes regulate the accumulation and production of active ingredients in medicinal plants (Tshikhudo et al., 2023; Hashem et al., 2023). Endophytes are a source of many bioactive metabolites, including alkaloids, saponins, quinones, flavonoids, and terpenoids. These compounds are highly bioactive and have attracted substantial attention in natural drug research (Gouda et al., 2016). Thus, identifying the endophytes present in medicinal plants and understanding their effect on the physiology and biochemistry of these plants are essential for discovering new bioactive compounds.
Alsophila spinulosa, a member of the genus Alsophila in the Cyatheaceae family, is the only woody fern and often referred to as “the king of ferns” (Ma et al., 2020). It is classified as a second-class protected species under the newly issued List of Wild Plants of National Key Protection. A. spinulosa has become a hotspot of research in diverse fields, such as paleontology, paleobotany, paleogeology, paleoclimate, and paleoenvironmental changes (Loiseau et al., 2020). Moreover, some metabolites found in A. spinulosa exhibit antitumor and antibacterial properties (Longtine and Tejedor, 2017). The stems of A. spinulosa, containing flavonoids, phenols, amino acids, and alkaloids, are used in traditional Chinese medicine (Huang et al., 2022). Traditionally, A. spinulosa has been valued for its medicinal properties that include removing dampness, strengthening tendons and bones, activating blood circulation, and clearing heat and toxins (Chiang et al., 1994; Chaparro-Hernández et al., 2022), earning it the nickname “Dragon’s Bone Wind.” Existing studies on A. spinulosa have mainly focused on its population distribution, physiological ecology, medicinal components, and phylogenetic relationships (Xiong et al., 2023; Yi et al., 2023). However, no study has yet examined the distribution patterns of its endophytic composition, which hinders our comprehensive understanding of interactions between A. spinulosa and its endophytes as well as efforts to conserve the species and enhance its quality.
In the present study, we isolated and identified endophytes from the leaves, petioles, roots, and stems of A. spinulosa. Using meta-amplicon sequencing, we systematically cataloged endophyte species and their diversity across various plant parts and determined the distribution characteristics of the endophyte population. The findings of this study provide a scientific basis for further exploration and use of endophytic resources in A. spinulosa.
Materials and methods
Sample collection
For microbiome research, A. spinulosa tissues were collected from the National Germplasm Resource Center (29.9°N, 103.14°E), located in Hongya County, Sichuan Province, China. Mature tissues of the roots (R), stems (S), leaves (L), and petioles (P) collected from three individual trees were used as biological replicates. The collected samples were first rinsed with running water and then subjected to surface sterilization. The samples were sterilized first with 75% ethanol for 1 min and then with 5% sodium hypochlorite for 5 min. Subsequently, the samples were rinsed twice with sterilized deionized water and dried using sterilized filter paper. The final rinse water was used to verify the sterility of tissue surfaces through both polymerase chain reaction (PCR) amplification of 16S rRNA and internal transcribed spacer (ITS) genes and the plate cultivation method (Seghers et al., 2004). The verified tissue samples were placed in 50-mL sterilized centrifuge tubes, immediately frozen in liquid nitrogen, and stored at −80°C for 1 week.
DNA extraction, library construction, and sequencing
We extracted total genomic DNA from A. spinulosa samples by using a HiPure Soil DNA Kit (Magen, Guangzhou, China) in accordance with the manufacturer’s instructions. DNA concentration and purity were assessed using a NanoDrop NC2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) and agarose gel electrophoresis, respectively. The extracted DNA was diluted to a concentration of 50 ng/μL with nuclease-free water. PCR amplification was performed using specific primer pairs for 16S rRNA (515F: GTGYCAGCMGCCGCGGTAA, 806R: GGACTACNVGGGTWTCTAAT) and ITS regions (ITS1F: CTTGGTCATTTAGAGGAAGTAA, ITS2: GCTGCGTTCTTCATC GATGC). The PCR reagents were obtained from New England Biolabs (USA). The initial PCR mixture consisted of 10 μL of Q5 reaction buffer (5×), 10 μL of Q5 High GC Enhancer (5×), 1.5 μL of 2.5 mM dNTPs, 1.5 μL of 10 μM each of forward and reverse primers, 0.2 μL of Q5 high-fidelity DNA polymerase, 1 μL of template DNA, and 24.3 μL of ddH2O. The PCR protocol involved an initial denaturation at 95°C for 5 min, followed by 30 cycles of denaturation at 95°C for 1 min, annealing at 60°C for 1 min, and extension at 72°C for 1 min, with a final extension at 72°C for 7 min. The PCR products were checked by gel electrophoresis (Supplementary Figures S1, S2) and purified from a 2% agarose gel using AMPure XP Beads (Beckman Coulter, Brea, CA, United States). DNA concentrations of the purified products were measured using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, United States), and these products were used as templates for the second round of PCR by using the same primers. The second-round PCR mixture comprised 5 μL of Q5 reaction buffer (5×), 1.5 μL of Q5 high GC enhancer (5×), 1.5 μL of 2.5 mM dNTPs, 1 μL of 10 mM each of index and universal PCR primers, 1 μL of Q5 high-fidelity DNA polymerase, and 1 μL of template DNA; 8 μL of ddH2O was added to make up the reaction volume to 20 μL. PCR protocol for the second round was as follows: initial denaturation at 95°C for 5 min, 12 cycles of denaturation at 95°C for 1 min, annealing at 60°C for 1 min, extension at 72°C for 1 min, and a final extension at 72°C for 7 min. The amplicons were purified, quantified using an ABI StepOnePlus Real-Time PCR System (Life Technologies, Foster City, United States), pooled in equal amounts, and subjected to high-throughput amplicon sequencing on an Illumina Novaseq 6,000 platform (Illumina, San Diego, CA, United States) to generate paired-end reads of approximately 250 bp. Totally, we obtained 1,526,330 and 1,533,219 raw reads for 16S rRNA and ITS sequencing, respectively (Supplementary Tables S1, S2).
Bioinformatics and statistical analysis
The obtained raw 16S rRNA and ITS gene sequences were processed using the Quantitative Insights into Microbial Ecology pipeline (QIIME; version 1.9.0) (Caporaso et al., 2010). Initially, the sequences that contained any ambiguous bases, had more than two mismatches to the primers, had one mismatch to the barcode, and were shorter than 200 bp or had an average quality score of <20 were discarded. After filtering out these sequences and removing chimeras, the operational taxonomic units (OTUs) were picked using USEARCH (version 7.0.1090) at a 97% sequence identity threshold. Singletons were filtered out, and taxonomic annotation for the representative 16S rRNA and ITS sequences was performed. This was based on the Greengenes database (version 13.5) and UNITE database (version 7.0) using the RDP Classifier’s Bayes algorithm, with a confidence threshold of 70% (Goyer et al., 2022). Alpha diversity, which measures the variety within a single sample, was calculated and visualized using these tools. Beta diversity, which indicates the similarity between microbial communities across different samples, was also calculated using QIIME (version 1.9.0). To determine significant differences in microbial taxa among different tissue types, we performed linear discriminant analysis (LDA) effect size (LEfSe) (Segata et al., 2011), with a cutoff for discriminative features set at a logarithmic LDA score of 3.5.
The number of unique and common endophytic bacterial/fungal OTUs across different tissues of A. spinulosa was compared through Venn analysis, which was conducted using the VennDiagram package (Chen and Boutros, 2011) in R (version 3.6.0) (R Core Team, 2019). This analysis provided a preliminary understanding of the species composition of each subgroup. Diversity indices, namely Chao1, ACE, Shannon, Simpson, Good’s coverage, and Pielou’s evenness, were calculated using QIIME (version 1.9.0). Alpha diversity indices of A. spinulosa endophytes were visualized through box line plotting in Origin (version 2022). To determine significant differences in these indices among different tissues, we used the package “best Normalize” (Peterson, 2021) in R (version 3.6.0) to transform the data normality, after passing the test of homogeneity of variances (Supplementary Tables S3, S4), we conducted LSD multiple comparisons in SPSS (version 27.0). We investigated the endophytic diversity across different tissues. A p < 0.05 indicated statistical significance. The community composition of different subgroups is displayed in a stacked bar plot, created using the ggplot2 package (Kahle and Wickham, 2013) in R (version 3.6.0). In addition, the hierarchical cluster analysis and principal coordinates analysis (PCoA) of Bray–Curtis distances were performed using the Vegan package (Huffman et al., 2002) in R (version 3.6.0). The results highlighting similarities and differences in community composition between the samples were plotted using the ggplot2 package (Kahle and Wickham, 2013) in R (version 3.6.0).
To simplify network complexity, we selected genera with a relative abundance of >1.0% in the entire dataset as well as in each tissue type of A. spinulosa. Accordingly, co-occurrence networks were constructed (Fan et al., 2019). The Pearson’s correlation coefficients and p-values were calculated using the corr.test function of the psych package (Revelle and Revelle, 2015) in R (version 3.6.0). A Pearson’s correlation coefficient between two genera was deemed statistically significant if it exceeded 0.6 (Zhu et al., 2019) and had a p < 0.05 (Zhang et al., 2024). The co-occurrence network was built using the “igraph” package (Zhu et al., 2019) in R (version 3.6.0). Network modularization was determine using the “cluster_fast_greedy” function in the igraph package (Zhu et al., 2019). The topological parameters of the network were exported and visualized using Cytoscape (version 3.10.1). In this network, each node represents a genus, and each edge indicates a strong and significant correlation between different genera.
Results
Structural distribution of endophytic bacterial and fungal communities in A. spinulosa tissues
After filtering and optimizing the 12 sequenced samples, we obtained 1,562,330 high-quality bacterial sequences and 1,427,157 high-quality fungal sequences. Clustering based on 97% sequence similarity revealed 1,247 bacterial OTUs across 35 phyla, 90 classes, 205 orders, 308 families, 512 genera, and 210 species. In addition, we identified 476 fungal OTUs spanning 11 phyla, 31 classes, 74 orders, 222 families, 247 genera, and 222 species.
The dilution curves for the bacterial and fungal sequences showed that the data plateaued at volumes of 8e+04 and 1e+05, respectively. This finding indicated that the sequencing data volume was adequate, ensuring the reliability and representativeness of the results for subsequent analyses (Supplementary Figure S3). Venn diagrams (Figure 1) present the distribution of endophytic bacterial and fungal OTUs across the samples. For endophytic bacteria, 481 OTUs were common across all four sample groups, with leaves containing the highest number of OTUs (n = 317), followed by roots (n = 277), petioles (n = 271), and stems (n = 223). Each plant part exhibited unique OTUs, with the root tissues exhibiting the highest number of unique species (n = 59), indicating a greater diversity of endophytic bacteria in roots (Figure 1A). Similarly, for endophytic fungi, 243 OTUs were common across the four groups, with roots again having the highest number of OTUs (n = 193), followed by leaves (n = 177), petioles (n = 168), and stems (n = 150). Each plant part had unique OTUs of fungi, with roots having the highest number of unique species (n = 26), suggesting a higher prevalence of endemic fungal species in roots (Figure 1B).
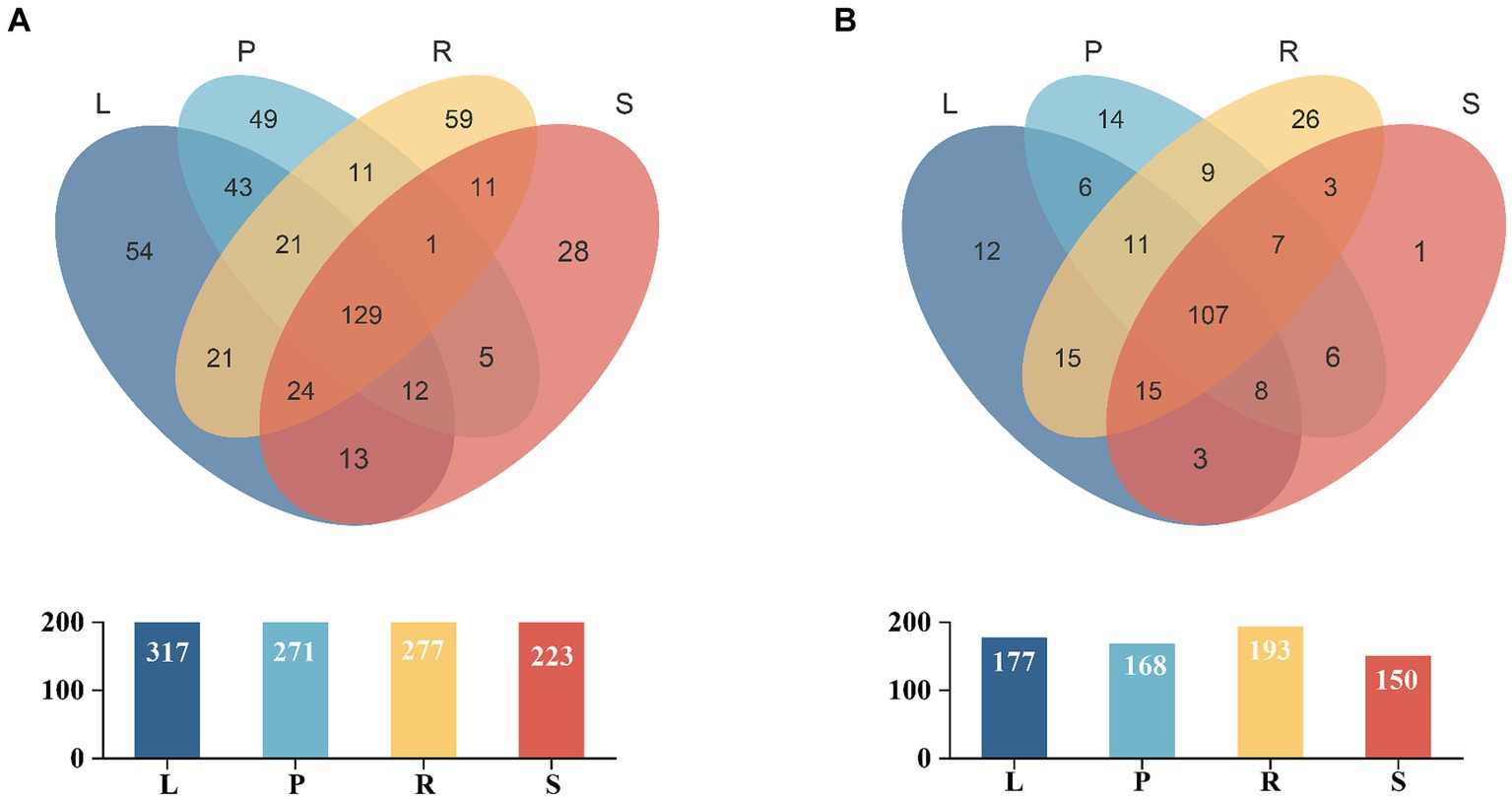
Figure 1. Venn diagram for the OTUs of bacterial (A) and fungal (B) populations in different parts of A. spinulosa. L, P, R, and S represent the leaves, petioles, roots, and stems of A. spinulosa, respectively. Different groups are represented by different colors, and numbers in the overlapping areas denote the number of species shared by the population.
Alpha diversity of endophytes in A. spinulosa
Alpha diversity is used to an indicator for species richness, evenness, and sequencing depth within a specific environment or ecosystem. Chao1 and Ace indices assess the richness of community microorganisms, with higher values indicating greater richness. Shannon and Simpson indices are used to evaluate the diversity of bacterial and fungal species, with higher values indicating highly diverse communities. Pielou index indicates the evenness or relative density of species, with higher values indicating greater evenness. Moreover, the Good’s coverage index assesses microbial coverage, with higher values suggesting a lower probability of missing new species in a sample. As shown in Figure 2, the sample coverage index for endophytic bacteria and fungi reached 1, suggesting that the sequencing depth covered nearly all species within the sample, which indicated that our data represent the actual community composition.
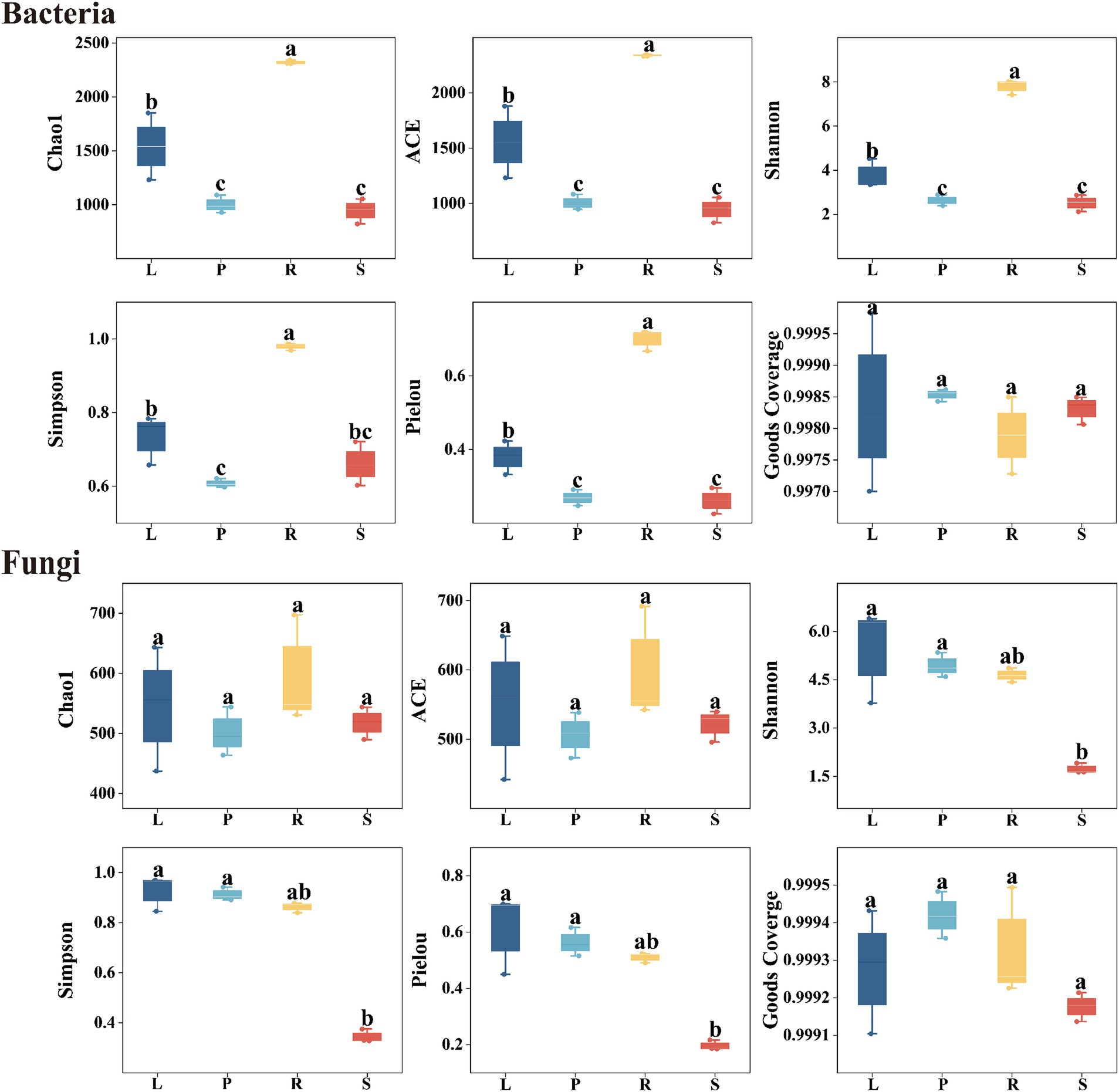
Figure 2. Alpha diversity of endophytic bacteria and fungi in different parts of A. spinulosa. L, P, R, and S represent the leaves, petioles, roots, and stems of A. spinulosa, respectively.
The Chao1 and Ace indices indicated that the richness of endophytic bacteria in A. spinulosa samples ranked in the order of roots > leaves > petioles > stems. This pattern suggests that the roots harbored the most endophytic bacteria, and stems had the least richness, with only slight differences in the bacterial abundance between petioles and stems. Similarly, the Shannon, Simpson, and Pielou indices for endophytic bacteria showed the sequence of roots > leaves > stems > petioles, indicating that the bacterial diversity and evenness were highest in the roots. By contrast, diversity and evenness in the leaves, petioles, and stems were similar but less pronounced. For endophytic fungi, the Chao1 and Ace indices did not differ significantly across the four tissue types of A. spinulosa (p > 0.05). However, the Shannon, Simpson, and Pielou indices revealed that the diversity and evenness of endophytic fungi in the leaves, petioles, and roots were similar but significantly higher than those in stems, indicating that stems have the lowest diversity and evenness of endophytic fungi.
Beta diversity of endophytes in A. spinulosa
The PCoA method was used to analyze the beta diversity of A. spinulosa endophytes, which elucidated the similarities and differences in the community composition among different plant tissues. For endophytic bacterial communities (Figure 3A), PCo1 and PCo2 accounted for 40.22 and 29.68% of the variance, respectively. The bacterial communities from the roots and stems were primarily found in the first and second quadrants, distinguishing them from those in other plant parts. By contrast, the bacterial communities in the leaves and petioles clustered more closely, indicating similar structural compositions between these two parts. As depicted in Figure 3B, the endophytic fungal communities in the leaves and petioles also showed close clustering. In addition, distinct clustering of the samples based on the tissue type indicated the significant effect of tissue type on the endophyte community structure.
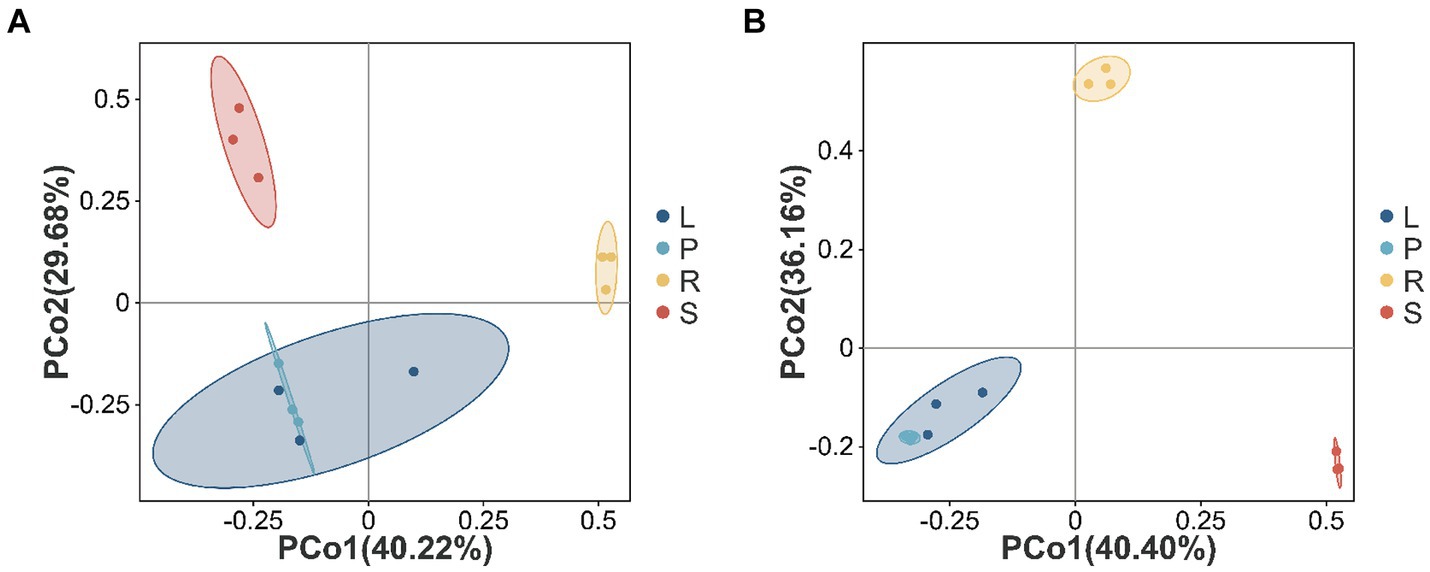
Figure 3. Beta diversity in different parts of A. spinulosa. The Bray–Curtis distance matrix for the principal coordinate analysis (A,B) of different endophytic bacterial and fungal communities in A. spinulosa. The distribution of endophytic bacterial (A) and fungal (B) communities. Each sample has three biological replicates. The values of the axes represent the percentage of variance that can be explained by each axis.
Composition of endophytic bacterial and fungal communities
The structure of the endophytic bacterial community in the four parts of A. spinulosa was analyzed at the genus level on the basis of OTU species nodes. The top 10 average richness in all samples were detailed, whereas the remaining species were uniformly categorized as “Other.” Tags that could not be classified at this level were grouped as “Unclassified” (Figure 4).

Figure 4. Histograms of relative richness at the genus level of bacteria (A) and fungi (B) in different parts of A. spinulosa.
At the genus level, the relative richness of Methylobacteria-methylorubrum was the highest in the leaves (11.618%), followed by that of Ralstonia (1.582%) and Bacillus (1.315%). In petioles, Methylobacteria-methylorubrum was the most abundant (1.676%), followed by Pseudomonas (0.694%) and Burkholderia-Caballeronia Paraburkholderia (0.332%). Root communities predominantly contained Bradyrhizobium (4.257%), Actinospica (3.919%), and Puia (3.686%). However, Pseudomonas (10.830%) and Bacillus (0.892%) were abundant in the stems. The primary endophytic bacterial groups in the leaves, petioles, roots, and stems were Methylobacteria-methylorubrum and Pseudomonas (Figure 4A).
At the genus level of endophytic fungi, in leaves, the relative richness of Cladosporium was the highest (9.405%), followed by that of Plenodomus (5.847%), Pestalotiopsis (5.052%), and Mycoleptodiscus (3.682%). In petioles, Pseudopithomyces was the most abundant (12.785%), followed by Cladosporium (8.140%) and Mycoleptodiscus (3.720%). Additionally, Pseudofabraea (34.050%), Delicatula (5.012%), and Colletotrichum (3.243%) were the main root endophytic fungi, whereas Cutaneotrichosporon (80.789%) was the predominant stem endophytic fungus. The main endophytic fungal groups in the leaves, petioles, roots, and stems belonged to Cutaneotrichosporon and Pseudofabraea (Figure 4B).
The characteristic endophytic bacteria and fungi in the four parts of A. spinulosa were identified through LEfSe analysis. According to LDA with a threshold of ≥3.5, five characteristic genera of endophytic bacteria and eight characteristic genera of endophytic fungi were identified (Supplementary Figure S4). The characterized genera of endophytic bacteria included Stenotrophomonas from stems, Pajaroellobacter from roots, Pelagibacteriu from petioles, and Methylobacterium_Methylorubrum and Sphingomonas from leaves. Endophytic characteristic genera of fungi were Pseudeurotium, Tulasnella, and Exophiala from roots; Periconia and Clonostachys from petioles; and Dichotomophthora, Cladosporium, and Coprinellus from leaves. No endophytic fungi characterizing genera with LDA (log10) > 3.5 were noted in A. spinulosa stems.
Network symbiotic structure of endophytic bacteria and fungi in A. spinulosa
The network structure of endophytic bacteria and fungi in A. spinulosa is characterized by modules, which are densely connected network areas where internal links outnumber external ones. After modularization, the endophytic bacterial community co-emergence network of the leaves and petioles was divided into 10 modules, whereas that of the roots and stems was divided into 8 and 7 modules, respectively. This finding suggested the presence of stronger connections among endophytic bacteria in the stems of A. spinulosa (Supplementary Figure S5). The endophytic fungal community co-emergence network of the petioles, roots, and stems had 11 modules each, whereas that of the leaves had eight modules, indicating stronger connections among endophytic fungi in the leaves of A. spinulosa (Supplementary Figure S6). Moreover, the modularity index of each symbiotic network exceeded 0.4, demonstrating that the endophytic bacterial and fungal networks in all parts of A. spinulosa exhibited a modular structure.
Analyzing the network nodes at the genus level according to phyla revealed the presence of 20, 16, 19, and 18 bacterial phyla in the community networks of the leaves, petioles, roots, and stems, respectively. Four phyla—Proteobacteria, Firmicutes, Bacteroidota, and Actinobacteriota—were particularly prevalent, accounting for 79.03, 84.77, 65.31, and 74.81% of all nodes in these communities, respectively (Figure 5). Similarly, the fungal community network of the leaves, petioles, roots, and stems was divided into 4, 16, 19, and 18 phyla, respectively. The phyla Basidiomycota and Ascomycota were predominantly distributed, accounting for 98.64, 97.7, 94.92, and 98.88% of all fungal nodes, respectively (Figure 6). These results indicate that the main endophytic bacteria and fungi were similarly distributed across the different parts of A. spinulosa.
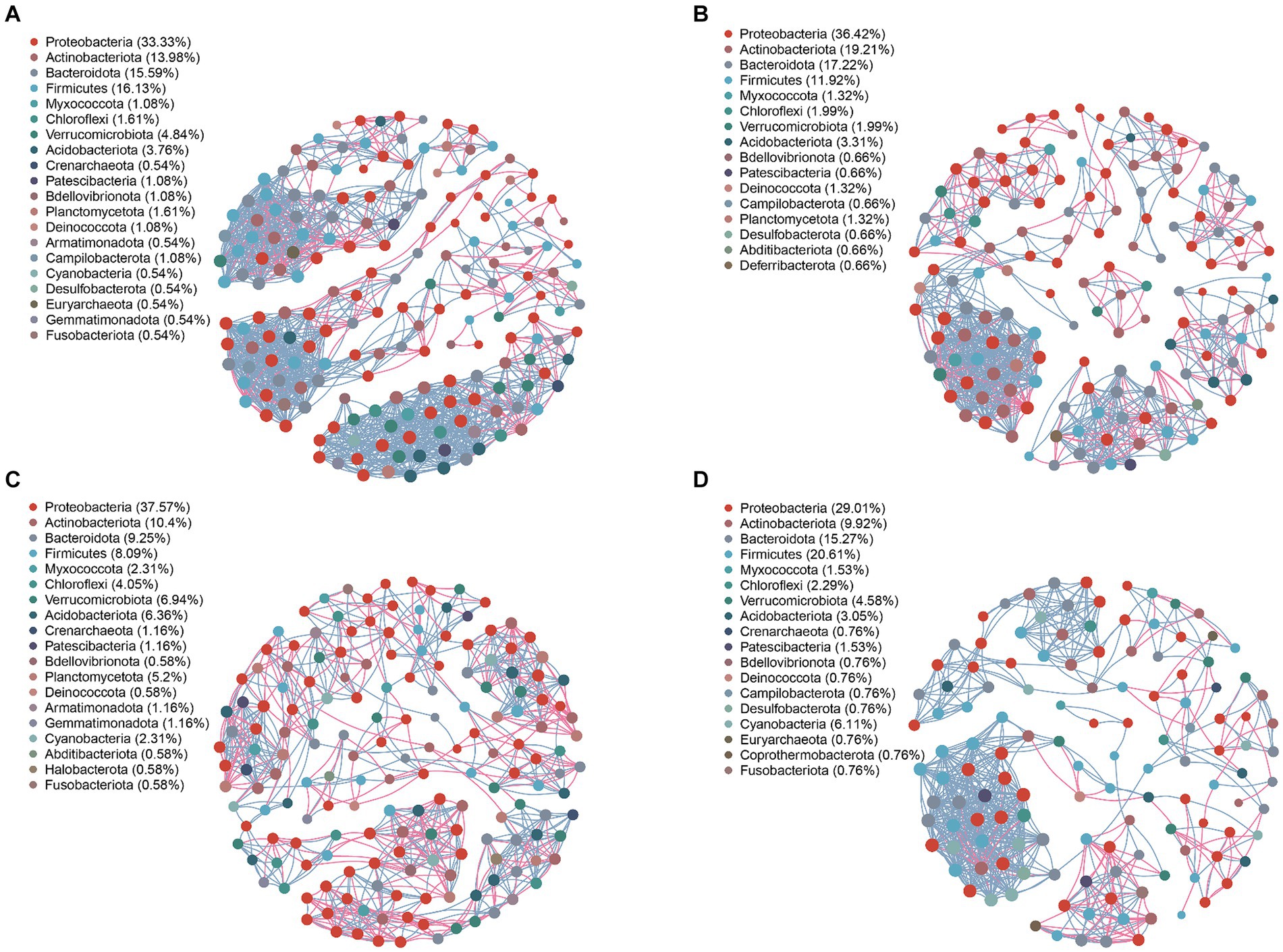
Figure 5. Symbiotic network characteristics of A. spinulosa endophytic bacteria in the leaves (A), petioles (B), roots (C), and stems (D). Genera with a relative abundance of >0.01 were selected, and the Pearson’s correlation coefficient between two genera was calculated. Based on correlation [Pearson’s coefficient >0.6 (correlation); p < 0.05 (significant)] relationships, a network map was drawn. The size of the node in the network is related to the connective degree of the point; the color represents the level of the phylum, and the red and sky blue lines represent the positive and negative correlations between the two nodes, respectively; the thicker the line, the stronger is the correlation. Different taxa are represented by different colors.
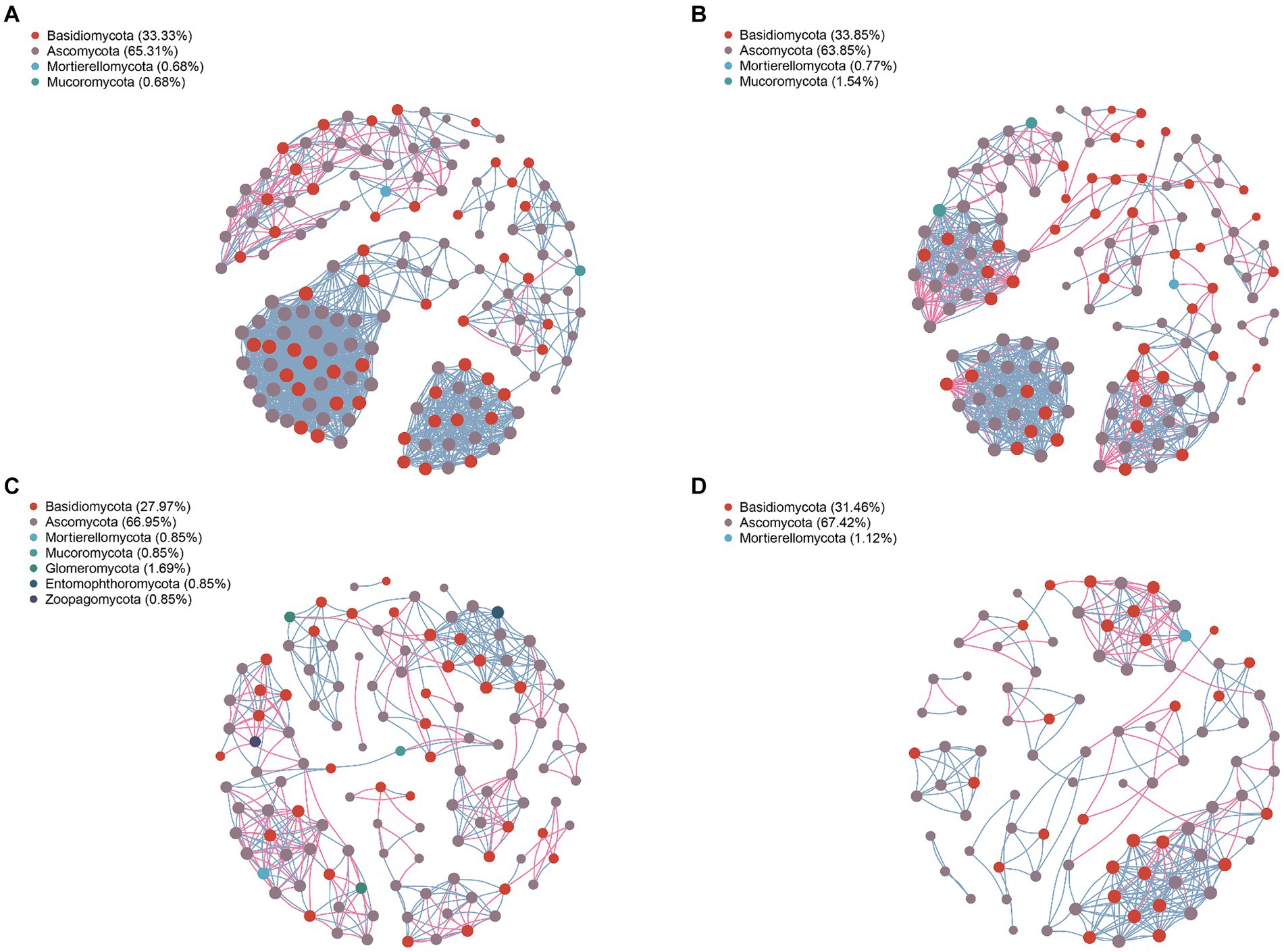
Figure 6. Symbiotic network characteristics of endophytic fungi in each part of A. spinulosa. (A) Leaves; (B) petioles; (C) roots; and (D) stems.
To further understand the co-occurrence patterns of endophytic bacteria in each part of A. spinulosa, separate bacterial and fungal networks were constructed for the four sample types, and their topological properties were analyzed (Table 1). These properties elucidated the complex patterns of node correlations within the networks. The analysis of co-occurrence network characteristics revealed that the leaf bacterial community network exhibited the highest complexity, whereas the stem network exhibited the least complexity, indicating that the leaf network had the highest number of nodes and connections, whereas the stem network had the least number of nodes and connections. Specifically, the leaf endophytic bacterial community network comprised 186 nodes (genera) and 1,381 connections, whereas the fungal network in the same part included 147 nodes and 1,346 connections. By contrast, the stem bacterial network contained only 131 nodes and 665 connections, and its fungal counterpart had 89 nodes and 300 connections. Furthermore, the leaf community displayed predominantly positive symbiotic relationships, as indicated by the highest ratios of positive to negative correlation connections (P/N) in both bacterial (5.057) and fungal (13.956) networks. The root networks, both bacterial and fungal, exhibited longer paths, greater diameters, and better modularity, suggesting a stronger topological structure. Furthermore, the leaf endophytic bacterial network showed the highest average clustering coefficient (avgCC) and average degree (avgK), indicating superior network connectivity compared with those of the other parts. By contrast, the root bacterial network had lower avgCC and avgK, suggesting less connectivity in the root tissues compared with the other tissue types.
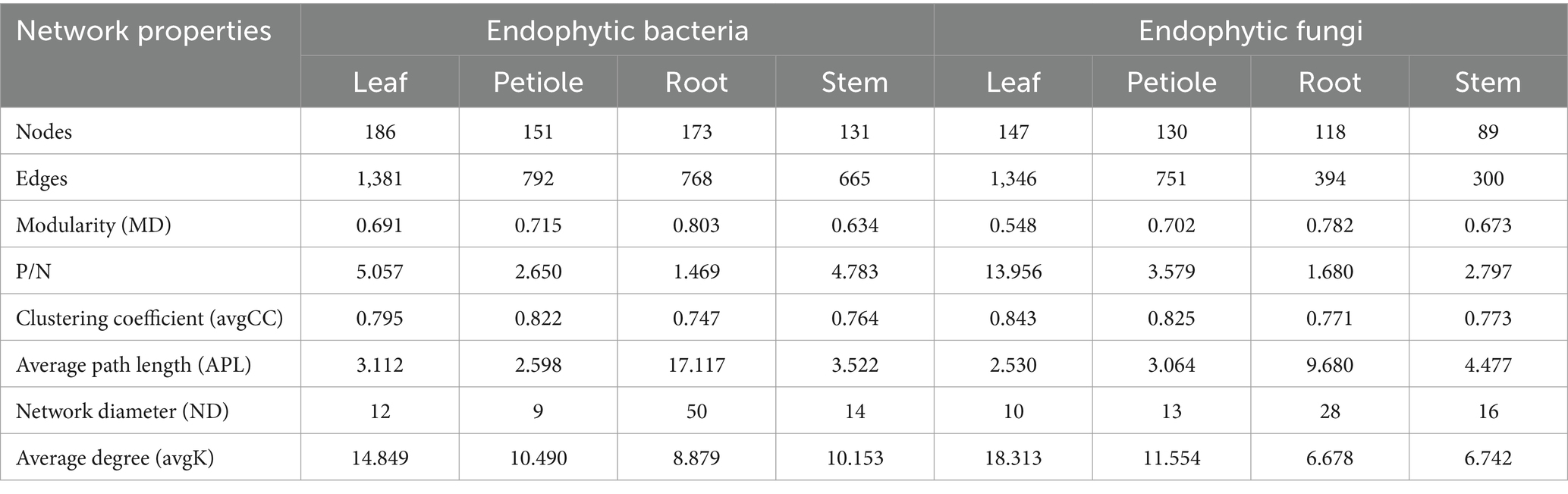
Table 1. Symbiotic network characteristics of endophytic bacterial and fungal communities in the leaf, petiole, root, and stem tissues of A. spinulosa.
Discussion
In this study, we used 16S rRNA and ITS high-throughput sequencing to perform a differential and comparative analysis of the composition and diversity of endophytes communities in the leaves, petioles, roots, and stems of A. spinulosa. This methodology eliminates the need to isolate microorganisms from purified cultures, offering unique advantages in characterizing microbial communities. This method enables us to obtain a more comprehensive understanding of endophytes by allowing direct extraction of total DNA from the sample environment and its sequencing, thereby circumventing the limitations of traditional culture methods. This approach is particularly suitable for studying various samples, such as plant microorganisms (Glenn et al., 2019; Quijada et al., 2020). In our study, we identified 481 shared OTUs among the four sample groups at the genus level of endophytic bacterial genera in A. spinulosa, with leaves containing the highest number of OTUs. This finding may be attributed to microenvironmental differences across plant tissues, which affect the survival of endophytic bacteria (Lin et al., 2022). Specifically, the leaf microenvironment in A. spinulosa appeared to be more conducive to the survival of diverse endophytic bacteria, leading to a higher number of endophytic bacterial OTUs in leaves than in the other tissues.
Our alpha diversity analysis revealed that the diversity of endophytic bacteria in A. spinulosa varied across different tissues. Specifically, the roots of A. spinulosa displayed higher endophytic bacterial diversity than its leaf blades, petioles, and stems; however, the stems exhibited the lowest diversity of endophytic fungi. Furthermore, beta diversity analyses indicated that the endophytic bacterial communities in the roots and stems were distinct from those in the other plant parts. By contrast, endophytic bacterial communities in leaf blades and petioles were closely clustered. Petioles and leaf blades constitute the leaf tissues. The petiole serves as the conduit for water and nutrient transfer across the leaf blades and stems, in addition to providing support to the leaf blades for optimal sunlight exposure. Thus, the petioles and leaf blades are interconnected, leading to similarities in the structure of endophyte communities. This observation aligns with the conclusions drawn by Wei et al. (2018) and Ferreira-Silva et al. (2021), highlighting the variation in the distribution of endophyte communities across different tissues. The genetic characteristics of A. spinulosa might affect the diversity and structure of endophyte communities. Factors such as genetic traits and growth characteristics can lead to differences in the physical structure, chemical composition, and nutrient content of various tissues, thereby affecting the diversity and structure of endophyte communities in plants.
The structure of endophyte communities in plants is dynamic and affected by various abiotic and biotic factors, including soil conditions, biogeography, plant species, microbe–microbe interactions, and plant–microbe interactions, operating at both local and larger scales (García-Guzmán and Heil, 2014). During evolution, endophytes of A. spinulosa have developed a mutually beneficial symbiotic relationship with their host plants. The host plant provides photosynthesized products and minerals to endophytes, which, in turn, support the host plant’s growth, development, and phylogenetic evolution. Our study revealed high diversity of the endophytic bacteria, belonging to 35 phyla, 90 classes, 205 orders, 308 families, 512 genera, and 210 species, in A. spinulosa. The identified endophytic fungi were found to belong to 11 phyla, 31 classes, 74 orders, 222 families, 247 genera, and 222 species. These findings provide insights into the composition of endophytic bacterial and fungal communities in A. spinulosa.
At the genus level, A. spinulosa harbored endophytic bacteria with high abundance, including Methylobacterium-Methylorubrum, Pseudomonas, and Bradyrhizobium. Previous studies have reported the unique one-carbon metabolic pathway of Methylobacterium, which plays a crucial role in the biometabolic pathways and evolutionary diversity (Chistoserdova et al., 1998). Pseudomonas plays a crucial role in biocontrol (Schlatter et al., 2017) by protecting plants from ion-induced oxidative stress through enhancing nutrient availability, reducing ion uptake, and triggering plant antioxidant responses (Islam et al., 2014). In our study, the genus Burkholderia-Caballeronia-Paraburkholderia was detected in the petioles of A. spinulosa, whereas numerous slow-growing rhizobacteria were found in its roots. These bacteria alleviate nutrient stress in the host plant by performing nitrogen fixation and secreting growth hormones, particularly under relatively infertile conditions, thereby enhancing plant growth (Hara et al., 2019; Dudeja et al., 2021). Moreover, certain endophytic bacterial populations, such as Pseudomonas and Bacillus, contribute to the host’s resistance against pathogens, drought, salt, and other biotic and abiotic stresses (Ali et al., 2014; Naveed et al., 2014). The enrichment of these endophytic bacterial flora substantially promotes plant health, growth, and development. However, the mechanisms underlying their effects on the growth of A. spinulosa remain to be fully elucidated.
Most endophytic fungi belonging to the genera Cutaneotrichosporon, Pseudofabraea, and Cladosporium were distributed across all tissues—leaves, petioles, roots, and stems—of the A. spinulosa samples. Cladosporium species/strains produce secondary metabolites, which serve as a rich source of bioactive compounds, and these compounds are widely used as antimicrobials, anthelmintics, and anticancer agents (Salvatore et al., 2021). Moreover, Cladosporium can produce indole-3-acetic acid and/or sterols, which play a role in enhancing plant growth. Thus, further genome analysis of Cladosporium species can lead to the discovery of new metabolites and metabolic pathways associated strongly with plant growth (Yang et al., 2023). Endophytic fungi offer numerous benefits for plant growth, including nitrogen fixation, phosphorus solubilization, phytohormone production, root morphology alteration, osmotic regulation, increased iron phosphate carrier production, enhanced solubilizing activity, and stomatal regulation. However, limited studies on the genera Cutaneotrichosporon and Pseudofabraea have hindered our understanding of whether an intrinsic link exists between these endophytic fungal taxa and the healthy growth of A. spinulosa as well as their metabolite activities.
Network analysis offers a unique and comprehensive perspective on microbial interactions by allowing clear visualization of the various forms of interactions between different taxa within a microbial community (Barberán et al., 2012; Ziegler et al., 2018; Banerjee et al., 2019). Modularity index values exceeding 0.4 indicate a typical modular structure (Newman, 2006). In our study, all modularity index values were higher than 0.4, indicating a robust modular structure in the networks constructed by endophytic flora at each site. A robust modular structure not only helps the endophytes of A. spinulosa in resisting environmental changes but also enhances the stability of the interaction network (Krause et al., 2003). Ecological networks are typically characterized by diverse biological interactions, where species display complex positive and negative associations (Chow et al., 2014). Positive correlations indicate cooperative relationships among biological populations, whereas negative correlations indicate competitive or predatory relationships. Our findings revealed that the nodes and connections of endophytic bacterial and fungal networks were most abundant in the leaves. Moreover, the symbiotic network of leaf endophyte communities displayed the highest number of positive and negative correlations, suggesting that the endophyte network in A. spinulosa leaves were most complex and had positive symbiotic relationships. Finally, our results align with those of previous studies, which have indicated that endophytic bacteria in most plants belong mainly to the phyla Proteobacteria, Firmicutes, Bacteroidota, and Actinobacteriota (Hardoim et al., 2015) and that the endophytic fungal community is dominated by Ascomycota and Basidiomycota (Marques et al., 2015).
Conclusion
The abundance, distribution, and diversity of endophytic bacteria in A. spinulosa are likely affected by both genetic characteristics and tissue types. Our study revealed the highest diversity of endophytic bacteria in A. spinulosa roots, and the diversity of endophytic fungi was lowest in the stems of A. spinulosa. Furthermore, leaf tissues showed the highest complexity of the endophyte network, whereas stems exhibited the lowest complexity of the mycorrhizal network. Some endophytes exhibit clear tissue preferences, unlike the endophytic bacterium Methylobacterium-Methylorubrum and the endophytic fungus Cladosporium, among others, which can parasitize various tissues. A limitation of the present study is the lack of a detailed examination of the practical application effects of endophytes in A. spinulosa. Identification of endophyte species in A. spinulosa can not only enhance our understanding of its microbial community composition and abundance but also provide insights into the symbiotic relationship between A. spinulosa and microorganisms. This study lays the theoretical groundwork for future research into its biological roles, particularly the biocontrol and antiretroviral properties of A. spinulosa.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
XC: Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Investigation, Methodology, Validation. MD: Writing – original draft, Writing – review & editing, Data curation, Methodology, Validation, Visualization. YL: Writing – review & editing, Resources, Validation, Visualization. JS: Resources, Software, Writing – review & editing. AZ: Methodology, Resources, Software, Writing – review & editing. XH: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Sichuan Province Innovative Talent Funding Project for Postdoctoral Fellows (grant no. 0312322999070 to XH), China Postdoctoral Science Foundation Funded Project (grant no. 2023M742510 to XH), and Natural Science Foundation of Sichuan Province (grant no. 22NSFSC0135 to XC).
Acknowledgments
We thank P. Song and X. Lu (Chengdu Homology Biotechnology Development Co., Ltd) for assistance with the sample collections.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1445315/full#supplementary-material
References
Abdalla, M. A., and Matasyoh, J. C. (2014). Endophytes as producers of peptides: an overview about the recently discovered peptides from endophytic microbes. Nat. Prod. Bioprospect. 4, 257–270. doi: 10.1007/s13659-014-0038-y
Ali, S., Charles, T. C., and Glick, B. R. (2014). Amelioration of high salinity stress damage by plant growth-promoting bacterial endophytes that contain ACC deaminase. Plant Physiol. Biochem. 80, 160–167. doi: 10.1016/j.plaphy.2014.04.003
Asad, S., Priyashantha, A. K. H., Tibpromma, S., Luo, Y., Zhang, J., Fan, Z., et al. (2023). Coffee-associated endophytes: plant growth promotion and crop protection. Biology 12:911. doi: 10.3390/biology12070911
Aye, M., Aung, H., Sein, M., and Armijos, C. (2019). A review on the phytochemistry, medicinal properties and pharmacological activities of 15 selected Myanmar medicinal plants. Molecules 24:293. doi: 10.3390/molecules24020293
Banerjee, S., Walder, F., Büchi, L., Meyer, M., Held, A. Y., Gattinger, A., et al. (2019). Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 13, 1722–1736. doi: 10.1038/s41396-019-0383-2
Barberán, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351. doi: 10.1038/ismej.2011.119
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chaparro-Hernández, I., Rodríguez-Ramírez, J., Barriada-Bernal, L. G., and Méndez-Lagunas, L. (2022). Tree ferns (Cyatheaceae) as a source of phenolic compounds – A review. J. Herbal Med. 35:100587. doi: 10.1016/j.hermed.2022.100587
Chen, H., and Boutros, P. C. (2011). Venn diagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinf. 12:35. doi: 10.1186/1471-2105-12-35
Chiang, H.-C., Lo, Y.-J., and Lu, F.-J. (1994). Xanthine oxidase inhibitors from the leaves of Alsophila Spinulosa (HOOK) Tryon. J. Enzym. Inhib. 8, 61–71. doi: 10.3109/14756369409040777
Chistoserdova, L., Vorholt, J. A., Thauer, R. K., and Lidstrom, M. E. (1998). C1 transfer enzymes and coenzymes linking methylotrophic bacteria and methanogenic archaea. Science 281, 99–102. doi: 10.1126/science.281.5373.99
Chow, C.-E. T., Kim, D. Y., Sachdeva, R., Caron, D. A., and Fuhrman, J. A. (2014). Top-down controls on bacterial community structure: microbial network analysis of bacteria, T4-like viruses and protists. ISME J. 8, 816–829. doi: 10.1038/ismej.2013.199
Dudeja, S. S., Suneja-Madan, P., Paul, M., Maheswari, R., and Kothe, E. (2021). Bacterial endophytes: molecular interactions with their hosts. J. Basic Microbiol. 61, 475–505. doi: 10.1002/jobm.202000657
Fan, K., Delgado-Baquerizo, M., Guo, X., Wang, D., Wu, Y., Zhu, M., et al. (2019). Suppressed N fixation and diazotrophs after four decades of fertilization. Microbiome 7:143. doi: 10.1186/s40168-019-0757-8
Ferreira-Silva, A., Hughes, F. M., Rosa, C. A., and Rosa, L. H. (2021). Higher turnover of endophytic fungal assemblages in the tissues of globose cactus Melocactus ernestii from Brazilian semi-arid biome. Symbiosis 85, 79–91. doi: 10.1007/s13199-021-00795-z
García-Guzmán, G., and Heil, M. (2014). Life histories of hosts and pathogens predict patterns in tropical fungal plant diseases. New Phytol. 201, 1106–1120. doi: 10.1111/nph.12562
Glenn, T. C., Pierson, T. W., Bayona-Vásquez, N. J., Kieran, T. J., Hoffberg, S. L., Thomas Iv, J. C., et al. (2019). Adapterama II: universal amplicon sequencing on Illumina platforms (TaggiMatrix). PeerJ 7:e7786. doi: 10.7717/peerj.7786
Gouda, S., Das, G., Sen, S. K., Shin, H.-S., and Patra, J. K. (2016). Endophytes: a treasure house of bioactive compounds of medicinal importance. Front. Microbiol. 7:1538. doi: 10.3389/fmicb.2016.01538
Goyer, C., Neupane, S., Zebarth, B. J., Burton, D. L., Wilson, C., and Sennett, L. (2022). Diverse compost products influence soil bacterial and fungal community diversity in a potato crop production system. Appl. Soil Ecol. 169:104247. doi: 10.1016/j.apsoil.2021.104247
Hara, S., Morikawa, T., Wasai, S., Kasahara, Y., Koshiba, T., Yamazaki, K., et al. (2019). Identification of nitrogen-fixing Bradyrhizobium associated with roots of field-grown sorghum by metagenome and proteome analyses. Front. Microbiol. 10:407. doi: 10.3389/fmicb.2019.00407
Hardoim, P. R., Van Overbeek, L. S., Berg, G., Pirttilä, A. M., Compant, S., Campisano, A., et al. (2015). The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 79, 293–320. doi: 10.1128/MMBR.00050-14
Hashem, A. H., Attia, M. S., Kandil, E. K., Fawzi, M. M., Abdelrahman, A. S., Khader, M. S., et al. (2023). Bioactive compounds and biomedical applications of endophytic fungi: a recent review. Microb. Cell Factories 22:107. doi: 10.1186/s12934-023-02118-x
Huang, X., Wang, W., Gong, T., Wickell, D., Kuo, L.-Y., Zhang, X., et al. (2022). The flying spider-monkey tree fern genome provides insights into fern evolution and arborescence. Nat. Plants 8, 500–512. doi: 10.1038/s41477-022-01146-6
Huffman, D. L., Huyett, J., Outten, F. W., Doan, P. E., Finney, L. A., Hoffman, B. M., et al. (2002). Spectroscopy of cu (II)-PcoC and the multicopper oxidase function of PcoA, two essential components of Escherichia coli Pco copper resistance operon. Biochemistry 41, 10046–10055. doi: 10.1021/bi0259960
Islam, F., Yasmeen, T., Ali, Q., Ali, S., Arif, M. S., Hussain, S., et al. (2014). Influence of pseudomonas aeruginosa as PGPR on oxidative stress tolerance in wheat under Zn stress. Ecotoxicol. Environ. Saf. 104, 285–293. doi: 10.1016/j.ecoenv.2014.03.008
Kahle, D., and Wickham, H. (2013). Ggmap: spatial visualization with ggplot2. The R J. 5:144. doi: 10.32614/RJ-2013-014
Krause, A. E., Frank, K. A., Mason, D. M., Ulanowicz, R. E., and Taylor, W. W. (2003). Compartments revealed in food-web structure. Nature 426, 282–285. doi: 10.1038/nature02115
Kushwaha, P., Kashyap, P. L., Bhardwaj, A. K., Kuppusamy, P., Srivastava, A. K., and Tiwari, R. K. (2020). Bacterial endophyte mediated plant tolerance to salinity: growth responses and mechanisms of action. World J. Microbiol. Biotechnol. 36:26. doi: 10.1007/s11274-020-2804-9
Ley, S. V., and Baxendale, I. R. (2002). New tools and concepts for modern organic synthesis. Nat. Rev. Drug Discov. 1, 573–586. doi: 10.1038/nrd871
Liang, X., Zhou, K., Li, P., Wan, D., Liu, J., Yi, X., et al. (2023). Characteristics of endophytic bacteria and active ingredients in the Eucommiae cortex from different origins. Front. Microbiol. 14:1164674. doi: 10.3389/fmicb.2023.1164674
Lin, H., Liu, C., Peng, Z., Tan, B., Wang, K., and Liu, Z. (2022). Distribution pattern of endophytic bacteria and fungi in tea plants. Front. Microbiol. 13:872034. doi: 10.3389/fmicb.2022.872034
Loiseau, O., Weigand, A., Noben, S., Rolland, J., Silvestro, D., Kessler, M., et al. (2020). Slowly but surely: gradual diversification and phenotypic evolution in the hyper-diverse tree fern family Cyatheaceae. Ann. Bot. 125, 93–103. doi: 10.1093/aob/mcz145
Lombardino, J. G., and Lowe, J. A. (2004). The role of the medicinal chemist in drug discovery—then and now. Nat. Rev. Drug Discov. 3, 853–862. doi: 10.1038/nrd1523
Longtine, C., and Tejedor, A. (2017). Antimicrobial activity of ethanolic and aqueous extracts of medicinally used tree ferns Alsophila cuspidata and Cyathea microdonta. Acta Bot. Malacitana 42, 119–123. doi: 10.24310/abm.v42i1.2885
Ma, Y., Jiao, P., Qi, Z., Jiang, Z., and Guan, S. (2020). Characterization of the complete chloroplast genome of Alsophila spinulosa, an endangered species endemic to China. Mitochondrial DNA Part B 5, 2262–2263. doi: 10.1080/23802359.2020.1772142
Mario Geysen, H., Schoenen, F., Wagner, D., and Wagner, R. (2003). Combinatorial compound libraries for drug discovery: an ongoing challenge. Nat. Rev. Drug Discov. 2, 222–230. doi: 10.1038/nrd1035
Marques, J. M., Da Silva, T. F., Vollú, R. E., De Lacerda, J. R. M., Blank, A. F., Smalla, K., et al. (2015). Bacterial endophytes of sweet potato tuberous roots affected by the plant genotype and growth stage. Appl. Soil Ecol. 96, 273–281. doi: 10.1016/j.apsoil.2015.08.020
Naveed, M., Mitter, B., Reichenauer, T. G., Wieczorek, K., and Sessitsch, A. (2014). Increased drought stress resilience of maize through endophytic colonization by Burkholderia phytofirmans PsJN and Enterobacter sp. FD17. Environ. Exp. Bot. 97, 30–39. doi: 10.1016/j.envexpbot.2013.09.014
Newman, M. E. J. (2006). Modularity and community structure in networks. Proc. Natl. Acad. Sci. 103, 8577–8582. doi: 10.1073/pnas.0601602103
Omomowo and Babalola (2019). Bacterial and fungal endophytes: tiny giants with immense beneficial potential for plant growth and sustainable agricultural productivity. Microorganisms 7:481. doi: 10.3390/microorganisms7110481
Peterson, R. A. (2021). Finding optimal normalizing transformations via best normalize. The R J. 13:310. doi: 10.32614/RJ-2021-041
Quijada, N. M., Hernández, M., and Rodríguez-Lázaro, D. (2020). High-throughput sequencing and food microbiology. Adv. Food Nutr. Res. 91, 275–300. doi: 10.1016/bs.afnr.2019.10.003
R Core Team (2019). R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing.
Rani, S., Kumar, P., Dahiya, P., Maheshwari, R., Dang, A. S., and Suneja, P. (2022). Endophytism: a multidimensional approach to plant–prokaryotic microbe interaction. Front. Microbiol. 13:861235. doi: 10.3389/fmicb.2022.861235
Salvatore, M. M., Andolfi, A., and Nicoletti, R. (2021). The genus Cladosporium: a rich source of diverse and bioactive natural compounds. Molecules 26:3959. doi: 10.3390/molecules26133959
Schlatter, D., Kinkel, L., Thomashow, L., Weller, D., and Paulitz, T. (2017). Disease suppressive soils: new insights from the soil microbiome. Phytopathology 107, 1284–1297. doi: 10.1094/PHYTO-03-17-0111-RVW
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Seghers, D., Wittebolle, L., Top, E. M., Verstraete, W., and Siciliano, S. D. (2004). Impact of agricultural practices on the Zea mays L. endophytic community. Appl. Environ. Microbiol. 70, 1475–1482. doi: 10.1128/AEM.70.3.1475-1482.2004
Tang, P., Shen, T., Wang, H., Zhang, R., Zhang, X., Li, X., et al. (2023). Challenges and opportunities for improving the drug ability of natural product: why need drug delivery system? Biomed. Pharmacother. 164:114955. doi: 10.1016/j.biopha.2023.114955
Tshikhudo, P. P., Ntushelo, K., and Mudau, F. N. (2023). Sustainable applications of endophytic bacteria and their physiological/biochemical roles on medicinal and herbal plants: review. Microorganisms 11:453. doi: 10.3390/microorganisms11020453
Wei, W., Zhou, Y., Chen, F., Yan, X., Lai, Y., Wei, C., et al. (2018). Isolation, diversity, and antimicrobial and immunomodulatory activities of endophytic actinobacteria from tea cultivars Zijuan and Yunkang-10 (Camellia sinensis var. assamica). Front. Microbiol. 9:1304. doi: 10.3389/fmicb.2018.01304
Xiong, Y., Zeng, Y., Liu, J., Chen, X., Li, Y., Zhang, X., et al. (2023). Gametophyte development and sporophyte regeneration of Alsophila spinulosa. J. Plant Growth Regul. 42, 7488–7499. doi: 10.1007/s00344-023-11026-8
Yan, K., Pei, Z., Meng, L., Zheng, Y., Wang, L., Feng, R., et al. (2022). Determination of community structure and diversity of seed-vectored endophytic fungi in Alpinia zerumbet. Front. Microbiol. 13:814864. doi: 10.3389/fmicb.2022.814864
Yang, N., Zhang, W., Wang, D., Cao, D., Cao, Y., He, W., et al. (2023). A novel endophytic fungus strain of Cladosporium: its identification, genomic analysis, and effects on plant growth. Front. Microbiol. 14:1287582. doi: 10.3389/fmicb.2023.1287582
Yi, H., Dong, S., Yang, L., Wang, J., Kidner, C., and Kang, M. (2023). Genome-wide data reveal cryptic diversity and hybridization in a group of tree ferns. Mol. Phylogenet. Evol. 184:107801. doi: 10.1016/j.ympev.2023.107801
Zhang, S., Song, F., Wang, J., Li, X., Zhang, Y., Zhou, W., et al. (2024). Gut microbiota facilitate adaptation of invasive moths to new host plants. ISME J. 18:wrae031. doi: 10.1093/ismejo/wrae031
Zhou, Z.-Y., Liu, X., Cui, J.-L., Wang, J.-H., Wang, M.-L., and Zhang, G. (2022). Endophytic fungi and their bioactive secondary metabolites in medicinal leguminosae plants: nearly untapped medical resources. FEMS Microbiol. Lett. 369:fnac052. doi: 10.1093/femsle/fnac052
Zhu, H. Z., Zhang, Z. F., Zhou, N., Jiang, C. Y., Wang, B. J., Cai, L., et al. (2019). Diversity, distribution and co-occurrence patterns of bacterial communities in a karst cave system. Front. Microbiol. 10:1726. doi: 10.3389/fmicb.2019.01726
Ziegler, M., Eguíluz, V. M., Duarte, C. M., and Voolstra, C. R. (2018). Rare symbionts may contribute to the resilience of coral–algal assemblages. ISME J. 12, 161–172. doi: 10.1038/ismej.2017.151
Keywords: Alsophila spinulosa, endophytic bacteria, endophytic fungi, high-throughput sequencing, co-occurrence network
Citation: Chen X, Dou M, Li Y, Su J, Zhao A and Huang X (2024) Elucidating the endophytic bacterial and fungal community composition and diversity in the tree fern Alsophila spinulosa through meta-amplicon sequencing. Front. Microbiol. 15:1445315. doi: 10.3389/fmicb.2024.1445315
Edited by:
Ajar Nath Yadav, Eternal University, IndiaReviewed by:
Modupe Stella Ayilara, North-West University, South AfricaShouke Zhang, Zhejiang Agriculture and Forestry University, China
Copyright © 2024 Chen, Dou, Li, Su, Zhao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiong Huang, aHVhbmd4aW9uZ0BzaWNhdS5lZHUuY24=
†These authors have contributed equally to this work