
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 25 June 2024
Sec. Microbe and Virus Interactions with Plants
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1431047
 Jingxin Zhang1,2,3*†
Jingxin Zhang1,2,3*† Dayuan Sun1,2,3†
Dayuan Sun1,2,3† Huifang Shen1,2,3
Huifang Shen1,2,3 Xiaoming Pu1,2,3
Xiaoming Pu1,2,3 Pingping Liu1,2,3
Pingping Liu1,2,3 Birun Lin1,2,3
Birun Lin1,2,3 Qiyun Yang1,2,3*
Qiyun Yang1,2,3*Bacterial soft rot caused by coinfection with Dickeya spp. and Pectobacterium spp. in hosts can cause successive changes in fields, and it is difficult to prevent the spread of and control the infection. Pectobacterium spp. are prevalent in the growing areas of tuberous crops, including taro and potato. Recently, Dickeya fangzhongdai has emerged as a virulent pathogen in taro. To determine the prevalence status of the causal agents and evaluate the potential spreading risks of D. fangzhongdai, screening and taxonomic classification were performed on phytopathogenic bacteria collected from different taro-growing areas in Guangdong Province, China, and biological and genomic characteristics were further compared among typical strains from all defined species. The causative agents were verified to be phytobacterial strains of D. fangzhongdai, Pectobacterium aroidearum and Pectobacterium colocasium. P. aroidearum and P. colocasium were found to form a complex preferring Araceae plants and show intensive genomic differentiation, indicating their ancestor had adapted to taro a long time prior. Compared with Pectobacterium spp., D. fangzhongdai was more virulent to taro corms under conditions of exogenous infection and more adaptable at elevated temperatures. D. fangzhongdai strains isolated from taro possessed genomic components of additional T4SSs, which were accompanied by additional copies of the hcp-vgrG genes of the T6SS, and these contributed to the expansion of their genomes. More gene clusters encoding secondary metabolites were found within the D. fangzhongdai strains than within the Pectobacterium complex; interestingly, distinct gene clusters encoding zeamine and arylpolyene were both most similar to those in D. solani that caused potato soft rot. These comparisons provided genomic evidences for that the newly emerging pathogen was potentially equipped to compete with other pathogens. Diagnostic qPCR verified that D. fangzhongdai was prevalent in most of the taro-growing areas and coexisted with the Pectobacterium complex, while the plants enriching D. fangzhongdai were frequently symptomatic at developing corms and adjacent pseudostems and caused severe symptoms. Thus, the emerging need for intensive monitoring on D. fangzhongdai to prevent it from spreading to other taro-growing areas and to other tuberous crops like potato; the adjustment of control strategies based on different pathopoiesis characteristics is recommended.
Soft rot Pectobacteriaceae (SRP), including bacteria from the genera Dickeya and Pectobacterium, are among the most important bacterial plant pathogens based on their scientific or economic importance (Mansfield et al., 2012). They have been found to infect a wide range of plants, including Gramineae crops (rice, corn, etc.), tuberous crops (potato, taro, carrot, etc.), fruits (banana, etc.), and ornamental plants (orchids, etc.) (Ma et al., 2007). Phytopathogenic bacteria of these two genera frequently coinfect potato plants and cause severe blackleg and tuber soft rot worldwide (van der Wolf et al., 2017; Ma et al., 2018; Rossmann et al., 2018). More recently, the emergence of the virulent pathogen Dickeya solani has affected the potato industry in Europe and raised global concerns (van der Wolf et al., 2014a). Under pressure from both local evolving SRP pathogens and invasive foreign pathogens, verified host plants are at risk of coinfection with Dickeya spp. and Pectobacterium spp. or evolved pathogens with increased virulence; moreover, the expansion of their host ranges poses a threat to potential hosts.
Taro (Colocasia esculenta) is a world widely cultivated aroid plant featured with large starchy edible corms, and is essential for food security, especially in Asia, Africa, and the Pacific islands; in 2016, this crop had a global production value of 4.1 billion dollars (Delude et al., 2022). This crop has been widely grown in the Yangtze River and Pearl River basins in China. In Guangdong Province, Zhangxi taro from Lechang, Shaoguan, and Wengang taro from Huadu, Guangzhou, are the most famous products with geographical indications; these taro products are characterized by large corms and are the best-selling products on the market, with an output value of more than 360,000 yuan/hm2. In recent years, bacterial soft rot has seriously affected this industry and is common in different growing areas, with incidences ranging from 3 to 15%. Especially in 2023, the incidences reached 40% at the core production areas in Lechang, Shaoguan. Like potato in China, taro is strongly affected by Pectobacterium spp. in different regions, including Guangdong, Guangxi, and Fujian. In 2021, Dickeya fangzhongdai was first reported to infect taro in China (Huang et al., 2021). This causative agent is also a major threat to orchids and is widely distributed in most orchid growing areas in Guangdong, China (Zhang et al., 2018). Other important crops, including pear (Tian et al., 2016), banana (Yang et al., 2022), and onion (Ma et al., 2019), are also seriously damaged by this virulent pathogen.
Dickeya spp. and Pectobacterium spp. were found to occasionally cause soft rot diseases in the same host, such as potato, vegetables, and flowers (Van Gijsegem et al., 2021). For a long time, P. atrosepticum and P. carotovorum were the major pathogenic agents for soft rot diseases in potato, and subsequently, other pathogens gradually emerged, including D. dianthicola, P. brasiliense, and P. wasabiae; in this process, interactions among the pathogenic bacteria could induce their succession, and the newly emerging pathogenic bacteria might gradually spread to larger territories and then become the dominant population (Gross et al., 1991; Yap et al., 2004; Kim et al., 2009). For the newly emerging pathogen on taro isolated in Zhangxi village of Lechan, Shaoguang, Guangdong (Huang et al., 2021), it was not clear whether D. fangzhongdai infected the new host by chance or had been spreading in fields.
In 2022, we again isolated D. fangzhongdai in another village (Mawu) of Lechan, as well as another causative agent, Pectobacterium aroidearum. However, the spread and prevalence of D. fangzhongdai in the taro-growing areas of Guangdong, as well as the possibility that this emerging pathogen could become the dominant pathogenic bacterium in taro fields, require further investigation. In this study, the phylogenetic determination of pathogenic bacteria isolated from different growing areas and the identification of different kinds of pathogens in plants were performed to determine the spreading status of D. fangzhongdai in the field, as well as the actual composition of causative agents for taro soft rot in fields. The biological and genomic characteristics of typical D. fangzhongdai strains isolated from taro were compared to those of D. fangzhongdai strains isolated from other hosts and Pectobacterium spp. isolated from taro to determine the potential for changes in dominant pathogens and the subsequent risks. The clarification of the prevalence status and possible succession between D. fangzhongdai and previously colonized Pectobacterium spp. not only provides a theoretical basis for the complex epidemic patterns most likely caused by pathogen interactions but also draws attention to the emergence of virulent pathogens such as D. fangzhongdai and helps in the development of strategies for the prevention of spread. This will also provide a reference for addressing the risks posed by the adaptation of Dickeya pathogens in other tuberous crops, for example, for infection in the important staple crop potato which was also reported to be strongly affected by Pectobacterium spp. in China (Zhang et al., 2012; Jiang et al., 2019; Cao et al., 2021; Li et al., 2021; Handique et al., 2022).
From 2022 to 2023, taro corms with typical soft rot symptoms were collected from 10 different locations in Guangdong Province, including three in Lechang city, one in Ruyuan County, two in Wujiang District, two in Renhua County, one in Huadu District, and one in Longmeng County. Small pieces of tissue were removed from the margins between diseased and healthy areas and subjected to surface sterilization. The sterilized sections were placed onto nutrient agar (3 g/L beef extract, 0.5 g/L yeast extract paste, 5 g/L peptone, and 10 g/L agar, pH 7.0) and cultured for 48 h at 32°C. Colonies were selected for hypersensitivity testing on tobacco and 16S rDNA sequencing, and the SRPs were further subcultured three times.
Genomic DNA was extracted from overnight bacterial cultures with a TIANamp Bacterial DNA Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s directions. Portions of the housekeeping genes (dnaX, leuS and recA) of SRP were sequenced according to the protocol provided by Portier et al. (2019). Sequences obtained by the Sanger method were manually checked and aligned, and each truncated sequence of three genes was concatenated to construct a phylogenetic tree using MEGA 11 with the maximum likelihood method and a bootstrapping test of 1,000 replicates.
Plants were grown from taro corms and maintained in a greenhouse. The pseudostems of 9- to 10-week-old plants were directly injected with 200 μL of 108 CFU/mL bacteria at 5 cm above ground level, and the corms near the basal pseudostems were also directly inoculated; 200 μL of sterile demineralized water served as the control. Each inoculation was repeated three times. Symptoms on the whole plant were observed during disease development.
To compare the initial infection ability of different SRPs on the taro corms under conditions that simulated exogenous infection, each pipette tip with 200 μL of 108 CFU/mL bacteria or sterile water was used to lightly stab the upper side of the corm, and the stab locations were subsequently wrapped with parafilm to prevent the pipette tip from falling and to prevent drying and leakage of the bacteria. Five corms were inoculated with each tested SRP strains, and five other corms were used as controls. The inoculated corms were kept in plastic bags to maintain humidity. The soft rot symptoms were observed and recorded after 5 d of inoculation.
Typical strains of D. fangzhongdai, P. aroidearum and Pectobacterium colocasium were cultured in LB broth overnight, and the cell densities were adjusted to 108 CFU/mL. Fifteen microliters of each tested strain were added to 15 mL of LB broth in a 50 mL centrifuge tube and cultured at 200 rpm and at different incubator temperatures. Each inoculation was repeated three times. The temperatures were set at three different levels for the growth of the SRP: 16°C, 30°C and 42°C. The growth curves of the tested strains were generated during 36 h of bacterial culture based on the OD600.
The genomic DNA of D. fangzhongdai J21-2, P. aroidearum B2-6 and T25-1, and P. colocasium MT27-1 was extracted from 2 mL of each bacterial suspension using a TIANamp Bacterial DNA Kit, and the quality of the extracted DNA was detected using a Qubit (Thermo Fisher Scientific, Waltham, MA) and Nanodrop (Thermo Fisher Scientific, Waltham, MA). Qualified genomic DNA was fragmented with G-tubes (Covaris, Woburn, MA, United States) and end-repaired. Repaired DNAs were selected by a Blue Pippin system to construct SMRTbell DNA libraries with fragment sizes >10 kb according to the manufacturer’s specifications (PacBio, CA, United States). A Qubit 2.0 fluorometer (Life Technologies, CA, United States) and a Bioanalyzer 2,100 (Agilent, CA, United States) were used to determine library quality and average fragment sizes, respectively. SMRT sequencing was performed on a Pacific Biosciences Sequel (PacBio) at Genedenovo (Guangzhou, China) according to standard protocols.
Qualified genomic DNA was also sonicated randomly and then end-repaired, A-tailed, and adaptor ligated using the NEBNext® ΜLtra™ DNA Library Prep Kit for Illumina (NEB, MA, United States) according to the manufacturer’s protocol. DNA fragments with lengths of 300–400 bp were enriched by PCR. These PCR products were purified using an AMPure XP system (Beckman Coulter, CA, United States) and sequenced on an Illumina NovaSeq 6,000 sequencer (Illumina CA, United States) using paired-end technology (PE 150).
Continuous long reads obtained from SMRT sequencing were de novo assembled using Falcon (version 0.3.0) (Chin et al., 2016). Clean reads generated from the Illumina platform were aligned to the assemblies from SMRT sequencing. Inconsistencies between PacBio assemblies and Illumina reads were corrected to improve the quality of the assembly and determine the final genome sequences using Pilon (version 1.23) (Walker et al., 2014). Genomic assembly of D. fangzhongdai J21-2, P. aroidearum B2-6 and T25-1, and P. colocasium MT27-1 revealed complete genomes of 5,191,363 bp (1024.03-fold genome coverage of qualified sequenced reads, GC content of 56.44%), 4,806,190 bp (595.06-fold, 51.82%), 4,976,432 bp (930.97-fold, 51.86%), and 4,759,205 bp (521.38-fold, 51.77%), respectively. Plasmid extraction and agarose gel electrophoresis indicated these sequenced bacteria did not contain the plasmids, as well as several other D. fangzhongdai strains isolated from taro and orchid (Supplementary Figure S1).
The NCBI prokaryotic genome annotation pipeline was used to predict open reading frames (ORFs) (Tatusova et al., 2016), and 4,441, 4,099, 4,309, and 4,049 ORFs were predicted within the above-mentioned genomes. Furthermore, gene islands, transposons, and prophages were identified using IslandPath-DIMOB (version 1.0.0) (Bertelli and Brinkman, 2018), TransposonPSI (version 20,100,822), and PHAST (version 2.0) (Fouts, 2006), respectively. Gene function was further predicted through alignment to genes deposited in different databases, including the National Center for Biotechnology Information (NCBI) nonredundant protein sequence (Nr), UniProt/Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), and Cluster of Orthologous Groups of proteins (COG) databases.
The amino acid sequences among the available genomes were compared by Diamond (version 2.0.7) (Buchfink et al., 2021), and similarity clustering was carried out by OrthoMCL (version 1.4) (Li et al., 2003) to obtain a list of clustered homologous genes. A single copy of a homologous gene was selected for multisequence alignment using MUSCLE (version 3.8.31), and a phylogenetic tree was constructed using IQTREE (version 1.6.3) (Nguyen et al., 2015) based on the concatenated single-copy gene sequences.
Collinearity analysis between genomes was performed on MUMmer (version 3.1) (Kurtz et al., 2004) software, and SyRI (version 1.4) (Goel et al., 2019) was used to confirm the local positional arrangement. Pyani (version 0.2.7) (Pritchard et al., 2016) was further used to calculate the average nucleotide identity (ANI) of the comparison regions of each of the two analyzed genomes, and GGDC 3.0 (Meier-Kolthoff et al., 2022) was applied to evaluate the values of in silico DNA–DNA hybridization (isDDH). Generally, an ANI of 0.96 and an isDDH of 0.70 are used as the classification thresholds to distinguish different species. Additional genomic comparisons were performed on specific functional features, such as gene clusters of secondary metabolites predicted by antiSMASH (version 4.1.0) (Blin et al., 2017; Chevrette et al., 2017), secretory proteins predicted by SignalP4.0 (Petersen et al., 2011), transmembrane proteins predicted by TMHMM (version 2.0) (Krogh et al., 2001), and TNSSs (type N secretion systems, I-VII types) retrieved from the KEGG and Nr annotations. The classification of predicted T4SSs were based on the mating-pair formation genes (mpf) (Guglielmini et al., 2014).
Nine large taro-growing fields, which are distributed in most of the important taro-growing areas in Guangdong Province, were selected for the investigation of soft rot disease. Forty-eight symptomatic or asymptomatic corms were collected and used for DNA extraction. The ground corm tissue was processed with a MiniBEST Plant Genomic DNA Extraction Kit (Takara, Beijing, China) according to the manufacturer’s instructions. Primers and probes specific for D. fangzhongdai (Tian et al., 2020) and Pectobacterium spp. (Arizala et al., 2022) and 100 ng of DNA were included in the reaction mixture with Premix Ex Taq™ (Probe qPCR) (Takara) at the recommended amounts, and qPCR was performed on a CFX96 real-time PCR instrument (Bio-Rad, United States) in triplicate. The cycling conditions were as follows: initial denaturation, 5 min at 95°C; extension, 40 cycles of 15 s at 95°C and 35 s at 60°C. Fluorescence was quantitated at the end of each extension step.
Previously, D. fangzhongdai and Pectobacterium spp. (P. aroidearum and P. colocasium) were detected in different fields. In 2022, we first isolated strains of D. fangzhongdai (J) and P. aroidearum (B) from the same field in Lechan, Shaoguang, and isolated P. aroidearum strains (T) from another field in Wujiang, Shaoguang (Figure 1). In 2023, we again sampled diseased taro plants with typical soft rot symptoms from seven different areas of Guangdong Province, China, and isolated 59 strains of Dickeya spp. and 26 strains of Pectobacterium spp. To determine the actual composition of causative agents for taro soft rot, these pathogenic bacteria were processed to taxonomic classification. Among them, the 59 Dickeya strains were all identified as D. fangzhongdai according to their close relationship with two D. fangzhongdai strains, ZXC1 and PL145, these phylogenetic related strains constructed a clade separating from other Dickeya species. The 26 Pectobacterium strains were placed into two subgroups, respectively, represented by P. aroidearum L6 and LJ2 and P. colocasium LJ1, and they were determined as strains from two individual species: P. aroidearum and P. colocasium (Figure 1). Thus, pathogenic strains of different Pectobacterium species occurred in fields, while the pathogens from Dickeya genus were validated as the same species, D. fangzhongdai. Notably, D. fangzhongdai was often isolated from fields where P. aroidearum and P. colocasium survived.
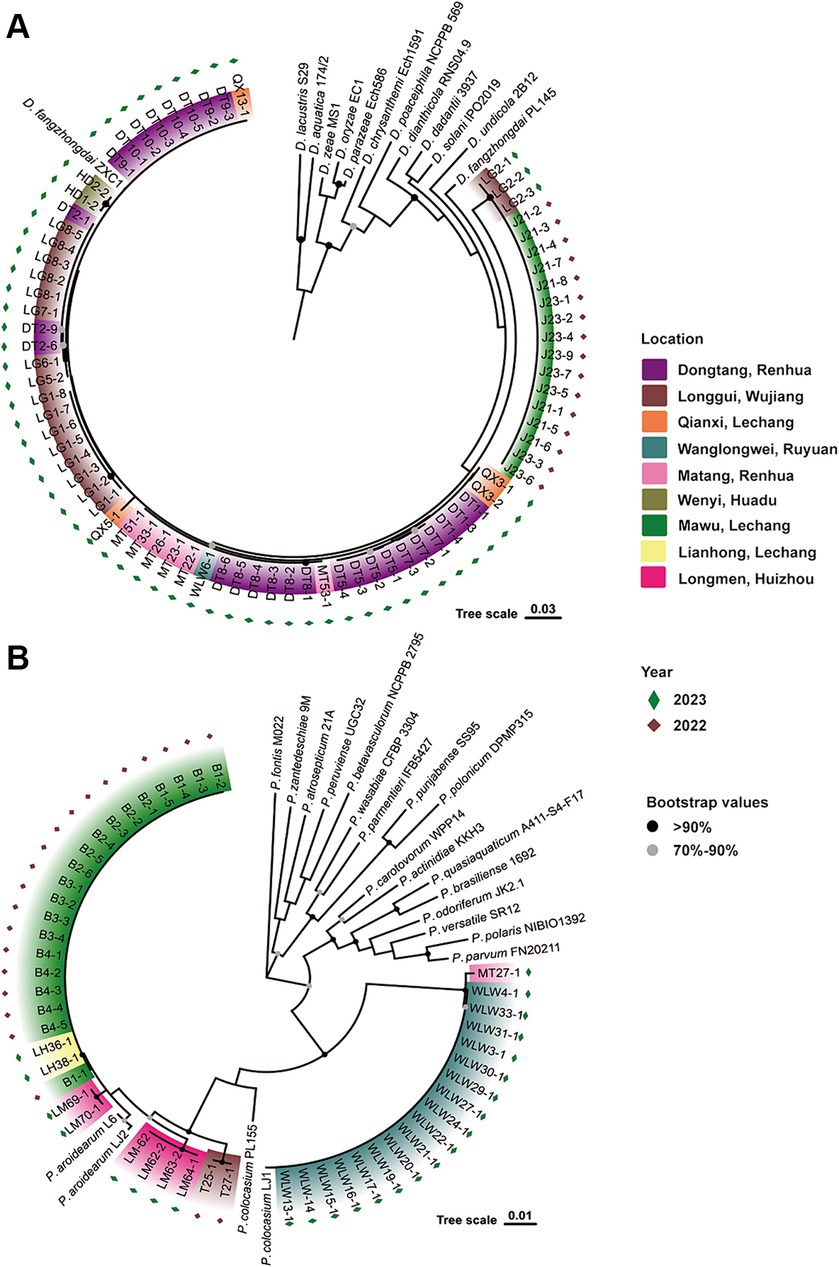
Figure 1. Phylogenetic determination of phytobacteria causing taro soft rot in Guangdong Province, China, from 2022 to 2023. (A) D. fangzhongdai strains along with typical strains of different species in the genus Dickeya. (B) P. aroidearum and P. colocasium strains along with typical strains of different species in the genus Pectobacterium.
Seed-borne pathogenesis is an important pathogenic mode of taro soft rot, and the typical symptoms of this disease are initiated at the mother tubers. The D. fangzhongdai and P. aroidearum strains were first isolated from the same plant in 2022 in this study; these strains were isolated from the developing corms near the ground and from the mother tuber downward, respectively. However, typical symptoms were observed only in the corm above rather than in the mother tuber, which was symptom free (Figure 2A), indicating a consequence of exogenous infection. Unlike SRPs, which cause both blackleg in potato stems and soft rot in tubers, the tested strains including D. fangzhongdai J21-2, P. aroidearum B2-6, and P. colocasium MT27-1 could not cause symptoms in the pseudostems of taro, while typical soft rot symptoms developed after direct injection into the interior of the corms, and invasion in the underground part was the only way in which the disease was initiated (Supplementary Figure S2). Under conditions that simulate exogenous infection in corms, D. fangzhongdai exhibited a much greater ability to decompose corm tissue than did Pectobacterium spp.; similarly, D. fangzhongdai presented much greater virulence on taro than did Dickeya strains isolated from other hosts, including orchids and banana (Figure 2B). Under favorable conditions for bacterial growth (30°C), both D. fangzhongdai and Pectobacterium spp. grew well. When they were grown at a lower temperature (16°C), four strains of P. aroidearum and P. colocasium displayed significantly greater growth density than did the D. fangzhongdai strain after 36 h of culture (p < 0.01), even though the D. fangzhongdai and Pectobacterium spp. strains grew slowly during the first 12 h under this nonoptimal condition (Figure 2C). However, they showed opposite growth differences at 42°C; D. fangzhongdai adapted well to the elevated temperature and could still grow to moderate bacterial densities in the stationary phase, while Pectobacterium spp. developed a much lower population under high-temperature conditions (Figure 2C). Thus, the newly emerging pathogen D. fangzhongdai was highly aggressive toward taro and strongly adaptive at elevated temperatures; therefore, this pathogen should receive increased attention.
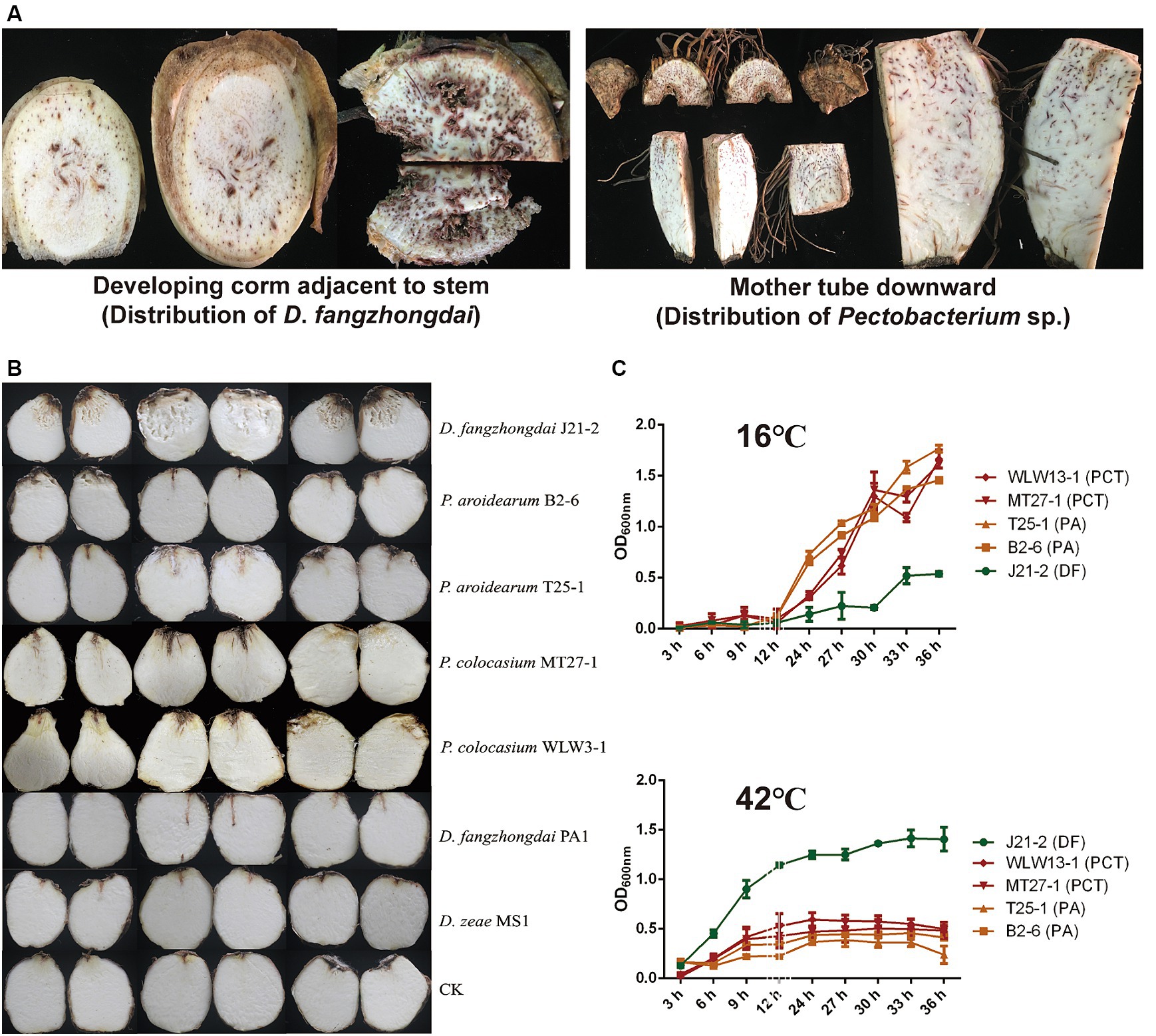
Figure 2. Differences in taro pathogenicity and temperature-dependent growth between D. fangzhongdai and Pectobacterium spp. (A) Symptoms observed in different parts of the same field-collected taro plant and between D. fangzhongdai and P. aroidearum were only isolated from tissues of developing corms and mother tubers, respectively. (B) Differences in virulence between taro corms under conditions of exogenous infection with different SRP strains. (C) Differences in growth of strains of D. fangzhongdai J21-2, P. aroidearum B2-6 and T25-1, and P. colocasium MT27-1 and WLW13-1 under different temperatures.
Validation of the composition of the collected phytobacteria suggested that two different Pectobacterium species (P. aroidearum and P. colocasium) were both important causative agents of the disease (Figure 1). According to phylogenetic analyses of strains from 20 Pectobacterium species based on 12 housekeeping genes (dnaX, dnaN, gyrB, recA, acnA, pgi, proA, mdh, fusA, gapA, rplB, and rpoS), typical strains of P. aroidearum and P. colocasium displayed the closest relationship according to their obvious divergence from 18 other Pectobacterium species (Figure 3A), indicating a combination of genetically close pathogens separating other Pectobacterium species. Since the strains within P. colocasium were only recorded on the taro plants and the species was named after the typical host (Colocasia esculenta), we further investigate their phylogenetical relationship with the closely related species, P. aroidearum. The values of ANI between pairs of P. aroidearum B2-6, LJ2, and T25-1 ranged 0.980–0.985 and values of isDDH ranged 0.825–0.870; P. colocasium MT27-3 and LJ1 presented 0.990 of ANI and 0.924 of isDDH (Figure 3B). However, the values of ANI and isDDH between P. colocasium PL155 from Hawaii and either P. colocasium MT27-1 or LJ1 were approximately 0.92 and 0.43, respectively, as indicated the dramatic dissimilarities in this taro-strain-only species (Figure 3B). According to the classification thresholds of different species (ANI: 0.96, isDDH: 0.70), PL155 from Hawaii could not be grouped into P. colocasium. However, those values were much lower than those between the PL155 and P. aroidearum strains, which were approximately 0.94 and 0.54, respectively (Figure 3B). PL155 presented as an intermediate type linking these two closely related Pectobacterium species and was proposed to be separated as a novel species. This was also supported by both phylogenetic analyses based on whole-genome sequences (Figure 3C). Thus, we suggested that these taro pathogens were grouped into a Pectobacterium complex consisted of three closely related species, which preferred Araceae plants (Nabhan et al., 2013).
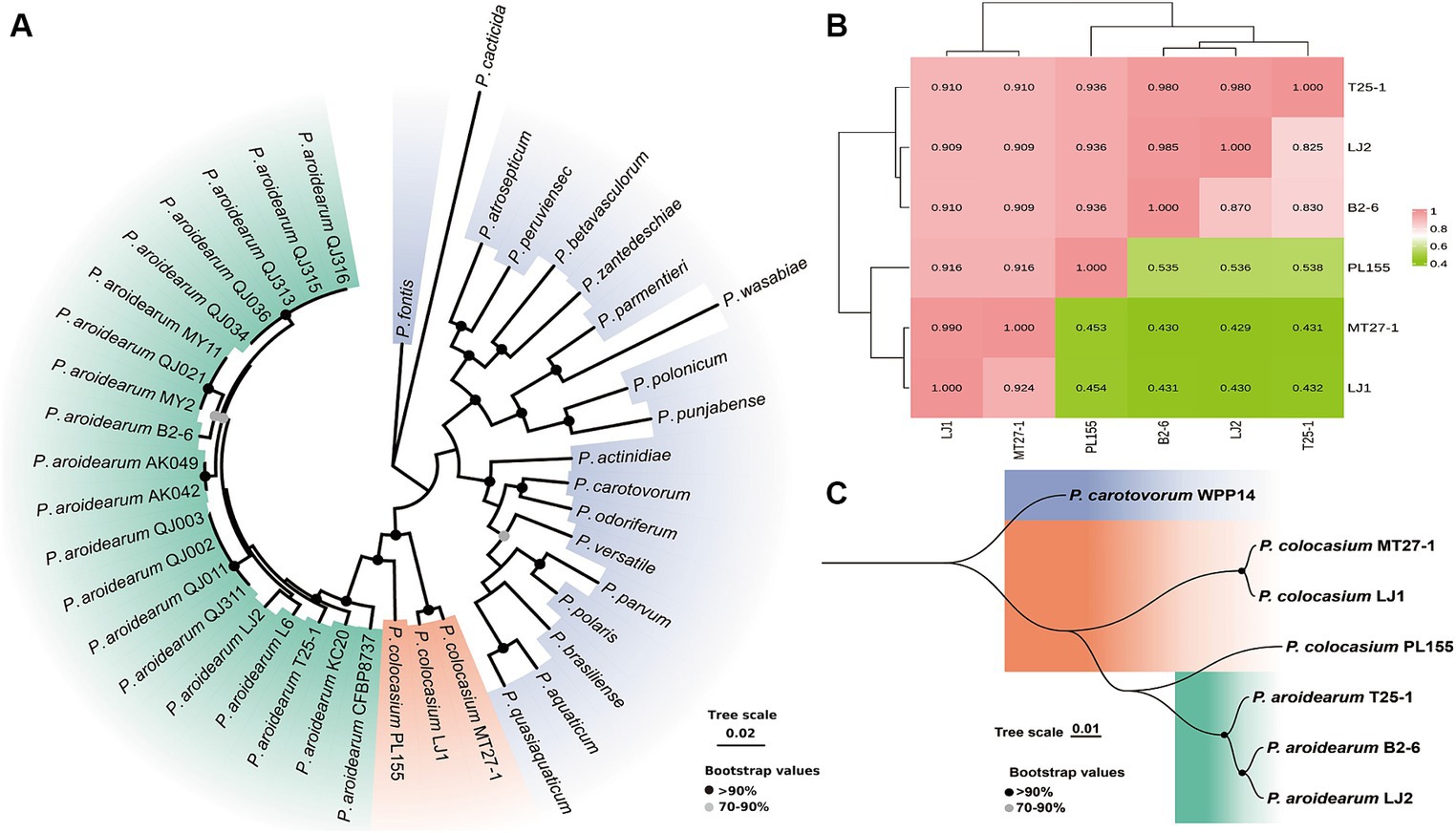
Figure 3. Phylogenetic determination and whole-genome similarities of the Araceae-preferring Pectobacterium complex. (A) Phylogenetic analyses of typical strains of P. aroidearum, P. colocasium and 18 other Pectobacterium species based on 12 housekeeping genes. (B) Heatmap of ANI (numbers on top of the incline) and isDDH values (numbers below the incline) of typical strains of P. aroidearum and P. colocasium. (C) Phylogenetic analyses of typical strains of P. aroidearum and P. colocasium based on whole-genome sequences.
The strong differentiation was not only occurred among P. colocasium, but also found among strains P. aroidearum. Typical strains of P. aroidearum isolated from taro, B2-6 and T25-1, were isolated in the same city but not grouped within a close cluster that could be distinguished from strains isolated from konjak (Amorphophallus konjac). Instead, they presented greater genetic distance than that among strains isolated from these different hosts (Supplementary Figure S3). Synteny analyses between B2-6 and T25-1 revealed a genomic arrangement at the majority of the genomic regions, and they presented only moderate similarities: 89.24% for T25-1 and 91.98% for B2-6 (Supplementary Figure S3). Thus, the pathogen infecting taro had differentiated intensively for a long time to develop a complex consisted of varied species, which however were close in phylogenetic relationship.
Using whole-genome sequences, D. fangzhongdai strains from taro (J21-2, ZXC1, and PL145) were distinct from the groups of typical strains isolated from either orchids or pear trees in the phylogenetic analysis (Figure 4A). The genomes of taro strains were much larger than those of the domestic strains from other hosts (Supplementary Table S1). Synteny alignment indicated that 94.99–95.28% of the genomes of orchid strains were aligned to 91.29–91.88% of the J21-2 genome, and 92.85% of the genomic sequences of pear strains were matched to 90.28% of the J21-2 genome (Supplementary Figure S4). Among the D. fangzhongdai strains available of genome sequences, specific genes within the strains from taro, J21-2, ZXC1, and PL145, were much greater than the numbers in other D. fangzhongdai strains (Supplementary Figure S5; Supplementary Table S2). These results indicated that the taro strains possessed expanded genomes and greatly differed from the domestic strains previously isolated from other hosts.
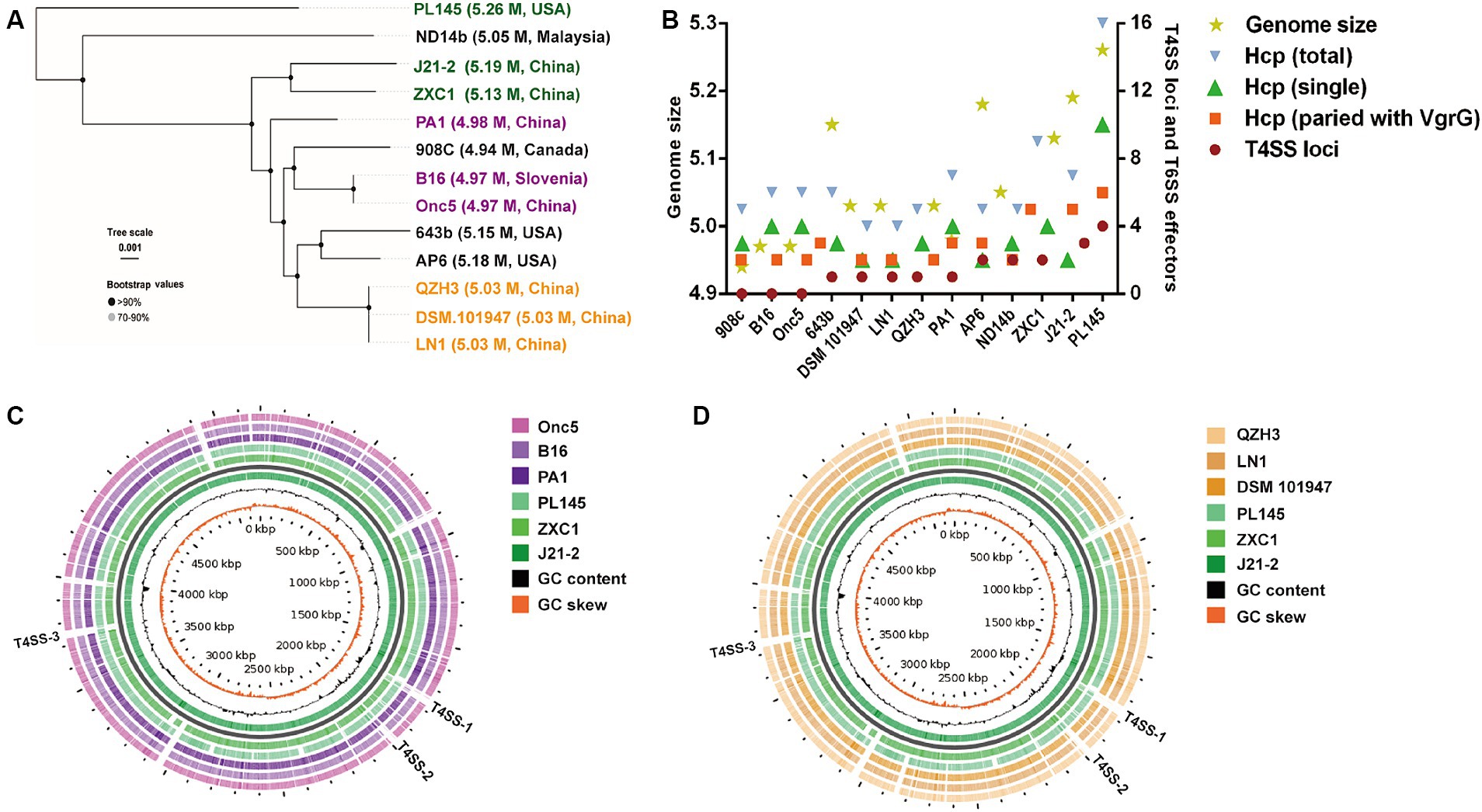
Figure 4. Phylogenetic analysis based on D. fangzhongdai genomes and the presentation of typical features for genomic comparison. (A) Phylogenetic tree of D. fangzhongdai strains isolated from different hosts and different areas. (B) Correlations between the number of T4SS loci and genomic size or the number of T6SS effectors (hcp, hcp-vgrG). (C) Genome BLAST-Atlas analysis between taro strains and orchid strains. (D) Genome BLAST-Atlas analysis between taro strains and pear strains.
Whole-genome comparisons revealed that the number of T4SS loci varied among the D. fangzhongdai genomes, and the correlation between genome size and the number of T4SS loci was significant (r = 0.8908, p < 0.0001) (Figure 4B). BLAST-Atlas analysis revealed that several T4SS gene clusters and accessory genomic components presented in the genomic regions exhibiting large dissimilarities between taro strains and both orchid strains (Figure 4C) and pear strains (Figure 4D). T4SS gene cluster of MPFT family (T4SS-1), virB11-virB1, was conserved among the taro strains (J21-2, ZXC1 and PL145) and three other strains from other hosts or from environment (PA1, AP6, and ND14b), along with mobC encoding relaxase and mobB encoding coupling protein (Figure 5). The taro strains had another T4SS gene cluster (virB11-virB1) of MPFT family at T4SS-2 locus (Figure 5); notably, genes within T4SS-2 locus showed sequence similarities with Pectobacterium strains. Additionally, a different T4SS gene cluster belonging to MPFT family, trbK-trbI, was also found within only taro strains at T4SS-3 locus, including J21-2 and PL145 (Figure 5). These three T4SS clusters of MPFT family were not found in the pear strains, but a T4SS clusters of MPFG family were harbored by these strains; this cluster (tfc) was also found among PL145, AP6 and 643b (Figure 6). More interestingly, a ten-gene array encoding type IV pilus proteins were inserted into this T4SS locus, except for the taro strain PL145 which also presented other difference in the genomic characteristics like the adjacent mobH gene and many specific genes (Figure 6).
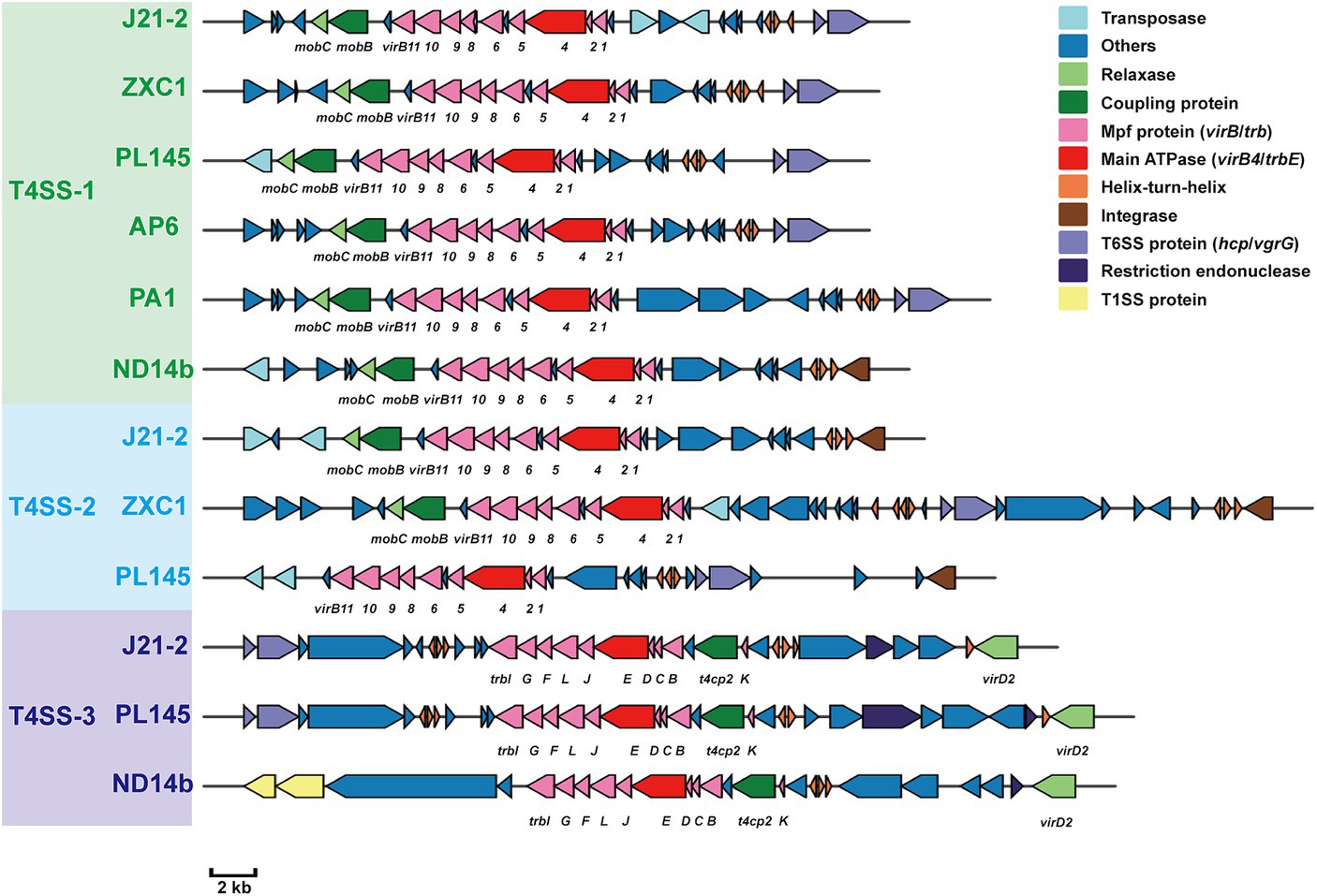
Figure 5. T4SS gene clusters of MPFT family presenting in D. fangzhongdai strains and their upstream and downstream genomic components. Green text of D. fangzhongdai strains indicates the strains containing gene clusters within T4SS-1 locus; blue text of D. fangzhongdai strains indicates the strains containing gene clusters within T4SS-2 locus; purple text of D. fangzhongdai strains indicates the strains containing gene clusters within T4SS-3 locus. Each gene array informs the constituents of MPFT T4SS and the genomic features at upstream and downstream, including elements of HGT and T6SS. The gene symbols below the gene arrays indicate the typical T4SS genes, including genes encoding Mpf proteins (VirB, Trb), main ATPase (VirB4), relaxases (MobC, VirD2/MobP) and coupling proteins (MobB, T4CP2).
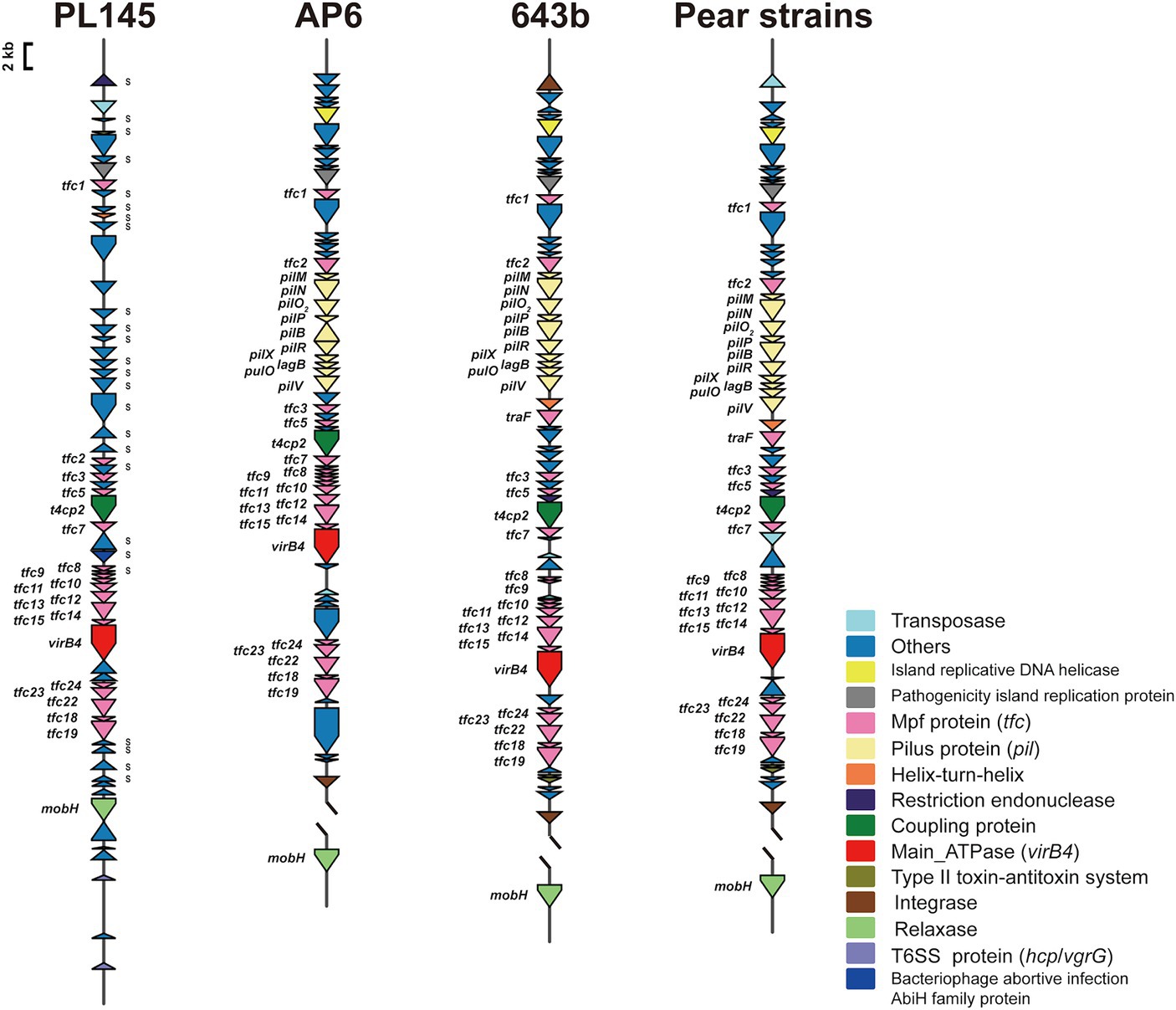
Figure 6. T4SS gene clusters of MPFG family presenting in D. fangzhongdai strains from taro and pear trees. Each gene array informs the constituents of MPFG T4SS and the genomic features at upstream and downstream, including elements of HGT and T6SS. The gene symbols below the gene arrays indicate the typical T4SS genes, including genes encoding Mpf proteins (Tfc), main ATPase (VirB4), relaxases (MobH) and coupling proteins (T4CP2). S attached to the gene cluster indicates specific genes for strain PL145 from taro. Array of genes encoding Pilus (pil) was found between tfc2 and tfc3 of MPFG T4SS of strains isolated from pears, as well AP6 and 643b.
Within this T4SS clusters of MPFG family and its neighborhood genes, PL145 presented a large proportion of specific genes, and 25 of the 61 genes (PL145_16600-PL145_16250) were not found among the other D. fangzhongdai strains (Figure 6). Moreover, 11 of these 25 specific genes were most similar to those in Pectobacterium strains (Supplementary Table S3). These foreign genomic components were probably acquired by horizontal gene transfer (HGT) from related microorganisms since genes encoding transposase, integrase, pathogenicity island replication protein, and bacteriophage abortive infection AbiH protein were distributed within this T4SS loci (Figure 6). Genomic features related to HGT were also common in the three other kinds of T4SS clusters (Figures 5, 6). In addition to PL145, the taro strain J21-2 had 29 IS transposase and 10 integrase genes among 207 specific genes in comparison with other nontaro strains (Supplementary Table S4), and 26 specific genes were also matched to the sequences of Pectobacterium strains, including P. aroidearum and P. colocasium. These results indicated that Pectobacterium strains are important genomic donors for expanded genomes. Variation in the T4SS cluster mediated by HGT could be an effective tool for acquisition of different foreign DNA fragments and bacterial interactions.
In addition to HGT features, T6SS effectors, such as hcp genes or hcp-vgrG pair genes, were found to be closely related to T4SS clusters (Figures 5, 6). We found that the number of hcp-vgrG pair genes was much greater in the taro strains that had the most T4SS loci (correlation r = 0.8397, p < 0.001), especially if they had T4SSs of MPFT family; the number of single hcp genes was also linked to the T4SS loci (Figure 4B). However, the T4SS of MPFG family did not increase the number of T6SS effectors, and we only detected the hcp gene near the T4SS cluster of MPFG family in strain PL145, which does not harbor pilus biogenesis genes (Figure 6).
According to the nine genomes of three different kinds of taro pathogens, more secondary metabolite biosynthesis-related gene clusters were detected within D. fangzhongdai strains (10 clusters) than within both P. aroidearum strains (6 clusters) and P. colocasium strains (5 clusters) by antiSMASH (Supplementary Table S5). Five clusters of P. colocasium were all predicted within P. aroidearum, and either the siderophore or betalactone cluster was missing in P. colocasium. Most of those clusters in P. aroidearum and P. colocasium were also found among D. fangzhongdai strains from taro, but the latter harbored other distinct types of secondary metabolite biosynthesis-related gene clusters, such as the zeamine, cyanobactin, and arylpolyene (APE) clusters. Zeamine clusters were conserved among all 13 analyzed D. fangzhongdai strains, and APE clusters were found only within the taro strains and two other non-host-isolated strains, ND14b and 908C. More interestingly, the zeamine clusters and APE clusters in the D. fangzhongdai strains were most similar to those in the rising pathogen Dickeya solani, which frequently infects the important tuberous crop potato and has high virulence (Figure 7). These indicated that taro strains of D. fangzhongdai was genetically close to another recently evolved pathogens from tuberous crops at typical genomic features, like zeamine and APE clusters.
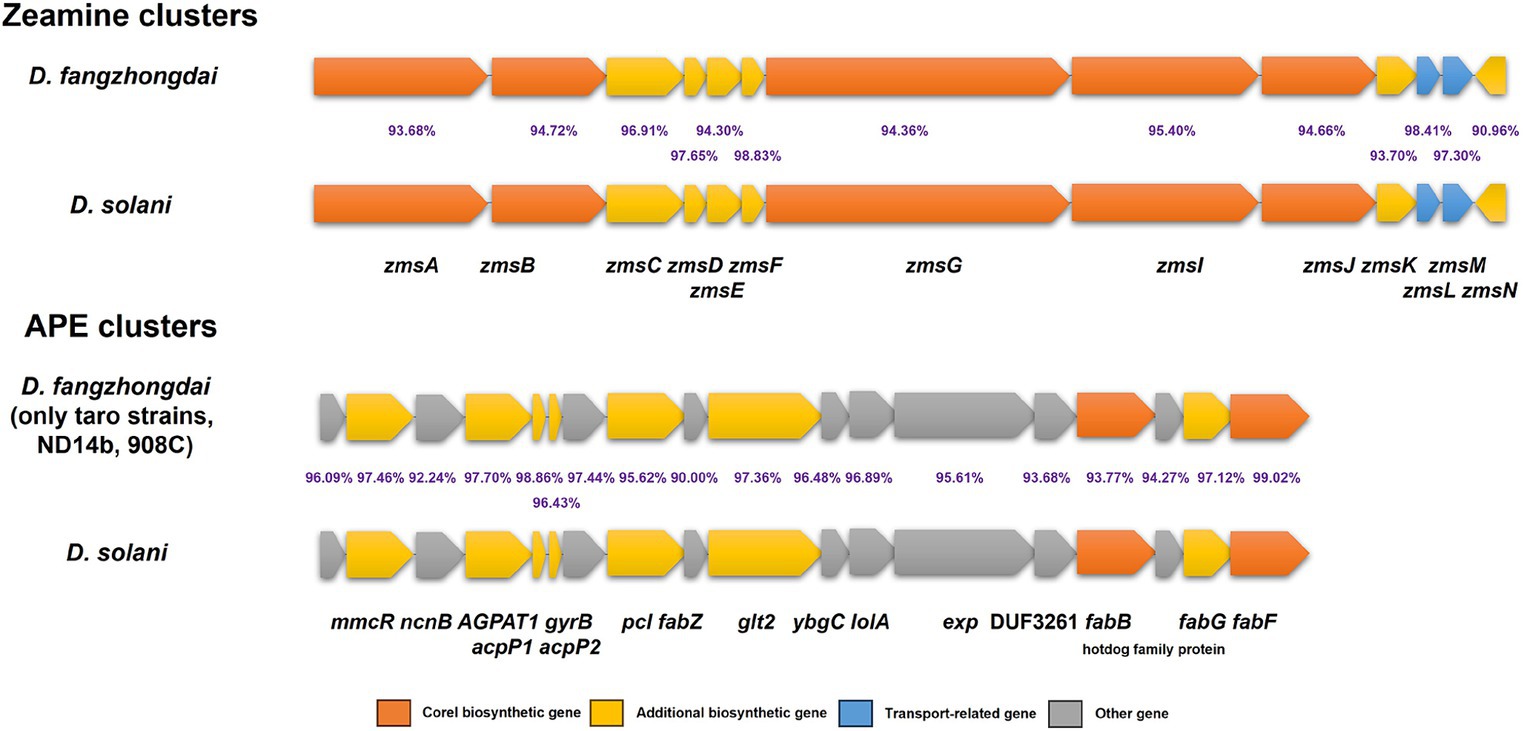
Figure 7. Genomic array of zeamine clusters and arylpolyene (APE) clusters within D. fangzhongdai and D. solani. The percentages between each pair of homologous genes indicate the similarities of protein sequences.
The occurrence of taro soft rot was prevalent in the growing areas, soft rot disease was observed in all nine investigated fields in 2023, and 48 symptomatic or asymptomatic corms were collected (Table 1). Using primers and probes specific to D. fangzhongdai, 24 corms from eight locations were found to contain this pathogen; using primers and probes specific to Pectobacterium spp., a positive reaction was found among 35 corms from eight locations. The newly emerging pathogen D. fangzhongdai, as well as Pectobacterium spp., is prevalent in the taro-growing areas in Guangdong Province. However, the occurrence of Pectobacterium spp. was still more frequent than that of D. fangzhongdai. Among the individual taro corms, we found 14 corms coinfected with D. fangzhongdai and Pectobacterium spp., despite the prevalence of both kinds of pathogens, indicating potential competition in the process of coinfection. Thus, D. fangzhongdai was identified as another important causative agent of taro soft rot but still struggled in the presence of Pectobacterium spp.
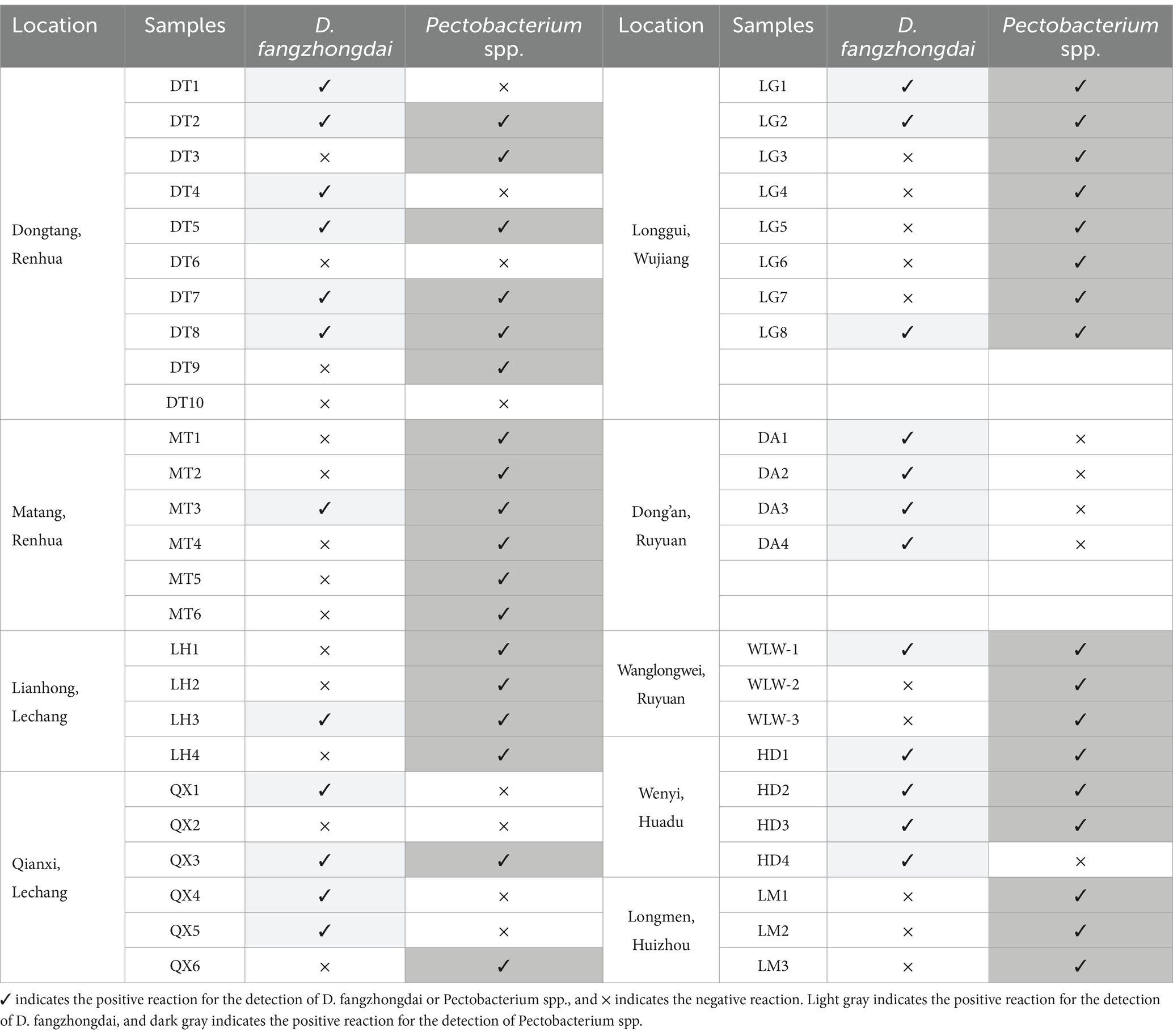
Table 1. qPCR detection of D. fangzhongdai and Pectobacterium spp. in taro corms collected from different growing areas.
Nevertheless, D. fangzhongdai caused soft rot symptoms at the developing corms and their adjacent basal pseudostems more frequently, rather than Pectobacterium spp. Comparison of typical symptoms appearing on the samples collected from Dongtang, Renhua and Longgui, Wujiang at June, 2023, revealed that plants enriching D. fangzhongdai showed severe symptoms at the developing corms adjacent to basal pseudostems with or without the presence of Pectobacterium spp., and the symptoms developed up to the basal pseudostems (Figure 8). In contrast, plants with Pectobacterium spp. were always found to be symptom-free at the developing corms and their adjacent pseudostems in the absence of D. fangzhongdai (Figure 8). These were consistent with the observation of their virulence difference under conditions of exogenous infection.

Figure 8. Symptoms on developing corms and mother tubes of taro plants collected from Renhua and Wujiang, Shaoguan city, in June 2023. DT indicates Dongtang in Renhua, LG indicates Longgui in Wujiang, D indicates detection of D. fangzhongdai pathogens using qPCR, and P indicates detection of Pectobacterium spp. using qPCR. + and – indicate positive and negative, respectively.
Soilborne bacterial diseases seriously affect the growth of various crops, and their causative agents could be combinations of different pathogenic bacteria. In many cases, these pathogens, including SRP pathogens, were found to simultaneously infect the same plant (Fitt et al., 2006). During the occurrence and epidemic spread of SRP, the increase in temperature is an essential driver of the colonization and propagation of emerging pathogens (Degefu et al., 2013; Potrykus et al., 2016). According to the Blue Book Climate Change in China 2023, the period from 2015 to 2022 was the warmest eight-year period on record, and the global average temperature in 2013–2022 increased by 0.05°C compared with that in 2011–2020. Previously, taro soft rot was usually observed in June in Guangdong Province, but the onset period of this disease has shifted to early May in recent years. We further found that the maximum temperatures in May in Shaoguan city, Guangdong, presented an increasing trend (Supplementary Figure S6). The newly emerging causative agent D. fangzhongdai grew much better at elevated temperatures than did Pectobacterium spp. (Figure 2C), indicating that D. fangzhongdai could be more adaptable to warmer climates. The increasing temperatures could drive the succession of these different pectinolytic bacteria and increase the possibility of D. fangzhongdai being a dominant phytobacterium in the field (Toth et al., 2011).
In addition to the seed-borne mode, exogenous infection and related virulence differences among different pathogens could be additional concerns associated with this emerging pathogen. Under simulated conditions of exogenous infection, D. fangzhongdai had a much greater ability to decompose corm tissue than did Pectobacterium spp. and caused typical soft rot symptoms with a soaked appearance (Figure 2B). This difference was consistent with the observations of plant samples collected in different fields, as supported that D. fangzhongdai was more likely to invade externally and cause symptoms on developing corms (Figure 8). In contrast, Pectobacterium spp. more commonly caused infection via the seed-borne mode than did D. fangzhongdai, but the Pectobacterium-positive mother tubers sometimes remained symptomless (Figure 8). Thus, caution should be taken when using D. fangzhongdai for treatment in the taro industry because of the frequency of exogenous infection and its ability to cause initial tissue breakage. Similarly, D. solani was more infectious and adaptable in Europe than were previously colonized Pectobacterium spp., and it was able to cause symptoms under conditions of lower bacterial density (van der Wolf et al., 2014b). D. solani has become the dominant pathogenic bacterium causing potato soft rot in Europe. D. fangzhongdai also causes great losses in the taro industry when present with Pectobacterium spp., and therefore, whether D. fangzhongdai has become the dominant phytobacterium and spread more widely should be evaluated.
SRP pathogens can survive in the reproductive organs or residues in plant soil and surface water and become “indigenous” pathogenic bacteria in local agroecosystems after long-term colonization (Czajkowski et al., 2010, 2011; Ge et al., 2021). In this study, we found typical strains of P. aroidearum and P. colocasium were closest in phylogenetic relationship, but they showed dramatic differentiation in each species. First, P. aroidearum strains have been found on many hosts, but most of them were reported to infect Araceae plants, including taro, konjak, Syngonium podophyllum, Pinellia ternate, and Zantedeschia aethiopica (genomic data from NCBI; Nabhan et al., 2013; Zhou et al., 2022). In particular, in China, P. aroidearum has prevailed in the growth areas of different Araceae tuberous crops, such as taro and konjak, and the genomic dissimilarities among strains from taro were even larger than those across different hosts. Second, the strains from taro-exclusive species, P. colocasium, varied greater in genomic similarities. PL155 from Hawaii should be classified into a novel species based on the species thresholds (ANI: 0.96, isDDH: 0.70), and the novel species was proposed as Pectobacterium hawaiiensis sp. nov. The dramatic genomic differentiation occurred among those causative agents of taro soft rot which were assigned to three species in a complex preferring Araceae plants, it was thus indicated that the ancestor of this Pectobacterium complex had adapted to the taro host for quite a long time and was thus can be considered “indigenous” pathogenic bacteria in taro fields. On the other hand, newly emerging pathogenic agents are considered foreign invaders and interact with the “indigenous” population in pursuit of colonization, as will determine the ability to geographically spread and the possibility of population dominance.
D. dianthicola was once the prevailing causative agent of soft rot in potato in Europe, and subsequently, D. solani emerged in the field; this foreign pathogen successfully colonized and coexisted with D. dianthicola through competitive interactions (Blin et al., 2021). This situation is common among SRP pathogens coinfecting the same host in the same region (Ge et al., 2021; Motyka-Pomagruk et al., 2021). In this study, D. fangzhongdai would also compete with the Pectobacterium complex in the process of infection onset and field colonization for wide-spreading, since they shared similar ecological niches and caused similar symptoms in the host (Czajkowski et al., 2011). However, stronger virulence did not imply that the pathogens were more competitive and would always dominate among coexisting pathogenic populations. The virulence of D. dianthicola was greater than that of P. parmentieri coexisting in the same region, while D. dianthicola was not able to suppress its competitor, and it became the minor pathogenic bacterium. The interactions of different pathogens infecting the same host are complicated, and their succession is not simply determined by differences in virulence. The genomic diversity and typical genomic features of different coexisting SRP pathogens have previously been found to be correlated with their prevalence and virulence (Yap et al., 2004; Kim et al., 2009), and in this study, the genomic comparisons of different kinds of coexisting SRP strains might provide a feasible way to evaluate the prevalence of D. fangzhongdai.
Genomic comparison of available genomes of D. fangzhongdai strains isolated from different hosts and different areas revealed that the acquisition of multiplex T4SSs via HGT increased the genome sizes of the taro strains (Figure 4). As an exceptionally versatile system, the T4SS mediates the transfer of conjugative DNA and protein substrates to recipient cells and DNA uptake from or release into the extracellular milieu (Fronzes et al., 2009) and is necessary for bacterial survival, host-pathogen interactions, and biotic and abiotic adhesion (Costa et al., 2024). The distinctive T4SS and the portable pilus cluster were found to reflect the motility, virulence and biofilm formation of D. fangzhongdai isolated from pear trees and might contribute to its adaptation to woody hosts (Chen et al., 2020). Compared with the typical hosts reported, taro could be considered a distinctive type of host since its vegetative reproduction occurs via the formation of tuberous organs abundant in starch, protein, crude fiber, and polysaccharides, and it was initially infected only in the underground part rather than in the pseudostem (Supplementary Figure S1). Diversification of the T4SS could also have assisted D. fangzhongdai in colonizing a distinctive type of host and introducing more genomic components. Like taro strain-specific T4SS-2 arrays, specific genes that were most similar to those for Pectobacterium strains were well distributed within the genomes of taro strains in comparison with non-taro strains in D. fangzhongdai. The closely related genus Pectobacterium were considered to be the important genomic donors for the delivery of DNA fragments to D. fangzhongdai. These provided evidence that primary infection and successful colonization by D. fangzhongdai involved bacterial interactions with Pectobacterium spp.
Unlike the interactions between phytopathogenic bacteria and host plants, the relationships among pathogenic bacteria coexisting within the same ecological niches have rarely been elucidated (Xin et al., 2018). Competition and cooperation are the main ways in which these pathogenic bacteria interact (Abdullah et al., 2017), and competition is more common than cooperation (Bauer et al., 2018). The production of antimicrobial compounds, contact-dependent growth inhibition (CDI), and quorum sensing are the most intensively studied mechanisms of bacterial competition (Hibbing et al., 2010). Interestingly, we found that the T4SS gene cluster of MPFT always carried T6SS effectors and obviously increased the number of copies of hcp-vgrG pair genes among D. fangzhongdai strains from taro. The T6SS provides an intercellular transport pathway for contact-dependent effectors and mediates bacterial interactions (Schwarz et al., 2010). Hcp and VgrG are substrate and structural components of the T6SS and are structurally related to the T4 bacteriophage tail proteins (Galan and Waksman, 2018); with the aid of this protein pair, various other active toxin effectors are delivered to neighbors directly to contribute to bacterial competition (Russell et al., 2014). For example, the recombination hotspot protein (Rhs) effector in D. dadantii exhibited nuclease activity at the C-terminal toxic domain and was proven to inhibit the growth of adjacent cells (Koskiniemi et al., 2013). A more diverse arsenal of Hcp-VgrG in D. fangzhongdai strains from taro could outperform competitors since they are more likely to transfer distinctive toxin effectors.
As a method of noncontact-dependent growth inhibition, the production of antimicrobial compounds to mediate competition between different species or between related strains of the same species has been verified (Hibbing et al., 2010). Bacteria produce arrays of antimicrobial nonribosomally synthesized peptides (NRPs) and polyketides (PKs), which are typically encoded by adjacent genes. Gene clusters encoding NRPSs and PKSs are common in the SRP genomes, and some of these genes produce metabolites with antibacterial activity (Minowa et al., 2007). Various bacteriocins (carbapenem, carocins, pectocin, pyocin, and carotovoricin) are secreted by Pectobacterium spp. and have been reported to directly inhibit a range of bacteria, including closely related bacteria in both the Pectobacterium genus and Dickeya genus (Van Gijsegem et al., 2021). However, genes encoding these bacteriocins were not predicted among the taro strains of the Pectobacterium complex, while gene arrays encoding other types of NRPSs and PKSs were relatively conserved (Supplementary Table S5). Instead, many more secondary metabolite biosynthesis gene clusters were predicted within D. fangzhongdai from taro, including the zeamine cluster, cyanobactin cluster and APE cluster. In the genus Dickeya, this zeamine cluster was first verified to be involved in the virulence of D. zeae strains from rice (Zhou et al., 2015) and is mainly distributed within D. zeae, D. fangzhongdai, and D. solani. The sequences of the zeamine clusters within D. fangzhongdai were most similar to those within D. solani, and the high GC content (≈61%) suggested that these clusters might have been acquired by HGT (Zhang et al., 2018). The zeamine clusters within D. fangzhongdai shared high similarities with those within D. solani, which might indicate a potential function of competitiveness in the infection onset on the tuberous crops, since D. solani has been documented to compete with previously colonized SRP pathogens when they were coinfecting potato plants. More interestingly, the APE clusters within the taro strains were also most similar to those in D. solani, while there was no homolog within D. fangzhongdai strains from other plants. APE pigments have been found to protect bacteria against oxidative stress and assist them in adapting to the surrounding environment (Grammbitter et al., 2019; Johnston et al., 2021). Thus, the production of secondary metabolites such as zeamine and APE might enhance the competitiveness and adaptation to typical crops and relevant surroundings.
In our experience, it was more difficult to isolate Pectobacterium pathogens from diseased samples than to isolate D. fangzhongdai, especially from samples that were decayed seriously and were parasitized with plenty of saprophytic bacteria, and Pectobacterium pathogens were often not obtained in subcultures since the chosen colonies always contained mixtures of endophytic and environmental bacteria, which could indicate relative competitive weakness in the Pectobacterium complex. Genomic comparison also provided insight into the possible competitive advantage of the newly emerging pathogen D. fangzhongdai in comparison with the “indigenous” population of Pectobacterium spp. Nevertheless, the composition of the pathogenic bacterial population and relative proportions of members were determined not only via evolutionary gains but also via the response to environmental signals. Different pathogens might form a stable community over the long term. In this study, both D. fangzhongdai and Pectobacterium spp. were prevalent in different important taro-growing areas in Guangdong Province, and Pectobacterium spp. were still common (Table 1). In northern Finland, D. solani was also found to coexist with four different Pectobacterium species, and Pectobacterium species also appeared to be more frequent (Degefu, 2021). It is highly likely that D. fangzhongdai and Pectobacterium spp. can coexist in fields for long periods, and the spread of D. fangzhongdai should be monitored and prevented because of its particularity in terms of environmental adaptation, invasion versatility, and evolved arsenal for bacterial interaction. In addition to the use of clean mother tubers, control procedures should be performed earlier, as should preventive measures to reduce external injury and cross-infection. Notably, our pathogen surveillance on potato in early 2024 indicated that P. aroidearum occurred in the fields of Enping City, Guangdong with previously reported causative agents of potato black leg, P. carotovorum (Zhang et al., 2012) and P. brasiliense (Jiang et al., 2019) (data not shown), suggesting a chance of spread of pathogenic bacteria from taro to potato. Thus, the risks of spread to potato cannot be ignored, and this study will also mitigate the risks posed by D. fangzhongdai.
In this study, actual composition of causative agents of taro soft rot were identified to be different SRP bacterial species, including D. fangzhongdai, P. aroidearum, and P. colocasium. The typical Pectobacterium strains varied greatly, exhibiting both intra- and interspecies differences, but they were still phylogenetically close and were considered to cluster into a typical species complex. Compared with these previous colonizing pathogens, D. fangzhongdai emerged later and displayed greater virulence on taro corms under conditions of exogenous infection and better growth at elevated temperature, warranting urgent attention as it could frequently set on developing corms and cause greater loss in fields. Through HGT, D. fangzhongdai strains from taro acquired genomic components of additional T4SSs accompanied by hcp-vgrG genes of the T6SS, which contributed to the expansion of the genome. Among the secondary metabolite biosynthesis gene clusters distinct from those in the Pectobacterium complex, the zeamine and arylpolyene clusters in the D. fangzhongdai strains were both most similar to those in D. solani. Benefiting from these genomic components, the D. fangzhongdai strains could be equipped to compete with the Pectobacterium complex in the field. Accordingly, detection in field samples indicated that D. fangzhongdai prevailed in most of the taro-growing areas in Guangdong Province and coexisted with the Pectobacterium complex. Thus, the newly emerging pathogen D. fangzhongdai requires close attention in different taro-growing areas and in different tuberous crops.
Publicly available datasets were analyzed in this study. This data can be found here: the sequencing read data of strains of D. fangzhongdai J21-2, P. aroidearum B2-6 and T25-1, and P. colocasium MT27-1 are under the BioProject accession no. PRJNA890429 / Sample accession no. SAMN31278287, PRJNA890439 / SAMN31278450, PRJNA890452 / SAMN31278767, PRJNA1050810 / SAMN38754036, respectively. Genome assembly and annotation strains of D. fangzhongdai J21-2, P. aroidearum B2-6 and T25-1, and P. colocasium MT27-1 were deposited in NCBI GenBank under accession nos. CP109768, CP109769, CP109770, and CP140126, respectively.
JZ: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. DS: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation. HS: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation. XP: Writing – review & editing, Writing – original draft, Visualization, Methodology, Investigation, Formal analysis, Data curation. PL: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation. BL: Writing – review & editing, Writing – original draft, Supervision, Project administration, Investigation, Funding acquisition, Conceptualization. QY: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Project administration, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (32372509), the Natural Science Foundation of Guangdong Province (2022A1515010082), and the Project of Collaborative Innovation Center of GDAAS (XTXM202202), and the Construction Project of Modern Agricultural Science and Technology Innovation Alliance of Guangdong Province, China (2023KJ112). The funders had no role in the design of this study, collection, analysis, interpretation of the data, or in writing the manuscript.
We thank Dr. Mohammad Arif from Department of Plant and Environmental Protection Sciences, University of Hawaii at Manoa, United States for the suggestion of pathogenicity tests on taro plants and diagnostic qPCR on collected samples.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1431047/full#supplementary-material
Abdullah, A. S., Moffat, C. S., Lopez-Ruiz, F. J., Gibberd, M. R., Hamblin, J., and Zerihun, A. (2017). Host-multi-pathogen warfare: pathogen interactions in co-infected plants. Front. Plant Sci. 8:1806. doi: 10.3389/fpls.2017.01806
Arizala, D., Dobhal, S., Babler, B., Crockford, A. B., Rioux, R. A., Alvarez, A. M., et al. (2022). Development of a multiplex TaqMan qPCR targeting unique genomic regions for the specific and sensitive detection of Pectobacterium species and P. parmentieri. J. Appl. Microbiol. 132, 3089–3110. doi: 10.1111/jam.15447
Bauer, M. A., Kainz, K., Carmona-Gutierrez, D., and Madeo, F. (2018). Microbial wars: competition in ecological niches and within the microbiome. Microb. Cell 5, 215–219. doi: 10.15698/mic2018.05.628
Bertelli, C., and Brinkman, F. (2018). Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics 34, 2161–2167. doi: 10.1093/bioinformatics/bty095
Blin, P., Robic, K., Khayi, S., Cigna, J., Munier, E., Dewaegeneire, P., et al. (2021). Pattern and causes of the establishment of the invasive bacterial potato pathogen Dickeya solani and of the maintenance of the resident pathogen D. dianthicola. Mol. Ecol. 30, 608–624. doi: 10.1111/mec.15751
Blin, K., Wolf, T., Chevrette, M. G., Lu, X., Schwalen, C. J., Kautsar, S. A., et al. (2017). antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41. doi: 10.1093/nar/gkx319
Buchfink, B., Reuter, K., and Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Cao, Y. N., Sun, Q. H., Feng, Z. W., Handique, U., Wu, J., Li, W. S., et al. (2021). First report of Pectobacterium parmentieri causing blackleg on potato in Inner Mongolia, China. Plant Dis. 105:2237. doi: 10.1094/PDIS-11-20-2502-PDN
Chen, B., Tian, Y. L., Zhao, Y. Q., Wang, Y. J., Chuan, J. C., Li, X., et al. (2020). Genomic characteristics of Dickeya fangzhongdai isolates from pear and the function of type IV pili in the chromosome. J. Integr. Agric. 19, 906–920. doi: 10.1016/S2095-3119(19)62883-2
Chevrette, M. G., Aicheler, F., Kohlbacher, O., Currie, C. R., and Medema, M. H. (2017). SANDPUMA: ensemble predictions of nonribosomal peptide chemistry reveal biosynthetic diversity across Actinobacteria. Bioinformatics 33, 3202–3210. doi: 10.1093/bioinformatics/btx400
Chin, C. S., Peluso, P. F., Sedlazeck, J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
Costa, T., Patkowski, J. B., Mace, K., Christie, P. J., and Waksman, G. (2024). Structural and functional diversity of type IV secretion systems. Nat. Rev. Microbiol. 22, 170–185. doi: 10.1038/s41579-023-00974-3
Czajkowski, R., de Boer, W. J., Velvis, H., and van der Wolf, J. M. (2010). Systemic colonization of potato plants by a soilborne, green fluorescent protein-tagged strain of Dickeya sp. biovar 3. Phytopathology. 100, 134–142. doi: 10.1094/PHYTO-100-2-0134
Czajkowski, R., Pérombelon, M. C. M., van Veen, J. A., and van derWolf, J. M. (2011). Control of blackleg and tuber soft rot of potato caused by Pectobacterium and Dickeya species: a review. Plant Pathology. 60, 999–1013. doi: 10.1111/j.1365-3059.2011.02470.x
Degefu, Y. (2021). Co-occurrence of latent Dickeya and Pectobacterium species in potato seed tuber samples from northern Finland. Agric. Food Sci. 30, 1–7. doi: 10.23986/afsci.101446
Degefu, Y., Potrykus, M., Golanowska, M., Virtanen, E., and Lojkowska, E. (2013). A new clade of Dickeya spp. plays a major role in potato blackleg outbreaks in North Finland. Ann. Appl. Biol. 162, 231–241. doi: 10.1111/aab.12020
DeLude, A., Wells, R., Boomla, S., Chuang, S. C., Urena, F., Shipman, A., et al. (2022). Loop-mediated isothermal amplification (LAMP) assay for specific and rapid detection of Dickeya fangzhongdai targeting a unique genomic region. Sci. Rep. 12:19193. doi: 10.1038/s41598-022-22023-4
Fitt, B. D., Huang, Y. J., van den Bosch, F., and West, J. S. (2006). Coexistence of related pathogen species on arable crops in space and time. Annu. Rev. Phytopathol. 44, 163–182. doi: 10.1146/annurev.phyto.44.070505.143417
Fouts, D. E. (2006). Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 34, 5839–5851. doi: 10.1093/nar/gkl732
Fronzes, R., Christie, P. J., and Waksman, G. (2009). The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7, 703–714. doi: 10.1038/nrmicro2218
Galan, J. E., and Waksman, G. (2018). Protein-injection Machines in Bacteria. Cell 172, 1306–1318. doi: 10.1016/j.cell.2018.01.034
Ge, T., Ekbataniamiri, F., Johnson, S. B., Larkin, R. P., and Hao, J. (2021). Interaction between Dickeya dianthicola and Pectobacterium parmentieri in potato infection under field conditions. Microorganisms 9:316. doi: 10.3390/microorganisms9020316
Goel, M., Sun, H., Jiao, W. B., and Schneeberger, K. (2019). SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20:277. doi: 10.1186/s13059-019-1911-0
Grammbitter, G., Schmalhofer, M., Karimi, K., Shi, Y. M., Schoner, T. A., Tobias, N. J., et al. (2019). An uncommon type II PKS catalyzes biosynthesis of aryl polyene pigments. J. Am. Chem. Soc. 141, 16615–16623. doi: 10.1021/jacs.8b10776
Gross, D. C., Powelson, M. L., Regner, K., and Radamaker, G. K. (1991). A bacteriophage-typing system for surveying the diversity and distribution of strains of Erwinia carotovora in potato fields. Phytopathology 81, 220–226. doi: 10.1094/phyto-81-220
Guglielmini, J., Bertrand, N., Abby, S. S., Pilar Garcillan-Barcia, M., de la Cruz, F., and Rocha, E. (2014). Key components of the eight classes of type IV secretion systems involved in bacterial conjugation or protein secretion. Nucleic Acids Res. 42, 5715–5727. doi: 10.1093/nar/gku194
Handique, U., Cao, Y., Wang, D., Zhang, R., Li, W., Sun, Q., et al. (2022). First report of Pectobacterium punjabense causing blackleg and soft rot on potato in Hebei and Fujian Province, China. Plant Dis. 106:1977. doi: 10.1094/PDIS-12-21-2731-PDN
Hibbing, M. E., Fuqua, C., Parsek, M. R., and Peterson, S. B. (2010). Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 8, 15–25. doi: 10.1038/nrmicro2259
Huang, S. F., Chen, Z. Q., Hu, M., Xue, Y., Liao, L. S., and Zhang, L. H. (2021). First report of bacterial soft rot disease on Taro caused by Dickeya fangzhongdai in China. Plant Dis. 105:3737. doi: 10.1094/PDIS-10-20-2225-PDN
Jiang, S. B., Lin, B. R., Yang, Q. Y., Zhang, J. X., Shen, H. F., Pu, X. M., et al. (2019). First report of bacterial soft rot of potato caused by Pectobacterium carotovorum subsp. brasiliense in Guangdong Province, China. Plant Dis. 103:2468. doi: 10.1094/PDIS-03-19-0653-PDN
Johnston, I., Osborn, L. J., Markley, R. L., McManus, E. A., Kadam, A., Schultz, K. B., et al. (2021). Identification of essential genes for Escherichia coli aryl polyene biosynthesis and function in biofilm formation. NPJ Biofilms Microbiomes 7:56. doi: 10.1038/s41522-021-00226-3
Kim, H. S., Ma, B., Perna, N. T., and Charkowski, A. O. (2009). Phylogeny and virulence of naturally occurring type III secretion system-deficient Pectobacterium strains. Appl. Environ. Microbiol. 75, 4539–4549. doi: 10.1128/AEM.01336-08
Koskiniemi, S., Lamoureux, J. G., Nikolakakis, K. C., T'Kint, D. R. C., Kaplan, M. D., Low, D. A., et al. (2013). Rhs proteins from diverse bacteria mediate intercellular competition. Proc. Natl. Acad. Sci. USA 110, 7032–7037. doi: 10.1073/pnas.1300627110
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Li, H. W., Lin, Z. J., Luo, W. B., Xu, G. C., Xu, Y. Q., Ji, R. C., et al. (2021). Identification and analysis of genetic diversity of the pathogen causing potato blackleg disease in Fujian Province. J. Plant Prot. 48, 305–313. doi: 10.13802/j.cnki.zwbhxb.2021.2020272
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Ma, X., Bonasera, J. M., Asselin, J. A. E., Beer, S. V., and Swingle, B. (2019). First report of Dickeya fangzhongdai causing soft rot of onion in New York state. Plant Dis. 104:1251. doi: 10.1094/PDIS-09-19-1940-PDN
Ma, B., Hibbing, M. E., Kim, H. S., Reedy, R. M., Yedidia, I., Breuer, J., et al. (2007). Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya. Phytopathology 97, 1150–1163. doi: 10.1094/PHYTO-97-9-1150
Ma, X., Schloop, A., Swingle, B., and Perry, K. L. (2018). Pectobacterium and Dickeya responsible for potato blackleg disease in New York state in 2016. Plant Dis. 102, 1834–1840. doi: 10.1094/PDIS-10-17-1595-RE
Mansfield, J., Genin, S., Magori, S., Citovsky, V., Sriariyanum, M., Ronald, P., et al. (2012). Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13, 614–629. doi: 10.1111/j.1364-3703.2012.00804.x
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., and Goker, M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Minowa, Y., Araki, M., and Kanehisa, M. (2007). Comprehensive analysis of distinctive polyketide and nonribosomal peptide structural motifs encoded in microbial genomes. J. Mol. Biol. 368, 1500–1517. doi: 10.1016/j.jmb.2007.02.099
Motyka-Pomagruk, A., Zoledowska, S., Sledz, W., and Lojkowska, E. (2021). The occurrence of bacteria from different species of Pectobacteriaceae on seed potato plantations in Poland. Eur. J. Plant Pathol. 159, 309–325. doi: 10.1007/s10658-020-02163-x
Nabhan, S., De Boer, S. H., Maiss, E., and Wydra, K. (2013). Pectobacterium aroidearum sp. nov., a soft rot pathogen with preference for monocotyledonous plants. Int. J. Syst. Evol. Microbiol. 63, 2520–2525. doi: 10.1099/ijs.0.046011-0
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Portier, P., Pedron, J., Taghouti, G., Fischer-Le, S. M., Caullireau, E., Bertrand, C., et al. (2019). Elevation of Pectobacterium carotovorum subsp. odoriferum to species level as Pectobacterium odoriferum sp. Nov., proposal of Pectobacterium brasiliense sp. Nov. and Pectobacterium actinidiae sp. Nov., emended description of Pectobacterium carotovorum and description of Pectobacterium versatile sp. Nov., isolated from streams and symptoms on diverse plants. Int. J. Syst. Evol. Microbiol. 69, 3207–3216. doi: 10.1099/ijsem.0.003611
Potrykus, M., Golanowska, M., Sledz, W., Zoledowska, S., Motyka, A., Kolodziejska, A., et al. (2016). Biodiversity of Dickeya spp. isolated from potato plants and water sources in temperate climate. Plant Dis. 100, 408–417. doi: 10.1094/PDIS-04-15-0439-RE
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/c5ay02550h
Rossmann, S., Dees, M. W., Perminow, J., Meadow, R., and Brurberg, M. B. (2018). Soft rot Enterobacteriaceae are carried by a large range of insect species in potato fields. Appl. Environ. Microbiol. 84, e00281–e00218. doi: 10.1128/AEM.00281-18
Russell, A. B., Peterson, S. B., and Mougous, J. D. (2014). Type VI secretion system effectors: poisons with a purpose. Nat. Rev. Microbiol. 12, 137–148. doi: 10.1038/nrmicro3185
Schwarz, S., Hood, R. D., and Mougous, J. D. (2010). What is type VI secretion doing in all those bugs? Trends Microbiol. 18, 531–537. doi: 10.1016/j.tim.2010.09.001
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tian, Y. L., Zhao, Y. Q., Chen, B. H., Chen, S., Zeng, R., Hu, B. S., et al. (2020). Real-time PCR assay for detection of Dickeya fangzhongdai causing bleeding canker of pear disease in China. J. Integr. Agric. 19, 898–905. doi: 10.1016/S2095-3119(19)62881-9
Tian, Y., Zhao, Y., Yuan, X., Yi, J., Fan, J., Xu, Z., et al. (2016). Dickeya fangzhongdai sp. nov., a plant-pathogenic bacterium isolated from pear trees (Pyrus pyrifolia). Int. J. Syst. Evol. Microbiol. 66, 2831–2835. doi: 10.1099/ijsem.0.001060
Toth, I. K., van der Wolf, J. M., Saddler, G., Lojkowska, E., Hélias, V., Pirhonen, M., et al. (2011). Dickeya species: an emerging problem for potato production in Europe. Plant Pathol. 60, 385–399. doi: 10.1111/j.1365-3059.2011.02427.x
van der Wolf, J. M., de Haan, E. G., Kastelein, P., Krijger, M., de Haas, B. H., Velvis, H., et al. (2017). Virulence of Pectobacterium carotovorum subsp. brasiliense on potato compared with that of other Pectobacterium and Dickeya species under climatic conditions prevailing in the Netherlands. Plant Pathol. 66, 571–583. doi: 10.1111/ppa.12600
van der Wolf, J. M., Kastelein, P., Förch, M. G., and Krijger, M. C. (2014b). “The risks of air-borne inoculum on infections of tubers with Dickeya solani and Pectobacterium wasabiae” in 2nd Euphresco II Dickeya and Pectobacterium meeting. Available at: https://edepot.wur.nl/330892
van der Wolf, J. M., Nijhuis, E. H., Kowalewska, M. J., Saddler, G. S., Parkinson, N., Elphinstone, J. G., et al. (2014a). Dickeya solani sp. nov., a pectinolytic plant-pathogenic bacterium isolated from potato (Solanum tuberosum). Int. J. Syst. Evol. Microbiol. 64, 768–774. doi: 10.1099/ijs.0.052944-0
Van Gijsegem, F., Hugouvieux-Cotte-Pattat, N., Kraepiel, Y., Lojkowska, E., Moleleki, L. N., Gorshkov, V., et al. (2021). “Molecular interactions of Pectobacterium and Dickeya with plants” in Plant diseases caused by Dickeya and Pectobacterium species, eds. F. Gijsegem Van, J. M. Wolf van der, and I. K. Toth Switzerland: Springer International Publishing, 85–147.
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Xin, X. F., Kvitko, B., and He, S. Y. (2018). Pseudomonas syringae: what it takes to be a pathogen. Nat. Rev. Microbiol. 16, 316–328. doi: 10.1038/nrmicro.2018.17
Yang, D., Du, C. J., Ye, Y. F., Pan, L. F., Zhang, J., and Fu, G. (2022). First report of Dickeya fangzhongdai causing peduncle soft rot of Banana in China. Plant Dis. 106:1980. doi: 10.1094/PDIS-11-21-2513-PDN
Yap, M. N., Barak, J. D., and Charkowski, A. O. (2004). Genomic diversity of Erwinia carotovora subsp. carotovora and its correlation with virulence. Appl. Environ. Microbiol. 70, 3013–3023. doi: 10.1128/AEM.70.5.3013-3023.2004
Zhang, J. X., Hu, J., Shen, H. F., Zhang, Y. C., Sun, D. Y., Pu, X. M., et al. (2018). Genomic analysis of the Phalaenopsis pathogen Dickeya sp. PA1, representing the emerging species Dickeya fangzhongdai. BMC Genomics 19:782. doi: 10.1186/s12864-018-5154-3
Zhang, J. X., Lin, B. R., Shen, H. F., Pu, X. M., Chen, Z. N., and Feng, J. J. (2012). First report of bacterial soft rot of potato caused by Pectobacterium carotovorum subsp. carotovorum in Guangdong Province of China. Plant Dis. 96:1819. doi: 10.1094/PDIS-07-12-0620-PDN
Zhou, J. N., Cheng, Y. Y., Lv, M. F., Liao, L. S., Chen, Y. F., Gu, Y. F., et al. (2015). The complete genome sequence of Dickeya zeae EC1 reveals substantial divergence from other Dickeya strains and species. BMC Genomics 16:571. doi: 10.1186/s12864-015-1545-x
Keywords: Dickeya fangzhongdai, Pectobacterium species complex, coinfection, virulence difference, genomic comparison
Citation: Zhang J, Sun D, Shen H, Pu X, Liu P, Lin B and Yang Q (2024) Dickeya fangzhongdai was prevalent and caused taro soft rot when coexisting with the Pectobacterium complex, with a preference for Araceae plants. Front. Microbiol. 15:1431047. doi: 10.3389/fmicb.2024.1431047
Edited by:
Ravinder Kumar, Indian Agricultural Research Institute (ICAR), IndiaReviewed by:
Huiqiao Pan, Carnegie Institution for Science, United StatesCopyright © 2024 Zhang, Sun, Shen, Pu, Liu, Lin and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingxin Zhang, Y2hvdWd1QDEyNi5jb20=; Qiyun Yang, eWFuZ3FpeXVuQGdkYWFzLmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.