 Marte Glambek1
Marte Glambek1 Steinar Skrede1,2
Steinar Skrede1,2 Audun Sivertsen3Bård Reiakvam Kittang2,4
Audun Sivertsen3Bård Reiakvam Kittang2,4 Alba Kaci5Christine Monceyron Jonassen5,6Hannah Joan Jørgensen7Norwegian Study Group on Streptococcus dysgalactiae
Alba Kaci5Christine Monceyron Jonassen5,6Hannah Joan Jørgensen7Norwegian Study Group on Streptococcus dysgalactiae Oddvar Oppegaard1,2*
Oddvar Oppegaard1,2*- 1Department of Medicine, Haukeland University Hospital, Bergen, Norway
- 2Department of Clinical Medicine 2, Department of Clinical Science, University of Bergen, Bergen, Norway
- 3Department of Microbiology, Haukeland University Hospital, Bergen, Norway
- 4Department of Internal Medicine, Haraldsplass Deaconess Hospital (HDS), Bergen, Norway
- 5Center for Laboratory Medicine, Østfold Hospital, Grålum, Norway
- 6Department of Virology, Division of Infection Control and Environmental Health, Norwegian Institute of Public Health, Oslo, Norway
- 7Norwegian Veterinary Institute, Ås, Norway
Background: Streptococcus dysgalactiae (SD) is an important pathogen in humans as well as in a broad range of animal species. Escalating rates of antibiotic resistance in SD has been reported in both human and veterinary clinical practice, but the dissemination of resistance determinants has so far never been examined in a One Health Perspective. We wanted to explore the occurrence of zoonotic transmission of SD and the potential for exchange of resistance traits between SD from different host populations.
Methods: We compared whole genome sequences and phenotypical antimicrobial susceptibility of 407 SD isolates, comprising all isolates obtained from human bloodstream infections in 2018 (n = 274) and available isolates associated with animal infections from the years 2018 and 2019 (n = 133) in Norway.
Results: Antimicrobial resistance genes were detected in 70 (26%), 9 (25%) and 2 (2%) of the isolates derived from humans, companion animals and livestock, respectively. Notably, distinct host associated genotypic resistomes were observed. The erm(A) gene was the dominant cause of erythromycin resistance in human associated isolates, whereas only erm(B) and lsa(C) were identified in SD isolates from animals. Moreover, the tetracycline resistance gene tet(O) was located on different mobile genetic elements in SD from humans and animals. Evidence of niche specialization was also evident in the phylogenetic analysis, as the isolates could be almost perfectly delineated in accordance with host species. Nevertheless, near identical mobile genetic elements were observed in four isolates from different host species including one human, implying potential transmission of antibiotic resistance between different environments.
Conclusion: We found a phylogenetic delineation of SD strains in line with host adapted populations and niche specialization. Direct transmission of strains or genetic elements carrying resistance genes between SD from different ecological niches appears to be rare in our geographical region.
Introduction
Streptococcus dysgalactiae (SD) causes a broad spectrum of human infections ranging from asymptomatic carriage via non-invasive soft tissue infections to life threatening conditions like necrotizing fasciitis and toxic shock syndrome (Brandt and Spellerberg, 2009). In the past decades there has been a significant increase in invasive infections in humans caused by SD in several geographical regions, and SD is currently among the most common pathogens detected in bloodstream infections in some countries (Couture-Cossette et al., 2018; UK Health Security Agency, 2022; Nevanlinna et al., 2023; Oppegaard et al., 2023).
SD is not a strict human pathogen, but capable of infecting a broad range of host species. It is recognized as a major cause of bovine mastitis, arthritis in swine and ovine lambs and necrotic ulcers in fish in aquaculture (Abdelsalam et al., 2010; Moreno et al., 2016; Zhang et al., 2018; Smistad et al., 2021). Moreover, SD is associated with a variety of infections in dogs, cats, and horses, underpinning the ecological versatility of this pathogen (Acke et al., 2015). At the same time, a phylogenetic diversity is evident within the SD taxon, and likely extends beyond the current delineation into the subspecies Streptococcus dysgalactiae subsp. dysgalactiae (SDSD) and Streptococcus dysgalactiae subsp. equisimilis (SDSE) (Ciszewski et al., 2016; Alves-Barroco et al., 2022). SDSD is, by definition, restricted to α-hemolytic group C strains, predominantly infecting cattle and sheep, whereas SDSE comprises all β-hemolytic strains (Vieira et al., 1998).
Penicillin remains a cornerstone in the treatment of infections caused by SD, and resistance to this antibiotic is exceedingly rare. Regarding second line alternatives, however, the situation is more alarming. Rising rates of macrolide, lincosamide and streptogramin (MLS) resistance have been noted, and in the United Kingdom MLS resistance is approaching 40% in SD isolates collected from humans (UK Health Security Agency, 2022). Even higher rates have been reported in bovine associated SD in China, as well as SD isolated from swine in South America (Moreno et al., 2016; Zhang et al., 2018). Moreover, increasing numbers of tetracycline resistant pyogenic streptococci are observed, and, as for the MLS resistance, this trend seems independent of host species (Abdelsalam et al., 2010; Ciszewski et al., 2016; Garch et al., 2020).
The main drivers for increasing antibiotic resistance are selection of resistant microbes by use and overuse of antibiotics, the possibility of horizontal genetic transfer of resistance traits between bacteria, and the spread of resistant bacteria between different geographic and ecological environments, both locally and globally. The complex ecological processes call for collaboration of multiple science fields in a “One health perspective” to approach and overcome emerging antibiotic resistance (McEwen and Collignon, 2018). However, our current knowledge on antimicrobial resistance in SD is predominantly based on studies limited to distinct host reservoirs, and data on dissemination of resistance determinants between ecological niches is scarce. Hence, there is a need for comparative studies on contemporary and spatially related isolates from different host species to investigate possible pathways for spreading of resistance traits.
We sought to explore antimicrobial resistance of SD in a One Health perspective and have examined clinically relevant SD isolates collected from various host species within a confined temporal and geographical setting. By dissecting phenotypic susceptibility patterns and whole genome sequences, we compared the resistomes associated with the different host specific reservoirs and attempted to elucidate possible routes for spread of resistance traits.
Methods
Bacterial isolates
All SD isolates identified in human blood cultures in Norway during 2018 were collected as part of the Norwegian surveillance program for antimicrobial resistance (NORM, 2019). SD isolates from bovine sources were randomly selected among isolates from bovine mastitis by TINE SA mastitis laboratory (Molde, Norway) in 2018. Additionally, all SD isolates from clinical samples from animals submitted to the Norwegian Veterinary Institute in 2018 and 2019, were included. Only one isolate per person and one isolate per animal flock or herd was included.
Species identification in the primary laboratories was based on colony morphology (hemolytic reaction on 5% sheep blood agar and colony size >0.5 mm after 24 h of incubation), serogroup specificity using rapid Lancefield agglutination test, and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). All isolates identified as SD were submitted to either Haukeland University Hospital, Bergen, or Østfold Hospital Trust, Grålum, for susceptibility testing and genomic characterization.
Susceptibility testing
All isolates were examined for susceptibility to benzylpenicillin, tetracycline, erythromycin, clindamycin, and trimethoprim-sulphamethoxazole according to the NORM protocol (NORM, 2019). Briefly, isolates were plated on Mueller-Hinton agar supplemented with defibrinated horse blood and β-NAD, and minimum inhibitory concentrations (MIC) were determined using MIC gradient strips. The Kirby-Bauer double disc diffusion method was used to assign the constitutive macrolide-lincosamide-streptogramin (cMLS), the inducible MLS (iMLS) and the macrolide (M) resistance phenotypes. Clinical breakpoints (version 14.0) set by the European Committee for Antimicrobial Susceptibility Testing (EUCAST) were used for interpretation of susceptibility.
Whole genome sequencing
Whole Genome Sequencing of 119 SD of human origin as well as all 133 SD from animals was performed at Haukeland University Hospital on an Illumina 4,000 HiSeq system to produce 150 bp paired end reads, as previously described (Oppegaard et al., 2017). The remaining 155 human isolates were sequenced at Østfold Hospital Trust by the Ion Torrent technology on an Ion S5XL system as previously described (Kaci et al., 2023).
In silico analysis
For data generated on the Illumina HiSeq system, reads were trimmed with Trimmomatic v0.39 (Bolger et al., 2014). For Ion Torrent generated data, reads were processed with the incorporated S5 software plug-ins. All trimmed reads from the sequenced isolates were de novo assembled by SPAdes v5.14 (Bankevich et al., 2012). Genome annotation was accomplished using RAST v1.073 (Aziz et al., 2008). Species identity was confirmed by 16S rDNA analysis. A core genome single-nucleotide polymorphism phylogeny was generated by CSI Phylogeny at the Center for Genomic Epidemiology1 using default settings and the SDSE type strain NCTC13762 as a reference. The resulting maximum likelihood phylogenetic tree was visualized and annotated using the Interactive Tree of Life platform, iTol v6 (Letunic and Bork, 2021).
Multilocus sequence typing (MLST) of the isolates was performed using the MLST 2.0 software available at the CGE webpage. The emm-database at the Centers for Disease Control and Prevention webpage was used to determine the emm-types.2 A minimum spanning phylogenetic tree using MLST types was constructed using the Phyloviz online tool,3 using triple-locus variant limitation for clustering.
RESfinder was used to screen for the presence of resistance genes (Florensa et al., 2022). Geneious Prime v 2022.2 was used to inspect the genetic context of the resistance genes, and screen for known mobilization genes from streptococcal mobile genetic elements using a database adapted form CONJdb.4 BLASTn was used to search for closest matches to putative mobile elements.
There are no validated methods for genotypic distinction between the two subspecies SDSE and SDSD. In accordance with the phenotypic definition proposed by Vieira et al. (1998), we defined SDSD in silico as genomes harboring the Lancefield group C-antigen operon, lacking the streptolysin S operon (corresponding to an α- or nonhemolytic reaction on blood agar), and lacking the streptokinase gene (inferring that streptokinase activity on human plasminogen does not occur). All other genomes were classified as SDSE.
Statistical analysis
Differences in resistance rates between the host associated SD populations were tested for statistical significance by Fisher’s exact test. A two-sided p-value <0.05 was considered significant. Due to the low number of isolates available from some host associated populations, the data were pooled into isolates derived from humans, companion animals (dog and horse) and livestock (cow, sheep, swine) for statistical analysis.
Results
A total of 407 SD isolates were included in the study (Supplementary Table S1). Of these, 274 were isolated from human blood cultures in 2018, constituting all SD isolates registered in the national surveillance program this year (NORM, 2019). Among the 133 animal associated isolates, 97 originated from livestock, including cattle (n = 74), sheep (n = 11) and swine (n = 12). The remaining 36 isolates were from dogs (n = 20) and horses (n = 16).
Whole genome sequencing and phylogenetic analyses
The draft genomes of the 407 SD isolates had an average assembly length of 2.10 Mb, GC content of 39.3%, 2,100 protein encoding genes, and a coverage of approximately 220x. Based on whole genome sequencing analysis, 83 out of 85 isolates from bovine and ovine sources were classified as SDSD, whereas all other isolates belonged to the subspecies equisimilis. This phylogenetic delineation was in line with the phenotypic species identification.
Phylogenetic analysis delineated the isolates largely in accordance with host species (Figure 1). A notable exception was the distinct cluster corresponding to SDSD, where the isolates derived from bovine and ovine hosts were phylogenetically inseparable. The large clade of human associated isolates was clearly demarcated from the SD isolated from different animals. Nevertheless, indications of SD isolates crossing species barriers were observed. One isolate obtained from a human blood culture clustered phylogenetically with dog associated isolates. Inversely, five isolates obtained from dogs were found scattered in the cluster of isolates from humans, comprising 25% of all dog associated isolates. The minimum spanning tree based on MLST types was congruent with the single nucleotide polymorphism phylogenetic tree (Supplementary Figure S1).
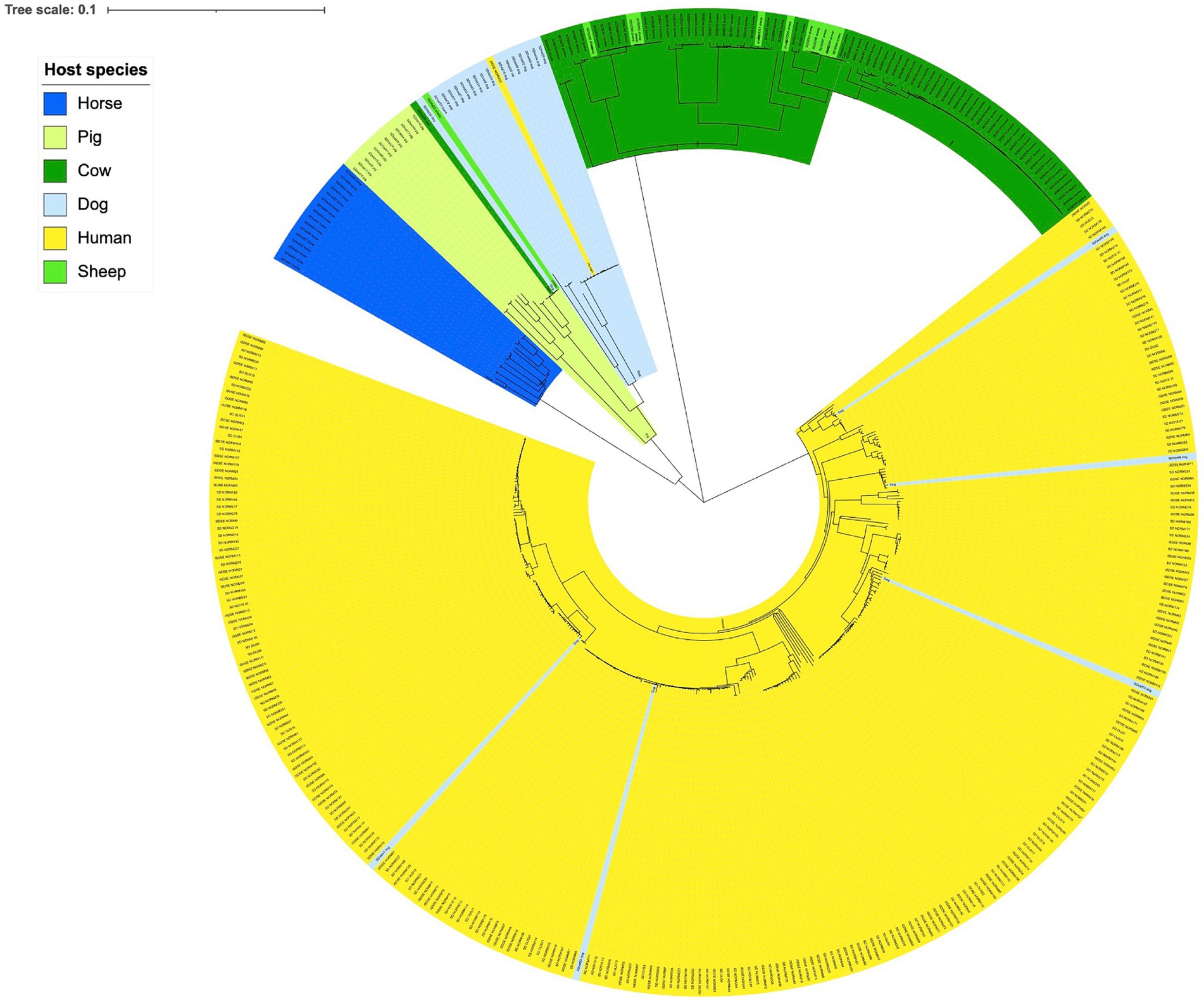
Figure 1. Genetic relationship between Streptococcus dysgalactiae isolates from different host species. Scale indicates substitutions per site. The phylogenetic tree is constructed based on a core genome single nucleotide polymorphism alignment using a maximum likelihood method.
Phenotypic susceptibility testing
The phenotypic antimicrobial resistance rates were relatively similar among isolates derived from humans, companion animals and livestock (Figure 2A). The only significant difference was a higher tetracycline resistance rate in SD from livestock (41%) compared to SD from companion animals (19%, p = 0.02). Resistance to erythromycin or clindamycin (MLS-resistance) was detected in 33 (12%), 3 (8%) and 8 (8%) of the isolates obtained from humans, companion animals and livestock, respectively. Of note, MLS-resistance in companion animals was derived from isolates from dogs only, and all MLS-resistance among livestock was detected in bovine associated isolates. All isolates in the study were susceptible to benzylpenicillin and trimethoprim-sulfamethoxazole.
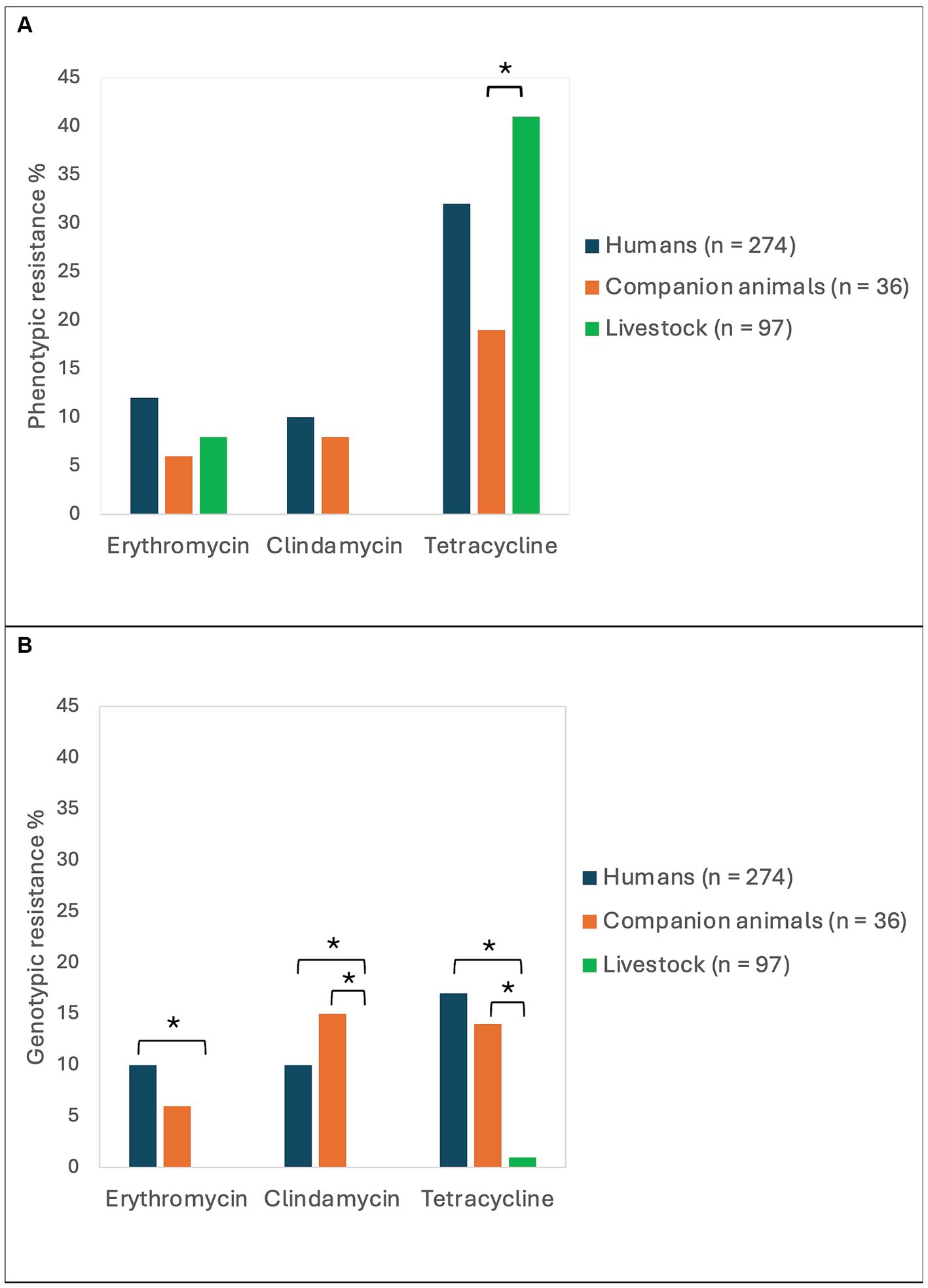
Figure 2. Antimicrobial resistance rates in Streptococcus dysgalactiae isolates from different host groups. The figure displays the prevalence of phenotypic (A) and genotypic (B) resistance to erythromycin, clindamycin, and tetracycline among Streptococcus dysgalactiae isolates procured from humans, companion animals and livestock animals. Asterisks mark significant differences in antimicrobial resistance levels.
Resistome analysis
Whole genome sequences of all the included bacterial strains were screened for antimicrobial resistance genes (Table 1). In total, resistance genes were detected in 70 (26%), 9 (25%), and 2 (2%) of the isolates derived from humans, companion animals and livestock, respectively. We almost exclusively detected genes encoding resistance to either tetracyclines or MLS-antibiotics, and tet(M), tet(O) and erm(A) were the most abundant. However, distinct host associated resistance gene profiles were noted; erm(A) accounted for 82% of the MLS resistance in human associated isolates, whereas only erm(B) and lsa(C) were identified in MLS resistant SD from companion animals. Moreover, the rates of genotypic resistance to macrolides, clindamycin and tetracycline were all significantly lower among isolates procured from livestock than from humans and companion animals (Figure 2B).
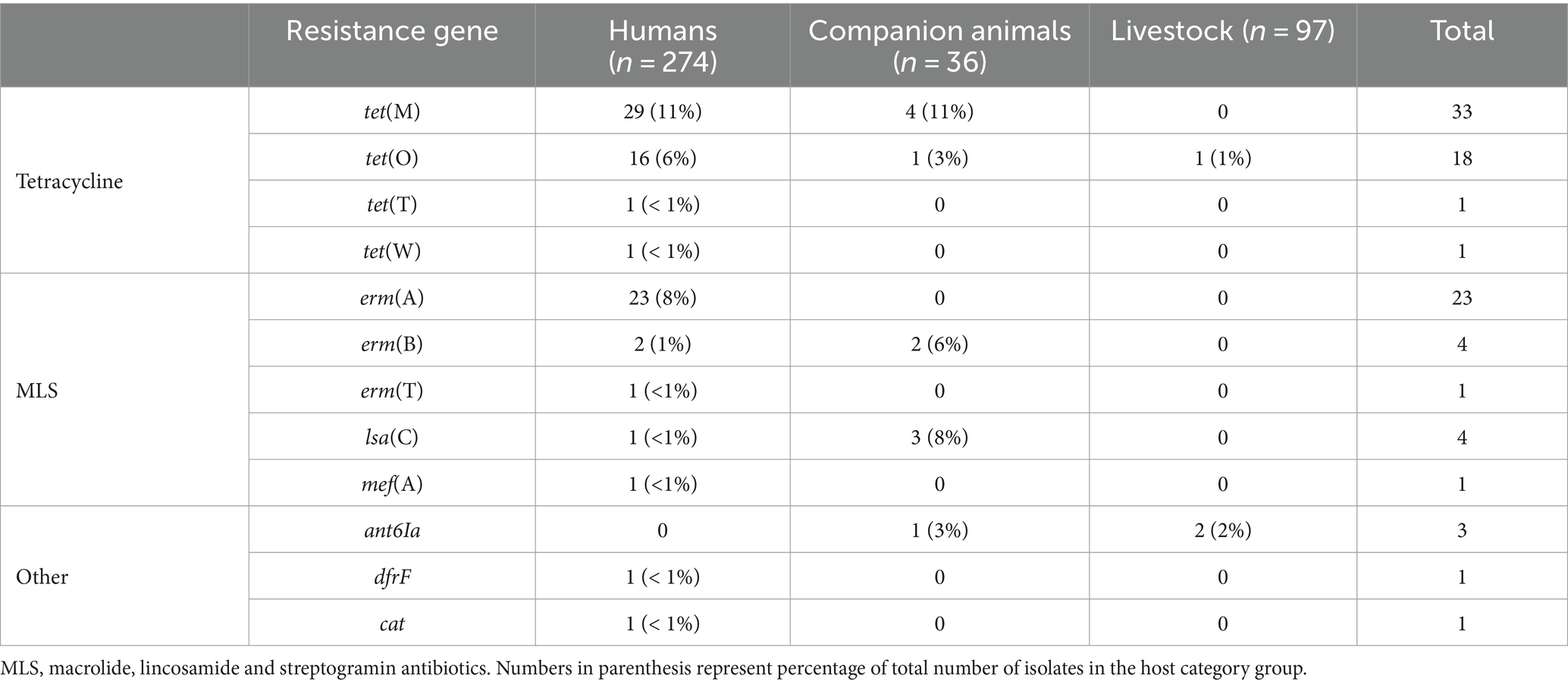
Table 1. Genotypic resistance found in isolates of Streptococcus dysgalactiae of different host origin.
The predicted genotypic resistance rates were substantially lower than the observed phenotypic resistance rates, particularly for tetracycline. Exploring this discrepancy, we found that the overall distribution of the tetracycline MIC values in our SD population appeared to be trimodal, and only the cluster with the highest MIC values correlated with isolates harboring genes encoding resistance to tetracycline (Figure 3A). The central cluster was intersected by the EUCAST tetracycline breakpoint, but the isolates lacked identifiable validated resistance genes. This resistance gene negative, low-grade resistant population included 39 of 40 livestock associated SD isolates with reduced susceptibility to tetracycline, but also comprised a distinct phylogenetic cluster of human associated strains (Figure 3B).
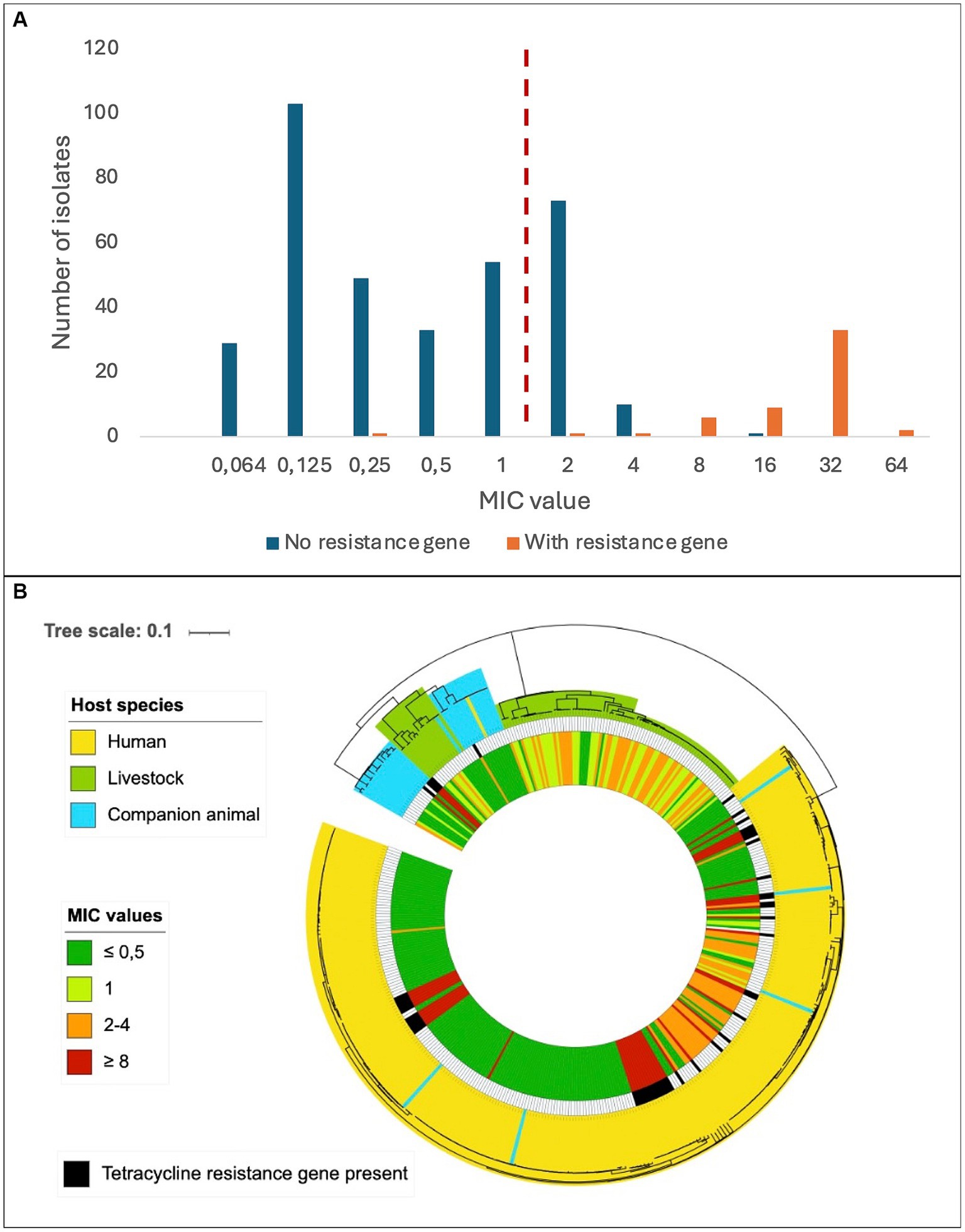
Figure 3. Distribution of tetracycline resistance in Streptococcus dysgalactiae isolates. (A) Minimum inhibitory concentrations of tetracycline among Streptococcus dysgalactiae isolates with or without a known tetracycline resistance gene. The dashed, red line represents the current EUCAST susceptibility breakpoint. (B) Phylogenetic tree of Streptococcus dysgalactiae isolates indicating host species (outer circle), presence of known tetracycline resistance gene (middle circle) and minimum inhibitory concentration of tetracycline (inner circle). Scale indicates substitutions per site.
Incongruence between phenotypic and genotypic resistance traits was also observed for other antimicrobial agents. All eight erythromycin resistant livestock associated isolates had MIC values just above the susceptibility breakpoint, and none of them harbored identifiable resistance genes. An identical pattern was observed for 7 of 33 human associated SD isolates displaying reduced susceptibility to erythromycin.
Inversely, a few isolates had identifiable resistance genes but displayed phenotypical susceptibility, including three isolates harboring lsa(C)-genes, and one strain with a truncated tet(M)-gene.
Mobile genetic elements and resistance genes
Analyses of flanking sequences of the detected resistance genes revealed a location on mobile genetic elements (MGEs) in almost all cases (Table 2). The one exception to this was a tet(M) gene located on a contig with a flanking sequence too short to determine the location with certainty. Integrative conjugative elements (ICEs) were the predominant form of MGEs detected, but the erm(T) gene was carried on a small p5580-like plasmid, and one dog isolate harbored a bacteriophage carrying an erm(B) gene. The major vector for MLS resistance was MGEs belonging to the ICESp2905 family. These ICEs harbored 85% of the MLS resistance genes, including lsa(C) genes in both human and animal associated isolates. Nevertheless, ICESp2905 elements in strains from different host sources displayed less than 95% sequence similarity based on core ICE conjugation genes.
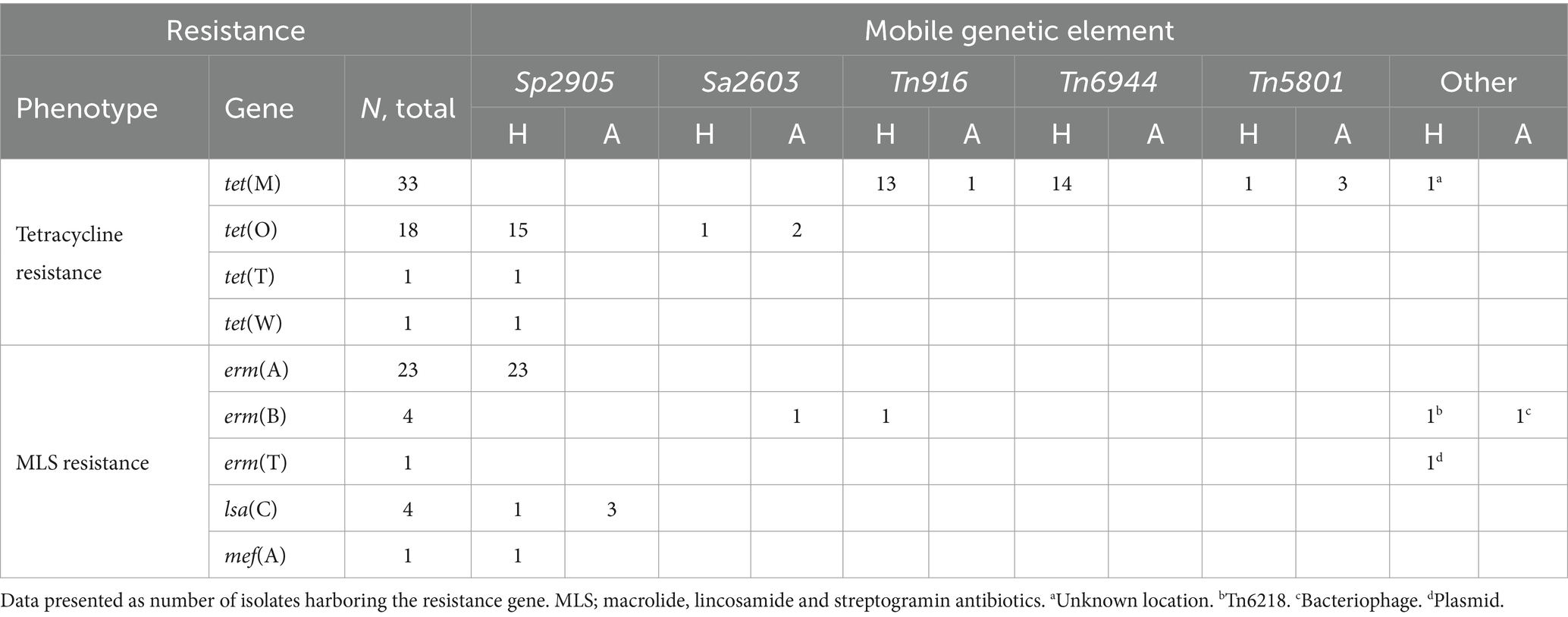
Table 2. Antimicrobial resistance genes and their associated mobile genetic elements among human (H) and animal (A) associated isolates of Streptococcus dysgalactiae.
On a similar note, the tet(O) genes, giving resistance to tetracycline, were located on ICEs belonging to the ICESp2905 and ICESa2603 family, but were distinctly associated with SD from human and animal sources, respectively. The location of tet(M) was more diverse, but the two major vectors in human associated SD isolates were the ICEs Tn916 and Tn6944.
The element Tn5801 was the most common harboring tet(M) in SD isolated from animal hosts. Tn5801 is divided into type A and B, where the type B variant is lacking two genes at the beginning of the element (León-Sampedro et al., 2016). Two isolates from horses and one from a dog carried the type A variant of Tn5801, whereas one human associated isolate harbored the type B. Notably, the Tn5801 element in the dog associated isolate SDVet48 clustered phylogenetically to the horse associated elements (Figure 4), even though the bacterial isolate itself phylogenetically resided among human isolates. By BLASTn search, both variants of the Tn5801 elements were detected in a range of streptococcal, enterococcal and staphylococcal strains, isolated from both human and animal sources. Several of these showed more than 99% overall sequence homology to the type A and B Tn5801 elements detected in our isolates.
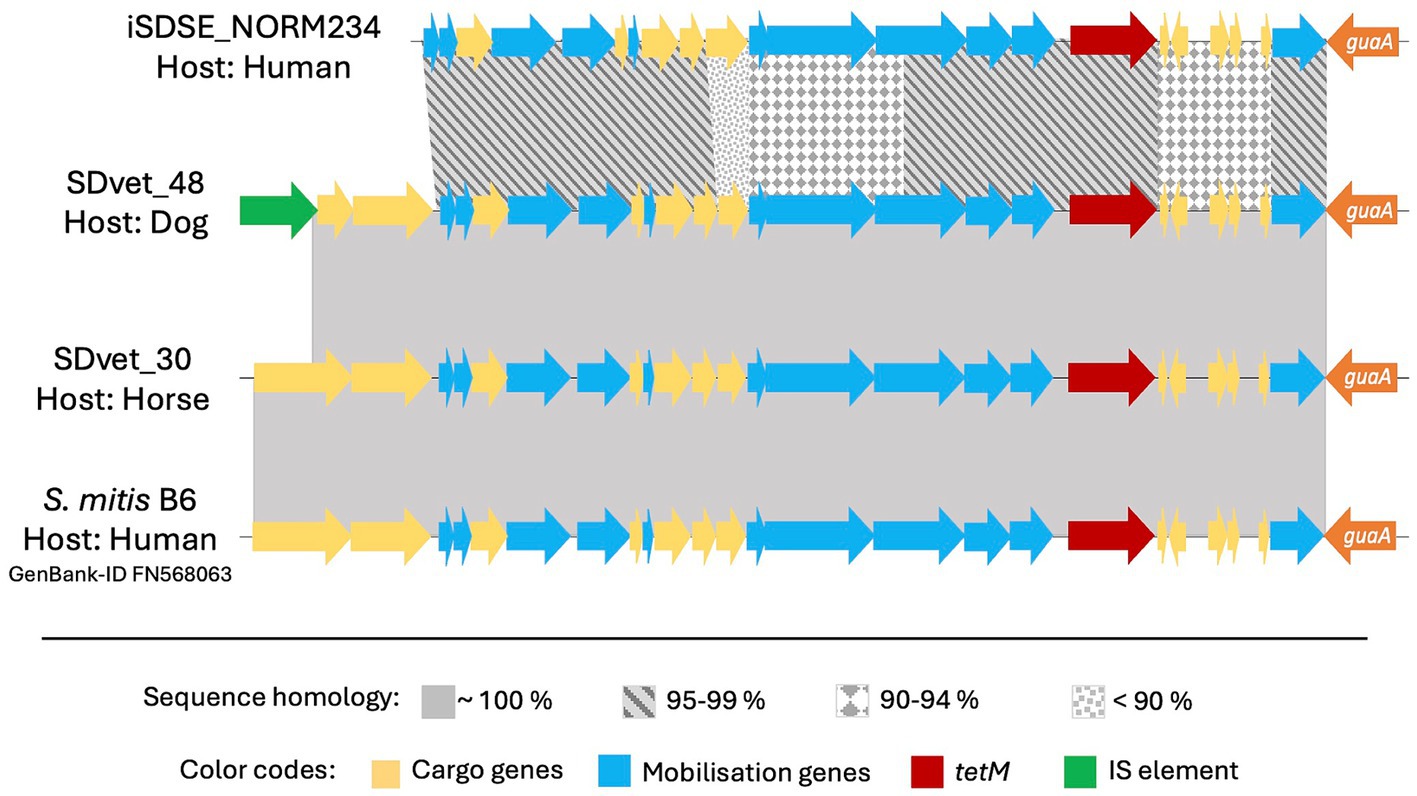
Figure 4. Comparative analyses of the tet(M) carrying mobile genetic element Tn5801 in different bacteria. Tn5801 was in all cases studied found integrated immediately downstream to the chromosomally located gene guaA. Among the Streptococcus dysgalactiae isolates included in this study, Tn5801 was present in four: one from a human, one from a dog and two from horses. The Tn5801 in the animal isolates had an inter-sequence homology close to 100%, while the human sequence was more divergent. This finding was not in line with the phylogenetic relationship of the bacterial isolates themselves, of which the isolate from the dog was more closely linked to the human isolate than to the horse isolates. By BLAST search we found Tn5801 also to be present in other species than S. dysgalactiae, here represented by an element located in an invasive human isolate of S. mitis, which interestingly is a closer match to the mobile genetic element in S. dysgalactiae of animal than of human origin.
Discussion
To our best knowledge, this is the first comparative analysis of antimicrobial resistance patterns in contemporary Streptococcus dysgalactiae (SD) isolates in a One Health perspective. Overall, we found that direct transmission of bacterial strains or genetic content between different ecological niches appears to be infrequent. This is underpinned by the observed host specialization among the SD isolates and predominantly a difference in the genetic recipe for resistance, inferring different origins of the expressed traits.
A delineation of SD populations in accordance with host species has also been reported previously (Porcellato et al., 2021; Alves-Barroco et al., 2022). Porcellato et al. found that SD isolates harbored several host specific virulence factors and appeared to have evolved through genetic exchange with other bacterial species residing within their ecological niche (Porcellato et al., 2021). Segregation into host adapted phylogenetic lineages is also seen in studies of Streptococcus agalactiae and Staphylococcus aureus, and such niche specialization potentially represents a barrier to cross-species transmission (Maeda et al., 2020; Matuszewska et al., 2020). Supporting this, epidemiological collections of whole genomes of SD isolates from Japan, Canada and Denmark all reveal a very low frequency of animal associated MLST-profiles among SD isolated from humans, suggesting that zoonotic transmission of this pathogen is rare (Lother et al., 2017; Rebelo et al., 2022; Shinohara et al., 2023).
Despite the apparent transmission barrier conferred by niche adaptation, we found evidence of some level of cross-species exchange of SD isolates, predominantly between humans and companion animals. This is not surprising considering the close contact that often exists between humans and their dogs, and to a certain degree also between humans and horses. Similarly, Pinho et al. found that two SD isolates from dog and horse, respectively, were clustered together with human isolates in a phylogenetic study based on MLST analysis (Pinho et al., 2016). An SD isolate with the same MLST sequence type as the dog isolate to which it is referred was sampled from a boy living in the same household as the dog, reinforcing the hypothesis of a cross-species transmission event (Schrieber et al., 2014). In Singapore, an SD isolate phylogenetically resembling piscine associated SD strains was identified in a skin infection in a fish handler (Koh et al., 2009). Taken together, these findings indicate that cross-species transmission does occur, but predominantly in situations with prolonged or extensive exposure, such as between humans and their companion animals. Moreover, transmission events in a human to animal direction appear to be more common than the opposite, but the numbers are too small to draw firm conclusions.
We found highly diverging resistomes in SD isolates of human and animal origin, including both resistance genes and their associated MGEs, inferring limited genetic exchange between the host associated populations. Nevertheless, we observed an almost identical resistance element, Tn5801, in two SD isolates derived from a dog and a horse, respectively (Figure 4). Moreover, a highly similar element from a human associated Streptococcus mitis isolate was deposited in GenBank, strongly supporting the presence of genetic transfer between different ecological niches. However, the resistance MGEs detected in human associated SD isolates predominantly displayed similarities to MGEs derived from other β-hemolytic streptococcal isolates from humans, including S. agalactiae and S. pyogenes. Thus, the bulk of conjugative transfer and transduction of resistance determinants likely occurs within the boundaries of the ecological niche.
Reports on antimicrobial resistance in SD are quite sparse and limited to SD infecting humans and cattle. Studies on antimicrobial susceptibility in SD from the past decade demonstrate MLS resistance rates varying from 1 to 48% among bovine associated SD (Zhang et al., 2018; Duse et al., 2021) and from 17 to 42% among invasive, human associated strains (Park et al., 2019; Rojo-Bezares et al., 2021). Regarding tetracycline resistance, available data from the same period has shown resistance rates varying in the ranges 33–100% and 30–56% for bovine and human associated isolates, respectively (Traverso et al., 2016; Zhang et al., 2018; Park et al., 2019; Shen et al., 2021). Compared to these numbers, our findings indicate a relatively low frequency of resistant SD strains of both human and animal origin in Norway, possibly reflecting the strict policy regarding the use of antibiotics in both human and veterinary medicine in our country. European surveillance data about veterinary antimicrobial consumption obtained from the European Medicines Agency shows an at least tenfold higher consumption of all antibiotics relevant to this study in most European countries compared to Norway (EMA, 2024).
We observed a substantial incongruence between phenotypic and genotypic susceptibility rates for tetracycline, particularly in bovine associated isolates. In January 2023, EUCAST lowered the MIC breakpoint for tetracycline resistance in SD by merging the “I” (susceptible, increased exposure) category into the “R” (resistant) group. This change had a great impact on our results, doubling the number of strains entering the resistant category relative to earlier versions of the EUCAST clinical breakpoint table. Notably, most of these low-grade resistant strains lacked identifiable validated resistance genes. A MIC distribution intersected by the current EUCAST breakpoint was also reported in a recent Scandinavian study examining oxytetracycline-susceptibility among 231 SD isolates of bovine origin (Jensen et al., 2024). They found a uniform distribution with a proposed tentative epidemiological cut off (TECOFF) of 8 mg/L, which is three dilution steps above the breakpoint. Retrospectively applying the current breakpoint to previously published reports on tetracycline susceptibility in bovine associated SD, a tetracycline MIC distribution encircling the novel breakpoint was observed also in studies from Canada, New Zealand, and Europe, suggesting that this is a widespread feature in this species (McDougall et al., 2014; Cameron et al., 2016; Garch et al., 2020). In the present study a low-grade tetracycline resistant subpopulation was also evident among SD isolates of human origin (Figure 3B), indicating that this phenomenon is not limited to isolates of bovine origin, nor related to the distinction between the two subspecies SDSE and SDSD.
Breakpoints intersecting defined bacterial populations is generally avoided by EUCAST, as inherent analytic variations in the susceptibility testing makes the susceptible/resistant categorization unreliable. The observed high proportion of SD isolates displaying tetracycline MIC values encircling the breakpoint could either reflect a breakpoint poorly adapted to the SD wild type or be the result of a so far unrecognized mechanism of low-grade resistance. The trimodal distribution of the low-grade tetracycline resistant strains in our material could infer the latter, and the genetic basis for this phenomenon should be subjected to scrutiny.
In our study, we have included SD from a relatively large selection of host species, which entail a multifaceted base for comparative studies. The collection of strains from all hosts from within the same temporal and geographical delimitated setting is also a strength of the study, enabling a real time comparison of SD in a One Health perspective.
A limitation of the study is the low prevalence of antimicrobial resistance in Norway, potentially underestimating the extent of cross-species transmission. Antimicrobial resistance in streptococci in a One Health perspective should therefore be explored also in regions with higher rates of antimicrobial resistance. Moreover, analyzing pooled resistance rates for companion animals and livestock does not reflect the phylogenetic diversity within the SD taxon, and could potentially obscure significant differences between these ecological niches. Another potential limitation is the use of MIC gradient strips instead of disc diffusion or broth dilution. Nevertheless, a large proportion of our isolates was also examined by disc diffusion, and the results were congruent (Supplementary Table S1). A potential confounder is the delimitation of included human associated strains to exclusively bloodborne isolates, whereas the animal associated strains predominantly are from non-invasive infections. Nevertheless, national surveillance data on antimicrobial susceptibility in non-invasive human associated isolates of SD from 2018 does not reveal major differences between invasive and non-invasive strains (NORM, 2019). Lastly, we only examined isolates from a confined temporal context, and dissemination of resistance traits over time could not be evaluated. Longitudinal collection of isolates from asymptomatic carriers in contemporary and spatially related animal and human populations would be interesting. However, the execution of such an investigation probably would entail ethical challenges regarding sampling procedure on healthy animals.
In conclusion, we found a phylogenetic delineation of SD strains in line with host adapted populations and niche specialization. Moreover, the resistome differed significantly between SD in these host associated groups both regarding the repertoire of circulating resistance genes and their associated mobile gene elements. Our findings indicate that direct transmission events of strains or genetic elements carrying resistance genes between SD from different ecological niches are rare in our geographic region.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found here: BioProject, accession number PRJEB74563, available at NCBI (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJEB74563).
Ethics statement
The studies involving humans were approved by Regional Committee for Medical Research Ethics in Western Norway (2021/63132). The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because samples from animals were collected from sick animals for diagnostic purposes by veterinarians in clinical practice, which does not require ethical approval.
Author contributions
MG: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. SS: Conceptualization, Funding acquisition, Writing – review & editing. AS: Writing – review & editing. BK: Conceptualization, Project administration, Writing – review & editing. AK: Writing – review & editing. CJ: Writing – review & editing. HJ: Writing – review & editing. OO: Conceptualization, Data curation, Investigation, Methodology, Project administration, Supervision, Writing – review & editing.
Members of the Norwegian Study Group on Streptococcus dysgalactiae
Aasmund Fostervold, Department of Clinical Microbiology, Stavanger University Hospital, Stavanger, Norway; Aleksandra Jakovljev, Department of Microbiology, St. Olav’s University Hospital, Trondheim, Norway; Nadine Durema Pullar, Department of Clinical Microbiology, Vestre Viken Hospital Trust, Bærum, Norway; Åshild Marvik, Department of Microbiology, Vestfold Hospital Trust, Tønsberg, Norway; Einar Nilsen, Department of Microbiology, Møre and Romsdal Hospital Trust, Ålesund, Norway; Fredrik Müller, Department of Microbiology, Oslo University Hospital, Oslo, Norway; Ghantous Milad Chedid, Laboratory for Clinical Microbiology, Fonna Hospital Trust, Haugesund, Norway; Gunnar Skov Simonsen, Department of Microbiology and Infection Control, University Hospital of North Norway, Tromsø, Norway; Elisabeth Sirnes, Department of Microbiology, Division of Medicine, District General Hospital of Førde, Førde, Norway; Roar Magne Bævre-Jensen, Department of Clinical Microbiology, Vestre Viken Hospital Trust, Drammen, Norway; Sandra Åsheim, Unit for Clinical Microbiology, Norland Hospital Trust, Bodø, Norway; Ståle Tofteland, Department of Clinical Microbiology, Sørlandet Hospital Trust, Agder, Norway; Rolf-Arne Sandnes, Department of Clinical Microbiology, Innlandet Hospital Trust, Lillehammer, Norway; Truls Michael Leegaard, Department of Microbiology and Infection Control, Akershus University Hospital, Lørenskog, Norway.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Norwegian surveillance program for antimicrobial resistance (NORM) (Grant number: 19/08, to OO and 19/16 to CJ) and Center for Laboratory Medicine at Østfold Hospital Trust. The genomic Core Facility (GCF) at the University of Bergen, which is part of the NorSeq consortium, provided services on the whole genome sequencing in this study; GCF is supported by major grants from the Research Council of Norway (grant no. 245979/F50) and Trond Mohn Foundation (grant no. BFS2017TMT04/BFS2017TMT08). This study was received also financial contributions from the Western Norway Regional Health Authority (grant no. 912231). SS received grants from the Norwegian Research Council under the frames of NordForsk (Project: 90456, PerAID) and ERA PerMed (Project: 2018-151, PerMIT).
Acknowledgments
We thank the staff at Microbiological Department at Haukeland University Hospital for excellent technical assistance and for providing access to their laboratory facilities. We acknowledge Haima Mylvaganam for her significant contribution to streptococcal research at Haukeland University Hospital during the past two decades, and for inspiration to continued scientific exploration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1423762/full#supplementary-material
Footnotes
References
Abdelsalam, M., Chen, S., and Yoshida, T. (2010). Phenotypic and genetic characterizations of Streptococcus dysgalactiae strains isolated from fish collected in Japan and other Asian countries. FEMS Microbiol. Lett. 302, 32–38. doi: 10.1111/j.1574-6968.2009.01828.x
Acke, E., Midwinter, A., Lawrence, K., Gordon, S., Moore, S., Rasiah, I., et al. (2015). Prevalence of Streptococcus dysgalactiae subsp. equisimilis and S. equi subsp. zooepidemicus in a sample of healthy dogs, cats and horses. N. Zealand Vet. J. 63, 265–271. doi: 10.1080/00480169.2015.1016133
Alves-Barroco, C., Brito, P. H., Santos-Sanches, I., and Fernandes, A. R. (2022). Phylogenetic analysis and accessory genome diversity reveal insight into the evolutionary history of Streptococcus dysgalactiae. Front. Microbiol. 13:952110. doi: 10.3389/fmicb.2022.952110
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brandt, C. M., and Spellerberg, B. (2009). Human infections due to Streptococcus dysgalactiae subspecies equisimilis. Clin. Infect. Dis. 49, 766–772. doi: 10.1086/605085
Cameron, M., Saab, M., Heider, L., McClure, J. T., Rodriguez-Lecompte, J. C., and Sanchez, J. (2016). Antimicrobial susceptibility patterns of environmental streptococci recovered from bovine Milk samples in the maritime provinces of Canada. Front. Vet. Sci. 3:79. doi: 10.3389/fvets.2016.00079
Ciszewski, M., Zegarski, K., and Szewczyk, E. M. (2016). Streptococcus dysgalactiae subsp. equisimilis isolated from infections in dogs and humans: are current subspecies identification criteria accurate? Curr. Microbiol. 73, 684–688. doi: 10.1007/s00284-016-1113-x
Couture-Cossette, A., Carignan, A., Mercier, A., Desruisseaux, C., Valiquette, L., and Pépin, J. (2018). Secular trends in incidence of invasive beta-hemolytic streptococci and efficacy of adjunctive therapy in Quebec, Canada, 1996-2016. PLoS One 13:e0206289. doi: 10.1371/journal.pone.0206289
Duse, A., Persson-Waller, K., and Pedersen, K. (2021). Microbial Aetiology, antibiotic susceptibility and pathogen-specific risk factors for udder pathogens from clinical mastitis in dairy cows. Animals 11:2113. doi: 10.3390/ani11072113
EMA (2024). European surveillance of veterinary antmicrobial consumption (ESVAC): 2009–2023. Available at: https://www.ema.europa.eu/en/veterinary-regulatory-overview/antimicrobial-resistance-veterinary-medicine/european-surveillance-veterinary-antimicrobial-consumption-esvac-2009-2023 (Accessed April 26, 2024).
Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D., and Aarestrup, F. M. (2022). ResFinder – an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. 8:000748. doi: 10.1099/mgen.0.000748
Garch, F. E., Youala, M., Simjee, S., Moyaert, H., Klee, R., Truszkowska, B., et al. (2020). Antimicrobial susceptibility of nine udder pathogens recovered from bovine clinical mastitis milk in Europe 2015–2016: VetPath results. Vet. Microbiol. 245:108644. doi: 10.1016/j.vetmic.2020.108644
Jensen, V. F., Damborg, P., Norström, M., Nonnemann, B., Slettemeås, J. S., Smistad, M., et al. (2024). Estimation of epidemiological cut-off values for eight antibiotics used for treatment of bovine mastitis caused by Streptococcus uberis and Streptococcus dysgalactiae subsp. dysgalactiae. Vet. Microbiol. 290:109994. doi: 10.1016/j.vetmic.2024.109994
Kaci, A., Jonassen, C. M., Skrede, S., Sivertsen, A., and Steinbakk, M.Norwegian Study Group on Streptococcus dysgalactiae, et al. (2023). Genomic epidemiology of Streptococcus dysgalactiae subsp. equisimilis strains causing invasive disease in Norway during 2018. Front. Microbiol. 14:1171913. doi: 10.3389/fmicb.2023.1171913
Koh, T. H., Sng, L.-H., Yuen, S. M., Thomas, C. K., Tan, P. L., Tan, S. H., et al. (2009). Streptococcal cellulitis following preparation of fresh raw seafood. Zoonoses Public Heal. 56, 206–208. doi: 10.1111/j.1863-2378.2008.01213.x
León-Sampedro, R., Novais, C., Peixe, L., Baquero, F., and Coque, T. M. (2016). Diversity and evolution of the Tn 5801-tet (M)-like integrative and conjugative elements among Enterococcus, Streptococcus, and Staphylococcus. Antimicrob. Agents Chemother. 60, 1736–1746. doi: 10.1128/aac.01864-15
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lother, S. A., Demczuk, W., Martin, I., Mulvey, M. R., Dufault, B., Lagacé-Wiens, P., et al. (2017). Clonal clusters and virulence factors of group C and G Streptococcus causing severe infections, Manitoba, Canada, 2012–2014. Emerg. Infect. Dis. 23, 1079–1088. doi: 10.3201/eid2307.161259
Maeda, T., Tsuyuki, Y., Fujita, T., Fukushima, Y., Goto, M., Yoshida, H., et al. (2020, 2019). Comparison of Streptococcus agalactiae isolates from humans and companion animals reveals genotypic and phenotypic differences. Jpn. J. Infect. Dis. 73:441. doi: 10.7883/yoken.jjid.2019.441
Matuszewska, M., Murray, G. G. R., Harrison, E. M., Holmes, M. A., and Weinert, L. A. (2020). The evolutionary genomics of host specificity in Staphylococcus aureus. Trends Microbiol. 28, 465–477. doi: 10.1016/j.tim.2019.12.007
McDougall, S., Hussein, H., and Petrovski, K. (2014). Antimicrobial resistance in Staphylococcus aureus, Streptococcus uberis and Streptococcus dysgalactiae from dairy cows with mastitis. N. Zealand Vet. J. 62, 68–76. doi: 10.1080/00480169.2013.843135
McEwen, S. A., and Collignon, P. J. (2018). Antimicrobial resistance: a one health perspective. Microbiol. Spectr. 6. doi: 10.1128/microbiolspec.arba-0009-2017
Moreno, L. Z., da Costa, B. L. P., Matajira, C. E. C., Gomes, V. T. M., Mesquita, R. E., Silva, A. P. S., et al. (2016). Molecular and antimicrobial susceptibility profiling of Streptococcus dysgalactiae isolated from swine. Diagn. Microbiol. Infect. Dis. 86, 178–180. doi: 10.1016/j.diagmicrobio.2016.07.020
Nevanlinna, V., Huttunen, R., Aittoniemi, J., Luukkaala, T., and Rantala, S. (2023). Incidence, seasonal pattern, and clinical manifestations of Streptococcus dysgalactiae subspecies equisimilis bacteremia; a population-based study. Eur. J. Clin. Microbiol. Infect. Dis. 42, 819–825. doi: 10.1007/s10096-023-04607-8
NORM (2019). NORM/NORM-VET 2018. Usage of Antimicrobial Agents and Occurrence of Antimicrobial Resistance in Norway. Tromsø/Oslo 2019. Available at: https://www.unn.no/4a79d7/siteassets/documents/kompetansetjenester--sentre-og-fagrad/norm---norsk-overvakingssystem-for-antibiotikaresistens-hos-mikrober/rapporter/norm-norm-vet-2018.pdf
Oppegaard, O., Glambek, M., Skutlaberg, D. H., Skrede, S., Sivertsen, A., and Kittang, B. R. (2023). Streptococcus dysgalactiae bloodstream infections, Norway, 1999–2021. Emerg. Infect. Dis. 29, 260–267. doi: 10.3201/eid2902.221218
Oppegaard, O., Mylvaganam, H., Skrede, S., Lindemann, P. C., and Kittang, B. R. (2017). Emergence of a Streptococcus dysgalactiae subspecies equisimilis st G62647-lineage associated with severe clinical manifestations. Sci. Rep. 7:7589. doi: 10.1038/s41598-017-08162-z
Park, J. H., Jung, J., Kim, M. J., Sung, H., Kim, M.-N., Chong, Y. P., et al. (2019). Incidence, clinical characteristics, and outcomes of Streptococcus dysgalactiae subspecies equisimilis bacteremia in a tertiary hospital: comparison with S. agalactiae bacteremia. Eur. J. Clin. Microbiol. Infect. Dis. 38, 2253–2258. doi: 10.1007/s10096-019-03667-z
Pinho, M. D., Erol, E., Ribeiro-Gonçalves, B., Mendes, C. I., Carriço, J. A., Matos, S. C., et al. (2016). Beta-hemolytic Streptococcus dysgalactiae strains isolated from horses are a genetically distinct population within the Streptococcus dysgalactiae taxon. Sci. Rep. 6:31736. doi: 10.1038/srep31736
Porcellato, D., Smistad, M., Skeie, S. B., Jørgensen, H. J., Austbø, L., and Oppegaard, O. (2021). Whole genome sequencing reveals possible host species adaptation of Streptococcusdysgalactiae. Sci. Rep. 11:17350. doi: 10.1038/s41598-021-96710-z
Rebelo, A. R., Ibfelt, T., Bortolaia, V., Leekitcharoenphon, P., Hansen, D. S., Nielsen, H. L., et al. (2022). One day in Denmark: Nationwide point-prevalence survey of human bacterial isolates and comparison of classical and whole-genome sequence-based species identification methods. PLoS One 17:e0261999. doi: 10.1371/journal.pone.0261999
Rojo-Bezares, B., Toca, L., Azcona-Gutiérrez, J. M., Ortega-Unanue, N., Toledano, P., and Sáenz, Y. (2021). Streptococcus dysgalactiae subsp. equisimilis from invasive and non-invasive infections in Spain: combining epidemiology, molecular characterization, and genetic diversity. Eur. J. Clin. Microbiol. Infect. Dis. 40, 1013–1021. doi: 10.1007/s10096-020-04119-9
Schrieber, L., Towers, R., Muscatello, G., and Speare, R. (2014). Transmission of Streptococcus dysgalactiae subsp. equisimilis between child and dog in an aboriginal Australian community. Zoonoses Public Heal. 61, 145–148. doi: 10.1111/zph.12057
Shen, J., Wu, X., Yang, Y., Lv, Y., Li, X., Ding, X., et al. (2021). Antimicrobial resistance and virulence factor of Streptococcus dysgalactiae isolated from clinical bovine mastitis cases in Northwest China. Infect. Drug Resist. 14, 3519–3530. doi: 10.2147/idr.s327924
Shinohara, K., Murase, K., Tsuchido, Y., Noguchi, T., Yukawa, S., Yamamoto, M., et al. (2023). Clonal expansion of multidrug-resistant Streptococcus dysgalactiae subspecies equisimilis causing bacteremia, Japan, 2005–2021. Emerg. Infect. Dis. 29, 528–539. doi: 10.3201/eid2903.221060
Smistad, M., Tollersrud, T. S., Austbø, L., Porcellato, D., Wolff, C., Asal, B., et al. (2021). Molecular detection and genotype characterization of Streptococcus dysgalactiae from sheep flocks with outbreaks of infectious arthritis. Vet. Microbiol. 262:109221. doi: 10.1016/j.vetmic.2021.109221
Traverso, F., Blanco, A., Villalón, P., Beratz, N., Nieto, J. A. S., Lopardo, H., et al. (2016). Molecular characterization of invasive Streptococcus dysgalactiae subsp. equisimilis. Multicenter study: Argentina 2011–2012. Rev. Argent. Microbiol. 48, 279–289. doi: 10.1016/j.ram.2016.07.001
UK Health Security Agency (2022). Laboratory surveillance of pyogenic and non-pyogenic streptococcal bacteraemia in England: 2021 update. Health Protect. Rep. 16:13.
Vieira, V. V., Teixeira, L. M., Zahner, V., Momen, H., Facklam, R. R., Steigerwalt, A. G., et al. (1998). Genetic relationships among the different phenotypes of Streptococcus dysgalactiae strains. Int J Syst Evol Micr 48, 1231–1243. doi: 10.1099/00207713-48-4-1231
Zhang, S., Piepers, S., Shan, R., Cai, L., Mao, S., Zou, J., et al. (2018). Phenotypic and genotypic characterization of antimicrobial resistance profiles in Streptococcus dysgalactiae isolated from bovine clinical mastitis in 5 provinces of China. J. Dairy Sci. 101, 3344–3355. doi: 10.3168/jds.2017-14031
Keywords: Streptococcus dysgalactiae, antibiotic resistance, One Health, whole genomic sequencing, horizontal genetic transfer, animal, human, Norway
Citation: Glambek M, Skrede S, Sivertsen A, Kittang BR, Kaci A, Jonassen CM, Jørgensen HJNorwegian Study Group on Streptococcus dysgalactiae and Oppegaard O (2024) Antimicrobial resistance patterns in Streptococcus dysgalactiae in a One Health perspective. Front. Microbiol. 15:1423762. doi: 10.3389/fmicb.2024.1423762
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceCopyright © 2024 Glambek, Skrede, Sivertsen, Kittang, Kaci, Jonassen, Jørgensen, Norwegian Study Group on Streptococcus dysgalactiae and Oppegaard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oddvar Oppegaard, by5vcHBlZ2FhcmRAdWliLm5v