 Pierre Grève
Pierre Grève Bouziane Moumen
Bouziane Moumen Didier Bouchon
Didier Bouchon- Université de Poitiers, Ecologie et Biologie des Interactions, UMR CNRS 7267, Poitiers, France
Introduction: Endosymbiotic bacteria in the genus Wolbachia have evolved numerous strategies for manipulating host reproduction in order to promote their own transmission. This includes the feminization of males into functional females, a well-studied phenotype in the isopod Armadillidium vulgare. Despite an early description of this phenotype in isopods and the development of an evolutionary model of host sex determination in the presence of Wolbachia, the underlying genetic mechanisms remain elusive.
Methods: Here we present the first complete genomes of the three feminizing Wolbachia (wVulC, wVulP, and wVulM) known to date in A. vulgare. These genomes, belonging to Wolbachia B supergroup, contain a large number of mobile elements such as WO prophages with eukaryotic association modules. Taking advantage of these data and those of another Wolbachia-derived feminizing factor integrated into the host genome (f element), we used a comparative genomics approach to identify putative feminizing factors.
Results: This strategy has enabled us to identify three prophage-associated genes secreted by the Type IV Secretion System: one ankyrin repeat domain-containing protein, one helix-turn-helix transcriptional regulator and one hypothetical protein. In addition, a latrotoxin-related protein, associated with phage relic genes, was shared by all three genomes and the f element.
Conclusion: These putative feminization-inducing proteins shared canonical interaction features with eukaryotic proteins. These results pave the way for further research into the underlying functional interactions.
Introduction
The central role of host-symbiont interactions in the biology, ecology and evolution of the biotic world is now well recognized, which has led to the popularization of the holobiont concept (McFall-Ngai et al., 2013; Bordenstein and Theis, 2015). There are therefore no macroorganisms that do not host symbionts. Among these symbionts is Wolbachia pipientis (hereafter Wolbachia), a bacterium originally described in the mosquito Culex pipiens (Hertig and Wolbach, 1924). Since then, many studies have shown that this maternally inherited intracellular symbiont, belonging to the order Rickettsiales of the Alphaproteobacteria, exhibited exceptional traits. It is the most widespread endosymbiont in the animal world, representing a very wide diversity of strains infecting ~50% of arthropods and several nematodes (Zug and Hammerstein, 2012; Kaur et al., 2021). This enormous genetic diversity is reflected in the classification of Wolbachia where at least 17 phylogenetic supergroups (named A-F, H-Q and S; Supergroups A and B being the most numerous) can be identified to date (Kaur et al., 2021). Furthermore, these bacteria, often referred to as reproductive parasites, have attracted attention for the diversity of phenotypes they induce in their terrestrial arthropod hosts. Indeed, one of the main characteristics of Wolbachia is its remarkable ability to interfere with the reproduction of its hosts, through four main phenotypes, to optimize its own transmission, which has earned it the title of master manipulator (Werren et al., 2008).
Cytoplasmic Incompatibility (CI) represents the most common Wolbachia-induced phenotype (Werren et al., 2008; Landmann, 2019), resulting in embryonic death in crosses between infected males and uninfected females. It has been observed in Insecta as well as in Acari and in Isopoda (Landmann, 2019). The molecular basis of CI has recently been demonstrated by the identification of two genes, cifA and cifB, from the Eukaryotic Association Module (EAM) of Wolbachia’s prophage WO (Shropshire and Bordenstein, 2019; Shropshire et al., 2020). The other three Wolbachia-induced phenotypes lead to sex ratio biases in the host. Male-killing (MK), first described in the ladybird Adalia bipunctata, results in males death favoring female survival (Hurst et al., 1999; Fukui et al., 2015). As for CI, a candidate MK gene (termed WO-mediated killing wmk) was identified in the EAM of WO prophage (Perlmutter et al., 2019). Wolbachia-induced parthenogenesis (PI) was first described in Trichogramma Hymenoptera (Stouthamer et al., 1990) in which infected females can produce daughters from unfertilized eggs (Ma and Schwander, 2017). Recently, two putative PI-inducing factors (PifA and PifB) has been identified, also localized in EAM (Fricke and Lindsey, 2024). Wolbachia-induced feminization, which consists in the feminization of genetic males, is a reproductive manipulation that has been widely described in isopods and also observed in two insect species (Hiroki et al., 2002; Negri et al., 2006; Bouchon et al., 2008). However, the mechanisms involved are different: in butterflies, the feminizing Wolbachia interact with the master regulator genes that control sex determination, whereas in crustaceans, they interact with the hormonal regulatory genes (Kageyama et al., 2017; Herran et al., 2021).
The first description of feminization in isopods was due to the pioneering work on the pill bug Armadillidium vulgare (Juchault et al., 1974). The identification of Wolbachia (wVulC strain) as the feminizing factor came later (Rousset et al., 1992; Bouchon et al., 1998). Two additional Wolbachia (wVulM and wVulP strains), have been identified in A. vulgare (Cordaux et al., 2004; Verne et al., 2007). In A. vulgare, genetically (ZZ) male embryos carrying Wolbachia inherited from the mother develop into functional females morphologically undistinguishable from genetic females (ZW). Several studies have suggested that feminization of genetic males results from inhibition of androgenic gland differentiation that produce the androgenic hormone [review in Bouchon et al. (2008) and Herran et al. (2020)]. As males inverted in females produce female-biased broods, all Wolbachia-infected females were ZZ individuals in natural populations (Juchault et al., 1993). This situation has given rise to strong genetic conflicts with major evolutionary consequences for interactions between Wolbachia and A. vulgare (Rigaud, 1997). In particular, it has been suggested that a feminizing factor called f element, derived from the wVulC Wolbachia strain, was at the origin of a new W-type chromosome (Juchault and Mocquard, 1993). This hypothesis has recently been verified, as the f element corresponds to the insertion of a large part of the Wolbachia wVulC chromosome into the A. vulgare genome (Leclercq et al., 2016). It has also been shown that wVulC and the f element never co-occur (Durand et al., 2023).
Until now, genomic data on Wolbachia endosymbionts from woodlice has been very fragmentary, with only two genome assemblies. One is wCon, known to induce CI in Cylisticus convexus (Moret et al., 2001; Badawi et al., 2018). The other one available in databases is an assembly of 10 contigs from the genome of wVulC. In this study, we performed a comparative genomic analysis of the three Wolbachia strains identified so far in A. vulgare. We obtained the complete genomes of wVulC, wVulM using both short and long read sequencing and wVulP strains using long read sequencing. Our data shed light on the evolution of Wolbachia strains in A. vulgare and highlighted putative candidate feminization genes.
Materials and methods
Host lineages and DNA extraction
Four A. vulgare lineages were used in this study: a Wolbachia-free lineage (called BF) collected in 1967 in Nice (France), a wVulC-infected lineage (called ZN) collected in 1991 in Celles-sur-Belle (France), a wVulM-infected lineage (called BI) collected in 1999 in Méry sur Cher (France) and a wVulP-infected lineage (called CP) collected in 2007 in Poitiers (France) (Cordaux et al., 2004; Verne et al., 2007). These lineages have since been stably maintained in the laboratory, at 20°C under natural photoperiod, with food ad libitum (dead lime leaves and carrots). Controlled rearing on a standard diet homogenizes the diversity of the gut microbiota and no other sex-parasitic bacteria have been identified (Dittmer et al., 2014; Dittmer and Bouchon, 2018).
The sex ratios observed in the lineages were recorded each year. The Wolbachia-free BF lineage is used as a control for genetic sex determination as attempts to cure individuals with antibiotics have been unsuccessful (Rigaud and Juchault, 1998). The proportion of males was determined in each brood over 5 years and visualized by boxplots using arcsine transformations. Comparison of the mean male ratio was performed in R version 4.3.2 (R Core Team, 2023) through a generalized linear mixed model with host lineage as fixed effect and clutch size and year as random effects. Assessment of the model was performed using the performance package (Lüdecke et al., 2021).
DNA extraction was carried out by homogenizing ovaries of 30 to 50 infected females with a Dounce tissue grinder B in a PBS solution supplemented with sucrose (0.25 M) and L-glutamine (5 mM) which allow the cells to be crushed but not the nuclei. Large fragments were removed by passing the solution through a 5 μm filter. The remaining nuclei were pelleted after centrifugation at 200 x g (4°C, 20 min.). The supernatant was then centrifuged at 4100 x g to pellet the bacteria (4°C, 20 min.). DNA purification was performed using the Qiagen DNeasy Blood and Tissue Kit as follows. The Wolbachia-enriched pellet was first resuspended in 180 μL ATL buffer plus 20 μL of proteinase K (10 mg/mL) and incubated for 1 h at 54°C. After treatment with RNase A (0.2 μg/μL, at 37°C for 15 min.), DNA was recovered following the manufacturer’s instructions. DNA quantification was performed using a Nanodrop 1000 spectrophotometer (Thermo Scientific) and the Qubit 2.0 fluorimeter (Invitrogen).
Genome sequencing, assembly, and annotation
Library preparation for nanopore sequencing was performed using the protocols for the SQK-LSK109 Ligation Sequencing Kit (Oxford Nanopore Technologies, UK). Libraries were sequenced on R9.4.1 flowcells on a MinION sequencer for 48 h. Base-calling was performed using the Guppy base-caller software v4.2.2 (Oxford Nanopore Technologies, UK) using high-accuracy mode, with a quality score cut-off of 9 and minimum read length filter set of 200. Adapters were trimmed with Porechop 0.2.41 on the basecalled reads.
Prior to Illumina sequencing, targeted genome enrichment was used for wVulC and wVulM strains as described in Geniez et al. (2012). Illumina libraries were prepared and samples were sequenced at HudsonAlpha Genome Sequencing Center (Hunstville AL35806, USA) on the Illumina HiSeq 2000 (Geniez, 2013).
These two sequencing strategies led to 2,001,609 and 2,581,285 Nanopore long reads and 72,894,720 and 64,625,816 Illumina reads for wVulC and wVulM, respectively. The wVulP genome was assembled from 1,795,327 Nanopore long reads.
Bacterial genome assembly was performed using the long reads assembler Flye v2.8.1 (Kolmogorov et al., 2019). The best assembly was chosen based on the expected size and circular status of the genome, after comparing meta and single mode assembly using several overlap parameters. The resulting assemblies were first polished with Nanopore reads using Nanopolish v0.14.0 (Loman et al., 2015). Additional Illumina polishing using Medaka v1.6.0 was performed on assemblies for wVulC and wVulM strains after assessing the quality of Illumina reads with FastQC v0.11.9 (Andrews, 2010) and removing adapters and quality filtering using Fastp v0.21.0 (Chen et al., 2018). Genome completeness was assessed by using the Benchmarking Universal Single-Copy Orthologs (BUSCO) pipeline v5.4.6 and the rickettsiales_odb10 database (Manni et al., 2021). The polished assemblies with the highest BUSCO score were selected for further analysis.
The genomes were functionally annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016).
Comparative genomics and phylogenomics
The three Wolbachia genomes were presented starting with dnaA gene as for wMel (Wu et al., 2004) and most other Wolbachia genomes. Comparisons between the three genomes were performed and visualized using FastANI v1.3.3 (Jain et al., 2018) implemented in the NanoGalaxy platform (de Koning et al., 2020) and Mauve v2.4.0 progressive alignments (Darling et al., 2010) using Circos software v0.69–9 (Krzywinski et al., 2009).
Comparisons of wVulC genome with a previous draft genome (GCA_001027565.1) and the wVulC inserts identified into the pill bug nuclear genome (Leclercq et al., 2016) were performed using FastANI v1.3.3 (Jain et al., 2018) implemented in the NanoGalaxy platform (de Koning et al., 2020).
Orthofinder v2.5.5 (Emms and Kelly, 2019) implemented in the NanoGalaxy platform (de Koning et al., 2020) was used to identify orthologous sequences and infer the species tree from 29 Wolbachia genomes belonging to the six A-F supergroups. Four hundred and fourteen single-copy proteins were aligned with MAFFT v. 7.505 (Katoh et al., 2017). The concatenated alignment was used for phylogenetic reconstruction by maximum-likelihood with the iQtree 2.1.2 (Minh et al., 2020) implemented in the NanoGalaxy platform (de Koning et al., 2020). The best model (JTT + F + R6) was selected by ModelFinder (Kalyaanamoorthy et al., 2017), implemented in IQ-TREE, based on Bayesian Information Criterion. Branch support was assessed using ultrafast bootstrap with 1,000 replicates. The resulting consensus tree was drawn using iTol v6.8.2 (Letunic and Bork, 2021).
Analyses of Wolbachia prophage regions, mobile elements, T4SS, effectors, and biotin operon
Prophage regions (WO prophages) in the three Wolbachia assemblies were estimated using the PHASTEST web server (Wishart et al., 2023). WO phage genomic maps were drawn using the gggenes package v.0.5.1 in R (Wilkins, 2023). EAMs (Bordenstein and Bordenstein, 2016; Bordenstein and Bordenstein, 2022) were identified by annotating CDSs flanking or located within the PHASTEST-predicted prophage regions through BLASTp queries using the NCBI clustered protein database. Alignment of the large serine recombinase of the WO prophages was generated with MAFFT v. 7.505 (Katoh et al., 2017) and the phylogeny was inferred by maximum likelihood using the iQtree server v1.6.12 (Trifinopoulos et al., 2016). The best substitution model (JTT + F + G4) was selected by ModelFinder (Kalyaanamoorthy et al., 2017) and tree topology was tested by ultrafast bootstrap (Minh et al., 2013) of 1,000 iterations.
Insertion sequence (IS) elements were determined in the three genomes using the ISEScan v1.7.2.3 (Xie and Tang, 2017) implemented in the NanoGalaxy platform (de Koning et al., 2020). Candidate intron sequences were identified using RASTtk pipeline (Brettin et al., 2015) and BLASTp searches against the Database for Bacterial Group II Introns (Candales et al., 2012). The comparative location of mobile elements between the three genomes was represented using Circos software v0.69–9 (Krzywinski et al., 2009).
Prediction of intact secretion systems and secreted proteins was performed using EffectiveDB queries v5.2 (Eichinger et al., 2016).
Genes of the biotin operon were identified by BLASTn using the sequences previously identified in the wVulC draft genome. Comparison of the structure of the operons in the three Wolbachia genomes was visualized using gggenes R package (Wilkins, 2023).
Results
Sex ratio bias in Wolbachia-infected host lineages
The Wolbachia strains sequenced in this study have been isolated from females belonging to three different lineages of A. vulgare, which exhibit sex ratio biases (Figure 1; Supplementary Table S1; Supplementary Figure S1). The male proportions (mean ± se) were 19.71 ± 0.03% for ZN lineage (infected with wVulC), 23.45 ± 0.03% for BI lineage (infected with wVulM), and 19.96 ± 0.03% for CP lineage (infected with wVulP) with no significant differences. The greater variance in male proportions in the infected lineages reflected variations in the Wolbachia transmission rate (Figure 1). In contrast, the uninfected BF control line showed a balanced sex ratio of close to 50% (49.9 ± 0.01%).
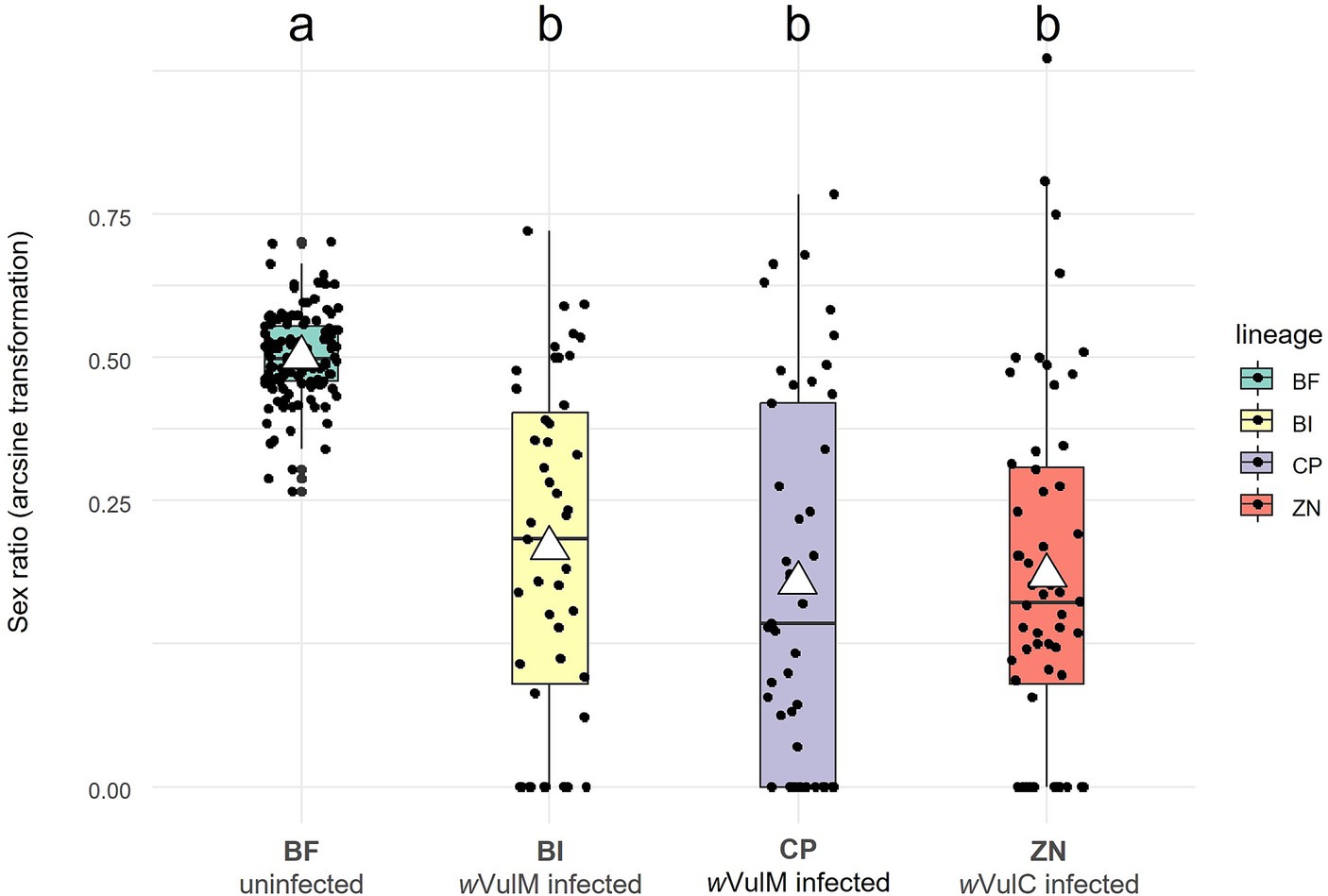
Figure 1. Boxplot showing sex ratios (proportion of males) of the four laboratory lineages of A. vulgare (BF, BI, CP, and ZN) over five years. BF lineage is uninfected whereas BI, CP, and ZN lineages are infected with wVulM, wVulP, and wVulC Wolbachia strains, respectively. Sex ratios have been arcsine transformed. White triangles correspond to mean values. Statistical analysis was performed using a Generalized Linear Mixed Model (conditional R-squared = 0.222).
Comparative genomics of the three feminizing strains
The three Wolbachia genomic sequences were assembled into single molecules of 1,711,483 bp for the wVulC strain, 1,638,198 bp for the wVulM strain and 1,566,000 bp for the wVulP strain, with a G + C content ranging from 34.8 to 34.9% (Figure 2). The two wVulC and wVulM genomes were circularized and even though wVulP was not completely closed, it was a genome of the same quality assembled into a single scaffold. The general features of the genomes are presented in Table 1. Their sizes were comparable to those of other Wolbachia strains inducing reproductive phenotypes (Sun et al., 2001), with each genome containing between 1,282 and 1,484 protein-coding genes (Table 1). The wVulC, wVulM, and wVulP assemblies were assessed by BUSCO, showing a very small number of fragmented and missing genes (Table 1), resulting in high scores (99.8, 99.5 and 98.3% for wVulC, wVulM, and wVulP, respectively). The three genomes were very similar as shown by the high ANI value obtained from pairwise comparisons (> 98%, Supplementary Table S2). However, numerous genomic rearrangements have been identified, particularly in the wVulP genome (Figures 2, 3; Supplementary Figures S2A–C). Orthofinder analysis of the three genomes revealed 1,094 orthogroups in common out of 1,259 (Supplementary Table S3). wVulC and wVulM shared 126 orthogroups whereas each of these strains shared only 15 and 10 orthogroups with wVulP. Finally, one to 11 specific orthogoups corresponded to four, three and twenty-four specific genes of wVulC, wVulM, and wVulP, respectively (Supplementary Table S3). The wVulC specific genes were two hypothetical protein paralogs (wVulc_000316 and wVulc_000382; 100% amino acid identity) and two DUF4815 domain-containing protein paralogs (wVulc_000708 and wVulc_001210; 99.7% amino acid identity). In wVulM, the three specific genes were paralogs of recombinase (wVulm_000691, wVulm_001174 and wVulm_001253; 100% amino acid identity) corresponding to the third recombinase in the core module prophage WOVulM1_2, WOVulM3_4, and WOvulM5_6 (see below). As to the wVulP-specific genes, five were duplicated mobile elements (IS and group II introns), one was a duplicated phage tail protein, one was a duplicated SET domain-containing protein, one was a duplicated ANK gene and three were duplicated hypothetical proteins. Each duplicated gene was 100% identical with the exception of the phage tail paralogs which were 77% identical (Supplementary Table S3).
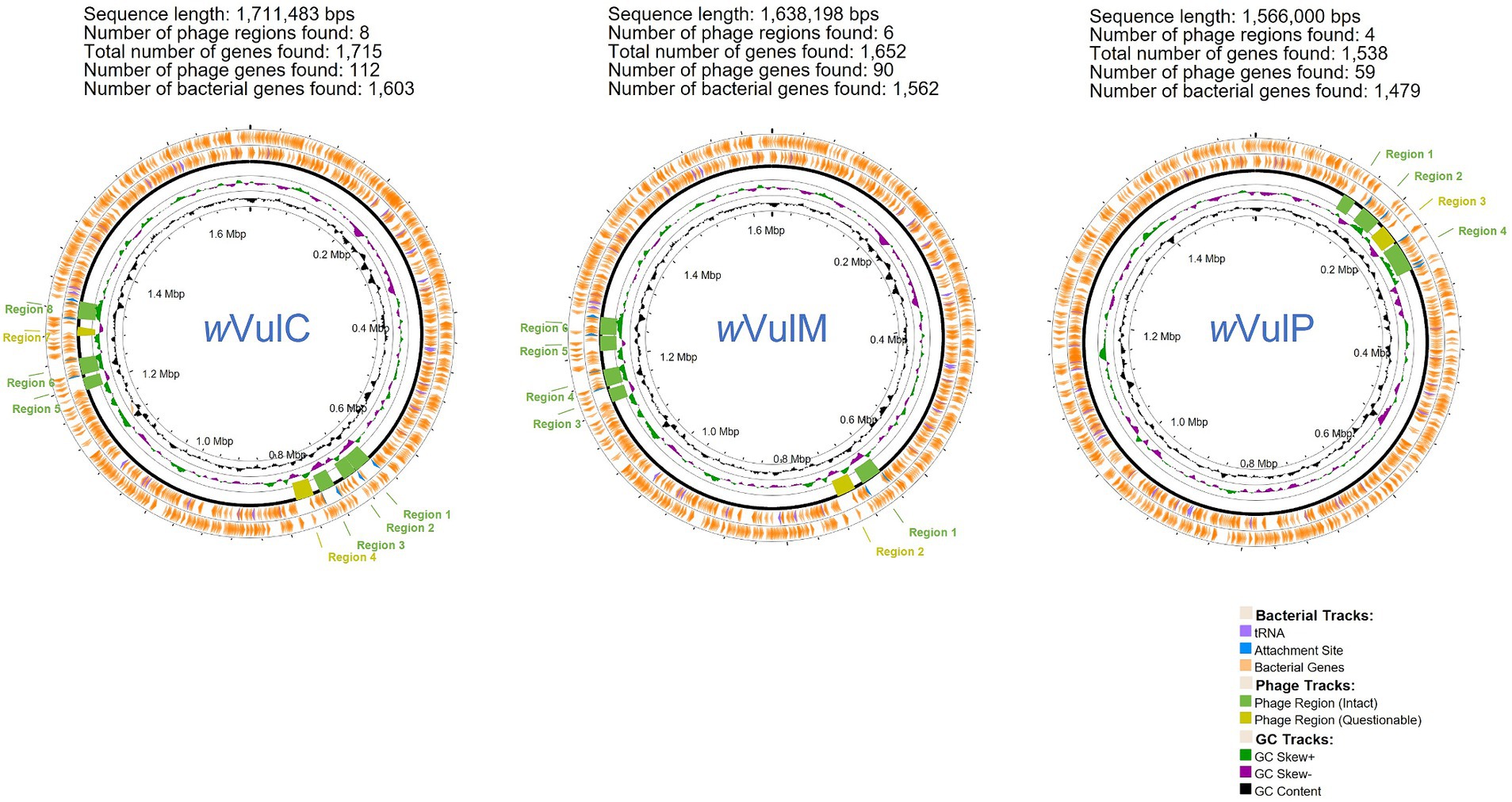
Figure 2. Graphical representation of the circular genome map of the three Wolbachia strains wVulC, wVulM, wVulP. PHASTEST description, from the outside to the center: (1) Predicted CDSs transcribed in the counterclockwise direction. (2) Predicted CDSs transcribed in the clockwise direction. (3) Predicted phage regions (intact and questionable). (4) GC skew. (5) GC content.
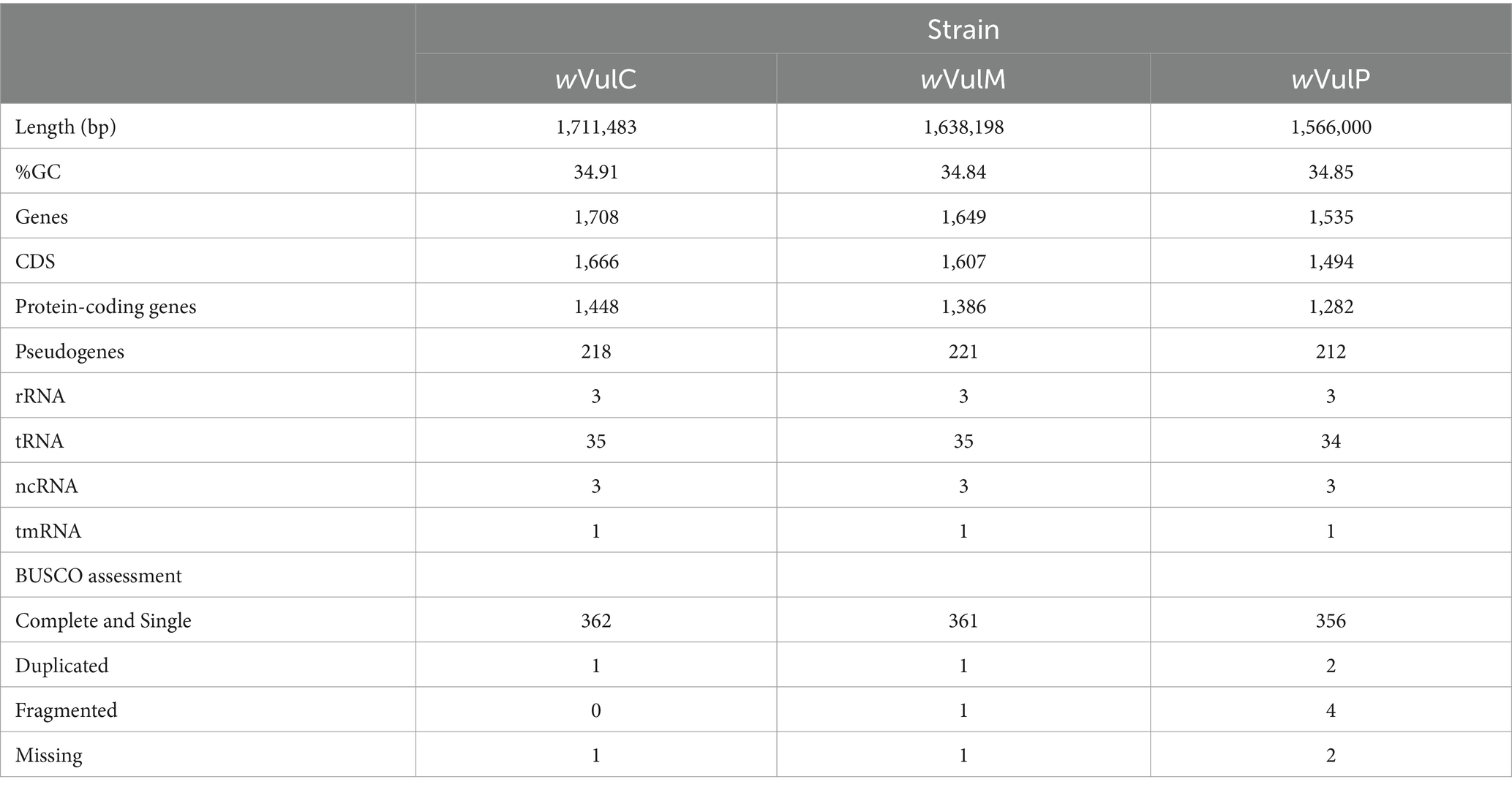
Table 1. Comparative statistics and BUSCO assessment of the three Wolbachia genomes wVulC, wVulM, and wVulP isolated from A. vulgare.
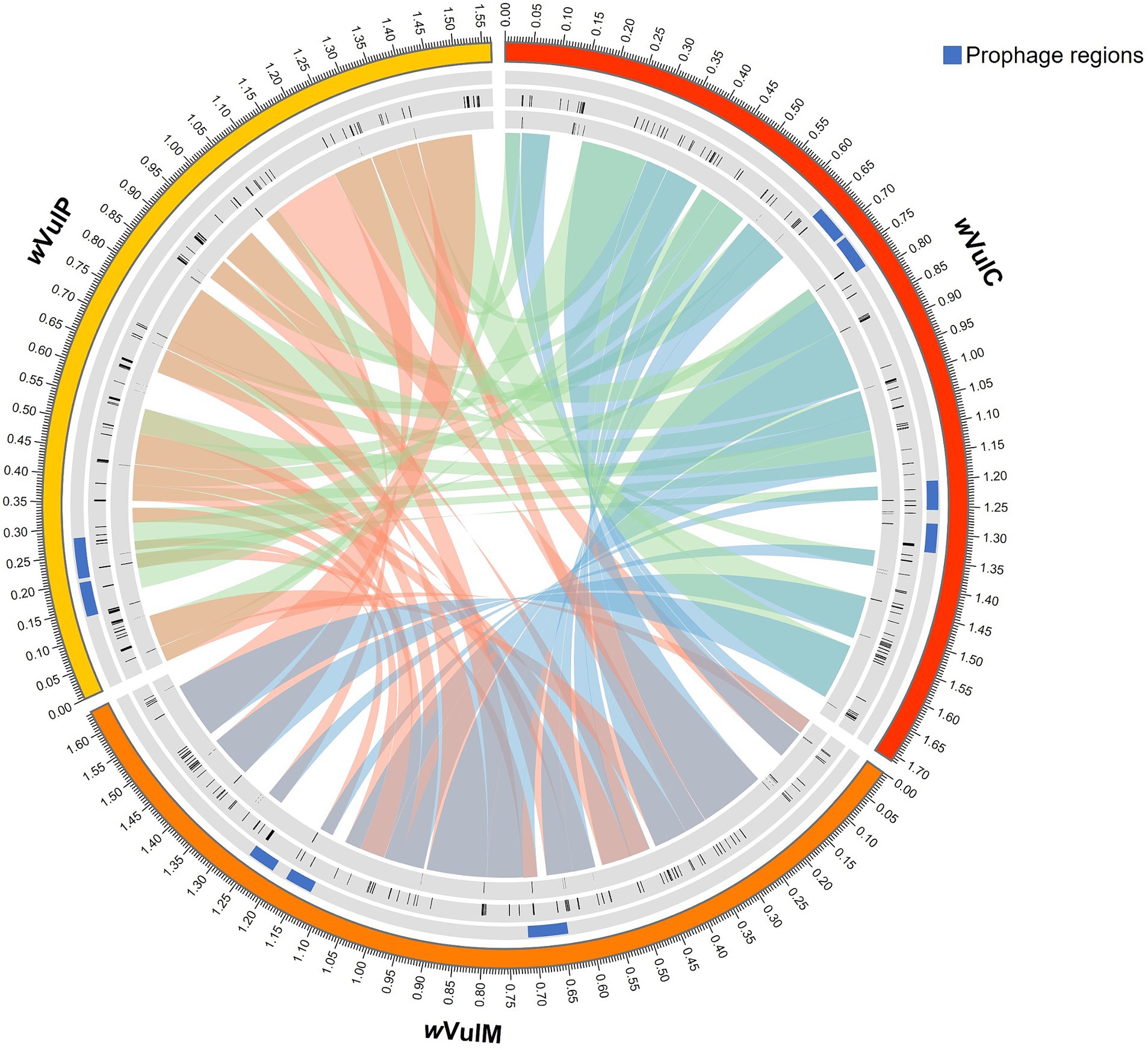
Figure 3. Circos representation of the progressive Mauve alignment and mobilome of the three Wolbachia strains (wVulC, wVulM, wVulP) of A. vulgare. First inner ring: WO prophage regions in blue. Second inner ring: complete IS elements in black. Third inner ring: group II introns in black.
Furthermore, comparison of the complete, closed wVulC genome with a previous draft genome obtained by Sanger sequencing showed a high ANI value of 99.9%. The non-collinearity between these two sequences was due to artificial joining of the 10 contigs in the Sanger draft assembly (Supplementary Figure S3). Comparison of the wVulC genome was also carried out with the f element (derived from wVulC) which comprised nine scaffolds spanning 3.13 Mb (Leclercq et al., 2016). This showed a high average nucleotide identity of 99.6% (Supplementary Figure S4) but revealed numerous genomic rearrangements indicating multiple insertion and duplication events of the f element in the A. vulgare genome.
Phylogenomic relationships
The phylogenomic position of the three Wolbachia from A. vulgare was determined by comparison with 26 publicly available and annotated Wolbachia genomes, including five from supergroup A, thirteen from supergroup B, three from supergroup C, three from supergroup D and one for supergroup E and F strains from various host species (Supplementary Table S4). A total of 415 single-copy gene ortholog clusters were used to reconstruct the phylogenomic tree (Figure 4; Supplementary Table S5). The three Wolbachia strains isolated from A. vulgare belonged to the supergroup B, forming a separated clade, with wVulC and wVulM being closely related (bootstrap support 100, Figure 4).
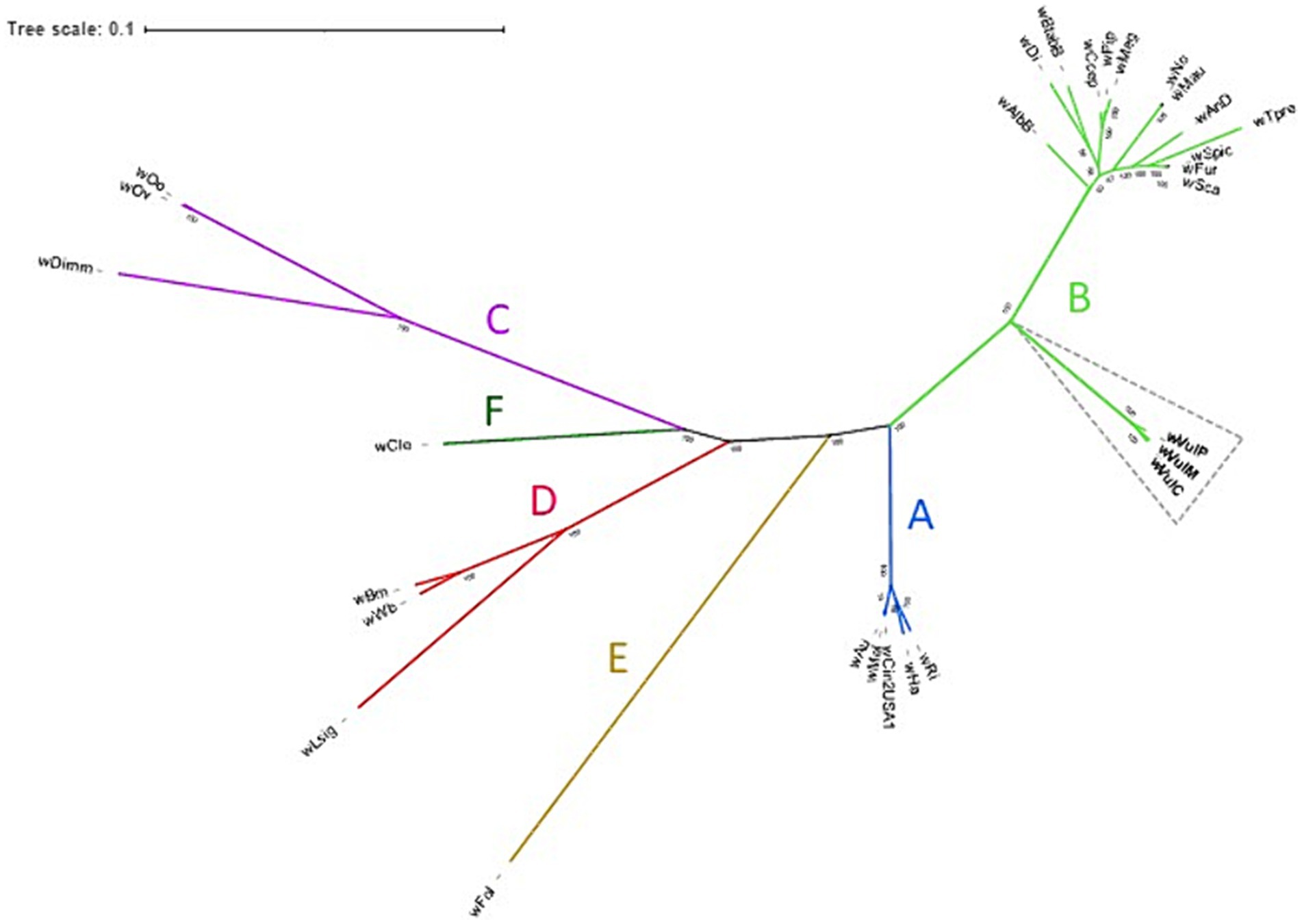
Figure 4. Unrooted maximum-likelihood consensus tree based on concatenated alignments of 415 single-copy orthologues in 29 Wolbachia genomes. Newly assembled genomes are shown in bold. Tree scale bar represents substitutions per site. Bootstrap support values (> 50%) from 1,000 replicates are shown at the edges.
Mobilome identification
ISEScan predicted 105, 103 and 77 complete IS elements from 10 different families in wVulC, wVulM and wVulP, respectively, representing around 8% of the genome size (Supplementary Table S6). The IS families most represented in the three genomes, when complete sequences were taken into account, were IS110, IS256, IS3, IS4, IS5 with the exception of wVulp where IS3 had only one representative. Specific IS clusters were ISNCY_191 for wVulC, IS110_236, new_343 and IS4_64 for wVulP (Supplementary Table S6). Many IS element positions coincided with regions where synteny breaks occurred (Figure 3), suggesting that IS elements may have contributed to the genomic rearrangements among the three genomes.
RASTtk annotations and BLASTp searches against the Database for Bacterial Group II Introns showed 29, 25 and 29 group II intron-associated genes in wVulC, wVulM and wVulP, respectively (Supplementary Table S7). The sum of the lengths of these genes was 17,892 bp, 14,556 bp, and 13,332 bp respectively, representing 0.9–1% of the genome size. The distribution of these introns was not homogeneous and regions of several successive genes were observed (Figure 3). As with IS elements, these group II introns are often located in regions of synteny breaks (Figure 3).
Prophage regions were identified by PHASTEST and BLASTp searches using annotated CDS, both in predicted and flanking regions. The results were also manually curated using the recently published WO prophage annotations (Bordenstein and Bordenstein, 2022). This strategy enabled us to extend the regions initially predicted by PHASTEST (Figure 2), notably by including EAMs (Bordenstein and Bordenstein, 2016). Functional annotations of these regions were shown in Supplementary Table S8.
We identified four complete prophage regions in wVulC measuring 70,719 bp (WOVulC1_2), 57,601 bp (WOVulC3_4), 55,840 bp (WOVulC5_6), and 64,755 bp (WOVulC7_8). WOVulC1_2 and WOVulC3_4 formed a continuous region with two sets of all core prophage modules separated by two EAMs (Figure 5A; Supplementary Table S8). Three complete prophage regions were identified in wVulM measuring 63,169 bp (WOVulM1_2), 55,822 bp (WOVulM3_4) and 63,644 bp (WOVulM5_6) (Figure 5B; Supplementary Table S8). In wVulP, only two prophage regions of 63,842 bp (WOVulP1_2) and 75,199 bp (WOVulP3_4) were found (Figure 5C; Supplementary Table S8). They formed a continuous prophage region separated by two EAMs. In addition, WOVulP3_4 is interrupted by an internal EAM. Overall, these prophages accounted for around 10% of each genome. The core genomic content of each prophage WO began with a large serine recombinase and ended with a patatin. With the exception of prophages WOVulC3_4 and WOVulP1_2, the majority of prophages contained two different serine recombinase genes, the second downstream of the first. In tandem to the latter, a third short recombinase was specifically present in all three prophage regions of wVulM (Figure 5B; Supplementary Table S8).

Figure 5. Annotation of prophage regions of wVulC, wVulM, wVulP by PHASTEST and manual curation. (A) wVulC harbors four intact prophage regions (70.7 kb, 57.6 kb, 55.8 kb and 64.8 kb). (B) wVulM harbors three intact prophage regions (63.2 kb, 55.8 kb, and 63.6 kb) and (C) wVulP harbors two intact prophage regions (63.8 kb and 75.2 kb). Lengths of the graphs are proportional to the size (bp) of the prophages. The legends indicate the color coding of the modules. EAM-like regions are highlighted. Locus tags of T4SS effectors are indicated.
Four distinct WO variants (sr1WO-sr4WO) have been described based on the large serine recombinase phylogeny and core module synteny (Bordenstein and Bordenstein, 2022). Based on the phylogeny of the first serine recombinase genes marking the start of the core modules, classification of the WO prophages of the three Wolbachia strains showed that they all belonged to the sr3WO group (Figure 6). The general structure of these prophages corresponded to the module synteny described for sr3WO variants, which includes an internal core prophage WO region flanked by EAM genes (Bordenstein and Bordenstein, 2022). Although the synteny of genes within each core module was consistent with the sr3WO organization (i.e., connector/baseplate, head, replication & repair, tail), variations between prophages were observed, some due to the presence of mobile elements (transposases and group II introns). These variations were consistent with the phylogenetic position established with the serine recombinase gene, with the closest prophages sharing a better synteny. In particular, strong collinearity is observed between the closely related core prophage modules WOVulC1_2 and WOVulM1_2, WOVulC5_6 and WOVulM_3_4 and WOVulC7_8 and WOVulM5_6 (Figure 6). In contrast, WOVulP3_4 had a particular structure with 3 EAM-like regions, including one region localized within the core prophage module (Figure 5C).
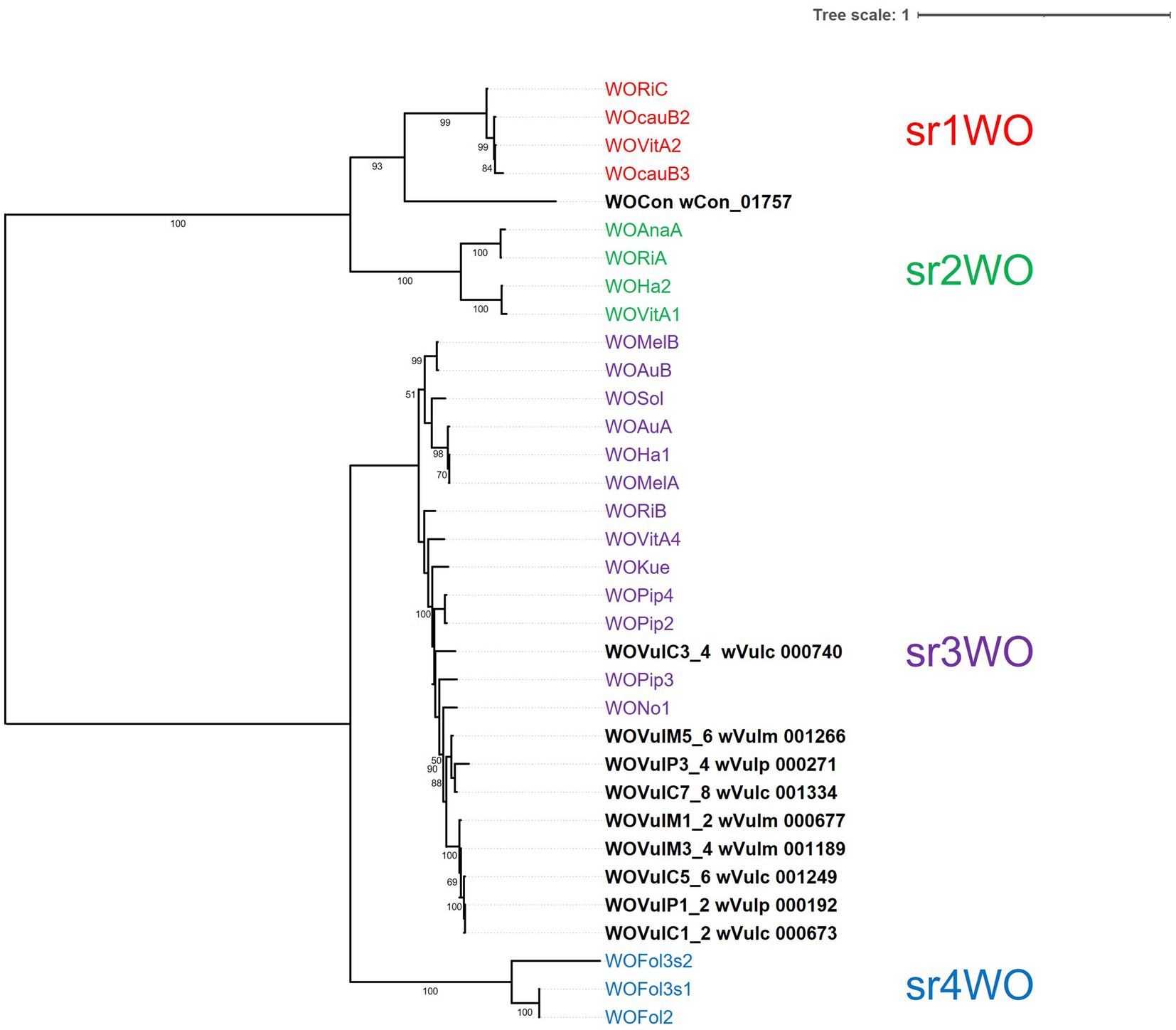
Figure 6. Maximum-likelihood phylogeny based on the large serine recombinase of Wolbachia prophage regions. The phylogeny was generated from 34 sequences (630 amino acid sites) using the JTT + F + G4 model. The tree was rooted at the mid-point. Tree scale bar represents substitutions per site.
Using PHASTEST, we also examined the wCon assembly of 237 contigs and identified one large serine recombinase (locus wCon_01757) in an incomplete prophage region due to the end of the contig. This sequence was added in the phylogenetic tree, showing a putative prophage belonging to sr1WO cluster (Figure 6). This could reflect different prophage features depending on the Wolbachia reproductive phenotype.
Genes encoding proteins containing an ankyrin repeat domain (ANK genes) were localized in each core prophage module with a highly conserved arrangement in the three Wolbachia genomes (Figures 5A–C). At the beginning of the module, two contiguous ANK genes were located just after a DUF2924 domain-containing protein downstream of the large serine recombinase, then toward the middle, one ANK located just after the phage terminase large subunit gene (except for WOVulP1_2) and at the end of the module, one ANK gene located between a patatin-like phospholipase and a holin-like protein constituting a lytic cassette (Bordenstein and Bordenstein, 2022).
Regions similar to EAMs previously reported in other Wolbachia genomes were also observed in wVulC, wVulM and wVulP prophage regions (Figures 5A–C). These EAM-like regions contained genes encoding transcription regulators (helix-turn-helix HTH domain-containing protein) and ankyrin repeat proteins that might be involved in the host manipulation (Bordenstein and Bordenstein, 2022). RadC genes encoding JAB domain-containing proteins were also identified in EAMs from WOVulC1_2, WOVulC3_4, WOVulM1_2, and WOVulP1_2 prophages. Finally, mobile elements (Group II introns and transposases) were also frequent in EAMs. Here again, there was a strong collinearity between the EAMs of closely related prophages, with most EAMs being very similar in composition and structure, with the exception of those of WOVulP3_4.
T4SS and phage-related putative feminizing effectors
The two operons characteristic of the T4SS were identified in the three genomes (Supplementary Table S9). The virB3-virB6 operon was constituted of virB3, virB4 and four virB6 genes. The virB8-virD4 operon was constituted of virB8, virB9, virB10, virB11 and virD4 genes. All these genes were highly conserved between the three genomes (96 to 100% of amino acid identity) with the exception of the second duplicated virB6 (83% amino acid identity).
The prediction of secreted proteins showed a high number of potential T4SS effectors among the total number of effectors: 97 (92 unique genes) out of 1,530 for wVulC, 100 (96 unique genes) out of 1,474 for wVulM and 83 (82 unique genes) out of 1,353 for wVulP (Supplementary Table S9). Among these T4SS effectors, particular attention was paid to genes present in the prophage regions and common to all three genomes and the f element. This led to the identification of genes encoding an ankyrin-repeat protein, an HTH-domain protein and a hypothetical protein.
Ten ANK genes were shared by all the three Wolbachia strains, including one which is localized in the core prophage modules. It is represented by two homologs (wVulc_000675 and wVulc_001247) in WOVulC1_2 and WOVulC5_6, two homologs (wVulm_000679 and wVulm_001187) in WOVulM1_2 and WOVulM3_4, and one homolog (wVulp_000190) in WOVulP1_2 (Figures 5A–C). These five ANK genes were highly conserved, from 98.9 to 100% amino-acid identity (Supplementary Table S9). Moreover, seven homologs of this gene were also present in the f insert (Wxf_00764, Wxf_00854, Wxf_01743, Wxf_02351, Wxf_02903, Wxf_0307 and, Wxf_03107) with an amino acid identity ranging from 77.2 to 97.4% (Supplementary Table S9).
A gene encoding a transcriptional regulator (HTH domain protein) shared by all three strains and the f element, was also predicted as a T4SS effector (Supplementary Table S9). This gene was localized in the EAMs of the WOVulC3_4 (wVulc_000790), WOVulM1_2 (wVulm_000729) and WOVulP3_4 (wVulp_000216) prophages (Figures 5A–C). Interestingly, five homologs (Wxf_00824, Wxf_00827, Wxf_00907, Wxf_01690 and Wxf_02404) of this gene were annotated in the largest scaffold of the f element. Conservation of residues of these sequences was low (around 37% of amino acid identity) but the HTH domain is still present.
Finally, 18 hypothetical proteins common to all three genomes were also predicted as T4SS effectors. Among them, one with 100% amino acid identity, was located in the WOVulC3_4, WOVulM1_2, and WOVulP1_2 (wVulc_000778, wVulm_000717 and wVulp_000155 respectively) (Supplementary Table S8; Figures 5A–C). This hypothetical protein was also identified in the f element, with a degree of conservation of around 70% (Supplementary Table S8; Wxf_00810, Wxf_00893, Wxf_01704, and Wxf_02390).
Another T4SS effector shared by the three genomes, a latrotoxin-related protein (wVulc_001131, wVulm_001070, and wVulp_000491) might be involved in host-Wolbachia interactions. This gene present a latrotoxin C-terminal domain characteristic of the latrotoxins of the black widow spider (Grishin, 1998). It was not located in the prophages but in a region containing phage relic, just two CDS upstream a phage terminase large subunit encoding gene. Interestingly, one homolog (Wxf_02470; 90.4% amino acid identity) was also present in the f element (Supplementary Table S9).
Conservation of the biotin pathway
A highly conserved biotin operon was identified in the three Wolbachia genomes (Figure 7), suggesting a potential supply of this vitamin by the bacteria, although this role is probably not essential given the non-obligatory nature of the association. This operon contained the six canonical biotin genes [bioB, bioF, bioH, bioC, bioD and bioA; (Gerth and Bleidorn, 2016)] which are highly conserved (99 to 100% amino acid sequence identity). This operon was inverted in the wVulP genome. The structure of the nearby flanking regions was also conserved, bordered by IS elements and containing two identical hypothetical proteins and one ankyrin gene (97 to 99% amino acid sequence identity) (Figure 7). In wVulC and wVulM, this region was bordered by identical IS5 elements belonging to two different clusters (IS5_226 and IS5_519) (Supplementary Table S6).
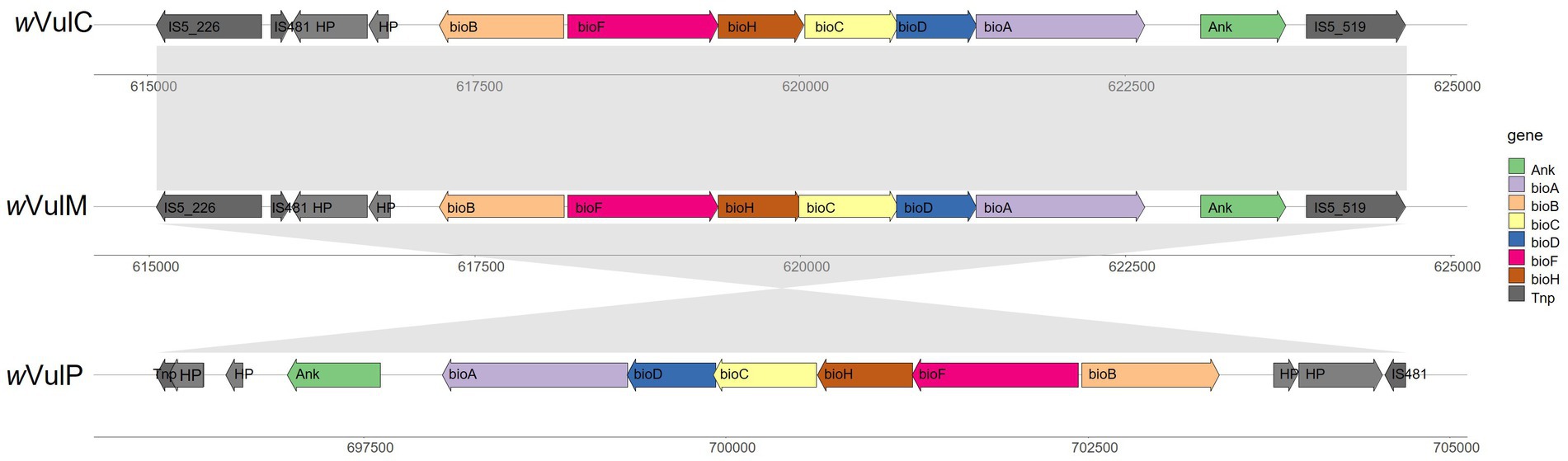
Figure 7. Comparison of the biotin operon of wVulC, wVulM, and wVulP and flanking regions showing collinearity.
Discussion
Although studies on Wolbachia have gained momentum since the late 1990s, the mechanisms involved in host-Wolbachia interactions are still poorly understood (Landmann, 2019). Indeed, the search for bacterial factors responsible for the different phenotypes induced by Wolbachia has long been the subject of intensive study (Rice et al., 2017; Carpinone et al., 2018). The sequencing of numerous Wolbachia genomes over the last twenty years has provided access to all the bacterium’s genetic information and opened up the way to comparative genomic analyses (Kaur et al., 2021). The presence of prophages in most of these genomes was of particular interest, as these mobile elements could carry effectors of the feminizing phenotype of this endosymbiont, in particular genes involved in mutualistic relationships or in the manipulation of host reproduction (Bordenstein and Bordenstein, 2016). Although in most Wolbachia genomes the prophages appeared to be sedentary, it has been shown in the parasitoid wasp Nasonia vitripennis that they can still be active and propagate (Bordenstein et al., 2006). In this Wolbachia strain, it was also shown that the sedentarized copies had undergone numerous rearrangements (Bordenstein and Bordenstein, 2022). In the end, these prophage analyses identified the cifA and cifB genes as well as the wmk gene, involved in CI and MK, respectively (LePage et al., 2017; Perlmutter et al., 2019). While the genome of the Wolbachia strain of Trichogramma contained only degenerated prophage regions (Lindsey et al., 2016), two putative PI factors were identified in a degraded EAM (Fricke and Lindsey, 2024).
In this study, we performed a comparative genomic analysis of the three Wolbachia strains wVulC, wVulM, and wVulP infecting A. vulgare, focusing on the identification of putative feminizing factors.
Since their collection from the wild, the three Wolbachia-infected lineages of A. vulgare were stably maintained in the laboratory. The sex-ratio of the progenies as well as the presence of the corresponding Wolbachia strains in female genitors were regularly monitored by diagnostic PCR. Although multiple strains may coexist in A. vulgare populations, no individual has been found to be multiinfected (Verne et al., 2007). Moreover, feminization led to cytoplasmic sex determination in natural populations where all infected Wolbachia-infected females were ZZ reversed males (Juchault et al., 1993; Rigaud et al., 1997). This implies variations in offspring sex ratios as a function of Wolbachia transmission rate, as it has been demonstrated in A. vulgare populations infected with either wVulC or wVulM (Cordaux et al., 2004). In the three laboratory lineages of A. vulgare, male biased ratio may be thus observed in some offspring of infected females. Far from being surprising, this confirms that these females were necessarily ZZ inverted males, which can produce male biased offspring when the Wolbachia transmission rate falls below 50%. In contrast, the balanced male ratio observed in the uninfected control line was the result of chromosomal sex determination with individuals of ZZ and WZ genotypes. All these observations confirmed the feminizing phenotype of the three Wolbachia strains of A. vulgare, leading to transmission rate-dependent cytoplasmic sex determination.
Consistent with previous single gene (16S rDNA, ftsZ, wsp, and GroE) phylogenies (Bouchon et al., 1998; Cordaux et al., 2001; Wiwatanaratanabutr et al., 2009), as well as multi-locus sequence typing data (Sicard et al., 2014), wVulC, wVulM and wVulP belonged to the supergroup B forming a separated clade in phylogenomic analyses (bootstrap support 100, Figure 3). This reflected a general pattern of Wolbachia from terrestrial isopods, which clustered in a monophyletic clade (referred as the Oni clade for Oniscidea; (Cordaux et al., 2001)) within supergroup B. This result also confirmed the genetic proximity of the wVulC and wVulM strains.
The wVulC, wVulM, and wVulP genomes were between 1.7 and 1.6 Mb in size and contained between 1,700 and 1,500 genes, corresponding to a coding density of 75 to 67% (Table 1). These characteristics correlated with genome size, wVulC being the largest and wVulP the smallest. They were also consistent with the features of the B supergroup of Wolbachia which are known to have a larger number of genes than the A supergroup (Vancaester and Blaxter, 2023). The genome length of the three strains was among the longest known for Wolbachia (Scholz et al., 2020). These sizes were highly correlated with the extent of the mobilome (including prophages, IS elements and group II introns), representing a total of 415,545 bp for wVulC, 338,395 bp for wVulM and 287,918 bp for wVulP, i.e., from 24 to 18% of the genome. The high density of mobile and repeated elements in the three genomes, in particular IS belonging to the IS110 and IS256 families, could explain the genomic rearrangements observed (Figure 3; Supplementary Figures S2A–C). The copy number of group II introns was also one of the highest reported in Wolbachia genomes (Leclercq et al., 2011). These features were, to varying degrees, common to Wolbachia reproductive parasites, as demonstrated by the first sequenced genomes (Wu et al., 2004; Klasson et al., 2008, 2009). Particular attention was paid to the wVulC strain, as a Sanger sequence assembly (GCA_001027565.1) and wVulC inserts identified into the pill bug nuclear genome are publicly available (Leclercq et al., 2016). Consistently, genomic comparisons have confirmed that locally collinear blocks are conserved, but that significant genomic rearrangements are probably due to the impact of the mobilome, in particular the IS and group II introns (Figure 3; Supplementary Figures S2A–C, S3, S4). This is particularly obvious in the comparison with the nine scaffolds of the f element where, despite a high average nucleic identity, collinearity remains low. This pattern was also evident when only the longest scaffold of 2,798,100 bp (LYUU01002088.1) is included in the comparison (Leclercq et al., 2016). It has long been known that Wolbachia can recombine with several implications on the evolution of these bacteria (Jiggins et al., 2001; Werren and Bartos, 2001). Based on single gene analyses, two recombination events have been shown in feminizing Wolbachia and more specifically in wVulP (Verne et al., 2007) and complete pathway for homologous recombination pathway was found in wVulC (Badawi et al., 2014). Wolbachia’s great ability for recombination could therefore explain the genomic plasticity observed.
A complete biotin operon has been identified in the three genomes, suggesting that these Wolbachia strains were capable of synthesizing B vitamins. It was also conserved in the f element inserted into the genome of A. vulgare. Although generally absent from most Wolbachia genomes, a complete biotin operon has been identified in around fifteen Wolbachia strains (Beliavskaia et al., 2023). However, it has been shown that only the Wolbachia wCle from the bedbug Cimex lectularius supplies B vitamins to its host (Nikoh et al., 2014). This beneficial symbiosis was not demonstrated in the other strains harboring the biotin operon. For example, biotin supplementation remains unlikely in the wCfeF strain of Ctenochephalides felis, since the bioB gene is frameshifted (Beliavskaia et al., 2023). Similarly, the wOo strain of Onchocerca ochengi has a completely disrupted biotin operon (Darby et al., 2012; Nikoh et al., 2014). It therefore seems that this operon undergoes evolutionary events leading to its degradation, presumably due to the absence of a selective advantage for the host. In the particular case of A. vulgare, although the biotin operon is complete in the three Wolbachia genomes and also in the f element, and may supply B vitamins as well, the absence of an obligate association with the host may indicate that this contribution is not essential. Finally, the presence of IS and transposases in the flanking regions of the operon is consistent with an acquisition by lateral gene transfer, supported by the lack of congruence between the phylogenies of the biotin genes and the Wolbachia (Nikoh et al., 2014; Driscoll et al., 2020; Beliavskaia et al., 2023). Accordingly, phylogenetic analyses support the hypothesis of at least three independent acquisition of the biotin operon by Rickettsiales (Driscoll et al., 2020; Lefoulon et al., 2020).
Multiple copies of the WO prophages were identified in the genomes of all three feminizing strains. However, copy number differs from genome to genome, wVulC having 4 copies representing a total of 248,915 bp (including the EAM-like regions), followed by wVulM with three copies representing 182,635 bp and wVulP with only 2 copies representing 139,041 bp. This copy number contributes to the size of the genomes, with wVulC being the largest and wVulP the smallest, representing between 9 and 10% of the total. Multiple phage infections have been observed in many Wolbachia strains from different arthropod host species (Gavotte et al., 2006). In particular, PCR amplification of the minor capsid gene orf7 showed the presence of 4 to 6 copies of the WO phage in the terrestrial isopods A. vulgare, Porcellionides pruinosus, and Porcellio dilatatus (Braquart-Varnier et al., 2005). In agreement with our genomic data, the four copies of the orf7 amplified in A. vulgare, corresponded to the capsid assembly proteins (wVulc_000689, wVulc_000754, wVulc_001230, and wVulc_0001319), annotated, respectively, in the core regions of prophages WOVulC1_2, WOVulC3_4, WOVulC5_6, and WOVulC7_8 of the wVulC genome.
The prophage loci were not restricted to the core modules of the prophage regions, and WO-like Islands (Bordenstein and Bordenstein, 2022) corresponding to “relic” prophages have been annotated in all three genomes. Due to the presence of numerous hypothetical proteins, it was difficult to define the precise boundaries of these regions. However, a general pattern seemed to emerge for the three genomes with two main WO-like islands. The first island comprised two portal proteins associated with two ankyrin repeat proteins in wVulC (from wVulc_000522 to wVulm_000527) and in wVulM (from wVulm_000522 to wVulm_000527) and only one portal protein and one ANK gene in wVulP (from wVulp_000412 to wVulp_000415). The second WO-like island had a structure comprising a terminase large subunit, an ANK gene, a hypothetical protein, a DNA modification methylase, a Holliday junction resolvase and a RhuM domain containing protein. This module was colinear in all three genomes, ranging from wVulc_000813 to wVulc_000818 in wVulC, from wVulm_000751 to wVulm_000756 in wVulM, and, with reverse synteny, from wVulp_000974 to wVulp_000969 in wVulP. These results were in line with those observed in various Wolbachia strains where RhuM virulence genes are located close to prophage genes encoding Holliday junction resolvase and DNA methylase proteins (Kent and Bordenstein, 2010; Fallon, 2020). Finally, single prophage genes encoding proteins of head, baseplate and tail, flanked either by hypothetical proteins or mobile elements (group II intron and IS) were annotated in the three genomes. These regions are too short to constitute genomic islands, but they testify to WO prophage degradation processes in Wolbachia genomes (Bordenstein and Bordenstein, 2022). Apart from this common pattern, a small cluster of prophage WO-like genes was identified in wVulP. This region (wVulp_000778 to wVulp_000780), flanked by two hypothetical proteins, contained genes encoding one head-tail connector protein, one DUF3168 domain-containing protein and one phage tail tube protein.
The T4SS is an efficient way for Wolbachia to transfer DNA and/or proteins to eukaryotic cells (Lindsey, 2020). Indeed, putative T4SS substrates have been already described in Wolbachia (Sheehan et al., 2016; Rice et al., 2017; Carpinone et al., 2018). This secretion system, consisting of virB and virD genes clustered at two loci, was conserved in Wolbachia reproductive parasites (Pichon et al., 2009). In line with this work, the presence of these two operons were confirmed in the three genomes. The ubiquitous presence of these operons in Wolbachia strains strongly suggested that they were functional, and led to an extensive search for potential substrates (Rice et al., 2017; Carpinone et al., 2018). Off all the hundred proteins identified as putatively secreted by the T4SS, three were located in the prophage regions of all three strains and had homologs in the f element: one ankyrin repeat domain-containing protein, one HTH domain-containing protein and one hypothetical protein. ANK motifs in bacterial proteins were thought to mimic eukaryotic protein–protein interactions, enabling bacteria to interact with host factors (Jernigan and Bordenstein, 2014). T4SS-secreted ANK proteins that interact with host cells has already been identified in bacteria such as Legionella pneumophila, Anaplasma phagocytophilum or Ehrlichia chaffeenis (Rikihisa and Lin, 2010; Yu et al., 2018). They were therefore ideal candidates as symbiotic factors in reproductive parasitism. In addition, protein–protein interactions were demonstrated between an MK inducer, Oscar, a Wolbachia protein containing ankyrin repeats, and the Masc protein involved in both masculinization and dosage compensation in the moth Ostrinia furnacalis (Katsuma et al., 2022). The induced reduction in Masc accumulation led to the inhibition of masculinization and the failure of dosage compensation, resulting in the death of male offspring (Katsuma et al., 2022). Another MK factor (SpAID), a protein with ANK repeats and a OTU (ovarian tumor) deubiquitinase domain, has been identified in Spiroplasma poulsonii (Harumoto and Lemaitre, 2018). The authors proposed a model in which the OTU domain induced nuclear localization, allowing SpAID to interact through its ANK domain with the Male Specific Lethal complex, thereby disrupting dosage compensation that led to male-killing phenotype. Interestingly, this is the only ANK protein identified in the Spiroplasma genome. In wMel genome, a MK gene candidate (wmk) encoding two HTH, XRE family DNA-binding domains has been identified in the EAM of the prophage WOMelB, next to the cifA and cifB genes that are involved in CI (Perlmutter et al., 2019). Homologs of wmk were also found in prophage EAMs from different MK strains. This protein likely interacts with DNA through its two HTH domains and could act as a transcriptional regulator potentially also targeting dosage compensation mechanisms. Based on this work, the ANK gene and the HTH-domain gene identified in this study were therefore be strong putative candidates for feminization. These genes were present, as expected, both in the feminizing Wolbachia genomes and in the f element, the latter also inducing a feminizing phenotype in the host into whose genome it has been inserted (Leclercq et al., 2016). In the three feminizing strains, the ANK gene was part of the WO prophage core modules whereas the HTH domain-containing gene was localized in an EAM, like the Wolbachia CI and MK factors (LePage et al., 2017; Shropshire et al., 2020). Nevertheless, putative factors inducing PI in parasitoid wasps have been identified in Wolbachia prophage relics (Fricke and Lindsey, 2024). This is why we also paid attention to a latrotoxin-related protein localized near isolated phage genes, evoking phage relics and present in all feminizing strains and the f element. After processing of the latrotoxin C-terminal domain, the toxin encoded by this gene may be able to form ion-permeable membrane pores leading to host cell lysis, as does the latrotoxin of widow spiders venom in the cells of their prey (Zhang et al., 2012). Spider latrotoxins appear to have been acquired by lateral transfer from a bacterial endosymbiont (Bordenstein and Bordenstein, 2016). Such toxins could also be involved in the feminizing phenotype. Indeed, previous data have shown that Wolbachia do not directly target the androgenic hormone neither its receptors, but more likely the nerve centres that control the activity of the receptors (Juchault and Legrand, 1985; Herran et al., 2021). The toxin could therefore target these nerve centers and disrupt androgen receptor function.
Overall, our study highlighted three strong candidates for feminization of A. vulgare males. Further experiments need to be performed to confirm that these candidates disrupt the androgenic hormone pathway. Indeed, these genes need to be expressed during embryonic development, at the stage where the androgenic hormone expression that induces male differentiation is inhibited in infected animals. Besides, the Wolbachia load increases just at this stage, defining the window of action enabling the bacteria to counteract the masculinizing effect of the androgen hormone and induce the development of male embryos into females (Herran et al., 2020). Monitoring the expression of these candidate genes during the development of the offspring of Wolbachia-infected females could provide further support for our hypotheses. To decipher the mechanisms of feminization, it will be necessary to identify host targets with which these candidates can interact.
Data availability statement
The Wolbachia genomes of A. vulgare recovered in this study were deposited in GenBank (https://www.ncbi.nlm.nih.gov/genbank) under BioProjects PRJNA1093130, PRJNA1093132, and PRJNA1093134 and the genomes are available under the accession numbers CP156068, CP156069, and CP156070.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because animals are invertebrates and are not subject to any ethical declaration.
Author contributions
PG: Conceptualization, Investigation, Methodology, Formal analysis, Writing – original draft, Writing – review & editing. BM: Data curation, Methodology, Formal analysis, Writing – review & editing. DB: Conceptualization, Funding acquisition, Investigation, Methodology, Formal analysis, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the 2015–2020 State-Region Planning Contracts (CPER), European Regional Development Fund (FEDER) (BiodivUP project, coordinator DB), and intramural funds from the Centre National de la Recherche Scientifique (CNRS) and the University of Poitiers.
Acknowledgments
We would like to thank Yann Dussert from the EBI laboratory for his invaluable help in using the Circos software and Alexandra Lafitte from the EBI laboratory for animal rearing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1416057/full#supplementary-material
Supplementary Table S1 | Sex ratio of progenies in four different laboratory lineages of A. vulgare over 5 years. BI, CP, and ZN were lineages naturally infected with wVulM, wVulP, and wVULC Wolbachia strains, respectively. BF was an uninfected lineage.
Supplementary Table S2 | Pairwise comparison of average nucleotide identity (ANI) values between wVulC, wVulM, and wVulP genomes (wVulC as reference).
Supplementary Table S3 | OrthoFinder analysis to identify orthologous groups of genes from the three Wolbachia genomes.
Supplementary Table S4 | Wolbachia genomes used in the phylogenomic analysis.
Supplementary Table S5 | Orthogroup analysis from 29 Wolbachia complete genomes.
Supplementary Table S6 | Annotations of IS elements predicted by ISEScan analysis.
Supplementary Table S7 | Annotations of group II intron-associated genes.
Supplementary Table S8 | Annotations of the phage regions.
Supplementary Table S9 | EffectiveDB predictions of Type IV Secretion Systems (T4SS) and potential effectors.
Supplementary Figure S1 | Plot of mean male ratio of progeny from the four laboratory lines of A. vulgare (BF, BI, CP, and ZN) per year over five-years. BF lineage is uninfected whereas BI, CP, and ZN lineages are infected with wVulM, wVulP, and wVulC Wolbachia strains, respectively. White circles correspond to mean values and whiskers represent standard error.
Supplementary Figure S2 | Alignment of the three Wolbachia genomes: (A) Collinearity between wVulC and wVulM. (B) Collinearity between wVulC and wVulP. (C) Collinearity between wVulM and wVulP. Pairwise comparisons were performed by BLASTn using FastANI 1.3 software. Each red line segment indicates a reciprocal match between two sequences. The colored bar indicates the percentage of identity. Homologous regions are indicated by segments of the same color.
Supplementary Figure S3 | Alignment of the complete wVulC genome obtained by ONT sequencing technology (this study) with the previous draft genome (GCA_001027565.1; unpublished) obtained by Sanger technology. Collinearity detected by BLASTn using FastANI 1.3 software. Each red line segment denotes a reciprocal match between two sequences. Homologous regions are indicated by segments of the same color. The colored bar indicates the percentage of identity.
Supplementary Figure S4 | Alignment of the complete wVulC genome with the concatenated eight scaffolds of the Wolbachia inserts (f element; Leclercq et al., 2016) in the nuclear genome of A. vulgare. Collinearity detected by BLASTn using FastANI 1.3 software. Each red line segment denotes a reciprocal match between two sequences. Homologous regions are indicated by segments of the same color. The colored bar indicates the percentage of identity.
Footnotes
References
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed January 23, 2023).
Badawi, M., Giraud, I., Vavre, F., Grève, P., and Cordaux, R. (2014). Signs of neutralization in a redundant gene involved in homologous recombination in Wolbachia endosymbionts. Genome Biol. Evol. 6, 2654–2664. doi: 10.1093/gbe/evu207
Badawi, M., Moumen, B., Giraud, I., Grève, P., and Cordaux, R. (2018). Investigating the molecular genetic basis of cytoplasmic sex determination caused by Wolbachia endosymbionts in terrestrial isopods. Genes (Basel) 9:290. doi: 10.3390/genes9060290
Beliavskaia, A., Tan, K.-K., Sinha, A., Husin, N. A., Lim, F. S., Loong, S. K., et al. (2023). Metagenomics of culture isolates and insect tissue illuminate the evolution of Wolbachia, rickettsia and bartonella symbionts in Ctenocephalides spp. fleas. Microb Genom 9:mgen001045. doi: 10.1099/mgen.0.001045
Bordenstein, S. R., and Bordenstein, S. R. (2016). Eukaryotic association module in phage WO genomes from Wolbachia. Nat. Commun. 7:13155. doi: 10.1038/ncomms13155
Bordenstein, S. R., and Bordenstein, S. R. (2022). Widespread phages of endosymbionts: phage WO genomics and the proposed taxonomic classification of Symbioviridae. PLoS Genet. 18:e1010227. doi: 10.1371/journal.pgen.1010227
Bordenstein, S. R., Marshall, M. L., Fry, A. J., Kim, U., and Wernegreen, J. J. (2006). The tripartite associations between bacteriophage, Wolbachia, and arthropods. PLoS Pathog. 2:e43. doi: 10.1371/journal.ppat.0020043
Bordenstein, S. R., and Theis, K. R. (2015). Host biology in light of the microbiome: ten principles of Holobionts and Hologenomes. PLoS Biol. 13:e1002226. doi: 10.1371/journal.pbio.1002226
Bouchon, D., Cordaux, R., and Grève, P. (2008). “Feminizing Wolbachia and the evolution of sex determination in isopods” in Insect Symbiosis. Contemporary Topics in Entomology, vol. 3 (Boca Raton, FL: CRC Press), 273–294.
Bouchon, D., Rigaud, T., and Juchault, P. (1998). Evidence for widespread Wolbachia infection in isopod crustaceans: molecular identification and host feminization. Proc. Royal Soc. London Series B Biol. Sci. 265, 1081–1090. doi: 10.1098/rspb.1998.0402
Braquart-Varnier, C., Grève, P., Félix, C., and Martin, G. (2005). Bacteriophage WO in Wolbachia infecting terrestrial isopods. Biochem. Biophys. Res. Commun. 337, 580–585. doi: 10.1016/j.bbrc.2005.09.091
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Candales, M. A., Duong, A., Hood, K. S., Li, T., Neufeld, R. A. E., Sun, R., et al. (2012). Database for bacterial group II introns. Nucleic Acids Res. 40, D187–D190. doi: 10.1093/nar/gkr1043
Carpinone, E. M., Li, Z., Mills, M. K., Foltz, C., Brannon, E. R., Carlow, C. K. S., et al. (2018). Identification of putative effectors of the type IV secretion system from the Wolbachia endosymbiont of Brugia malayi. PLoS One 13:e0204736. doi: 10.1371/journal.pone.0204736
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cordaux, R., Michel-Salzat, A., and Bouchon, D. (2001). Wolbachia infection in crustaceans: novel hosts and potential routes for horizontal transmission. J. Evol. Biol. 14, 237–243. doi: 10.1046/j.1420-9101.2001.00279.x
Cordaux, R., Michel-Salzat, A., Frelon-Raimond, M., Rigaud, T., and Bouchon, D. (2004). Evidence for a new feminizing Wolbachia strain in the isopod Armadillidium vulgare: evolutionary implications. Heredity 93, 78–84. doi: 10.1038/sj.hdy.6800482
Darby, A. C., Armstrong, S. D., Bah, G. S., Kaur, G., Hughes, M. A., Kay, S. M., et al. (2012). Analysis of gene expression from the Wolbachia genome of a filarial nematode supports both metabolic and defensive roles within the symbiosis. Genome Res. 22, 2467–2477. doi: 10.1101/gr.138420.112
Darling, A. E., Mau, B., and Perna, N. T. (2010). Progressive mauve: multiple genome alignment with gene gain, loss and rearrangement. Plos One 5:e11147. doi: 10.1371/journal.pone.0011147
de Koning, W., Miladi, M., Hiltemann, S., Heikema, A., Hays, J. P., Flemming, S., et al. (2020). Nano galaxy: nanopore long-read sequencing data analysis in galaxy. GigaScience 9:giaa105. doi: 10.1093/gigascience/giaa105
Dittmer, J., Beltran-Bech, S., Lesobre, J., Raimond, M., Johnson, M., and Bouchon, D. (2014). Host tissues as microhabitats for Wolbachia and quantitative insights into the bacterial community in terrestrial isopods. Mol. Ecol. 23, 2619–2635. doi: 10.1111/mec.12760
Dittmer, J., and Bouchon, D. (2018). Feminizing Wolbachia influence microbiota composition in the terrestrial isopod Armadillidium vulgare. Sci. Rep. 8:6998. doi: 10.1038/s41598-018-25450-4
Driscoll, T. P., Verhoeve, V. I., Brockway, C., Shrewsberry, D. L., Plumer, M., Sevdalis, S. E., et al. (2020). Evolution of Wolbachia mutualism and reproductive parasitism: insight from two novel strains that co-infect cat fleas. PeerJ 8:e10646. doi: 10.7717/peerj.10646
Durand, S., Lheraud, B., Giraud, I., Bech, N., Grandjean, F., Rigaud, T., et al. (2023). Heterogeneous distribution of sex ratio distorters in natural populations of the isopod Armadillidium vulgare. Biol. Lett. 19:20220457. doi: 10.1098/rsbl.2022.0457
Eichinger, V., Nussbaumer, T., Platzer, A., Jehl, M.-A., Arnold, R., and Rattei, T. (2016). EffectiveDB—updates and novel features for a better annotation of bacterial secreted proteins and type III, IV, VI secretion systems. Nucleic Acids Res. 44, D669–D674. doi: 10.1093/nar/gkv1269
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Fallon, A. M. (2020). Computational evidence for antitoxins associated with RelE/ParE, RatA, fic, and AbiEii-family toxins in Wolbachia genomes. Mol. Gen. Genomics. 295, 891–909. doi: 10.1007/s00438-020-01662-0
Fricke, L. C., and Lindsey, A. R. I. (2024). Identification of parthenogenesis-inducing effector proteins in Wolbachia. Genome Biol. Evol. :evae036. doi: 10.1093/gbe/evae036
Fukui, T., Kawamoto, M., Shoji, K., Kiuchi, T., Sugano, S., Shimada, T., et al. (2015). The endosymbiotic bacterium Wolbachia selectively kills male hosts by targeting the masculinizing gene. PLoS Pathog. 11:e1005048. doi: 10.1371/journal.ppat.1005048
Gavotte, L., Henri, H., Stouthamer, R., Charif, D., Charlat, S., Bouletreau, M., et al. (2006). A survey of the bacteriophage WO in the endosymbiotic bacteria Wolbachia. Mol. Biol. Evol. 24, 427–435. doi: 10.1093/molbev/msl171
Geniez, S. (2013). Investigation of Wolbachia symbiosis in isopods and filarial nematodes by genomic and interactome studies. Poitiers: Université de Poitiers.
Geniez, S., Foster, J. M., Kumar, S., Moumen, B., LeProust, E., Hardy, O., et al. (2012). Targeted genome enrichment for efficient purification of endosymbiont DNA from host DNA. Symbiosis 58, 201–207. doi: 10.1007/s13199-012-0215-x
Gerth, M., and Bleidorn, C. (2016). Comparative genomics provides a timeframe for Wolbachia evolution and exposes a recent biotin synthesis operon transfer. Nat. Microbiol. 2, 1–7. doi: 10.1038/nmicrobiol.2016.241
Grishin, E. V. (1998). Black widow spider toxins: the present and the future. Toxicon 36, 1693–1701. doi: 10.1016/S0041-0101(98)00162-7
Harumoto, T., and Lemaitre, B. (2018). Male-killing toxin in a drosophila bacterial symbiont. Nature 557, 252–255. doi: 10.1038/s41586-018-0086-2
Herran, B., Geniez, S., Delaunay, C., Raimond, M., Lesobre, J., Bertaux, J., et al. (2020). The shutting down of the insulin pathway: a developmental window for Wolbachia load and feminization. Sci. Rep. 10:67428. doi: 10.1038/s41598-020-67428-1
Herran, B., Houdelet, C., Raimond, M., Delaunay, C., Cerveau, N., Debenest, C., et al. (2021). Feminising Wolbachia disrupt Armadillidium vulgare insulin-like signalling pathway. Cell. Microbiol. 23:e13381. doi: 10.1111/cmi.13381
Hertig, M., and Wolbach, S. B. (1924). Studies on rickettsia-like micro-organisms in insects. J. Med. Res. 44:7.
Hiroki, M., Kato, Y., Kamito, T., and Miura, K. (2002). Feminization of genetic males by a symbiotic bacterium in a butterfly, Eurema hecabe (lepidoptera: Pieridae). Naturwissenschaften 89, 167–170. doi: 10.1007/s00114-002-0303-5
Hurst, G. D. D., Jiggins, F. M., von der Schulenburg, J. H. G., Bertrand, D., West, S. A., Goriacheva, I. I., et al. (1999). Male-killing Wolbachia in two species of insect. Proc. Biol. Sci. 266:735. doi: 10.1098/rspb.1999.0698
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jernigan, K. K., and Bordenstein, S. R. (2014). Ankyrin domains across the tree of life. PeerJ 2:e264. doi: 10.7717/peerj.264
Jiggins, F. M., Schulenburg, J. H. G. V. D., Hurst, G. D. D., and Majerus, M. E. N. (2001). Recombination confounds interpretations of Wolbachia evolution. Proc. R. Soc. Lond. B 268, 1423–1427. doi: 10.1098/rspb.2001.1656
Juchault, P., and Legrand, J. J. (1985). Contribution to the study of refractory state mechanism in the androgenic hormone in Armadillidium vulgare Latr (Crustaceae, isopoda, Oniscoida) sheltering a feminizing bacteria. Gen. Comp. Endocrinol. 60, 463–467. doi: 10.1016/0016-6480(85)90082-6
Juchault, P., Legrand, J.-J., and Martin, G. (1974). Action interspecifique du facteur epigenetique feminisant responsable de la thelygenie et de l’intersexualite du crustace Armadillidium vulgare (isopode oniscoide). Ann. Embryol. Morphogenese 7, 265–276.
Juchault, P., and Mocquard, J. P. (1993). Transfer of a parasitic sex factor to the nuclear genome of the host: a hypothesis on the evolution of sex-determining mechanisms in the terrestrial isopod Armadillidium vulgare Latr. J. Evol. Biol. 6, 511–528. doi: 10.1046/j.1420-9101.1993.6040511.x
Juchault, P., Rigaud, T., and Mocquard, J.-P. (1993). Evolution of sex determination and sex ratio variability in wild populations of Armadillidium vulgare (Latr.) (crustacea, isopoda): a case study in conflict resolution. Acta Oecol. Int. J. Ecol. 14, 547–562.
Kageyama, D., Ohno, M., Sasaki, T., Yoshido, A., Konagaya, T., Jouraku, A., et al. (2017). Feminizing Wolbachia endosymbiont disrupts maternal sex chromosome inheritance in a butterfly species. Evol. Lett. 1, 232–244. doi: 10.1002/evl3.28
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Katsuma, S., Hirota, K., Matsuda-Imai, N., Fukui, T., Muro, T., Nishino, K., et al. (2022). A Wolbachia factor for male killing in lepidopteran insects. Nat. Commun. 13:6764. doi: 10.1038/s41467-022-34488-y
Kaur, R., Shropshire, J. D., Cross, K. L., Leigh, B., Mansueto, A. J., Stewart, V., et al. (2021). Living in the endosymbiotic world of Wolbachia: a centennial review. Cell Host Microbe 29, 879–893. doi: 10.1016/j.chom.2021.03.006
Kent, B. N., and Bordenstein, S. R. (2010). Phage WO of Wolbachia: lambda of the endosymbiont world. Trends Microbiol. 18, 173–181. doi: 10.1016/j.tim.2009.12.011
Klasson, L., Walker, T., Sebaihia, M., Sanders, M. J., Quail, M. A., Lord, A., et al. (2008). Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol. Biol. Evol. 25, 1877–1887. doi: 10.1093/molbev/msn133
Klasson, L., Westberg, J., Sapountzis, P., Näslund, K., Lutnaes, Y., Darby, A. C., et al. (2009). The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc. Natl. Acad. Sci. USA 106, 5725–5730. doi: 10.1073/pnas.0810753106
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Krzywinski, M. I., Schein, J. E., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. doi: 10.1101/gr.092759.109
Landmann, F. (2019). The Wolbachia endosymbionts. Microbiol Spectr 7:2019. doi: 10.1128/microbiolspec.BAI-0018-2019
Leclercq, S., Giraud, I., and Cordaux, R. (2011). Remarkable abundance and evolution of Mobile group II introns in Wolbachia bacterial endosymbionts. Mol. Biol. Evol. 28, 685–697. doi: 10.1093/molbev/msq238
Leclercq, S., Thézé, J., Chebbi, M. A., Giraud, I., Moumen, B., Ernenwein, L., et al. (2016). Birth of a W sex chromosome by horizontal transfer of Wolbachia bacterial symbiont genome. Proc. Natl. Acad. Sci. 113, 15036–15041. doi: 10.1073/pnas.1608979113
Lefoulon, E., Clark, T., Borveto, F., Perriat-Sanguinet, M., Moulia, C., Slatko, B. E., et al. (2020). Pseudoscorpion Wolbachia symbionts: diversity and evidence for a new supergroup S. BMC Microbiol. 20:188. doi: 10.1186/s12866-020-01863-y
LePage, D. P., Metcalf, J. A., Bordenstein, S. R., On, J., Perlmutter, J. I., Shropshire, J. D., et al. (2017). Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 543, 243–247. doi: 10.1038/nature21391
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lindsey, A. R. I. (2020). Sensing, Signaling, and secretion: a review and analysis of Systems for Regulating Host Interaction in Wolbachia. Genes (Basel) 11:813. doi: 10.3390/genes11070813
Lindsey, A. R. I., Werren, J. H., Richards, S., and Stouthamer, R. (2016). Comparative genomics of a parthenogenesis-inducing Wolbachia symbiont. G3 (Bethesda) 6, 2113–2123. doi: 10.1534/g3.116.028449
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
Lüdecke, D., Ben-Shachar, M., Patil, I., Waggoner, P., and Makowski, D. (2021). Performance: an R package for assessment, comparison and testing of statistical models. JOSS 6:3139. doi: 10.21105/joss.03139
Ma, W.-J., and Schwander, T. (2017). Patterns and mechanisms in instances of endosymbiont-induced parthenogenesis. J. Evol. Biol. 30, 868–888. doi: 10.1111/jeb.13069
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., and Zdobnov, E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654. doi: 10.1093/molbev/msab199
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C. G., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Minh, B. Q., Nguyen, M. A. T., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Moret, Y., Juchault, P., and Rigaud, T. (2001). Wolbachia endosymbiont responsible for cytoplasmic incompatibility in a terrestrial crustacean: effects in natural and foreign hosts. Heredity (Edinb) 86, 325–332. doi: 10.1046/j.1365-2540.2001.00831.x
Negri, I., Pellecchia, M., Mazzoglio, P. J., Patetta, A., and Alma, A. (2006). Feminizing Wolbachia in Zyginidia pullula (Insecta, Hemiptera), a leafhopper with an XX/X0 sex-determination system. Proc. R. Soc. B Biol. Sci. 273, 2409–2416. doi: 10.1098/rspb.2006.3592
Nikoh, N., Hosokawa, T., Moriyama, M., Oshima, K., Hattori, M., and Fukatsu, T. (2014). Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc. Natl. Acad. Sci. USA 111, 10257–10262. doi: 10.1073/pnas.1409284111
Perlmutter, J. I., Bordenstein, S. R., Unckless, R. L., LePage, D. P., Metcalf, J. A., Hill, T., et al. (2019). The phage gene wmk is a candidate for male killing by a bacterial endosymbiont. PLoS Pathog. 15:e1007936. doi: 10.1371/journal.ppat.1007936
Pichon, S., Bouchon, D., Cordaux, R., Chen, L., Garrett, R. A., and Greve, P. (2009). Conservation of the type IV secretion system throughout Wolbachia evolution. Biochem. Biophys. Res. Commun. 385, 557–562. doi: 10.1016/j.bbrc.2009.05.118
R Core Team (2023). R: A language and environment for statistical Computing_. Vienna, Austria: R Foundation for Statistical Computing.
Rice, D. W., Sheehan, K. B., and Newton, I. L. G. (2017). Large-scale identification of Wolbachia pipientis effectors. Genome Biol. Evol. 9, 1925–1937. doi: 10.1093/gbe/evx139
Rigaud, T. (1997). “Inherited microorganisms and sex determination of arthropod hosts” in Influential passengers: Inherited microorganisms and arthropod reproduction (Oxford: Oxford University Press), 81–101.
Rigaud, T., and Juchault, P. (1998). Sterile intersexuality in an isopod induced by the interaction between a bacterium (Wolbachia) and the environment. Can. J. Zool. 76, 493–499. doi: 10.1139/z97-216
Rigaud, T., Juchault, P., and Mocquard, J. (1997). The evolution of sex determination in isopod crustaceans. Bio Essays 19, 409–416. doi: 10.1002/bies.950190508
Rikihisa, Y., and Lin, M. (2010). Anaplasma phagocytophilum and Ehrlichia chaffeensis type IV secretion and Ank proteins. Curr. Opin. Microbiol. Host 13, 59–66. doi: 10.1016/j.mib.2009.12.008
Rousset, F., Bouchon, D., Pintureau, B., Juchault, P., and Solignac, M. (1992). Wolbachia endosymbionts responsible for various alterations of sexuality in arthropods. Proc. Royal Soc. London Series B Biol. Sci. 250, 91–98. doi: 10.1098/rspb.1992.0135
Scholz, M., Albanese, D., Tuohy, K., Donati, C., Segata, N., and Rota-Stabelli, O. (2020). Large scale genome reconstructions illuminate Wolbachia evolution. Nat. Commun. 11:5235. doi: 10.1038/s41467-020-19016-0
Sheehan, K. B., Martin, M., Lesser, C. F., Isberg, R. R., and Newton, I. L. G. (2016). Identification and characterization of a candidate Wolbachia pipientis type IV effector that interacts with the actin cytoskeleton. MBio 7, e00622–e00616. doi: 10.1128/mBio.00622-16
Shropshire, J. D., and Bordenstein, S. R. (2019). Two-by-one model of cytoplasmic incompatibility: synthetic recapitulation by transgenic expression of cifA and cifB in drosophila. PLoS Genet. 15:e1008221. doi: 10.1371/journal.pgen.1008221
Shropshire, J. D., Rosenberg, R., and Bordenstein, S. R. (2020). The impacts of cytoplasmic incompatibility factor (cifA and cifB) genetic variation on phenotypes. Genetics 217:iyaa007. doi: 10.1093/genetics/iyaa007
Sicard, M., Bouchon, D., Ceyrac, L., Raimond, R., Thierry, M., Le Clec’h, W., et al. (2014). Bidirectional cytoplasmic incompatibility caused by Wolbachia in the terrestrial isopod Porcellio dilatatus. J. Invertebr. Pathol. 121, 28–36. doi: 10.1016/j.jip.2014.06.007
Stouthamer, R., Luck, R. F., and Hamilton, W. D. (1990). Antibiotics cause parthenogenetic Trichogramma (Hymenoptera/Trichogrammatidae) to revert to sex. Proc. Natl. Acad. Sci. USA 87, 2424–2427. doi: 10.1073/pnas.87.7.2424
Sun, L. V., Foster, J. M., Tzertzinis, G., Ono, M., Bandi, C., Slatko, B. E., et al. (2001). Determination of Wolbachia genome size by pulsed-field gel electrophoresis. J. Bacteriol. 183, 2219–2225. doi: 10.1128/JB.183.7.2219-2225.2001
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A., and Minh, B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256
Vancaester, E., and Blaxter, M. (2023). Phylogenomic analysis of Wolbachia genomes from the Darwin tree of life biodiversity genomics project. PLoS Biol. 21:e3001972. doi: 10.1371/journal.pbio.3001972
Verne, S., Johnson, M., Bouchon, D., and Grandjean, F. (2007). Evidence for recombination between feminizing Wolbachia in the isopod genus Armadillidium. Gene 397, 58–66. doi: 10.1016/j.gene.2007.04.006
Werren, J. H., Baldo, L., and Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Werren, J. H., and Bartos, J. D. (2001). Recombination in Wolbachia. Curr. Biol. 11, 431–435. doi: 10.1016/S0960-9822(01)00101-4
Wilkins, D. (2023). Gggenes: draw gene arrow maps in “ggplot2”. R package version 0.1. Available online at: https://wilkox.org/gggenes/ (Accessed January 23, 2023).
Wishart, D. S., Han, S., Saha, S., Oler, E., Peters, H., Grant, J. R., et al. (2023). PHASTEST: faster than PHASTER, better than PHAST. Nucleic Acids Res. 51, W443–W450. doi: 10.1093/nar/gkad382
Wiwatanaratanabutr, I., Kittayapong, P., Caubet, Y., and Bouchon, D. (2009). Molecular phylogeny of Wolbachia strains in arthropod hosts based on groE-homologous gene sequences. Zool. Sci. 26, 171–177. doi: 10.2108/zsj.26.171
Wu, M., Sun, L. V., Vamathevan, J., Riegler, M., Deboy, R., Brownlie, J. C., et al. (2004). Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by Mobile genetic elements. PLoS Biol. 2:e69. doi: 10.1371/journal.pbio.0020069
Xie, Z., and Tang, H. (2017). ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 33, 3340–3347. doi: 10.1093/bioinformatics/btx433
Yu, X., Noll, R. R., Romero Dueñas, B. P., Allgood, S. C., Barker, K., Caplan, J. L., et al. (2018). Legionella effector AnkX interacts with host nuclear protein PLEKHN1. BMC Microbiol. 18:5. doi: 10.1186/s12866-017-1147-7
Zhang, D., de Souza, R. F., Anantharaman, V., Iyer, L. M., and Aravind, L. (2012). Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol. Direct 7:18. doi: 10.1186/1745-6150-7-18
Keywords: Wolbachia, feminization, Armadillidium vulgare, genomics, isopod crustacean, effectors, f element
Citation: Grève P, Moumen B and Bouchon D (2024) Three feminizing Wolbachia strains in a single host species: comparative genomics paves the way for identifying sex reversal factors. Front. Microbiol. 15:1416057. doi: 10.3389/fmicb.2024.1416057
Edited by:
Naser Safaie, Tarbiat Modares University, IranReviewed by:
Astrid Collingro, University of Vienna, AustriaSergio López-Madrigal, Indiana University Bloomington, United States
Copyright © 2024 Grève, Moumen and Bouchon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Didier Bouchon, ZGlkaWVyLmJvdWNob25AdW5pdi1wb2l0aWVycy5mcg==