 Jie Fan1,2†
Jie Fan1,2† Hui Kang1,3†
Hui Kang1,3† Meiqi Lv2†
Meiqi Lv2† Yuhuan Zhai1,3
Yuhuan Zhai1,3 Yangyang Jia4
Yangyang Jia4 Zixin Yang3
Zixin Yang3 Chengcheng Shi2
Chengcheng Shi2 Changhao Zhou2Lin Diao2
Changhao Zhou2Lin Diao2 Jingsuo Li2Xiaowei Jin5Shanshan Liu2
Jingsuo Li2Xiaowei Jin5Shanshan Liu2 Karsten Kristiansen6,7
Karsten Kristiansen6,7 Peijun Zhang3*
Peijun Zhang3* Jianwei Chen2,6,7*
Jianwei Chen2,6,7* Songhai Li3,8
Songhai Li3,8- 1College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 2BGI Research, Qingdao, China
- 3Marine Mammal and Marine Bioacoustics Laboratory, Institute of Deep-sea Science and Engineering, Chinese Academy of Sciences, Sanya, China
- 4BGI Research, Shenzhen, China
- 5China National Environmental Monitoring Centre, Beijing, China
- 6Qingdao Key Laboratory of Marine Genomics, and Qingdao-Europe Advanced Institute for Life Sciences, BGI Research, Qingdao, China
- 7Laboratory of Genomics and Molecular Biomedicine, Department of Biology, University of Copenhagen, Copenhagen, Denmark
- 8The Innovation Research Center for Aquatic Mammals, and Key Laboratory of Aquatic Biodiversity and Conservation of the Chinese Academy of Sciences, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China
Cetaceans play a crucial role in marine ecosystems; however, research on their gastrointestinal microbiota remains limited due to sampling constraints. In this study, we collected hindgut samples from 12 stranded cetaceans and performed 16S rRNA gene amplicon sequencing to investigate microbial composition and functional potentials. Analysis of ZOTUs profiles revealed that the phyla Firmicutes, Proteobacteria, and Bacteroidetes dominated all hindgut samples. However, unique microbial profiles were observed among different cetacean species, with significant separation of gut microbiota communities according to biological evolutionary lineages. Different genera that contain pathogens were observed distinguishing delphinids from physeteroids/ziphiids. Delphinid samples exhibited higher abundances of Vibrio, Escherichia, and Paeniclostridium, whereas physeteroid and ziphiid samples showed higher abundances of Pseudomonas, Enterococcus, and Intestinimonas. Functional analysis indicated convergence in the gut microbiota among all cetaceans, with shared bacterial infection pathways across hindgut samples. In addition, a comparison of the gastrointestinal microbial composition between a stranded short-finned pilot whale (Globicephala macrorhynchus) and a stranded rough-toothed dolphin (Steno bredanensis) using 16S rRNA gene sequencing revealed distinct microbial community structures and functional capacities. To the best of our knowledge, this study represents the first report on the gastrointestinal microbiota of the pantropical spotted dolphin (Stenella attenuata), Blainville’s beaked whale (Mesoplodon densirostris), and rough-toothed dolphin, with various comparisons conducted among different cetacean species. Our findings enhance the understanding of microbial composition and diversity in cetacean gastrointestinal microbiota, providing new insights into co-evolution and complex interactions between cetacean microbes and hosts.
Introduction
Cetaceans, the largest known marine mammals, have evolved into approximately 90 species including whales, dolphins, and porpoises (Fordyce, 2018). As apex predators in the ocean, they play vital roles in marine ecosystems (Zhang and Goodman, 2023; Zhang et al., 2023a,b). Although the population of cetaceans has garnered global attention, their health status is facing multiple threats, including overfishing, habitat destruction, marine pollution, and pathogenic infections (Nachtsheim et al., 2021; Yuri and Findlay, 2021). These factors contribute to an uncertain outlook for these marine mammals. Recent studies even indicate that cetaceans under human care can live longer than their wild counterparts (Kirk-Cohen et al., 2023; Tidière et al., 2023). Previous research highlights the substantial influence of the mammalian gastrointestinal tract microbiota on various physiological processes including digestion, metabolism, tissue differentiation, pathogen resistance, and immunity (Sanders et al., 2015; Bik et al., 2016; Ost and Round, 2018; Yin et al., 2023). While extensive studies have explored the gastrointestinal microbiota of humans (Lin et al., 2023) and terrestrial mammals such as mice (Kieser et al., 2022), pigs (Hu et al., 2024), cattle (Xie et al., 2021), and giant pandas (Guo et al., 2020), research on cetacean gut microbiota remains limited (Sanders et al., 2015; Bik et al., 2016), particularly concerning microbial diversity and composition across different regions of their gastrointestinal tracts (Li et al., 2021).
Preliminary studies on the diversity, structure, and function of gut microbiota in cetaceans have revealed a highly diverse microbial community comprising more than 60 prokaryotic phyla with diverse functional profiles (Higgins, 2000; Waltzek et al., 2012). Over 400 prokaryotic species have been identified in the cetacean gut to date. The phyla Firmicutes, Proteobacteria, Bacteroidetes, and Fusobacteria are among the most abundant species in the cetacean gut microbiota, showing distinct differences from fish, terrestrial mammals, and seawater microbiota (Higgins, 2000; Waltzek et al., 2012). Significant variations in the diversity and composition of gut microbiota have been observed across different cetacean families. Specifically, the previous analysis revealed that toothed whales, alongside the North Pacific right whales (Eubalaena japonica), and different baleen whale species, exhibited markedly distinct abundances of Mycobacterium, Cetobacterium, Bacillus, and Coprobacillus, highlighting the diversity and specificity of microbial communities across these cetacean groups (Sanders et al., 2015). In addition, the gut microbiota of certain baleen whale species, such as the Pacific humpback whales (Megaptera novaeangliae) and bowhead whales (Balaena mysticetus), play critical roles in amino acid metabolism and synthesis, as well as lipid digestion (Sanders et al., 2015; Miller et al., 2020). Intestinal dysbiosis in cetaceans, triggered by factors, such as injury, exposure to organic pollutants, and bacterial infections, can result in host malnutrition, stranding events, and mortality (Nachtsheim et al., 2021; Yuri and Findlay, 2021). Pathogenic infections play a significant role in cetacean stranding, often indicating underlying health issues within these marine mammals (Waltzek et al., 2012; Di Guardo et al., 2018). Notable pathogens, such as Brucella (Guzmán-Verri et al., 2012), Aeromonas (Pérez et al., 2015), and Vibrio (Buck and Spotte, 1986), are significant contributors to poor health in these animals, often associated with cetacean stranding. The cetacean gut microbiota is crucial for maintaining host health, particularly influencing disease susceptibility and immune system function (Ley et al., 2008; Krajmalnik-Brown et al., 2012; Hanning and Diaz-Sanchez, 2015; Lloyd-Price et al., 2019). However, current research on the cetacean gut microbiota remains constrained by small sample sizes of fewer than 10 individuals and limited species diversity. These limitations impede large-scale surveys and comprehensive comparative studies.
Compared to studies on the gut microbiota of cetaceans, research on the microbial composition of gastrointestinal tract tissues is even more infrequent due to the scarcity of anatomical samples from cetaceans. Only a few publications have documented the microbial composition of the stomach and foregut in cetaceans (Sanders et al., 2015; Monteiro et al., 2021; Wan et al., 2021, 2023). Preliminary analyses using amplicon sequencing and metagenomic approaches have revealed differences in microbial communities across different digestive tract regions of cetaceans, showing close functional interactions with their hosts (Erwin et al., 2017; Bai et al., 2022). For instance, studies have identified over 30 prokaryotic phyla in the gastrointestinal tracts of the common bottlenose dolphin (Tursiops truncatus), including two novel phyla, one named Delphibacteria, which suggests denitrification capabilities (Bik et al., 2016; Dudek et al., 2017). Moreover, Firmicutes predominated throughout the gastrointestinal tract of stranded pygmy sperm whales (Kogia breviceps) and dwarf sperm whales (Kogia sima), whereas Proteobacteria and Fusobacteria were abundant in the proximal gut, and Bacteroidetes emerged in the large intestine (Erwin et al., 2017; Li et al., 2021). Distinct core microbiota compositions have been reported for stomachs compared to the intestinal microbiota, featuring genera such as Fusobacterium, Cetobacterium, Peptostreptococcus, Lactococcus, and Actinobacillus, which are the dominant genera in the cetacean intestine (Miller et al., 2020).
In this study, we collected 19 gastrointestinal samples from 12 cetacean individuals representing 9 species. These included previously unreported species such as the pantropical spotted dolphin (Stenella attenuata), Blainville’s beaked whale (Mesoplodon densirostris), and rough-toothed dolphin (Steno bredanensis), alongside six species previously documented: Indo-Pacific humpback dolphin (Sousa chinensis), Risso’s dolphin (Grampus griseus), pygmy sperm whale (Kogia breviceps), dwarf sperm whale (Kogia simus), sperm whale (Physeter macrocephalus), and short-finned pilot whale (Globicephala macrorhynchus). We conducted 16S rRNA gene amplicon sequencing to explore microbial composition, diversity, and functional potential in the hindgut samples of cetaceans. Our investigation encompassed multiple perspectives, including host evolutionary relationships, stranding geographic locations, and different gastrointestinal tract tissues. Furthermore, we performed 16S and 18S rRNA gene amplicon sequencing on stomach, foregut, and hindgut samples from rough-toothed dolphins and short-finned pilot whales, providing new insights into the composition of prokaryotic and eukaryotic communities.
These data help address gaps in our understanding of the microbiota and its functions within the gastrointestinal tract of various cetacean species. They demonstrate how food sources may influence the gastrointestinal microbiota of cetaceans, supporting future research aimed at constructing a more comprehensive microbial overview for a larger number of cetacean species. Moreover, analysis of the gastrointestinal microbiota from multiple stranded cetaceans provides new insights into pathogenic detection and clinical health assessments of stranded individuals (Obusan et al., 2015).
Materials and methods
Sample collection
Samples were collected from 12 stranded individuals of nine cetacean species in China between 2014 and 2019 (Figure 1 and Table 1). These individuals had been stranded in seven different cities. Four Indo-Pacific humpback dolphins, two from Qinzhou (Guangxi Zhuang Autonomous Region) and two from Zhanjiang (Guangdong Province), were found deceased and subsequently dissected for autopsies, from which hindgut samples were collected. The remaining eight cetaceans were initially alive when stranded in southern China but succumbed despite rescue efforts. The twelve stranded individuals were categorized into two groups based on their biological evolutionary lineages: the delphinid group, comprising eight Delphinidae individuals, and the physeteroid and ziphiid group, which included three Physeteroidea individuals and one Ziphiidae individual (Table 1). Standard post-mortem analyses were conducted on the deceased cetaceans, following established protocols. Hindgut samples were collected during necropsies for all individuals, while gastrointestinal samples, including foregut contents and stomach contents from a short-finned pilot whale and a rough-toothed dolphin, were also obtained and included in this study. All collected specimens were stored at −80°C until DNA extraction. Ethical approval for the collection of gastrointestinal samples in this study was granted by the Institute of Deep-sea Science and Engineering, Chinese Academy of Sciences, under ethical statement number SIDSSE-SYLL-MMMBL-01 and the Institutional Review Board of BGI (NO. BGI-R052-3 and NO. FT17160). All procedures adhered to ethical guidelines and legal requirements in China.
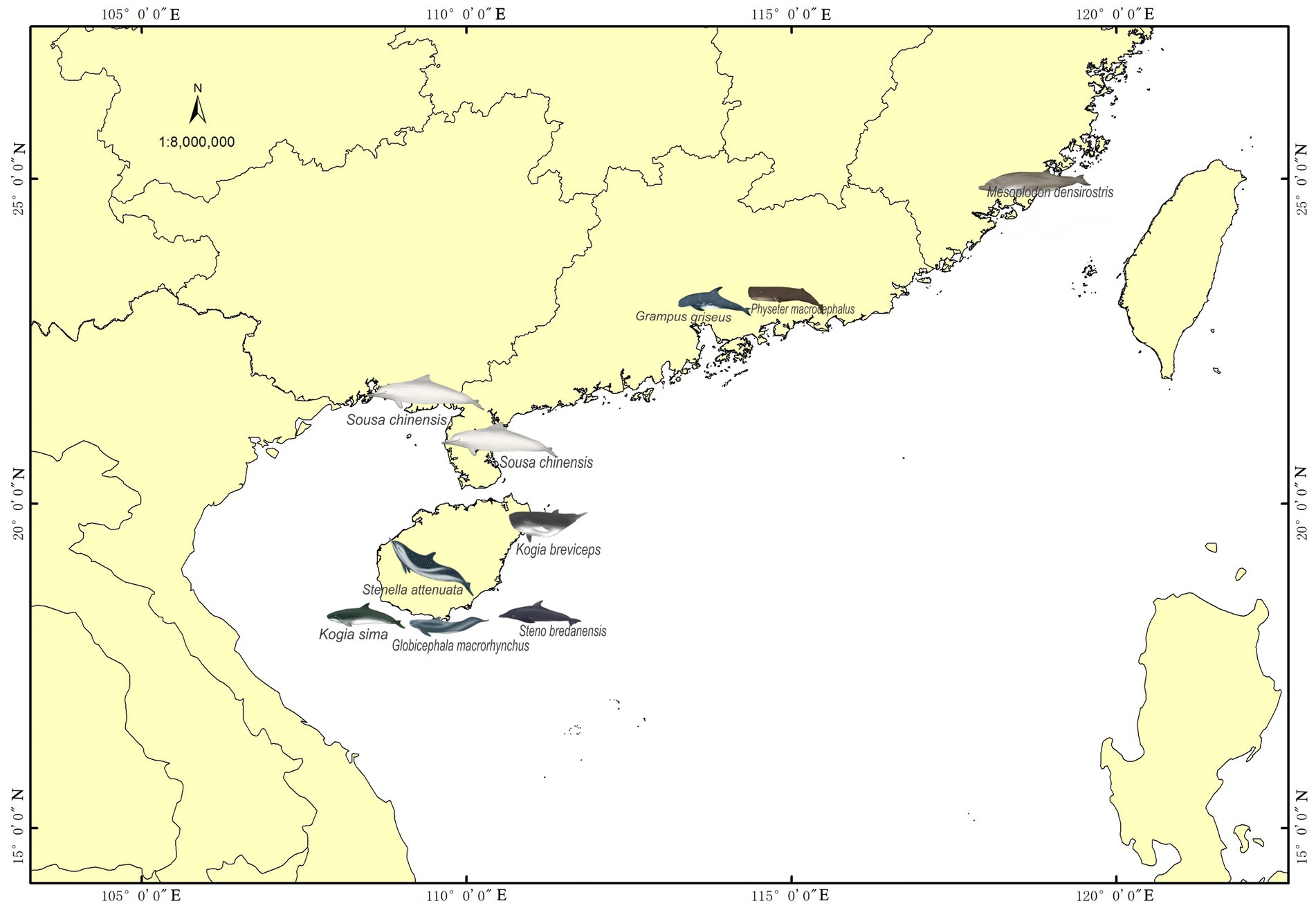
Figure 1. Stranded cetaceans sampling area. The red dots indicate the sampling locations in the coastal areas of southern China, mainly distributed in the Hainan Provinces, Guangdong Provinces, Fujian Provinces, and Guangxi Zhuang Autonomous Region.
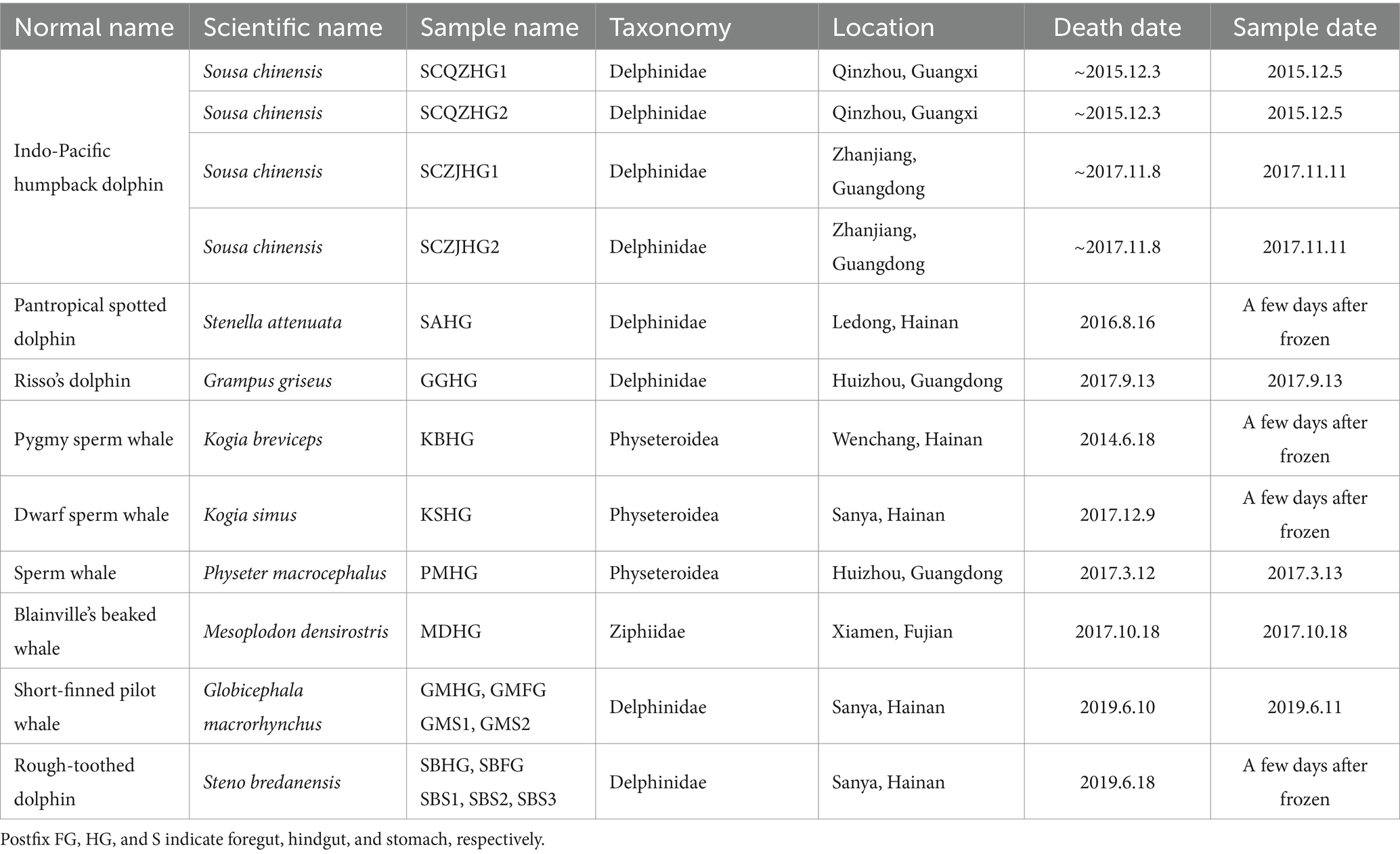
Table 1. Sampling information of 12 stranded cetaceans.
DNA extraction and 16S rRNA gene amplicon sequencing of gastrointestinal samples
Approximately 10 g of gastrointestinal tract contents were collected from each cetacean individual. Each sample was homogenized and divided into three parts for DNA extraction. DNA extraction followed a modified CTAB protocol, and quantification was performed using a Qubit 3.0 Fluorometer. Due to the low microbial biomass in the intestinal contents of stranded cetaceans, each extraction did not meet the amplification requirements. Therefore, the three separate extractions were combined into one sample for subsequent PCR amplification. PCR amplification was carried out using the universal primers 515F-806R targeting the V4 region of the 16S rRNA gene (515F: GTGCCAGCMGCCGCGGTAA, 806R: GGACTACHVGGGTWTCTAAT). The PCR reactions were conducted using KAPA HiFi HotStart ReadyMix (2x) with primers and templates under the following conditions: initial denaturation at 95°C for 5 min, followed by 15 cycles of denaturation at 98°C for 20 s, annealing at 58°C for 30 s, extension at 72°C for 30 s, and a final extension at 72°C for 10 min (Jia et al., 2022). Subsequently, the purification of PCR products was performed using the GeneJET Gel Extraction Kit (Thermo Scientific). The PCR products with target bands were purified and pooled in equal masses to construct 16S amplicon libraries using the MGIEasy Universal DNA Library Prep kit v1.0 (PN: 1000006986), following the standard DNA nanoball (DNB) library construction protocol. All libraries were sequenced on the DNBSEQ-G400 platform with a paired-end sequencing mode with 200 bp read length at BGI Research (Qingdao, China) (Zhang Z. et al., 2023).
Bioinformatic analysis of 16S amplicon data
The raw amplicon sequencing of paired-end reads was conducted using the EasyAmplicon pipeline (v1.0) (Liu Y. X. et al., 2023). Initially, FLASH (v1.2.11) (Magoč and Salzberg, 2011) merged the paired-end reads into tags with parameters “-min-overlap 10 -max-mismatch-density 0.1.” Primer sequences were then removed, and low-quality reads were filtered out using VSEARCH (v2.13.1) (Rognes et al., 2016) to generate clean tags. These clean tags were processed further to create zero-radius operational taxonomic units (ZOTUs) using the Unoise3 algorithm from USEARCH (Edgar and Flyvbjerg, 2015) for denoising (Liu G. et al., 2023). Taxonomic annotation of ZOTU representative sequences was performed using the sintax algorithm (Edgar, 2018) against the RDP training set (v18), employing a 0.8 confidence cutoff value. The abundance profile of ZOTUs was generated using the USEARCH “-otutab” command and normalized based on a standard sequence count derived from the sample with the average number of sequences, using the “-otutab_rare” command. Alpha diversity indices and beta diversity distances were computed using the vegan package (v2.6.4) within R software (v4.0.2). ZOTU sequences were aligned using PyNAST software, and a phylogenetic tree was constructed using FastTree (v2.1.5) (Price et al., 2010). Visualization of the phylogenetic tree was performed using iTOL (v5) (Letunic and Bork, 2019). The feature table used for linear discriminant analysis effect size (LEfSe) analysis was transformed by the EasyAmplicon pipeline. Subsequently, the LEfSe analysis was conducted and visualized by using the online tool ImageGP (Chen et al., 2022). The COI sequences of the nine cetaceans were downloaded from NCBI and then aligned by using MUSCLE (v3.8.31). The phylogenetic tree and distance matrix were measured by using Fasttree (v2.1.10). Finally, the Mantel test between the evolutionary distance matrix and microbial Bray Curtis distance matrix was conducted using the “mantel” function of the R package “vegan.”
Functional predictions for prokaryotic communities
PICRUSt2 (Douglas et al., 2020) was employed to predict prokaryotic KEGG functions based on the taxonomy of 16S rRNA gene sequences, generating abundance profiles of KEGG Orthologs (KOs) and metabolic pathways. Statistical testing to discern significant differential KEGG pathways and enzymes among different cetaceans was performed using the edgeR package in R software (v4.0.2), and the results were visualized using OriginPro and ImageGP. Simultaneously, the Functional Annotation of PROkaryotic TAXa (FAPROTAX) database (Louca et al., 2016) was utilized to identify potentially pathogenic groups and detect experimentally verified pathogens from 16S rRNA gene sequences. Pathogens posing threats to human, aquatic animal, or plant welfare were categorized, including “animal parasites or symbionts,” “intracellular parasites,” “invertebrate parasites,” “human pathogens all,” “human pathogens diarrhea,” “human pathogens gastroenteritis,” “human pathogens meningitis,” “human pathogens nosocomial,” “human pathogens pneumonia,” “human pathogens septicemia,” “plant pathogen,” and “fish parasites.”
Results
Prokaryotic community overview of hindgut samples
For the 12 hindgut samples collected from all stranded cetaceans, we obtained a total of 771,308 clean tags, with an average of 64,275 per sample. This analysis resulted in 535 prokaryotic ZOTUs, including 6 Archaea ZOTUs and 529 Bacteria ZOTUs (Figure 2, Supplementary Tables S1, S2). The rarefaction curves for each sample reached saturation, indicating that our sequencing strategy effectively captured the microbial taxa in all samples (Supplementary Figure S1). We identified 19 core ZOTUs, primarily from Firmicutes (10 ZOTUs) and Proteobacteria (6 ZOTUs), present in ≥75% of the hindgut samples (Supplementary Table S1). In addition, 184 specific ZOTUs, constituting approximately 34.39% of all ZOTUs, appeared in only one of the twelve samples, with the majority (116/535, 21.68%) being unclassified Firmicutes (Supplementary Table S1). The 19 core ZOTUs were also the most abundant across all hindgut samples, with an average relative abundance of 23.81% for each ZOTU, whereas the 183 specific ZOTUs were rare, exhibiting extremely low abundances (average ~ 0.01%). This indicates a significant overlap in the core gut microbiota among different cetaceans.
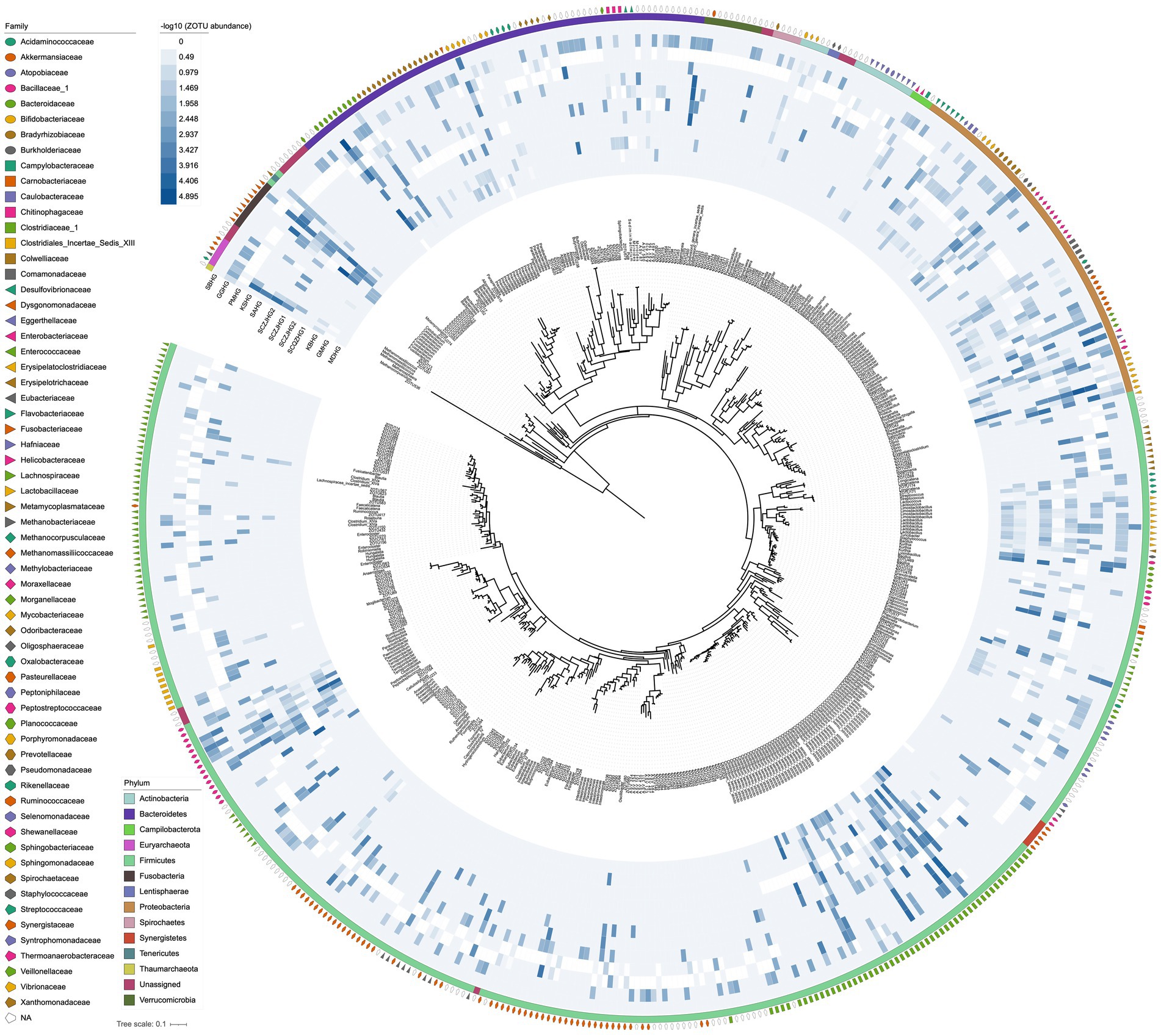
Figure 2. Phylogenetic tree of prokaryotic ZOTUs and distribution of relative abundances in the 12 hindgut samples. Relative abundance heatmap values are −log10 transformed. The outside circles represent the phylum and family taxonomic classification of the ZOTUs.
The gut microbial compositions of the three cetacean species, sequenced for the first time, varied significantly (Figure 2). In the Blainville’s beaked whale, the dominant genus was Bacteroides (76.8%), followed by Clostridium sensu stricto (7.43%), Vibrio (5.13%), Parabacteroides (4.58%), Morganella (2.46%), and Providencia (2.11%). In the pantropical spotted dolphin, the most abundant genera were Peptostreptococcus (41.2%), Clostridium sensu stricto (20.1%), Kurthia (12.7%), Enterococcus (4.74%), and Actinobacillus (2.64%), along with a substantial fraction of unclassified microbes (16.3%). The rough-toothed dolphin exhibited a distinct microbial profile compared to the pantropical spotted dolphin, with the top five genera being Vibrio (39.5%), Bacteroides (28.1%), Cetobacterium (13.9%), Clostridium sensu stricto (7.47%), and Paeniclostridium (2.70%).
Gut microbiota differences between the delphinids, and physeteroids, and ziphiid
We conducted principal coordinate analysis (PCoA) based on the Bray Curtis β-diversity indices for the hindgut samples. The PCoA results revealed a separation in microbial profiles aligned with the evolutionary relationships of the cetacean species. Specifically, the hindgut microbiota communities of delphinid samples (Delphinidae cetaceans) significantly differed from those of physeteroid and ziphiid samples (Physeteroidea and Ziphiidae cetaceans) (ANOSIM test: 999 permutations, p < 0.05) (Figure 3A). To reveal the relationship between the distance between hindgut microbial samples and the evolutionary distance of the cetaceans, we constructed the COI phylogenetic tree of the nine cetacean species, and then, we observed two separated clades between the delphinids and the physeteroids and ziphiid (Supplementary Figure S2). Mantel test analysis between the evolutionary distance matrix and hindgut microbial Bray Curtis distance matrix revealed phylosymbiosis signatures of the hindgut microbial composition across different cetacean species (R = 0.51, p = 0.04). The gut microbiota of the ziphiid Blainville’s beaked whale (MDHG) was more similar to that of physeteroids, differing from delphinids. Conversely, the gut microbial compositions of the pantropical spotted dolphin (SAHG) and rough-toothed dolphin (SBHG) were similar to those of other delphinids. We also calculated the α-diversity indices to evaluate microbial community diversity in the hindgut samples, finding that the Chao1 index was notably higher in physeteroid samples (average = 165.80) compared to delphinid samples (average = 94.88), although this difference was not statistically significant (ANOVA test, adjusted p > 0.05) (Figure 3B).
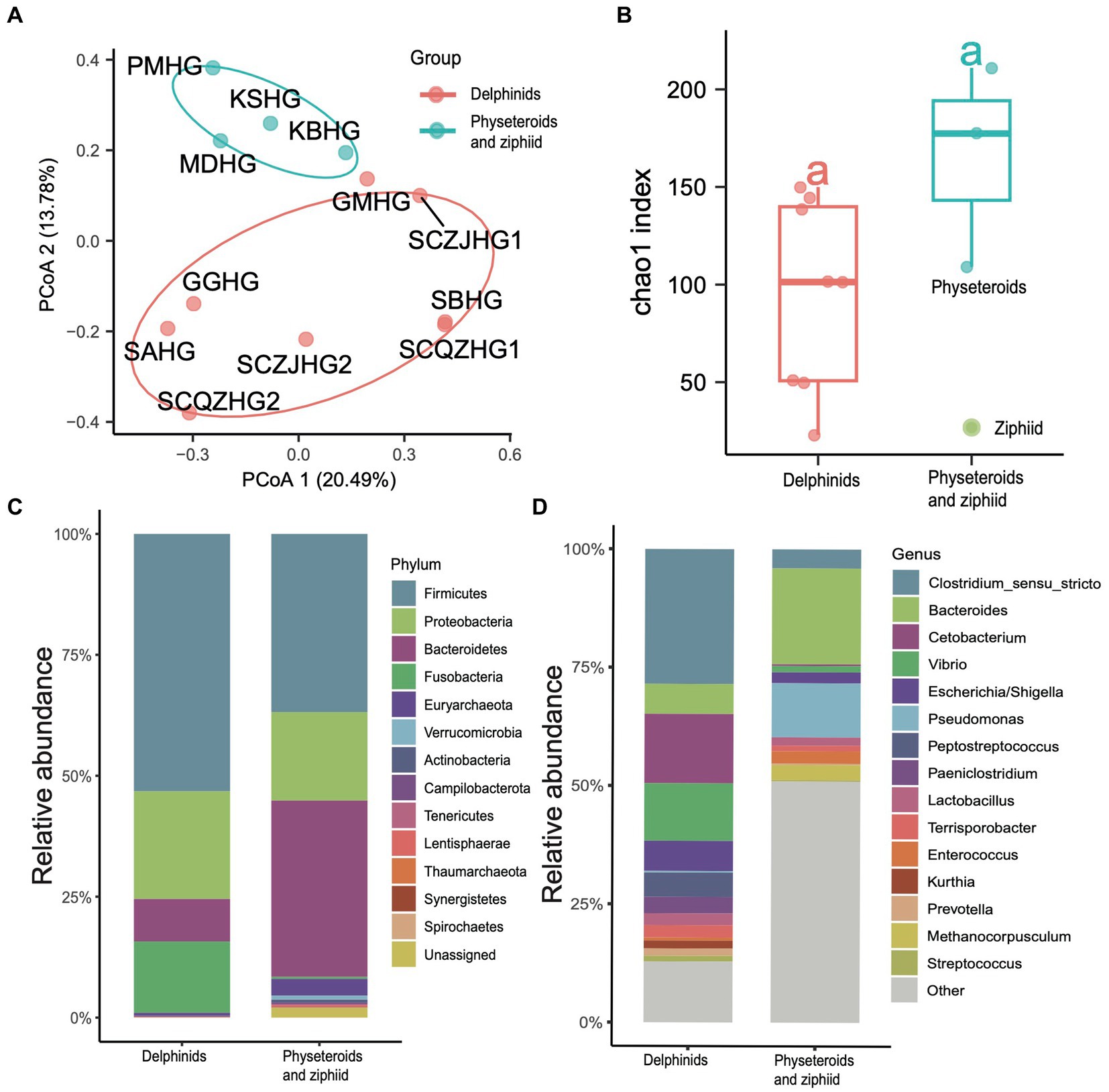
Figure 3. Gut microbiota composition differs among cetacean individuals. (A) PCoA of 12 hindgut samples at the ZOTU level based on Bray Curtis distances. (B) Boxplot showing the comparison of the α-diversity indices Chao1 index between the eight delphinid samples and four physeteroid and ziphiid samples. (C,D) The stacked bar chart shows the average prokaryotic relative abundance of hindgut samples at the phylum and genus level of the two cetacean lineages. (C) A comparison of the microbiota of delphinids, physeteroids, and ziphid and the phylum level. A total of 13 phyla were detected, while the unclassified taxa were named “Unassigned.” (D) A comparison of the microbiota of delphinids, physeteroids, and ziphid and the genus level. A total of 128 genera were detected, only the top 15 most abundant genera are shown here, and the remaining genera including unclassified taxa were grouped as “Other”.
The major phyla identified in the three physeteroid hindgut samples and one ziphiid sample were Firmicutes (36.70%), Bacteroidetes (36.58%), Proteobacteria (17.81%), and Euryarchaeota (3.42%). In contrast, the eight delphinid samples were predominantly composed of Firmicutes (53.04%), Proteobacteria (20.84%), Fusobacteria (14.67%), and Bacteroidetes (8.72%) (Figure 3C). Physeteroid and ziphiid samples exhibited a higher abundance of Bacteroidetes, whereas the delphinid samples demonstrated a greater prevalence of Firmicutes and Fusobacteria. Despite differences in microbial distributions at the phylum level, both groups showed a relatively high proportion of Proteobacteria. At the genus level, the dominant genera in all cetacean hindgut samples were Clostridium sensu stricto (20.40%), Bacteroides (10.96%), and Cetobacterium (9.83%) (Figure 3D). However, the genera Vibrio, Escherichia, and Paeniclostridium were more abundant in delphinids, with average relative abundances of 12.28, 6.25, and 3.44%, respectively, compared to 1.43, 2.20, and 0.04% in the physeteroid and ziphiid samples. In contrast, the physeteroid and ziphiid samples had a higher abundance of Pseudomonas (11.15%), Enterococcus (1.14%), and Intestinimonas (0.74%) than the delphinids, which had average relative abundances of 0.32, 0.59, and 0.88%, respectively. In addition, unclassified prokaryotic communities accounted for an average of 12.28% of the total relative abundance at the genus level, indicating that much remains unknown about the cetacean gut microbiota.
Microbial functional convergence in cetacean hindgut samples
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed on the 12 hindgut samples to generate the KEGG functional abundance profile. The results revealed a total of 162 pathways across all cetacean samples. Notably, pathways related to the biosynthesis of ansamycins and fatty acid biosynthesis were abundant in all hindgut samples. PCoA indicated that the KEGG pathway abundances in cetacean microbiota were convergent between the delphinid and the physeteroid/ziphiid samples (PERMANOVA test: 999 permutations, p > 0.05) (Figure 4A). Only nine of the 162 pathways showed significant differences in abundance between the two cetacean lineages. Specifically, pathways, such as glycosaminoglycan degradation, glycine, serine, threonine, and histidine metabolism and sphingolipid metabolism, were significantly enriched in the physeteroid and ziphiid samples, while pathways, such as glycerophospholipid metabolism, phosphotransferase system (PTS), and D-alanine metabolism, were significantly enriched in the delphinids (p < 0.05) (Supplementary Table S3). Furthermore, the abundance profile of enzymes revealed significant variation between samples (PERMANOVA test: 999 permutations, p < 0.05) (Figure 4B). We identified a total of 15 enzymes that were more abundant in the delphinid samples, including ATP phosphoribosyltransferase and beta-N-acetylhexosaminidase, which are related to energy and carbohydrate metabolism. Conversely, 28 enzymes, such as diadenylate cyclase and ribosomal-protein-alanine N-acetyltransferase, which are related to nucleic acid and protein metabolism, were more abundant in the physeteroid and ziphiid samples (p < 0.05) (Figure 4C).
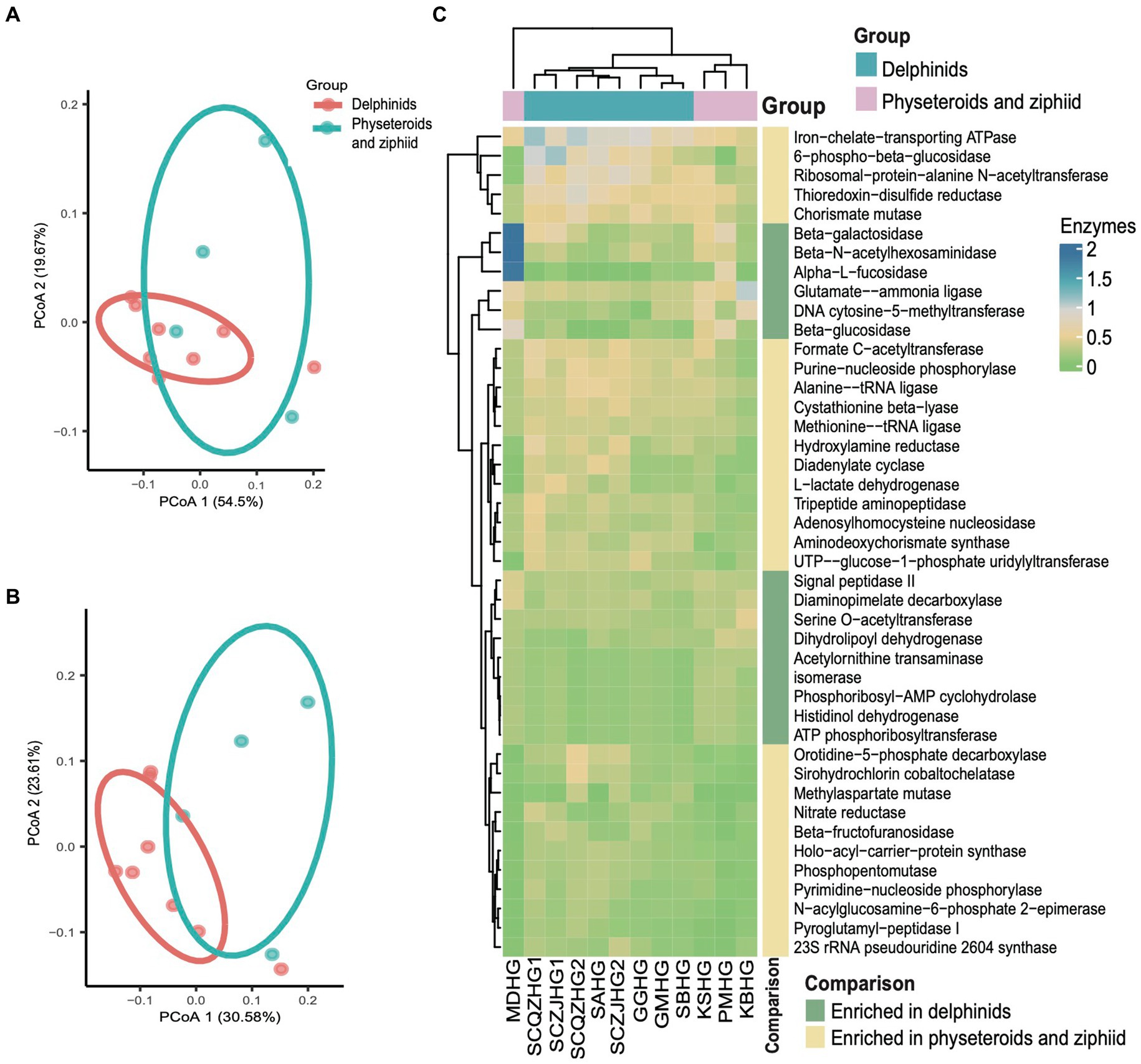
Figure 4. Predicted functional potential of the cetacean hindgut microbiota. (A,B) PCoA based on the Bray Curtis distance of KEGG pathways and enzyme functional abundance profiles, respectively. (C) A total of 43 enzymes were over-represented for the two cetacean lineages.
Moreover, six KEGG pathways, including those related to Vibrio cholerae infection and the Vibrio cholerae pathogenic cycle, were found to have a combined total abundance of 7.04% across all hindgut samples (Supplementary Figure S3). In addition, we identified 24 ZOTUs with a relative abundance of approximately 12.21% that were assigned to potential pathogenic groups classified as “animal parasites or symbionts” and “human pathogens all” according to the FAPROTAX database. These potentially pathogenic ZOTUs included species from Prevotella (14 ZOTUs), Actinobacillus (4 ZOTUs), other Pasteurellaceae (3 ZOTUs), Stenotrophomonas (2 ZOTUs), Escherichia (2 ZOTUs), and Roseburia (1 ZOTU) (Supplementary Table S4). The potential pathogenic Prevotella and Stenotrophomonas were widespread across all hindgut samples of sampled cetaceans, with a notably higher relative abundance in sample ZJBHT1, which is a sample of Sousa chinensis.
Sousa chinensis stranded at two locations harbor distinct microbial communities
We further analyzed the hindgut microbial composition of the four Indo-Pacific humpback dolphins (Sousa chinensis) stranded in Zhanjiang City (Leizhou Bay colony) and Qinzhou City (Sanniang Bay colony). We observed significant differences in the Chao1 indices between the two locations (ANOVA test, adjusted p < 0.05) (Figure 5A), and although the Shannon indices of Indo-Pacific humpback dolphins stranded in Zhanjiang were higher than those in Qinzhou, the difference was not statistically significant (Figure 5B). We also calculated the relative abundances at the phylum and genus levels to provide detailed information on the microbial community composition. At the phylum level, samples from Zhanjiang City were predominantly composed of Firmicutes (~72.20%) with a notable presence of Bacteroidetes (~6.76%), while samples from Qinzhou City were dominated by Firmicutes (~56.55%) and Fusobacteria (~30.25%) with remarkably fewer Bacteroidetes (~0.01%) (Figure 5C). The common abundant genus across all four samples was Clostridium sensu stricto (average ~ 42.60%). The genera Cetobacterium (30.25% vs. 5.8%), Escherichia (12.10% vs. 4.52%), and Paeniclostridium (11.54% vs. 0.18%) were more dominant in samples from Qinzhou City, whereas Terrisporobacter (3.90% vs. 5.16%) and Lactobacillus (0.02% vs. 8.34%) were more abundant in samples from Zhanjiang City (Figure 5D). Regarding α-diversity analysis (richness and Shannon indices), both indices were notably higher in the gut microbiota of dolphins from Zhanjiang City compared to those from Qinzhou City. We also found several genera that were abundant in Zhanjiang dolphins, such as Prevotella, Actinobacillus, Streptococcus, Proteus, and Vibrio, each with a relative abundance of over 2%, but these were rarely or not observed in Qinzhou dolphins (Figure 5D).
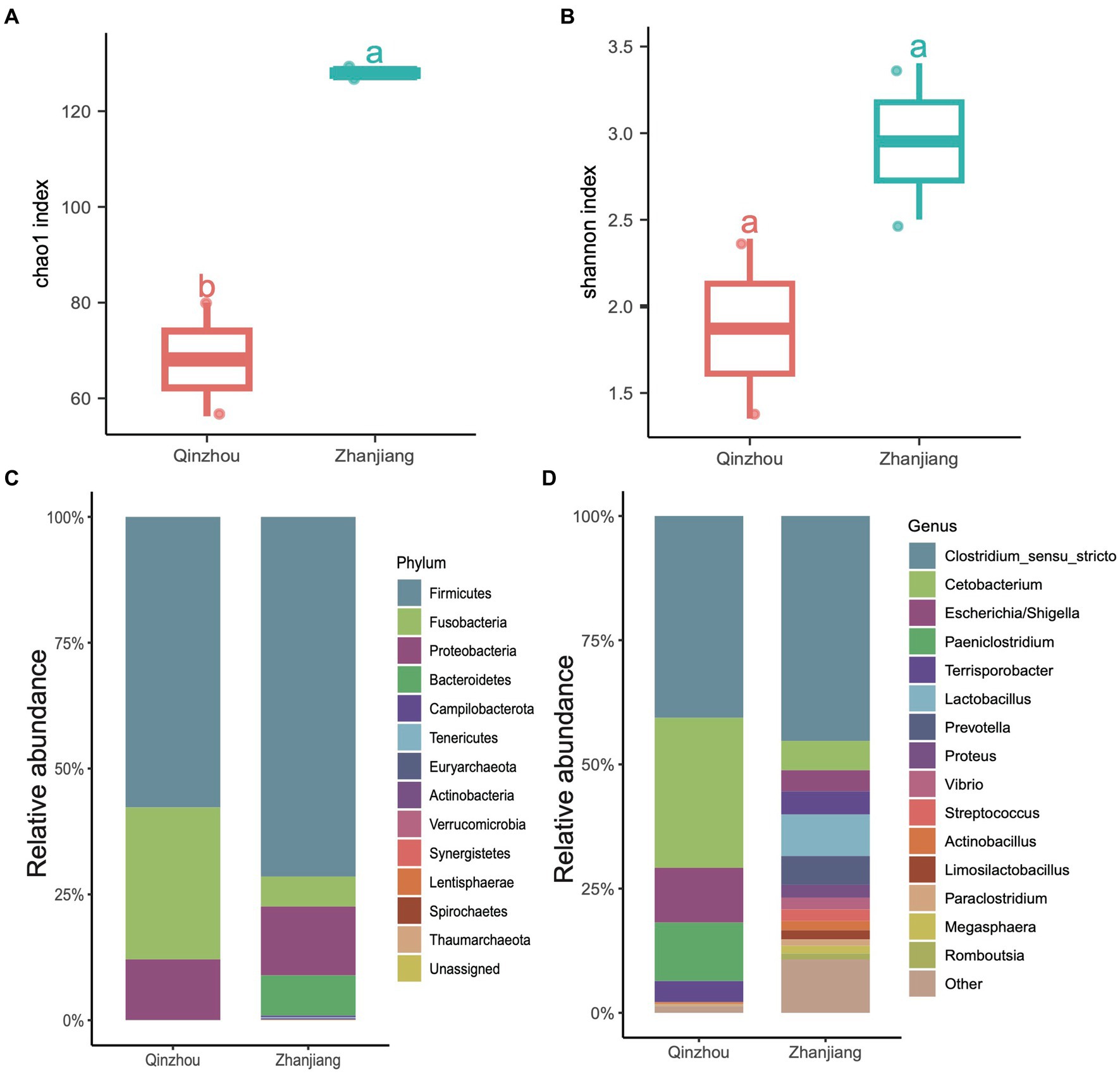
Figure 5. Taxonomic composition of four Indo-Pacific humpback dolphins. (A,B) Comparisons of α-diversity indices, including Chao1 index (A) and Shannon index (B), of the four Indo-Pacific humpback dolphins stranded in Qinzhou City and Zhanjiang City. (C,D) Stacked bar chart showing the average relative abundance at the phylum (C) and genus (D) levels for the four Indo-Pacific humpback dolphin hindgut samples.
Microbial community differentiation in the gastrointestinal tract of two different delphinids
In addition to examining the differences in the microbial communities of hindgut samples among different cetaceans, we investigated the community composition and structure of the stomach and foregut microbiota in two delphinid species: the rough-toothed dolphin (Steno bredanensis, SB) and the short-finned pilot whale (Globicephala macrorhynchus, GM). After quality assessment, we obtained a total of 443,154 clean tags from five gastrointestinal tract samples from rough-toothed dolphins and four from short-finned pilot whales, resulting in the identification of 139 ZOTUs (Supplementary Tables S1, S2). We calculated the α-diversities of microbial communities in the gastrointestinal tracts of these two Delphinidae species. The Chao1 and Shannon indices indicated that the α-diversity of the gastrointestinal microbiota in GM was consistently higher than in SB, although this difference was not statistically significant (ANOVA test, adjusted p > 0.05) (Figures 6A,B). However, PCoA based on Bray Curtis distances revealed distinct separation between the two species, suggesting they harbor different gastrointestinal microbial communities (PERMANOVA test: 999 permutations, SumsOfSqs = 7.79, F = 21.50, p < 0.05) (Figure 6C).
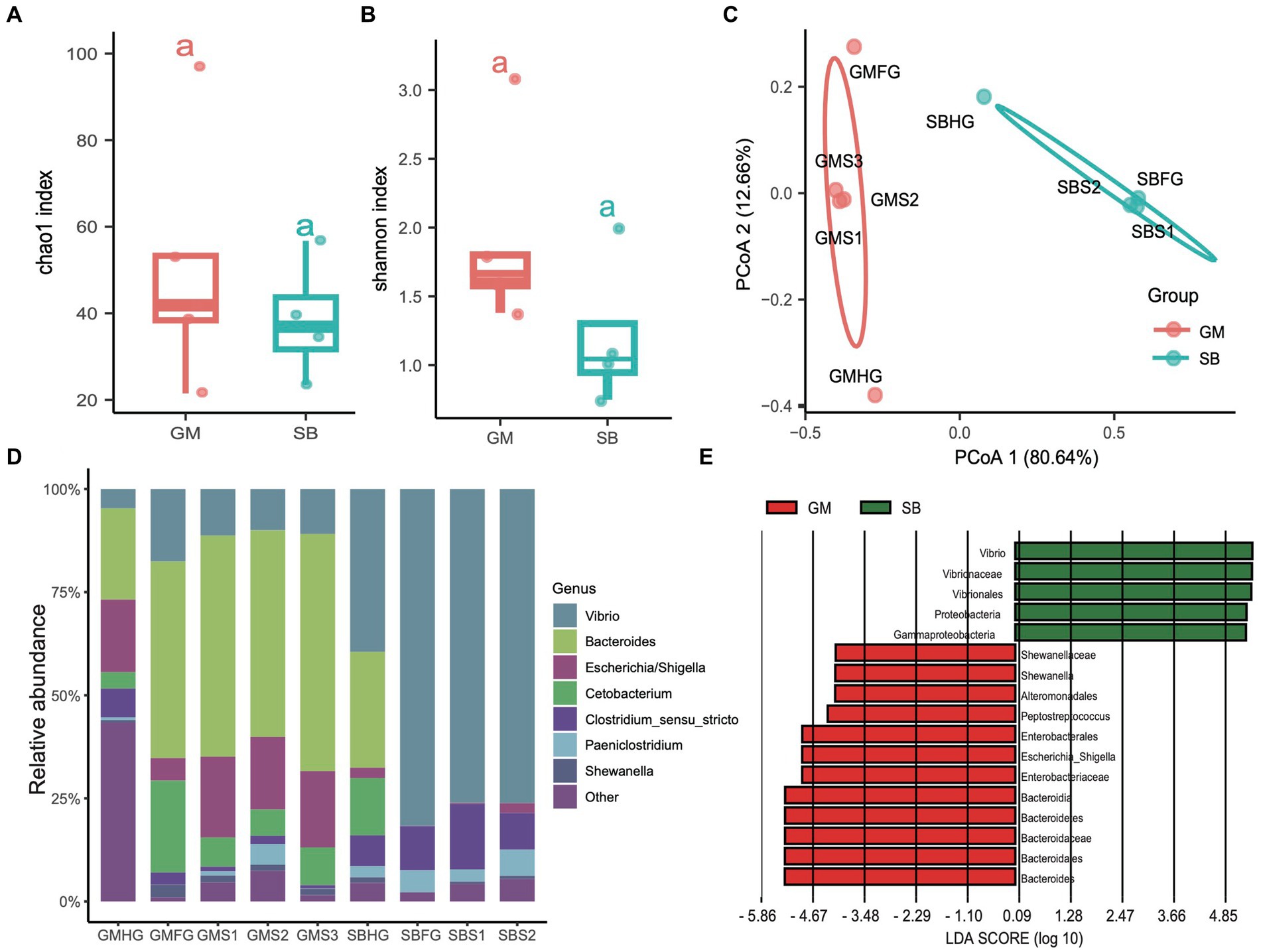
Figure 6. Comparisons of the prokaryotic diversity and composition in the gastrointestinal tract of rough-toothed dolphin and short-finned pilot whales. (A,B) Boxplots showing the α-diversity indices comparisons, including Chao1 index (A) and Shannon index (B), of the gastrointestinal tract samples from rough-toothed dolphin (SB) and four from the short-finned pilot whale (GM). (C) PCoA based on the Bray Curtis distance to visualize the structure of prokaryotic communities in the stomach (GMS1, GMS2, and GMS3), foregut (GMFG), and hindgut (GMHG) of GM samples and stomach (SBS1 and SBS2), foregut (SBFG), and hindgut (SBHG) of SB samples. (D) Stacked bar chart showing the prokaryotic relative abundance in the stomach, foregut, and hindgut samples of GM and SB at the genus level. (E) Prokaryotic biomarkers significantly enriched in the GM and SB gastrointestinal tract samples determined by the linear discriminant analysis effect size (LEfSe) analysis.
The genera Bacteroides (range 28.20 to 55.00%), Vibrio (10.10 to 39.60%), and Escherichia (2.40 to 19.90%) were predominant in the GM samples, whereas the SB samples were dominated by the genera Vibrio (4.69 to 81.40%), Clostridium sensu stricto (6.28 to 16.20%), and Paeniclostridium (0.81 to 6.03%) (Figure 6D and Supplementary Table S1). We also conducted LEfSe analysis to identify significant biomarkers distinguishing the two delphinid species. The result revealed that genera or families related to Vibrio and Escherichia, which are known to potentially cause infections and diseases (Di Renzo et al., 2017), were particularly abundant in SB and GM, respectively (Figure 6E).
KEGG pathways predicted by PICRUSt2 revealed a total of 141 pathways across the nine gastrointestinal samples. Functional profiling indicated that the hindgut samples from the two delphinid species exhibited similar compositions, as did the foregut samples. However, we observed significant differences in the functional composition of the gastrointestinal samples between the two delphinid species based on the KEGG pathway abundance profile (PERMANOVA test: 999 permutations, SumsOfSqs = 0.028, F = 11.54, p < 0.05) and the enzyme abundance profile (PERMANOVA test: 999 permutations, SumsOfSqs = 0.089, F = 11.37, p < 0.05), and the stomach samples between the two species were significantly separated (Figures 7A,B). Using edgeR, we identified significant pathways that differed significantly in abundance between stomach samples from the two species (p < 0.05). The analysis revealed that 32 pathways, including glycerolipid metabolism and glycerophospholipid metabolism, were enriched in the GM stomach samples. In contrast, 23 pathways, such as the Vibrio cholerae pathogenic cycle and carbon fixation pathways in prokaryotes, were enriched in the SB stomach samples (Figure 7C).
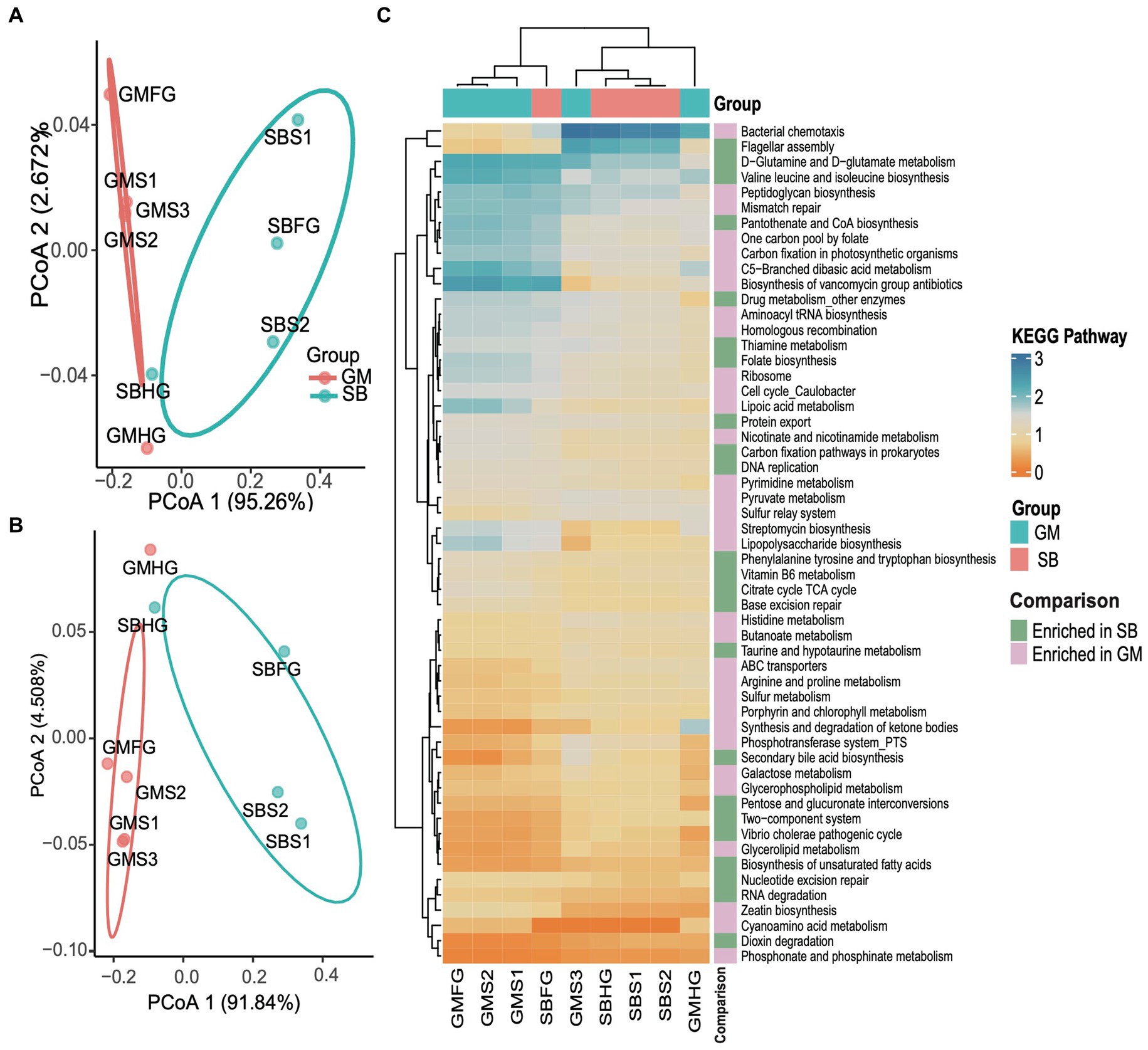
Figure 7. Comparative analyses of potential functions in the stomach, foregut, and hindgut of the short-finned pilot whale (GM) and rough-toothed dolphin (SB) gastrointestinal samples. (A) PCoA based on the Bray Curtis distance of the KEGG pathway functional profiles of the two delphinids. (B) PCoA based on the Bray Curtis distance of the enzyme profiles. (C) Relative abundance heatmap of 55 KEGG pathways significantly enriched in stomach samples from the two delphinids.
Discussion
Cetaceans, as top predators, play a crucial role in marine ecosystems, and their distribution can indicate prey availability and overall ocean health. However, many cetacean species face threats and are highly sensitive to anthropogenic influences such as bycatch in fishing gear, ship strikes, hunting, chemicals, noise pollution, and environmental changes across their habitats (Coombs et al., 2019). The composition and structure of their gastrointestinal microbiota are often species- and tissue-specific and are closely related to the physiological conditions. The microbiota can vary significantly in response to the health status of the host, and even minor disruptions can lead to microbial dysfunction and potentially result in host illness (Li C. et al., 2022). To better characterize the gastrointestinal microbiota of different cetaceans, we conducted 16S rDNA amplicon sequencing on 19 gastrointestinal samples from 12 stranded cetaceans. Consistent with previous studies (Li et al., 2019, 2021; Wan et al., 2021), Firmicutes, Bacteroidetes, Proteobacteria, and Fusobacteria were the dominant phyla in cetacean intestines (Figure 3C). The genera Cetobacterium, Lactobacillus, Pseudomonas, and Clostridium sensu stricto were the most prevalent commensal bacteria across all hindgut samples (Figure 3D). These results provide insights into the gastrointestinal microbiota of cetaceans, highlighting the dominant bacterial phyla and prevalent symbiotic genera. Understanding these microbial communities can help assess cetacean health and the impact of environmental changes on marine ecosystems.
In addition to normal commensals, we identified a large number of potentially pathogenic bacteria, including Paeniclostridium, Escherichia, and Pasteeurellaceae, as well as pyogenic bacteria such as Pseudomonas, Vibrio, and Streptococcus, which are commonly associated with inflammation. These genera comprise several known potential pathogens or pro-inflammatory bacteria. For example, Paeniclostridium sordellii, Pseudomonas aeruginosa, Vibrio cholerae, and Vibrio vulnificus, which are widely distributed in soil, water, plants, and animals, are bacterial pathogens that may cause serious, even lethal infections in humans (Lin et al., 2023). In addition, the family Pasteeurellaceae and the genus Streptococcus comprise some opportunistic pathogens, such as the species Proteus mirabilis, Proteus vulgaris, and Proteus penneri, from Pasteeurellaceae, and the species S. pyogenes, S. faecalis, and S. suis from Streptococcus. These species are occasionally found in the gastrointestinal tract of humans, pigs, and different mammalian species causing diarrhea or inflammation (Lin et al., 2023; Hu et al., 2024). Notably, a previous study indicated that Pasteurellaceae and Paeniclostridium were abundant in the intestines of dwarf sperm whales, potentially acting as opportunistic pathogens causing acute infections (Li X. et al., 2022). Furthermore, Pseudomonas and Vibrio were possibly contributing to infections that may lead to stranding or death (Waltzek et al., 2012; Di Guardo et al., 2018). The presence of both pathogenic and pyogenic bacteria has been linked to the compromised health of cetaceans, and their dominance in the samples suggests a possible link to the unhealthy condition of the stranded cetaceans (Waltzek et al., 2012; Di Guardo et al., 2018). A comprehensive understanding of pathogenic bacteria in cetacean intestinal microbiota is pivotal for effective disease prevention and treatment strategies, thereby forming a critical foundation for cetacean conservation efforts.
Functional predictions indicated that even though different cetacean lineages have different hindgut microbiota composition (Figures 2, 3A), their functional potential was similar without significant separation between different cetacean lineages, indicating that hindgut bacterial functions in cetaceans exhibit marked similarities (Figures 4A,B). The KEGG pathways in hindgut samples from delphinids, physeteroids, and ziphiids showed convergence, with only a few pathways differing between the groups. Similar results were observed in four-stranded Indo-Pacific humpback dolphins from two different locations (Qinzhou vs. Zhanjiang) (Figure 5). Lipids are crucial diagnostic indicators of the metabolic and physiological status of cetaceans, and lipid-related metabolic functions observed consistently in all hindgut samples may reflect the digestion of lipid-rich preys and the absorption of wax (Tang et al., 2018; Miller et al., 2020; Monteiro et al., 2021). Physeteroid and ziphiid hindgut samples exhibited higher abundances of pathways related to glycosaminoglycan degradation and histidine metabolism, whereas glycerophospholipid metabolism was significantly enriched in delphinid samples (Figure 4C). Our results suggest that not only terrestrial mammals but also marine mammals have developed persistent and specific interactions with the hindgut microbiota during evolution. We also investigated the spatial microbial composition and function along the gastrointestinal tracts of a rough-toothed dolphin (SB) and a short-finned pilot whale (GM). Although significant differences in the gastrointestinal microbiota were observed between individuals (Figures 6D,E), the functional analysis indicated that while there were distinct functional profiles in the stomach and foregut samples, the hindgut samples exhibited similar functions (Figures 7A,B). The different functional potential of the stomach microbiota possibly reflects the ingestion of different food resources. In addition, except for different food sources, these differences may also be attributable to the distinct physiological roles of the digestive tract in the two species (Krajmalnik-Brown et al., 2012).
Due to challenges in studying the natural feeding habits of cetaceans, metabarcoding sequencing of the gastrointestinal tracts from stranded cetaceans has become a primary method for exploring their diets (Zhang X. et al., 2023). Previous studies have shown that pilot whales typically consume at least 18 species of cephalopods (including those from the families Octopodidae and Ommastrephidae), as well as fish, crustaceans, and other mollusks. In contrast, rough-toothed dolphins tend to feed on cephalopods and large fish such as saury, needlefish, and mahimahi (Barros et al., 2004; Santos et al., 2014; Beasley et al., 2019). We performed 18S rDNA and 12S rDNA sequencing on the gastrointestinal tract samples from the GM and SB to analyze the composition of eukaryotic taxa as previously described (Supplementary material S1) (Zou et al., 2020; Chen et al., 2021, 2024). Our results indicated that Ochrophyta was relatively abundant in both the stomach and gut samples of these cetaceans, along with various candidate food species, including marine fish, mollusks, and cephalopods (Supplementary Figure S4 and Supplementary Table S5). Most of the detected species appeared to be from natural sources rather than artificial foods, indicating that they are likely part of the natural diet of these delphinid species. However, further comparative tests are necessary to substantiate this notion. The differences in gastrointestinal eukaryotes likely reflect the host’s diet and may also influence their microbial communities.
The composition and function of gastrointestinal microbiota in cetaceans play a crucial role in maintaining host health, particularly in relation to disease manifestation and the proper functioning of the immune system. An imbalance in the intestinal micro-ecology and the production of harmful microbial metabolites can lead to host inflammation, impaired immune responses, malnutrition, and various diseases (Li et al., 2019; Nachtsheim et al., 2021). The microorganisms in the cetacean digestive tract may not only provide valuable insights into host health but also reflect immune status, evolutionary history, habitat, and dietary habits, all of which can influence the composition and distribution of the gastrointestinal microbiota (Bik et al., 2016; Bai et al., 2021; Levin et al., 2021). Our study offers insights into the composition and function of the gastrointestinal microbiota in cetaceans, addressing significant gaps in our knowledge of the gut microbiota across various whale species. However, the current understanding of this field remains limited. To deepen our comprehension of the structure and assembly mechanisms of cetacean gastrointestinal microbial communities, future research should focus on examining the spatiotemporal distribution of these microbiota across multiple individuals of the same cetacean species, as well as among different cetacean species.
Since our study was based on stranded cetaceans (12 individuals) and relied on gut microbiota samples that may not represent healthy, free-ranging populations, our study has some limitations. Some data were collected shortly after attempted rescue and exposure to artificial diets and antibiotics, which could alter the microbiota and may not reflect natural conditions. The SB and GM individuals were subjected to attempted rescue and were given artificial foods and antibiotics for approximately 10 days before samples were collected immediately after their deaths. The reliance on 16S rDNA sequencing constrains our ability to explore functional genomics and other microbial communities such as viruses and fungi. The absence of long-term, spatiotemporal sampling restricts understanding of microbiota dynamics over time and across different habitats. Future research should include larger, diverse sample sizes, incorporate healthy control groups, and extend to longitudinal studies.
Data availability statement
The data that support the findings of this study have been deposited into the China National GeneBank Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession numbers CNP0005144.
Ethics statement
The animal study was approved by the Institutional Review Board of the Institute of Deep-sea Science and Engineering Chinese Academy of Sciences (No. SIDSSE-SYLL-MMMBL-01) and BGI (NO. BGI-R052-3 and NO. FT17160). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JF: Formal analysis, Validation, Visualization, Writing – original draft. HK: Data curation, Writing – review & editing. ML: Formal analysis, Writing – review & editing. YZ: Data curation, Writing – review & editing. YJ: Funding acquisition, Writing – review & editing. ZY: Data curation, Writing – review & editing. CS: Formal analysis, Writing – review & editing. CZ: Formal analysis, Visualization, Writing – review & editing. LD: Methodology, Writing – review & editing. JL: Formal analysis, Writing – review & editing. XJ: Methodology, Writing – review & editing. ShL: Methodology, Writing – review & editing. KK: Supervision, Writing – review & editing. PZ: Funding acquisition, Supervision, Writing – review & editing. JC: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. SoL: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was partly supported by the National Key R&D Program of China (Grant Nos. 2022YFF1301601, 2021YFC2800304, and 2021YFC2800403), the Joint Funds of the National Natural Science Foundation of China (Grant No. U2106228), the National Natural Science Foundation of China (Grant No. 32100047), the “One Belt and One Road” Science and Technology Cooperation Special Program of the International Partnership Program of the Chinese Academy of Sciences (Grant No. 183446KYSB20200016), and the Hainan Provincial Natural Science Foundation of China (Grant No. 422RC744).
Acknowledgments
We thank all staff and students from the Marine Mammal and Marine Bioacoustics Laboratory, Institute of Deep-sea Science and Engineering, Chinese Academy of Sciences for their assistance in collecting specimens. The amplicon sequencing of this study was supported by the China National GeneBank, and we are thankful to the China National GeneBank for producing the sequencing data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1394745/full#supplementary-material
References
Bai, S., Zhang, P., Zhang, C., Du, J., Du, X., Zhu, C., et al. (2021). Comparative study of the gut microbiota among four different marine mammals in an aquarium. Front. Microbiol. 12:769012. doi: 10.3389/fmicb.2021.769012
Bai, S., Zhang, P., Zhang, X., Yang, Z., and Li, S. (2022). Gut microbial characterization of melon-headed whales (Peponocephala electra) stranded in China. Microorganisms 10:572. doi: 10.3390/Microorganisms10030572
Barros, N. B., Jefferson, T. A., and Parsons, E. (2004). Feeding habits of indo-Pacific humpback dolphins (Sousa chinensis) stranded in Hong Kong. Aquat. Mamm. 30, 179–188. doi: 10.1578/AM.30.1.2004.179
Beasley, I., Cherel, Y., Robinson, S., Betty, E., Hagihara, R., and Gales, R. (2019). Stomach contents of long-finned pilot whales, Globicephala melas mass-stranded in Tasmania. PLoS One 14:e0206747. doi: 10.1371/journal.pone.0206747
Bik, E. M., Costello, E. K., Switzer, A. D., Callahan, B. J., Holmes, S. P., Wells, R. S., et al. (2016). Marine mammals harbor unique microbiotas shaped by and yet distinct from the sea. Nat. Commun. 7:10516. doi: 10.1038/ncomms10516
Buck, J. D., and Spotte, S. (1986). The occurrence of potentially pathogenic vibrios in marine mammals. Mar. Mamm. Sci. 2, 319–324. doi: 10.1111/j.1748-7692.1986.tb00142.x
Chen, J., Chen, Z., Liu, S., Guo, W., Li, D., Minamoto, T., et al. (2021). Revealing an invasion risk of fish species in Qingdao underwater world by environmental DNA metabarcoding. J. Ocean Univ. China 20, 124–136. doi: 10.1007/s11802-021-4448-2
Chen, T., Liu, Y. X., and Huang, L. (2022). ImageGP: an easy-to-use data visualization web server for scientific researchers. iMeta 1:e5. doi: 10.1002/imt2.5
Chen, J., Peng, L., Zhou, C., Li, L., Ge, Q., Shi, C., et al. (2024). Datasets of fungal diversity and pseudo-chromosomal genomes of mangrove rhizosphere soil in China. Sci. Data. In press. doi: 10.1038/s41597-024-03748-5
Coombs, E. J., Deaville, R., Sabin, R. C., Allan, L., O'Connell, M., Berrow, S., et al. (2019). What can cetacean stranding records tell us? A study of UK and Irish cetacean diversity over the past 100 years. Mar. Mamm. Sci. 35, 1527–1555. doi: 10.1111/mms.12610
Di Guardo, G., Centelleghe, C., and Mazzariol, S. (2018). Cetacean host-pathogen interaction (s): critical knowledge gaps. Front. Immunol. 9:2815. doi: 10.3389/fimmu.2018.02815
Di Renzo, L., Di Francesco, G., Profico, C., Di Francesco, C. E., Ferri, N., Averaimo, D., et al. (2017). Vibrio parahaemolyticus- and V. alginolyticus-associated meningo-encephalitis in a bottlenose dolphin (Tursiops truncatus) from the Adriatic coast of Italy. Res. Vet. Sci. 115, 363–365. doi: 10.1016/j.rvsc.2017.06.023
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Dudek, N. K., Sun, C. L., Burstein, D., Kantor, R. S., Aliaga Goltsman, D. S., Bik, E. M., et al. (2017). Novel microbial diversity and functional potential in the marine mammal oral microbiome. Curr. Biol. 27, 3752–3762.e6. doi: 10.1016/j.cub.2017.10.040
Edgar, R. C. (2018). Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ 6:e4652. doi: 10.7717/peerj.4652
Edgar, R. C., and Flyvbjerg, H. (2015). Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31, 3476–3482. doi: 10.1093/bioinformatics/btv401
Erwin, P. M., Rhodes, R. G., Kiser, K. B., Keenan-Bateman, T. F., McLellan, W. A., and Pabst, D. A. (2017). High diversity and unique composition of gut microbiomes in pygmy (Kogia breviceps) and dwarf (K. sima) sperm whales. Sci. Rep. 7:7205. doi: 10.1038/s41598-017-07425-z
Fordyce, R. E. (2018). “Cetacean evolution” in Encyclopedia of marine mammals. eds. B. Würsig, J. G. M. Thewissen, and K. M. Kovacs (Amsterdam: Elsevier), 180–185.
Guo, M., Liu, G., Chen, J., Ma, J., Lin, J., Fu, Y., et al. (2020). Dynamics of bacteriophages in gut of giant pandas reveal a potential regulation of dietary intake on bacteriophage composition. Sci. Total Environ. 734:139424. doi: 10.1016/j.scitotenv.2020.139424
Guzmán-Verri, C., González-Barrientos, R., Hernández-Mora, G., Morales, J.-A., Baquero-Calvo, E., Chaves-Olarte, E., et al. (2012). Brucella ceti and brucellosis in cetaceans. Front. Cell. Infect. Microbiol. 2:3. doi: 10.3389/fcimb.2012.00003
Hanning, I., and Diaz-Sanchez, S. (2015). The functionality of the gastrointestinal microbiome in non-human animals. Microbiome 3:51. doi: 10.1186/s40168-015-0113-6
Hu, J., Chen, J., Ma, L., Hou, Q., Zhang, Y., Kong, X., et al. (2024). Characterizing core microbiota and regulatory functions of the pig gut microbiome. ISME J. 18. doi: 10.1093/ismejo/wrad037
Jia, Y., Zhao, S., Guo, W., Peng, L., Zhao, F., Wang, L., et al. (2022). Sequencing introduced false positive rare taxa lead to biased microbial community diversity, assembly, and interaction interpretation in amplicon studies. Environ. Microbiome 17:43. doi: 10.1186/s40793-022-00436-y
Kieser, S., Zdobnov, E. M., and Trajkovski, M. (2022). Comprehensive mouse microbiota genome catalog reveals major difference to its human counterpart. PLoS Comput. Biol. 18:e1009947. doi: 10.1371/journal.pcbi.1009947
Kirk-Cohen, G., Tidière, M., and Zhang, P. (2023). Marine mammal longevity study reveals remarkable advances in animal welfare in zoological institutions. Innov. Geo. 1:100041. doi: 10.59717/j.xinn-geo.2023.100041
Krajmalnik-Brown, R., Ilhan, Z. E., Kang, D. W., and DiBaise, J. K. (2012). Effects of gut microbes on nutrient absorption and energy regulation. Nutr. Clin. Pract. 27, 201–214. doi: 10.1177/0884533611436116
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Levin, D., Raab, N., Pinto, Y., Rothschild, D., Zanir, G., Godneva, A., et al. (2021). Diversity and functional landscapes in the microbiota of animals in the wild. Science 372:eabb5352. doi: 10.1126/science.abb5352
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Li, X., He, L., Luo, J., Zheng, Y., Zhou, Y., Li, D., et al. (2022). Paeniclostridium sordellii hemorrhagic toxin targets TMPRSS2 to induce colonic epithelial lesions. Nat. Commun. 13:4331. doi: 10.1038/s41467-022-31994-x
Li, J., Hu, A., Bai, S., Yang, X., Sun, Q., Liao, X., et al. (2021). Characterization and performance of lactate-feeding consortia for reductive Dechlorination of Trichloroethene. Microorganisms 9:751. doi: 10.3390/microorganisms9040751
Li, C., Tan, X., Bai, J., Xu, Q., Liu, S., Guo, W., et al. (2019). A survey of the sperm whale (Physeter catodon) commensal microbiome. PeerJ 7:e7257. doi: 10.7717/peerj.7257
Li, C., Xie, H., Sun, Y., Zeng, Y., Tian, Z., Chen, X., et al. (2022). Insights on gut and skin wound microbiome in stranded indo-Pacific finless porpoise (Neophocaena phocaenoides). Microorganisms 10:1295. doi: 10.3390/microorganisms10071295
Lin, X., Hu, T., Chen, J., Liang, H., Zhou, J., Wu, Z., et al. (2023). The genomic landscape of reference genomes of cultivated human gut bacteria. Nat. Commun. 14:1663. doi: 10.1038/s41467-023-37396-x
Liu, Y. X., Chen, L., Ma, T., Li, X., Zheng, M., Zhou, X., et al. (2023). EasyAmplicon: an easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research. iMeta 2:e83. doi: 10.1002/imt2.83
Liu, G., Li, T., Zhu, X., Zhang, X., and Wang, J. (2023). An independent evaluation in a CRC patient cohort of microbiome 16S rRNA sequence analysis methods: OTU clustering, DADA2, and Deblur. Front. Microbiol. 14:1178744. doi: 10.3389/fmicb.2023.1178744
Lloyd-Price, J., Arze, C., Ananthakrishnan, A. N., Schirmer, M., Avila-Pacheco, J., Poon, T. W., et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. doi: 10.1038/s41586-019-1237-9
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Miller, C. A., Holm, H. C., Horstmann, L., George, J. C., Fredricks, H. F., Van Mooy, B. A., et al. (2020). Coordinated transformation of the gut microbiome and lipidome of bowhead whales provides novel insights into digestion. ISME J. 14, 688–701. doi: 10.1038/s41396-019-0549-y
Monteiro, J. P., Maciel, E., Melo, T., Flanagan, C., Urbani, N., Neves, J., et al. (2021). The plasma phospholipidome of Tursiops truncatus: from physiological insight to the design of prospective tools for managed cetacean monitorization. Lipids 56, 461–473. doi: 10.1002/lipd.12307
Nachtsheim, D. A., Viquerat, S., Ramírez-Martínez, N. C., Unger, B., Siebert, U., and Gilles, A. (2021). Small cetacean in a human high-use area: trends in harbor porpoise abundance in the North Sea over two decades. Front. Mar. Sci. 7:606609. doi: 10.3389/fmars.2020.606609
Obusan, M. C. M., Aragones, L. V., Salibay, C. C., Siringan, M. A. T., and Rivera, W. L. (2015). Occurrence of human pathogenic bacteria and toxoplasma gondii in cetaceans stranded in the Philippines: providing clues on ocean health status. Aquat. Mamm. 41, 149–166. doi: 10.1578/AM.41.2.2015.149
Ost, K. S., and Round, J. L. (2018). Communication between the microbiota and mammalian immunity. Ann. Rev. Microbiol. 72, 399–422. doi: 10.1146/annurev-micro-090817-062307
Pérez, L., Abarca, M. L., Latif-Eugenín, F., Beaz-Hidalgo, R., Figueras, M. J., and Domingo, M. (2015). Aeromonas dhakensis pneumonia and sepsis in a neonate Risso’s dolphin Grampus griseus from the Mediterranean Sea. Dis. Aquat. Org. 116, 69–74. doi: 10.3354/dao02899
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahe, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Sanders, J. G., Beichman, A. C., Roman, J., Scott, J. J., Emerson, D., McCarthy, J. J., et al. (2015). Baleen whales host a unique gut microbiome with similarities to both carnivores and herbivores. Nat. Commun. 6:8285. doi: 10.1038/ncomms9285
Santos, M. B., Monteiro, S. S., Vingada, J. V., Ferreira, M., López, A., Martínez Cedeira, J. A., et al. (2014). Patterns and trends in the diet of long-finned pilot whales (Globicephala melas) in the Northeast Atlantic. Mar. Mamm. Sci. 30, 1–19. doi: 10.1111/mms.12015
Tang, C. H., Lin, C. Y., Tsai, Y. L., Lee, S. H., and Wang, W. H. (2018). Lipidomics as a diagnostic tool of the metabolic and physiological state of managed whales: a correlation study of systemic metabolism. Zoo Biol. 37, 440–451. doi: 10.1002/zoo.21452
Tidière, M., Colchero, F., Staerk, J., Adkesson, M. J., Andersen, D. H., Bland, L., et al. (2023). Survival improvements of marine mammals in zoological institutions mirror historical advances in human longevity. Proc. R. Soc. B 290:20231895. doi: 10.1098/rspb.2023.1895
Waltzek, T. B., Cortes-Hinojosa, G., Wellehan, J. F. Jr., and Gray, G. C. (2012). Marine mammal zoonoses: a review of disease manifestations. Zoonoses Public Health 59, 521–535. doi: 10.1111/j.1863-2378.2012.01492.x
Wan, X., Li, J., Ao, M., McLaughlin, R. W., Fan, F., Wang, D., et al. (2023). The intestinal microbiota of a Risso’s dolphin (Grampus griseus): possible relationships with starvation raised by macro-plastic ingestion. Int. Microbiol. 26, 1001–1007. doi: 10.1007/s10123-023-00355-z
Wan, X., Li, J., Cheng, Z., Ao, M., Tian, R., Mclaughlin, R. W., et al. (2021). The intestinal microbiome of an indo-Pacific humpback dolphin (Sousa chinensis) stranded near the Pearl River estuary, China. Integr. Zool. 16, 287–299. doi: 10.1111/1749-4877.12477
Xie, F., Jin, W., Si, H., Yuan, Y., Tao, Y., Liu, J., et al. (2021). An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 9:137. doi: 10.1186/s40168-021-01078-x
Yin, J., Li, Y., Tian, Y., Zhou, F., Ma, J., Xia, S., et al. (2023). Obese Ningxiang pig-derived microbiota rewires carnitine metabolism to promote muscle fatty acid deposition in lean DLY pigs. Innovation (Camb) 4:100486. doi: 10.1016/j.xinn.2023.100486
Yuri, M., and Findlay, K. (2021). Whales of the Southern Ocean: biology, whaling and perspectives of population recovery. Mar. Mamm. Sci. 37, 366–367. doi: 10.1007/978-3-030-29252-2
Zhang, P. J., and Goodman, S. J. (2023). Learning from the heaviest ancient whale. Innovation 4:100501. doi: 10.1016/j.xinn.2023.100501
Zhang, P. J., Goodman, S., O'Connell, M., Bai, S. J., and Li, S. H. (2023a). Marine mammal genomes: important resources for unravelling adaptation and evolution in the marine environment. Innov. Geosci. 1:100022. doi: 10.59717/j.xinn-geo.2023.100022
Zhang, X., Luo, D., YuU, R. Q., and Wu, Y. (2023). Multilocus DNA metabarcoding diet analyses of small cetaceans: a case study on highly vulnerable humpback dolphins and finless porpoises from the Pearl River estuary, China. Integr. Zool. 18, 183–198. doi: 10.1111/1749-4877.12640
Zhang, P. J., Zhang, X. F., Wang, S., Vallette, P., Sirugue, C., Ounais, N., et al. (2023b). The international aquarium network – a powerful supporter for the UN decade of ocean science. Innovation Life 1:100021. doi: 10.59717/j.xinn-life.2023.100021
Zhang, Z., Zhu, J., Ghenijan, O., Chen, J., Wang, Y., and Jiang, L. (2023). Prokaryotic taxonomy and functional diversity assessment of different sequencing platform in a hyper-arid Gobi soil in Xinjiang Turpan Basin, China. Front. Microbiol. 14:1211915. doi: 10.3389/fmicb.2023.1211915
Keywords: stranded cetaceans, delphinids, physeteroids and ziphiid, gut microbiota, gastrointestinal microbiota, functional potentials, food digestion
Citation: Fan J, Kang H, Lv M, Zhai Y, Jia Y, Yang Z, Shi C, Zhou C, Diao L, Li J, Jin X, Liu S, Kristiansen K, Zhang P, Chen J and Li S (2024) Taxonomic composition and functional potentials of gastrointestinal microbiota in 12 wild-stranded cetaceans. Front. Microbiol. 15:1394745. doi: 10.3389/fmicb.2024.1394745
Edited by:
Ngoc Tuan Tran, Shantou University, ChinaReviewed by:
Yafei Duan, South China Sea Fisheries Research Institute, ChinaYaotong Hao, Ocean College, Hebei Agricultural University, China
Tiffanie Maree Nelson, The University of Newcastle, Australia
Copyright © 2024 Fan, Kang, Lv, Zhai, Jia, Yang, Shi, Zhou, Diao, Li, Jin, Liu, Kristiansen, Zhang, Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianwei Chen, Y2hlbmppYW53ZWlAZ2Vub21pY3MuY24=; Peijun Zhang, cGp6aGFuZ0BpZHNzZS5hYy5jbg==
†These authors have contributed equally to this work