 Diana Molina1
Diana Molina1 Julio C. Carrión–Olmedo
Julio C. Carrión–Olmedo Gabriela N. Tenea
Gabriela N. Tenea- 1Biofood and Nutraceutics Research and Development Group, Faculty of Engineering in Agricultural and Environmental Sciences, Universidad Técnica del Norte, Ibarra, Ecuador
- 2Laboratorio de Secuenciamiento de Ácidos Nucleicos, Dirección de Innovación, Instituto Nacional de Biodiversidad (INABIO), Quito, Ecuador
Introduction: Foodborne infections, which are frequently linked to bacterial contamination, are a serious concern to public health on a global scale. Whether agricultural farming practices help spread genes linked to antibiotic resistance in bacteria associated with humans or animals is a controversial question.
Methods: This study applied a long–read Oxford Nanopore MinION-based sequencing to obtain the complete genome sequence of a multi-drug resistant Escherichia coli strain (L1PEag1), isolated from commercial cape gooseberry fruits (Physalis peruviana L.) in Ecuador. Using different genome analysis tools, the serotype, Multi Locus Sequence Typing (MLST), virulence genes, and antimicrobial resistance (AMR) genes of the L1PEag1 isolate were determined. Additionally, in vitro assays were performed to demonstrate functional genes.
Results: The complete genome sequence of the L1PEag1 isolate was assembled into a circular chromosome of 4825.722 Kbp and one plasmid of 3.561 Kbp. The L1PEag1 isolate belongs to the B2 phylogroup, sequence type ST1170, and O1:H4 serotype based on in silico genome analysis. The genome contains 4,473 genes, 88 tRNA, 8 5S rRNA, 7 16S rRNA, and 7 23S rRNA. The average GC content is 50.58%. The specific annotation consisted of 4,439 and 3,723 genes annotated with KEEG and COG respectively, 3 intact prophage regions, 23 genomic islands (GIs), and 4 insertion sequences (ISs) of the ISAs1 and IS630 families. The L1PEag1 isolate carries 25 virulence genes, and 4 perfect and 51 strict antibiotic resistant gene (ARG) regions based on VirulenceFinder and RGI annotation. Besides, the in vitro antibiotic profile indicated resistance to kanamycin (K30), azithromycin (AZM15), clindamycin (DA2), novobiocin (NV30), amikacin (AMK30), and other antibiotics. The L1PEag1 isolate was predicted as a human pathogen, matching 464 protein families (0.934 likelihood).
Conclusion: Our work emphasizes the necessity of monitoring environmental antibiotic resistance, particularly in commercial settings to contribute to develop early mitigation techniques for dealing with resistance diffusion.
Introduction
Foodborne illnesses, often attributed to bacterial contamination, represent a significant worldwide risk to public health. The role of agricultural farming systems in transferring antibiotic resistance genes to humans or animals is actively debated (Cheng et al., 2016; Tiedje et al., 2023). Escherichia coli is an important indicator organism in food and water contamination (Reid et al., 2020). A query to NCBI shows that whole genome sequences of E. coli from animals and associated meats are frequent, while sequences from food products, like fruits and vegetables, are rare. Food products may acquire drug-resistant E. coli from animal manure fertilizers, contaminated irrigation water, and other sources (Possas and Pérez-Rodríguez, 2023). Among the different strains of E. coli, certain pathogenic strains are known to cause gastrointestinal infections, ranging from mild to severe, and can lead to life-threatening complications (Thomas et al., 2024). Understanding their transmission, virulence factors, and mechanisms of pathogenesis is crucial for effective prevention and control strategies (Melo and Quintas, 2023).
Emergent antibiotic resistance among Enterobacter species is a source of significant concern, compromising the effectiveness of antimicrobial treatment options (Zurita et al., 2020). The inappropriate use of antibiotics in human and veterinary medicine, as well as in agriculture, has contributed to the selection and spread of resistant strains (Mann et al., 2021). Bacteria found in the human gut, such as E. coli, are frequently shown to be the root cause of drug-resistant infections (Manges and Johnson, 2012). The human gut microbiota is largely influenced by food, and this ecosystem is also a hotspot for horizontal gene transfer (HGT), where genetic components of antibiotic resistance may be traded between commensals and opportunistic infections (Coque et al., 2023). Even though almost all therapeutically significant antimicrobial medications are effective on E. coli, this pathogen has a strong probability of acquiring resistance genes, mainly via HGT (Paitan, 2018; Poirel et al., 2018; Arbab et al., 2022). The development of resistance mechanisms to extended-spectrum β-lactamases (ESBLs), which confer resistance to broad-spectrum cephalosporins, Metallo-β-lactamases enzymes (MBLs), which confer resistance to carbapenems, 16S rRNA methylases, which confer pan-resistance to aminoglycosides, and mcr genes, which confer resistance to polymyxins are among the most problematic mechanisms in E. coli (Poirel et al., 2018).
Among the many plant foods, fruits are a significant source of the vital elements needed for a healthy diet. Additionally, they offer bioactive phytochemicals, including flavonoids and phenolic compounds, which have been linked to several health advantages (Melo and Quintas, 2023). The microbiological safety of fruits has raised significant concerns in the food industry and public health agencies, and it is one of the most rapidly expanding sectors in the last years (Mostafidi et al., 2020). Due to the lack of preventive microbiological techniques to ensure the elimination or destruction of pathogens before consumption, such food products may expose consumers to risk of contracting foodborne infections (Pérez-Lavalle et al., 2020). Several reports have indicated the presence of pathogenic strains of Salmonella enterica, E. coli, and Listeria monocytogenes in minimally processed foods (Solomon et al., 2002; Strawn et al., 2011; Yoo et al., 2015; Pérez-Lavalle et al., 2020).
Escherichia coli-related outbreaks, associated with the consumption of fresh produce, have been increasing in frequency worldwide, as this microorganism is particularly plastic and resilient in its adaptations to survive on a wide variety of fruits, vegetables, environments, and sanitation methods (Luna-Guevara et al., 2019). Enteric pathogens such as E. coli, that are often involved in these outbreaks, have been understudied, emphasizing notable gaps in our understanding on the physiology and adaptations of human enteric pathogens, that are active or viable on agricultural produce (Lynch et al., 2008). A previous study recorded the presence of tetracycline resistant E. coli isolated from ready-to-eat, store-bought, mixed salad, arugula, and cilantro from two German cities (Reid et al., 2020). A recent study in Ecuador described the potential of irrigation water systems and agricultural products as a source of beta-lactam resistant E. coli, with 11% of the sampled vegetables positive for E. coli and 58% of the 165 E. coli laboratory cultures with the ESBL phenotype (Montero et al., 2021). Salazar-Llorente et al. (2021) and Cárdenas et al. (2024) surveyed microbial presence on hundreds of street market food samples from Ecuador, including fruits, their findings suggest poor sanitation and lack of clean water for food processing.
Ecuador has a high rate of antibiotic resistance in poultry and water systems (Molina et al., 2024), which requires the creation of regulations and guidelines for the use of antibiotics (Amancha et al., 2023). To better track the use of antibiotics and changing resistance patterns, the current surveillance system must be enhanced. The bacteriological safety of fresh fruits in Ecuadorian agricultural farms (Montero et al., 2021) and low-cost markets was recently assessed (Tenea et al., 2023); since this produce is usually eaten raw, there is a greater chance that bacteria will survive and spread to the human gut, than when cooked food is consumed (Melo and Quintas, 2023).
Cape gooseberry (Physalis peruviana), locally known as uvilla or uchuva, is a super nutritive and exquisite fruit that requires attention, as it is contaminated with Staphylococcus spp. and several Enterobacteriaceae, including E. coli (Tenea et al., 2023). Among a pool of antibiotic resistant isolates, one isolate annotated L1PEag1 was selected, and the whole genome sequenced. Through various in silico tools, the isolate was typed, serotyped, and its evolutionary relationship assessed. With the assistance of web server tools, we predicted the presence and characteristics of antibiotic-resistant genes, virulence factors, CRISPR sequences, pathogenicity factors, and fimH alleles. In vitro assays (antibiotic sensitivity, hemolysis, gelatinase activity) were applied to confirm antibiotic resistance and pathogenicity. Besides exploring, characterizing, annotating, and describing a new genome of E. coli native to Ecuador, our research contributes to current efforts on drawing attention on food contamination with pathogenic bacteria and to the search for solutions to combat cross-contamination, helping to promote public health and ensure safer food systems.
Materials and methods
DNA extraction and genome sequencing
The isolate L1PEag1 was isolated from fruit peel (exocarp) of cape gooseberry, purchased from the local market of Ibarra city, northern Ecuador, and following a standard procedure (Tenea et al., 2023). The purified isolate was grown overnight on Luria Bertani (LB) agar (Oxoid, UK) at 37°C. Genomic DNA was extracted using a high molecular weight DNA extraction kit (Wizard Genomic DNA purification kit), and following the manufacturer indications (Promega, USA). The DNA was quantified, using a NanoDrop™ 2000 Spectrophotometer (Thermo Fisher, USA). The library was prepared using a Ligation Sequencing kit V14 (SQK-LSK114) (Oxford Nanopore Technologies Ltd., UK). High molecular weight DNA was nick–repaired and end–prepared with the NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (E7180S). The end–preparation protocol phosphorylated 5’ends and dA-tailed 3′ ends of gDNA fragments. A magnetic bead clean-up of enzymes was performed before ligation. Sequencing adapter ligation was performed with the NEBNext® Quick Ligation Module (E6056). After ligation, a second magnetic bead clean–up was performed before sequencing. The library was sequenced on a MinION Mk1C sequencer and a Flow Cell version R10.4.1 (FLO-MIN114, Oxford Nanopore Technologies Ltd., UK). Sequencing run took 48 h and generated 998.45 k reads at an estimated coverage of 400x.
Genome assembly
Raw sequence data was high-accuracy basecalled into fastq data (HAC) under the dna_r10.4.1_e8.2_400bps_hac@v4.2.0 model with the Dorado 0.2.1 package (Hariharan et al., 2020). Quality control was performed with NanoPlot version 1.41.3 (Coster et al., 2018) and FastQC version 0.12.11. To reduce errors in assembly, we applied filtering and trimming with NanoFilt 2.8.0 (Coster et al., 2018). Reads with less than a quality value of Q14 were filtered out for the assembly. To allow for plasmid assembly, short reads were not filtered out. Trimming was performed on the first 20 bps for each end of the reads to eliminate adapters. A subsequent round of quality control was performed after the filtering and trimming. De novo assembly was made according to the long–read assembly pipeline and the Trycycler tool by Wick et al. (2021), these consist in estimating a final consensus assembly from multiple independent assemblies as input. The reduction of error during the final assembly process, assisted by the Trycycler pipeline and its algorithms, relies on the fact that multiple independent subset assemblies provide alternative sources of evidence that compensate previous assembly biases. In brief: (1) By randomly subsampling the original read set, sequence data was subsampled into 12 independent read sets. To maximize the independence of assembly results, each subset was independently assembled with different assembling tools. The assemblers used for this step were (a) the repeat graph assembler FLYE 2.9.2-b1786 (Kolmogorov et al., 2019), (b) Minipolish (Wick and Holt, 2020), and (c) Raven (Vaser and Šikić, 2021). (2) The different assemblies were clustered based on their k-mer content. For the case of the L1PEag1 isolate, clusters recovered two replicons that consisted of the main circular genome and one accompanying plasmid. (3) The clustered sequences (contigs) were then reconciled by normalizing their directionality or orientation and aligned to each other to repair circularization issues such as missing bases or overlapping bases. (4) Each cluster of sequences was rotated to a common starting sequence, preparing them for multiple sequence alignment. (5) A global multiple sequence alignment was produced for each cluster of sequences. (6) A definitive consensus sequence for each cluster was obtained based on the minimum total Hamming distance to optional or variable regions in the consensus and the best total alignment score. (7) Finally, the consensus sequence was polished according to the Medaka v.1.1.1.3 algorithms2.
Typing and evolutionary relationship
The genome FASTA sequence was uploaded to the Type (Strain) Genome Server (TYGS) to conduct a complementary genome-based taxonomic study and a phylogenetic relationship inference (Meier-Kolthoff and Göker, 2019). The closest strain genome type was determined by comparing the L1PEag1 genome to all accessible strain genomes in the TYGS database using the MASH algorithm (Meier-Kolthoff and Göker, 2019). Strains with the shortest MASH distance were selected automatically. Thus, the precise distance was calculated using the Genome BLAST distance phylogeny (GBDP) approach under the “coverage” algorithm and distance formula (Meier-Kolthoff et al., 2013). Phylogroups were determined using the in silico Clermont Phylotyper EzClermont3 (Waters et al., 2020). In addition, multi-locus sequence typing (MLST)4 (Larsen et al., 2012) was used for accurate subtyping, with the E. coli # 1 (Wirth et al., 2006), and E. coli # 2 (Jaureguy et al., 2008) schemes. Further, SerotypeFinder 2.05 was used to identify the serotype (Joensen et al., 2015), using 95% sequence identity and 60% sequence coverage setup. To enrich the phylogenetic contrast, an additional genome search for similar E. coli sequences from Ecuador was made at NCBI’s databases and guided by previous published work (Montero et al., 2021; Rothstein et al., 2023). While genome sequences from earlier research have been made available at NCBI, these are either assembly-level contigs or raw sequences (SRA experiments in NCBI terminology), which do not meet the requirements for phylogenetic comparisons of the type carried out in this work on the whole assembled genome.
Gene prediction and functional annotation
The L1PEag1 genome was annotated using the PROKKA tool (Seemann, 2014). Plasmid annotation was performed with pLannotate (McGuffie and Barrick, 2021). The predictions of CDS, rRNA, tRNA/tmRNA, signal leader peptide, and noncoding RNA were performed using Prodigal (Hyatt et al., 2010), RNAmmer6, Aragorn7, Signal IP8 and Infernal (Nawrocki and Eddy, 2013). Additionally, the predicted genes were annotated through the Global Catalog of Microorganisms genome annotation project pipeline9 and based ondatabases SwissProt10, MetaCyc11 (this is a database that contains pathways responsible for both primary and secondary metabolism) (Caspi et al., 2018), VFDB (a virulence factor database12) (Liu B. et al., 2019), PHI: The Pathogen-Host Interaction database13, KEGG (Kyoto Encyclopedia of Genes and Genomes), Orthology Database of Prokaryotes, COG (Clusters of Orthologous Groups of proteins), and pFam14 (Wu and Ma, 2019). A circular map was generated by importing the FASTA sequence into the PROKSEE server15 (Grant et al., 2023).
Prediction of CRISPR sequences, prophage, and mobile elements
CRISPRFinder16 and PHAge Search Tool Enhanced Release (PHASTER)17 (Arndt et al., 2016) were used to detect CRISPR, Cas sequences, truncated Cas sequences, and prophage sequences. Only predicted prophages with intact completeness in PHASTER, as defined by Arndt et al. (2017), were considered for report. In addition, mobileOG-DB was used to annotate the mobile elements (Brown et al., 2022).
Antibiotic-resistant genes, putative virulence genes, and pathogenicity prediction
The Comprehensive Antibiotic Resistance Database (CARD)18 (Jia et al., 2017) and the Resistance Gene Identifier tool (RGI) were used to detect the antibiotic resistance genes by importing the contig files in FASTA format to the database (Zankari et al., 2012). The ResFinder 4.3.3 server19 was used to identify acquired antimicrobial resistance genes (Bortolaia et al., 2020). The putative virulence factors were predicted using the VirulenceFinder 2.0 web server20 (Cosentino et al., 2013). The bacterial pathogenicity was predicted using the PathogenFinder web server21 (Cosentino et al., 2013). Whereas FimTyper 1.022 was used to type de fimH alleles (Roer et al., 2017). The detection standard parameters were set at 90% sequence identity and 60% sequence coverage for VirulenceFinder and ResFinder.
Identification of genomic islands (GIs) and insertion sequences (ISs)
GIs were predicted with the webserver IslandViewer 4, using E. coli SMS-3-5 as a reference strain (Bertelli et al., 2017). To search for ISs, the ISfinder tool (ISsaga V.2.0) was used (Siguier et al., 2006).
In vitro assays
Hemolysis and gelatinase
For hemolysin production, plates containing 5% human blood agar was inoculated with the L1PEag1 isolate and incubated at 37°C for 24 h (Tabasi et al., 2015). The presence of partial or complete hemolysis was assessed on the plates. For gelatinase production, gelatin nutrient agar was used (Mittal et al., 2014). An overnight-old L1PEag1 inoculum was applied to the plate. Following observation of the organism’s growth, mercuric chloride solution was poured onto the plate. When a colony is flooded with mercuric chloride solution, the medium becomes opaque, and a clearing zone forms around it. This indicates that the colony is liquefying gelatin and is positive for gelatinase. As control, a human E. coli Ec1 (human enteropathogenic donated by Saint Vincente de Paul Clinical Hospital, Ibarra) and a human non-pathogenic E. coli ATCC25922 strains were also used.
Antibiotic profile
Antibiotic susceptibility was determined using the Muller-Hilton (MH) agar disk diffusion procedure, and according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (EFSA Panel on Additives and Products or Substances used in Animal Feed et al., 2018). Briefly, 100 μL of inoculum (107–108 CFU/mL) was streaked onto MH plates. The commercial antibiotic disks (Merck, USA) were chosen as recommended by the National Plan for Surveillance and Control of Contaminants in Primary Production (NTE INEN 1529–2-2013)23. The disks were plated on MH agar plates, and incubated at 37°C for 48 h. The diameter of each clear zone was measured in millimeters by scanning the plates with a microplate reader (SCAN500, Interscience, Fr). E. coli ATCC25922 and E. coli UTNEc1 were used for quality control and comparison. The microbiological breakpoints reported by the FEEDAP standards were used to categorize E. coli as susceptible, intermediate, or resistant (EFSA Panel on Additives and Products or Substances used in Animal Feed et al., 2018).
Beta-lactamase resistance and virulence genes detection
Genomic DNA was extracted from the L1PEag1 isolate using the Wizard Genomic DNA purification Kit (# 1120 Promega, USA). The DNA concentration and purity were determined in a NanoDrop™ (Thermo Fisher Scientific, USA) at 230, 260, and 280 nm. The primers for blaTEM, blaSHV, blaCTXM-2, blaCTXM-9, blaCTXM-8/25, blaNDM, blaKPC, blaVIM, and blaOXA-48/181 genes were used at a concentration of 0.3 μM (Hernández-Alomía et al., 2023). The genetic determinants for virulence evaluated in this study were those that code for type 1 fimbriae (fimH), pili associated with pyelonephritis (pap), and S fimbriae (sfa) (Dadi et al., 2020). The sequences of these primers are listed in the Supplementary Table S1. The amplification was performed in reactions of 25 μL containing 2X GoTaq® Green Master Mix (#7132, Promega, USA) and the PCR reaction was performed in a Genemax Thermal Cycler (IQM, Olso, Norway). The amplification conditions for antibiotic resistance genes were as follows: denaturation step 2 min at 94°C, followed by 35 cycles of 1 min (denaturation) at 94°C, 1 min (annealing) at 50–60°C and 1.5 min (extension) at 72°C, and 1 cycle of 10 min (final step extension) at 72°C. For virulence genes, the amplification conditions were denaturation step for 4 min at 95°C, followed by 35 cycles of 40 s (denaturation) at 95°C, 45 s (annealing) at 53–65°C and 1 min (extension) at 72°C, and 1 cycle of 5 min (final step extension) at 72°C. The PCR products were separated by electrophoresis on 1% agarose gels in 1 x Tris-Borate EDTA (TBE, pH 8.0) buffer (Sigma-Aldrich Co., USA). Gels were stained in TBE buffer containing 0.5 μg/mL ethidium bromide. The results were registered as plus / minus for the presence of each amplicon.
Results and discussion
L1PEag1 isolate typing and phylogenetic relationship
An estimated genome size of 4825.72 Kbp (400X coverage) and a plasmid of 3.561 Kbp (400X coverage) were generated by the sequence analysis. Species matching resulted in a 100% hit on the entire contig for Escherichia coli. The results of the whole genome analysis placed the L1PEag1 strain on the same lineage as E. coli DSM30083 (Figure 1). Based on the BLASTN analysis, the plasmid pL1PEag1 showed 99.96% sequence identity with plasmid pECQ4552_IHU08 from Klebsiella quasipneumoniae subsp. similipneumoniae, strain IF3SW-P1 (CP092122.1); 99.89% identity with an unnamed plasmid from Staphylococcus aureus strain, Alexandria 2020–19 (CP113245.1); and 99.81% sequence identity with plasmid pECQ4552_IHU08 from E. coli strain Q4552 (CP077071.1).
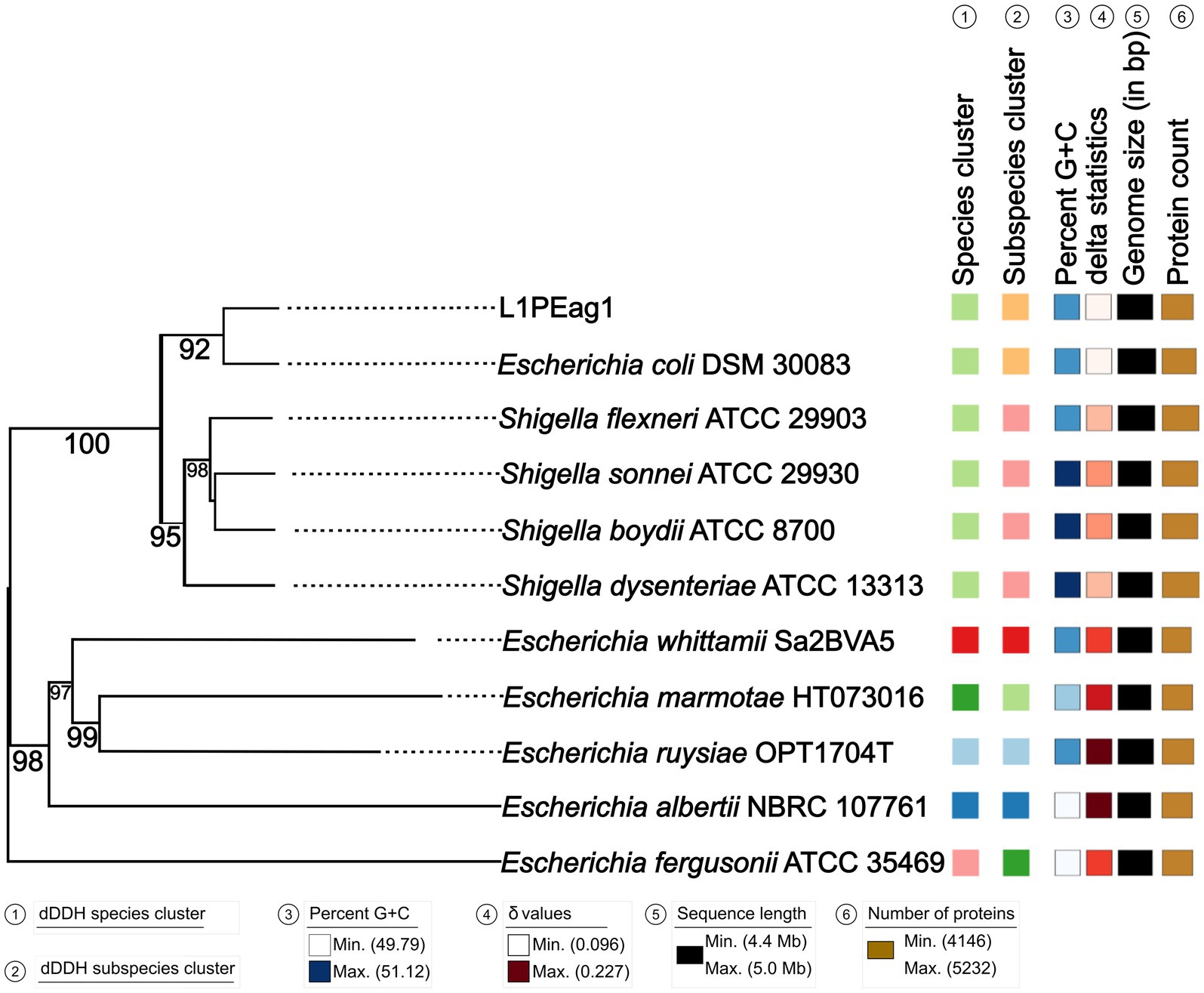
Figure 1. Phylogenetic tree based on TYGS results for the L1PEag1 whole-genome data set. Branch lengths are scaled in terms of GBDP (genome BLAST distance phylogeny method) distance; numbers below branches are GBDP pseudo-bootstrap support values from 100 replications. Leaf labels are annotated by affiliation to species clusters; subspecies clusters; genomic G + C content (min 49.79 – max 51.12); δ values (min 0.096 – max 0.227); overall genome sequence length (4,416,104 – 5,037,933); and number of proteins (min 4,146 – max 5,232).
Phylogroup typing, MLST, and serotyping
Based on the Clermont Escherichia-typing method (Waters et al., 2020), the L1PEag1 isolate belongs to phylogroup B2 (TspE4 +, chu +, yjaA +, and arpA –). Generally, B2 phylogroup strains are associated with Extraintestinal pathogenic E. coli (ExPEC) (Xia et al., 2022). A comparison of several E. coli strains prevalent in inflammatory bowel disease indicated that the B2 phylogroup strains share a specific metabolism and carry virulence genes enabling them to colonize intestinal mucosa (Fang et al., 2018). In E. coli, MLST requires assessment of 7 housekeeping genes (adk, fumC, gyrB, icd, mdh, purA and, recA) to assign sequence types (ST) genes (Wirth et al., 2006); thus, by using the E. coli #1 scheme, L1PEag1 was identified as the ST1170 sequence type. By using the E. coli #2 scheme (by the assessment of genes dinB, icdA, pabB, polB, putP, trpA, trpB, and uidA), no sequence type was discovered; nonetheless, the closest ST518 and ST966 were recommended (Supplementary Table S2). Several studies indicated the presence of specific STs in foods, whether of plant (ST2847, ST973) (Liu and Song, 2019; Song et al., 2020; Montero et al., 2021) or animal origin (ST1170) (Castellanos et al., 2017). E. coli isolates with phylogroup B2 and sequence type ST1170 were detected in wastewater from pig transport trucks (Savin et al., 2021) and from nosocomial and ambulatory infections in Germany (Pietsch et al., 2017). The ST966 type was identified in Klebsiella pneumoniae isolated from animal feed (Wu et al., 2022), suggesting environmental contamination. Cilantro-isolated E. coli samples, from a market in Germany, were classified as phylogroup B1 with multi-locus sequence types ST6186, ST165, ST58, and ST641 (Reid et al., 2020). Several E. coli strains, isolated from strawberries and banana obtained from agricultural farms in Ecuador, were classified into phylogroup B1, with the most frequent multi-locus sequence types being ST453, ST847, and ST6598 (Montero et al., 2021). Observed patterns of sequence types and phylogroups may be related with country- or producer-specific production methods, such as those pertaining to antimicrobial use, hygiene, and irrigation water systems (Molina et al., 2024). Based on the fimH typing, the L1PEag1 serotype ST1170 was classified as fimH20, with 100% sequence identity. Moreover, the subtyping results indicated the presence of the fliC gene, which matches the H-type H4 (100% identity with E. coli U1-41, AJ605764.1) (Beutin et al., 2005) and wzy genes (98.92% identity with the E. coli O1,H12 variant and 99.14% identity with the O1,H20 variant, KY115223.1). The presence of these variant alleles provided evidence for classifying the L1PEag1 isolate into the O1:H4 serotype. A single structural subunit (flagellin) encoded by the fliC gene defines the H antigen of E. coli (Reid et al., 1998), whereas the wzy gene encodes the O-antigen polymerase, which plays an important role in the synthesis of the lipopolysaccharide of bacteria (Zuo et al., 2019). The E. coli O1:H12 variant was detected in pig feces (Delannoy et al., 2017).
Gene prediction and annotation
The genome contains 4,473 genes, 88 tRNAs, and 22 rRNA (8 copies of 5S rRNA, 7 copies of 16S rRNA, and 7 copies of 23S rRNA). A physical genomic map is shown in Figure 2A. Prokka was used to predict the location, while BLAST was used to infer the function and identification of assembled sequences against nucleotide and protein sequence databases. Predicted genes in the previous step were aligned with several databases to obtain their corresponding annotations with the aligners (Table 1). The number of genes associated with COG (3,723 genes) and KEGG (4,439 genes) functional annotation categories are shown in Supplementary Figures S1A,B. Plasmid annotation (Figure 2B) consists of the following features: (1) R–region: an endolysin with trans glycosylase activity that degrades host peptidoglycans and participates with the holin and spanin proteins in the sequential events which lead to the programmed host cell lysis releasing the mature viral particles; (2) Rz: Component of the spanin complex that disrupts the host outer membrane and participates in cell lysis during virus exit (Berry et al., 2013; Rajaure et al., 2015); (3) bor-not known function, is expressed during lysogeny in E. coli (bacteriophage); (4) ydfO-putative protein (COG5562), 74.9% similarity with E. coli K-12 strain; (5) S- [Isoform Antiholin]: Counteracts the aggregation of the holin molecules and therefore pore formation; (6) cos: lambda cos site; allows packaging into phage lambda particles; (7) ybcW: Protein inferred from homology (E. coli O157:H7); (8) ylcI: unknown function, protein predicted in the reference genome of E. coli K12.
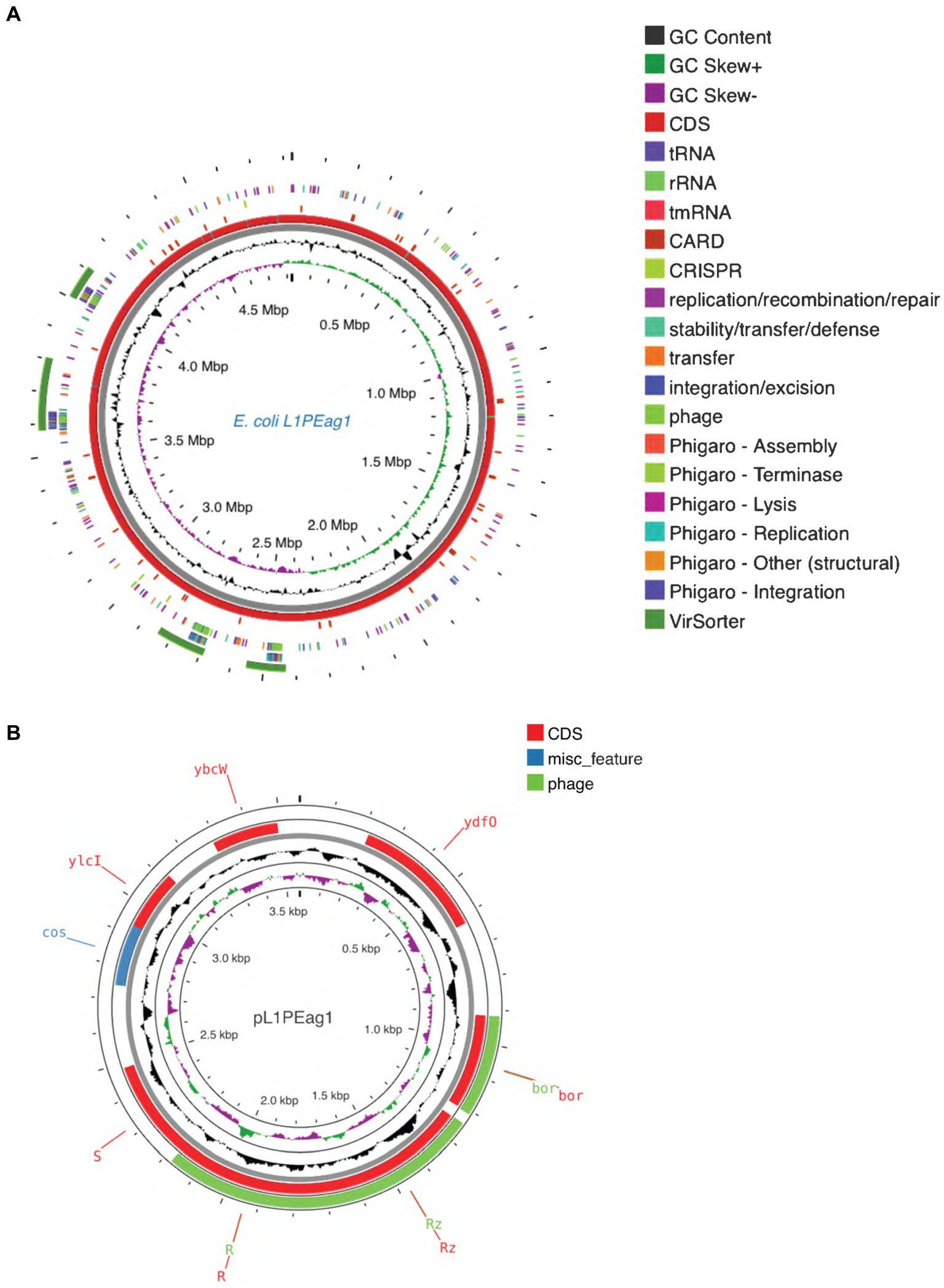
Figure 2. (A) Circular maps of the L1PEag1 genome predicted with the PROKSEE viewer (https://beta.proksee.ca/projects; Accessed on November 30th, 2023). The contents are arranged in feature rings. Starting with the outermost ring: Ring 1, prophage prediction with VirSorter annotation (mustard color); ring 2, mobile genetic elements (MGE) annotation with Alien Hunter predicting HGT (Horizontal Genetic Transfer) events (in red color); ring 3, prophage prediction with Phigaro annotation (genes involved in integration/replication/lysis/terminase are marked in different colors); ring 4, MGE annotation with Mobile OG DB marking the hsdR gene involved in stability/transfer/defense; rings 5–6 show the CDS (protein-coding genes) with Prokka annotation (blue color), tRNA, rRNA, and tmRNA are marked; ring 7 displays the GC content plot (black); ring 8 displays G/C skew information in the (+) strand (green color) and (−) strand (purple color). (B) Plasmid map as predicted by pLannotate (http://plannotate.barricklab.org).
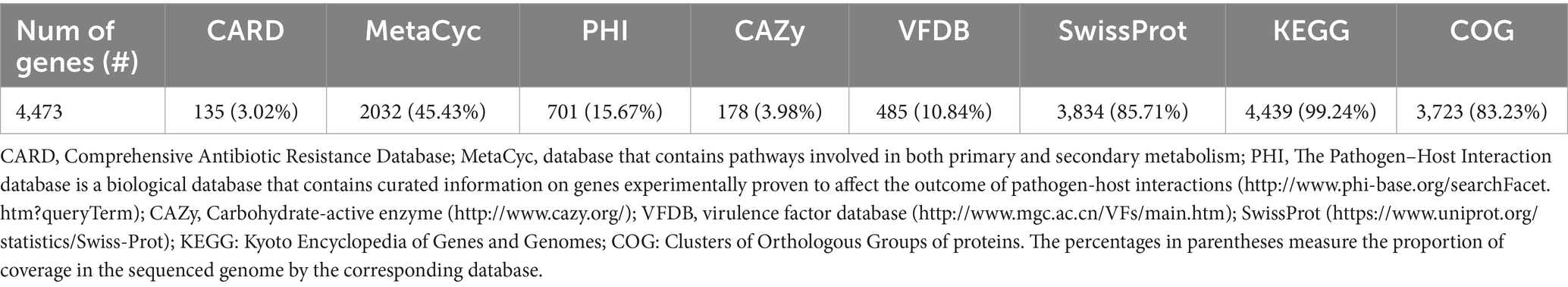
Table 1. Gene annotation summary.
Prediction of CRISPR elements, prophage, virulence factors (VFs), genomic islands, insertion sequences, and pathogenicity
Two sequences with CRISPR were found within the L1PEag1 genome (beginning at position165680 and ending at 1656883 or at position 4,818,020 and ending at 4818143, respectively). Both encompass a short spacer sequence, spanning between degenerate repeats (consensus DRs) with 95.65 and 96.87% conserved repeats respectively, and 100% spacer conservation. No Cas elements were detected. There are seven identified prophage regions; of which three are complete, three are incomplete, and one is questionable. The Enterobacteria phages mEp460, SfV, and cdtI were the most frequently occurring intact phages (Table 2). Previous studies indicated the presence of Enterobacteria phage mEp460 in three E. coli strains isolated from seawater and marine sediment, indicating its high adaptability to different environments (Lai et al., 2017). Phage SfV is a temperate serotype-converting phage of Shigella flexneri and encodes the factors involved in type V O-antigen modification (Allison et al., 2002). Shigella phage SfIV and Enterobacteria phage cdtI (cytolethal distending toxins) were detected in the human gut microbiome and were associated with diarrhea (Federici et al., 2023). Additionally, a total of 23 GIs of 238,133 bp were predicted with IslandViewer using as a reference the multi-drug resistant E. coli SMS-3-5 genome (Fricke et al., 2008). A total of 229 hypothetical proteins, virulence genes, and antibiotic resistance genes were annotated in the GIs. Due to their frequent rearrangements, excisions, transfers, and further acquisition of additional DNA, these genomic regions play a significant role in the rapid evolution, diversification, and adaptation of E. coli. Most of them encode several proteins involved in the defense mechanism, virulence factors, iron-acquisition systems, and toxins (Supplementary Table S3). Our study included both in vitro and in silico approaches to analyze the pathogenicity of L1PEag1. The virulence factors detected in silico in the L1PEag1 genome are shown in Table 3. Among 25 genes from the virulence database, 8 genes showed perfect identity match (ID % = 100 match for the given gene, covering the entire length of the virulence genes in the database, 100% alignment), 12 were non-perfect matches (ID% < 100%, covering the entire length of the virulence genes in the database, 100 alignment), and 5 were non-perfect matches (ID < 85%, input sequence length is shorter than the virulence gene length, 60% alignment). It has been shown that virulence-associated regions are specific to carriage patterns, generally corresponding with specific E. coli STs. Out of 120 selected E. coli strains, 96% showed the presence of fimbrial adhesin gene fimH, whereas 63% contained glutamate decarboxylase gene gad (Reid et al., 2020). In our study, both virulence factors, fimH and gad, were detected with 100% identity. fimH is a type 1 fimbriae detected in more than 80% of E. coli strains and plays a significant role in the virulence of E. coli extraintestinal enteropathogenic strains (Poole et al., 2017). The enzyme GAD (glutamate decarboxylase) has been reported to be limited to pathogenic E. coli, being a virulence marker for contaminated food and water (Grant et al., 2001). In addition, the yfcV gene encodes a fimbrinal protein, the OmpT gene encodes an outer membrane protease (protein protease 7), the fyuA encodes a siderophore receptor, and sitA encodes an iron transport protein. In the L1PEag1 genome, all three genes, OmpT, fyuA, and fimH, were found with 100% match identity, within the island picks and were previously detected in urinary infections (Spurbeck et al., 2012; He et al., 2015). Although not found with a 100% match, the capsular polysaccharide gene kpsM, serine protease autotransporter vat, and increased serum survival iss, which has been previously associated with ExPEC infection (Ravan and Amandadi, 2015), were detected in the target L1PEag1 genome. In addition, the virulence fimA, fimC, fimD, fimH, fimG, focC, kpsM, and papC genes were annotated with Prokka. Complementary PCR analysis confirmed that L1PEag1 harbored a gene that encodes for type 1 fimbriae (fimH) and pap (pili associated with pyelonephritis) but not for S fimbriae (sfa) (Supplementary Table S1). This virulence gene profile was previously detected in E. coli isolated from urine culture of a patient with urine infection, and they belonged to phylogroup B2 (Rahdar et al., 2015).
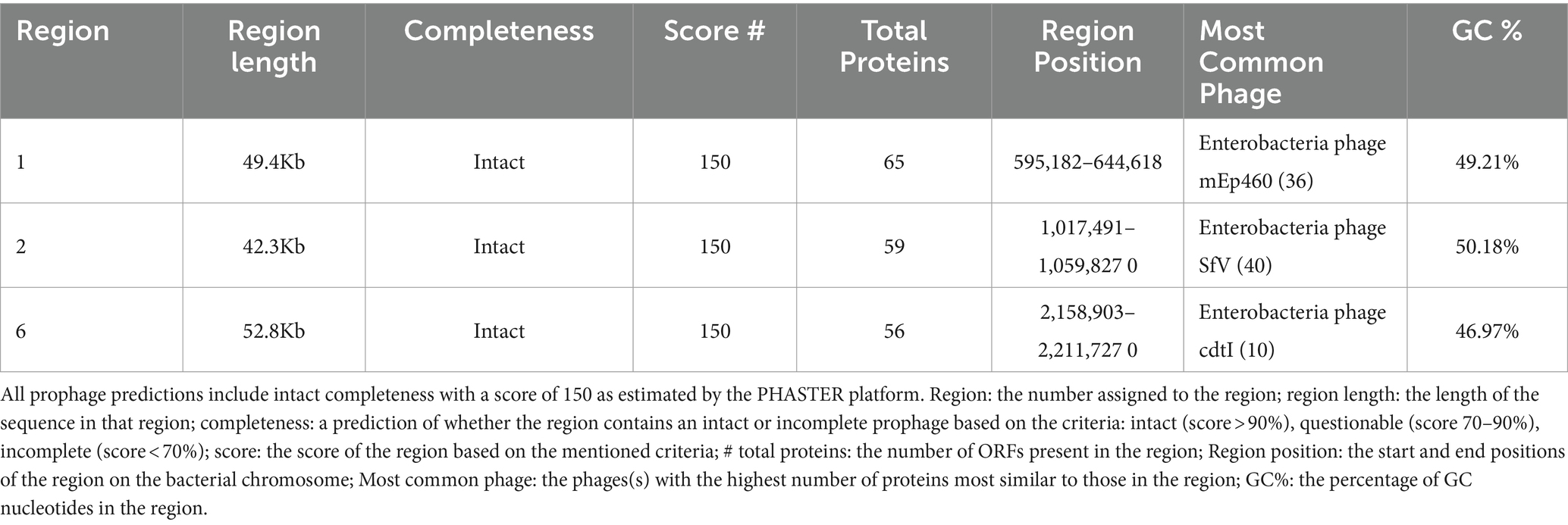
Table 2. Prophage prediction with intact completeness, from genome analysis of the L1PEag1 isolate.
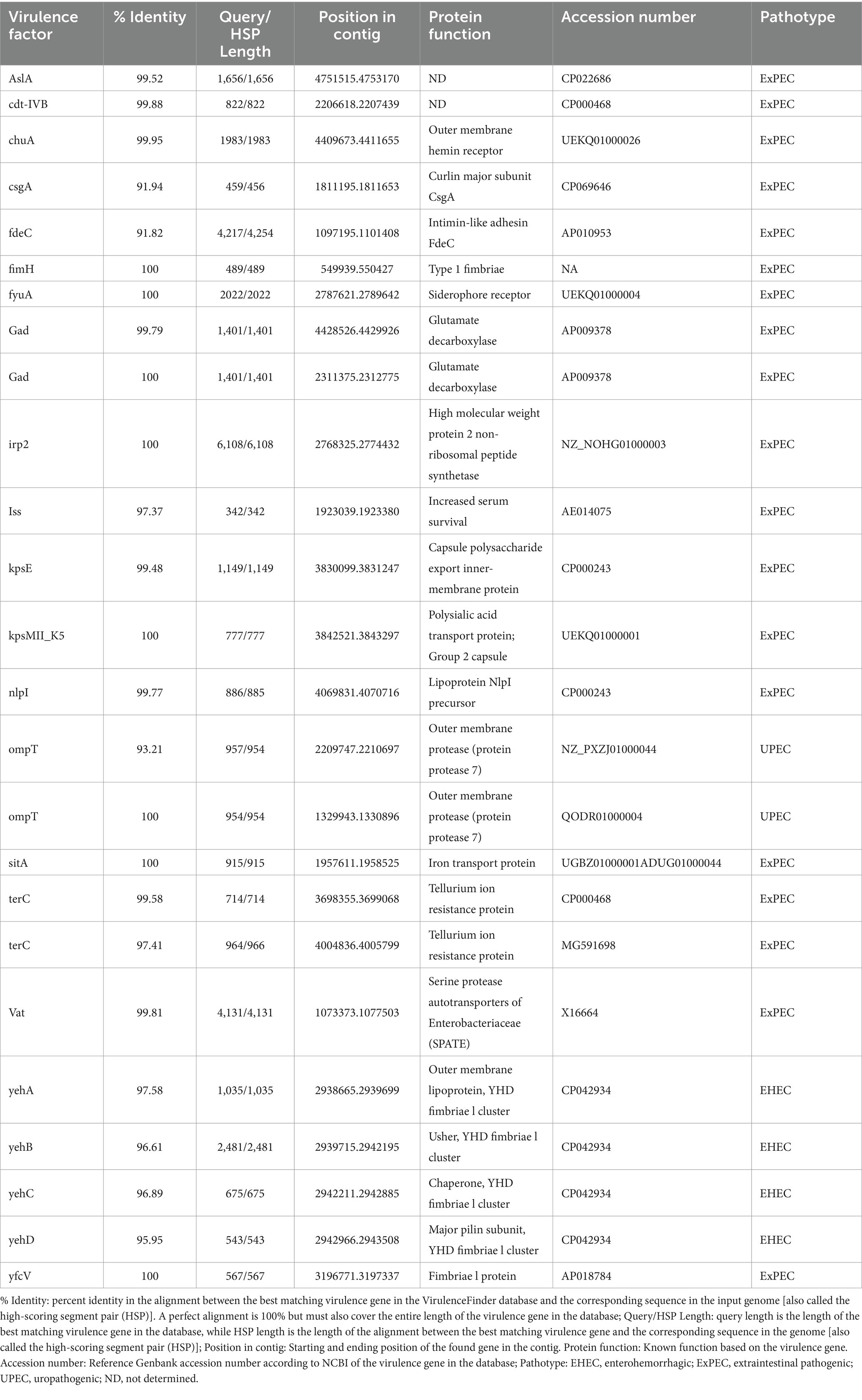
Table 3. In silico characteristics of virulence factors detected in the L1PEag1 genome.
A total of two insertion sequences (ISs) and two miniature inverted-repeat transposable elements (MITEs) were grouped into 2 families (ISAs1 and IS630) and were retrieved with the ISfinder web tool. These genomic datasets can provide information on the ecological strength and adaptability of strains as well as their function in various environments. Besides, a total of 399 mobile elements were predicted with mobileOG-db (Brown et al., 2022), from which 50 were insertion/excision, 117 were replication/recombination/repair, 141 belonged to phage, 46 were stability/transfer/defense and 45 were transfer. Based on the 464 matched pathogenic families (10.44% of proteome), the results showed that the L1PEag1 isolate is a human pathogen (0.934 likelihood). Prokka annotation indicated the presence of the hlyE hemolysis gene in the L1PEag1 genome. The BLASTP analysis indicated a 100% identity to the hemolysin E protein family of the E. coli strain AR_451 (a clinical isolate, AWZ82741.1). Hemolysin hlyE is a novel pore-forming toxin and was first discovered in E. coli K-12 (Wyborn et al., 2004). This result was also complemented by the inhibition zone on blood agar media with the effect of hemolytic activity (data not shown). Although gelatinase is a less significant virulence factor in E. coli (Shruthi et al., 2012); in the current investigation, the gelatinase enzyme was produced by both L1PEag1 and UTNEc1. These results concur with earlier research, which showed that many E. coli isolates from urinary infections were gelatinase positive (Sharma et al., 2007).
Antibiotic profile
Previous studies indicated that E. coli is a significant source of resistance genes that could be the cause of treatment failures in veterinary and human medicine. An increasing number of resistance genes have been found in E. coli isolates, and many of these resistance genes were acquired through horizontal gene transfer (Poirel et al., 2018). The putative antibiotic resistance genes identified in the genome of L1PEag1 is shown in Figure 3. A total of 4 perfect and 51 strict hits were detected, and the CARD resistant gene identifier report is shown in Supplementary Table S4. Within the former set of hits, the following antibiotic resistance genes were detected cpxA, mdtH, H-NS, and evgA, these confer resistance to aminoglycoside, aminocoumarin, fluoroquinolone and macrolide, cephalosporin, cephamycin, penam, and tetracycline. Previous research in E. coli indicated that Cpx-regulated genes are centrally involved in cell energetics, mediated envelope stress adaptation, and antibiotic resistance (Raivio et al., 2013). The mdtH (multidrug transport) gene is one of the 35 efflux pumping encoding genes detected in E. coli. Overexpression of cloned mdtD in the E. coli K-12 strain, which lacks the major efflux pump permease AcrB (E. coli KAM3) (Hirakawa et al., 2003a), results in a two-fold increase in resistance to norfloxacin and enoxacin (compared to the mutant parent) but does not impact the resistance to a range of other toxic compounds (dyes, detergents, antibiotics, and others) (Nishino and Yamaguchi, 2001). H-NS is a histone-like protein involved in global gene regulation in Gram-negative bacteria (Nishino and Yamaguchi, 2004). It is a repressor of the membrane fusion protein genes acrE, mdtE, and emrK as well as nearby genes of many intrinsic multidrug exporters (Nishino and Yamaguchi, 2004). EvgA, when phosphorylated, is a positive regulator for efflux protein complexes emrKY and mdtEF (Hirakawa et al., 2003b) which confer resistance to erythromycin, cloxacillin, tetracycline, oxacillin, and norfloxacin. Notably, an SNP mutation (R234F) was found in the E. coli elongation factor EF-Tu, which confers resistance to pulvomycin. The E. coli glpT gene (E448K) confers resistance to fosfomycin. The UhpT (E350Q) carries a mutation that confers resistance to fosfomycin; The PBP3 (D350N, S357N) gene, similar to Haemophilus influenzae, carries a mutation that confers resistance to beta-lactam antibiotics; and E. coli AcrAB-TolC bears MarR mutations (Y137H, G103S), which confer resistance to ciprofloxacin and tetracycline (Supplementary Table S4). The resistance to azithromycin (AZM15), clindamycin (DA2), novobiocin (NV30), amikacin (AMK30), oxacillin (OX1), erythromycin (E15), bacitracin, and other antibiotics was confirmed by the disk assay method; out of the 21 antibiotics tested, the target strain showed resistance to 14 antibiotics belonging to several classes as shown in Figure 4. Antibiotic resistance was previously evaluated in several E. coli strains isolated from lettuce, cabbage, cucumbers, and tomatoes (Rahman et al., 2021). Besides, 100% of the E. coli isolates obtained from fruits and vegetables collected from various regions in Ecuador, exhibited resistance to ampicillin, cefazolin, cefotaxime, and tetracycline (Montero et al., 2021). The strains isolated from fresh produce have demonstrated variable resistance to colistin (CST), AMK, E15, cefotaxime (CTX), ceftazidime (CAZ), AMP, gentamycin (GEN), and amoxicillin (AMX) (Shah et al., 2015; Gómez-Aldapa et al., 2016). In addition, E. coli isolated from fresh vegetables (i.e., pak choi and lettuce) were resistant to kanamycin, levofloxacin, doxycycline, fosfomycin (FOS), CTX, AMP, GEN, NA, TE, and colistin (CST) (Liu B-T et al., 2019; Manageiro et al., 2020). PCR analysis results confirmed the resistance to tetracycline (blaTEM-1) and carbapenemase (blaKPC) (Supplementary Table S1). The gene blaTEM-1 is widely found in animal-derived E. coli and encodes for β-lactamases with a narrow spectrum that are capable of inactivating aminopenicillins and penicillins (Poirel et al., 2018). In the study of Montero et al. (2021) there were differences in a subset of ESBL-E. coli isolates that tested positive for certain genes by PCR but negative by whole genome sequencing (WGS); these were 12 isolates with blaTEM, nine isolates with blaSHV, and one isolate with blaCTX-M. Furthermore, WGS revealed blaSHV and blaTEM in two isolates, but PCR results were negative. A recent study revealed a relatively high prevalence of blaKPC–carrying, carbapenemase-resistant Enterobacteriaceae in fecal isolates, among food handlers in Kuwait (Moghnia et al., 2021). Nonetheless, non-specific amplification products were detected for blaVIM (Verona integron-encoded metallo-β-lactamase), oxacillinases (OXA48/181), and cefotaxime (blaCTXM-9). This might be produced by primers binding to seemingly random locations in the sample DNA. No amplification was detected for the other tested β-lactam genes (Supplementary Table S1).
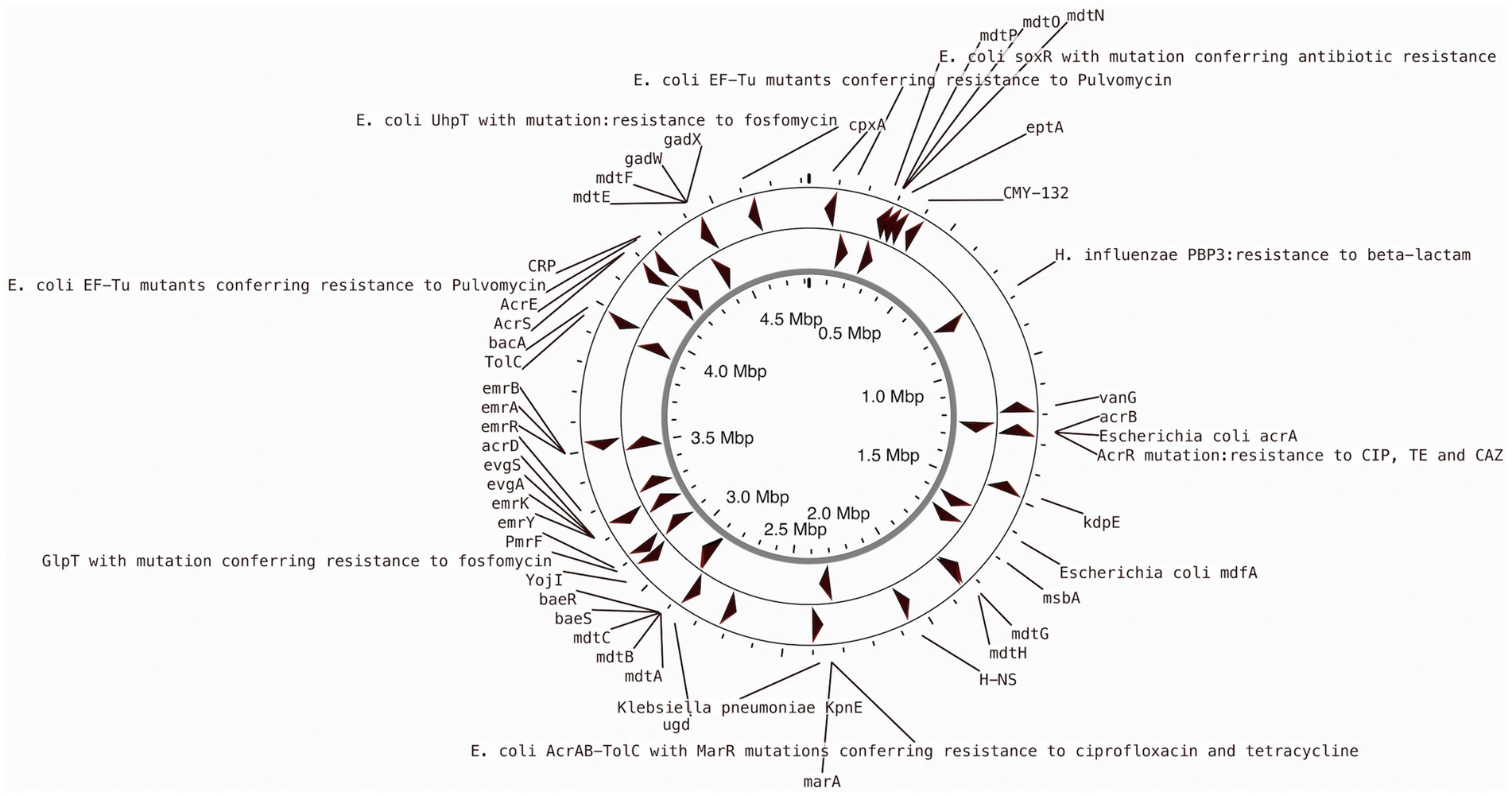
Figure 3. CARD annotation of the antibiotic resistance genes in the L1PEag1 genome.
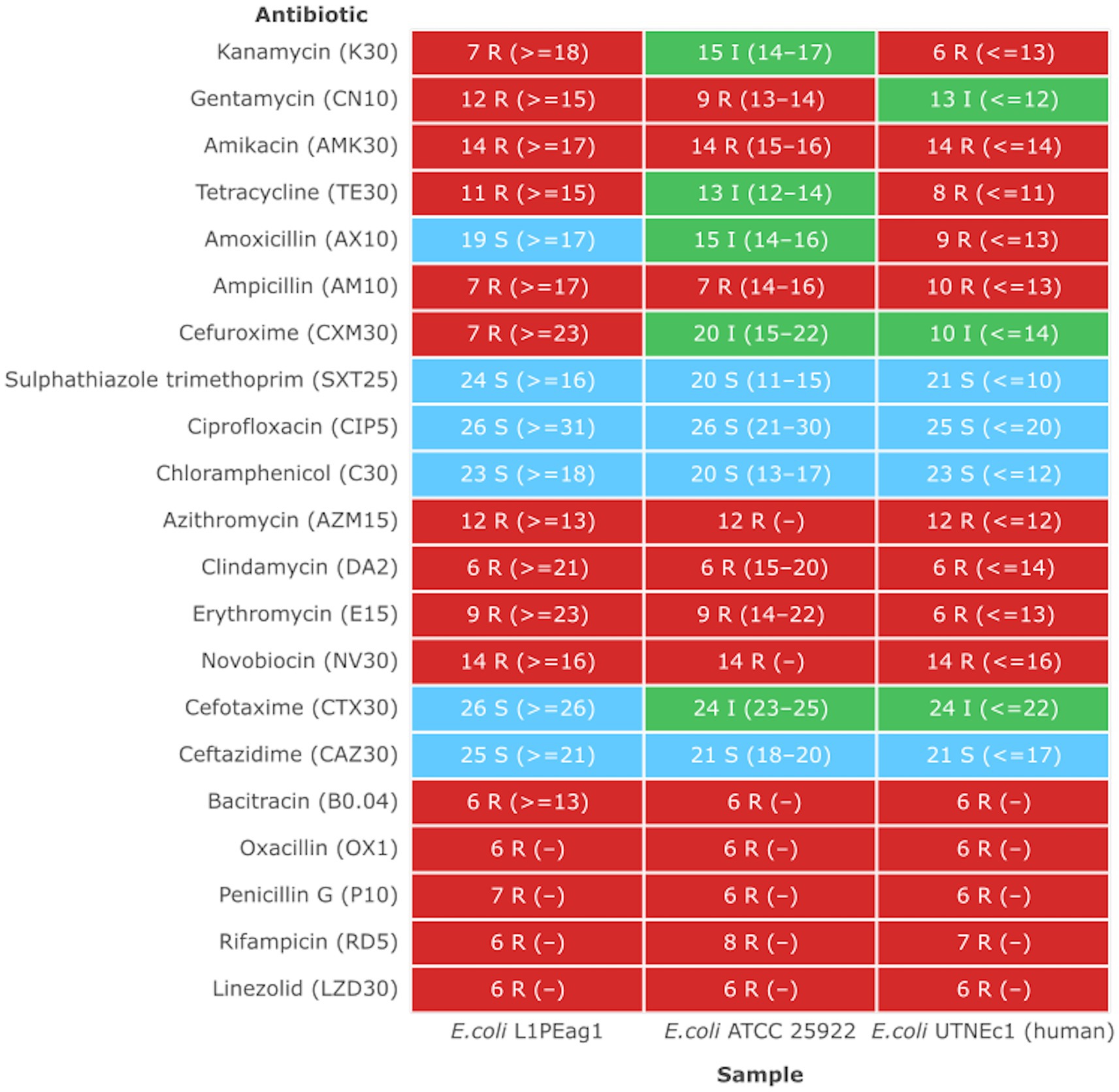
Figure 4. Antimicrobial heatmap profile. The strains showing a MIC higher than the EFSA breakpoint were considered resistant (EFSA Panel on Additives and Products or Substances used in Animal Feed et al., 2018). Susceptible (S, green): a bacterial strain is defined as susceptible when it is inhibited at a concentration of a specific antimicrobial equal to or lower than the established cut-off value (S ≤ x mg/L). Resistant (R, red): a bacterial strain is defined as resistant when it is not inhibited at a concentration of a specific antimicrobial above the established cut-off value (R > x mg/L). Intermediate (I, green): a bacterial strain is defined as intermediate to a given antibiotic when it is inhibited in vitro by a concentration of this drug that is associated with an uncertain therapeutic effect. (−) No CLSI standard inhibition zone determined.
Based on RGI analysis, the sitABCD genes were detected in the genome of the L1PEag1 isolate (i.e., sitA: a periplasmic binding protein, sitB: an ATP-binding component, sitC: an inner membrane component, and sitD: an inner membrane component, which conferred resistance to hydrogen peroxide). Along with other ion transport systems, SitABCD is an iron and manganese transporter that may confer oxidative stress resistance and bactericidal effects. These genes showed 97.31% identity with a sitABCD gene located on the virulence plasmid pAPEC-1 of an avian pathogenic E. coli strain (AY598030.2) (Sabri et al., 2006).
Conclusion
To the best of our knowledge, this is the first genome characterization of an E. coli isolate L1PEag1, originating from cape gooseberry ready-to-eat fruit, and that also shows antibiotic resistance. The L1PEag1 isolate belongs to the B2 phylogroup, sequence type ST1170, and serotype O1:H4 based on in silico genome analysis. L1PEag1 genome harbored several virulence factors and was predicted as a human pathogen. Based on the results we speculate that this isolate could cross contaminate the fruit at the studied market sites. It illustrates the possible contribution of the fruit environment to the spread of pathogenic E. coli strains. The potential risks to public health posed by the presence of antibiotic resistance genes in ready-to-eat cape gooseberry are unknown. This emphasizes the need for a comprehensive strategy that addresses pre- and post-harvest processing as well as produce distribution. Our work emphasizes the necessity of monitoring environmental antibiotic resistance, particularly in commercial settings, since this would help in developing early mitigation techniques for dealing with resistance diffusion.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
DM: Investigation, Writing – review & editing. JCC-O: Writing – review & editing. PJ-V: Software, Writing – review & editing. GNT: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Centre of Research of the Universidad Técnica del Norte, GNT Grant No. 7874/2023.
Acknowledgments
The authors are thankful to the Centre of Research of the Universidad Técnica del Norte, Ecuador for funding this research. We thank the Korea International Cooperation Agency (KOICA) and the National Institute of Biological Resources of Korea (NIBR) for providing support in genome-producing capabilities to authors JC-O and PJ-V and for partially covering the APC costs.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1392333/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Mapping of the COG (A) and KEGG (B) proteins according to their function. The number of genes in each category is shown.
Footnotes
1. ^http://www.bioinformatics.babraham.ac.uk/projects/fastqc
2. ^https://github.com/nanoporetech/medaka
4. ^https://cge.food.dtu.dk/services/MLST
5. ^https://cge.food.dtu.dk/services/SerotypeFinder
6. ^http://www.cbs.dtu.dk/services/RNAmmer
7. ^http://www.ansikte.se/ARAGORN
8. ^http://www.cbs.dtu.dk/services/SignalP
10. ^https://www.uniprot.org/statistics/Swiss-Prot
12. ^http://www.mgc.ac.cn/VFs/main.htm
13. ^http://www.phi-base.org/searchFacet.htm?queryTerm
14. ^http://xfam.org
15. ^https://proksee.ca/projects/new
16. ^https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index
18. ^http://arpcard.mcmaster.ca
19. ^https://cge.cbs.dtu.dk/services/ResFinder/
20. ^https://cge.food.dtu.dk/services/VirulenceFinder/
21. ^http://cge.cbs.dtu.dk/services/PathogenFinder/
22. ^https://cge.food.dtu.dk/services/FimTyper/
23. ^https://www.normalizacion.gob.ec/buzon/normas/1529–2-1R.pdf
References
Allison, G. E., Angeles, D., Tran-Dinh, N., and Verma, N. K. (2002). Complete genomic sequence of SfV, a serotype-converting temperate bacteriophage of Shigella flexneri. J. Bacteriol. 184, 1974–1987. doi: 10.1128/jb.184.7.1974-1987.2002
Amancha, G., Celis, Y., Irazabal, J., Falconi, M., Villacis, K., Thekkur, P., et al. (2023). High levels of antimicrobial resistance in Escherichia coli and Salmonella from poultry in Ecuador. Rev. Panam. Salud Pública 47:e15. doi: 10.26633/RPSP.2023.15
Arbab, S., Ullah, H., Wang, W., and Zhang, J. (2022). Antimicrobial drug resistance against Escherichia coli and its harmful effect on animal health. Vet. Med. Sci. 8, 1780–1786. doi: 10.1002/vms3.825
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Arndt, D., Marcu, A., Liang, Y., and Wishart, D. S. (2017). PHAST, PHASTER and PHASTEST: tools for finding prophage in bacterial genomes. Brief. Bioinform. 20, 1560–1567. doi: 10.1093/bib/bbx121
Berry, J. D., Rajaure, M., and Young, R. (2013). Spanin function requires subunit homodimerization through intermolecular disulfide bonds. Mol. Microbiol. 88, 35–47. doi: 10.1111/mmi.12167
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Beutin, L., Strauch, E., Zimmermann, S., Kaulfuss, S., Schaudinn, C., Männel, A., et al. (2005). Genetical and functional investigation of fliC genes encoding flagellar serotype H4 in wildtype strains of Escherichia coli and in a laboratory E. coli K-12 strain expressing flagellar antigen type H48. BMC Microbiol. 5:4. doi: 10.1186/1471-2180-5-4
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Brown, C. L., Mullet, J., Hindi, F., Stoll, J. E., Gupta, S., Choi, M., et al. (2022). mobileOG-db: a manually curated database of protein families mediating the life cycle of bacterial Mobile genetic elements. Appl. Environ. Microbiol. 88:e0099122. doi: 10.1128/aem.00991-22
Cárdenas, B. D., Llorente, E. S., Gu, G., Nou, X., Ortiz, J., Maldonado, P., et al. (2024). Microbial composition and diversity of high-demand street-vended foods in Ecuador. J. Food Prot. 87:100247. doi: 10.1016/j.jfp.2024.100247
Caspi, R., Billington, R., Fulcher, C. A., Keseler, I. M., Kothari, A., Krummenacker, M., et al. (2018). The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46, D633–D639. doi: 10.1093/nar/gkx935
Castellanos, L. R., Donado-Godoy, P., León, M., Clavijo, V., Arevalo, A., Bernal, J. F., et al. (2017). High heterogeneity of Escherichia coli sequence types Harbouring ESBL/AmpC genes on IncI1 plasmids in the Colombian poultry chain. PLoS One 12:e0170777. doi: 10.1371/journal.pone.0170777
Cheng, W., Li, J., Wu, Y., Xu, L., Su, C., Qian, Y., et al. (2016). Behavior of antibiotics and antibiotic resistance genes in eco-agricultural system: a case study. J. Hazard. Mater. 304, 18–25. doi: 10.1016/j.jhazmat.2015.10.037
Coque, T. M., Cantón, R., Pérez-Cobas, A. E., Fernández-de-Bobadilla, M. D., and Baquero, F. (2023). Antimicrobial resistance in the Global Health network: known unknowns and challenges for efficient responses in the 21st century. Microorganisms 11:1050. doi: 10.3390/microorganisms11041050
Cosentino, S., Larsen, M. V., Aarestrup, F. M., and Lund, O. (2013). PathogenFinder - distinguishing friend from foe using bacterial whole genome sequence data. PLoS One 8:e77302. doi: 10.1371/journal.pone.0077302
Coster, W. D., D’Hert, S., Schultz, D. T., Cruts, M., and Broeckhoven, C. V. (2018). NanoPack: visualizing and processing long read sequencing data. bioRxiv :237180. doi: 10.1101/237180
Dadi, B. R., Abebe, T., Zhang, L., Mihret, A., Abebe, W., and Amogne, W. (2020). Distribution of virulence genes and phylogenetics of uropathogenic Escherichia coli among urinary tract infection patients in Addis Ababa, Ethiopia. BMC Infect. Dis. 20:108. doi: 10.1186/s12879-020-4844-z
Delannoy, S., Beutin, L., Mariani-Kurkdjian, P., Fleiss, A., Bonacorsi, S., and Fach, P. (2017). The Escherichia coli serogroup O1 and O2 lipopolysaccharides are encoded by multiple O-antigen gene clusters. Front. Cell. Infect. Microbiol. 7:30. doi: 10.3389/fcimb.2017.00030
EFSA Panel on Additives and Products or Substances used in Animal FeedRychen, G., Aquilina, G., Azimonti, G., Bampidis, V., de L, B. M., et al. (2018). Guidance on the characterization of microorganisms used as feed additives or as production organisms. EFSA J. 16:e05206. doi: 10.2903/j.efsa.2018.5206
Fang, X., Monk, J. M., Mih, N., Du, B., Sastry, A. V., Kavvas, E., et al. (2018). Escherichia coli B2 strains prevalent in inflammatory bowel disease patients have distinct metabolic capabilities that enable colonization of intestinal mucosa. BMC Syst. Biol. 12:66. doi: 10.1186/s12918-018-0587-5
Federici, S., Kviatcovsky, D., Valdés-Mas, R., and Elinav, E. (2023). Microbiome-phage interactions in inflammatory bowel disease. Clin. Microbiol. Infect. 29, 682–688. doi: 10.1016/j.cmi.2022.08.027
Fricke, W. F., Wright, M. S., Lindell, A. H., Harkins, D. M., Baker-Austin, C., Ravel, J., et al. (2008). Insights into the environmental resistance Gene Pool from the genome sequence of the multidrug-resistant environmental isolate Escherichia coli SMS-3-5. J. Bacteriol. 190, 6779–6794. doi: 10.1128/JB.00661-08
Gómez-Aldapa, C. A., Cerna-Cortes, J. F., Rangel-Vargas, E., Torres-Vitela, M. R., Villarruel-López, A., Gutiérrez-Alcántara, E. J., et al. (2016). Presence of multidrug-resistant Shiga toxin-producing Escherichia coli, Enteropathogenic E. Coli and Enterotoxigenic E. coli, on raw Nopalitos (Opuntia ficus-indica L.) and in Nopalitos salads from local retail Markets in Mexico. Foodborne Pathog. Dis. 13, 269–274. doi: 10.1089/fpd.2015.2065
Grant, J. R., Enns, E., Marinier, E., Mandal, A., Herman, E. K., Chen, C., et al. (2023). Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51, W484–W492. doi: 10.1093/nar/gkad326
Grant, M. A., Weagant, S. D., and Feng, P. (2001). Glutamate decarboxylase genes as a prescreening marker for detection of pathogenic Escherichia coli groups. Appl. Environ. Microbiol. 67, 3110–3114. doi: 10.1128/aem.67.7.3110-3114.2001
Hariharan, J., Wright, K., and Passalacqua, P. (2020). Dorado: a Python package for simulating passive particle transport in shallow-water flows. J. Open Source Softw. 5:2585. doi: 10.21105/joss.02585
He, X. L., Wang, Q., Peng, L., Qu, Y.-R., Puthiyakunnon, S., Liu, X.-L., et al. (2015). Role of uropathogenic Escherichia coli outer membrane protein T in pathogenesis of urinary tract infection. Pathog. Dis. 73:ftv006. doi: 10.1093/femspd/ftv006
Hernández-Alomía, F., Bastidas-Caldes, C., Ballesteros, I., Tenea, G. N., Jarrín, V. P., Molina, C. A., et al. (2023). Beta-lactam antibiotic resistance genes in the microbiome of the public transport system of Quito, Ecuador. Int. J. Environ. Res. Public Heal. 20:1900. doi: 10.3390/ijerph20031900
Hirakawa, H., Nishino, K., Hirata, T., and Yamaguchi, A. (2003a). Comprehensive studies of drug resistance mediated by overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J. Bacteriol. 185, 1851–1856. doi: 10.1128/jb.185.6.1851-1856.2003
Hirakawa, H., Nishino, K., Yamada, J., Hirata, T., and Yamaguchi, A. (2003b). β-Lactam resistance modulated by the overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J. Antimicrob. Chemother. 52, 576–582. doi: 10.1093/jac/dkg406
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Jaureguy, F., Landraud, L., Passet, V., Diancourt, L., Frapy, E., Guigon, G., et al. (2008). Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genomics 9:560. doi: 10.1186/1471-2164-9-560
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Joensen, K. G., Tetzschner, A. M. M., Iguchi, A., Aarestrup, F. M., and Scheutz, F. (2015). Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 53, 2410–2426. doi: 10.1128/JCM.00008-15
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Lai, J. Y. H., Zhang, H., Chiang, M. H. Y., Lun, C. H. I., Zhang, R., and Lau, S. C. K. (2017). The putative functions of lysogeny in mediating the survivorship of Escherichia coli in seawater and marine sediment. FEMS Microbiol. Ecol. 94, 1–10. doi: 10.1093/femsec/fix187
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of Total-genome-sequenced Bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Liu, B.-T., Li, X., Zhang, Q., Shan, H., Zou, M., and Song, F.-J. (2019). Colistin-resistant mcr-positive Enterobacteriaceae in fresh vegetables, an increasing infectious threat in China. Int. J. Antimicrob. Agents 54, 89–94. doi: 10.1016/j.ijantimicag.2019.04.013
Liu, B.-T., and Song, F.-J. (2019). Emergence of two Escherichia coli strains co-harboring mcr-1 and blaNDM in fresh vegetables from China. Infect. Drug Resist. 12, 2627–2635. doi: 10.2147/IDR.S211746
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Luna-Guevara, J. J., Arenas-Hernandez, M. M. P., de la Peña, C. M., Silva, J. L., and Luna-Guevara, M. L. (2019). The role of Pathogenic E. coli in fresh vegetables: behavior, contamination factors, and preventive measures. Int. J. Microbiol 2019:2894328. doi: 10.1155/2019/2894328
Lynch, M. F., Tauxe, R. V., and Hedberg, C. W. (2008). The growing burden of foodborne outbreaks due to contaminated fresh produce: risks and opportunities. Epidemiol. Infect. 137, 307–315. doi: 10.1017/s0950268808001969
Manageiro, V., Jones-Dias, D., Ferreira, E., and Caniça, M. (2020). Plasmid-mediated Colistin resistance (mcr-1) in Escherichia coli from non-imported fresh vegetables for human consumption in Portugal. Microorganisms 8:429. doi: 10.3390/microorganisms8030429
Manges, A. R., and Johnson, J. R. (2012). Food-borne origins of Escherichia coli causing Extraintestinal infections. Clin. Infect. Dis. 55, 712–719. doi: 10.1093/cid/cis502
Mann, A., Nehra, K., Rana, J. S., and Dahiya, T. (2021). Antibiotic resistance in agriculture: perspectives on upcoming strategies to overcome upsurge in resistance. Curr. Res. Microb. Sci. 2:100030. doi: 10.1016/j.crmicr.2021.100030
McGuffie, M. J., and Barrick, J. E. (2021). pLannotate: engineered plasmid annotation. Nucleic Acids Res. 49, W516–W522. doi: 10.1093/nar/gkab374
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10:2182. doi: 10.1038/s41467-019-10210-3
Melo, J., and Quintas, C. (2023). Minimally processed fruits as vehicles for foodborne pathogens. AIMS Microbiol. 9, 1–19. doi: 10.3934/microbiol.2023001
Mittal, S., Sharma, M., and Chaudhary, U. (2014). Study of virulence factors of uropathogenic Escherichia coli and its antibiotic susceptibility pattern. Indian J. Pathol. Microbiol. 57:61. doi: 10.4103/0377-4929.130899
Moghnia, O. H., Rotimi, V. O., and Al-Sweih, N. A. (2021). Preponderance of blaKPC-carrying Carbapenem-resistant Enterobacterales among fecal isolates from community food handlers in Kuwait. Front. Microbiol. 12:737828. doi: 10.3389/fmicb.2021.737828
Molina, C. A., Quiroz-Moreno, C., Jarrín, V. P., Díaz, M., Yugsi, E., Pérez-Galarza, J., et al. (2024). Bacterial community assessment of drinking water and downstream distribution systems in highland localities of Ecuador. J. Water Health 22, 536–549. doi: 10.2166/wh.2024.290
Montero, L., Irazabal, J., Cardenas, P., Graham, J. P., and Trueba, G. (2021). Extended-Spectrum Beta-lactamase producing-Escherichia coli isolated from irrigation Waters and produce in Ecuador. Front. Microbiol. 12:709418. doi: 10.3389/fmicb.2021.709418
Mostafidi, M., Sanjabi, M. R., Shirkhan, F., and Zahedi, M. T. (2020). A review of recent trends in the development of the microbial safety of fruits and vegetables. Trends Food Sci Tech 103, 321–332. doi: 10.1016/j.tifs.2020.07.009
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Nishino, K., and Yamaguchi, A. (2001). Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 183, 5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001
Nishino, K., and Yamaguchi, A. (2004). Role of histone-like protein H-NS in multidrug resistance of Escherichia coli. J. Bacteriol. 186, 1423–1429. doi: 10.1128/JB.186.5.1423-1429.2004
Paitan, Y. (2018). Escherichia coli, a Versatile Pathogen. Curr. Top. Microbiol. Immunol. 416, 181–211. doi: 10.1007/82_2018_110
Pérez-Lavalle, L., Carrasco, E., and Valero, A. (2020). Strategies for microbial decontamination of fresh blueberries and derived products. Food Secur. 9:1558. doi: 10.3390/foods9111558
Pietsch, M., Eller, C., Wendt, C., Holfelder, M., Falgenhauer, L., Fruth, A., et al. (2017). Molecular characterisation of extended-spectrum β-lactamase (ESBL)-producing Escherichia coli isolates from hospital and ambulatory patients in Germany. Vet. Microbiol. 200, 130–137. doi: 10.1016/j.vetmic.2015.11.028
Poirel, L., Madec, J.-Y., Lupo, A., Schink, A.-K., Kieffer, N., Nordmann, P., et al. (2018). Antimicrobial Resistance in Escherichia coli. Microbiol. Spectr. 6, 1–27. doi: 10.1128/microbiolspec.ARBA-0026-2017
Poole, N. M., Green, S. I., Rajan, A., Vela, L. E., Zeng, X.-L., Estes, M. K., et al. (2017). Role for FimH in Extraintestinal pathogenic Escherichia coli invasion and translocation through the intestinal epithelium. Infect. Immun. 85, 1–17. doi: 10.1128/iai.00581-17
Possas, A., and Pérez-Rodríguez, F. (2023). New insights into cross-contamination of fresh-produce. Curr. Opin. Food Sci. 49:100954. doi: 10.1016/j.cofs.2022.100954
Rahdar, M., Rashki, A., Miri, H. R., and Rashki Ghalehnoo, M. (2015). Detection of pap, sfa, afa, foc, and fim adhesin-encoding operons in uropathogenic Escherichia coli isolates collected from patients with urinary tract infection. Jundishapur J. Microbiol. 8:e22647. doi: 10.5812/jjm.22647
Rahman, M., Alam, M.-U., Luies, S. K., Kamal, A., Ferdous, S., Lin, A., et al. (2021). Contamination of fresh produce with antibiotic-resistant Bacteria and associated risks to human health: a scoping review. Int. J. Environ. Res. Public Heal. 19:360. doi: 10.3390/ijerph19010360
Raivio, T. L., Leblanc, S. K. D., and Price, N. L. (2013). The Escherichia coli Cpx envelope stress response regulates genes of diverse function that impact antibiotic resistance and membrane integrity. J. Bacteriol. 195, 2755–2767. doi: 10.1128/jb.00105-13
Rajaure, M., Berry, J., Kongari, R., Cahill, J., and Young, R. (2015). Membrane fusion during phage lysis. Proc. Natl. Acad. Sci. 112, 5497–5502. doi: 10.1073/pnas.1420588112
Ravan, H., and Amandadi, M. (2015). Analysis of yeh Fimbrial gene cluster in Escherichia coli O157:H7 in order to find a genetic marker for this serotype. Curr. Microbiol. 71, 274–282. doi: 10.1007/s00284-015-0842-6
Reid, C. J., Blau, K., Jechalke, S., Smalla, K., and Djordjevic, S. P. (2020). Whole genome sequencing of Escherichia coli from store-bought produce. Front. Microbiol. 10:3050. doi: 10.3389/fmicb.2019.03050
Reid, S. D., Selander, R. K., and Whittam, T. S. (1998). Sequence diversity of flagellin (fliC) alleles in pathogenic Escherichia coli. J. Bacteriol. 181, 153–160. doi: 10.1128/jb.181.1.153-160.1999
Roer, L., Tchesnokova, V., Allesøe, R., Muradova, M., Chattopadhyay, S., Ahrenfeldt, J., et al. (2017). Development of a web tool for Escherichia coli subtyping based on fimH alleles. J. Clin. Microbiol. 55, 2538–2543. doi: 10.1128/JCM.00737-17
Rothstein, A. P., Jesser, K. J., Feistel, D. J., Konstantinidis, K. T., Trueba, G., and Levy, K. (2023). Population genomics of diarrheagenic Escherichia coli uncovers high connectivity between urban and rural communities in Ecuador. Infect Genet Evol. 113:105476. doi: 10.1016/j.meegid.2023.105476
Sabri, M., Léveillé, S., and Dozois, C. M. (2006). A SitABCD homologue from an avian pathogenic Escherichia coli strain mediates transport of iron and manganese and resistance to hydrogen peroxide. Microbiology 152, 745–758. doi: 10.1099/mic.0.28682-0
Salazar-Llorente, E., Morales, M., Sornoza, I., Mariduena-Zavala, M. G., Gu, G., Nou, X., et al. (2021). Microbiological quality of high-demand food from three major cities in Ecuador. J. Food Prot. 84, 128–138. doi: 10.4315/jfp-20-271
Savin, M., Bierbaum, G., Kreyenschmidt, J., Schmithausen, R. M., Sib, E., Schmoger, S., et al. (2021). Clinically relevant Escherichia coli isolates from process Waters and wastewater of poultry and pig slaughterhouses in Germany. Microorganisms 9:698. doi: 10.3390/microorganisms9040698
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shah, M. S., Eppinger, M., Ahmed, S., Shah, A. A., Hameed, A., and Hasan, F. (2015). Multidrug-resistant diarrheagenic E. coli pathotypes are associated with ready-to-eat salad and vegetables in Pakistan. J. Korean Soc. Appl. Biol. Chem. 58, 267–273. doi: 10.1007/s13765-015-0019-9
Sharma, S., Bhat, G., and Shenoy, S. (2007). Virulence factors and drug resistance in Escherichia coli isolated from extraintestinal infections. Indian J. Med. Microbiol. 25, 369–373. doi: 10.1016/S0255-0857(21)02053-3
Shruthi, N., Kumar, R., and Kumar, R. (2012). Phenotypic study of virulence factors in Escherichia coli isolated from antenatal cases, catheterized patients, and faecal flora. J. Clin. Diagn. Res. 6, 1699–1703. doi: 10.7860/jcdr/2012/4669.2634
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference Centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Solomon, E. B., Yaron, S., and Matthews, K. R. (2002). Transmission of Escherichia coli O157:H7 from contaminated manure and irrigation water to lettuce plant tissue and its subsequent internalization. Appl. Environ. Microbiol. 68, 397–400. doi: 10.1128/aem.68.1.397-400.2002
Song, J., Oh, S.-S., Kim, J., and Shin, J. (2020). Extended-spectrum β-lactamase-producing Escherichia coli isolated from raw vegetables in South Korea. Sci. Rep. 10:19721. doi: 10.1038/s41598-020-76890-w
Spurbeck, R. R., Dinh, P. C., Walk, S. T., Stapleton, A. E., Hooton, T. M., Nolan, L. K., et al. (2012). Escherichia coli isolates that carry vat, fyuA, chuA, and yfcV efficiently colonize the urinary tract. Infect. Immun. 80, 4115–4122. doi: 10.1128/iai.00752-12
Strawn, L. K., Schneider, K. R., and Danyluk, M. D. (2011). Microbial safety of tropical fruits. Crit. Rev. Food Sci. Nutr. 51, 132–145. doi: 10.1080/10408390903502864
Tabasi, M., Karam, M. R. A., Habibi, M., Yekaninejad, M. S., and Bouzari, S. (2015). Phenotypic assays to determine virulence factors of Uropathogenic Escherichia coli (UPEC) isolates and their correlation with antibiotic resistance pattern. Res. Pers. 6, 261–268. doi: 10.1016/j.phrp.2015.08.002
Tenea, G. N., Reyes, P., Molina, D., and Ortega, C. (2023). Pathogenic microorganisms linked to fresh fruits and juices purchased at low-cost Markets in Ecuador, potential carriers of antibiotic resistance. Antibiotics 12:236. doi: 10.3390/antibiotics12020236
Thomas, G. A., Gil, T. P., Müller, C. T., Rogers, H. J., and Berger, C. N. (2024). From field to plate: how do bacterial enteric pathogens interact with ready-to-eat fruit and vegetables, causing disease outbreaks? Food Microbiol. 117:104389. doi: 10.1016/j.fm.2023.104389
Tiedje, J. M., Fu, Y., Mei, Z., Schäffer, A., Dou, Q., Amelung, W., et al. (2023). Antibiotic resistance genes in food production systems support one health opinions. Curr. Opin. Environ. Sci. Heal. 34:100492. doi: 10.1016/j.coesh.2023.100492
Vaser, R., and Šikić, M. (2021). Time- and memory-efficient genome assembly with raven. Nat. Comput. Sci. 1, 332–336. doi: 10.1038/s43588-021-00073-4
Waters, N. R., Abram, F., Brennan, F., Holmes, A., and Pritchard, L. (2020). Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. 2:acmi000143. doi: 10.1099/acmi.0.000143
Wick, R. R., and Holt, K. E. (2020). Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Research. 8:2138. doi: 10.12688/f1000research.21782.4
Wick, R. R., Judd, L. M., Cerdeira, L. T., Hawkey, J., Méric, G., Vezina, B., et al. (2021). Trycycler: consensus long-read assemblies for bacterial genomes. Genome Biol. 22, 1–17. doi: 10.1186/s13059-021-02483-z
Wirth, T., Falush, D., Lan, R., Colles, F., Mensa, P., Wieler, L. H., et al. (2006). Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x
Wu, X., Liu, J., Feng, J., Shabbir, M. A. B., Feng, Y., Guo, R., et al. (2022). Epidemiology, environmental risks, virulence, and resistance determinants of Klebsiella pneumoniae from dairy cows in Hubei, China. Front. Microbiol. 13:858799. doi: 10.3389/fmicb.2022.858799
Wu, L., and Ma, J. (2019). The global catalogue of microorganisms (GCM) 10K type strain sequencing project: providing services to taxonomists for standard genome sequencing and annotation. Int. J. Syst. Evol. Microbiol. 69, 895–898. doi: 10.1099/ijsem.0.003276
Wyborn, N. R., Clark, A., Roberts, R. E., Jamieson, S. J., Tzokov, S., Bullough, P. A., et al. (2004). Properties of haemolysin E (HlyE) from a pathogenic Escherichia coli avian isolate and studies of HlyE export. Microbiology 150, 1495–1505. doi: 10.1099/mic.0.26877-0
Xia, F., Jiang, M., Wen, Z., Wang, Z., Wang, M., Xu, Y., et al. (2022). Complete genomic analysis of ST117 lineage extraintestinal pathogenic Escherichia coli (ExPEC) to reveal multiple genetic determinants to drive its global transmission: ST117 E. coli as an emerging multidrug-resistant foodborne ExPEC with zoonotic potential. Transbound. Emerg. Dis. 69, 3256–3273. doi: 10.1111/tbed.14678
Yoo, B. K., Liu, Y., Juneja, V., Huang, L., and Hwang, C.-A. (2015). Growth characteristics of Shiga toxin-producing Escherichia coli (STEC) stressed by chlorine, sodium chloride, acid, and starvation on lettuce and cantaloupe. Food Control 55, 97–102. doi: 10.1016/j.foodcont.2015.02.040
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zuo, J., Tu, C., Wang, Y., Qi, K., Hu, J., Wang, Z., et al. (2019). The role of the wzy gene in lipopolysaccharide biosynthesis and pathogenesis of avian pathogenic Escherichia coli. Microb. Pathog. 127, 296–303. doi: 10.1016/j.micpath.2018.12.021
Keywords: cape gooseberry, antibiotic resistance, Escherichia coli, whole genome, food pathogens, beta-lactamase, fimH alleles
Citation: Molina D, Carrión–Olmedo JC, Jarrín–V P and Tenea GN (2024) Genome characterization of a multi-drug resistant Escherichia coli strain, L1PEag1, isolated from commercial cape gooseberry fruits (Physalis peruviana L.). Front. Microbiol. 15:1392333. doi: 10.3389/fmicb.2024.1392333
Edited by:
Ioana Cristina Marinas, University of Bucharest, RomaniaReviewed by:
William Calero-Cáceres, Technical University of Ambato, EcuadorSusan Leonard, United States Food and Drug Administration, United States
Copyright © 2024 Molina, Carrión–Olmedo, Jarrín–V and Tenea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela N. Tenea, Z250ZW5lYUB1dG4uZWR1LmVj