
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 April 2024
Sec. Infectious Agents and Disease
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1387208
 Adrian C. Paskey1,2Kevin L. Schully3
Adrian C. Paskey1,2Kevin L. Schully3 Logan J. Voegtly1,2
Logan J. Voegtly1,2 Catherine E. Arnold1,4
Catherine E. Arnold1,4 Regina Z. Cer1Kenneth G. Frey1Paul W. Blair3,5Danielle V. Clark3,5
Regina Z. Cer1Kenneth G. Frey1Paul W. Blair3,5Danielle V. Clark3,5 Hong Ge6
Hong Ge6 Allen L. Richards6
Allen L. Richards6 Christina M. Farris6Kimberly A. Bishop-Lilly1*
Christina M. Farris6Kimberly A. Bishop-Lilly1*Infection with either Rickettsia prowazekii or Orientia tsutsugamushi is common, yet diagnostic capabilities are limited due to the short window for positive identification. Until now, although targeted enrichment had been applied to increase sensitivity of sequencing-based detection for various microorganisms, it had not been applied to sequencing of R. prowazekii in clinical samples. Additionally, hybridization-based targeted enrichment strategies had only scarcely been applied to qPCR of any pathogens in clinical samples. Therefore, we tested a targeted enrichment technique as a proof of concept and found that it dramatically reduced the limits of detection of these organisms by both qPCR and high throughput sequencing. The enrichment methodology was first tested in contrived clinical samples with known spiked-in concentrations of R. prowazekii and O. tsutsugamushi DNA. This method was also evaluated using clinical samples, resulting in the simultaneous identification and characterization of O. tsutsugamushi directly from clinical specimens taken from sepsis patients. We demonstrated that the targeted enrichment technique is helpful by lowering the limit of detection, not only when applied to sequencing, but also when applied to qPCR, suggesting the technique could be applied more broadly to include other assays and/or microbes for which there are limited diagnostic or detection modalities.
The Rickettsiaceae comprise a family of gram-negative obligate intracellular bacteria that has areas of endemicity on every continent except Antarctica. Rickettsia prowazekii, a member of the globally distributed typhus group of rickettsiae, is the etiologic agent of epidemic typhus, vectored by the human body louse (Pediculus humanus). Orientia tsutsugamushi, the etiologic agent of scrub typhus, is transmitted by trombiculid mites (particularly by the larva stage known as chiggers) and is historically distributed across approximately the Tsutsugamushi Triangle, an area spanning 13,000,000 km2 from South Asia in the west to Eastern Russia and Japan in the east and south to Australia. More recently, scrub typhus has been confirmed outside of the Tsutsugamushi Triangle including South America, Sub-Saharan Africa, and the Middle East with etiologic agents such as Candidatus Orientia chiloensis and Candidatus Orientia chuto, significantly expanding the distribution and risk of scrub typhus (Kelly et al., 2002; Abdad et al., 2018; Jiang et al., 2022).
Due to the broad geographic distribution of these pathogens and their vectors, billions of people are at risk for infection. Historically, outbreaks of epidemic typhus have occurred in overcrowded or unhygienic environments, such as jails and other resource-limited settings including war, societal collapse, or natural disasters (Perine et al., 1992; Badiaga and Brouqui, 2012; Nyatanyi et al., 2016). Following an incubation period of 7–14 days, R. prowazekii infection causes a high fever and severe headache. Patients may also present with myalgias, dry cough, and delirium. A key feature is a dull red rash that begins on the trunk and spreads peripherally, sparing the soles and palms. If left untreated, mortality can be as high as 60% (Akram et al., 2021). O. tsutsugamushi is responsible for nearly one-quarter of febrile illnesses in endemic areas and mortality can be as high as 30% without proper treatment (Chattopadhyay and Richards, 2007; Sriwongpan et al., 2013; Griffith et al., 2014). Recent evidence of the acute manifestation of scrub typhus is characterized by sudden onset of fever approximately 6–21 days after exposure with chills, headache, backache and myalgia, profuse sweating, vomiting and enlarged lymph nodes. In some patients, an eschar may develop at the site of arthropod feeding, often located at the interface of two skin surfaces, such as axilla, groin, and inguinal areas (Devine, 2003).
Serial serology (acute and convalescent titers) remains the diagnostic gold standard for rickettsial infections but is not clinically useful for guiding acute treatment. Agents that cause rickettsial diseases are obligate intracellular pathogens and isolation requires cell culture techniques that are not widely performed and can require weeks of incubation for growth, identification, and characterization (Gouriet et al., 2005). Therefore, the intracellular lifestyle of Rickettsiaceae results in a narrow diagnostic window that makes detection difficult (Luce-Fedrow et al., 2015), and treatment generally begins before confirming a positive test result. Culture is further complicated by R. prowazekii’s designation as a Select Agent and the biocontainment requirements for most rickettsiae and orientiae. While qPCR can be used to confirm rickettsial infections more quickly than serology or culture, sensitivity is low with standard techniques (Angelakis et al., 2012). High-throughput sequencing (HTS) has the potential to aid not only in a more rapid diagnosis of rickettsial diseases but also in predicting antibiotic susceptibility and virulence. However, one of the limitations of using molecular approaches (i.e., quantitative real-time polymerase chain reaction (qPCR), whole genome sequencing) is that this pathogen is generally present in the blood at very low levels and for a relatively short period (Chattopadhyay et al., 2005; Luce-Fedrow et al., 2015; Paris et al., 2015). Applying a targeted enrichment strategy to clinical specimens could lower the limit of detection (LoD) for qPCR or HTS and achieve more rapid and reliable diagnosis of rickettsial diseases.
It has previously been demonstrated that a variety of in-solution hybridization-based enrichment (Gnirke et al., 2009) designs can increase the sensitivity of HTS for the detection of viruses or bacteria in complex sample matrices (Christiansen et al., 2014; Matranga et al., 2014; Bonsall et al., 2015; Briese et al., 2015; Mate et al., 2015; Wylie et al., 2015; Brown et al., 2016; O'Flaherty et al., 2018; Lim et al., 2019; Paskey et al., 2019; Furtwängler et al., 2020; Li et al., 2020; Elliott et al., 2021; Wylezich et al., 2021; Kuchinski et al., 2022) although there can also be challenges in terms of false positive identification of pathogens and the bioinformatic methods involved (Kapel et al., 2023). Hybridization enrichment sequencing has also been demonstrated to enrich the sensitivity of detecting antimicrobial resistance genes in wastewater samples (Baba et al., 2023). Here, we extend the application of this strategy to investigate the possibility that a whole genome, targeted enrichment approach could widen the diagnostic window for R. prowazekii and O. tsutsugamushi and, unlike previous studies, including an O. tsutsugamushi study (Elliott et al., 2021) we apply this approach not just to whole genome sequence-based detection and characterization, but also to qPCR-based detection. Biotinylated 120-mer oligonucleotides were synthesized based on the publicly available R. prowazekii and O. tsutsugamushi complete genome sequences and used to enrich pathogen genomic DNA present in clinical-type specimens containing known copy number of pathogen genomes. The enriched samples were then subjected to qPCR to detect highly specific pathogen-derived genetic elements, as well as subjected to HTS to detect and characterize pathogen sequences. While targeted enrichment dramatically improved both qPCR and HTS detection of R. prowazekii and O. tsutsugamushi genomes in spiked, clinical-type specimens, we also demonstrated that the HTS approach enabled both detection and characterization of O. tsutsugamushi genomes from real world sepsis patient samples in which only sporadic detection of one or two Orientia genes per sample via metagenomic sequencing was previously reported. This underscores the full potential of this technique, which achieved coverage of the entire Orientia genome – a more straightforward positive result that would also allow for characterization (e.g., antimicrobial susceptibility).
A series of contrived samples were created with known concentrations of pathogen DNA. For samples intended for sequencing, human genomic DNA was extracted from 200 μL of whole blood (Reprocell; Beltsville, MD) using the Qiagen DNeasy kit (Qiagen; Valencia, CA) and normalized to 100 ng/sample. Genomic DNA from O. tsutsugamushi str. Karp and R. prowazekii str. Breinl from the Naval Medical Research Command’s collection (Bishop-Lilly et al., 2013) was isolated using Zymo Quick-DNA Miniprep kit (Zymo Research; Irvine, CA) and spiked into the human gDNA at 0, 12, 25, and 250 genome copies per sample. For samples intended for qPCR testing, plasmid oligonucleotide targets (Eurofins; Louisville, KY) were spiked into the human gDNA at a range from 0–20,000 copies. For the R. prowazekii assay, a pET24a vector containing the sequence fragment A of the ompB gene (Jiang et al., 2003) was used and for the O. tsutsugamushi plasmid assay, a VR1012 vector targeting a 118 base pair (bp) sequence of the 47 kDa antigen gene (hrtA) was used (Watt et al., 2005; Jiang et al., 2013).
Whole human blood was obtained from the Austere environments Consortium for Enhanced Sepsis Outcomes (ACESO) observational sepsis cohort in Cambodia under study NMRC.2013.0019 that has been previously described (Rozo et al., 2020) and approved by Naval Medical Research Command Institutional Review Board. Briefly, adult patients with a suspected infection were enrolled following written informed consent. Enrollment and exclusion criteria have been described previously, but in brief Adult patients (≥18 years) admitted within the last 48 h and who had a suspected infection (as judged by the attending physician) and met at least two of three clinical criteria (thermodysregulation defined as temperature > 38°C or < 36°C, tachypnea defined as Respiratory rate > 20/min and tachycardia defined as heart rate > 90 bpm) were considered for inclusion (Rozo et al., 2020). Multiple biospecimens were collected throughout the patients’ hospitalization and an exhaustive effort was undertaken to identify the infecting pathogens (Rozo et al., 2020). In addition to collecting other study-specific samples, one milliliter of whole blood drawn into a Na-Citrate blood tube was mixed with one milliliter of DNA/RNA Shield (Zymo Research; Irvine, CA). DNA was extracted from whole blood using the Zymo Quick-DNA Miniprep kit (Zymo Research; Irvine, CA). We leveraged our previously published work that was performed to identify infecting pathogens and chose a subset of these samples for inclusion in this current study. The previous data leveraged included serological testing and metagenomic analyses of total RNA isolated from the peripheral blood of sepsis patients, that ultimately identified O. tsutsugamushi infection in 16 patients. The samples selected were specifically chosen to assess the clinical utility of our enrichment approach and therefore consisted of three groups. The first group included blood samples in which few O. tsutsugamushi sequences were detected by unbiased shotgun sequencing and the samples were IgM+ by serological analysis (n = 8). The second group included samples from two patients negative for O. tsutsugamushi by unbiased shotgun sequencing and who were IgM+ serologically. The third group included samples from ten patients who were negative for O. tsutsugamushi by unbiased shotgun sequencing and serology, but positive for another pathogen as identified by culture (n = 7) or qPCR (n = 3) (Rozo et al., 2020).
The Agilent SureSelect design process was used to generate 120-mer RNA oligonucleotide probes to complement the entire length of each target genome. Custom probes for the Agilent SureSelect XT HS library kit (Agilent Technologies; Santa Clara, CA) were designed using the R. prowazekii and O. tsutsugamushi genomes listed in Table 1. The genomes were analyzed using a multiple-sequence alignment (MSA) and visualized using Mauve v2.3.1 to determine areas of genome collinearity and SNPs (Figures 1, 2; Supplementary materials) (Darling et al., 2004). To exclude probes with areas of low-complexity (i.e., homopolymer regions or di-nucleotide repeats), the probe set was filtered using DUST v1 (Morgulis et al., 2006). To further reduce redundancy, probes with greater than 95% similarity were excluded. As a final step, the probe set was aligned to the genomes of Mus musculus and Homo sapiens and complementary sequences were excluded from the probe set in order to prevent off-target binding due to cross-reactivity with host sequences. The resultant enrichment set comprises of 57,000 probes of varying depth along the target genomes (Supplementary Figures S3,S4 demonstrate alignment of probes to target genomes and the Supplementary Table 1 contains probe sequences and coordinates on target genomes used in design).
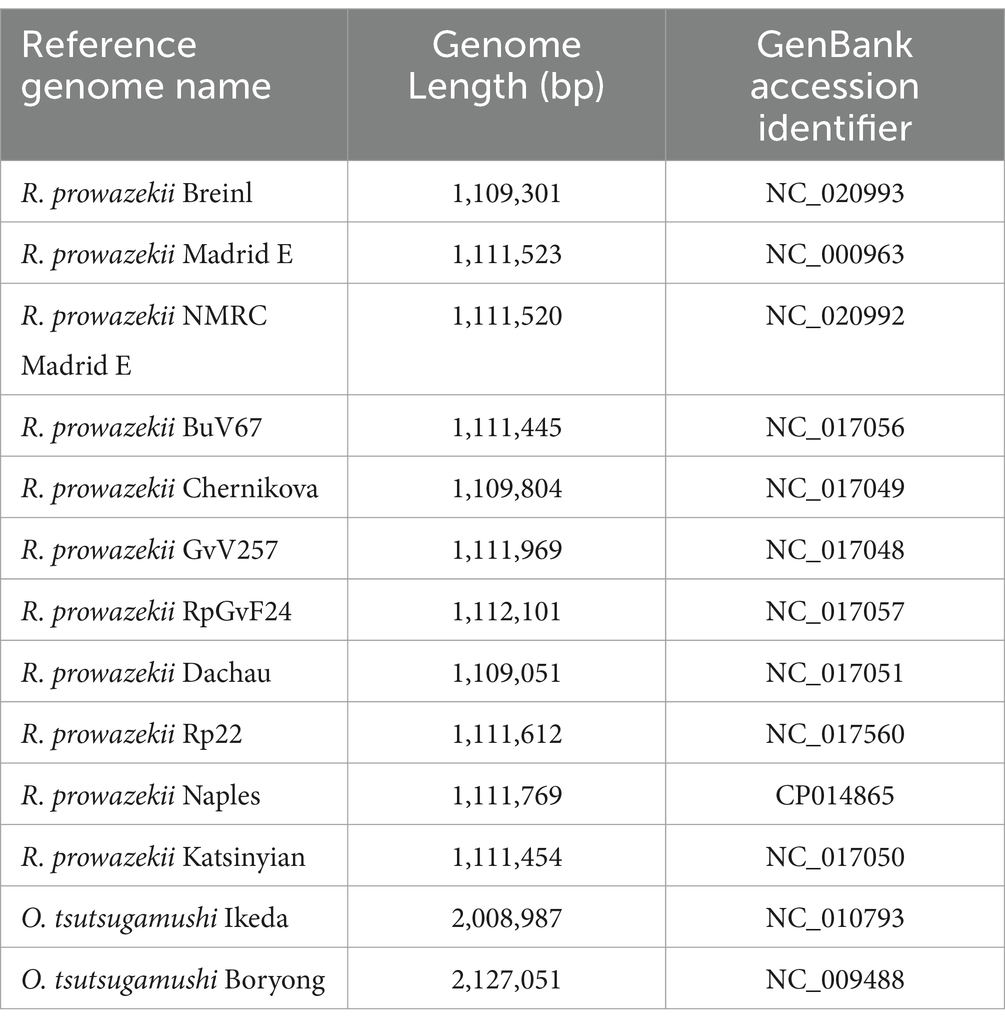
Table 1. Summary of NCBI genome data used in probe design.
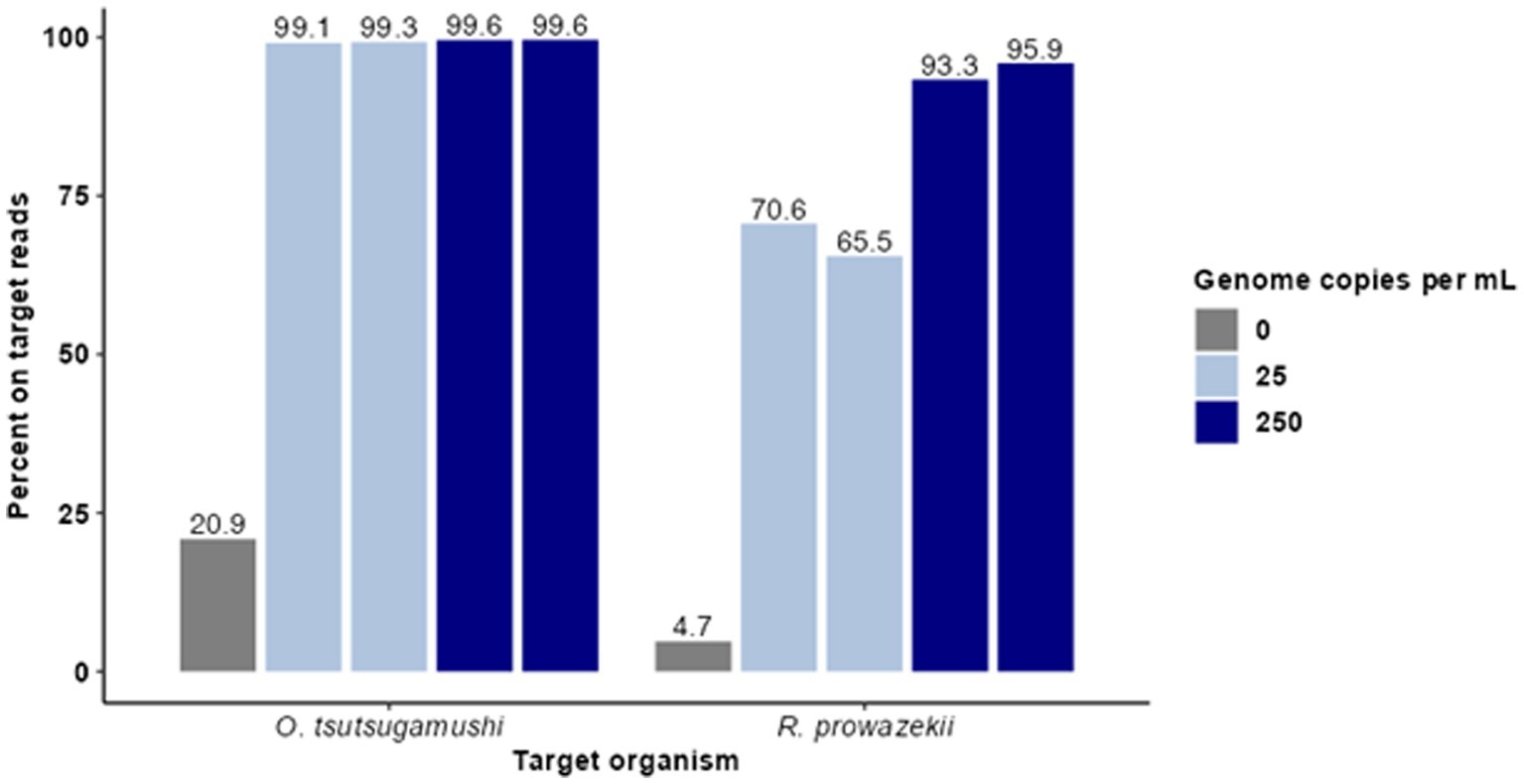
Figure 1. Hybridization-based enrichment of rickettsial sequences from complex samples. Known concentrations of gDNA from O. tsutsugamushi and R. prowazekii were spiked into complex matrices at 25 and 250 genome copies per mL. Sequence data from enriched samples were mapped to NCBI reference genomes O. tsutsugamushi Karp and R. prowazekii Breinl. The percent of trimmed reads mapped for each sample is graphed for each replicate.
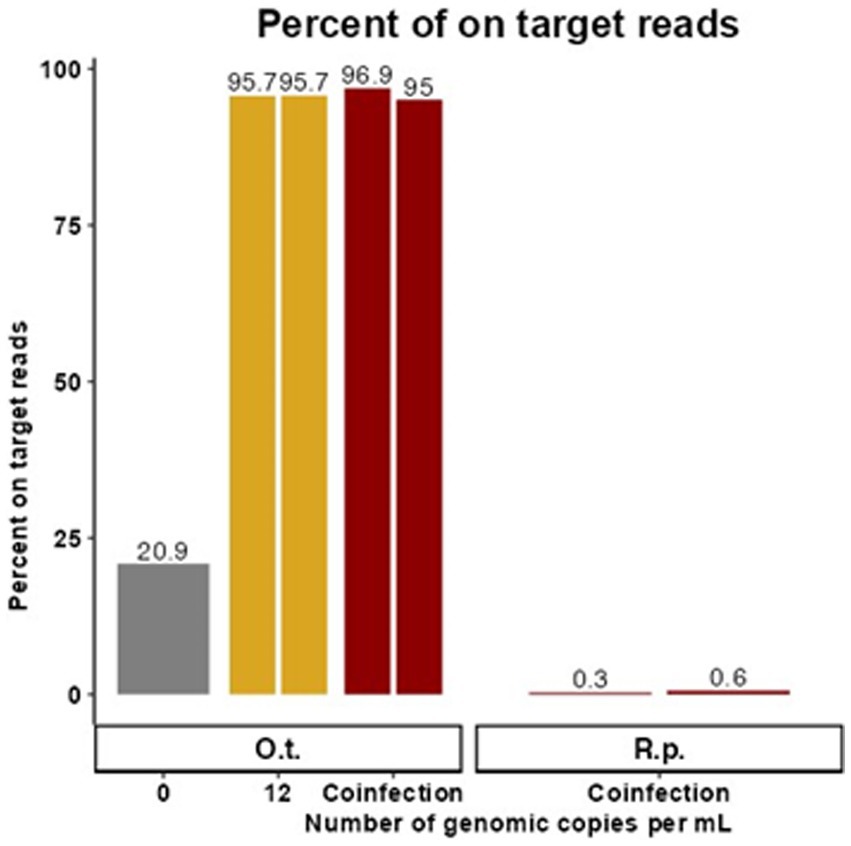
Figure 2. Targeted enrichment sequencing is effective in synthetic coinfection samples. These data represent samples containing target genetic material at the LoD of the existing qPCR assay for O. tsutsugamushi in the presence of R. prowazekii and human gDNA. The proportion of trimmed reads that were on target for O. tsutsugamushi is graphed for each replicate with 0 or 12 genome copies per mL of O. tsutsugamushi spiked in. Duplicate contrived coinfection samples also containing R. prowazekii genomic DNA are labeled as “Coinfection.” The proportion of R. prowazekii reads in the coinfection sample are represented in red bars on the right of the figure. There was no replicate for 0 gc/mL.
100 ng of gDNA from each sample was sheared to 450 bp fragments using a Covaris Ultra-Focused Sonicator M220 following manufacturer’s protocol (Covaris; Woburn, MA). Sequencing libraries were prepared using the Agilent SureSelect XT HS library kit per manufacturer’s protocol (Agilent Technologies; Santa Clara, CA). Hybridization was performed using the custom Agilent SureSelect probe set described above. Libraries were pooled in sets of 10–16 samples at a concentration of 10–12 pM and sequenced using MiSeq v3 chemistry (Illumina; San Diego, CA) for 600 cycles.
Quantitative polymerase chain reaction (qPCR) assays targeting htrA, the 47 kDa antigen gene of Orientia tsutsugamushi, and Rprow, the outer membrane protein B (ompB) of Rickettsia prowazekii, were performed as previously described using a StepOnePlus Real-Time PCR System (Applied Biosytems; Foster City, CA) to determine copy number (Jiang et al., 2003; Dasch et al., 2004). For htrA, each 25 μL reaction contained 1 μL of template DNA or negative control, 0.1 μM of each primer, 0.2 μM of probe, 0.5 μL of ROX Reference Dye (Invitrogen; Waltham, MA), 12.5 μL 2x Platinum Quantitative SuperMix-UDG (Invitrogen; Waltham, MA), and a final concentration of 5 mM MgCl2. For Rprow, each 25 μL reaction contained 1 mL of template DNA, 0.2 μM of each primer, 0.2 μM of probe, 0.5 μL of ROX Reference Dye, 12.5 μL 2x Platinum Quantitative SuperMix-UDG, and final concentration of 6 mM of MgCl2. A minimum of two replicates was performed per condition, as indicated in results tables.
The resultant sequence reads were trimmed, filtered to remove host and laboratory contaminant reads, then mapped to the available NCBI reference genome closest to the spike-in strain (Karp (Accession LS398548) Breinl (Accession NC_020993) using default parameters requiring a minimum of half the length of the read to map with 80% identity in CLC Genomics Workbench v23 (QIAGEN; CA, United States).; Karp (Accession LS398548) Breinl (Accession NC_020993). Results were visualized using R package ggplot2 (Wickham, 2016; R Core Team, 2022).
Sequences were quality controlled using bbduk v38.84 (Bushnell, 2014) with Q10 filtering and Q20 trimming, then assembled using SPAdes v3.15.2 (Bankevich et al., 2012). The resultant contigs were submitted to pubMLST (Jolley et al., 2018) specifically for the organism O. tsutsugamushi. The resulting allele numbers for each respective gene were used to determine the potential Sequence Type.
Type Surface Antigen 56 (tsa56) and sucB genes were selected for phylogenetic analysis based on the rationale that the tsa56 gene has historically been used for genotyping O. tsutsugamushi, and the sucB gene has been used by pubMLST for sequence typing. The full length of sucB (1,278 nt) and a 338 nt region of tsa56 were used for phylogenetic analysis. The nucleotide sequences of these genes were extracted from the SPAdes contigs by first using Bandage v0.8.1 (Wick et al., 2015) and BLAST (Altschul et al., 1990) to identify the contig that contains the genes and then using CLC Genomics Workbench 22 (CLC; QIAGEN; CA, USA) to extract the gene sequence. A short fragment of the 1,590 bp TSA56 was used because the full-length gene was not represented in the sequence data from every sample. For both genes, a multi-sequence alignment was generated using CLC. A maximum likelihood (ML) tree was generated using CLC with GTR + G + T model and 100 bootstrap and the resulting trees were visualized by FigTree v1.4.4 (Rambaut, 2009). The reference Boryong strain was omitted from sucB analysis due to a highly divergent sequence that created a long branch with little support.
Antimicrobial resistance (AMR) genes were identified using the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) (Olson et al., 2023) database for the eight existing references of O. tsutsugamushi: Boryong, Gilliam, Ikeda, Karp, Kato, TA686, UT176, and UT76. Contigs from each sample were screened for AMR genes using tBLASTn in CLC with a minimum threshold of 60% length of the gene.
Clinical features of 62 patients enrolled in an observational study of sepsis in Cambodia were analyzed. Of these patients, 54 were previously determined to be infected with pathogens other than O. tsutsugamushi (Rozo et al., 2020). Fisher’s exact tests were used on contingency tables to determine p-values. For clinical parameters, a Mann–Whitney U test was used to determine p-values. All statistical calculations were performed using GraphPad Prism 8.3.1 (GraphPad Software Inc.; San Diego, CA, United States).
For this proof of concept in detecting rickettsial pathogens using enrichment, we first sought to understand the limit of detection (LoD), specificity, cross-reactivity, depth, and breadth of targeted enrichment with rickettsial genomes using O. tsutsugamushi and R. prowazekii as representative species. To do so, contrived clinical samples were prepared with the pathogen genomic DNA spiked in at clinically relevant titers (measured in genomic copies (gc)/mL). Human gDNA was first extracted from 200 μL of whole blood and normalized to 100 ng/sample. Genomic DNA from O. tsutsugamushi and R. prowazekii was spiked in at 25 and 250 genome copies per mL. Agilent SureSelect probes targeting the O. tsutsugamushi and R. prowazekii genomes were used to enrich the contrived samples in duplicate, and enriched samples were sequenced using an Illumina MiSeq. Targeted enrichment performed well for each organism tested, recovering 85–88% of the O. tsutsugamushi genome and 88–91% of the R. prowazekii genome. Enriched samples were found to have >65% on-target reads (Figure 1). The average read coverage depth was 249x for O. tsutsugamushi and 165x for R. prowazekii; a depth of coverage more than adequate to perform strain level identification and SNP analysis in most cases. Minimal cross-reactivity producing two short regions of non-specific binding by human sequences was observed (visible in clinical results presented later, see Figure 3).
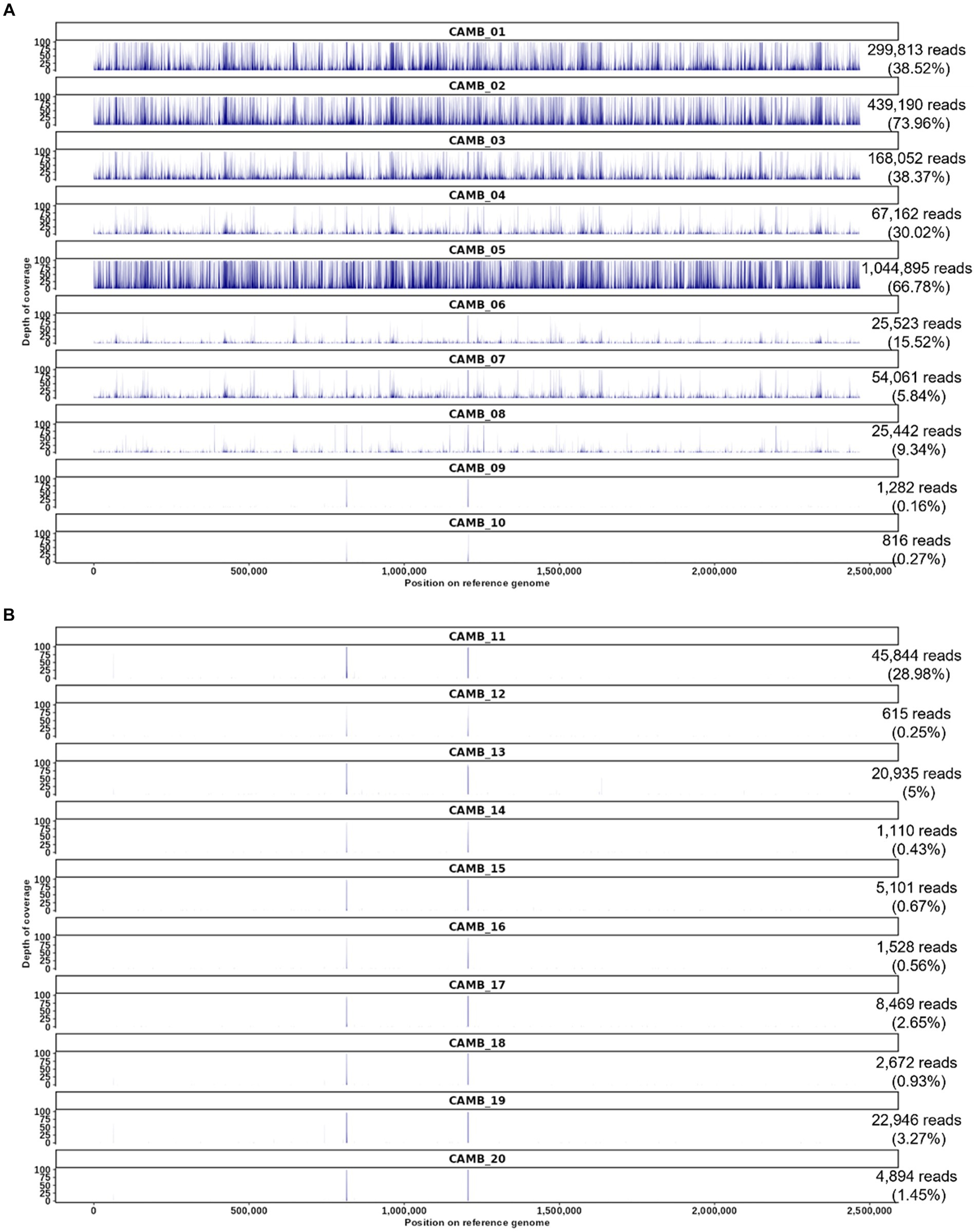
Figure 3. O. tsutsugamushi targeted enrichment applied to whole blood patient samples derived from an observational study of sepsis in Cambodia. (A) Patients who were positive by serology and weakly positive for O. tsutsugamushi sequences by unbiased shotgun sequencing and patient samples who were serology positive but whose samples did not contain O. tsutsugamushi sequences according to unbiased shotgun sequencing results (CAMB_09, CAMB_10). (B) Patient samples that were serology negative and unbiased shotgun sequencing negative for O. tsutsugamushi sequences, but positive for other pathogens by culture or qPCR in the prior study (Rozo et al., 2020). The number of reads mapped for each sample is shown on the right with the proportion of trimmed reads mapped in parentheses. The reference used in this read mapping is O. tsutsugamushi isolate Karp genome, accession LS398548, length 2,469,803 nucleotides.
Due to overlapping areas of endemicity and shared vectors, rickettsial co-infection has been observed although it is rare, and it poses a potential challenge for molecular diagnostics (Nogueras et al., 2015; Abreu et al., 2019; Kim et al., 2019). Therefore, replicate samples containing target genetic material at 12 gc/mL, the LoD of the existing qPCR assay for O. tsutsugamushi in the presence of R. prowazekii, were created in a background of human gDNA to simulate co-infected samples (and a control sample containing 0 gc/mL of O. tsutsugamushi gDNA was also included). Cleaned reads were mapped to the available NCBI reference genome closest to the spike-in strain (Karp or Breinl), and the proportion of on target reads calculated (Figure 2). For each replicate, 10,922 (0.3%) and 5,745 (0.6%) reads specific to R. prowazekii were enriched in the coinfected sample, compared to 3,766,741 (96.9%) and 918,840 (95%) reads specific to O. tsutsugamushi. Although we observed preferential enrichment for O. tsutsugamushi in the background of a low titer rickettsial coinfection, there was still a sufficient breadth of reads covering both genomes to provide evidence to determine the species and possibly proceed to further analyses (Supplementary Figure S1). The higher efficiency of enrichment by the O. tsutsugamushi probes as compared to the R. prowazekii probes in this 12-genome copy per pathogen per mL coinfection scenario was consistent with what we observed in singularly spiked, monoinfection samples at 25 gc/mL, whereas at higher titers, the probes perform similarly (Figure 1; Supplementary Figure S1). The presence of R. prowazekii genomic DNA did not impact the recovery of O. tsutsugamushi DNA, as similar proportions of “on target” (e.g., O. tsutsugamushi-specific) reads were recovered from samples with and without R. prowazekii spiked in.
We previously utilized unbiased shotgun sequencing to identify O. tsutsugamushi sequences directly from clinical specimens obtained from an observational study of sepsis in Cambodia as described in the methods section and elsewhere (Rozo et al., 2020), but in that prior work the genome sequences produced by shotgun sequencing were partial sequences. Therefore we resequenced those same samples here and found that our targeted enrichment sequencing assay produced whole genome sequence data from all eight samples that were previously known to be positive for O. tsutsugamushi (labeled CAMB_01 through _08 in Figure 3) (Rozo et al., 2020). While the previous results were weakly positive for O. tsutsugamushi from shotgun sequencing data, with reads mapping only to one or two genes (typically rRNA genes), targeted enrichment sequencing resulted in reads covering the entire O. tsutsugamushi genome (Figure 3A). Two samples that were previously IgM positive for O. tsutsugamushi but negative for O. tsutsugamushi by unbiased shotgun sequencing, CAMB_09 and _10, remained negative for O. tsutsugamushi using the targeted enrichment sequencing assay (Figure 3A), recapitulating the previously published findings. The culture and sequence-negative samples were still negative using the hybridization enrichment assay (Figure 3B), suggesting they may have had a titer below the LoD for the assay or the infection may already have been cleared from peripheral blood in these patients by the time these blood samples were drawn. Minimal cross-reactivity producing two short regions of non-specific binding by human sequences was observed (Figure 3). Finally, all samples that were positive for another pathogen as determined by culture or qPCR were also negative for O. tsutsugamushi sequences using targeted enrichment (Figure 3B). These results demonstrate that targeted enrichment provides increased specificity and sensitivity for detection and genetic characterization from real world clinical samples.
The depth and breadth of coverage obtained from O. tsutsugamushi positive clinical samples was considerably higher than was reported using shotgun metagenomic analysis of transcriptomic data from the same patients (Rozo et al., 2020). Therefore, we leveraged our newly enhanced dataset to analyze the characteristics of these pathogens circulating in Cambodia. Taxonomic classification and analysis of antibiotic resistance markers was performed as described in the methods.
Type Surface Antigen 56 (TSA56), derived from the 56-kDa gene (tsa56), has been the standard for typing O. tsutsugamushi. Similarly, dihydrolipoamide acetyltransferase component or Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex (sucB) is a gene used in multi-locus sequence typing (MLST) for O. tsutsugamushi (Jolley et al., 2018). Based on analysis of a 338 bp fragment of the gene tsa56, four samples (CAMB_01, CAMB_02, CAMB_04, and CAMB_05) were more closely related to the Kato strain whereas two samples (CAMB_03 and CAMB_06) were more closely related to Karp, UT76, and UT176 strains (Supplementary Figure S2). The tsa56 gene was not present in the assemblies for samples CAMB_07 and CAMB_08 and therefore those two samples could not be included in the analysis. Phylogenetic analysis of sucB revealed that the samples grouped with various representative sequences from the public database: CAMB_02 was most closely related to UT76 strain; sample CAMB_08 grouped with the Karp strain; samples CAMB_01, CAMB_4, CAMB_05, and CAMB_07 grouped with the Ikeda strain; and samples CAMB_03 and CAMB_06 were more closely related to TA686 and Gilliam strains (Figure 4). Note that samples CAMB_08 and CAMB_07 are only partial sequences and their placement is not well supported.
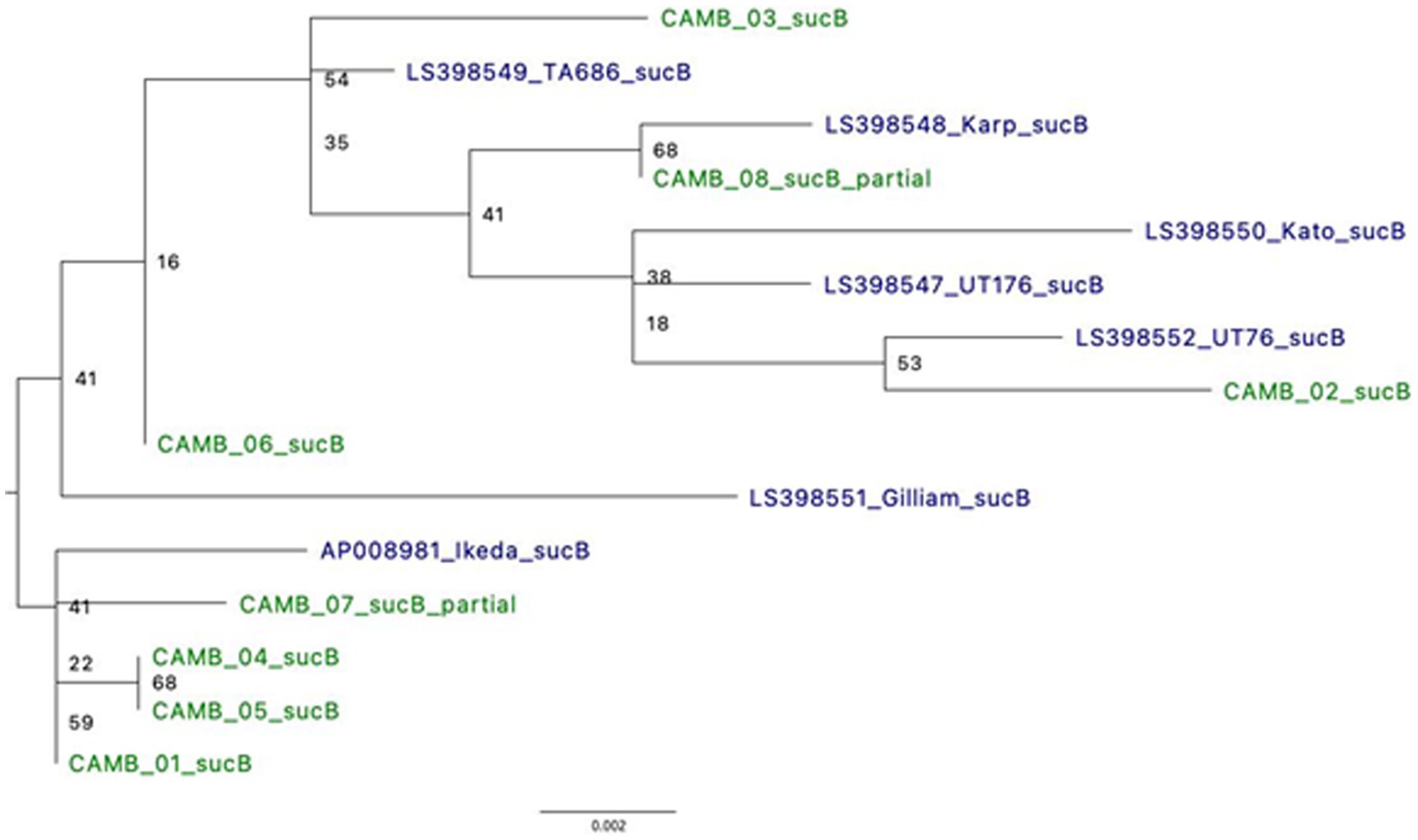
Figure 4. Phylogeny using full length sucB sequences demonstrates relatedness to known O. tsutsugamushi strains. Where possible, alignments of full sucB sequences were used to generate a Maximum Likelihood tree. There were two samples for which only a partial sucB gene sequence was available for analysis. *UT76 and UT176 are clinical isolates similar to the Karp strain (Paris et al., 2009).
In addition to conducting phylogenetic analyses, we also screened the dataset for 20 common antimicrobial resistance genes as described in the methods (Table 2). Of note, two genes that confer resistance to rifampicin (rpoB and rpoC), a treatment option for O. tsutsugamushi, were universally absent from each of our clinical isolates (Table 2) (Eremeeva and Dasch, 2002; el Sayed et al., 2018). These results demonstrate the clinical utility of HTS following targeted enrichment for the identification and characterization of O. tsutsugamushi directly from clinical specimens.
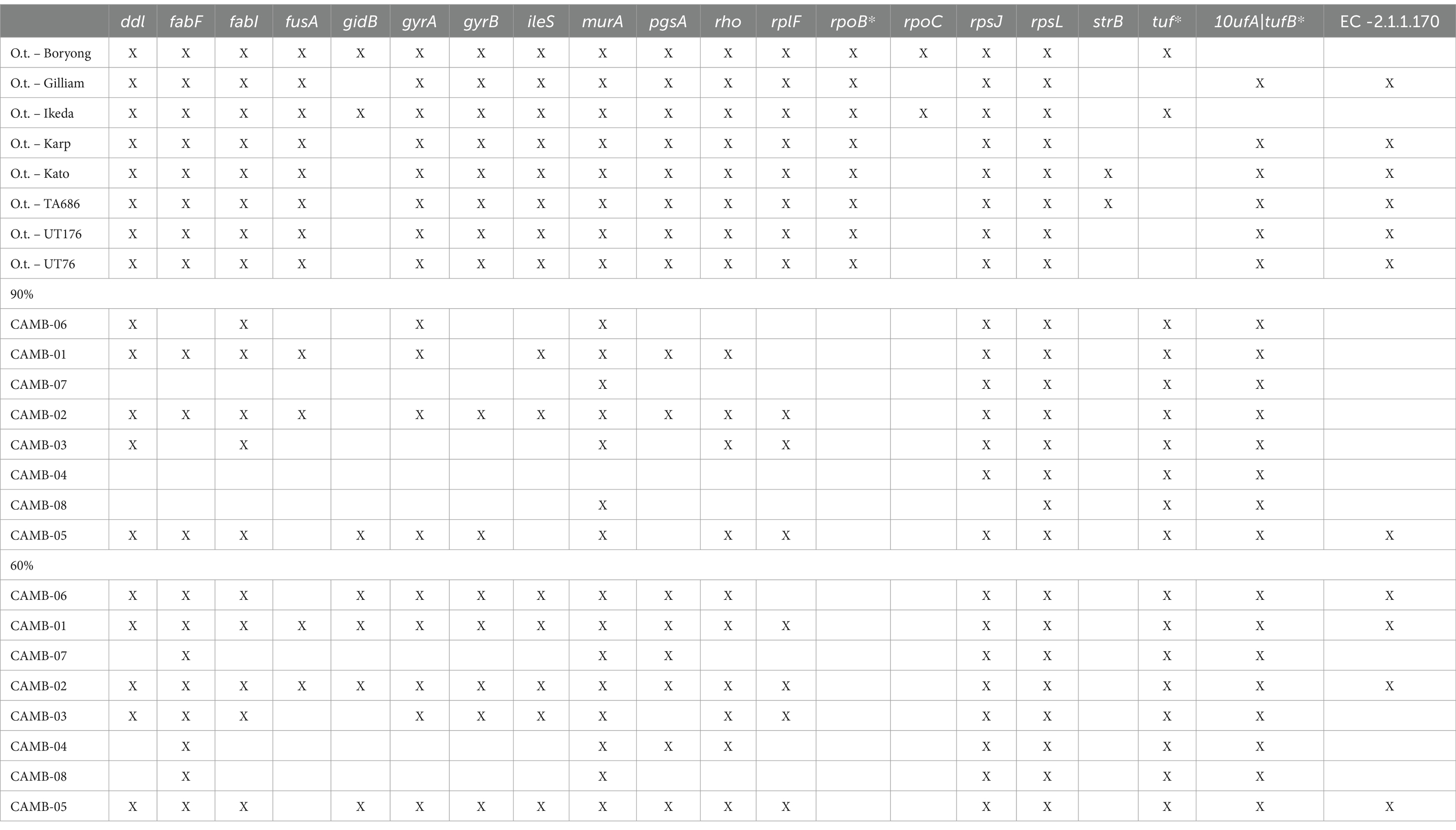
Table 2. Genes that confer antimicrobial resistance (AMR) present in O. tsutsugamushi positive clinical specimens.
HTS may not be available or appropriate in every case and qPCR remains a viable alternative for molecular diagnostics. One limitation to qPCR, as with HTS, is that the number of genomic copies in blood samples is extremely low in rickettsial infections. In fact, one study found the median titer was the equivalent of 13 copies per mL of blood, but as low as zero in many cases (Sonthayanon et al., 2009). To assess if targeted enrichment could increase the performance of qPCR for the detection of rickettsial genomes, we first sought to determine the limits of detection (LoD) of qPCR for R. prowazekii Breinl in contrived clinical samples both with and without targeted enrichment. Dilutions of either plasmid DNA containing fragment A of the ompB gene, or R. prowazekii Breinl genomic DNA were prepared ranging from 20,000 copies to ~1.5 copies, as described in the methods. A qPCR assay targeting the ompB gene of R. prowazekii was performed in duplicate as published by Jiang et al. (2003) and Dasch et al. (2004). While one replicate of qPCR detected ~6 copies of R. prowazekii Breinl genomic DNA, the lower LoD without targeted enrichment was ~12 copies (Table 3). We therefore selected 12.5, 25 and 50 genome copies for further evaluation. To confirm the baseline LoD, 20 replicates of each of the three target copy numbers were assayed using qPCR without prior enrichment. Without enrichment, the 12.5 genome copies per mL set was only detected in 17 of 20 replicates (85%), however, the target was reliably detected at both 25 and 50 copies per mL for all 20 replicates. Therefore, we determined that the LoD was 25 genome copies per mL for this qPCR assay (Table 4).
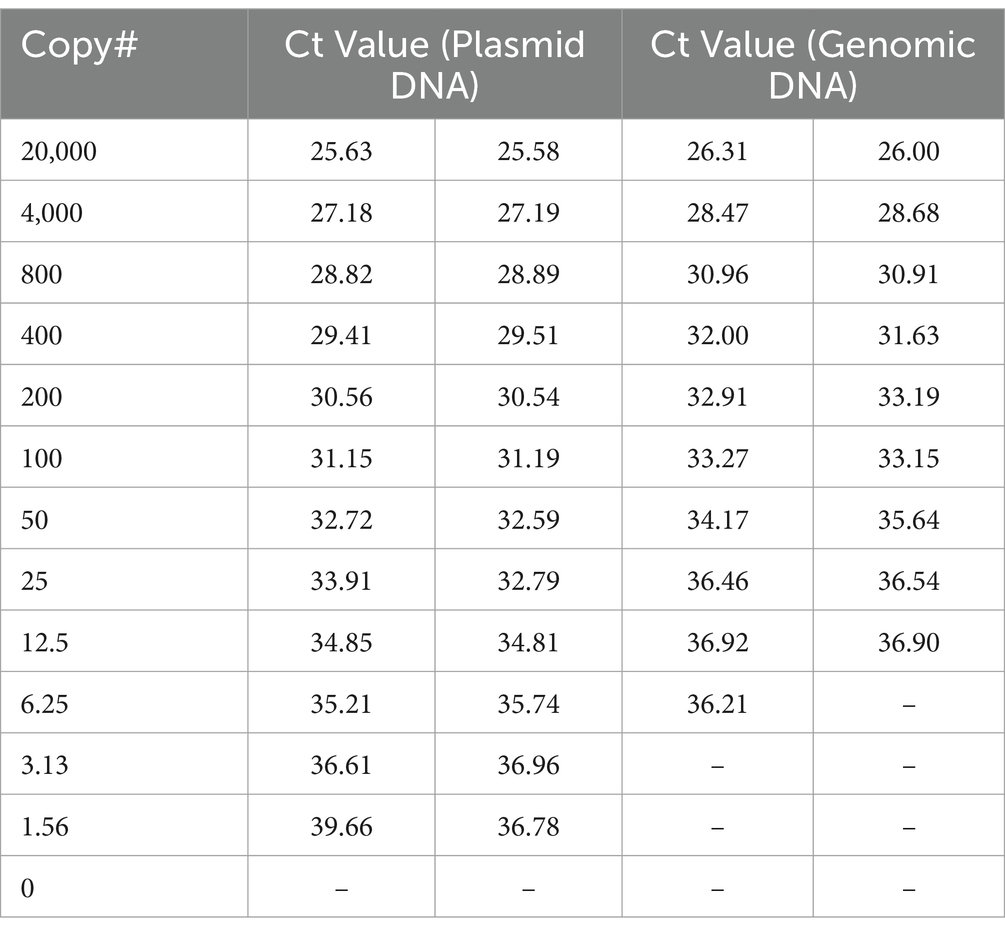
Table 3. qPCR of standard curve for determination of LoD (without enrichment).
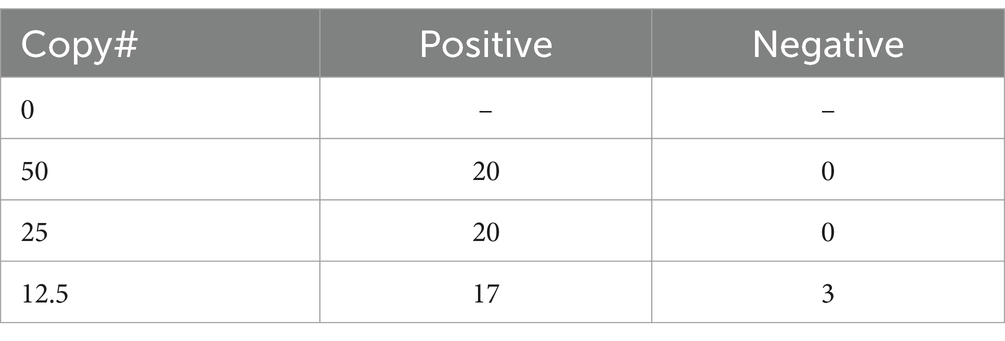
Table 4. Confirmation of LoD based on 20 replicates for each copy number (without enrichment).
Once the LoD for unenriched qPCR was established, we assessed if targeted enrichment coupled with the same qPCR assay could reduce the LoD by detecting even fewer genomic equivalents. To accomplish this, enriched sequencing libraries with starting material ranging from approximately 1.5 to 50 genome copies were produced in triplicate for each copy number and qPCR was performed in duplicate on an aliquot from each library, resulting in six replicates for each copy number being tested. Post-enrichment qPCR data allowed for positive detection down to one copy across six replicates, indicating the success of applying enrichment to increase sensitivity of the qPCR assay (Table 5). The Ct values for the 15 sequencing libraries after enrichment strategies (Supplementary Table S2) were consistent with the Ct values equivalent to greater than 4,000 genome copies (Table 3), an observation reinforced by copy number determination of the enriched sequence libraries (Supplementary Table S2). Taken together, these data demonstrate that genome-enriched samples performed better in qPCR assays than non-enriched samples.
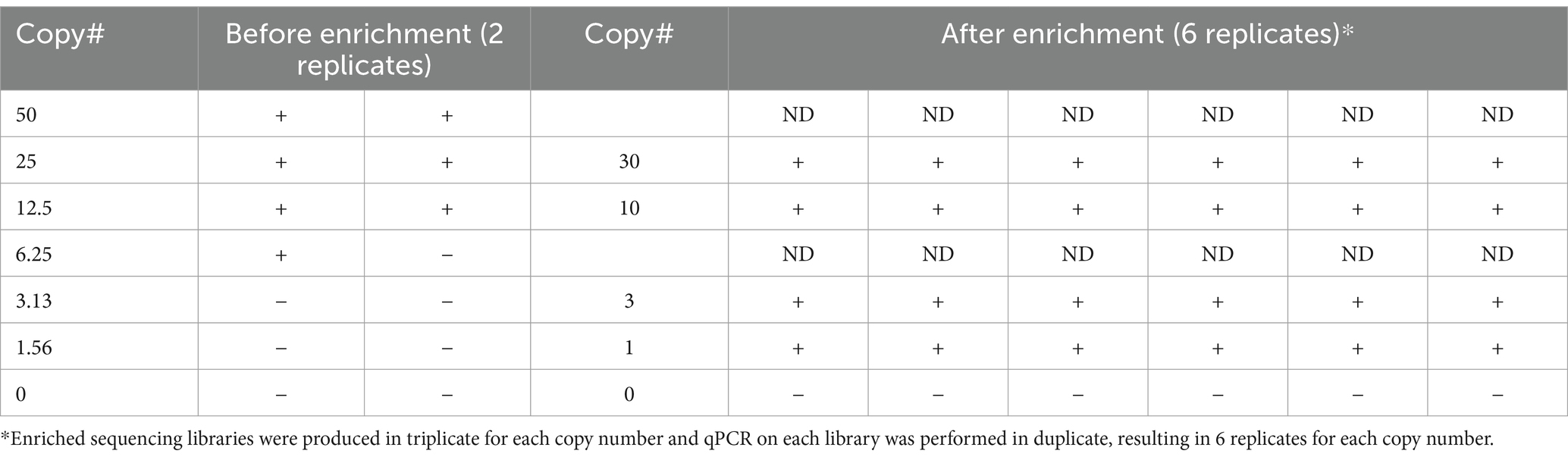
Table 5. Comparison of qPCR data generated before and after enrichment.
For both bacterial and viral pathogens, pathogen genomic sequencing is used to trace outbreaks routinely. For instance, a recent fatal outbreak of Burkholderia pseudomallei within the United States among patients with no travel history was traced to aroma therapy products using genomics. These four cases generated clinical isolates that were sequenced (Gee et al., 2022). However, in some cases, there are constraints that prevent clinical isolates from being grown from the primary patient samples (in particular when there are many patients or the organism is hard to culture). The genomic enrichment method would be valuable in cases where no clinical isolate is available and pathogen genetic variations are being used to perform molecular epidemiology. For instance, other examples of pathogen genomic sequencing that have been conducted in our own laboratory for similar use cases include that of SARS-CoV-2 (Cer et al., 2022; Lizewski et al., 2022) as well as methicillin-resistant Staphyloccus aureus [MRSA: (Millar et al., 2017, 2019)]. In addition to our own use of whole genome sequencing to trace transmission chains of SARS-CoV-2 in military settings, others have used whole genome sequencing to trace transmission chains in the hospital setting (Keehner et al., 2024) and the same has been done for Dengue virus in household transmission settings (Berry et al., 2021). These are just a few examples out of many that clearly demonstrate outbreaks for a variety of pathogens, both viral and bacterial, can be traced to identify source and transmission chains by use of whole genome sequencing data. Therefore, it is quite possible that in the future similar methods could be applied if there were an outbreak of a rickettsial organism.
A limitation of using unbiased HTS of metagenomic samples is that it suffers from a lack of sensitivity for pathogens in complex sample types that contain both host and commensal sequences in addition to the pathogen of interest (i.e., human clinical samples or environmental samples). Other complicating factors that affect the ability to detect an organism of interest include the organism’s genome size compared to other organisms present in the matrix (particularly for viral genomes), sample complexity (the total range of organisms present within the sample matrix), and titer. These factors influence the contribution of organism-specific versus host- and/or commensal-specific reads, and hence the LoD. We evaluated probe-based targeted enrichment as a strategy to augment both sequencing and molecular diagnostics (e.g., qPCR) for the detection of the intracellular pathogens R. prowazekii and O. tsutsugamushi, which generally have narrow diagnostic windows. This method has been applied previously for sequencing of some microbes in clinical samples (for example C. trachomatis in (Christiansen et al., 2014) and O. tsutsugamushi in Elliott et al., 2021), but to our knowledge not for sequencing Rickettsia spp. specifically, and only in very scarcely few examples in the literature for PCR assays of any pathogens until now (Yang et al., 2016; Bai et al., 2019), and not for PCR for rickettsial pathogens. Using this approach, we report improved sensitivity over traditional diagnostic methods – LoD as low as one genomic copy (Table 5) was detected using qPCR post enrichment, as compared to 25 genome copies without enrichment. We have also expanded the sensitivity of whole genome sequencing data that can be generated from clinical specimens, covering nearly the entire genome of the targeted organism as compared to the detection of only one or two pathogen genes without enrichment. Taken together, we demonstrated that this strategy allows for both identification and characterization of rickettsial diseases directly from clinical specimens, dramatically increasing the potential applications of this method. As compared to traditional diagnostic methods (i.e., culture-based assays, serology, qPCR), a HTS-based detection approach in general provides more information than other diagnostic assays through its ability to garner information beyond identification of the agent to include strain level typing and presence/absence of antimicrobial resistance genes and/or virulence factors.
Our data demonstrate that genome enrichment greatly enhances the sensitivity of both qPCR and sequencing for direct detection of pathogen nucleic acids. The previous sequence data we obtained for these clinical samples via unbiased shotgun (e.g., metagenomic) sequencing and reported in Rozo et al. (2020) consisted of sporadic detection of one or two Orientia genes per sample (typically rRNA genes), whereas now with the genome enrichment sequencing strategy reported in the current study, we have achieved coverage of the entire Orientia genome from these same samples (Figure 3). Obtaining from a patient sample a few partial gene sequences with homology to the pathogen of interest’s genome at low depth of coverage and within the milieu of reads deriving from commensal organisms and background might be considered an equivocal result as compared to obtaining an entire pathogen genome from a patient sample, which would be much more straightforward and would also allow for characterization (e.g., antimicrobial susceptibility). In other words, though both unbiased metagenomic sequencing and enrichment sequencing “detected” the pathogen, enrichment sequencing data are much more conclusive and leave much less room for doubt. In addition to demonstrating the increased sensitivity when genome enrichment is applied to sequencing-based detection, we also demonstrated in this study that genome enrichment greatly increased the sensitivity of detection by qPCR – reducing the limit of detection down from 25 genome copies per mL without enrichment to 1 genome copy per mL with enrichment. Therefore, we have demonstrated a much more sensitive direct detection assay.
We proved the clinical utility of this method by enriching patient blood samples obtained from an observational study of sepsis in Cambodia (Rozo et al., 2020). While rickettsial infections respond to early treatment with antibiotics, rickettsial infections are challenging to identify clinically. Eschars, while associated with a number of rickettsial infections, are often absent or overlooked and clinical laboratory features are nonspecific (Abdad et al., 2018). Multiple features consistent with a diagnosis of scrub typhus were observed among the eight patients from whom O. tsutsugamushi genomes were detected (Supplementary Tables S3,S4). Specifically, symptoms of fever and shortness of breath were uniformly present. Notable laboratory abnormalities that were present in most but not all O. tsutsugamushi positive patients included liver function test elevation and thrombocytopenia. These features are supportive of the diagnosis of scrub typhus and consistent with what has been reported in prior literature (Sriwongpan et al., 2013). Epidemiologic clues including the occupation of farming were also present (Rozo et al., 2020) but could be of limited clinical utility in predominantly agrarian societies where most patients may have risk factors for mite exposure. For an infection that is treated based on clinical suspicion, the lack of pathognomonic features escalates the need for improved clinical diagnostics – a gap that could be filled via the enrichment-augmented assays described in this study.
Clinical experience and animal models demonstrate that the diagnostic window for detection of O. tsutsugamushi from blood by qPCR is short lived, and the initial day for detection is dose related (Jiang et al., 2003; Chattopadhyay et al., 2005; Watt et al., 2005; Paris et al., 2015). While targeted enrichment increases the sensitivity in both contrived and clinical specimens, we were unable to draw definite conclusions regarding diagnostic window. Two of the septic patient samples we attempted to characterize using targeted enrichment were IgM positive at the time of sample collection. However, targeted enrichment failed to produce meaningful coverage of the O. tsutsugamushi genome. IgM antibodies can be nonspecific and could represent false positives due to cross-reactivity or could be derived from a previous infection, since their longevity can last up to a year from infection. A sufficiently powered study utilizing well-characterized O. tsutsugamushi patient samples will be necessary to draw specific conclusions with regard to diagnostic window. Taking into consideration the reduction of sensitivity down to a single genome copy per sample, we hypothesize that an expanded diagnostic window would result.
In addition to the identification of O. tsutsugamushi in patient samples, hybridization enrichment allowed us to detect antibiotic resistance markers and to perform MLST, neither of which was possible in the previous analysis of these same samples that used metagenomic sequencing without enrichment (Rozo et al., 2020). While sequence types were identified for four of the eight clinical specimens, there were not enough data to determine the sequence types for the other four because not all loci were found in contigs. We expected this result due to the variety of confounding features that could limit the completeness of the genomes including unknown duration of infection and previous antimicrobial therapy. We generated Maximum Likelihood trees using the standard O. tsutsugamushi classification regions tsa56 and sucB (Figure 4) and tsa56 (Supplementary Figure S2). In a congregate setting, such as many military settings like ships and recruit training centers, pathogen strain relatedness can be used to assess if there was one or multiple introductions of a pathogen into the congregate setting, and that can inform force health protection decision-making about what infection control measures should or should not be put in place to prevent further spread. This can apply to other congregate settings as well, such as hospitals and school dormitories. Also, if antimicrobial resistance markers are detected that would render a particular treatment ineffective that particular therapy could be avoided reducing treatment delay and improving clinical outcomes.
In the literature, resistance to tetracycline has been hypothesized as a mechanism underlying delays in clinical improvement for almost 30 years. It is not currently known what the true prevalence of antibiotic resistance is in O. tsutsugamushi, but it remains as a formal possibility [reviewed in (Lu et al., 2021)]. Thus, we also verified the presence of traditional O. tsutsugamushi antimicrobial resistance markers. Incorporating this approach with traditional diagnostic and treatment algorithms could both reduce diagnostic delay and lessen the need for empiric antimicrobial therapy. Given that the diagnostic window of R. prowazekii and O. tsutsugamushi by currently available assays is rather narrow and so treatment is often based on suspicion rather than positive tests, development of a diagnostic tool that could simultaneously provide a positive species- or strain-level identification simultaneously with information as to antimicrobial sensitivity could be expected to result in more timely and accurate diagnoses. Treatment generally begins before confirming a positive test result via culture, which could take weeks. The time estimation for sequencing via this method, not including DNA extraction, is 11 h: 8 h of library preparation, 1.5 h of quality control, and 1.5 h to load the sequencer. The sequencer would then run for 1–3 days (end-user can decide how long) and then a quick read mapping-based analysis could be completed within an hour. The result could be that more patients get treated adequately with the correct antimicrobials, with less delay. As such, we posit that this assay could allow for a novel sequence finding that would predict functional resistance faster than clinical treatment failure, and that there is an intrinsic benefit to developing assays that help avoid clinical treatment failure.
We have provided evidence that this technique is effective for the detection, identification, and characterization of R. prowazekii and O. tsutsugamushi in clinical-type samples and we are currently exploring some of the many potential applications for expansion of this enrichment technique. It is likely that these probes could be used to enhance sensitivity for detection of related organisms from almost any complex sample type. For instance, we performed sequence analyses that indicate these probes would work similarly for detection of the other typhus group rickettsia, Rickettsia typhi, which has a significant similarity at the nucleotide level (Supplementary Table S5) and overall genome synteny (Supplementary Figure S3). Minor modifications to the 120-mer probes could produce a pan-rickettsial enrichment panel. Given that the probes work in co-infection scenarios, it would be most economical to expand to a pan-rickettsial test that can detect multiple possible etiological agents. Additionally, deeper multiplexing can be used to decrease cost. Beyond rickettsial applications, probes for targeted enrichment can be designed against any relevant pathogen, resulting in combinations of relevant probes that could produce regionally specific panels, biothreat panels, and environmental panels to increase the LoD for pathogens of interest. Commercial vendors provide both freely-available tools to design custom probes, as well as make probe design available as part of ordering options. Enriched products can be characterized by qPCR simply for pathogen identification or via HTS for in-depth genetic characterization. The benefit of generating enough sequence data for genetic characterization, as demonstrated by our data, is that genome enrichment greatly enhances the ability to further characterize a clinical sample that is outside of the standard diagnostic window. We conclude this work demonstrates the following proof of concept: This method increased the sensitivity of both qPCR and sequencing for direct detection of pathogen nucleic acids that not only allowed for the species level identification but also virulence genes and AMR markers. Specifically, we demonstrated that this method enables the detection and characterization of O. tsutsugamushi genomes directly from sepsis patient samples.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA902390.
The studies involving humans were approved by Naval Medical Research Command Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
AP: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. KS: Investigation, Writing – original draft, Writing – review & editing. LV: Investigation, Writing – review & editing. CA: Formal analysis, Writing – review & editing. RC: Investigation, Supervision, Writing – original draft, Writing – review & editing. KF: Investigation, Writing – review & editing. PB: Investigation, Writing – review & editing. DC: Investigation, Writing – review & editing. HG: Investigation, Writing – review & editing. AR: Investigation, Writing – review & editing. CF: Investigation, Writing – review & editing. KB-L: Conceptualization, Formal analysis, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by Defense Threat Reduction Agency through CB1040 to KB-L and by Navy WUN A1417.
The authors would like to thank Haven Miner for assistance with formatting and editing.
AP and LV were employed by Leidos.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the U.S. Department of Health and Human Services, the U.S. Department of Defense, the U.S. Navy, the U.S. government or of the institutions and companies affiliated with the authors. Several of the authors are U.S. government employees. This work was prepared as part of their official duties. Title 17 U.S.C. § 105 states, “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1387208/full#supplementary-material
Abdad, M. Y., Abou Abdallah, R., Fournier, P. E., Stenos, J., and Vasoo, S. (2018). A concise review of the epidemiology and diagnostics of rickettsioses: Rickettsia and Orientia spp. J. Clin. Microbiol. 56:e01728-17. doi: 10.1128/JCM.01728-17
Abreu, D. P. B., Peixoto, M. P., Luz, H. R., Zeringóta, V., Santolin, Í. D. A. C., Famadas, K. M., et al. (2019). Two for the price of one: co-infection with Rickettsia bellii and spotted fever group Rickettsia in Amblyomma (Acari: Ixodidae) ticks recovered from wild birds in Brazil. Ticks Tick Borne Dis. 10:101266. doi: 10.1016/j.ttbdis.2019.101266
Akram, S. M., Ladd, M., and King, K. C. (2021). “Rickettsia Prowazekii” in StatPearls (Treasure Island (FL): StatPearls Publishing).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Angelakis, E., Richet, H., Rolain, J. M., la Scola, B., and Raoult, D. (2012). Comparison of real-time quantitative PCR and culture for the diagnosis of emerging rickettsioses. PLoS Negl. Trop. Dis. 6:e1540. doi: 10.1371/journal.pntd.0001540
Baba, H., Kuroda, M., Sekizuka, T., and Kanamori, H. (2023). Highly sensitive detection of antimicrobial resistance genes in hospital wastewater using the multiplex hybrid capture target enrichment. mSphere 8:e0010023. doi: 10.1128/msphere.00100-23
Badiaga, S., and Brouqui, P. (2012). Human louse-transmitted infectious diseases. Clin. Microbiol. Infect. 18, 332–337. doi: 10.1111/j.1469-0691.2012.03778.x
Bai, Y., Cui, Y., Suo, Y., Shi, C., Wang, D., and Shi, X. (2019). A rapid method for detection of Salmonella in Milk based on extraction of mRNA using magnetic capture probes and RT-qPCR. Front. Microbiol. 10:770. doi: 10.3389/fmicb.2019.00770
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Berry, I. M., Melendrez, M. C., Pollett, S., Figueroa, K., Buddhari, D., Klungthong, C., et al. (2021). Precision tracing of household dengue spread using inter- and intra-host viral variation data, Kamphaeng Phet, Thailand. Int. Conf. Emerg. Infect. Dis. 27, 1637–1644. doi: 10.3201/eid2706.204323
Bishop-Lilly, K. A., Ge, H., Butani, A., Osborne, B., Verratti, K., Mokashi, V., et al. (2013). Genome sequencing of four strains of Rickettsia prowazekii, the causative agent of epidemic typhus, including one flying squirrel isolate. Genome Announc. 1:e00399-13. doi: 10.1128/genomeA.00399-13
Bonsall, D., Ansari, M. A., Ip, C., Trebes, A., Brown, A., Klenerman, P., et al. (2015). Ve-SEQ: robust, unbiased enrichment for streamlined detection and whole-genome sequencing of HCV and other highly diverse pathogens. F1000Res 4:1062. doi: 10.12688/f1000research.7111.1
Briese, T., Kapoor, A., Mishra, N., Jain, K., Kumar, A., Jabado, O. J., et al. (2015). Virome capture sequencing enables sensitive viral diagnosis and comprehensive Virome analysis. MBio 6, e01491–e01415. doi: 10.1128/mBio.01491-15
Brown, J. R., Roy, S., Ruis, C., Yara Romero, E., Shah, D., Williams, R., et al. (2016). Norovirus whole-genome sequencing by SureSelect target enrichment: a robust and sensitive method. J. Clin. Microbiol. 54, 2530–2537. doi: 10.1128/JCM.01052-16
Bushnell, B. (2014). “BBMap: a fast, accurate, splice-aware aligner” in Lawrence Berkeley national lab (United States: (LBNL), Berkeley, CA)
Cer, R. Z., Voegtly, L. J., Adhikari, B. N., Pike, B. L., Lueder, M. R., Glang, L. A., et al. (2022). Genomic and virologic characterization of samples from a shipboard outbreak of COVID-19 reveals distinct variants within limited temporospatial parameters. Front. Microbiol. 13:960932. doi: 10.3389/fmicb.2022.960932
Chattopadhyay, S., Jiang, J., Chan, T. C., Manetz, T. S., Chao, C. C., Ching, W. M., et al. (2005). Successful induction of humoral and cellular immune responses in cynomolgus monkeys to the scrub typhus vaccine candidate, Kp r56. Infect. Immun. 73, 5039–5047. doi: 10.1128/IAI.73.8.5039-5047.2005
Chattopadhyay, S., and Richards, A. L. (2007). Scrub typhus vaccines: past history and recent developments. Hum. Vaccin. 3, 73–80. doi: 10.4161/hv.3.3.4009
Christiansen, M. T., Brown, A. C., Kundu, S., Tutill, H. J., Williams, R., Brown, J. R., et al. (2014). Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples. BMC Infect. Dis. 14:591. doi: 10.1186/s12879-014-0591-3
Christiansen, M. T., Kaas, R. S., Chaudhuri, R. R., Holmes, M. A., Hasman, H., and Aarestrup, F. M. (2014). Genome-wide high-throughput screening to investigate essential genes involved in methicillin-resistant Staphylococcus aureus sequence type 398 survival. PLoS One 9:e89018. doi: 10.1371/journal.pone.0089018
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dasch, G. A., Ching, W. M., Richards, A. L., Jiang, J., Chan, T. C., and Temenak, J. J. (2004). Development of a quantitative real-time polymerase chain reaction assay specific for Orientia tsutsugamushi. Am. J. Trop. Med. Hyg. 70, 351–356. doi: 10.4269/ajtmh.2004.70.351
Devine, J. (2003). A review of scrub typhus management in 2000-2001 and implications for soldiers. J. Rural Remote Environ. Health 2, 14–20.
el Sayed, I., Liu, Q., Wee, I., and Hine, P. Cochrane Infectious Diseases Group (2018). Antibiotics for treating scrub typhus. Cochrane Database Syst. Rev. 2018:CD002150. doi: 10.1002/14651858.CD002150.pub2
Elliott, I., Thangnimitchok, N., de Cesare, M., Linsuwanon, P., Paris, D. H., Day, N. P. J., et al. (2021). Targeted capture and sequencing of Orientia tsutsugamushi genomes from chiggers and humans. Infect Genet Evol. 91:104818. doi: 10.1016/j.meegid.2021.104818
Eremeeva, M. E., and Dasch, G. A. (2002). “Rickettsia and orientia” in Molecular medical microbiology. ed. M. Sussman (Philadelphia, PA, USA: Elsevier), 2177–2217.
Furtwängler, A., Neukamm, J., Böhme, L., Reiter, E., Vollstedt, M., Arora, N., et al. (2020). Comparison of target enrichment strategies for ancient pathogen DNA. BioTechniques 69, 455–459. doi: 10.2144/btn-2020-0100
Gee, J. E., Bower, W. A., Kunkel, A., Petras, J., Gettings, J., Bye, M., et al. (2022). Multistate outbreak of Melioidosis associated with imported aromatherapy spray. N. Engl. J. Med. 386, 861–868. doi: 10.1056/NEJMoa2116130
Gnirke, A., Melnikov, A., Maguire, J., Rogov, P., LeProust, E. M., Brockman, W., et al. (2009). Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189. doi: 10.1038/nbt.1523
Gouriet, F., Fenollar, F., Patrice, J. Y., Drancourt, M., and Raoult, D. (2005). Use of shell-vial cell culture assay for isolation of bacteria from clinical specimens: 13 years of experience. J. Clin. Microbiol. 43, 4993–5002. doi: 10.1128/JCM.43.10.4993-5002.2005
Griffith, M., Peter, J. V., Karthik, G., Ramakrishna, K., Prakash, J. A., Kalki, R. C., et al. (2014). Profile of organ dysfunction and predictors of mortality in severe scrub typhus infection requiring intensive care admission. Indian J. Crit. Care. Med. 18, 497–502. doi: 10.4103/0972-5229.138145
Jiang, J., Martínez-Valdebenito, C., Weitzel, T., Farris, C. M., Acosta-Jamett, G., Abarca, K., et al. (2022). Development of a new genus-specific quantitative real-time PCR assay for the diagnosis of scrub typhus in South America. Front. Med. 9:831045. doi: 10.3389/fmed.2022.831045
Jiang, J., Paris, D. H., Blacksell, S. D., Aukkanit, N., Newton, P. N., Phetsouvanh, R., et al. (2013). Diversity of the 47-kD HtrA nucleic acid and translated amino acid sequences from 17 recent human isolates of Orientia. Vector Borne Zoonotic Dis. 13, 367–375. doi: 10.1089/vbz.2012.1112
Jiang, J. U., Temenak, J. J., and Richards, A. L. (2003). Real-time PCR duplex assay for Rickettsia prowazekii and Borrelia recurrentis. Ann. N. Y. Acad. Sci. 990, 302–310. doi: 10.1111/j.1749-6632.2003.tb07380.x
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Kapel, N., Kalimeris, E., Lumley, S., Decano, A., Rodger, G., Lopes Alves, M., et al. (2023). Evaluation of sequence hybridization for respiratory viruses using the twist bioscience respiratory virus research panel and the OneCodex respiratory virus sequence analysis workflow. Microb. Genom. 9:001103. doi: 10.1099/mgen.0.001103
Keehner, J., Abeles, S. R., Longhurst, C. A., Horton, L. E., Myers, F. E., Riggs-Rodriguez, L., et al. (2024). Integrated genomic and social network analyses of severe acute respiratory syndrome coronavirus 2 transmission in the healthcare setting. Clin. Infect. Dis. doi: 10.1093/cid/ciad738
Kelly, D. J., Richards, A. L., Temenak, J., Strickman, D., and Dasch, G. A. (2002). The past and present threat of rickettsial diseases to military medicine and international public health. Clin. Infect. Dis. 34, S145–S169. doi: 10.1086/339908
Kim, S. W., Kim, C. M., Kim, D. M., and Yun, N. R. (2019). Case report: coinfection with Rickettsia monacensis and Orientia tsutsugamushi. Am. J. Trop. Med. Hyg. 101, 332–335. doi: 10.4269/ajtmh.18-0631
Kuchinski, K.S., Loos, K.D., Suchan, D.M., Russell, J.N., Sies, A.N., Kumakamba, C., et al., Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats. eLife, (2022). 11:e79777. doi: 10.7554/eLife.79777.
Li, B., Si, H. R., Zhu, Y., Yang, X. L., Anderson, D. E., Shi, Z. L., et al. (2020). Discovery of bat coronaviruses through surveillance and probe capture-based next-generation sequencing. mSphere 5:e00807-19. doi: 10.1128/mSphere.00807-19
Lim, X. F., Lee, C. B., Pascoe, S. M., How, C. B., Chan, S., Tan, J. H., et al. (2019). Detection and characterization of a novel bat-borne coronavirus in Singapore using multiple molecular approaches. J. Gen. Virol. 100, 1363–1374. doi: 10.1099/jgv.0.001307
Lizewski, R. A., Sealfon, R. S. G., Park, S. W., Smith, G. R., Porter, C. K., Gonzalez-Reiche, A. S., et al. (2022). SARS-CoV-2 outbreak dynamics in an isolated US military recruit training center with rigorous prevention measures. Epidemiology 33, 797–807. doi: 10.1097/EDE.0000000000001523
Lu, C. T., Wang, L. S., and Hsueh, P. R. (2021). Scrub typhus and antibiotic-Resistantorientia tsutsugamushi. Expert Rev. Anti-Infect. Ther. 19, 1519–1527. doi: 10.1080/14787210.2021.1941869
Luce-Fedrow, A., Mullins, K., Kostik, A. P., St John, H. K., Jiang, J., and Richards, A. L. (2015). Strategies for detecting rickettsiae and diagnosing rickettsial diseases. Future Microbiol. 10, 537–564. doi: 10.2217/fmb.14.141
Mate, S. E., Kugelman, J. R., Nyenswah, T. G., Ladner, J. T., Wiley, M. R., Cordier-Lassalle, T., et al. (2015). Molecular evidence of sexual transmission of Ebola virus. N. Engl. J. Med. 373, 2448–2454. doi: 10.1056/NEJMoa1509773
Matranga, C. B., Andersen, K. G., Winnicki, S., Busby, M., Gladden, A. D., Tewhey, R., et al. (2014). Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 15:519. doi: 10.1186/s13059-014-0519-7
Millar, E. V., Rice, G. K., Elassal, E. M., Schlett, C. D., Bennett, J. W., Redden, C. L., et al. (2017). Genomic characterization of USA300 methicillin-resistant Staphylococcus aureus (MRSA) to evaluate Intraclass transmission and recurrence of skin and soft tissue infection (SSTI) among high-risk military trainees. Clin. Infect. Dis. 65, 461–468. doi: 10.1093/cid/cix327
Millar, E. V., Rice, G. K., Schlett, C. D., Elassal, E. M., Cer, R. Z., Frey, K. G., et al. (2019). Genomic epidemiology of MRSA infection and colonization isolates among military trainees with skin and soft tissue infection. Infection 47, 729–737. doi: 10.1007/s15010-019-01282-w
Morgulis, A., Gertz, E. M., Schäffer, A. A., and Agarwala, R. (2006). WindowMasker: window-based masker for sequenced genomes. Bioinformatics 22, 134–141. doi: 10.1093/bioinformatics/bti774
Nogueras, M. M., Roson, B., Lario, S., Sanfeliu, I., Pons, I., Anton, E., et al. (2015). Coinfection with "Rickettsia sibirica subsp. mongolotimonae" and Rickettsia conorii in a human patient: a challenge for molecular diagnosis tools. J. Clin. Microbiol. 53, 3057–3062. doi: 10.1128/JCM.00457-15
Nyatanyi, T., Koama, J. B., Rwunganira, S., Condo, J., Umulisa, I., Galgalo, T., et al. (2016). A mixed outbreak of epidemic typhus fever and trench fever in a youth rehabilitation center: risk factors for illness from a case-control study, Rwanda, 2012. Am. J. Trop. Med. Hyg. 95, 452–456. doi: 10.4269/ajtmh.15-0643
O'Flaherty, B. M., Li, Y., Tao, Y., Paden, C. R., Queen, K., Zhang, J., et al. (2018). Comprehensive viral enrichment enables sensitive respiratory virus genomic identification and analysis by next generation sequencing. Genome Res. 28, 869–877. doi: 10.1101/gr.226316.117
Olson, R. D., Assaf, R., Brettin, T., Conrad, N., Cucinell, C., Davis, J. J., et al. (2023). Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR, Nucl. Acids Res. 51, D678– D689. doi: 10.1093/nar/gkac1003
Paris, D. H., Aukkanit, N., Jenjaroen, K., Blacksell, S. D., and Day, N. P. J. (2009). A highly sensitive quantitative real-time PCR assay based on the groEL gene of contemporary Thai strains of Orientia tsutsugamushi. Clin. Microbiol. Infect. 15, 488–495. doi: 10.1111/j.1469-0691.2008.02671.x
Paris, D. H., Chattopadhyay, S., Jiang, J., Nawtaisong, P., Lee, J. S., Tan, E., et al. (2015). A nonhuman primate scrub typhus model: protective immune responses induced by pKarp47 DNA vaccination in cynomolgus macaques. J. Immunol. 194, 1702–1716. doi: 10.4049/jimmunol.1402244
Paskey, A. C., Frey, K. G., Schroth, G., Gross, S., Hamilton, T., and Bishop-Lilly, K. A. (2019). Enrichment post-library preparation enhances the sensitivity of high-throughput sequencing-based detection and characterization of viruses from complex samples. BMC Genomics 20:155. doi: 10.1186/s12864-019-5543-2
Perine, P. L., Chandler, B. P., Krause, D. K., McCardle, P., Awoke, S., Habte-Gabr, E., et al. (1992). A clinico-epidemiological study of epidemic typhus in Africa. Clin. Infect. Dis. 14, 1149–1158. doi: 10.1093/clinids/14.5.1149
R Core Team. (2022). R: a language and environment for statistical computing. R Foundation for Statistical Computing.
Rambaut, A., FigTree. Tree figure drawing tool. Available at: http://tree.bio.ed.ac.uk/software/figtree/, (2009).
Rozo, M., Schully, K. L., Philipson, C., Fitkariwala, A., Nhim, D., Som, T., et al. (2020). An observational study of Sepsis in Takeo Province Cambodia: an in-depth examination of pathogens causing severe infections. PLoS Negl. Trop. Dis. 14:e0008381. doi: 10.1371/journal.pntd.0008381
Sonthayanon, P., Chierakul, W., Wuthiekanun, V., Phimda, K., Pukrittayakamee, S., Day, N. P., et al. (2009). Association of high Orientia tsutsugamushi DNA loads with disease of greater severity in adults with scrub typhus. J. Clin. Microbiol. 47, 430–434. doi: 10.1128/JCM.01927-08
Sriwongpan, P., Krittigamas, P., Tantipong, H., Patumanond, J., Tawichasri, C., and Namwongprom, S. (2013). Clinical risk-scoring algorithm to forecast scrub typhus severity. Risk. Manag. Healthc. Policy 7, 11–17. doi: 10.2147/RMHP.S55305
Watt, G., Singhsilarak, T., Wongchotigul, V., Jiang, J., Leowattana, W., Looareesuwan, S., et al. (2005). Short report: detection of Orientia tsutsugamushi in clinical samples by quantitative real-time polymerase chain reaction. Am. J. Trop. Med. Hyg. 72, 640–641. doi: 10.4269/ajtmh.2005.72.640
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wylezich, C., Calvelage, S., Schlottau, K., Ziegler, U., Pohlmann, A., Höper, D., et al. (2021). Next-generation diagnostics: virus capture facilitates a sensitive viral diagnosis for epizootic and zoonotic pathogens including SARS-CoV-2. Microbiome 9:51. doi: 10.1186/s40168-020-00973-z
Wylie, T. N., Wylie, K. M., Herter, B. N., and Storch, G. A. (2015). Enhanced virome sequencing using targeted sequence capture. Genome Res. 25, 1910–1920. doi: 10.1101/gr.191049.115
Keywords: rickettsial pathogens, targeted enrichment, detection, characterization, qPCR, high throughput sequencing
Citation: Paskey AC, Schully KL, Voegtly LJ, Arnold CE, Cer RZ, Frey KG, Blair PW, Clark DV, Ge H, Richards AL, Farris CM and Bishop-Lilly KA (2024) A proof of concept for a targeted enrichment approach to the simultaneous detection and characterization of rickettsial pathogens from clinical specimens. Front. Microbiol. 15:1387208. doi: 10.3389/fmicb.2024.1387208
Edited by:
Manuel Rodriguez-Iglesias, University of Cádiz, SpainReviewed by:
Lina Gutiérrez, Pontifical Bolivarian University, ColombiaCopyright © 2024 Paskey, Schully, Voegtly, Arnold, Cer, Frey, Blair, Clark, Ge, Richards, Farris and Bishop-Lilly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberly A. Bishop-Lilly, S2ltYmVybHkuYS5iaXNob3AtbGlsbHkuY2l2QGhlYWx0aC5taWw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.