
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 09 April 2024
Sec. Food Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1375384
This article is part of the Research TopicInteractions between Bioactive Food Ingredients and Intestinal Microbiota, volume IIView all 16 articles
 Zhenghui Lan†Xueling Zhang†Meng Xu
Zhenghui Lan†Xueling Zhang†Meng Xu Junkai KongXuancheng ZuoYixuan WangChenxi WangYingdi Teng
Junkai KongXuancheng ZuoYixuan WangChenxi WangYingdi Teng Yongqing Ni*
Yongqing Ni* Yan Zhang*
Yan Zhang*Introduction: As prebiotics, oligosaccharides are frequently combined with Bifidobacterium to develop synbiotic products. However, a highly diverse gene repertoire of Bifidobacterium is involved in sugar catabolism, and even phylogenetically close species may differ in their sugar utilization capabilities. To further explore the mechanism underlying the differences in Bifidobacterium animalis subsp. lactis oligosaccharide metabolism.
Methods: This study screened strains with differential oligosaccharide metabolism. Subsequently, these strains were subjected to genome-wide resequencing and RT-qPCR.
Results: The resequencing results indicated that the subspecies of B. animalis subsp. lactis had a high genome similarity. The RT-qPCR results revealed that glycosidase genes exhibited consistency in the phenotype of metabolism at the transcriptional level; the better the growth of the strains on the oligosaccharides, the higher was the expression of glycosidase genes related to the oligosaccharides. Our results suggested that the differences in the gene transcription levels led to intraspecies differences in the ability of the strains to metabolize oligosaccharides even when they belonged to the same subspecies.
Discussion: Future studies with more sample size could generalizable the conclusion to all B. animalis subsp. lactis strains, thus would lay the theoretical foundation for the utilization of the B. animalis subsp. lactis strain as probiotics and the development of synbiotic products.
The members of the genus Bifidobacterium are common inhabitants of the gastrointestinal tracts of humans and other mammals, where they ferment several diet-derived carbohydrates that cannot be digested by their hosts (O'connell et al., 2013). Until date, most studies have investigated Bifidobacterium and product development, such as the transformation of plant isoflavones by Bifidobacterium, the production of Bifidobacterium-fermented milk, and microencapsulation of Bifidobacterium (Gaya et al., 2017; Lohrasbi et al., 2020; Yan et al., 2020). The strains of B. animalis subsp. lactis are well-known health-promoting probiotics that are used commercially (Bunesova et al., 2017). The B. animalis subsp. lactis genome also contains a large number of transporters that transport complex carbohydrates, as well as genes for intracellular or extracellular hydrolases that degrade the glycosidic bonds in complex carbohydrates, which, in turn, facilitates the cell’s uptake of these complex carbon-hydrates and provides the bacterium with the energy required for growth, thereby adapting to a complex environment. The genome sequence of B. animalis subsp. lactis Bl-04 reveals putative prebiotic transport and catabolic pathways, as reported elsewhere (Paineau et al., 2008), thereby implying that the bacterium is highly adapted to the gastrointestinal tract (GIT) and can utilize diet-derived complex oligosaccharides. Certain indigestible carbon sources (oligosaccharides) consumed daily stimulate the growth of Bifidobacterium in the gut, are commonly used as the main prebiotics (Hopkins et al., 1998). Changes in the dietary composition may affect health; therefore, it is important to study the ability of Bifidobacterium to utilize carbohydrates as a theoretical guide for the rational design of new symbiotic products (“prebiotic-probiotic”).
Oligosaccharides are carbohydrates formed by 2–10 monosaccharide molecules connected by glycosidic bonds. Owing to the rich variety of monosaccharide molecules that constitute oligosaccharides and the variety of intermolecular connection types and connection positions, there are several types of oligosaccharides in the nature. They can be broadly categorized into common oligosaccharides and functional oligosaccharides based on their biological functions. Common oligosaccharides mainly include lactose, sucrose, and maltose, which are easily digested and absorbed by the intestinal tract and thereby provide energy for the body’s metabolism, growth, and development (Vieira et al., 2020; Liu et al., 2023). Functional oligosaccharides, also known as non-digestible oligosaccharides, are difficult to digest and absorb because of the lack of an enzyme system in the human intestinal tract that directly degrades these functional oligosaccharides. The functional oligosaccharides developed so far mainly include xylo-oligosaccharides (XOS), galacto-oligosaccharides (GOS), fructo-oligosaccharides (FOS), inulin, and resistant starch (Tomás-Barberán and Mine, 2013; Belorkar and Gupta, 2016). Because functional oligosaccharides are not hydrolyzed by intestinal digestive enzymes but can be decomposed, absorbed, and utilized by beneficial microorganisms in the intestinal tract, they are often used in combination with Bifidobacteria to develop symbiotic products.
However, the main challenge of using Bifidobacterium cultures directly in the food is that different matrices and environments hinder their growth and survival in food (Moradi et al., 2021). To increase the survival rate of Bifidobacterium, combining probiotics and prebiotics (symbiotic) has become a research hotspot. Prebiotics are substrates that are selectively used by host microbes to confer a health benefit (Sanders et al., 2019). However, probiotic bacteria selectively ferment prebiotics comprising mainly non-digestible oligosaccharides (Abou et al., 2013). Therefore, research was needed to understand the factors and mechanisms that the Bifidobacterium exhibits intraspecies differences in oligosaccharide utilization The application of symbiotic products is limited by the fact that not all oligosaccharides are appropriate for all Bifidobacterium. The efficacy of specific combination warrants validation before use. A past study has shown that Bifidobacterium exhibits interspecies differences in oligosaccharide utilization (Garrido et al., 2015). For example, B. longum has been found to consume most oligosaccharides, and the B. bifidum strain utilizes GOS with degrees of polymerization (DP) of 2 and 3 and exhibits very limited use of FOS (Li et al., 2017). In addition, on examining three Bifidobacterium strains, the strains exhibited specificity in utilizing human milk oligosaccharides (LoCascio et al., 2007). The B. longum subsp. infantis are more inclined to have better capabilities to metabolize human milk oligosaccharides. The XOS, FOS, soya bean oligosaccharides, and malto-oligosaccharides are more effective in promoting the growth of B. adolescentis. However, surprisingly, the oligosaccharide utilization ability varies between different isolates of the same species (Garrido et al., 2015). The presence of intraspecies differences in Bifidobacterium creates further challenges when combining symbiotic products and using Bifidobacterium. Ang-Xin Song et al. recently reported strain-specific utilization of wheat arabinoxylan by B. longum (Song et al., 2020). Furthermore, a study demonstrated that several endogalactanase-positive (GalA+) B. breve strains can utilize purified GOS (PGOS) components with a high DP, whereas endogalactanase-negative (GalA−) strains cannot. This intraspecies difference has been ascribed to galA present in B. breve strains (O'Connell Motherway et al., 2013). Bifidobacterium strain specifically utilizes oligosaccharides; such discoveries have generated substantial interest in the scientific community. The characteristics of the strains must thus be considered while producing symbiotic products. While synthesizing symbiotic products, specific prebiotics must be designed to pair with specific probiotics that will consume them and exhibit improved functional outcomes. Therefore, revealing the mechanisms underlying Bifidobacterium intraspecies differences in the metabolism of oligosaccharides and explaining the regularity of these differences are highly warranted for the development of effective synbiotics.
Most of the current studies are based on genotype–phenotype to investigate interspecies differences in the metabolism of functional oligosaccharides by Bifidobacterium (Bottacini et al., 2018; He et al., 2021). In contrast, the genomic structures of B. animalis subsp. lactis strains are similar, therefore there is a lack of differences in the metabolism of functional oligosaccharides by Bifidobacterium that can be analyzed from the transcriptional level.
The present study focused on B. animalis subsp. lactis obtained from human fecal samples of two regions with significant dietary differences. Then, we investigated their strain specificity in terms of the oligosaccharide-utilization capacity at the subspecies level. Using genome resequencing technology explored the key loci associated with oligosaccharide metabolism, combined phenotypic and genotypic data, and identified gene sequences significantly associated with differences in the oligosaccharide metabolism. RT-qPCR was performed to determine the differential gene expression during the growth of B. animalis subsp. lactis on oligosaccharides. The purpose of this study was to explore differences in the utilization phenotypes of different carbon sources, and to associate possible gene clusters for revealing the strain metabolism difference of functional oligosaccharides in B. animalis at the subspecies level and provides a theoretical basis for the study of B. animalis subsp. lactis and the application of synbiotic products.
FOS, GOS, XOS, isomalto-oligosacchrides (IMO), raffinose, stachyos, inulin, resistant starch (RS), and glucose (Glu) were all purchased from Shanghai Yuanye Bio-Technology Co., Ltd. All other chemicals used were of analytical grade.
In sterile containers, we collected the fecal samples of mothers and their infants residing in Hotan, Xinjiang, China and Hainan, China. The samples were collected at the participants’ homes and immediately frozen (−20°C) in a portable freezer. These samples were then transported to the laboratory and processed immediately upon receipt. The participants were not on any special diet, and, as instructed, had not taken probiotics or antibiotics for 2 months before sampling.
The samples were serially diluted (10−1 to 10−4) in an anaerobic chamber, and 0.1 mL of aliquot of the 10−2, 10−3, and 10−4 dilutions were plated on Bifidobacterium specific mMRS agar. The medium (per liter) was composed of yeast extract, 5 g; peptone, 10 g; beef paste, 10 g; Tween 80, 1 mL; sodium acetate anhydrous, 5 g; ammonium citrate dibasic, 2 g; K2HPO4, 2 g; MgSO4, 0.58 g; MnSO4, 0.25 g; glucose, 20 g; L-cysteine, 0.5 g (Blotopped, China); mupirocin, 0.05 g (Bomei, Hefei, China); nystatin, 0.025 g (Bomei, China), and powdered agar, 20 g (Ventura et al., 2006; Linares-Morales et al., 2022). All other reagents involved in the composition of the culture medium were purchased from Sinopharm Chemical Reagent Beijing Co., Ltd., China. These plates were subsequently incubated under an atmosphere of 10% H2, 10% CO2, and 80% N2 in an anaerobic chamber (DG520; DWS UK) and incubated for 48 h at 37°C (Shahin et al., 2003). All colonies appearing on the dilution plates were picked in succession and inoculated into mMRS broth.
The sample DNA was isolated from 1 mL of culture by using the FastPure Bacteria DNA Isolation Mini Kit (Vazyme, Nanjing, China) according to the manufacturer’s protocol. The purity and concentration of isolated DNA were measured using a micro nucleic acid quantitative instrument (Thermo Scientific, United States). DNA with a 260/280 nm ratio of >1.8 was used as the template DNA, and its integrity was determined using 1.2% agarose gels. Next, the strains were identified by conducting molecular biology assays, including 16S rRNA gene sequencing of genomic DNA and Bifidobacterium subspecies-specific PCR (aptD gene). PCR amplification of the 16S rRNA gene was performed with primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492r (5′-CTACGGCTACCTTGTTACGA-3′) (Nomoto et al., 2017). To further identify the isolated strains, atpD was PCR amplified with primers atp-1 (5′-CACCCTCGAGGTCGAAC-3′) and atp-2 (5′-CTGCATCTTGTGCCACTTC-3′) (Ventura et al., 2004). The PCR-amplified products were detected through 1.2% agarose gel electrophoresis and sequenced using GENEWIZ (Suzhou Jinweizhi Biotechnology Co., Ltd., China). The sequencing results were submitted to the GenBank of the National Center for Biotechnology Information (NCBI) for BLAST homology comparison. A phylogenetic tree was constructed using MAGE 7.0 software. For repetitive-element PCR (rep-PCR), the strain was grown on MRS broth under anaerobic conditions for 48 h, and DNA was extracted as described earlier. Rep-PCR was conducted to generate genomic amplification products of the test strains. Specifically, rep-PCR amplification was performed using the Tc-512 PCR gene amplification instrument (Techne, British) and BOXAIR primers (Masco et al., 2003). The gel was visualized and photographed under a UV transilluminator (Quantum CX5, France). The separation of rep-patterns was processed with CelCompar ‖ version 6.6 (Applied Maths, Sint-Matenslatem, Belgium) and analyzed using the Pearson’s correlation coefficient and the unweighted pair group method with arithmetic mean (UPGMA) clustering algorithm.
After 48 h of culture, the Bifidobacterium cells were centrifuged at 5000 xg for 5 min and washed twice with 0.9% normal saline. The cell suspensions (approximately 106 CFU/mL) were used to prepare bacterial inoculants for the carbohydrate fermentation experiments.
The in vitro utilization experiment was conducted using oligosaccharides (FOS, GOS, XOS, IMO, raffinose, stachyos, inulin, RS) as the unique carbon source in a 96-well microplate (Kan et al., 2020). The carbohydrate solution with 1% final concentration was added to the medium, thereby replacing glucose as a carbon source. Four microliters of each resulting suspension were inoculated into 200 μL of the modified medium. The experiment was conducted thrice with triplicate wells (He et al., 2021). The medium in which glucose was added instead of oligosaccharides was used as the positive control, whereas the medium without any carbon source was used as the negative control. Moreover, 0.05% (w/v) L-cysteine was added to the medium to consume free oxygen and promote strain growth and reproduction. The cells were incubated at 37°C in an anaerobic chamber for 48 h. Each carbon source fermentation experiment was monitored by assessing OD at 600nm by using a microplate reader (BioTek Instruments Inc., United States).
The optical density (OD) obtained for each strain grown on different substrates was compared with that obtained for each strain in the absence of a sugar source. This difference in OD (ΔOD) was used as a parameter for evaluating each strain’s ability to grow on different substrates.
The strains were cultured under anaerobic conditions in MRS broth containing 0.05% [v/w] L-cysteine hydrochloride at 37°C for 36 h. Chromosomal DNA was extracted from pure bacterial cultures by using the FastPure Bacteria DNA Isolation Mini kit. After DNA was extracted, DNA quantity and purity were assessed using the NanoDrop ND-2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, United States).
Then, 1 μg of genomic DNA was used to generate the sequencing libraries for each strain, followed by sequencing with the Illumina NovaSeq PE150 (Shanghai Biotree Biomedical Technology Co., Ltd.). The sequencing libraries were generated using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s recommendations, and the index codes were incorporated to attribute sequences to each sample. Briefly, the DNA sample was fragmented through sonication to achieve a size of 350 bp. The DNA fragments were then end-polished, A-tailed, and ligated with the full-length adaptor for Illumina sequencing, followed by PCR amplification. Finally, PCR products were purified (AMPure XP system). Libraries were analyzed for size distribution by using the Agilent 2,100 Bioanalyzer and quantified through real-time PCR.
After sequencing, sequence adapters, low-quality bases from paired reads, and reads with an average quality of <20 (<Q20) were first trimmed and filtered using fastp. Then, reads that passed quality-control filtering were aligned and assembled using SPAdes v3.15.3 with default k-mer sizes. Each assembly was annotated using prokka. The pairwise average nucleotide identity (ANI) values were calculated using pyANI v0.2.9 with default BLASTN+ settings (cut-off: ≥95% identity). Roary v.3.12.0 was used to obtain core genome data and perform multiple sequence alignment. The core genome phylogenetic tree was constructed using FastTree v2.1.91 with the GTR model having 1,000 bootstrap iterations. Whole-genome sequence alignments for single nucleotide polymorphisms (SNPs) and indel identifications were performed using Snippy v3.13. Publicly available gene sequences of B. animalis type strains were downloaded from the NCBI Genome database. All Bifidobacterium genomes were input into dbCAN2 v2.0.1 [47] to annotate carbohydrate utilization-related genes based on the carbohydrate-active enzymes database (amino acid identity ≥30%, E-value ≤1 × 10−5). The t-test function implemented in SPSS v26 was used to calculate statistically significant differences between the average numbers of glycosyl hydrolase (GH) genes belonging to the predominant GH families (p < 0.05).
Total RNA was isolated from 1 mL of B. animalis subsp. lactis cells (approximately 1 × 108 CFU/mL) in the log-phase cultures grown on oligosaccharides or glucose as the sole carbohydrate source. The samples were centrifuged at 8,000 rpm for 1 min at 4°C on a bench centrifuge to collect cell pellets. The pellets were washed twice with 500 μL of DEPC-treated ddH2O to remove the culture medium and centrifuged at 10,000 rpm. To disrupt the collected cell pellets, they were ground in liquid nitrogen. The Spin Column Bacteria Total RNA Purification Kit (Sangon Biotech) was used for extracting the total RNA. The extracted RNA was eluted in DEPC-treated ddH2O. RNA degradation and contamination were monitored through 1.2% agarose gel electrophoresis. The electrophoretic results of the extracted RNA revealed that the bands of 23S and 16S rRNAs were bright and clear and that of 5S rRNA was darker. This finding indicated that the RNA extracted from the strains had good integrity, was less degraded, and could be used for the follow-up experiments. The RNA sample concentration was measured using the micro nucleic acid quantitative instrument to calculate the volume required for reverse transcription. The RNA samples were then immediately stored at −80°C until further use.
Reverse transcription was conducted using the PerfectStart® Uni RT&qPCR Kit (TransGen Biotech, China). The expression of oligosaccharide metabolism-related differential genes was detected through three RT-PCR replicates. Specific primers for each gene (Table 1) were designed using Primer Premier 6 software. The experiments were conducted using the Real-Time Q-PCR System (MX3000P, Stratagene, United States) in combination with the PerfectStart® Uni RT&qPCR Kit and the PerfectStart® Green qPCR SuperMix (TransGen Biotech, China).
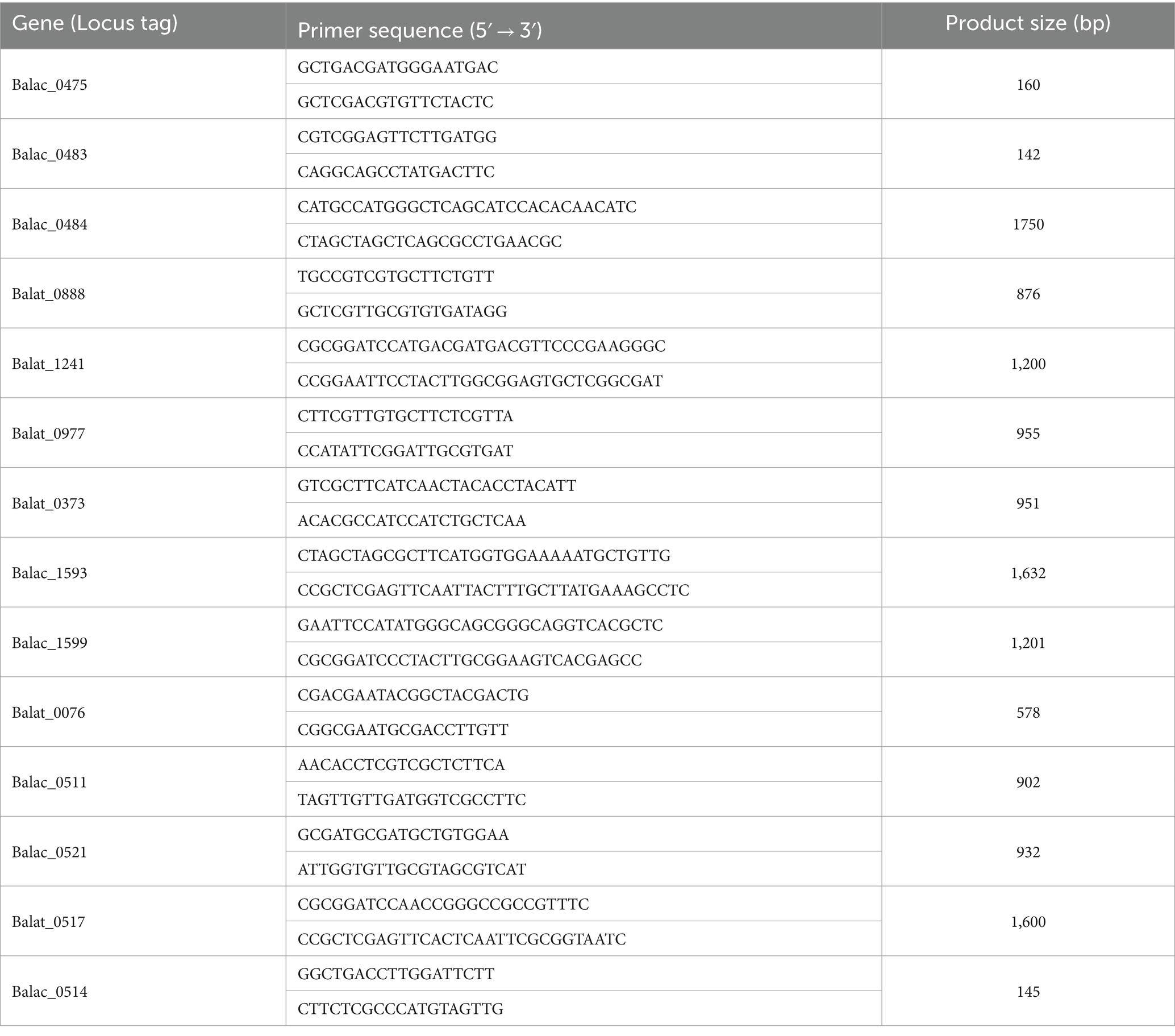
Table 1. Target gene oligonucleotide primers for RT-qPCR.
The gene expression was normalized using the ∆∆CT method, and groEL was used as the reference gene for the calculations. The control group consisted of strains cultured in MRS medium containing glucose as the carbon source, while the experimental group consisted of strains cultured in MRS medium containing each oligosaccharide as the carbon source. The experimental results were visualized by plotting heatmaps.
All experiments were three biological replicates. The data were statistically analyzed using SPSS V26 (IBM, Armonk, NY, United States). Data for all variables were normally distributed and allowed for parametric tests of significance. Equal variance data were analyzed by ANOVA, followed by Duncan’s multiple range test with 95% confidence intervals; the differences were considered significant at p < 0.05. Evolutionary trees were analyzed by MEGA7.0 software. A heatmap and the principal component analysis (PCA) were constructed in R software (ver 4.0.4).
In total, 68 Bifidobacterium strains were obtained from different sources of fecal samples (Hotan Prefecture and Hainan Province) and 9 strains were screened through the rep-PCR and carbohydrate utilization, and the 16S rRNA gene sequence of 9 isolates was determined and compared with the published sequences obtained from the GenBank nucleotide database by using the BLAST algorithm. All 16S rRNA gene sequences reported in this study have been deposited at GenBank (Figure 1). Because our phylogenetic analysis based on 16S rRNA gene sequences revealed that all strains possessed high sequence similarities (99.5%) to B. animalis subsp. lactis, the isolates were tentatively identified as B. animalis subsp. lactis.
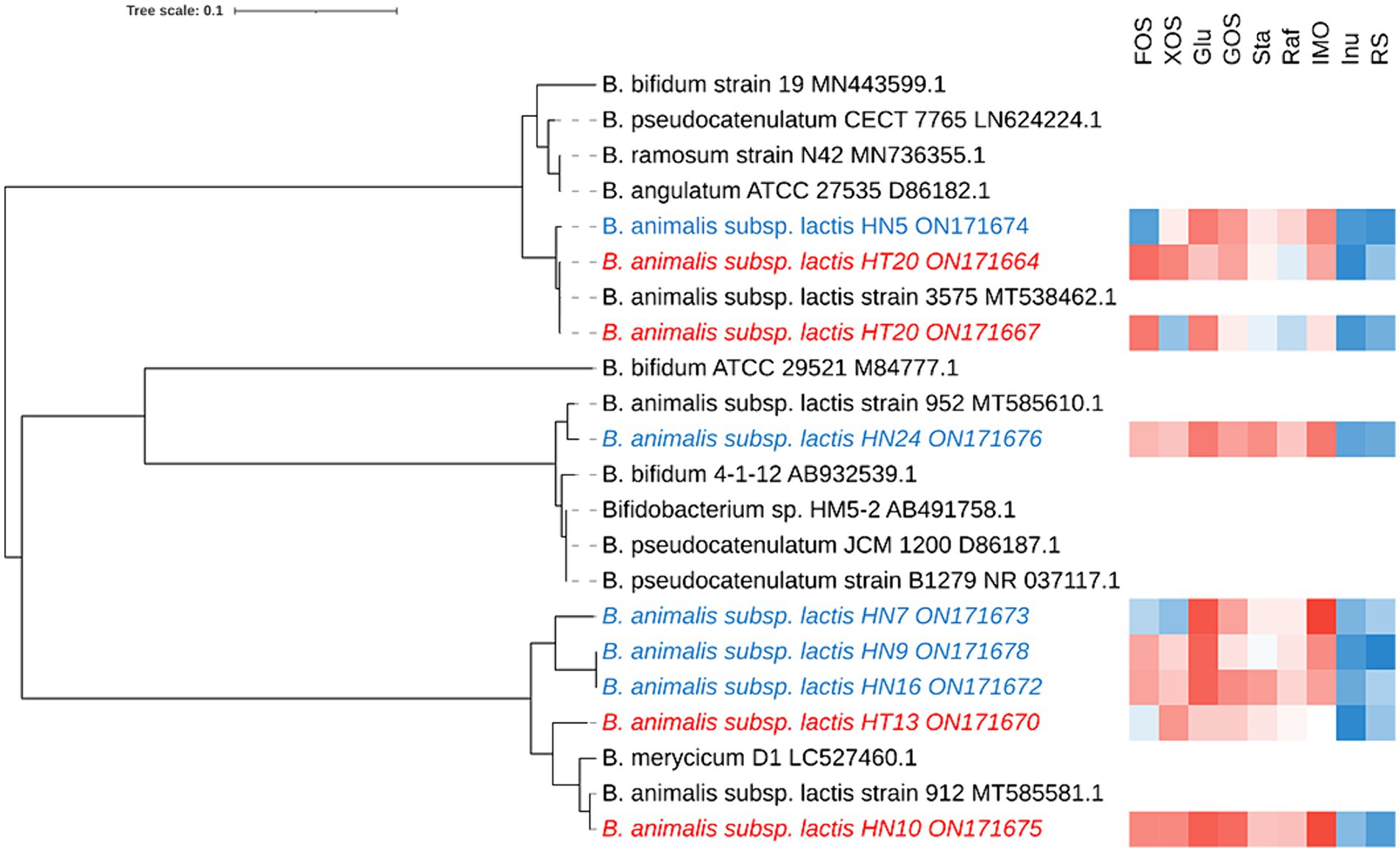
Figure 1. Phylogenetic tree and the functional oligosaccharide metabolism of B. animalis subsp. lactis strains. The color tending to red indicates that the strain grown better in the associated oligosaccharides, while that tending to blue indicates grown poorly in the associated oligosaccharides.
To investigate the intraspecies differences in oligosaccharide metabolism, we monitored the growth of B. animalis subsp. lactis strains on modified Rogosa medium containing 1% different oligosaccharides as the sole carbohydrate source. The results are shown in Figure 1, where strain-specificity exists in the utilization of sugars by different strains, but the general trend is that all strains exhibited vigorous growth on media containing FOS, XOS, Glu, GOS, raffinose, stachyose, and IMO as the sole carbon source. The higher growth performances of B. animalis subsp. lactis strains were noted on the media containing FOS, XOS, GOS, and IMO. HN5 exhibited poor growth on the medium containing FOS as the sole carbon source. HT20 and HN7 exhibited poor growth on the XOS-containing medium, whereas the other strains reached a high OD. When compared with FOS, XOS, GOS, and IMO, the strains grew moderately in raffinose and stachyose. Although most strains could utilize oligosaccharides, two exceptions were the utilization patterns of inulin and RS, where low or poor growth was observed in a few cases. HN5 and HN9 failed to grow in the media containing inulin and RS. All isolates from Hotan failed to grow in the inulin-containing medium. HN10 failed to grow in the RS-containing medium.
In order to assess whether there were area differences in oligosaccharide metabolism among the resequenced strains, PCA analyzed experimental data on carbohydrate utilization. As depicted in the PCA diagram (Figure 2), the strains were clustered depending on the area. The results demonstrated that the selected strains from different areas exhibited differences in metabolizing functional oligosaccharides. The strains from Hainan (HN9, HN10, HN16, and HN24) and the strains from Hotan (HT12, HT13, and HT20) were clustered separately.
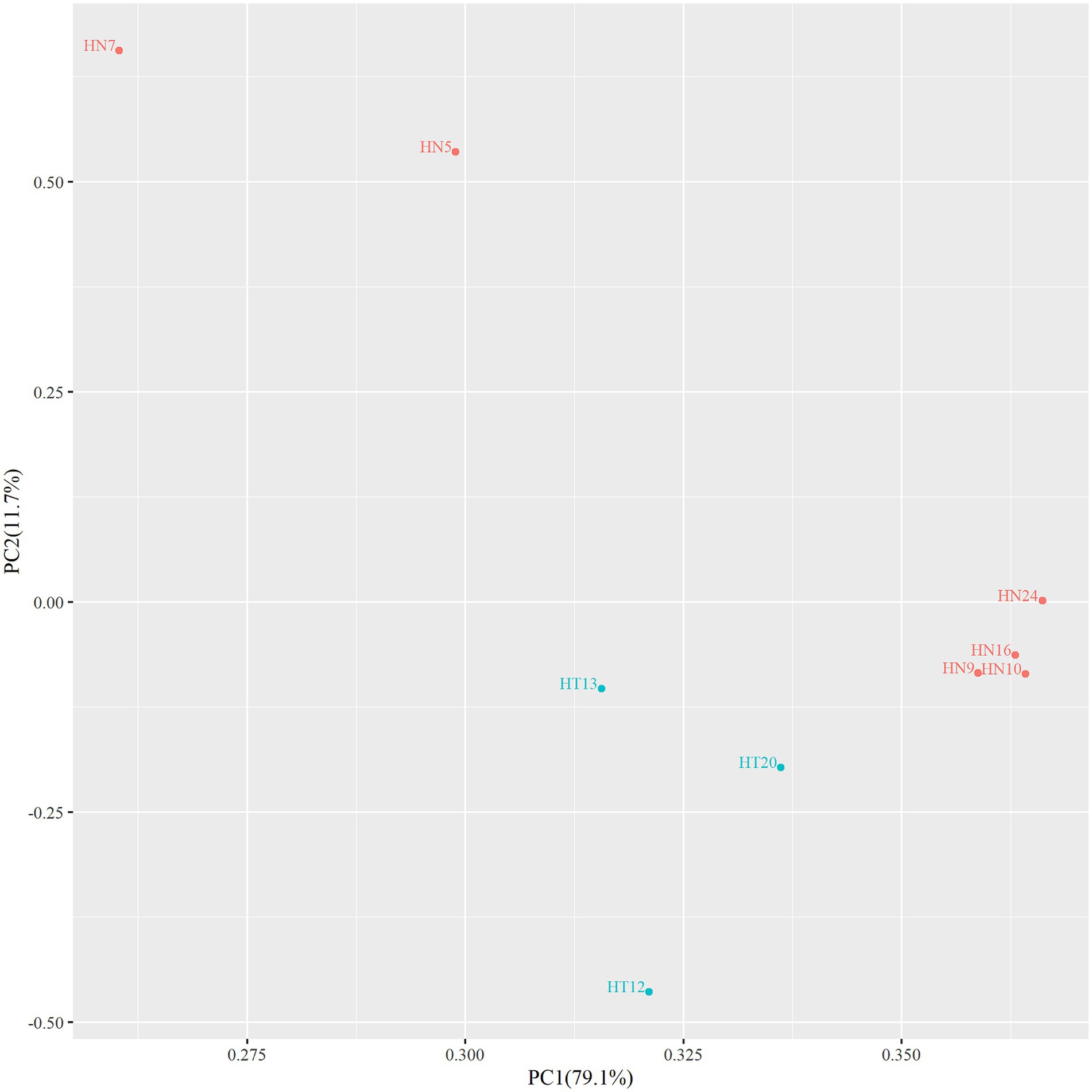
Figure 2. PCA diagram of functional oligosaccharide metabolism of resequenced strains.
To better comprehend the differences in the B. animalis subsp. lactis genomes, we resequenced 9 strains from China representing various geographical regions, including 3 strains from Hotan and 6 from Hainan. The results are shown in Table 2, and the genome size predicted for the strains ranged from 1.90 to 2.21 Mb. The genomes had an average G + C% content of 60.32%, an average predicted CDS number of 1,576, and the number of tRNA genes ranging from 53 to 59. In total, 1,452 core genes, 23 shell genes, and 171 cloud genes were identified in the B. animalis subsp. lactis genomes. The core genes, softcore genes, shell genes, and cloud genes were present in 99–100%, 95–99%, 15–95%, and 0–15% of the strains, respectively. The average nucleotide identity values were defined as the average base similarity between homologous segments of two microbial genomes. Nucleotide-level genomic similarities between the 9 genomes were determined using pyANI. The results indicated that B. animalis subsp. lactis ANI values ranged from 99.98 to 99.99% (Supplementary Table S1), indicating higher levels of sequence identity among all strains.
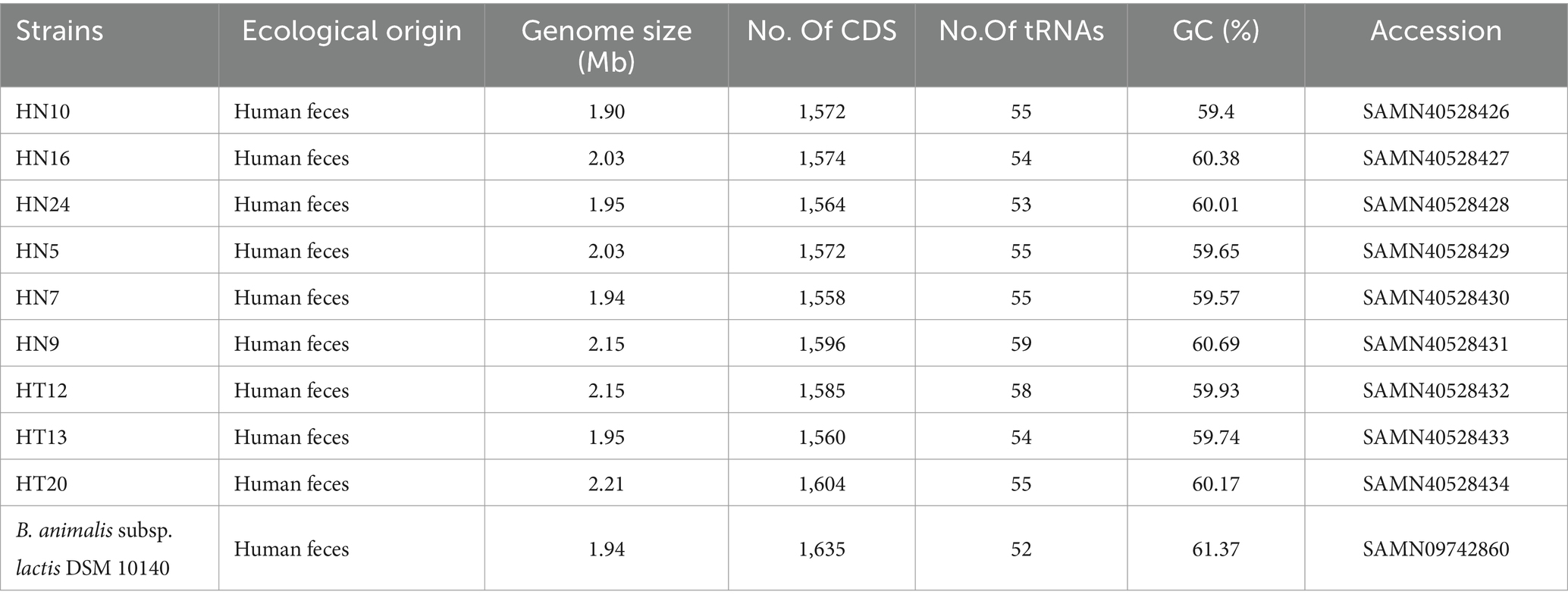
Table 2. Genome information for publicly available B. animalis subsp. lactis DSM 10140 and nine B. animalis subsp. lactis strains used in this study.
We further searched for the CAZymes profiles in B. animalis subsp. lactis genome. Of the 1736 gene families in the pangenome, 233 were determined to encode CAZymes, constituting 13.42% of functionally annotated genes, which reflects the saccharolytic character of the B. animalis subsp. lactis. These CAZymes included 315 glycosyl hydrolase (GH), 180 glycosyltransferases (GT), 36 carbohydrate esterases (CE), and 18 carbohydrate-binding motif-containing proteins (Figure 3). The most abundant CAZymes were GH13 (12 ± 0% GH genes per genome), followed by GT2 (8 ± 0% GH genes per genome), and GH43 and GH3 (3 ± 0% GH genes per genome). All these CAZymes encode enzymes associated with the hydrolysis plant-derived poly- and oligosaccharides, such as stachyose, raffinose and xylans, etc. And these poly- and oligosaccharides are major components of the adult diet. Studies have shown that the CAZymes profiles in Bifidobacteria genome are species and/or strain-specific, however, according to bioinformatic predictions, strain-specific CAZymes were not observed in this study (Genomics of the Genus Bifidobacterium Reveals Species-Specific Adaptation to the Glycan-Rich Gut Environment). On the contrary, regardless of the isolation source, all tested strains shared the same CAZyme repertoires.
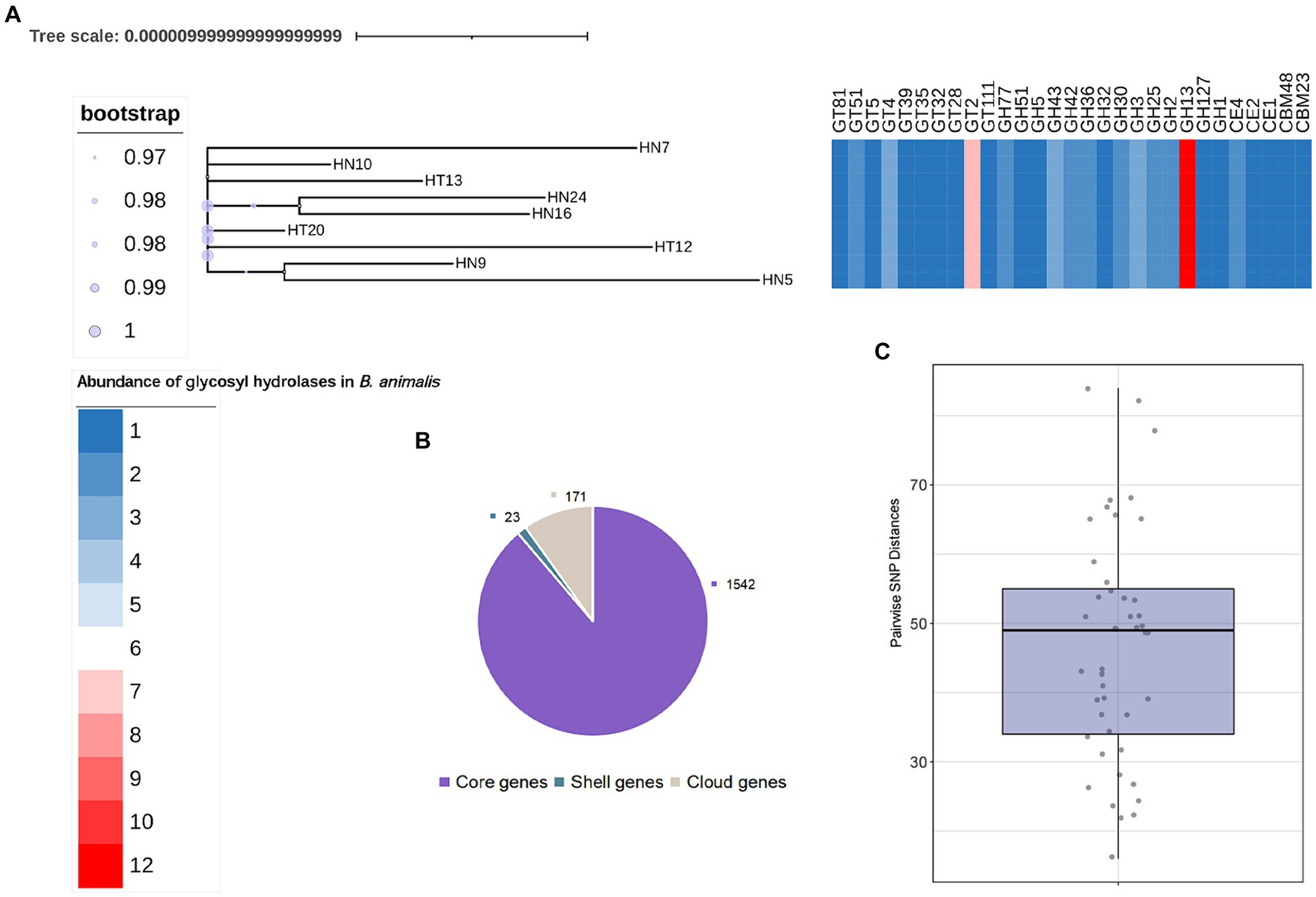
Figure 3. General Features of B. animalis subsp. lactis Genomes. (A) The maximum-likelihood trees were constructed based the core-genome alignment employing ‘GTR’model with 1,000 bootstraps. Heatmap represents abundance of specific GH families. See also Supplementary Table S2. (B) Pan- and core genes of B. animalis subsp. lactis. (C) Pairwise SNP Distances between B. animalis subsp. lactis. Strains.
We then explored the nucleotide-level differences in GH genes between the B. animalis subsp. lactis strains. The SNP analysis was conducted to compare the genomes of the 9 B. animalis subsp. lactis strains and B. animalis subsp. lactis DSM10140. Upon comparison of these 10 complete genome sequences, 493 validated SNPs were identified, and found that the B. animalis subsp. lactis strains exhibited low strain diversity, with the respective mean pairwise SNP distances of 46.73 ± 16.4 SNPs. Subsequently, these SNPs were functionally annotated. The results revealed that these SNPs were significantly associated with the transport and the metabolisms of carbohydrates (oligosaccharide). However, we found no significant SNPs that may cause functional changes in CAZyme genes. Interestingly, the experimental strains exhibited phenotypic differences in metabolizing different oligosaccharides. Therefore, we predicted that these SNPs were noncoding variants. Transcription factors play crucial roles in gene regulation by harboring these SNPs (Li et al., 2020).
Thus, to reveal the phenotypic differences in the B. animalis subsp. lactis strains in metabolizing different oligosaccharides at the transcription level, total RNA isolated from the experimental strains was reverse-transcribed into cDNA, and then, the expression of relevant key enzyme genes was detected through real-time quantitative PCR (RT-qPCR). In the culture medium containing different functional oligosaccharides, the expression of the carbohydrate transporter gene changed to varying degrees at the mRNA level (Figure 4). The composition of oligosaccharides and monosaccharides are similar. However, their carbon chain lengths are different, and thus, they can induce the expression of the same or similar genes. On the other hand, a substrate can induce the expression of several carbohydrate metabolism genes. In this study, The results showed that the OD600nm values of HN9, HN10, and HN16 strains were greater than 0.8 after 48 h of incubation in medium with FOS as the carbon source, and their relative expression of glycosidase genes (Balat_1241 and Balac_0888) related to FOS was higher than that of HN7, HT13, and HN5 (the OD600nm values were 0.5 ± 0.04, 0.61 ± 0.01, and 0.26 ± 0.02, respectively). For GOS metabolism, the HT12 and HT13 strains exhibited a strong ability to metabolize than HN9, HT20, and the expression of genes (Balat_0483, Balat_0484, Balat_0475) related GOS metabolism higher than HN9, HT20 strains. The OD600nm vanlues of HT12, HN24, HN7 and HT20 in the medium with XOS as carbon source were 1.05 ± 0.06, 0.87 ± 0.02, 0.42 ± 0.05 and 0.42 ± 0.05 respectively, and the expression levels of XOS-relevant metabolic genes (Balat_0517, Balac_0511, Balac_0514, and Balac_0521) of HT12 and HN24 higher than HN7 and HT20. However, the sample size of the resequencing and reverse transcription validation experimental strains in this study was small, and if further confirmation of intraspecies differences in metabolized oligosaccharides within the B. animalis subsp. lactis caused by transcriptional rather than the genetic levels is desired, the sample size of the experiments should be increased in subsequent studies in order to overcome the limitations caused by small sample sizes. And generalization of the conclusions to all strains of B. animalis subsp. lactis.
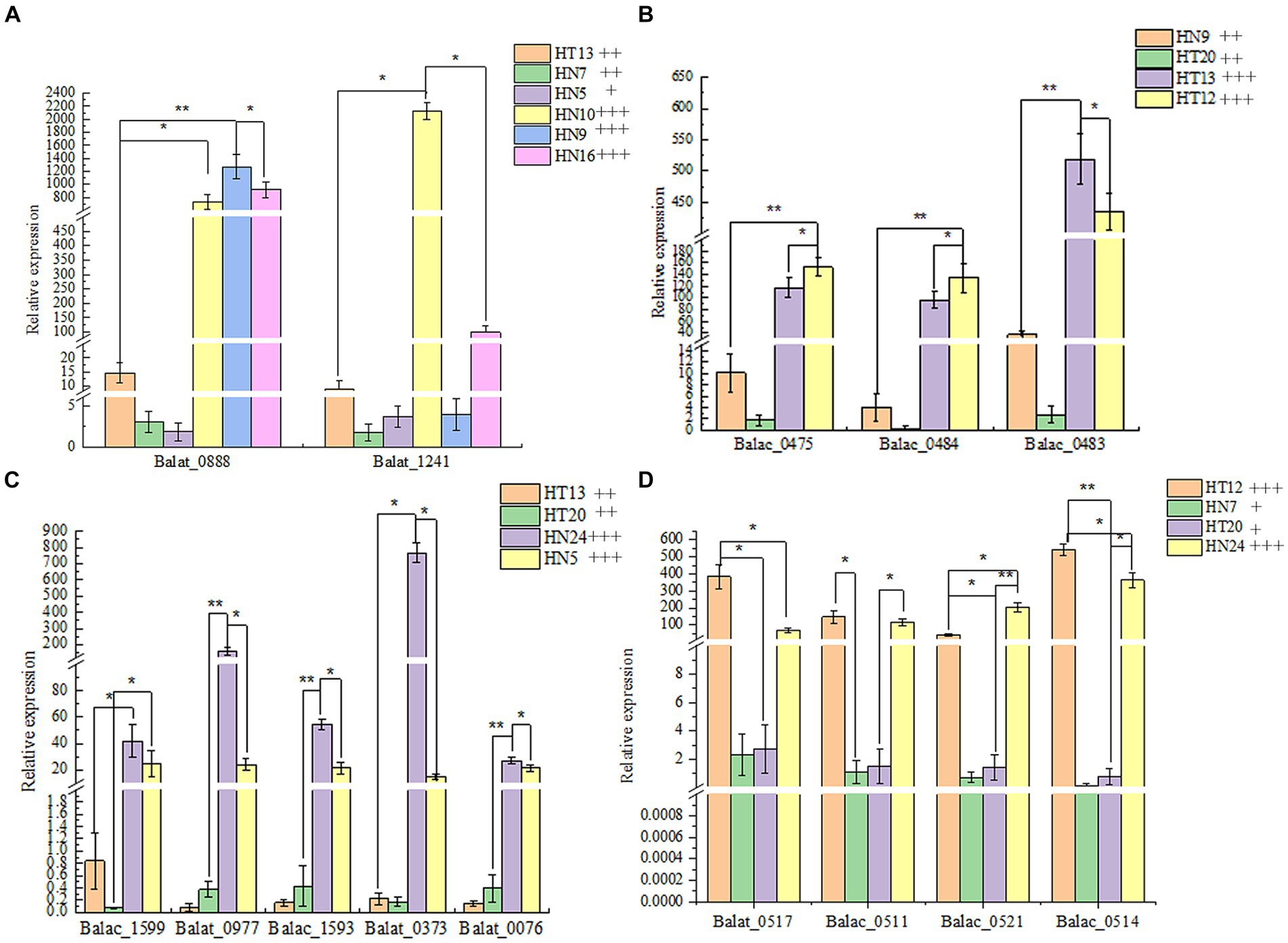
Figure 4. The relative expression of glycosidase genes in four oligosaccharides as the unique carbon source. (A–D): Metabolic phenotypes of FOS, GOS, IMO and XOS and mRNA transcription levels of related genes by strains, respectively. The phenotype of metabolism was classified by the following labels: “+” OD600nm 0.25–0.5, low- level growth; “++” OD600nm 0.5–0.8, medium-level growth; “+++” OD600nm >0.8, high-level growth.
B. animalis subsp. lactis are widely distributed in humans and other mammalian guts and are widely supplemented to multiple functional foods. Furthermore, B. animalis subsp. lactis possesses a saccharolytic lifestyle as well as the ability to utilize multiple oligosaccharides from a variety of host and dietary sources. Multi-omics techniques and the increased number of published Bifidobacterium sequences are beneficial for analyzing different strains of genomic multiplicity and the difference in functionality (Andersen et al., 2013; O'Connell Motherway et al., 2013; Nomoto et al., 2017). In this study, we explored the differences in the functional oligosaccharide metabolism of B. animalis subsp. lactis from three levels: phenotypic characteristics, gene level, and transcription level, and then analyzed the preference and utilization of different oligosaccharides by different strains so as to provide a theoretical reference for the development of suitable synbiotic (“probiotics-prebiotic”) products.
In this study, B. animalis subsp. lactis were recovery from Hotan, Xinjiang, China and Hainan, China. The results of PCA analysis showed regional variability in the utilization of polysaccharides by B. animalis subsp. lactis isolated from Hotan and Hainan regions. It could be due to the growth of the Bifidobacterium strains may be affected by the environment and eating habits. According to the accumulating evidence, intraspecies differences might occur because of the human gastrointestinal environment (Holscher, 2020). Diet is considered the main environmental contributor to the gastrointestinal microbiota structure and function in humans and animals; therefore, can markedly influence the functionality of indigenous microbiota (Gibson et al., 2004; Hansen et al., 2018). This study resequenced analyzing the strains to explore the mechanism of metabolic oligomerization differences in B. animalis subsp. lactis.
Studies have shown that about 14% of the genes of Bifidobacteria genome are involved in carbohydrate metabolism (Genomics of the Genus Bifidobacterium Reveals Species-Specific Adaptation to the Glycan-Rich Gut Environment). The predominant CAZymes class found in this study were GH43, GT2, GH3, and GT13, which were responsible for the metabolism of multiple plant-derived carbohydrates (Lugli et al., 2019; Liang et al., 2023; Srivastava et al., 2023). In line with this result, our in vitro experiments showed that B. animalis subsp. lactis could assimilate several plant-derived carbohydrates, such as IMO, GOS, and XOS, which may explain why B. animalis subsp. lactis could efficiently colonize several animal guts. According to B. animalis subsp. lactis glycobiome, all of the tested strains shared the same CAZymes profile. Nevertheless, our analysis detected strain-specific variation in carbohydrate utilization within 9 B. animalis subsp. lactis.
However, oligosaccharide utilization capacities of the B. animalis subsp. lactis strains differed because of the difference in the gene transcription level. In several studies, the Bifidobacterium genome functional annotation results revealed that most of the functions encoded by genes were related to carbohydrate metabolism (Milani et al., 2016). The oligosaccharide metabolism experiment combined with the PCR validation of functional genes indicated that the experimental strains could use 5–6 types of oligosaccharides and have genes encoding key enzymes involved in functional oligosaccharide metabolism. This finding suggests that the carbohydrate-utilizing ability of Bifidobacterium is crucial for its life activities. The study results are similar to those reported by Janer et al. (2004). In this past study, all experimental strains (B. animalis subsp. lactis) had genes encoding key enzymes involved in inulin metabolism, as determined through PCR amplification of the gene for β-fructosidase. However, almost none of the strains could metabolize inulin. This finding suggests that although strains possess key enzyme genes for metabolizing functional oligosaccharides, this finding does not confirm that they can surely metabolize relevant oligosaccharides, which could be because the associated genes are not expressed or less expressed. Global gene expression profiles were obtained for 9 B. animalis subsp. lactis, growing on 4 potential prebiotic oligosaccharides (FOS, GOS, XOS and IMO). RT-qPCR results unveiled that differentiation of gene expression regulation influences metabolic capabilities. HN9, HN10 and HN16 upregulated the expression of glycosyltransferase (Balat_0888) and β-fructofuranosidase (Balat_1241) which are allows to uptake FOS. HN24 and HN5 were observed to utilize IMO via upregulated ABC transporter (Balac_1599), isoamylase (Balat_0977), GH13_311, 6-α-Glucosidase (Balac_1593), 4-α-glucanotransferase (Balat_0373), and glucan phosphorylase (Balat_0076). Transporter differentially up regulated on XOS was also found in HT24, HN7, HT12 and HT20. Glycosidase genes exhibited consistency in terms of phenotype of functional oligosaccharide metabolism at the transcriptional level.
Similarly, the 16th International Symposium on Probiotics and Health reported that probiotics B. animalis subsp. lactis Probio-M8 and V9 shared similar carbohydrate metabolism-related genes, albeit their phenotypes of carbohydrate metabolism varied remarkably. As noted in the aforementioned report, our study results suggest that all experimental strains (B. animalis subsp. lactis) had similar genes related to the key enzymes involved in functional oligosaccharide metabolism, but their phenotypes of functional oligosaccharide metabolism were different. The difference in the phenotypes of metabolism may be caused by the difference in the gene transcription level and the protein translation level of relevant key enzymes. Differences were noted in the gene transcription of Saccharomyces cerevisiae in the presence of different long-chain fatty acids (Han Li et al., 2019). Moreover, the addition of glycerol effectively increased the transcription level of the lactone ring synthesis-related gene in Streptomyces ambofaciens. The relative transcript level of related genes unveiled that the increase in the oxygen supply level effectively promoted the relative transcription level of the forosamine synthesis-related gene. Thus, it was confirmed that the external growth environment can affect gene expression.
In this study, B. animalis subsp. lactis strains were isolated from human fecal samples collected from participants from across Hotan and Hainan. Comparison and analyses of their abilities were performed to utilize 8 types of functional oligosaccharides as well as their differences at the gene and transcription levels. The results indicated that, on one hand, the selected strains from different areas exhibited differences in metabolizing functional oligosaccharides. On the other hand, the ability of the strains to utilize the oligosaccharides was consistent with the transcript levels of the related glycosidase genes. This study provided a case for understanding functional diversity due to small gene expression changes within the B. animalis subsp. lactis. And the synbiotic formulations could be optimized through uncovered gene expression impacts phenotypic variations in B. animalis subsp. lactis. Subsequent studies could design targeted interventions through understanding the regulatory pathways involved in gene expression, thus developing more effective synbiotic products. This study may provide new ideas for the development of targeted bifidogenic functional oligosaccharides, for specific probiotic strains.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
ZL: Formal analysis, Writing – original draft, Writing – review & editing, Validation, Visualization. XZh: Formal analysis, Writing – original draft, Writing – review & editing, Data curation, Software. MX: Data curation, Methodology, Writing – original draft. JK: Writing – review & editing, Investigation. XZu: Investigation, Writing – review & editing. YW: Writing – review & editing, Data curation. CW: Writing – review & editing, Validation. YT: Writing – review & editing, Formal analysis. YN: Writing – review & editing, Resources. YZ: Resources, Writing – review & editing, Conceptualization, Funding acquisition, Project administration.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science of China (Project No. 32260568).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1375384/full#supplementary-material
Abou, H. M., Andersen, J. M., Barrangou, R., Møller, M. S., and Fredslund, F. (2013). Recent insight into oligosaccharide uptake and metabolism in probiotic bacteria. Biocatal. Biotransformation 31, 226–235. doi: 10.3109/10242422.2013.828048
Andersen, J. M., Barrangou, R., Abou Hachem, M., Lahtinen, S. J., Goh, Y. J., Svensson, B., et al. (2013). Transcriptional analysis of oligosaccharide utilization by Bifidobacterium lactis Bl-04. BMC Genomics 14:312. doi: 10.1186/1471-2164-14-312
Belorkar, S. A., and Gupta, A. K. (2016). Oligosaccharides: a boon from nature’s desk. Amb Express 6, 11. doi: 10.1186/s13568-016-0253-5
Bottacini, F., Morrissey, R., Esteban-Torres, M., James, K., Van Breen, J., Dikareva, E., et al. (2018). Comparative genomics and genotype-phenotype associations in Bifidobacterium breve. Sci. Rep. 8:10633. doi: 10.1038/s41598-018-28919-4
Bunesova, V., Killer, J., Javurkova, B., Vlkova, E., Tejnecky, V., Musilova, S., et al. (2017). Diversity of the subspecies Bifidobacterium animalis subsp lactis. Anaerobe 44, 40–47. doi: 10.1016/j.anaerobe.2017.01.006
Garrido, D., Ruiz-Moyano, S., Lemay, D. G., Sela, D. A., German, J. B., and Mills, D. A. (2015). Comparative transcriptomics reveals key differences in the response to milk oligosaccharides of infant gut-associated bifidobacteria. Sci. Rep. 5:13517. doi: 10.1038/srep13517
Gaya, P., Peirotén, Á., and Landete, J. M. (2017). Transformation of plant isoflavones into bioactive isoflavones by lactic acid bacteria and bifidobacteria. J. Funct. Foods 39, 198–205. doi: 10.1016/j.jff.2017.10.029
Gibson, G. R., Probert, H. M., Loo, J. V., Rastall, R. A., and Roberfroid, M. B. (2004). Dietary modulation of the human colonic microbiota: updating the concept of prebiotics. Nutr. Res. Rev. 17, 259–275. doi: 10.1079/NRR200479
Han Li, H. L., Li Lei, L. L., Yang HouRong, Y. H., He PeiXin, H. P., and Huang Shen, H. S. (2019). Transcriptomic sequence analysis of the effect of three fatty acids with different chain length on gene transcription in Saccharomyces cerevisiae.Pdf. Food Sci. Technol. 40, 106–112. doi: 10.7506/spkx1002-6630-20180316-221
Hansen, L. B., Roager, H. M., Søndertoft, N. B., Gøbel, R. J., Kristensen, M., Vallès-Colomer, M., et al. (2018). A low-gluten diet induces changes in the intestinal microbiome of healthy Danish adults. Nat. Commun. 9:4630. doi: 10.1038/s41467-018-07019-x
He, Z., Yang, B., Liu, X., Ross, R. P., Stanton, C., and Zhao, J. (2021). Short communication: genotype-phenotype association analysis revealed different utilization ability of 2′-fucosyllactose in Bifidobacterium genus. J. Dairy Sci. 104, 1518–1523. doi: 10.3168/jds.2020-19013
Holscher, H. D. (2020). Diet affects the gastrointestinal microbiota and health. J. Acad. Nutr. Diet. 120, 495–499. doi: 10.1016/j.jand.2019.12.016
Hopkins,, Cummings,, and Macfarlane, (1998). Inter-species differences in maximum specific growth rates and cell yields of bifidobacteria cultured on oligosaccharides and other simple carbohydrate sources. J. Appl. Microbiol. 85, 381–386. doi: 10.1046/j.1365-2672.1998.00524.x
Janer, C., Rohr, L. M., Peláez, C., Laloi, M., Cleusix, V., Requena, T., et al. (2004). Hydrolysis of Oligofructoses by the recombinant β-Fructofuranosidase from Bifidobacterium lactis. Syst. Appl. Microbiol. 27, 279–285. doi: 10.1078/0723-2020-00274
Kan, Z. Y., Luo, B. L., Cai, J. J., Zhang, Y., Tian, F. W., and Ni, Y. Q. (2020). Genotyping and plant-derived glycan utilization analysis of Bifidobacterium strains from mother-infant pairs. BMC Microbiol. 20:277. doi: 10.1186/s12866-020-01962-w
Li, M., Yao, S., Duan, Y. Y., Zhang, Y. J., Guo, Y., Niu, H. M., et al. (2020). Transcription factor enrichment analysis in enhancers identifies EZH2 as a susceptibility gene for osteoporosis. J. Clin. Endocrinol.Metab. 105, e1152–e1161. doi: 10.1210/clinem/dgz270
Li, Q., Zhang, Y., Shi, J.-L., Wang, Y.-L., Zhao, H.-B., Shao, D.-Y., et al. (2017). Mechanism and anticancer activity of the metabolites of an endophytic Fungi from Eucommia ulmoides Oliv. Anti Cancer Agents Med. Chem. 17, 982–989. doi: 10.2174/1871520616666160923094814
Liang, T. T., Jiang, T., Liang, Z., Zhang, N., Dong, B., Wu, Q. P., et al. (2023). Carbohydrate-active enzyme profiles of Lactiplantibacillus plantarum strain 84-3 contribute to flavor formation in fermented dairy and vegetable products. Food Chem. X 20:101036. doi: 10.1016/j.fochx.2023.101036
Linares-Morales, J. R., Salmerón-Ochoa, I., Rivera-Chavira, B. E., Gutiérrez-Méndez, N., Pérez-Vega, S. B., and Nevarez-Moorillón, G. V. (2022). Influence of culture media formulated with Agroindustrial wastes on the antimicrobial activity of lactic acid Bacteria. J. Microbiol. Biotechnol. 32, 64–71. doi: 10.4014/jmb.2107.07030
Liu, N., Shen, H. B., Zhang, F., Liu, X., Xiao, Q. R., Jiang, Q., et al. (2023). Applications and prospects of functional oligosaccharides in pig nutrition: A review. Animal Nutrition 13, 206–215. doi: 10.1016/j.aninu.2023.02.002
LoCascio, R. G., Ninonuevo, M. R., Freeman, S. L., Sela, D. A., Grimm, R., Lebrilla, C. B., et al. (2007). Glycoprofiling of bifidobacterial consumption of human milk oligosaccharides demonstrates strain specific, preferential consumption of small chain glycans secreted in early human lactation. J. Agric. Food Chem. 55, 8914–8919. doi: 10.1021/jf0710480
Lohrasbi, V., Abdi, M., Asadi, A., Rohani, M., Esghaei, M., and Talebi, M. (2020). The effect of improved formulation of chitosan-alginate microcapsules of Bifidobacteria on serum lipid profiles in mice. Microb. Pathog. 149:104585. doi: 10.1016/j.micpath.2020.104585
Lugli, G. A., Mancino, W., Milani, C., Duranti, S., Mancabelli, L., Napoli, S., et al. (2019). Dissecting the evolutionary development of the species Bifidobacterium animalis through comparative genomics analyses. Appl. Environ. Microbiol. 85:16. doi: 10.1128/AEM.02806-18
Masco, L., Huys, G., Gevers, D., Verbrugghen, L., and Swings, J. (2003). Identification of Bifidobacterium species using rep-PCR fingerprinting. Syst. Appl. Microbiol. 26, 557–563. doi: 10.1078/072320203770865864
Milani, C., Turroni, F., Duranti, S., Lugli, G. A., Mancabelli, L., and Ferrario, C. (2016). Genomics of the genus Bifidobacterium reveals species-specific adaptation to the glycan-rich gut environment. Appl. Environ. Microbiol. 82, 980–991. doi: 10.1128/AEM.03500-15
Moradi, M., Molaei, R., and Guimarães, J. T. (2021). A review on preparation and chemical analysis of postbiotics from lactic acid bacteria. Enzym. Microb. Technol. 143:109722. doi: 10.1016/j.enzmictec.2020.109722
Nomoto, R., Takano, S., Tanaka, K., Tsujikawa, Y., Kusunoki, H., and Osawa, R. (2017). Isolation and identification of Bifidobacterium species from feces of captive chimpanzees. Biosci. Microbiota Food Health 36, 91–99. doi: 10.12938/bmfh.16-027
O'Connell Motherway, M., Kinsella, M., Fitzgerald, G. F., and Van Sinderen, D. (2013). Transcriptional and functional characterization of genetic elements involved in galacto-oligosaccharide utilization by Bifidobacterium breve UCC 2003. Microb. Biotechnol. 6, 67–79. doi: 10.1111/1751-7915.12011
O'connell, K. J., Motherway, M. O., O'callaghan, J., Fitzgerald, G. F., Ross, R. P., Ventura, M., et al. (2013). Metabolism of four α-Glycosidic linkage-containing oligosaccharides by Bifidobacterium breve UCC 2003. Appl. Environ. Microbiol. 79, 6280–6292. doi: 10.1128/AEM.01775-13
Paineau, D., Carcano, D., Leyer, G., Darquy, S., Alyanakian, M. A., Simoneau, G., et al. (2008). Effects of seven potential probiotic strains on specific immune responses in healthy adults: a double-blind, randomized, controlled trial. FEMS Immunol. Med. Microbiol. 53, 107–113. doi: 10.1111/j.1574-695X.2008.00413.x
Sanders, M. E., Merenstein, D. J., Reid, G., Gibson, G. R., and Rastall, R. A. (2019). Probiotics and prebiotics in intestinal health and disease: from biology to the clinic. Nat. Rev. Gastroenterol. Hepatol. 16, 605–616. doi: 10.1038/s41575-019-0173-3
Shahin, M., Jamal, W., Verghese, T., and Rotimi, V. O. (2003). Comparative evaluation of Anoxomat and conventional anaerobic gas Pak jar systems for the isolation of anaerobic bacteria. Med. Princ. Pract. 12, 81–86. doi: 10.1159/000069116
Song, A.-X., Li, L.-Q., Yin, J.-Y., Chiou, J.-C., and Wu, J.-Y. (2020). Mechanistic insights into the structure-dependant and strain-specific utilization of wheat arabinoxylan by Bifidobacterium longum. Carbohydr. Polym. 249:116886. doi: 10.1016/j.carbpol.2020.116886
Srivastava, S., Bombaywala, S., Jakhesara, S. J., Patil, N. V., Joshi, C. G., Purohit, H. J., et al. (2023). Potential of camel rumen derived Bacillus subtilis and Bacillus velezensis strains for application in plant biomass hydrolysis. Mol. Gen. Genomics. 298, 361–374. doi: 10.1007/s00438-022-01987-y
Tomás-Barberán, F. A., and Mine, Y. (2013). A Key to Understanding the Effects of Food Bioactives in Health, Gut Microbiota. Journal of Agricultural and Food Chemistry 61, 9755–9757. doi: 10.1021/jf404354f
Ventura, M., Canchaya, C., Del Casale, A., Dellaglio, F., Neviani, E., Fitzgerald, G. F., et al. (2006). Analysis of bifidobacterial evolution using a multilocus approach. Int. J. Syst. Evol. Microbiol. 56, 2783–2792. doi: 10.1099/ijs.0.64233-0
Ventura, M., Canchaya, C., Van Sinderen, D., Fitzgerald, G. F., and Zink, R. (2004). Bifidobacterium lactis DSM 10140: identification of the atp (atp BEFHAGDC) operon and analysis of its genetic structure, characteristics, and phylogeny. Appl. Environ. Microbiol. 70, 3110–3121. doi: 10.1128/AEM.70.5.3110-3121.2004
Vieira, T. F., Corrêa, R. C. G., Peralta, R. A., Peralta-Muniz-Moreira, R. F., Bracht, A., and Peralta, R. M. (2020). An Overview of Structural Aspects and Health Beneficial Effects of Antioxidant Oligosaccharides. Current Pharmaceutical Design 26, 1759–1777. doi: 10.2174/1381612824666180517120642
Yan, S., Yang, B., Ross, R. P., Stanton, C., Zhang, H., and Zhao, J. (2020). Bifidobacterium longum subsp. longum YS108R fermented milk alleviates DSS induced colitis via anti-inflammation, mucosal barrier maintenance and gut microbiota modulation. J. Funct. Foods 73:104153. doi: 10.1016/j.jff.2020.104153
Keywords: Bifidobacterium animalis subsp. lactis, oligosaccharide metabolism, genotype comparison, resequencing, real-time PCR
Citation: Lan Z, Zhang X, Xu M, Kong J, Zuo X, Wang Y, Wang C, Teng Y, Ni Y and Zhang Y (2024) Whole-genome resequencing and transcriptional profiling association analysis revealed the intraspecies difference response to oligosaccharides utilization in Bifidobacterium animalis subsp. lactis. Front. Microbiol. 15:1375384. doi: 10.3389/fmicb.2024.1375384
Edited by:
Xiaodong Xia, Dalian Polytechnic University, ChinaReviewed by:
Corina-Diana Ceapă, National Autonomous University of Mexico, MexicoCopyright © 2024 Lan, Zhang, Xu, Kong, Zuo, Wang, Wang, Teng, Ni and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongqing Ni, bml5cWx6dUBzaW5hLmNvbQ==; Yan Zhang, emhhbmd5YW56cWdoQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.