
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 15 February 2024
Sec. Infectious Agents and Disease
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1359402
This article is part of the Research TopicAcute/Chronic Pathogen Infections: Roles of Resident, Probiotic, and Live Biotherapeutic Microbes in Limiting or Preventing DiseaseView all 26 articles
 Lucky Ronald Runtuwene1,2
Lucky Ronald Runtuwene1,2 Prince Kofi Parbie3,4
Prince Kofi Parbie3,4 Taketoshi Mizutani2,5
Taketoshi Mizutani2,5 Aya Ishizaka5Saori Matsuoka1Christopher Zaab-Yen Abana3Dennis Kushitor3
Aya Ishizaka5Saori Matsuoka1Christopher Zaab-Yen Abana3Dennis Kushitor3 Evelyn Yayra Bonney3Sampson Badu Ofori6
Evelyn Yayra Bonney3Sampson Badu Ofori6 Hiroshi Kiyono5,7,8Koichi Ishikawa1
Hiroshi Kiyono5,7,8Koichi Ishikawa1 William Kwabena Ampofo3
William Kwabena Ampofo3 Tetsuro Matano1,5*
Tetsuro Matano1,5*Human immunodeficiency virus (HIV) 1 infection is known to cause gut microbiota dysbiosis. Among the causes is the direct infection of HIV-1 in gut-resident CD4+ T cells, causing a cascade of phenomena resulting in the instability of the gut mucosa. The effect of HIV infection on gut microbiome dysbiosis remains unresolved despite antiretroviral therapy. Here, we show the results of a longitudinal study of microbiome analysis of people living with HIV (PLWH). We contrasted the diversity and composition of the microbiome of patients with HIV at the first and second time points (baseline_case and six months later follow-up_case, respectively) with those of healthy individuals (baseline_control). We found that despite low diversity indices in the follow-up_case, the abundance of some genera was recovered but not completely, similar to baseline_control. Some genera were consistently in high abundance in PLWH. Furthermore, we found that the CD4+ T-cell count and soluble CD14 level were significantly related to high and low diversity indices, respectively. We also found that the abundance of some genera was highly correlated with clinical features, especially with antiretroviral duration. This includes genera known to be correlated with worse HIV-1 progression (Achromobacter and Stenotrophomonas) and a genus associated with gut protection (Akkermansia). The fact that a protector of the gut and genera linked to a worse progression of HIV-1 are both enriched may signify that despite the improvement of clinical features, the gut mucosa remains compromised.
Human immunodeficiency virus (HIV) is still a worldwide health problem, with 38.4 million living with HIV worldwide. Approximately 1.5 million people acquired HIV, and 650,000 people died from HIV-related causes in 2021 (HIV.gov, 2022). There are still no vaccines available to prevent the disease. Nevertheless, life expectancy and health outcomes in people living with HIV (PLWH) have improved with the availability of combination antiretroviral therapy (ART) that controls HIV replication. HIV incidence has declined 32% since 2010 (HIV.gov, 2022). However, a cure is still not attainable due to the ability of the virus to persist in long-lived cells. These cells cannot be targeted with current ART or HIV-specific immune responses.
Persistence may also be caused by the gut microbiome and its metabolites (Koay et al., 2018; Crakes and Jiang, 2019; Ishizaka et al., 2021). They have been shown to promote inflammation and immune activation in HIV-infected adults. This inflammation and immune activation are correlated with the disruption of gut mucosal integrity. HIV targets gut-associated lymphoid tissues as major sites of viral transmission, replication and seeding, and CD4+ T-cell depletion (Khan et al., 2017). In the gut, altered T-cell homeostasis, particularly of CD4+ Th17 cells, coincides with disruption of the intestinal barrier, where tightly opposed enterocytes lose their adhesion to adjoining cells (Dandekar et al., 2010). When the intestinal barrier is disrupted, microbial products travel from the lumen into systemic circulation, accumulating in the liver and brain tissue and activating the immune system (Estes et al., 2010).
Furthermore, the effect of HIV infection on the dysbiosis of the gut microbiome remains unresolved despite ART therapy (Shacklett and Anton, 2010). HIV replication is effectively suppressed by ART, but HIV in the reservoirs cannot be eradicated and CD4+ T cells cannot be fully restored in the gut or peripheral tissues. The restoration of CD4+ T cells in the gut is delayed compared to those in the peripheral blood (Guadalupe et al., 2003). Initiation of ART fails to reduce chronic immune activation and markers of microbial translocation, such as lipopolysaccharide (LPS) and soluble CD14 (sCD14) (Wallet et al., 2010). The interaction between the host and microbe at the gut mucosal interface is critical in preventing microbial translocation and immunity activation in HIV-infected individuals receiving suppressive ART.
Nevertheless, the elucidation of gut microbial interaction with immune activation is not an easy task, as gut microbiome composition is different all over the world (de Filippo et al., 2010; Brito et al., 2016; He et al., 2018). We previously added to the collective knowledge the results of the microbiome analysis in healthy Ghanaians (Parbie et al., 2021a) and compared it with that in Ghanaian PLWH (Parbie et al., 2021b). Through microbiome analysis on our cohort, we found that our results were mostly in line with previous reports, that is, a significant increase in the abundance of Proteobacteria but a decrease in the abundances of Firmicutes and Bacteroidetes at the phylum level, similar to previous observations in other countries (McHardy et al., 2013; Vujkovic-Cvijin et al., 2013; Monaco et al., 2016). However, some bacteria belonging to Firmicutes, namely, Streptococcus, Dorea, and Blautia, are enriched in PLWH (Parbie et al., 2021b). We note that this is different from several other reports, in which the abundance of Lachnospiraceae, including Dorea and Blautia, is decreased in HIV-1-infected individuals (Mutlu et al., 2014; Vázquez-Castellanos et al., 2015). Furthermore, we found a depletion of Prevotella in PLWH in Ghana, whereas previous studies on the gut microbiome have described enrichment of Prevotella in HIV-1-infected populations (Lozupone et al., 2013; Dillon et al., 2014).
To further complement the data, here, we describe the results of a longitudinal microbiome analysis of PLWH in Ghana.
Data on HIV-1-negative (HIV-) and HIV-1-positive (HIV+) status at baseline (baseline_control and baseline_case, respectively) were described in Table 1. We enrolled matching pairs of HIV+ and HIV- individuals. Participants in the study were HIV+ individuals attending the Eastern Regional Hospital, Koforidua (RHK) in Ghana. A community health screening in these communities recruited HIV- individuals. We matched seronegative individuals by age (± 2 years), sex, and community of residence to serve as controls. Only adults above 18 years old were enrolled in this study. Seronegative participants who took antibiotics within four weeks of sample collection were not enrolled. Approximately 6 months after their baseline visit, stool samples were collected from HIV+ individuals, which represent the follow-up_case. CD4+ T-cell count and viral load were not measured on their second visit.
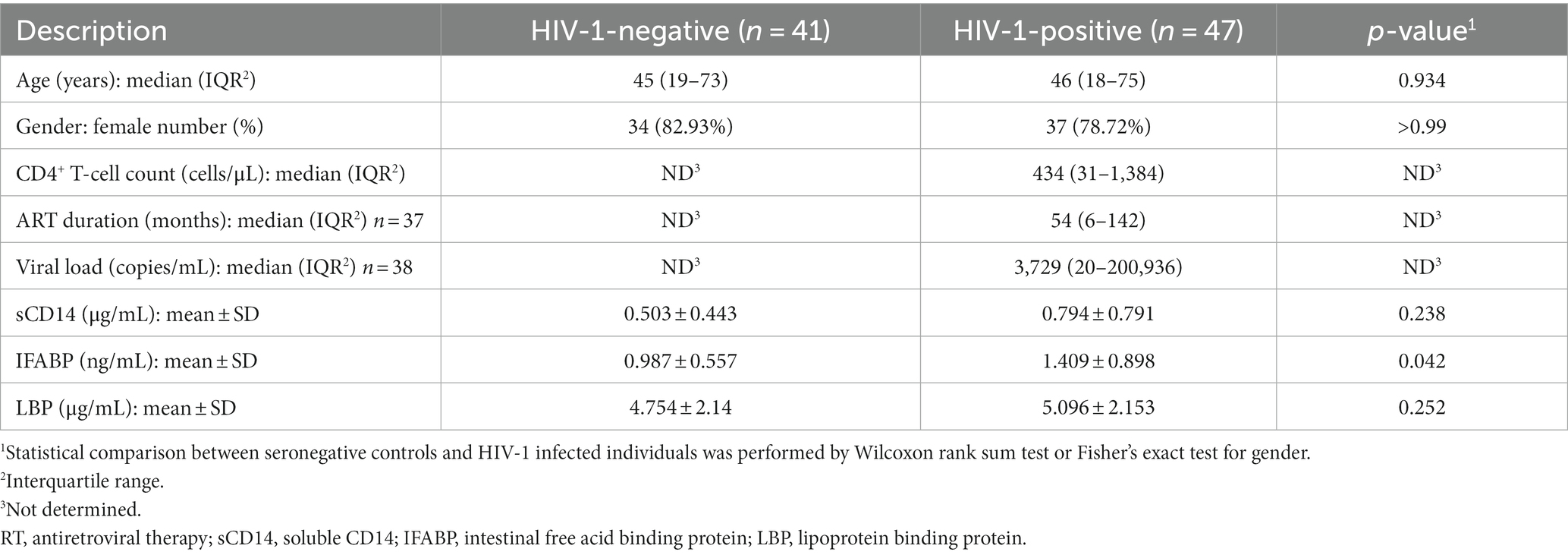
Table 1. Clinical and demographic characteristics.
We collected venous blood and stool samples from enrolled participants. The total of 135 samples includes 41 samples from HIV- individuals (baseline_controls), and 94 samples from HIV+ individuals (n = 47) at baseline (baseline_case) and six months later at follow-up time point (follow-up_case). In the 24 h following their collection, all biological samples were transported to the Noguchi Memorial Institute for Medical Research (NMIMR) in Ghana, where they were processed for storage. We prepared plasma and peripheral blood mononuclear cells from venous blood. Stool samples were collected and stored at −80°C until DNA extraction.
One gram of stool was washed with 3 mL of SM-plus buffer (100 mM NaCl, 50 mM Tris–HCl [pH 7.4], 8 mM MgSO4-7H2O, 5 mM CaCl2-2H2O, 0.01% [w/v] gelatin) and centrifuged at 6,000 × g for 5 min. This process was done for additional two times. Pellets were resuspended in 20 mL of SM-plus buffer and filtered through a 100-μm cell strainer (Corning, Tokyo, Japan). One milliliter of the filtered 20 mL of bacterial suspension was used for DNA extraction.
DNA was extracted from the fecal sample-derived bacterial fraction as previously described (Kim et al., 2013). Gene libraries for the hypervariable V3-V4 region of 16S rRNA were prepared as previously described (Parbie et al., 2021b) according to the 16S Metagenomic Sequencing Library Preparation guide (Illumina, San Diego, United States; Part # 15044223 Rev. B). Sequencing was performed on the Illumina MiSeq using MiSeq Reagent Kit v3 (600-cycle) with a 20% PhiX (Illumina) spike-in at Noguchi Memorial Institute for Medical Research, Ghana. Sequencing was performed in five batches.
Sequences were quality filtered, denoised, and analyzed with Quantitative Insights Into Microbial Ecology (QIIME 2 version 2022.2) (Bolyen et al., 2019). Briefly, raw paired-end reads were imported, demultiplexed, and quality filtered using the q2-demux plugin followed by denoising with DADA2 (Callahan et al., 2016) (via q2-dada2). Taxonomy was assigned to the resulting amplicon sequence variants against the SILVA database (release 138) (Quast et al., 2013) (via q2-feature-classifier). A phylogeny was constructed with the q2-phylogeny plugin. Batch effect was removed with an R package, MMUPHin (Ma et al., 2022) with the adjust_batch() function. Principal coordinate analysis calculated with Curtis-Bray, Unweighted UniFrac, and Weighted UniFrac distances, alpha diversity and microbial composition analyses were conducted with phyloseq (McMurdie and Holmes, 2013). Differentially abundant taxa by HIV-1 status were identified using the linear discriminant analysis (LDA) effect size (LEfSe) method (Segata et al., 2011) and the Analysis of Composition of Microbiome (ANCOM) (Mandal et al., 2015) approach with default parameters. Metagenomic pathway prediction was performed with Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) (Douglas et al., 2020) with default parameters. We analyzed only results associated with the MetaCyc database (Caspi et al., 2020).
CD4+ T-cell count was performed with BD FACSCount CD4+ reagents kit following manufacturer’s instructions. Viral load was measured with AmpliPrep/ COBAS TaqMan HIV-1 Test V2.0 following manufacturer’s instructions.
An enzyme-linked immunosorbent assay (ELISA) was used to measure plasma lipopolysaccharide-binding protein (LBP), soluble CD14 (sCD14), and intestinal fatty acid-binding protein (I-FABP) levels (R&D Systems, Minneapolis, MN, United States).
Data were obtained from 2017 to 2019. R 3.6.0 packages were used for statistical analyses. Comparison between categorical variables between groups was performed with the Wilcoxon using an R package, ggpubr with geom_pwc() function (Kassambara, 2023). p values less than 0.05 were considered significant. To test associations involving bacterial taxa showing significant differences in abundance by HIV-1 status and clinical and immunological markers, correlation analysis was performed using the R package Microbiome Multivariable Associations with Linear Models (MaAsLin2) (Mallick et al., 2021) using the following modifiers: analysis_method = “NEGBIN”, normalization = “NONE”, transform = “NONE”, max_significance = 0.5, min_prevalence = 0.5, min_abundance = 0.0001. PICRUSt2 data were visualized with Statistical Analysis of Taxonomic and Functional Profiles (STAMP) (Parks et al., 2014). Significant pathways between two groups (baseline_case vs. baseline_control and baseline_case vs. follow-up_case) were calculated with two-sided Welch’s t test and Bonferroni correction with a p value filter of 0.05.
The total of 135 samples includes 41 samples from HIV- individuals (baseline_control), and 94 samples from HIV+ individuals (n = 47) at baseline (baseline_case) and six months later at follow-up time point (follow-up_case). Beta diversity shows that each condition-timepoint has a characteristic expression. Unweighted UniFrac shows that the species mostly overlap at all condition-timepoints. Weighted UniFrac, on the other hand, shows that the abundance of the species in follow-up_case is more similar to baseline_control than baseline_case (Figure 1A). We measured the alpha diversity of these condition-timepoints. Compared with baseline_control, all indices had lower diversity in baseline_case. Richness and Shannon were statistically insignificant, but two indices, i.e., Fisher and Faith’s phylogenetic diversity (PD) (Figure 1B). In the follow-up case, the microbial diversity indices were similar to those in the baseline case. Nevertheless, Faith’s PD shows a significant reduction at the second time point (Figure 1B). Compositional plot, which shows the microbial composition for each condition-timepoint, revealed that several genera were up-or downregulated in the follow-up_case compared to the baseline_case, with relative amounts comparable to the baseline_control (Figure 1C). For example, the genera Subdoligranulum, Dorea, and Blautia were less abundant in the follow-up case. On the other hand, the genus Faecalibacterium were more abundant in the follow-up case.
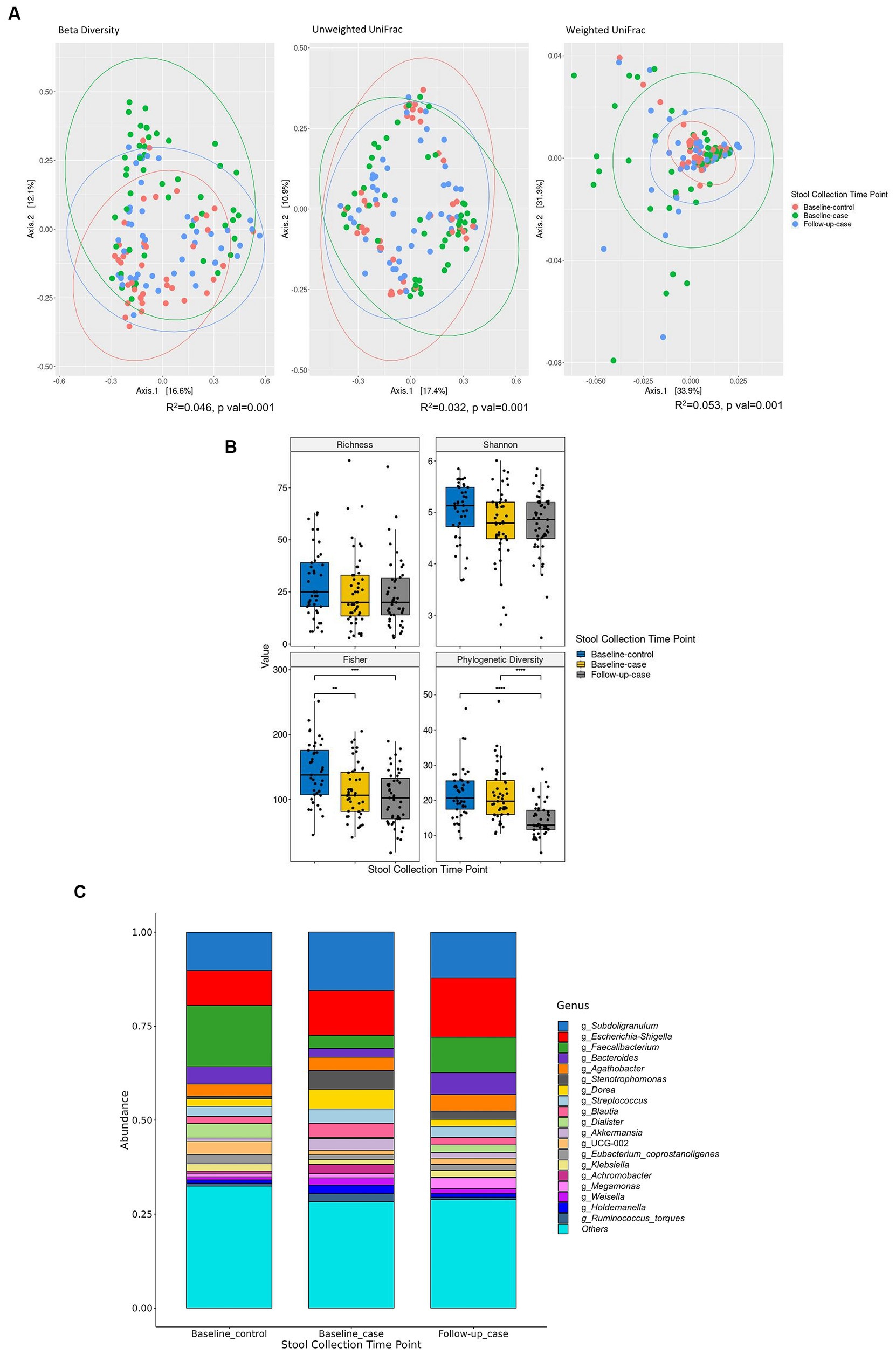
Figure 1. Alpha and beta diversity of the cohort. Principal component analysis shows that the microbial abundance of each of the three condition-timepoints can be clustered together. Each dot represents each sample’s taxon abundance in two dimensions. Beta diversity is measured using Curtis-Bray, Unweighted UniFrac, and Weighted UniFrac distances (A). (B) Shows the diversity indices between condition-timepoint. The relative abundance of the 19 highest expressed genera in each condition-timepoint is shown in (C). Statistical significance is calculated by the Wilcoxon test. **p < = 0.01, ***p < = 0.001, ****p < = 0.0001.
We employed the linear discriminant analysis (LDA) effect size (LEfSe) method to identify the taxa characterizing each condition-timepoint (Segata et al., 2011). The phyla Bacteriodota and Euryarchaeota (archaea) were enriched in healthy individuals (baseline_control), while Verucomicrobiota was abundant in PLWH (baseline_case) (Figure 2A). Within the phylum Firmicutes (or Bacillota), there was abundant diversity. For example, the class Negativicutes was enriched in the baseline_control, while Bacilli was abundant in the baseline_case. Furthermore, there seems to be an intersection of abundance of baseline_control and follow-up_case, i.e., the phylum Bacteriodota and class Negativicutes (Figure 2B), which are abundant in both baseline_control and follow-up_case.
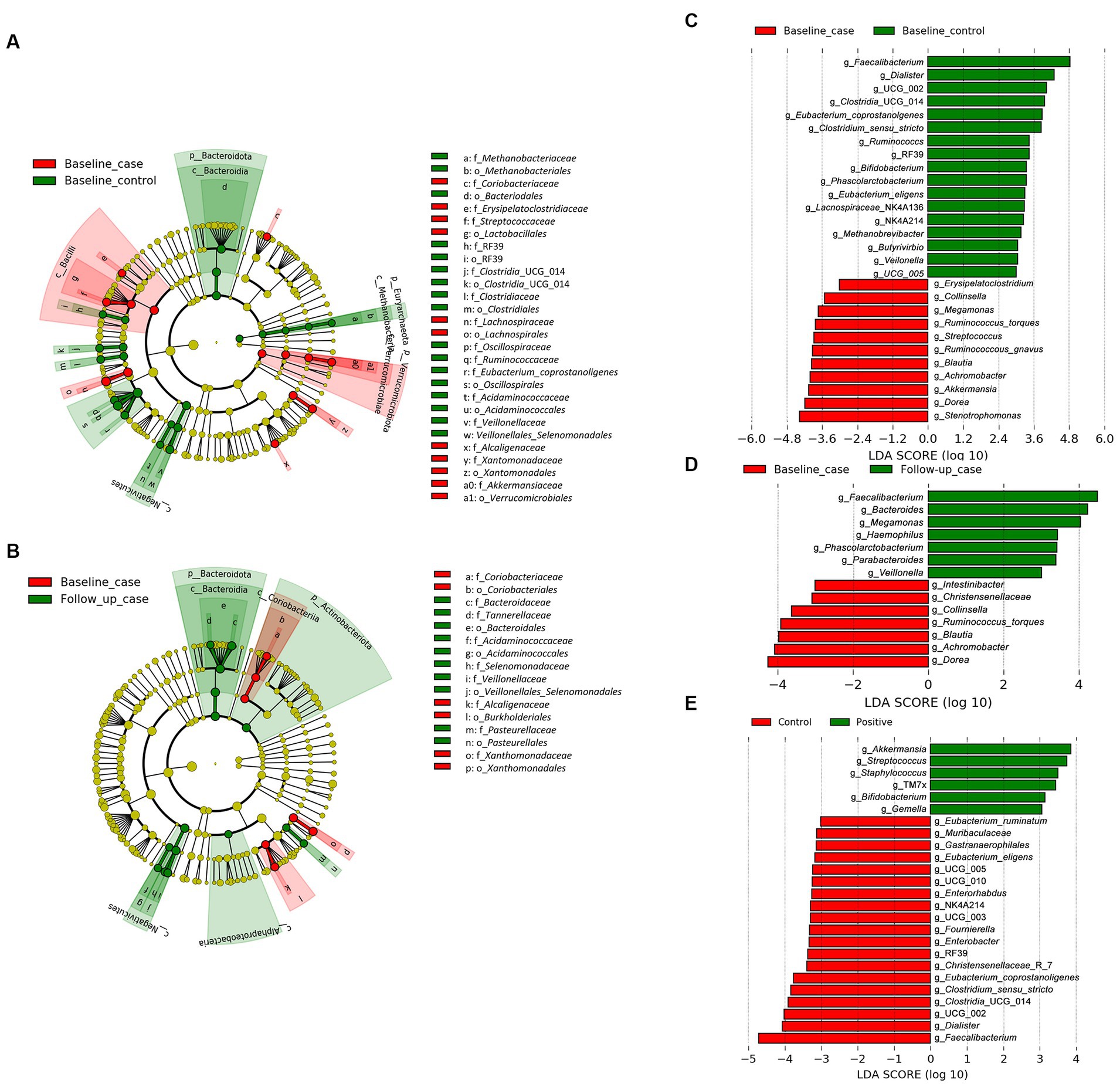
Figure 2. The result of linear discriminant analysis (LDA) effect size (LEfSe) analyses. LEfSe analysis between baseline_case and baseline_control and baseline_case and follow-up_case is shown in (A,B), respectively. The difference in genera abundance between baseline_case and baseline_control and baseline_case and follow-up_case is shown in (C,D), respectively. The difference in genera abundance between HIV negative and positive is shown in (E). LDA threshold is 2.5.
The genera Dorea and Blautia were increased in PLWH at baseline_case (Figure 2C), as well as Achromobacter and Stenotrophomonas, compared with baseline_control. Some genera in healthy people (baseline_control), such as Faecalibacterium and Phascolarctobacterium (Figure 2C) were also abundant in the follow-up_case (Figure 2D), corroborating the apparent reversion of the abundance of these genera. Nevertheless, some genera were highly abundant in PLWH regardless of time points, such as Streptococcus, Staphylococcus, and Gemella (Figure 2E). Analysis with ANCOM corroborates the LEfSe results, showing that the abundance in the baseline case and follow-up case was different: the genera Achromobacter and Phascolarctobacterium were abundant in the baseline case and follow-up case, respectively (Supplementary Figure 1).
The relative abundance of significantly differentially abundant genera of each condition-timepoint are presented in detail in Figure 3. A reversal in the abundance of the Faecalibacterium from baseline_case to follow-up_case and a decrease in the abundance of the Dorea in PLWH at baseline and follow-up_case was observed. Further, the persistence abundance of Streptococcus and Staphylococcus in HIV-positive despite the time point.
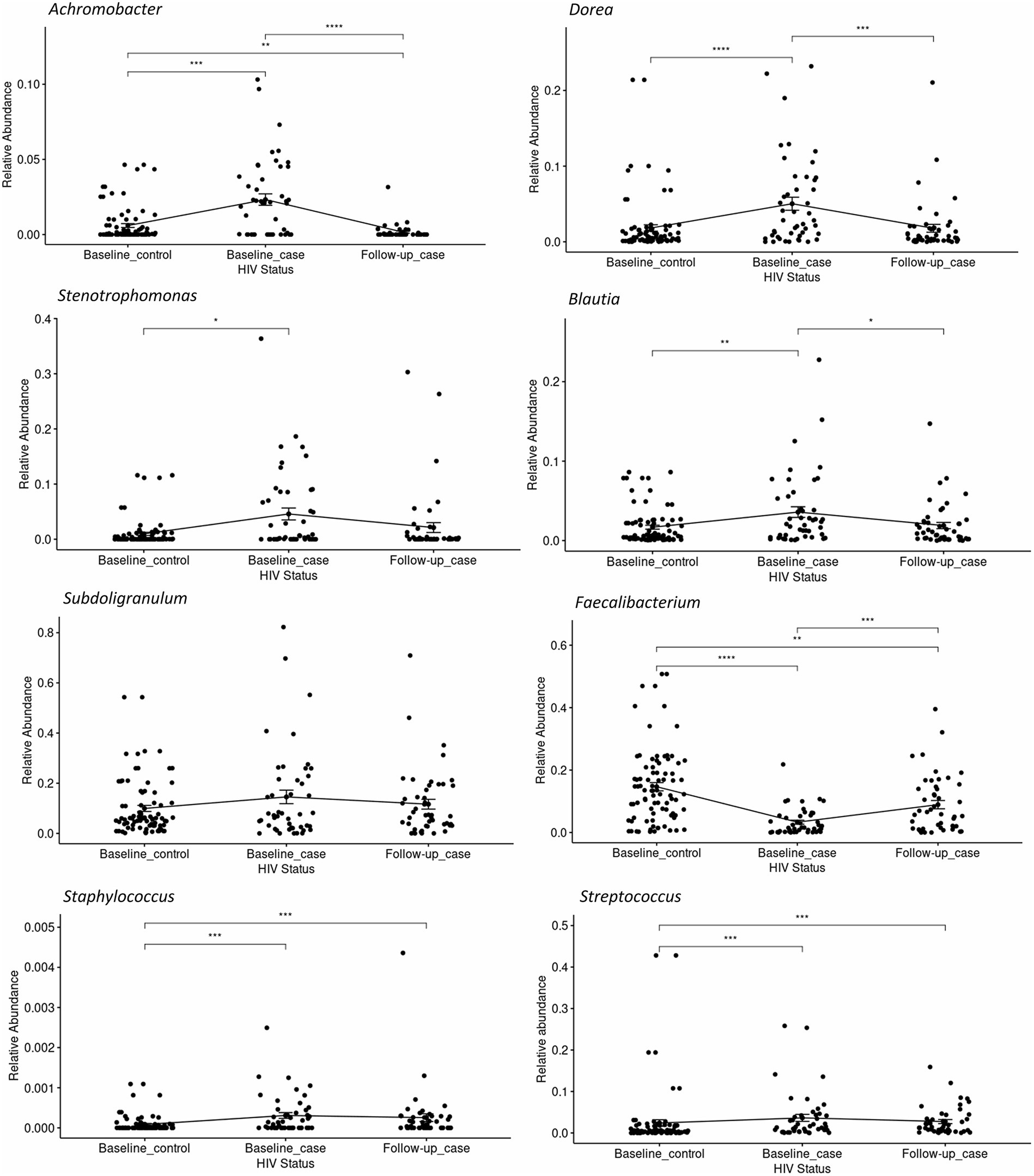
Figure 3. The relative abundance of select genera in each condition-timepoint. The genera Achromobacter, Dorea, Stenotrophomonas, Blautia, and Subdoligranulum shows low abundance in the baseline _control, high abundance in the baseline_case, and low abundance in the follow-up_case, albeit a non-significance in the genus Subdoligranulum. In contrast, the genus Faecalibacterium has a high abundance in the baseline_control, a reduction in the baseline_case, and a rebound in the follow-up_case. The genera Staphylococcus and Streptococcus, however, have high abundance in positive cases regardless of time point. Statistical significance is calculated by the Wilcoxon test. **p < = 0.01, ***p < = 0.001, ****p < = 0.0001.
The correlation between clinical characteristics and alpha diversity at baseline continued to be examined. We conducted a comparative analysis of alpha diversity, splitting the PLWH population into two groups based on the median values of each of the six parameters, which were CD4+ T-cell count, viral load, ART duration, sCD14, IFABP, and LPS levels. The patients with high CD4+ T-cell counts showed significantly higher alpha diversity than the patients with low counts at baseline (Figure 4A). In contrast, a high sCD14 level showed significantly lower alpha diversity (Figure 4B). Longer ART duration was correlated with lower diversity, although it was not significant in our cohort (Supplementary Figure 2A). Higher IFABP levels were not significantly correlated with lower diversity (Supplementary Figure 2B). Viral load and LPS level did not seem to be correlated with diversity indices (Supplementary Figures 2C,D).
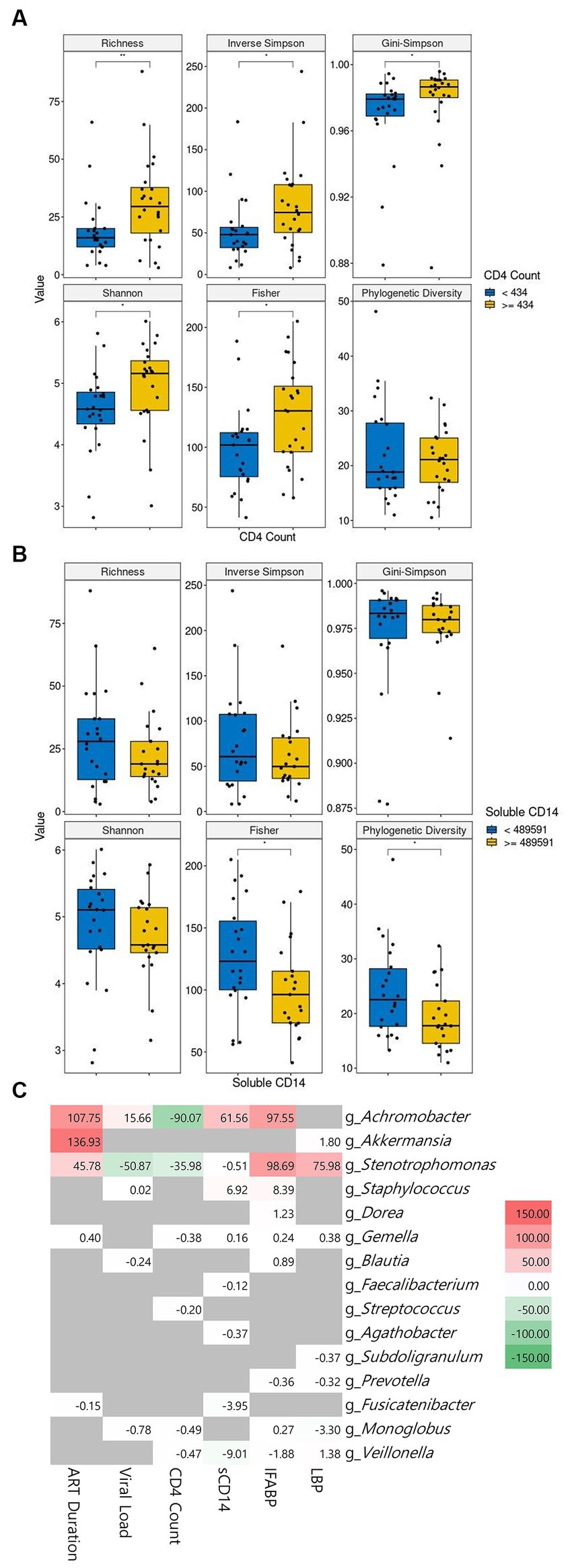
Figure 4. The correlation of microbiome abundance with clinical features. The diversity indices between low and high CD4+ cell count are shown in (A). The diversity indices between low and high soluble CD14 level are shown in (B). The cutoff value between high and low is the median value. CD4+ T-cell count is in cells/μL of blood and soluble CD4 is in pg/mL of blood. The correlation between genera and clinical features is observable in (C). Statistical significance is calculated by the Wilcoxon test. **p < = 0.01, ***p < = 0.001, ****p < = 0.0001. ART: antiretroviral, sCD14: soluble CD14, IFABP: intestinal fatty acid binding protein, LBP: lipopolysaccharide-binding protein.
We then sought to link the abundance of microbial genera to the clinical features. Our cohort showed a positive correlation of Achromobacter with ART duration, viral load, sCD14, and IFABP and its inverse correlation with CD4+ T-cell count. Likewise, Stenotrophomonas followed the same pattern as Achromobacter, albeit with an inverse correlation with viral load. Staphylococcus showed a positive correlation with sCD14 and IFABP. Other genera did not show a meaningful correlation with the clinical features (Figure 4C). Interestingly, the correlation of ART duration with Achromobacter and Sternotrophomonas was positive. The abundance of the genus Akkermansia was also highly significantly related to this parameter.
To evaluate the metabolic pathways, counting the functional genes directly following a shotgun metagenome sequencing is ideal. However, this needs a sequencer with a high sequencing capacity. In a situation where research limitations exist, we may use a prediction software. PICRUSt2 software was developed to predict the functional potential of a bacterial community based on marker gene sequencing profiles (Douglas et al., 2020). Utilizing this software, we compared the functional pathways based on the MetaCyc database (Caspi et al., 2020). Analysis revealed that bacterial pathways predicted a significant increase in the number of functional genes in baseline cases compared to baseline controls, but a decrease in the number of functional genes in follow-up cases compared to baseline cases (Figure 5). Exploring the MetaCyc database revealed the expected taxonomic range to possess the predicted pathways (Supplementary Table 1). While some of the pathways are not specific to bacteria, e.g., NAD biosynthesis II from tryptophan pathway is also found in eukaryotes, some pathways are found to be specific in bacteria, for instance ketogluconate degradation I, 2-nitrobenzoate degradation I, and fatty acid salvage pathways. Furthermore, some upregulated pathways are possessed by groups of bacteria known as pathogenic and opportunistic. Catechol degradation II, L-histidine degradation II, and nicotinate degradation I are pathways possessed by the phylum Pseudomonadota, whose members include the families Neisseriaceae, Campylobacteriaceae, Enterobacteriaceae, Klebsiella, Escherichia, Burkholderiaceae, and Pseudomonadaceae (Leão et al., 2023).
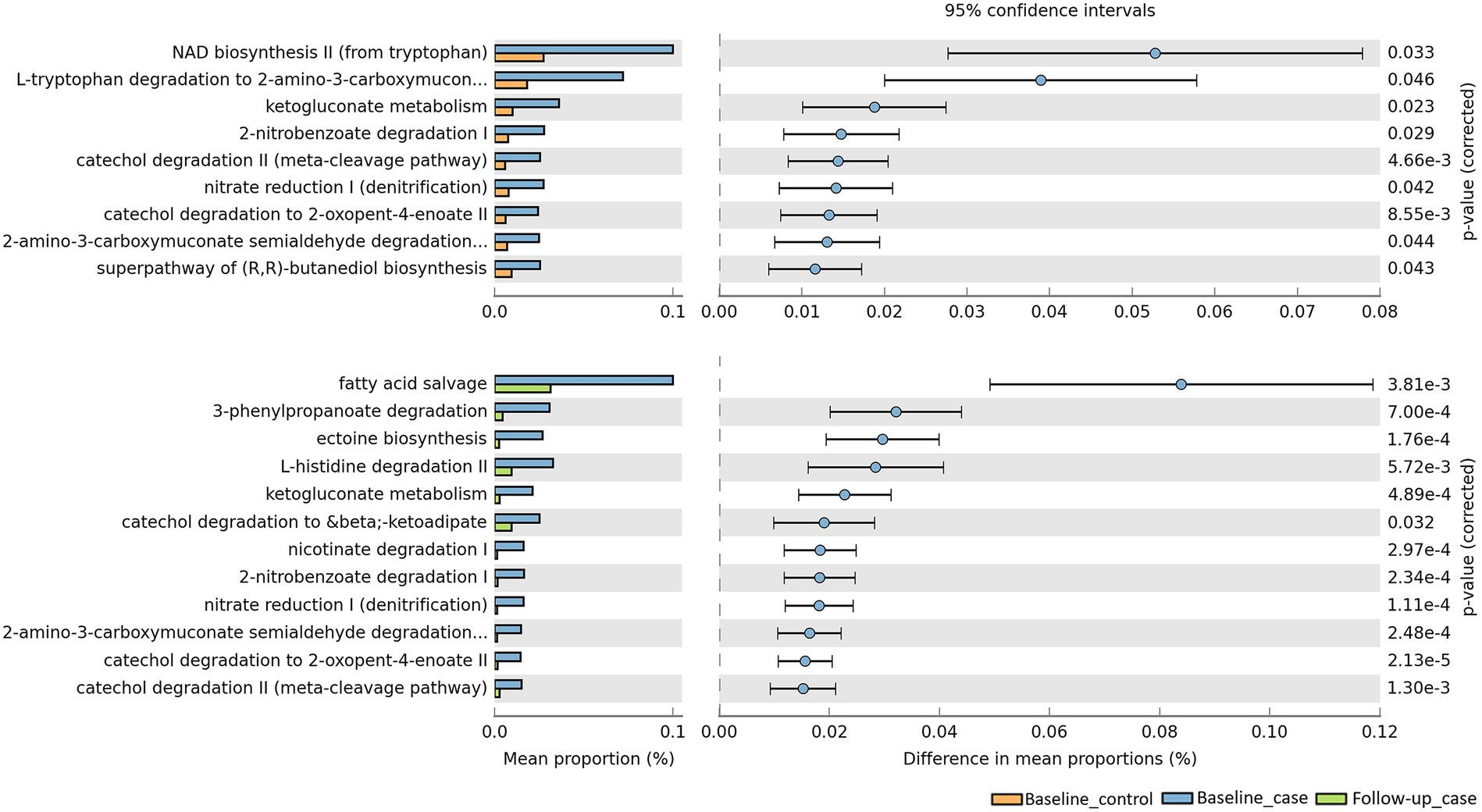
Figure 5. Predicted pathways upregulated/downregulated in our cohort. PICRUSt2 predicts that baseline_case samples have pathways that are significantly enriched compared to baseline_control and follow-up_case.
Our finding that the abundance of both beneficial and pathogenic bacteria genera is highly correlated with the duration of ART motivated us to perform additional longitudinal analysis based on treatment duration. Here, we classified our cohort into ART-naïve, PLWH with ART treatment < 36 months, and PLWH with treatment ≥ 36 months. We found that the correlation between bacterial abundance and clinical features was stronger at treatment ≥36 months than at any other treatment duration (Supplementary Figure 3). CD4+ T-cell count showed that there was no change in cell number in all treatment duration (Figure 6A). Nevertheless, the median CD4+ cell counts of 434 cells/μL was well above 200 cells/μL for all treatment duration. Despite non-significance, viral load was in a decreasing trend with a median value of 3,729 copies/μL (Figure 6B). sCD14 and IFABP showed a higher median value than naïve at early treatment duration but was reduced at late therapy (sCD14, Figure 6C) or no change (LBP, Figure 6D). The median LBP value was of no change (Figure 6E). Nevertheless, sCD14, IFABP, and LBP values showed an increasing trend. Alpha diversity indices were lower in PLWH on treatment at baseline, with more profound significant reduction at late treatment period (Figure 6F). At follow-up, there was no index difference between the treatment conditions. Nevertheless, in comparison to baseline, diversity at follow-up was more significantly reduced in ART-naïve individuals (Figure 6G). LDA LEfSe showed abundance of genus Faecalibacterium and class Negativicutes on follow up at both <36 and ≥ 36 months ART treatment. The analysis could not identify a taxon with a strong effect size at follow-up in ART-naïve (Figure 7). Collectively these results revealed a leaky gut phenomenon at late treatment period despite relatively controlled disease progression with a mix of beneficial and pathogenic bacterial environment.
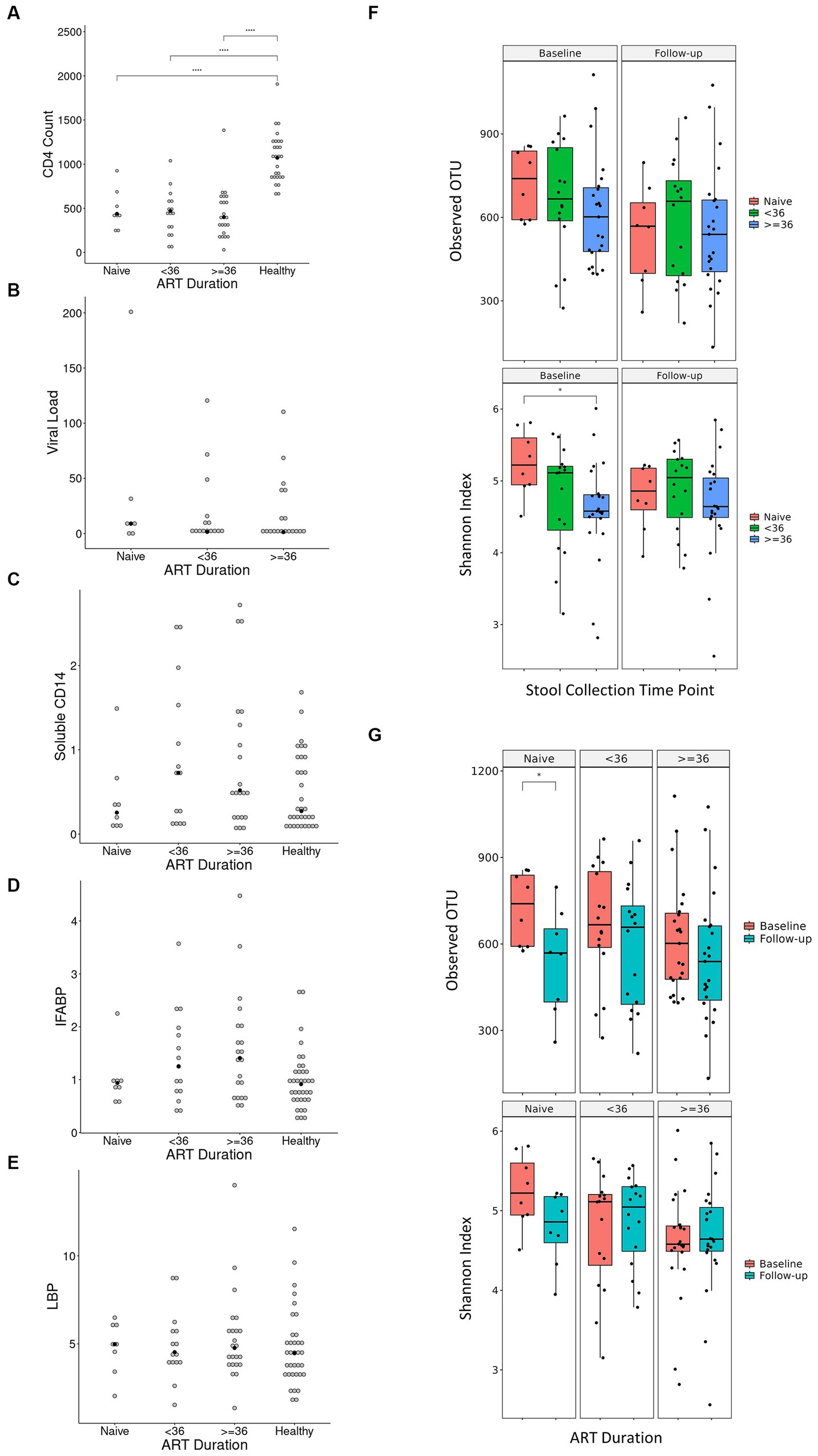
Figure 6. Diversity indices and clinical features in regard to treatment duration. Values of CD4+ T-cell count, viral load, soluble CD14, IFABP, and LBP in regard to treatment duration is shown in (A–E), respectively. Antiretroviral (ART) duration is in months, CD4+ T-cell count is in cells/μL of blood, viral load is in copies/μL of blood, soluble CD14 is in μg/mL of blood, intestinal fatty acid binding protein (IFABP) is in ng/mL of blood, and lipopolysaccharide-binding protein (LBP) is in μg/mL of blood. Each white dot is the value of a sample while the black dots show the median value. Diversity indices in regard to duration treatment is shown in (F,G). Statistical significance is calculated by the Wilcoxon test. **p < = 0.01, ***p < = 0.001, ****p < = 0.0001.
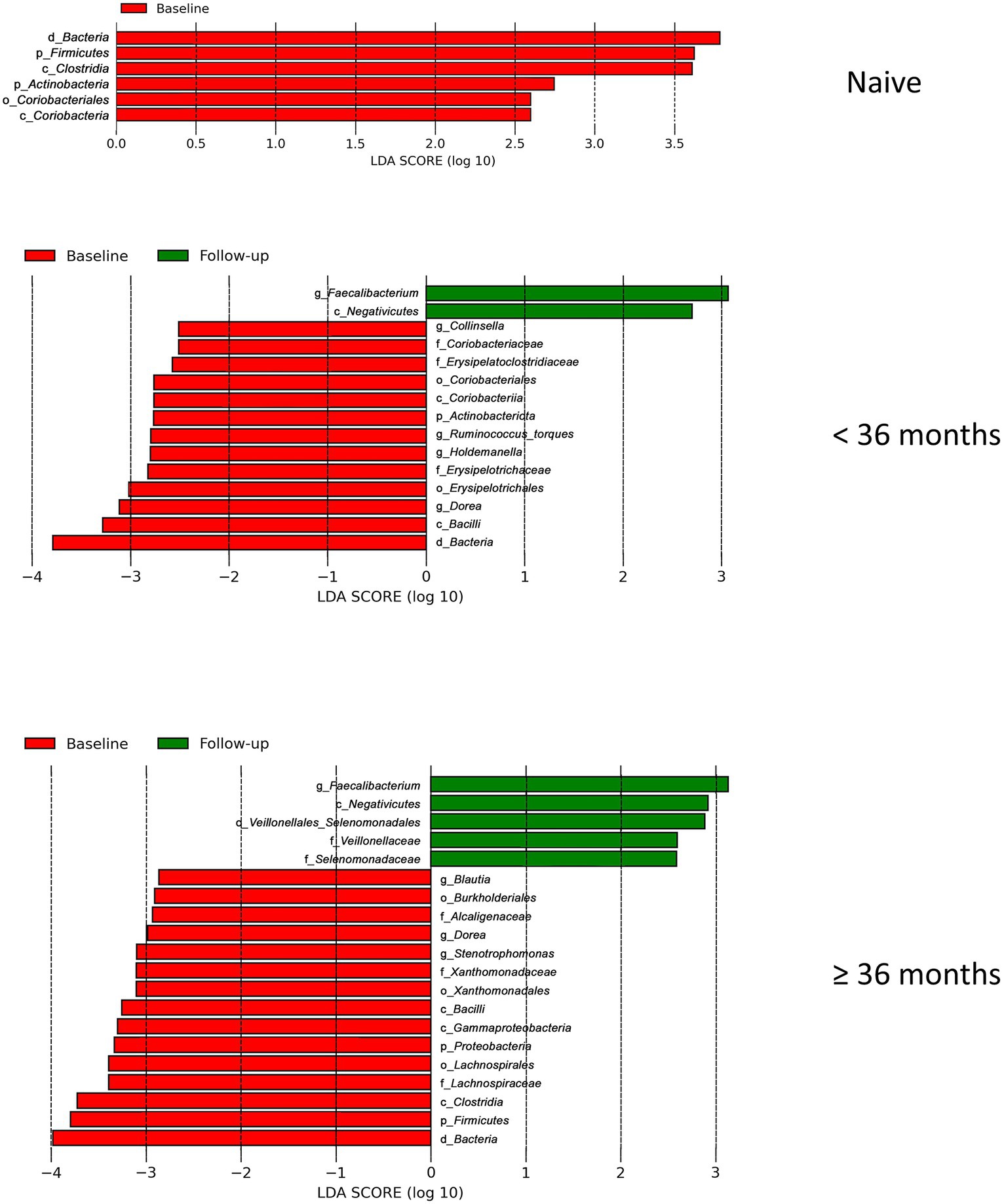
Figure 7. Linear discriminant analysis (LDA) effect size (LEfSe) in regard to treatment duration. LEfSe analysis between baseline_case and baseline_control and baseline_case and follow-up_case in regard to treatment duration is shown here. There is no follow-up result in naive due to its effect size is lower than the threshold 2.5.
Our previous work confirmed that the gut microbiome composition of Ghanaian PLWH is mostly similar to other reports, with some discrepancies (Parbie et al., 2021a). Faecalibacterium as well as Prevotella and Bacteroides were not included in the top 10 abundant taxa in HIV+ compared to healthy individuals, while the genera Achromobacter, Dorea, Blautia, Stenotrophomonas, and Streptococcus were abundant, indicating loss of critical commensals in HIV-1-infected adults and proliferation of opportunistic pathogens (Parbie et al., 2021b). One discrepancy was that depletion of Prevotella in chronic HIV infection was not reported previously (Lozupone et al., 2013; Dillon et al., 2014; Mutlu et al., 2014; Vázquez-Castellanos et al., 2015). In addition, the abundance of Dorea and Blautia was not reported by the aforementioned authors. The discrepancies observed between this study and others may be explained by differences in inclusion and exclusion criteria, duration of ART treatment, sampling methods and sites, sample sizes, sequencing techniques, and statistical methods (Monaco et al., 2016). For example, the abundance of Prevotella is usually linked to MSM community, regardless of HIV status (Kelley et al., 2017; Armstrong et al., 2018; Vujkovic-Cvijin et al., 2020). This is in contrast to our cohort being mostly female.
Lower diversity indices in HIV positive samples have been reported before (Mutlu et al., 2014; Vázquez-Castellanos et al., 2015; Noguera-Julian et al., 2016), while some reports have disagreements (Dillon et al., 2014; Ling et al., 2016). We observed lower Shannon and Fisher indices in our PLWH cohort. These indices are related to low microbial diversity, which lead to the loss of gut integrity and increased microbial migration (Koay et al., 2018). The low microbial diversity was maintained at the second time point in this longitudinal analysis, with Faith’s PD index displaying an even lower value than the first time point.
Compositional plot, on the other hand, revealed a different story. At the second time point, the microbial composition showed a partial shift similar to that of healthy Ghanaians. This included abundance rebound of the phylum Bacteroidota, class Negativicutes, and genus Faecalibacterium as well as depletion of genera Achromobacter, Dorea, and Blautia. Some genera, such as Streptococcus, Staphylococcus, and Gemella, did not show this reversal and maintained their high abundance.
The genera Faecalibacterium, Prevotella, and Bacteroides are commensals commonly found in healthy individuals and are correlated with good health (Wexler, 2007; Lopez-Siles et al., 2017; Yeoh et al., 2022). On the other hand, the genera Achromobacter, Streptococcus, and Stenotrophomonas are opportunistic pathogens (Gordon et al., 2002; Calza et al., 2003; Tatro et al., 2014). Dorea and Blautia from the phylum Firmicutes and their correlation to good health are somewhat controversial (Vacca et al., 2020). We hypothesized that the reversal may be contributed by CD4+ T cells and sCD14, as these two clinical markers are correlated significantly with microbial diversity. Indeed, a lower CD4+ T-cell count in HIV+ patients is associated with gut dysbiosis, a reduced abundance of healthy Ruminococcaceae bacteria in the gut, and elevated serum proinflammatory cytokine levels (Guillén et al., 2019; Lu et al., 2021). sCD14, which is a marker of monocyte activation, is indirectly correlated with microbial translocation (Marchetti et al., 2013). Lower diversity in samples with high sCD14 levels may indicate the translocation of gut microbes to the bloodstream. In accordance to this, we found that a higher CD4+ T-cell count and lower sCD14 levels were correlated with higher diversity indices in this cohort.
Further, the genera Achromobacter and Sternotrophomonas were found to be highly correlated with the clinical features. We found that with the improvement of the markers, the abundance of these genera was significantly reduced. Indeed, lower CD4+ T-cell percentages, greater CD4+ T-cell activation, and higher sCD14 are correlated with the immune response to Achromobacter xylosoxidans (Tatro et al., 2014). The repletion of CD4+ regulatory T cells in gut-associated lymphoid tissue may help with the localization of these bacteria in Peyer’s patches and mesenteric lymph nodes. Sternotrophomonas maltophilia has been shown to induce T-cell exhaustion, which may further increase T-cell apoptosis (Wang et al., 2021). It is likely that the improvement of the immune system limits the vicious cycle caused by the bacteria through an unknown mechanism. We also found that the abundance of one genus, Akkermansia, was very significantly correlated with the antiretroviral duration. Considering the role of these bacteria in maintaining the health of the gut mucous (Ouyang et al., 2020), the abundance of Akkermansia is a sign that the bacteria are working to preserve the stability of the gut microbiome during disease progression. However, Achromobacter and Stenotrophomonas were also positively correlated with antiretroviral duration, corroborating the finding that gut mucous integrity is not consistently restored despite the improvement in plasma CD4+ T-cell count and reduction in anti-inflammatory cytokines (Li et al., 2016). One causing factor is CD4+ T-cell depletion in the terminal ileum, including Peyer’s patches, which is accompanied by irreversible fibrosis that disrupts normal gut mucosal function (Shacklett and Anton, 2010).
We also predicted the bacterial pathways likely to be enriched using prediction software. Comparison between baseline_control and follow-up_case to baseline_case reveals that samples from baseline_case likely have enrichment of pathways related to bacteria, some even exclusive to the phylum Pseudomonadota. The members of this phylum are known to be pathogens of the respiratory tract (Achromobacter, Klebsiella, Pseudomonas, Sternotrophomonas), gastrointestinal tract (Aeromonas, Campylobacter, Escherichia, Helicobacter), and blood (Brevundimonas, Neisseria, Serratia, Stenotrophomonas), among other conditions (Citrobacter, Enterobacter, Haemophilus, Proteus) (Leão et al., 2023). This implies that the activity of pathogenic bacteria was high in the baseline case. The activity seemed to be reduced to be similar to control subjects in the follow-up_case, although not completely.
Classifying our cohort into ART-naïve, PLWH with ART treatment <36 months, and ART treatment ≥36 months gives a deeper insight about our cohort. First, we were able to observe that the Ghanaian PLWH might have a relatively good response to therapy. Despite non-significance, CD4+ T-cell count seemed on the increasing trend with a median value above WHO cutoff of 200 cells/μL. Viral load was also on the decreasing trend with a median of 3,729 copies/μL, despite non-significance. Microbial translocation markers were on the increasing trend. This suggests that despite a somewhat controlled viremia and disease progression, leaky gut was occurring. This was corroborated with lower diversity in late treatment period and on follow-up of ART-naïve subjects. LDA LEfSe showed abundance of the genus Faecalibacterium and class Negativicutes on follow-up at both <36 and ≥ 36 months ART treatment, showing a mixed bacterial environment. Negativicutes is a Gram-negative class belonging to the phylum Firmicutes that is enriched in the gut microbiota of HIV-infected subjects with CD4+ T cell recovery higher than 500 cell/μL. Its abundance was positively correlated with several inflammation markers that mediate an LPS-induced immune response (Baltazar-Díaz et al., 2023).
Taken together, all of these results show that at the second time point, the gut microbial activity partially recovered but not completely, similar to that of healthy subjects. This includes the reduction and the enrichment of some genera correlated to worse and better HIV progression, respectively. This may suggest that the regiment therapy is somewhat effective in our cohort, although not optimal. Additionally, the significant correlation of Akkermansia, Achromobacter, and Stenotrophomonas with antiretroviral duration implies that gut integrity may still be compromised despite the improvement in CD4+ T-cell count and sCD14 during the treatment period. However, a longer ART duration does not necessarily mean a better gut microbial environment as we also found an increasing trend of microbial translocation markers at the late treatment period which resulting in a mixed bacterial environment in the gut.
The limitation of this research is that we could only analyze the correlation of clinical features with bacterial composition at one time point (baseline_case) because we could not collect clinical information, such as CD4+ T-cell count and viral load from the follow-up_case samples due to logistical problems. The reason was because we could not import the required reagents for the analysis in time. Furthermore, although the viral load range is very large, but the median viral load of 3,729 copies/mL may suggest that this data may only be applicable to PLWH with relatively low viral burden. Our data are also limited to perform additional analyses of bacterial composition in high and low values of the clinical features. Another concern is we could not rule out the time of infection to further classify the PLWH as recently or chronically infected. This limits the understanding that the time to treatment or the antiretroviral itself may have a direct effect in modifying the gut microbiome. At least one study showed that nucleoside reverse-transcriptase inhibitor (NRTI)-based antiretroviral therapy has a more suppressive impact on microbiota composition and diversity in the gut (Imahashi et al., 2021). Lastly, in the publications prior to this analysis, we found a strong correlation of ART treatment with antibiotic usage (Parbie et al., 2021b). As antibiotics have a direct effect in modifying gut microflora, we could not rule out the possibility of antibiotics reduced the diversity at late treatment period observed here.
In conclusion, we have presented here a longitudinal analysis of the gut microbiome composition in Ghanaian PLWH six months apart. We have shown that at the second time point, gut microbial activity was partially recovered, similar to that of healthy subjects, but not completely. Despite the relatively controlled disease, we found that genera correlated with both worse and better gut microbiomes were enriched and highly correlated with the duration of treatment, likely suggesting a volatile gut environment. This may support the theory that the gut microbiome does not recover despite controlled disease.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ddbj.nig.ac.jp/, DRA010770 https://www.ddbj.nig.ac.jp/, DRA017623.
The studies involving humans were approved by the Institutional Review Board of Noguchi Memorial Institute for Medical Research (NMIMR) and the Ethical Committee of National Institute of Infectious Diseases (NIID). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
LR: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. PP: Data curation, Investigation, Methodology, Writing – review & editing. TaM: Data curation, Investigation, Methodology, Supervision, Writing – review & editing. AI: Writing – review & editing. SM: Formal analysis, Writing – review & editing. CA: Data curation, Writing – review & editing. DK: Data curation, Writing – review & editing. EB: Data curation, Writing – review & editing. SO: Data curation, Writing – review & editing. HK: Writing – review & editing. KI: Data curation, Methodology, Supervision, Writing – review & editing. WA: Funding acquisition, Supervision, Writing – review & editing. TeM: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Japan Agency for Medical Research and Development (AMED) (grant numbers: JP23fk0410035, JP23wm0125012, JP18fk0410003, JP20fk0410011, JP20fk0108125, JP20fk0108139, JP19kk0205024, JP20jk0210002, JP21am0401029, 223fa627003h0001, and JP21ae0121040), AMED-JICA (the Science and Technology Research Partnership for Sustainable Development [SATREPS]; JP20jm0110012), and the Ministry of Education, Culture, Sports, Science, and Technology in Japan (18H02666). The Kumamoto University International Scholarship Program generously provided travel support during this study.
The authors thank the staff of the Virology Department of the Noguchi Memorial Institute for Medical Research, University of Ghana in Accra, Ghana; Eastern Regional Hospital in Koforidua, Ghana; the Ghana Health Service; the Ghana National AIDS Control Program; and the AIDS Research Center of the National Institute of Infectious Diseases in Japan for their administrative and technical support. The authors are sincerely grateful to all the healthy uninfected and HIV-1-infected individuals who consented to participate in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1359402/full#supplementary-material
Armstrong, A. J. S., Shaffer, M., Nusbacher, N. M., Griesmer, C., Fiorillo, S., Schneider, J. M., et al. (2018). An exploration of Prevotella-rich microbiomes in HIV and men who have sex with men. Microbiome 6, 1–16. doi: 10.1186/s40168-018-0580-7
Baltazar-Díaz, T. A., Amador-Lara, F., Andrade-Villanueva, J. F., González-Hernández, L. A., Cabrera-Silva, R. I., Sánchez-Reyes, K., et al. (2023). Gut bacterial communities in HIV-infected individuals with metabolic syndrome: effects of the therapy with integrase strand transfer inhibitor-based and protease inhibitor-based regimens. Microorganisms 11:951, 1–21. doi: 10.3390/microorganisms11040951
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Brito, I. L., Yilmaz, S., Huang, K., Xu, L., Jupiter, S. D., Jenkins, A. P., et al. (2016). Mobile genes in the human microbiome are structured from global to individual scales. Nature 535, 435–439. doi: 10.1038/nature18927
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Calza, L., Manfredi, R., and Chiodo, F. (2003). Stenotrophomonas (Xanthomonas) maltophilia as an emerging opportunistic pathogen in association with HIV infection: a 10-year surveillance study. Infection 31, 155–161. doi: 10.1007/s15010-003-3113-6
Caspi, R., Billington, R., Keseler, I. M., Kothari, A., Krummenacker, M., Midford, P. E., et al. (2020). The MetaCyc database of metabolic pathways and enzymes-a 2019 update. Nucleic Acids Res. 48, D445–D453. doi: 10.1093/nar/gkz862
Crakes, K. R., and Jiang, G. (2019). Gut microbiome alterations during HIV/SIV infection: implications for HIV cure. Front. Microbiol. 10, 1–10. doi: 10.3389/fmicb.2019.01104
Dandekar, S., George, M. D., and Bäumler, A. J. (2010). Th17 cells, HIV and the gut mucosal barrier. Curr. Opin. HIV AIDS 5, 173–178. doi: 10.1097/COH.0b013e328335eda3
de Filippo, C., Cavalieri, D., di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S. A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
Dillon, S. M., Lee, E. J., Kotter, C. V., Austin, G. L., Dong, Z., Hecht, D. K., et al. (2014). An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 7, 983–994. doi: 10.1038/mi.2013.116
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Estes, J. D., Harris, L. D., Klatt, N. R., Tabb, B., Pittaluga, S., Paiardini, M., et al. (2010). Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 6, 49–50. doi: 10.1371/journal.ppat.1001052
Gordon, S. B., Chaponda, M., Walsh, A. L., Whitty, C. J. M., Gordon, M. A., Machili, C. E., et al. (2002). Pneumococcal disease in HIV-infected Malawian adults: acute mortality and long-term survival. AIDS 16, 1409–1417. doi: 10.1097/00002030-200207050-00013
Guadalupe, M., Reay, E., Sankaran, S., Prindiville, T., Flamm, J., McNeil, A., et al. (2003). Severe CD4+ T-cell depletion in gut lymphoid tissue during primary HIV-1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 77, 11708–11717. doi: 10.1128/JVI.77.21.11708-11717.2003
Guillén, Y., Noguera-Julian, M., Rivera, J., Casadellà, M., Zevin, A. S., Rocafort, M., et al. (2019). Low nadir CD4+ T-cell counts predict gut dysbiosis in HIV-1 infection. Mucosal Immunol. 12, 232–246. doi: 10.1038/s41385-018-0083-7
He, Y., Wu, W., Zheng, H. M., Li, P., McDonald, D., Sheng, H. F., et al. (2018). Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 24, 1532–1535. doi: 10.1038/s41591-018-0164-x
HIV.gov . The global HIV/AIDS epidemic. (2022). Available at: https://www.hiv.gov/hiv-basics/overview/data-and-trends/global-statistics/. (Accessed: July 16, 2023).
Imahashi, M., Ode, H., Kobayashi, A., Nemoto, M., Matsuda, M., Hashiba, C., et al. (2021). Impact of long term antiretroviral therapy on gut and oral microbiotas in HIV-1-infected patients. Sci. Rep. 11, 1–10. doi: 10.1038/s41598-020-80247-8
Ishizaka, A., Koga, M., Mizutani, T., Parbie, P. C., Prawisuda, D., Yusa, N., et al. (2021). Unique gut microbiome in HIV patients on antiretroviral therapy (ART) suggests association with chronic inflammation. Microbiol. Spectr. 9, e00708–e00721. doi: 10.1128/Spectrum.00708-21
Kelley, C. F., Kraft, C. S., de Man, T. J. B., Duphare, C., Lee, H. W., Yang, J., et al. (2017). The rectal mucosa and condomless receptive anal intercourse in HIV-negative MSM: implications for HIV transmission and prevention. Mucosal Immunol. 10, 996–1007. doi: 10.1038/mi.2016.97
Khan, S., Telwatte, S., Trapecar, M., Yukl, S., and Sanjabi, S. (2017). Differentiating immune cell targets in gut-associated lymphoid tissue for HIV cure. AIDS Res. Hum. Retrovir. 33, S-40–S-58. doi: 10.1089/aid.2017.0153
Kim, S. W., Suda, W., Kim, S., Oshima, K., Fukuda, S., Ohno, H., et al. (2013). Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 20, 241–253. doi: 10.1093/dnares/dst006
Koay, W. L. A., Siems, L. V., and Persaud, D. (2018). The microbiome and HIV persistence: implications for viral remission and cure. Curr. Opin. HIV AIDS 13, 61–68. doi: 10.1097/COH.0000000000000434
Leão, I., de Carvalho, T. B., Henriques, V., Ferreira, C., Sampaio-Maia, B., and Manaia, C. M. (2023). Pseudomonadota in the oral cavity: a glimpse into the environment-human nexus. Appl. Microbiol. Biotechnol. 107, 517–534. doi: 10.1007/s00253-022-12333-y
Li, S. X., Armstrong, A. J. S., Neff, C. P., Shaffer, M., Lozupone, C. A., and Palmer, B. E. (2016). Complexities of gut microbiome dysbiosis in the context of HIV infection and antiretroviral therapy. Clin. Pharmacol. Ther. 99, 600–611. doi: 10.1002/cpt.363
Ling, Z., Jin, C., Xie, T., Cheng, Y., Li, L., and Wu, N. (2016). Alterations in the fecal microbiota of patients with HIV-1 infection: an observational study in a Chinese population. Sci. Rep. 6:30673. doi: 10.1038/srep30673
Lopez-Siles, M., Duncan, S. H., Garcia-Gil, L. J., and Martinez-Medina, M. (2017). Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J. 11, 841–852. doi: 10.1038/ismej.2016.176
Lozupone, C. A., Li, M., Campbell, T. B., Flores, S. C., Linderman, D., Gebert, M. J., et al. (2013). Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe 14, 329–339. doi: 10.1016/j.chom.2013.08.006
Lu, D., Zhang, J., Wang, Y., Geng, S., Zhang, Z., Xu, Y., et al. (2021). Association between CD4+ T cell counts and gut microbiota and serum cytokines levels in HIV-infected immunological non-responders. BMC Infect. Dis. 21, 1–11. doi: 10.1186/s12879-021-06491-z
Ma, S., Shungin, D., Mallick, H., Schirmer, M., Nguyen, L. H., Kolde, R., et al. (2022). Population structure discovery in meta-analyzed microbial communities and inflammatory bowel disease using MMUPHin. Genome Biol. 23, 1–31. doi: 10.1186/s13059-022-02753-4
Mallick, H., Rahnavard, A., McIver, L. J., Ma, S., Zhang, Y., Nguyen, L. H., et al. (2021). Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, 1–27. doi: 10.1371/journal.pcbi.1009442
Mandal, S., Van Treuren, W., White, R. A., Eggesbø, M., Knight, R., Peddada, S. D., et al. (2015). Analysis of composition of microbiomes, a novel method for studying microbial composition. Microb. Ecol. Health Dis. 26, 1–7. doi: 10.3402/mehd.v26.27663
Marchetti, G., Tincati, C., and Silvestri, G. (2013). Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin. Microbiol. Rev. 26, 2–18. doi: 10.1128/CMR.00050-12
McHardy, I. H., Li, X., Tong, M., Ruegger, P., Jacobs, J., Borneman, J., et al. (2013). HIV infection is associated with compositional and functional shifts in the rectal mucosal microbiota. Microbiome 1, 1–12. doi: 10.1186/2049-2618-1-26
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Monaco, C. L., Gootenberg, D. B., Zhao, G., Handley, S. A., Ghebremichael, M. S., Lim, E. S., et al. (2016). Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe 19, 311–322. doi: 10.1016/j.chom.2016.02.011
Mutlu, E. A., Keshavarzian, A., Losurdo, J., Swanson, G., Siewe, B., Forsyth, C., et al. (2014). A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog. 10:e1003829. doi: 10.1371/journal.ppat.1003829
Noguera-Julian, M., Rocafort, M., Guillén, Y., Rivera, J., Casadellà, M., Nowak, P., et al. (2016). Gut microbiota linked to sexual preference and HIV infection. EBioMedicine 5, 135–146. doi: 10.1016/j.ebiom.2016.01.032
Ouyang, J., Lin, J., Isnard, S., Fombuena, B., Peng, X., Marette, A., et al. (2020). The bacterium Akkermansia muciniphila: a sentinel for gut permeability and its relevance to HIV-related inflammation. Front. Immunol. 11, 1–9. doi: 10.3389/fimmu.2020.00645
Parbie, P. K., Mizutani, T., Ishizaka, A., Kawana-Tachikawa, A., Runtuwene, L. R., Seki, S., et al. (2021a). Fecal microbiome composition in healthy adults in Ghana. Jpn. J. Infect. Dis. 74, 42–47. doi: 10.7883/yoken.JJID.2020.469
Parbie, P. K., Mizutani, T., Ishizaka, A., Kawana-Tachikawa, A., Runtuwene, L. R., Seki, S., et al. (2021b). Dysbiotic fecal microbiome in HIV-1 infected individuals in Ghana. Front. Cell. Infect. Microbiol. 11, 1–13. doi: 10.3389/fcimb.2021.646467
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Shacklett, B. L., and Anton, P. A. (2010). HIV infection and gut mucosal immune function: updates on pathogenesis with implications for management and intervention. Curr. Infect. Dis. Rep. 12, 19–27. doi: 10.1007/s11908-009-0072-9
Tatro, E. T., Purnajo, I., Richman, D. D., Smith, D. M., and Gianella, S. (2014). Antibody response to Achromobacter xylosoxidans during HIV infection is associated with lower CD4 levels and increased lymphocyte activation. Clin. Vaccine Immunol. 21, 46–50. doi: 10.1128/CVI.00553-13
Vacca, M., Celano, G., Calabrese, F. M., Portincasa, P., Gobbetti, M., and de Angelis, M. (2020). The controversial role of human gut Lachnospiraceae. Microorganisms 8, 1–25. doi: 10.3390/microorganisms8040573
Vázquez-Castellanos, J. F., Serrano-Villar, S., Latorre, A., Artacho, A., Ferrús, M. L., Madrid, N., et al. (2015). Altered metabolism of gut microbiota contributes to chronic immune activation in HIV-infected individuals. Mucosal Immunol. 8, 760–772. doi: 10.1038/mi.2014.107
Vujkovic-Cvijin, I., Dunham, R. M., Iwai, S., Maher, M. C., and Albright, R. G. (2013). Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci. Transl. Med. 5, 1–7. doi: 10.1126/scitranslmed.3006438
Vujkovic-Cvijin, I., Sortino, O., Verheij, E., Sklar, J., Wit, F. W., Kootstra, N. A., et al. (2020). HIV-associated gut dysbiosis is independent of sexual practice and correlates with noncommunicable diseases. Nat. Commun. 11:2448. doi: 10.1038/s41467-020-16222-8
Wallet, M. A., Rodriguez, C. A., Yin, L., Saporta, S., Chinratanapisit, S., Hou, W., et al. (2010). Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T cell activation following therapy. AIDS 24, 1281–1290. doi: 10.1097/QAD.0b013e328339e228
Wang, M., Yin, S., Qin, Q., Peng, Y., Hu, Z., Zhu, X., et al. (2021). Stenotrophomonas-maltophilia inhibits host cellular immunity by activating PD-1/PD-L1 signaling pathway to induce T-cell exhaustion. Mol. Immunol. 130, 37–48. doi: 10.1016/j.molimm.2020.12.019
Wexler, H. M. (2007). Bacteroides: the good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 20, 593–621. doi: 10.1128/CMR.00008-07
Keywords: HIV, PLWH, Ghana, longitudinal analysis, gut microbiome dysbiois
Citation: Runtuwene LR, Parbie PK, Mizutani T, Ishizaka A, Matsuoka S, Abana CZ-Y, Kushitor D, Bonney EY, Ofori SB, Kiyono H, Ishikawa K, Ampofo WK and Matano T (2024) Longitudinal analysis of microbiome composition in Ghanaians living with HIV-1. Front. Microbiol. 15:1359402. doi: 10.3389/fmicb.2024.1359402
Edited by:
Alina Maria Holban, University of Bucharest, RomaniaReviewed by:
Chunyi Zhou, University of Nebraska Medical Center, United StatesCopyright © 2024 Runtuwene, Parbie, Mizutani, Ishizaka, Matsuoka, Abana, Kushitor, Bonney, Ofori, Kiyono, Ishikawa, Ampofo and Matano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tetsuro Matano, bWF0YW5vQGltcy51LXRva3lvLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.