 Elvira Rey Redondo
Elvira Rey Redondo Yangbing Xu
Yangbing Xu Charmaine Cheuk Man Yung
Charmaine Cheuk Man Yung
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 May 2024
Sec. Aquatic Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1358574
This article is part of the Research Topic Insights in Aquatic Microbiology: 2023 View all 10 articles
Mamiellophyceae are dominant marine algae in much of the ocean, the most prevalent genera belonging to the order Mamiellales: Micromonas, Ostreococcus and Bathycoccus, whose genetics and global distributions have been extensively studied. Conversely, the genus Mantoniella, despite its potential ecological importance, remains relatively under-characterised. In this study, we isolated and characterised a novel species of Mamiellophyceae, Mantoniella tinhauana, from subtropical coastal waters in the South China Sea. Morphologically, it resembles other Mantoniella species; however, a comparative analysis of the 18S and ITS2 marker genes revealed its genetic distinctiveness. Furthermore, we sequenced and assembled the first genome of Mantoniella tinhauana, uncovering significant differences from previously studied Mamiellophyceae species. Notably, the genome lacked any detectable outlier chromosomes and exhibited numerous unique orthogroups. We explored gene groups associated with meiosis, scale and flagella formation, shedding light on species divergence, yet further investigation is warranted. To elucidate the biogeography of Mantoniella tinhauana, we conducted a comprehensive analysis using global metagenomic read mapping to the newly sequenced genome. Our findings indicate this species exhibits a cosmopolitan distribution with a low-level prevalence worldwide. Understanding the intricate dynamics between Mamiellophyceae and the environment is crucial for comprehending their impact on the ocean ecosystem and accurately predicting their response to forthcoming environmental changes.
Mamiellophyceae, an early-branching class of photosynthetic picoeukaryotes within the Chlorophyta (Marin and Melkonian, 2010), dominate various ocean regions (Wang et al., 2019; Yung et al., 2022), particularly coastal waters (Lopes dos Santos et al., 2017; Tragin and Vaulot, 2018). Among the Mamiellophyceae, the order Mamiellales, which includes the families Mamiellaceae and Bathycoccaceae, encompasses the most extensively studied marine Mamiellophyceae genera: Micromonas (Mamiellaceae), Ostreococcus and Bathycoccus (Bathycoccaceae), and more recently, Mantoniella (Mamiellaceae).
Understanding the biogeography and diversity of Mamiellophyceae is crucial due to their important role in primary production, impact on ocean ecosystems and sensitivity to environmental disturbances (Monier et al., 2016). Distinguishing Mamiellophyceae species based on morphology alone is challenging, as evidenced by the similarities among previously identified Mantoniella species (Yau et al., 2020b). Therefore, molecular methods are essential for accurate species identification and unravelling the relationship between their distribution and environmental factors. Previous studies have revealed genetic adaptations and distinct ecological niches within different Mamiellophyceae clades and species (Lovejoy et al., 2007; Foulon et al., 2008; Demir-Hilton et al., 2011; Subirana et al., 2013; Hu et al., 2016; Simmons et al., 2016; Simon et al., 2017; Tragin and Vaulot, 2019). For example, M. antarctica (Marchant et al., 1989), M. baffinensis and M. beaufortii (Balzano et al., 2012; Yau et al., 2020b) were exclusively found in cold, polar waters, while M. squamata is the only named Mantoniella species known to have a distribution beyond polar regions (Tragin and Vaulot, 2019; Belevich et al., 2021).
The predominant approach in Mamiellophyceae biogeography studies is metabarcoding, which involves sequencing marker genes such as the hypervariable regions of 18S ribosomal RNA (rRNA) (Monier et al., 2016; Tragin and Vaulot, 2019; Belevich et al., 2021). Metabarcoding has inherent limitations in true quantification and suffers from biases stemming from PCR recovery, strain polymorphisms, gene copy number variations and sequencing artifacts (Monier et al., 2016), and can underestimate the diversity of microbial eukaryotes (Piganeau et al., 2011a). For instance, Bathycoccus 18S rRNA sequences were found to be identical in different strains, masking huge variations in other genomic regions (Leconte et al., 2020).
To address the limitations of metabarcoding, metagenome read mapping and metagenome-assembled genomes (MAGs) have emerged as valuable tools for biogeographic studies. Mapping metagenomic reads to reference genomes provides a more accurate and sensitive method for biogeographic quantification, avoiding targeted amplicon biases and gene content variations, and enabling higher strain resolution. This approach has been successfully applied to six Mamiellales genomes from the well-studied genera (Micromonas, Ostreococcus and Bathycoccus) using global metagenomic data from the Tara Oceans Expedition (Leconte et al., 2020). However, the lack of reference genomes from cultured strains has made achieving genome-resolved metagenomic mapping with species resolution challenging for Mantoniella. Although a Mantoniella MAG was identified in one Tara metagenomic sample from the southwestern Pacific Ocean (Delmont et al., 2022), the binning of this MAG was based on marker genes, resulting in an incomplete MAG.
Existing research on the global biogeography of Mamiellophyceae has primarily focused on regions outside the western Pacific Ocean, particularly overlooking subtropical coastal South Asia. Localised studies conducted in the West Philippine Sea (Dela Peña et al., 2021), Bohai Sea (Xu et al., 2017), South China Sea (Wu et al., 2014; Lin et al., 2017a, 2021) and East China Sea (Lin et al., 2017b, 2022) have highlighted the importance of factors like distance to the coast and depth in shaping the distribution patterns of Mamiellophyceae species. Despite these efforts, limited information exists on the occurrence of the genus Mantoniella in subtropical West Pacific waters, with studies indicating its presence at certain sites but without detailed species identification (Lin et al., 2017b, 2021, 2022). Notably, no cultures of Mantoniella have been isolated from the Pacific Ocean. Globally, only a few large-scale metabarcoding studies have investigated the biogeography of Mantoniella (Lovejoy et al., 2007; Tragin and Vaulot, 2019; Yau et al., 2020b).
Our study addresses research gaps on the biogeography of Mamiellophyceae, focusing specifically on the genus Mantoniella. We present a newly identified species, Mantoniella tinhauana, isolated from the subtropical coastal waters of the South China Sea. This represents the first-ever whole genome project for Mantoniella. By analysing the genome of this novel species and exploring its global distribution using metagenome datasets, we provide insights into its morphology, phylogeny, genome characteristics, evolutionary relationships and ecological niches. Our findings contribute to the understanding of the ecological significance and biogeography of Mamiellophyceae, furthering our knowledge of these important photosynthetic organisms in marine ecosystems.
Coastal surface seawater samples (2 m depth) were collected from Lau Fau Shan in the Pearl River Estuary (22°28′09.0′N 113°58′50.1′E) in June 2020. The collected samples were sequentially filtered through 50 μm mesh and 1 μm polycarbonate filters (Whatman) to remove large multicellular organisms and capture the suitable size fraction for Mamiellophyceae (Yung et al., 2022). The filtrates were grown in flasks (SPL Life Sciences) in L1 medium (Guillard and Hargraves, 1993) and cultured at room temperature (21–23°C) in a shaking incubator (100 rpm) under a 12-h light/dark cycle (30 μmol photons m−2 s−1). After 2–3 weeks, collected algal pellets were used for 18S rRNA gene amplification (see below) to obtain initial taxonomic classification. Samples containing Mamiellophyceae were purified through serial dilution and treated with antibiotics. The novel Mantoniella strain obtained from this process was subcultured.
For transmission electron microscopy (TEM), culture was centrifuged, the pellet resuspended and fixed with glutaraldehyde (2.5% in 0.2 μm-filtered seawater), rinsed with Na cacodylate buffer (0.1 mol l−1) and fixed in OsO4 (1%), then dehydrated with a graded series of acetone solutions. After resin embedding and polymerisation, ultrathin sections were cut using a Leica EM UC7 Ultramicrotome and stained with uranyl acetate and lead citrate. The stained samples were examined under a Hitachi HT7700 Transmission Electron Microscope. Cell diameters were measured using the TEM images and scales as viewed on ImageJ (Collins, 2007).
For phylogenetic analysis, DNA was extracted from dense Mantoniella culture by centrifugation, resuspension and incubation at 98°C for 10 min, based on Saulnier et al. (2009) and Jahn et al. (2014). The 18S region was amplified following the protocol described by Hadziavdic et al. (2014) and Sanger sequenced by Tech Dragon (Hong Kong).
For whole genome sequencing, DNA was extracted from Mantoniella pellets using a Qiagen DNeasy Plant Pro Kit. Short-read Illumina sequencing was conducted by Novogene (Hong Kong) on the Novaseq 6,000 PE150 platform. Long-read DNA was extracted using a modified CTAB protocol (Stark et al., 2020) and sequenced on the PacBio SMRT Sequel platform by Novogene. To improve assembly contiguity, genetic material was extracted for proximity ligation (Hi-C) based on the protocol by Lafontaine et al. (2021). The chromatin was cross-linked following the instructions provided by Phase Genomics (United States) and subsequently subjected to Illumina Hi-C sequencing.
To improve gene prediction, a time-series of samples was collected from an exponentially growing culture every 3 h over a 24-h period. RNA was extracted using a Direct-zol RNA Miniprep Kit (Zymo Research) to capture the gene activity throughout the day. The RNA from the 8 timepoints was pooled and sequenced using Illumina PE150 by Novogene.
Paired-end Illumina reads were trimmed with trimmomatic v0.39 (Bolger et al., 2014) and quality controlled with fastp v0.23.2 (Chen et al., 2018). The short Illumina reads and long PacBio reads were combined to generate an initial hybrid assembly using MaSuRCA v4.0.9 (Zimin et al., 2013). Subsequently, the assembly was refined using POLCA (Zimin and Salzberg, 2020) for calling alternatives and improving accuracy.
For contig scaffolding and misassembly correction, Hi-C reads were aligned to the draft genome assembly using bwa-mem2 v2.0 (Li and Durbin, 2009). The resulting alignments were input into SALSA2 v2.3 (with default parameters and “-e GATC --clean”) (Ghurye et al., 2019).
Anvi’o v7.1 (Eren et al., 2015) was used to create a contigs database for removing extraneous sequences in the draft genome. Contigs were split into 2000 nt lengths, clustered and binned. HMM modelling and functional annotation against NCBI’s COGs database (Tatusov et al., 1997) were performed. Annotated bins and GC contents were visually examined, and genome bins not belonging to eukaryotic nuclear DNA were manually removed (Delmont and Eren, 2016).
To further enhance the draft genome, the initial trimmed and quality-controlled Illumina reads were realigned to the decontaminated draft genome using Bowtie2 v2.2.5 (Langmead and Salzberg, 2012). The resulting alignment underwent gap fixing, misassembly identification, variant calling, and other improvements using Pilon v1.24 (Walker et al., 2014).
Mamiellophyceae 18S rRNA genes and Mamiellaceae ITS region sequences were aligned onto the novel Mantoniella tinhauana genome assembly using Geneious Prime v2022.2.2. The resulting 18S gene sequence obtained from this mapping approach was 1,784 bp in length and showed 100% identity to the amplicon sequencing-derived sequence, with greater completeness (deposited to GenBank: OR835992). To extract the ITS2 gene, the novel Mantoniella ITS region (deposited to GenBank: OR835993) was submitted to the University of Würzburg ITS2 Database (Merget et al., 2012) online platform using the Viridiplantae model and default parameters, resulting in a 253 bp-long ITS2 gene sequence.
The novel Mantoniella ITS2 structure was predicted using Vienna files without gaps from Yau et al. (2020b) as templates on the ITS2 Database (Merget et al., 2012). The resulting Vienna file was exported and aligned with other Mamiellophyceae ITS2 structure files (including gaps) using ClustalW v2.1 (Aiyar, 1999) in 4SALE v1.7.1 (Seibel et al., 2006) (Supplementary Table S1). The novel ITS2 structure was visualised using the ViennaRNA web service forna (Gendron et al., 2001). Each base pair in the helix structures was compared to those of other Mantoniella and Mamiellophyceae ITS2 using the 4SALE alignment and manually labelled. Individual RNA helix sequences were extracted using Geneious Prime and also drawn on the ViennaRNA forna platform.
To construct the phylogenetic tree based on the 18S marker gene, the M. tinhauana 18S rRNA gene sequence was blasted against the NCBI nucleotide database in Geneious Prime (Supplementary Table S2). The top 10 matches, along with other complete Mantoniella 18S sequences (Tragin and Vaulot, 2019; Yau et al., 2020b) and representatives of other Mamiellophyceae 18S sequences were used to create the phylogenetic tree.
A total of 41 Mamiellophyceae 18S sequences, including the novel strain, were aligned using MAFFT v7.453 (Katoh and Standley, 2013). Low-quality positions containing gaps in over 50% of the sequences were removed with Goalign clean sites v0.3.5 (Lemoine and Gascuel, 2021). Maximum likelihood (ML) trees were built using IQ-TREE v2.2.0 (Nguyen et al., 2015) using the LG + F + R4 model of substitution and generating Shimodaira-Hasegawa (SH)-like approximate likelihood ratio test (aLRT) branch support values from 1,000 replicates. Markov chain Monte Carlo iterations were performed on the alignments for 1,000,000 generations sampling every 100 generations with 100,000 burn-in length using MrBayes v3.2.6 (Ronquist et al., 2012) as implemented on Geneious Prime, generating Bayesian posterior probability values for each node. The resulting tree was visualised using Interactive Tree Of Life (iTOL) v5 (Letunic and Bork, 2021).
To construct the ITS2 marker gene-based phylogenetic trees, gapped ITS2 sequences from 15 Mamiellophyceae species were aligned. ML and Bayesian trees were generated using the same methods as above.
To assess the completeness of the draft genome and Mamiellophyceae reference genomes, BUSCO v5.4.3 (Simão et al., 2015) was run against the Chlorophyta universal single-copy marker database in “genome” mode. Scaffold ends were manually examined for telomeric tandem repeats and scaffold and whole genome GC contents were computed using Geneious Prime.
The ploidy of the novel Mantoniella genome was estimated by aligning Illumina reads against the draft genome using ploidyNGS v3.1.3 (Corrêa dos Santos et al., 2017).
Transposable elements in the draft genome were identified using RepeatModeler v2.0.4 (Flynn et al., 2020), followed by masking of these elements, interspersed repeats and low complexity DNA sequences using RepeatMasker v4.1.4 (Smit et al., 2015) with RMBlast v2.13.0. Transcript reads from the transcriptome time-series were trimmed with trimmomatic, quality controlled with fastp as before, and aligned to the RepeatMasker-masked genome using STAR v2.1.10 (Dobin et al., 2013). The masked genome and the RNA-seq STAR alignment were input into BRAKER v3.0.2 (Brůna et al., 2021) for protein coding gene structure prediction in the novel genome using GeneMark and AUGUSTUS (Lukashin and Borodovsky, 1998; Keller et al., 2011), following BRAKER pipeline B.
The transcript reads were ribodepleted with SortMeRNA v4.3 (Kopylova et al., 2012) and assembled into a transcriptome with rnaSPAdes v3.15.4 (Bushmanova et al., 2019). Assembly quality was assessed using BUSCO in “transcriptome” mode against the Chlorophyta database, resulting in 94% complete BUSCOs. Open reading frames (ORFs) within the transcriptome were identified with Transdecoder v5.7.0 (Haas, 2023).
The unmasked genome, ab initio predictions from GeneMark and AUGUSTUS, output from BRAKER, and transcriptome ORF predictions from Transdecoder were combined in EVidenceModeler v2.0.0 (Haas et al., 2008) to generate a consensus proteome and a gene predictions file. The whole novel proteome was functionally annotated using HMMER v3.3 (Eddy, 2011) to generate Pfam (Mistry et al., 2021) domain annotations.
Mamiellophyceae proteomes (excluding O. mediterraneus) were obtained from NCBI. Gene synteny comparison was performed using the M. tinhauana gene predictions and proteome outputs from EVidenceModeler, along with those of the five other Mamiellophyceae species. The six proteomes were concatenated and subjected to all-against-all blastp analysis. MCScanX (Wang et al., 2012) was used to compare syntenic gene blocks with a minimum block size of five genes and a maximum gap size of 25 genes (Cho et al., 2023). The collinearity output was visualised using SynVisio in Multi-Level Analysis Tree View mode (Bandi et al., 2022).
The Big Outlier Chromosome (BOC), the mating type locus (MT) region within each BOC as delimited by Benites et al. (2021), and the Small Outlier Chromosome (SOC) annotated sequences were extracted from each Mamiellophyceae proteome. Gene block synteny analysis was performed using MCScanX as described above, comparing the BOC/MT/SOC regions of each Mamiellophyceae species against the entire Mantoniella proteome.
Orthogroups in the six proteomes were compared using Orthofinder v2.5.4 (Emms and Kelly, 2019) and visualised using UpSetR (Conway et al., 2017). Orthogroup information and the species tree produced by Orthofinder (made ultrametric) were used for gene family expansions and contractions analysis in CAFE v5 (Mendes et al., 2021) with a p-value cutoff of 0.05. The expansion and contraction tree was drawn using cafe5_draw_tree.py and pie charts for each tree node were added using meta-chart.com. Significantly expanded families in M. tinhauana were functionally annotated using eggNOG-mapper v2.1.9 (Cantalapiedra et al., 2021) with DIAMOND (Buchfink et al., 2021) and the eggNOG v5.0 database (Huerta-Cepas et al., 2019). GO enrichment analysis was performed following the published protocol (Zhou, 2022) in R and plotted using ggplot2 (Wickham, 2006).
Metagenomic reads from the Tara Oceans dataset were obtained from two projects (PRJEB4352-global and PRJEB9691-polar) through the European Nucleotide Archive in 2023. The samples were collected from the ocean surface at a depth of 5 m. The size fraction of interest was >0.8 μm (>0.8 μm, 0.8–5 μm or 0.8–20 μm samples were collected from different sites). In cases of site duplication, preference was given to >0.8 μm samples. A total of 124 metagenomic samples were mapped onto the full M. tinhauana genome and the Micromonas pusilla CCMP1545 genome (Worden et al., 2009) as a control using BBMap v38.96 (Bushnell, 2016) with a minimum identity of 95%. The mapping values were compiled using pileup.sh from the BBMap suite (Malfertheiner et al., 2022). To account for variations in total read numbers and genome lengths, Reads per Kilobase per Million (RPKM) values were computed as follows:
The coordinates and RPKM were collated. Custom code in R, along with the ggplot2 (Wickham, 2006) and scatterpie (Yu and Yu, 2018) packages, were utilised to plot the data onto world maps. Median values for environmental measurements for each sample site were downloaded (Supplementary Table S3). Spearman’s rank correlation coefficients (SRCC) were calculated in R for each pair of environmental variables, and half the variables with more than 0.5 pairwise correlation were removed. The variables that were not highly collinear (only temperature, salinity and nitrate) were used for SRCC calculation with the M. tinhauana RPKM and plotted with ggpairs (GGally) (Schloerke et al., 2018). Some nitrate values were missing and so those samples were excluded from the nitrate-RPKM correlation study.
In an effort to isolate Mantoniella species from coastal waters in the Western Pacific, a sweep of green algae from water samples were collected along the coast of Hong Kong in different seasons and subsequent purification using serial dilutions was performed. To assess their evolutionary distance compared to other cultured Mamiellophyceae, we sequenced and compared their partial 18S rRNA gene with algal sequences in GenBank. We discovered that one algal culture exhibited a remarkably high similarity to Mantoniella squamata, as evidenced by a 99.7% nucleotide identity.
To determine the taxonomic classification of this newly isolated alga, we constructed a phylogenetic tree based on the complete 18S rRNA gene. This analysis positioned the alga on the outer boundary of the Mantoniella genus, showing a close affinity to Micromonas (Figure 1). We named this novel species Mantoniella tinhauana. To aid in classification, Mantoniella strains were assigned to clades based on this phylogeny and previous studies (Tragin and Vaulot, 2019; Yau et al., 2020b). The novel strain had a very high shared identity (up to 99.9%) with several partial 18S sequences, for example strains assigned to clade B for which only the 18S V4 region was available (Tragin and Vaulot, 2019). As complete 18S sequences were lacking for certain strains (these were excluded), the novel Mantoniella could not be assigned to any known clade based on the whole 18S gene. As discussed before, relying solely on the 18S or any single marker gene for phylogenetic assessment may not be entirely reliable, especially in unicellular eukaryotes. This limitation is particularly evident in a genus like Mantoniella, which only has a few cultured and extensively studied species. To address this, we also examined the ITS2 marker.
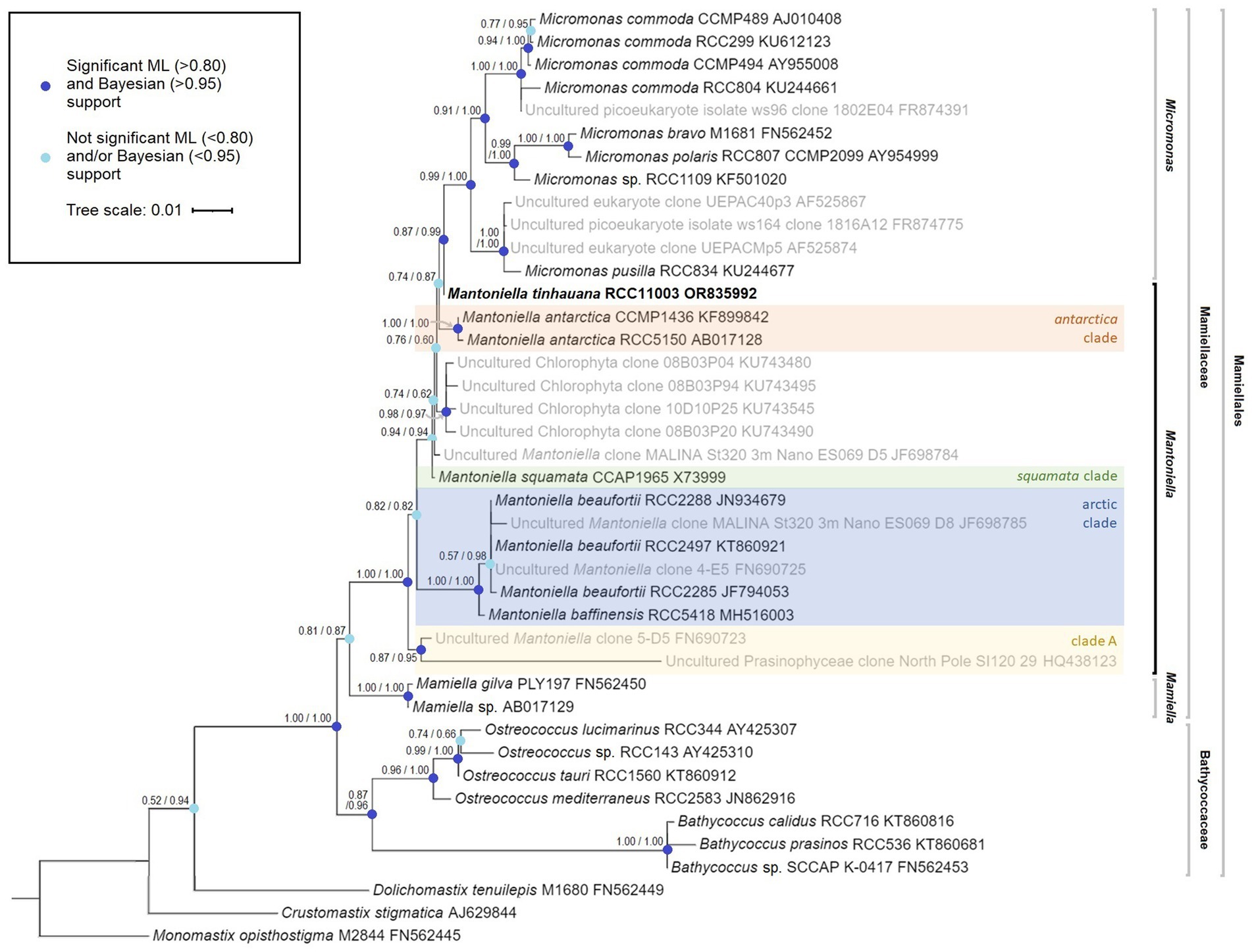
Figure 1. Mamiellophyceae 18S rRNA gene maximum likelihood tree, featuring species name, species code and 18S GenBank code (Monomastix opisthostigma as the outgroup). The numbers represent node support values, with the SH-like support values followed by the Bayesian posterior probability values. Nodes with enough support from both values (SH>0.80 and Bayes>0.95) are indicated in dark blue whereas nodes lacking sufficient support from either or both values are marked in light blue. Uncultured strains, obtained through blast search and previous studies, are depicted in grey where a complete 18S sequence was available. Mantoniella clades, if known, are labelled in different colours.
The analysis of the ITS2 sequences in M. tinhauana revealed the presence of eukaryotic universal hallmark motifs (Mai and Coleman, 1997; Schultz et al., 2005), namely a Y-Y (U–U) mismatch in helix 2 and a YRRY (UGGU) motif in helix 3. These motifs were consistently identified in all ITS2 sequences (highlighted in yellow in Figure 2A and Supplementary Figures S1, S2). We further examined the folding patterns of their ITS2 molecules to differentiate between base differences that have structural effects and compensatory base changes shared with related Mamiellophyceae species. In helix 2 (Figure 2A), we observed that approximately half of the bases were identical in all Mamiellophyceae (white, yellow), while the other half exhibited variations that were also present in other Mantoniella species (blue). Additionally, two base pairs were exclusive to M. tinhauana and Micromonas or Mamiella species (pink). Similar differences were observed in the other three helices (Supplementary Figures S1, S2). Based on the ITS2 helix folding structures, M. tinhauana shares a mix of base pair variants characteristic of arctic Mantoniella species and M. squamata, placing it firmly within the Mantoniella genus. Interestingly, it also possesses some structural base pairs that are unique to Micromonas or Mamiella strains.
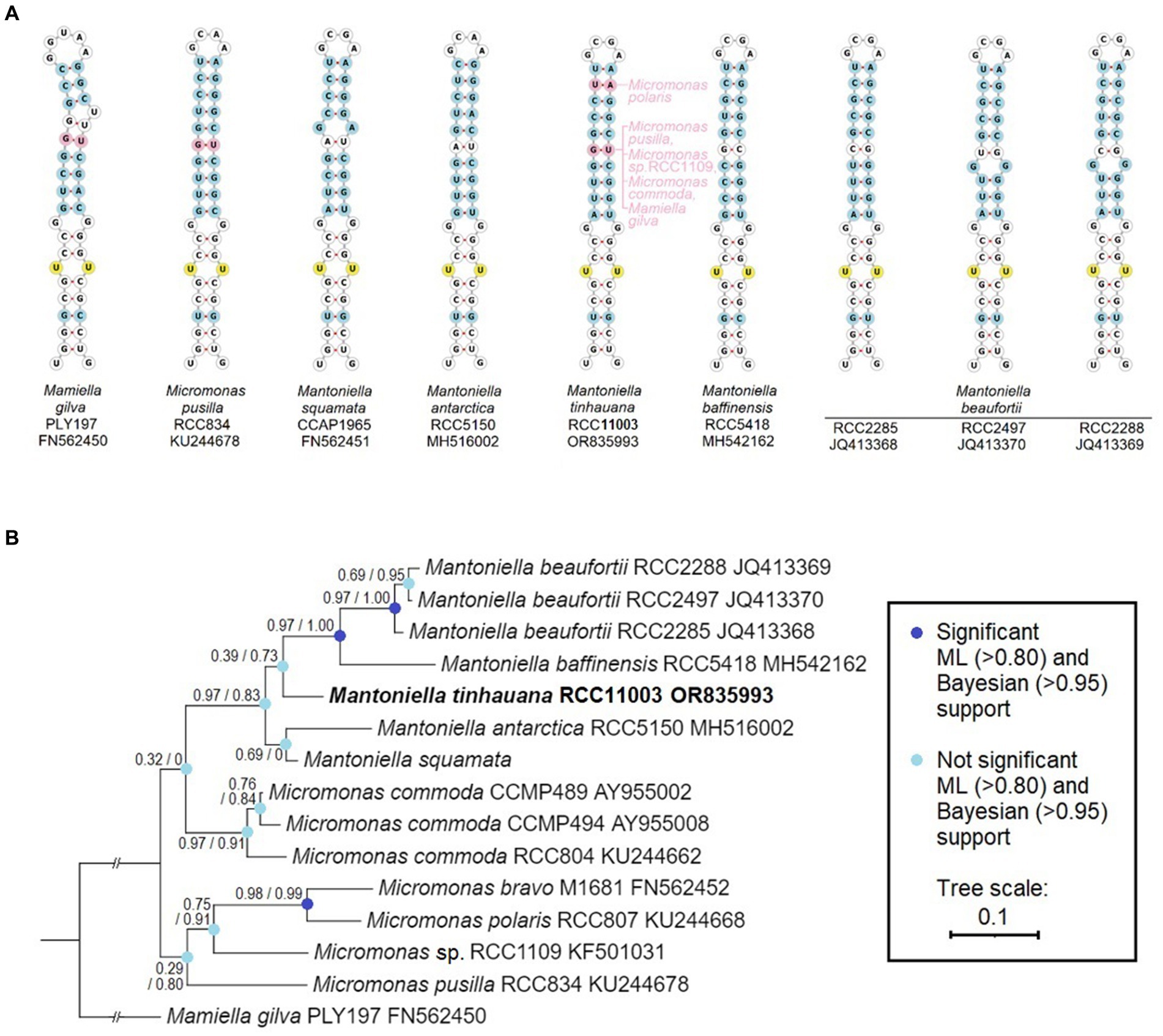
Figure 2. ITS2 RNA helix structural and base comparison, and phylogeny of Mamiellaceae. (A) ViennaRNA forna ITS2 folding structure of helix 2 (the other helices and complete ITS2 structure can be found in Supplementary Figures S1, S2), base-by-base comparison of novel Mantoniella tinhauana to other Mantoniella and Mamiellaceae strains, displayed in ITS2-based phylogenetic order. Only one representative Micromonas species and one Mamiella are shown. Yellow: universal eukaryote ITS2 motifs. Blue: site of nucleotide variant present in other Mantoniella species with structural effect. Pink: site of nucleotide variant absent in other Mantoniella species but present in other Mamiellophyceae species. (B) Mamiellaceae phylogenetic tree based on the structurally-informed comparison of ITS2 sequences, with branch support values as described.
In addition to visualising and comparing the structural differences in ITS2, we constructed a phylogenetic tree based on the structurally-informed ITS2 alignments (Figure 2B). This ITS2 tree differs significantly from the phylogeny based on the 18S gene, although branch support values are predominantly low. The discrepancies between the ITS2 and 18S trees can be attributed to varying evolutionary rates and histories, hybridisation events, and selection pressures (Baldwin et al., 1995; Hershkovitz and Lewis, 1996).
We conducted transmission electron microscopy (TEM) analysis of M. tinhauana, revealing cells larger than typical Ostreococcus, Bathycoccus and Micromonas cells – with cell sizes increasing, respectively, from 0.6 to 3 μm, as reported in previous research (Manton and Parke, 1960; Eikrem and Throndsen, 1990; Chrétiennot-Dinet et al., 1995; Kuroiwa et al., 2004). The cells of M. tinhauana measured between 1.8 and 4.5 μm, with characteristic features commonly observed in cells of the Mantoniella genus (Figure 3). No features unique to the new species Mantoniella tinhauana were identified, similar to previous observations of M. baffinensis and M. beaufortii (Yau et al., 2020b). These species exhibited slight differences in size but overall resembled M. squamata. One exception was the radiating pattern on the spiderweb scales, which allowed differentiation of M. beaufortii from the other two species based on the number of radial spokes. M. tinhauana exhibited spiderweb-like octaradial scale symmetry just like M. squamata and M. baffinensis. In any case, morphological differences among Mantoniella species are minor, prompting us to investigate genetic differences as the next step in our research.
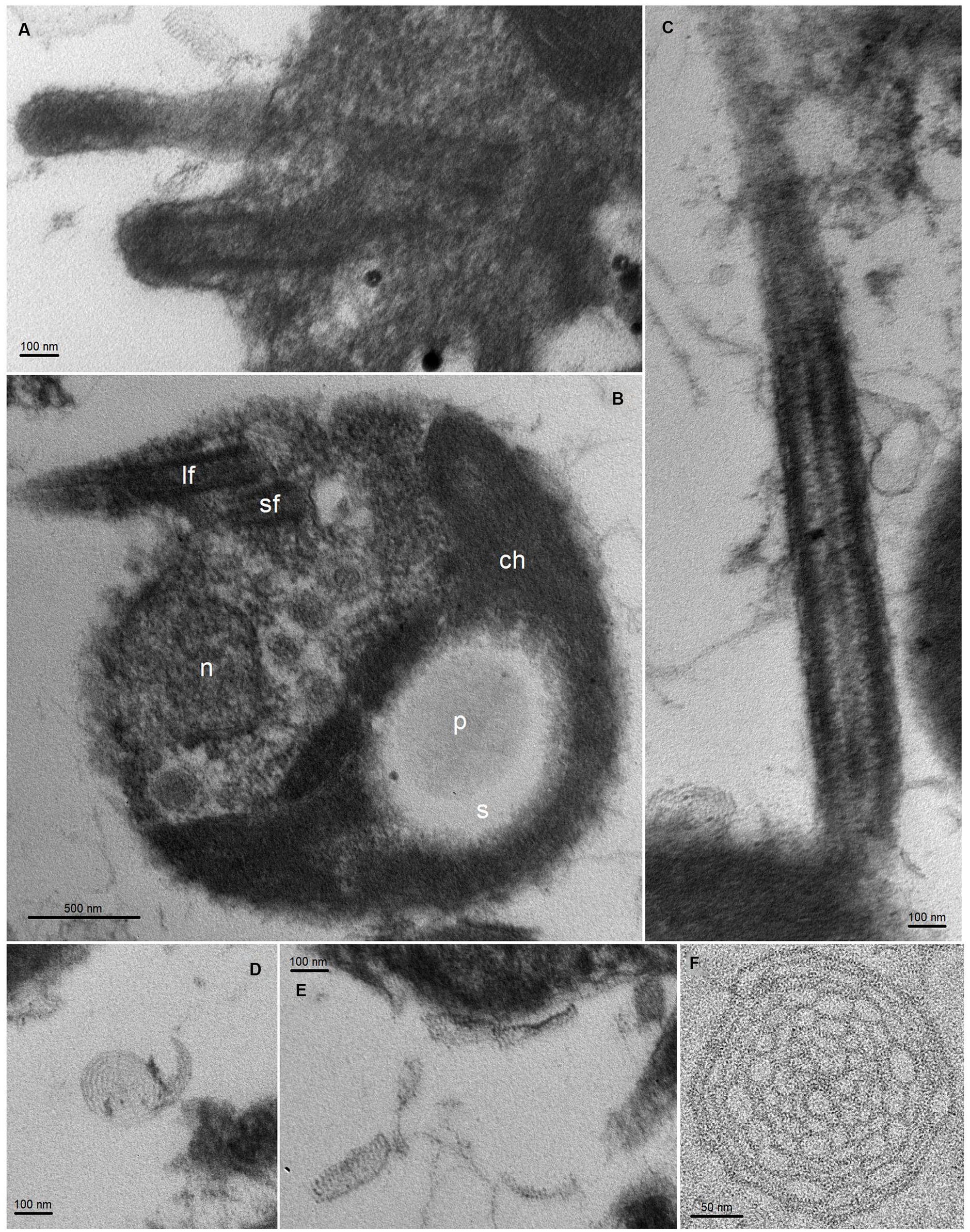
Figure 3. TEM thin sections of Mantoniella tinhauana. Cells were measured to be approximately 3 μm in diameter (10 cells measured, standard deviation 0.7 μm, min size 1.8 μm, max size 4.5 μm, median 3.1 μm), slightly smaller than but around the range of M. squamata (3–6.5 μm) (Manton and Parke, 1960), M. antarctica (2.8–5 μm) (Marchant et al., 1989), M. beaufortii (2.9–5 μm) and M. baffinensis (3.5–5.7 μm) (Yau et al., 2020b). (A) Bases of the long and short flagella. (B) Whole cell view with detail of organellar structures: n = nucleus, ch = chloroplast, s = starch granule, p = pyrenoid, lf = long flagellum, sf = short flagellum. (C) Detail of flagellum covered in scales (body and hair scales). (D–F) Body scales with octaradial spiderweb structure.
The first-ever Mantoniella strain whole genome was generated using short read paired-end sequencing, PacBio SMRT sequencing, and Hi-C sequencing techniques. The completeness of the assembly was assessed using BUSCO analysis against the Chlorophyta database, which indicated a high completeness value of 97.1% (Supplementary Table S4), comparable to other Mamiellophyceae reference genomes (Table 1). The de novo assembly yielded a 40 Mb genome composed of 27 scaffolds, with longer scaffolds compared to other Mamiellophyceae species (Supplementary Table S5a). However, only a low percentage of M. tinhauana scaffolds contained telomeres. Specifically, 11 scaffolds had telomeric tandem repeats on one end [CCCTAAA or the reverse complement TTTAGGG, the same as other Mamiellophyceae and plants (Richards and Ausubel, 1988)], and only four scaffolds had telomeres on both ends. This indicates imperfect genome assembly, a common issue observed in other reference genomes such as B. prasinos (Table 1). Despite the larger genome size and increased number of scaffolds, ploidy prediction confirmed that M. tinhauana is haploid, consistent with other known Mamiellophyceae species. Repeat masking revealed that 14.9% of the genome was composed of repeat elements (Supplementary Table S6), a significantly higher proportion (double) than seen in other Mamiellophyceae (Xu et al., 2022).
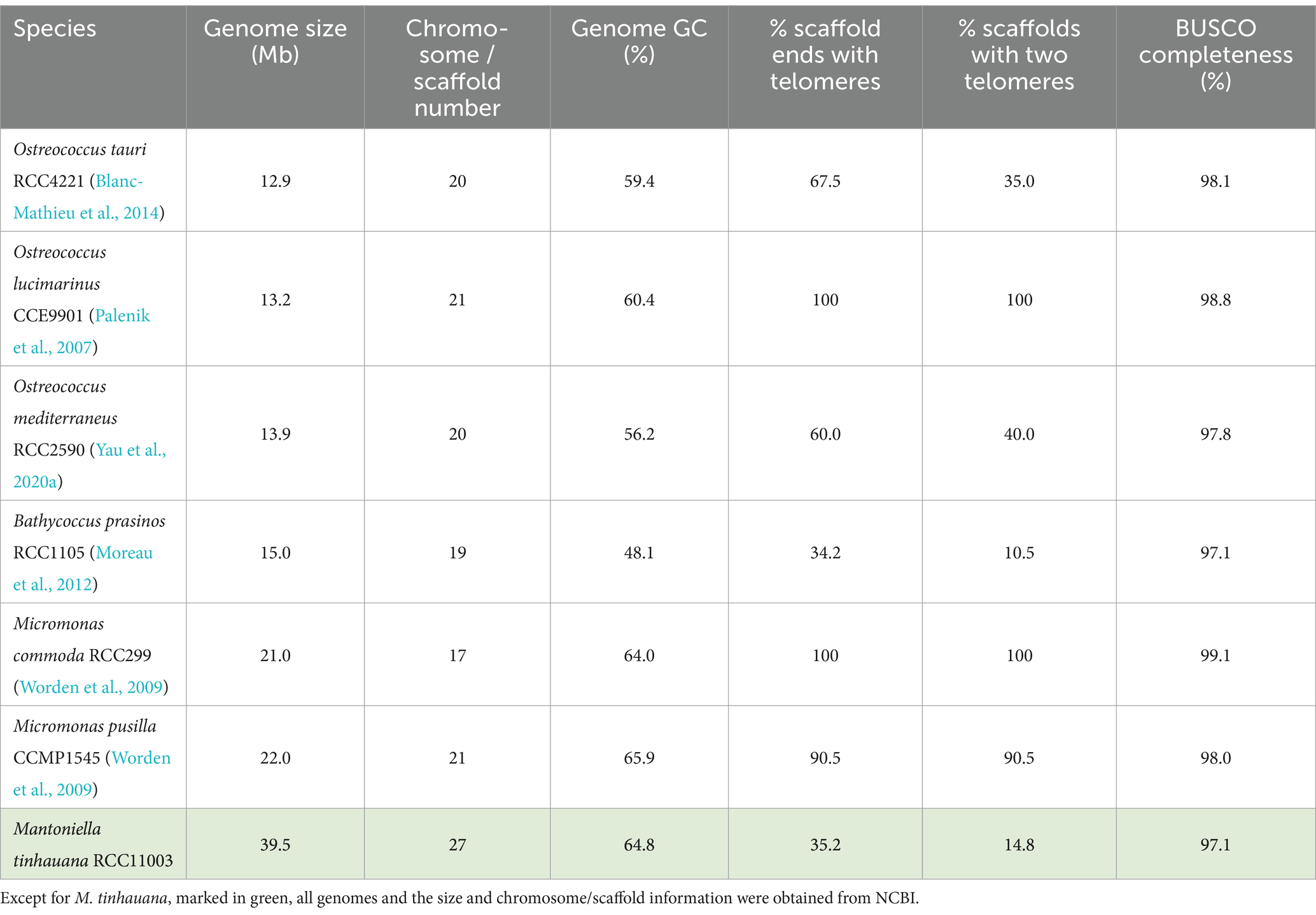
Table 1. Mamiellales genomes used in this study and their characteristics.
Mantoniella tinhauana represents the species with the largest recorded genome size within the Mamiellophyceae class to date. This species also exhibits larger cell sizes compared to other Mamiellales species, prompting the question of a possible correlation between genome size and cell size within the Mamiellophyceae class. Indeed, Ostreococcus species have the smallest cells (Chrétiennot-Dinet et al., 1995) and correspondingly small genomes. This trend continues with Bathycoccus species exhibiting slightly larger cells and genomes (Eikrem and Throndsen, 1990), and Micromonas following with even larger dimensions (Manton and Parke, 1960), as shown in Table 1. However, the number of scaffolds or chromosomes does not appear to follow a similar trend. A study contrasting the cell and genome sizes of O. tauri and a red algal species suggested that cell and genome sizes are not interdependent (Kuroiwa et al., 2004), but the comparison involved two phylogenetically distant species, potentially obscuring any correlation due to vastly different genomic structures and contents. To date, a systematic comparison of cell and genome sizes within Mamiellophyceae has not been undertaken. Preliminary observations hint at a significant correlation within this group. To test this hypothesis, further genomic sequencing and assembly of larger-celled Mamiellophyceae species, particularly from the genera Mantoniella or Mamiella (Alonso-González et al., 2014), would help validate this relationship between genome and cell sizes in this phylogenetic group.
Pairwise gene block synteny among the different Mamiellophyceae species is depicted in Figure 4. Notably, M. tinhauana exhibits similar grouping and colocalisation patterns of gene blocks as those of Micromonas species, but with longer gene blocks, indicating gene block duplications and expansions. O. lucimarinus and O. tauri have nearly identical chromosome and gene block order. In contrast, Micromonas commoda and Micromonas pusilla, despite belonging to the same genus, display more frequent chromosomal order and gene block rearrangements, similar to those observed between Micromonas pusilla and M. tinhauana. These findings suggest that there have been more rearrangements and less consistent arrangement of syntenic blocks in the Mamiellaceae compared to the Bathycoccaceae, and M. tinhauana follows this trend. Bathycoccaceae chromosomes tend to be shorter, resulting in fewer gene blocks corresponding to different chromosomes, whereas the Mamiellaceae have longer chromosomes, increasing the likelihood of rearrangements. Given that M. tinhauana possesses the longest chromosomes, it is unsurprising that gene blocks from multiple chromosomes in other species correspond to syntenic blocks on a single chromosome in M. tinhauana, and vice versa. Importantly, 75.7% of genes across all species exhibited collinearity, which suggests that, despite the rearrangement of gene blocks among different species, there is a substantial conservation of both gene content and, to a certain extent, the order of these blocks are correctly assembled in the novel species genome. This degree of collinearity provides substantial support to the accuracy of our genome assembly. Additionally, most M. tinhauana scaffolds with telomeres, particularly those with telomeres at both ends, demonstrate a higher level of synteny with other Mamiellophyceae species.
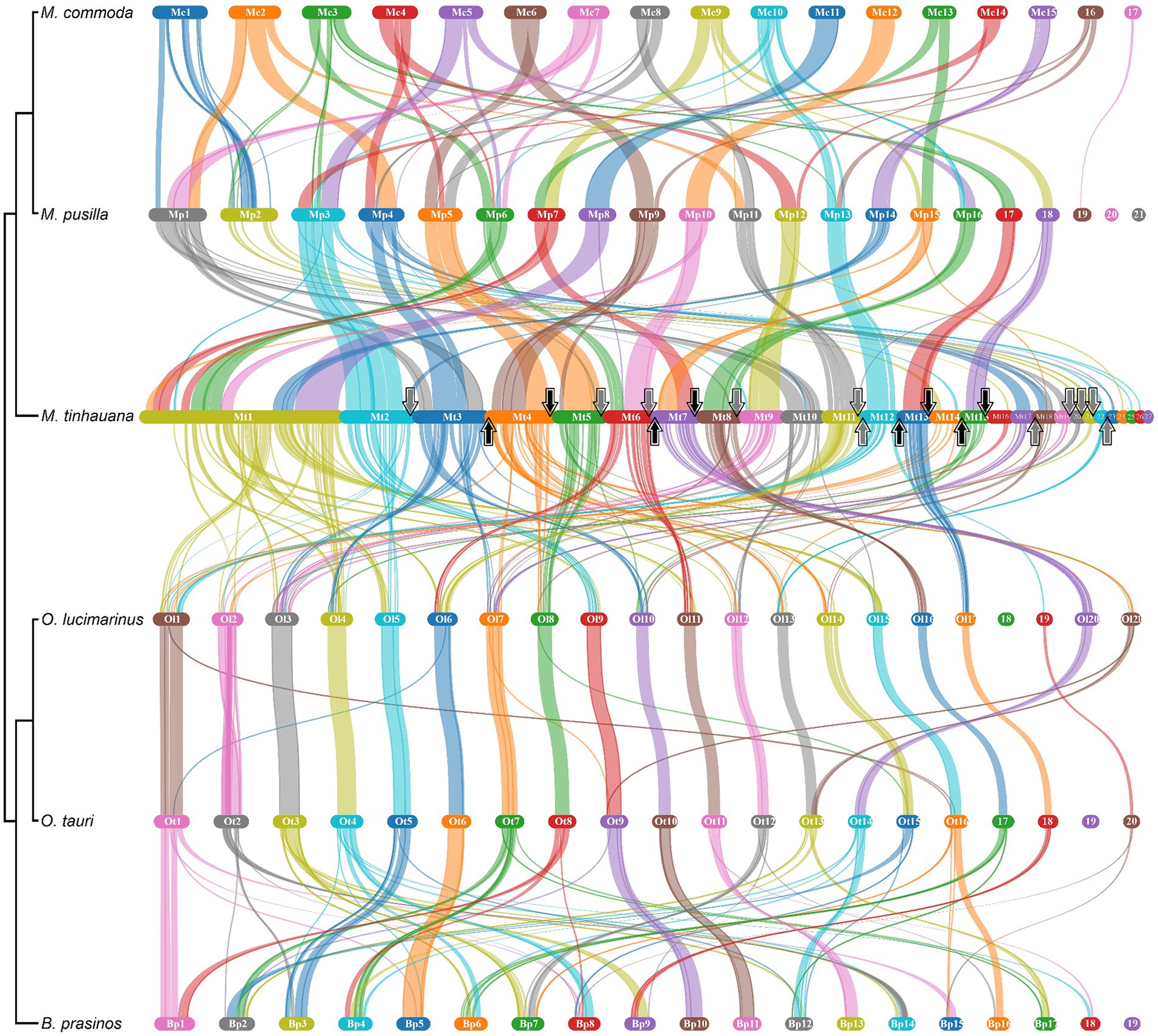
Figure 4. Pairwise gene block synteny of Mamiellales proteomes computed with MCScanX and visualised using SynVisio. Species are ordered by 18S-based phylogeny, with a simplified phylogenetic tree and names labelled on the left. Scaffolds or chromosomes for each species are shown in separate sections of different colours (random repeating 10 colour series), in order (largest to smallest), labelled. Collinear gene blocks between each pair of species are connected with ribbons of varying thicknesses denoting gene block lengths, in the colour corresponding to the source scaffold. For M. tinhauana, telomeres at the beginning (upwards arrow) or end (downwards arrow) of a scaffold are marked with arrows. The four scaffolds which have telomeres at both ends have black arrows.
In order to uncover the factors contributing to the larger genome of M. tinhauana, we conducted a comprehensive analysis of species orthogroups using Orthofinder. The overall and species-specific results of Orthofinder analysis can be found in Supplementary Table S7, with a graphical representation in Figure 5. The novel Mantoniella proteome exhibited notable characteristics, including the highest level of unassigned genes (1,288 or 11.6%), species-specific orthogroups (141 or 1.8%) and genes in species-specific orthogroups (394 or 3.5%). These findings were expected, considering that M. tinhauana is the first proteome in its genus and a newly discovered species with a significantly larger genome. The functional annotation of genes in Mantoniella tinhauana-specific orthogroups (188) is listed in Supplementary Table S8, which encompass a variety of roles, including motor proteins, carbohydrate metabolism and chromosome associated proteins. We further explored these species-specific orthogroups through Gene Ontology (GO) Enrichment Analysis.
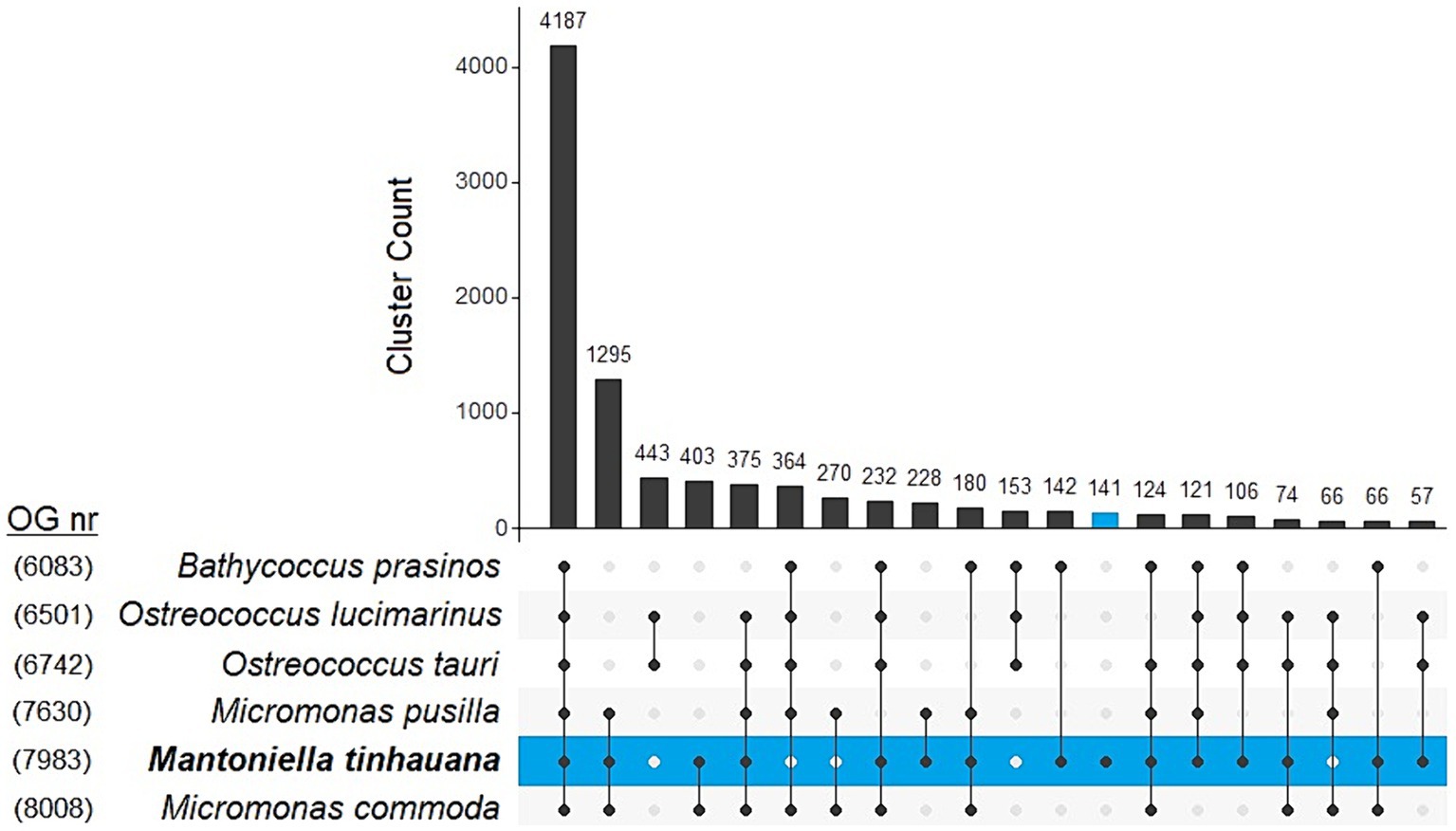
Figure 5. UpSet plot comparison of top 20 clustering unique and shared orthogroups in 6 Mamiellophyceae species. Number of orthogroups per species is labelled on the left.
To gain a comprehensive understanding of the orthogroup differences between species, particularly those with assignable functions, we performed expansions and contractions analysis. This analysis sought to elucidate the evolutionary history and relationship of M. tinhauana and its close relatives. Interestingly, M. tinhauana exhibited numerous gene family expansions, and even more contractions (Figure 6A). Additionally, GO enrichment analysis (Figure 6B) provided insights into the biological functions associated with the most significant expansions.
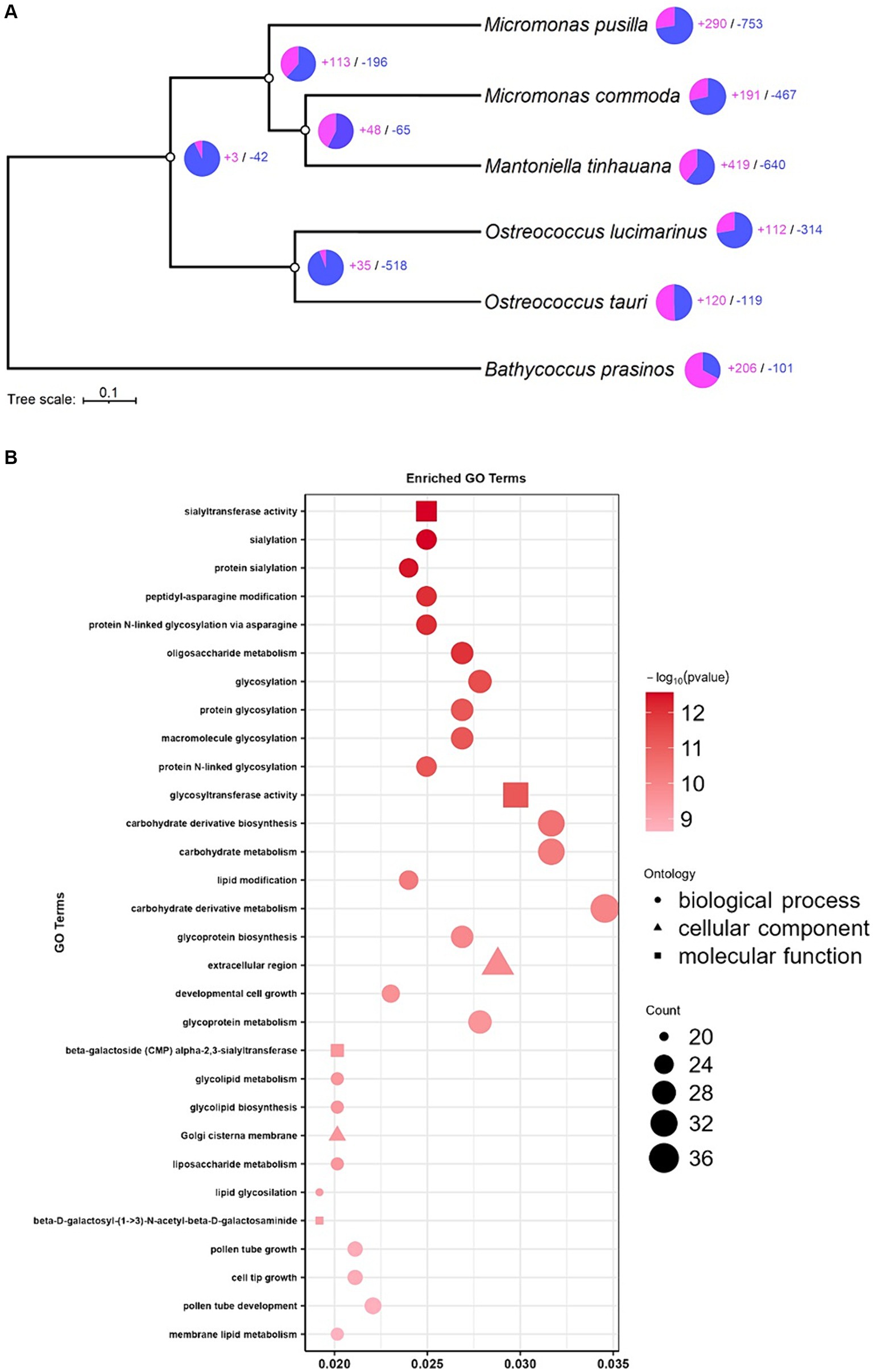
Figure 6. Expansion and contraction analysis and enrichment analysis. (A) Tree of expanded (purple) and contracted (blue) gene families. (B) GO enrichment analysis of significantly expanded gene families in M. tinhauana.
GO enrichment analysis revealed strong expansions in glycosylation gene families, particularly sialylation, which may be involved in the development of M. tinhauana’s characteristic scales and will be discussed below. It also unveiled significant expansions in Golgi associated genes, which is where sialylation takes place in the cell (Schauer and Kamerling, 2018). Expanded gene families associated with carbohydrate metabolism and cell growth (Raven and Beardall, 2003; Mathieu-Rivet et al., 2020) suggest that M. tinhauana is a fast-growing species well-adapted to eutrophic waters, where it was isolated.
Previously sequenced genomes of Mamiellophyceae in the genera Micromonas, Ostreococcus and Bathycoccus have a relatively high overall GC content and two lower GC content outlier chromosomes (Derelle et al., 2006; Piganeau et al., 2011b; Grimsley et al., 2015). These outlier chromosomes contain a higher proportion of transposable elements, leading to faster gene evolution and the presence of more species-specific, non-orthologous genes (Jancek et al., 2008). The BOC has distinct blocks with heterogeneous GC content, characterised by a section of lower GC flanked by normal, higher GC content. The low GC region suppresses recombination and contains one of two alternate mating type loci, which is hypothesised to predate Mamiellophyceae speciation (Blanc-Mathieu et al., 2017). In contrast, the SOC has low GC content throughout, and shows high variability in gene content even within the same species due to frequent duplications and internal rearrangements (Grimsley et al., 2015; Blanc-Mathieu et al., 2017). The SOC is associated with viral resistance (Moreau et al., 2012), as species possessing this variable chromosome demonstrate high viral sensitivity and the ability to rapidly develop resistance to viruses. SOC length is inversely correlated with viral susceptibility (Blanc-Mathieu et al., 2017). In unpublished experiments mentioned by Moreau et al. (2012), it was observed that two Mamiella and Mantoniella species do not exhibit the high viral sensitivity seen in the seven whole-genome sequenced species. These findings support the hypothesis that these species and potentially the entire genera may lack the SOC, although this has yet to be confirmed.
In the current study of the novel Mantoniella genome, no chromosomes with significantly lower GC content were identified (Supplementary Table S5b). Lower GC content is typically associated with suppressed recombination, which is a characteristic of sexual reproduction. However, no such regions were found in M. tinhauana, and direct observation of the process is virtually impossible. To confirm whether M. tinhauana undergoes sexual reproduction, we examined the presence of core meiosis genes (Derelle et al., 2006; Worden et al., 2009; Joli et al., 2017; Li et al., 2020), including the RWP-RK family of transcription factors known to be involved in gametogenesis in algae (Chardin et al., 2014). The findings demonstrate key meiosis genes are present at the expected levels in M. tinhauana (Supplementary Table S9), providing robust evidence supporting the existence of a sexual stage in this species.
In a study by Benites et al. (2021), the mean GC contents of coding sequences in Mamiellophyceae transcriptomes were compared to those of mating gene family coding sequences. Interestingly, some transcriptomes, including two Mantoniella transcriptomes, did not exhibit lower GC content in their mating gene family genes. In fact, certain species such as Dolichomastix even have a higher GC content in their mating genes. These findings support the possibility that earlier branching Mamiellophyceae gametologs are not confined to regions of lower GC content. It is conceivable that the genus Mantoniella, or at least M. tinhauana, never evolved to have recombination suppression in their mating gene regions, unlike Bathycoccus, Ostreococcus and Micromonas species. This could be because their mating genes are not clustered together on a single chromosome.
To investigate further, we analysed the synteny of five Mamiellophyceae BOCs in comparison to the novel Mantoniella scaffolds. The gene blocks did not align with a single scaffold, but with several (Figure 7A). Notably, M. tinhauana shares syntenic blocks with all five species BOCs, which is not the case when comparing other Mamiellaceae to Bathycoccaceae BOCs (Figure 7C). This suggests that M. tinhauana has gene blocks in common with both groups, further supporting its phylogenetic placement between the two. However, these syntenic blocks are located on separate scaffolds, indicating a distinct genomic organisation. The BOC has previously been identified as emerging before the speciation of Mamiellophyceae (Blanc-Mathieu et al., 2017), and thus it should be present in all Mamiellophyceae species. The absence of the BOC in the novel Mantoniella genome could indicate three possibilities: (a) translocation or rearrangement after the branching of the Mantoniella genus, (b) a more recent evolutionary origin of the BOC than previously thought, or (c) assembly errors.
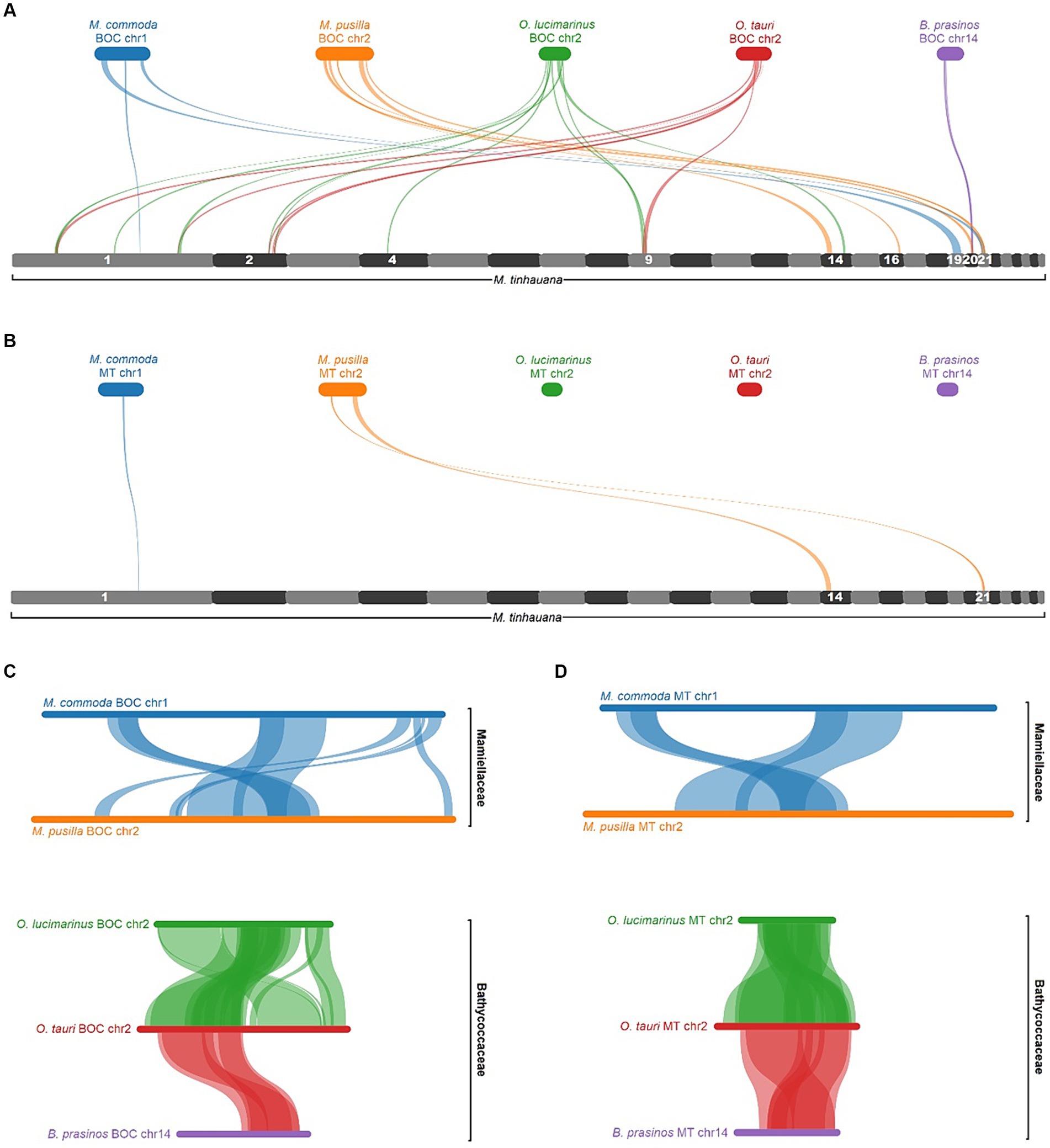
Figure 7. MCScanX pairwise alignment of BOCs and MTs. (A) BOC alignments to the full Mantoniella tinhauana genome. M. tinhauana scaffolds with syntenic blocks are numbered. (B) MT alignments to the Mantoniella tinhauana genome. (C) BOC alignments to one another. (D) MT alignments to one another. Collinear gene blocks (of at least 5 genes with a max gap of 25) are connected with ribbons of different thicknesses denoting gene block lengths, in the same colour as the source species.
To determine if the syntenic BOC blocks were associated with mating, we examined gene block synteny between the MT loci within each BOC and the Mantoniella proteome (Figure 7B). The analysis revealed that only a few Micromonas MT gene blocks were found in M. tinhauana, proving that the majority of syntenic blocks between Mamiellophyceae BOCs and M. tinhauana are unrelated to mating. When comparing BOC gene blocks among Mamiellophyceae, the syntenic blocks primarily correspond to the MT loci (Figure 7D). These findings suggest that in M. tinhauana, the mating loci are not clustered and have undergone rearrangements and/or genetic divergence compared to its phylogenetic relatives.
Attempts to align the distinct and variable sequences of SOC in Mamiellophyceae with the Mantoniella genome were unsuccessful. Consequently, the SOC can only be identified based on chromosome length and GC content. Moreau et al. (2012) proposed that Mantoniella may lack a SOC due to its purported lower sensitivity to viruses. The absence of the SOC in this study supports this hypothesis, although SOC-lacking species could very well interact with viruses in different ways. We are currently studying the viral susceptibility of M. tinhauana. Further investigations should also aim to identify virus resistance genes present in other Mamiellophyceae and determine whether the same or orthologous genes are present in the M. tinhauana genome, with special attention to their location if present.
The gene family expansion analysis uncovered a significant expansion in sialylation and other glycosylation gene families in M. tinhauana. To investigate these expansions further, we conducted a targeted analysis on four protein families previously found to be expanded in Bathycoccus prasinos hypothesised to be involved in scale formation (Moreau et al., 2012). We searched for these families in the Pfam-annotated proteome of M. tinhauana and found similar copy number expansions in two out of the four families: glycosyltransferase family 29 and neuraminidase/sialidase (Supplementary Table S10). Another study by van Baren et al. (2016) also identified expansions in only two out of the four families in both scaled B. prasinos and D. tenuilepis compared to non-scaled species. Our findings confirm theirs, supporting the hypothesis that glycosyltransferase family 29 and sialidases are involved in scale development.
The novel and other Mantoniella species possess two flagella, unlike other Mamiellophyceae genera that have one flagellum (Micromonas) or none (Bathycoccus, Ostreococcus). This difference in flagellar number should be reflected in their genomic characteristics, but gene family expansion analysis indicated no motor associated expansions. We analysed the Pfam-annotated proteome using a list of published flagella-related genes (Li et al., 2020). Comparison to other Mamiellophyceae revealed that M. tinhauana had higher copy numbers of certain microtubule synthesis and assembly protein domains compared to other species (Supplementary Table S11). A few cilia-, flagella- and motility-associated domains also showed a modest expansion in M. tinhauana compared to monoflagellated Micromonas. Marin and Melkonian (2010) proposed that the last common ancestor of the Mamiellophyceae had two flagella, as monoflagellate Monomastix and Micromonas species retain two basal bodies despite having only one flagellum. The identified gene expansions could potentially explain how M. tinhauana retained two flagella, but further functional studies are necessary to identify the genes involved in flagellar number and length control, as well as environmentally-stimulated and synchronous flagellar beating. These areas remain largely unexplored in Mamiellophyceae. Examining the expression of key genes, and not only their copy number, would provide further insights.
Metagenomic reads obtained from the Tara Oceans Expeditions were used to investigate the presence and abundance of M. tinhauana. Considering the size range of M. tinhauana cells (>1.8 μm) and Micromonas pusilla cells (>1 μm) (Manton and Parke, 1960), reads from the over 0.8 μm size fraction were used. These reads were mapped onto the novel and control genomes, and the relative abundance (RPKM) was calculated (Figure 8 and Supplementary Table S3).
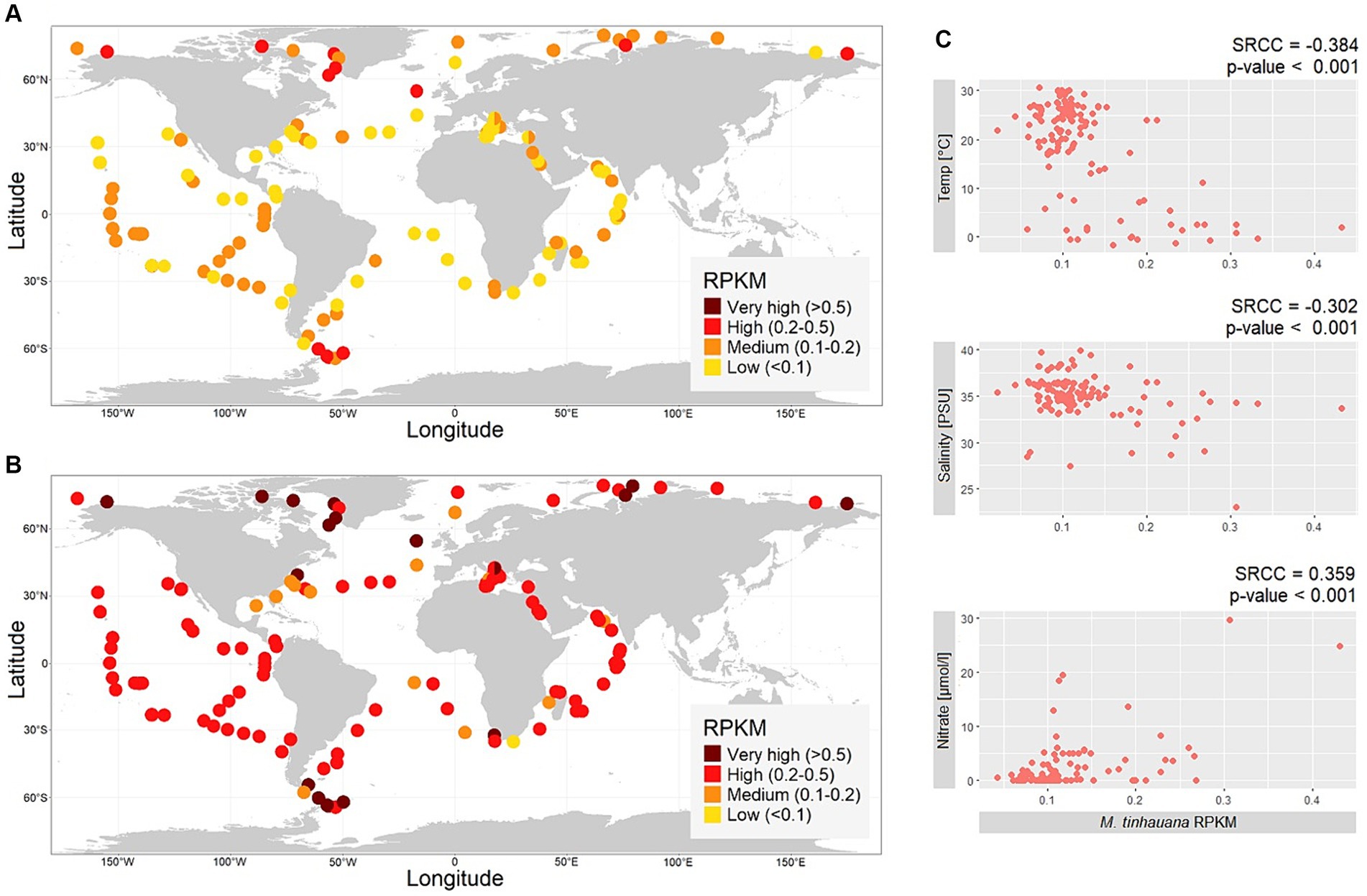
Figure 8. Biogeographic distribution of Mamiellophyceae. (A,B) Maps of global metagenomic RPKM read mapping of over 0.8 μm size fraction reads sampled at the ocean surface to (A) Mantoniella tinhauana genome, (B) Micromonas pusilla CCMP1545 genome. (C) Correlations between not highly collinear environmental variables and M. tinhauana RPKM values.
Our analysis revealed the widespread occurrence of Mantoniella tinhauana reads across the tested sites in the surface of the global ocean. The abundance of M. tinhauana reads was consistently low, indicating that it is a cosmopolitan species with a low-level prevalence, except for polar regions where it was higher. The mean worldwide RPKM value of the tested sites was 0.12, with a maximum of 0.43 RPKM (1.73% of reads). The mapping to the control Micromonas pusilla CCMP1545 genome (global mean 0.36 RPKM, maximum 1.54 RPKM or 3.38% of reads) showed similar levels as reported previously (Leconte et al., 2020) using similar methods.
To understand the environmental factors influencing the abundance of M. tinhauana, we examined the correlation between read abundance and various environmental variables (Figure 8C). Although statistically significant, the individual relationships were not strongly pronounced, with the most correlated factor being temperature. Nitrate showed a positive correlation with RPKM, while temperature and salinity exhibited negative correlations. These findings support the hypothesis that M. tinhauana is adapted to growing in diverse nutrient levels, and in environments rich in nutrients, for instance with high nitrate, it can readily thrive and proliferate. The negative correlation between the abundance of M. tinhauana and temperature, and its higher presence in polar regions suggest a preference for cooler habitats within this genus. All species within the genus, with the exception of M. squamata and M. tinhauana, have been isolated exclusively from polar sites. This intriguing observation presents an exciting opportunity to conduct genomic comparisons between the non-polar and polar species, with the aim of unravelling the underlying genomic traits that contribute to their distinct adaptations to cold and higher temperatures, respectively.
Illustrated in Figure 3. Round cells measuring 3 μm in diameter (standard deviation 0.7 μm, min size 1.8 μm, max 4.5 μm, median 3.1 μm), slightly smaller but around the range of other Mantoniella strains (Yau et al., 2020b). Biflagellate, with one long and one short flagellum. Cell body covered in octaradial spiderweb scales. One large chloroplast, occupying half the cell, with pyrenoid within starch granule. 18S and ITS2 rRNA sequences are species-specific (18S and ITS region GenBank accessions: OR835992 and OR835993, respectively).
Isolated on the 11th June 2020 (10:30 am) from surface water (2 m depth) that was 30.5°C via peristaltic pump at the Lau Fau Shan coast of Hong Kong, in the Pacific Ocean (22°28′09.0′N 113°58′50.1′E). Culture deposited in The Roscoff Culture Collection under accession RCC11003.
Tin Hau is the Cantonese name for the goddess of the sea revered in Hong Kong.
Cosmopolitan species in surface water, with higher dominance in polar regions. Growth positively correlated with nitrate level and negatively correlated with temperature and salinity.
We have discovered a novel species of Mamiellophycean green alga, Mantoniella tinhauana, from coastal surface water in the Western Pacific. Morphologically, it closely resembles other Mantoniella species, except for its smaller cell size. Through comparison of its 18S rRNA sequence and ITS2 structure with other Mamiellophyceae, we confirmed it to be a distinct species. Phylogenetically, it falls between the genus Micromonas and other Mantoniella strains.
The genome of M. tinhauana, the first draft genome of a Mantoniella species, was assembled with high completeness and annotated. It differs significantly from other Mamiellophyceae species, with a larger genome size (concurrent with larger cell size), more repeat elements and numerous unique genes. The larger genome size of the novel strain cannot be solely explained by the observed expansions. Comparative analysis with other Mamiellophyceae genomes revealed gene block duplications, expansions, as well as regions of no synteny and known function. To gain further insights, RNAseq should be conducted to determine the functions of more actively expressed genes.
Notably, M. tinhauana lacks the two low-GC outlier chromosomes found in all other studied Mamiellales, which are linked to mating and viral resistance. Despite the absence of synteny with BOC and MT loci seen in other species, the presence of meiosis genes in M. tinhauana suggests the existence of similar mating types and sexual reproduction mechanisms. These findings pose intriguing questions regarding the ancestry and evolution of Mamiellophyceae, which can only be hypothesised with a single divergent genome. Ongoing research on the viral infection dynamics of M. tinhauana may shed light on the absence of SOC and viral sensitivity in this algal class.
The analysis of expanded gene families revealed high glycosylation activity involved in scale formation. This and the two flagella in Mantoniella prompted a comparison of targeted gene groups between the novel species and other proteomes. While some investigated genes partially explained these structural differences, further study is required to fully understand the underlying mechanisms of scale patterns, flagella production and synchronised movement. The availability of this first whole genome of a biflagellate scaled Mamiellophycean species will undoubtedly contribute to future studies in these areas and beyond.
Future efforts should be focused on assembling and analysing the whole genomes of additional Mamiellophyceae species, including other Mantoniella strains and members of the understudied genus Mamiella. The challenges encountered in assembling the divergent de novo genome of M. tinhauana highlight the need for more cultures and complete genomes to enhance assembly accuracy and efficiency. Additionally, it would be valuable to assemble the plastid (chloroplast and mitochondrial) sequences of M. tinhauana and compare them to those of related species.
This study represents the first exploration of the distribution of a Mantoniella strain using non-metabarcoding or MAG-based means. M. tinhauana distribution was cosmopolitan at low level and with higher prevalence at high latitudes. Temperature and salinity were negatively correlated with M. tinhauana RPKM values. Nitrate concentration was the most positively correlated environmental variable, although further studies on growth rate and nutrient deficiencies would be informative. To further understand Mamiellophyceae distribution, future metagenomic studies should use expanded datasets to explore depth variations, seasonality and potentially conduct a more long-term analysis of global warming. It is important to understand the precise relationship between the environment and Mamiellophyceae dominance, as these organisms play a crucial role in ocean primary productivity. Such knowledge is essential for comprehending the dynamics of the ocean as a whole and its future implications in the context of climate change.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
ER: Writing – original draft, Writing – review & editing. YX: Writing – original draft, Writing – review & editing. CY: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We gratefully acknowledge the financial support provided by the Research Grants Council of Hong Kong (Early Career scheme: 26100521).
We thank Adriana Lopes dos Santos, Sheree Yau, and Kirill Konovalov for their generous help with RNA structures, as well as Wan Siu Hei for his dedicated efforts in maintaining the algae culture, ensuring its vitality throughout the course of this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1358574/full#supplementary-material
Aiyar, A. (1999). “The use of CLUSTAL W and CLUSTAL X for multiple sequence alignment” in Bioinformatics methods and protocols. eds. S. Misener and S. A. Krawetz (Totowa, NJ: Humana Press).
Alonso-González, A., Orive, E., David, H., García-Etxebarria, K., Garrido, J. L., Laza-Martínez, A., et al. (2014). Scaly green flagellates from Spanish Atlantic coastal waters: molecular, ultrastructural and pigment analyses. Bot. Mar. 57, 379–402. doi: 10.1515/bot-2013-0108
Baldwin, B. G., Sanderson, M. J., Porter, J. M., Wojciechowski, M. F., Campbell, C. S., and Donoghue, M. J. (1995). The ITS region of nuclear ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann. Mo. Bot. Gard. 82, 247–277. doi: 10.2307/2399880
Balzano, S., Gourvil, P., Siano, R., Chanoine, M., Marie, D., Lessard, S., et al. (2012). Diversity of cultured photosynthetic flagellates in the north East Pacific and Arctic oceans in summer. Biogeosci. Discuss. 9, 4553–4571. doi: 10.5194/bgd-9-6219-2012
Bandi, V., Gutwin, C., Siri, J. N., Neufeld, E., Sharpe, A., and Parkin, I. (2022). Visualization tools for genomic conservation. New York, NY: Springer US.
Belevich, T. A., Milyutina, I. A., Abyzova, G. A., and Troitsky, A. V. (2021). The pico-sized Mamiellophyceae and a novel Bathycoccus clade from the summer plankton of Russian Arctic seas and adjacent waters. FEMS Microbiol. Ecol. 97:fiaa 251. doi: 10.1093/femsec/fiaa251
Benites, L. F., Bucchini, F., Sanchez-Brosseau, S., Grimsley, N., Vandepoele, K., and Piganeau, G. (2021). Evolutionary genomics of sex-related chromosomes at the base of the Green lineage. Genome Biol. Evol. 13:evab 216. doi: 10.1093/gbe/evab216
Blanc-Mathieu, R., Krasovec, M., Hebrard, M., Yau, S., Desgranges, E., Martin, J., et al. (2017). Population genomics of picophytoplankton unveils novel chromosome hypervariability. Sci. Adv. 3:e1700239. doi: 10.1126/sciadv.1700239
Blanc-Mathieu, R., Verhelst, B., Derelle, E., Rombauts, S., Bouget, F.-Y., Carré, I., et al. (2014). An improved genome of the model marine alga Ostreococcus tauri unfolds by assessing Illumina de novo assemblies. BMC Genomics 15:1103. doi: 10.1186/1471-2164-15-1103
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl. 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M., and Borodovsky, M. (2021). BRAKER2: automatic eukaryotic genome annotation with gene mark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinforma. 3:lqaa 108. doi: 10.1093/nargab/lqaa108
Buchfink, B., Reuter, K., and Drost, H.-G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Bushmanova, E., Antipov, D., Lapidus, A., and Prjibelski, A. D. (2019). rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8:giz100. doi: 10.1093/gigascience/giz100
Bushnell, B. (2016). BBMap short read aligner. 2023. Available at: http://sourceforge.net/projects/bbmap
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, Orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
Chardin, C., Girin, T., Roudier, F., Meyer, C., and Krapp, A. (2014). The plant RWP-RK transcription factors: key regulators of nitrogen responses and of gametophyte development. J. Exp. Bot. 65, 5577–5587. doi: 10.1093/jxb/eru261
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cho, C. H., Park, S. I., Huang, T.-Y., Lee, Y., Ciniglia, C., Yadavalli, H. C., et al. (2023). Genome-wide signatures of adaptation to extreme environments in red algae. Nat. Commun. 14:10. doi: 10.1038/s41467-022-35566-x
Chrétiennot-Dinet, M.-J., Courties, C., Vaquer, A., Neveux, J., Claustre, H., Lautier, J., et al. (1995). A new marine picoeucaryote: Ostreococcus tauri gen. Et sp. nov. (Chlorophyta, Prasinophyceae). Phycologia 34, 285–292. doi: 10.2216/i0031-8884-34-4-285.1
Conway, J. R., Lex, A., and Gehlenborg, N. (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. doi: 10.1093/bioinformatics/btx364
Corrêa dos Santos, R. A., Goldman, G. H., and Riaño-Pachón, D. M. (2017). Ploidy NGS: visually exploring ploidy with next generation sequencing data. Bioinformatics 33, 2575–2576. doi: 10.1093/bioinformatics/btx204
Dela Peña, L., Tejada, A. J. P., Quijano, J. B., Alonzo, K. H., Gernato, E. G., Caril, A., et al. (2021). Diversity of marine eukaryotic Picophytoplankton communities with emphasis on Mamiellophyceae in northwestern Philippines. Philipp. J. Sci. 150, 27–42. doi: 10.56899/150.01.03
Delmont, T. O., and Eren, A. M. (2016). Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ 4:e1839. doi: 10.7717/peerj.1839
Delmont, T. O., Gaia, M., Hinsinger, D. D., Frémont, P., Vanni, C., Fernandez-Guerra, A., et al. (2022). Functional repertoire convergence of distantly related eukaryotic plankton lineages abundant in the sunlit ocean. Cell Genomics 2:100123. doi: 10.1016/j.xgen.2022.100123
Demir-Hilton, E., Sudek, S., Cuvelier, M. L., Gentemann, C. L., Zehr, J. P., and Worden, A. Z. (2011). Global distribution patterns of distinct clades of the photosynthetic picoeukaryote Ostreococcus. ISME J. 5, 1095–1107. doi: 10.1038/ismej.2010.209
Derelle, E., Ferraz, C., Rombauts, S., Rouzé, P., Worden, A. Z., Robbens, S., et al. (2006). Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc. Natl. Acad. Sci. 103, 11647–11652. doi: 10.1073/pnas.0604795103
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl. 29, 15–21. doi: 10.1093/bioinformatics/bts635
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Eikrem, W., and Throndsen, J. (1990). The ultrastructure of Bathycoccus gen. Nov. and B. Prasinos sp. nov., a non-motile picoplanktonic alga (Chlorophyta, Prasinophyceae) from the Mediterranean and Atlantic. Phycologia 29, 344–350. doi: 10.2216/i0031-8884-29-3-344.1
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Eren, A. M., Esen, Ö. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). Repeat modeler 2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Foulon, E., Not, F., Jalabert, F., Cariou, T., Massana, R., and Simon, N. (2008). Ecological niche partitioning in the picoplanktonic green alga Micromonas pusilla: evidence from environmental surveys using phylogenetic probes. Environ. Microbiol. 10, 2433–2443. doi: 10.1111/j.1462-2920.2008.01673.x
Gendron, P., Lemieux, S., and Major, F. (2001). Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 308, 919–936. doi: 10.1006/jmbi.2001.4626
Ghurye, J., Rhie, A., Walenz, B. P., Schmitt, A., Selvaraj, S., Pop, M., et al. (2019). Integrating hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput. Biol. 15:e1007273. doi: 10.1371/journal.pcbi.1007273
Grimsley, N., Yau, S., Piganeau, G., and Moreau, H. (2015). Typical features of genomes in the Mamiellophyceae. Tokyo: Springer Japan.
Guillard, R. R. L., and Hargraves, P. E. (1993). Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 32, 234–236. doi: 10.2216/i0031-8884-32-3-234.1
Haas, B. J. (2023). Trans decoder. Available at: https://github.com/TransDecoder/TransDecoder (accessed October 3, 2023).
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Hadziavdic, K., Lekang, K., Lanzen, A., Jonassen, I., Thompson, E. M., and Troedsson, C. (2014). Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS One 9:e87624. doi: 10.1371/journal.pone.0087624
Hershkovitz, M. A., and Lewis, L. A. (1996). Deep-level diagnostic value of the rDNA-ITS region. Mol. Biol. Evol. 13, 1276–1295. doi: 10.1093/oxfordjournals.molbev.a025693
Hu, Y. O. O., Karlson, B., Charvet, S., and Andersson, A. F. (2016). Diversity of Pico- to Mesoplankton along the 2000 km salinity gradient of the Baltic Sea. Front. Microbiol. 7:679. doi: 10.3389/fmicb.2016.00679
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Jahn, M. T., Schmidt, K., and Mock, T. (2014). A novel cost effective and high-throughput isolation and identification method for marine microalgae. Plant Methods 10:26. doi: 10.1186/1746-4811-10-26
Jancek, S., Gourbière, S., Moreau, H., and Piganeau, G. (2008). Clues about the genetic basis of adaptation emerge from comparing the proteomes of two Ostreococcus ecotypes (Chlorophyta, Prasinophyceae). Mol. Biol. Evol. 25, 2293–2300. doi: 10.1093/molbev/msn168
Joli, N., Monier, A., Logares, R., and Lovejoy, C. (2017). Seasonal patterns in Arctic prasinophytes and inferred ecology of Bathycoccus unveiled in an Arctic winter metagenome. ISME J. 11, 1372–1385. doi: 10.1038/ismej.2017.7
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keller, O., Kollmar, M., Stanke, M., and Waack, S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763. doi: 10.1093/bioinformatics/btr010
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Kuroiwa, T., Nozaki, H., Matsuzaki, M., Misumi, O., and Kuroiwa, H. (2004). Does cell size depend on the nuclear genome size in ultra-small algae such as Cyanidioschyzon merolae and Ostreococcus tauri? Cytologia (Tokyo) 69, 93–96. doi: 10.1508/cytologia.69.93
Lafontaine, D. L., Yang, L., Dekker, J., and Gibcus, J. H. (2021). Hi-C 3.0: improved protocol for genome-wide chromosome conformation capture. Curr. Protoc. 1:e198. doi: 10.1002/cpz1.198
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leconte, J., Benites, L. F., Vannier, T., Wincker, P., Piganeau, G., and Jaillon, O. (2020). Genome resolved biogeography of Mamiellales. Genes 11:66. doi: 10.3390/genes11010066
Lemoine, F., and Gascuel, O. (2021). Gotree/Goalign: toolkit and go API to facilitate the development of phylogenetic workflows. NAR Genomics Bioinforma. 3:lqab 075. doi: 10.1093/nargab/lqab075
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L., Wang, S., Wang, H., Sahu, S. K., Marin, B., Li, H., et al. (2020). The genome of Prasinoderma coloniale unveils the existence of a third phylum within green plants. Nat. Ecol. Evol. 4, 1220–1231. doi: 10.1038/s41559-020-1221-7
Lin, Y.-C., Chiang, K.-P., and Kang, L.-K. (2017a). Community composition of picoeukaryotes in the South China Sea during winter. Cont. Shelf Res. 143, 91–100. doi: 10.1016/j.csr.2017.04.009
Lin, Y.-C., Chin, C.-P., Chen, W.-T., Huang, C.-T., Gong, G.-C., Chiang, K.-P., et al. (2022). The spatial variation in chlorophyte community composition from coastal to offshore waters in a subtropical continental shelf system. Front. Mar. Sci. 9:865081. doi: 10.3389/fmars.2022.865081
Lin, Y.-C., Chung, C.-C., Chen, L.-Y., Gong, G.-C., Huang, C.-Y., and Chiang, K.-P. (2017b). Community composition of photosynthetic Picoeukaryotes in a subtropical coastal ecosystem, with particular emphasis on Micromonas. J. Eukaryot. Microbiol. 64, 349–359. doi: 10.1111/jeu.12370
Lin, L., Gu, H., Luo, Z., and Wang, N. (2021). Size-dependent spatio-temporal dynamics of eukaryotic plankton community near nuclear power plant in Beibu gulf, China. J. Oceanol. Limnol. 39, 1910–1925. doi: 10.1007/s00343-020-0248-6
Lopes dos Santos, A., Gourvil, P., Tragin, M., Noël, M.-H., Decelle, J., Romac, S., et al. (2017). Diversity and oceanic distribution of prasinophytes clade VII, the dominant group of green algae in oceanic waters. ISME J. 11, 512–528. doi: 10.1038/ismej.2016.120
Lovejoy, C., Vincent, W. F., Bonilla, S., Roy, S., Martineau, M.-J., Terrado, R., et al. (2007). Distribution, phylogeny, and growth of cold-adapted Picoprasinophytes in Arctic seas. J. Phycol. 43, 78–89. doi: 10.1111/j.1529-8817.2006.00310.x
Lukashin, A. V., and Borodovsky, M. (1998). Gene mark. Hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115. doi: 10.1093/nar/26.4.1107
Mai, J. C., and Coleman, A. W. (1997). The internal transcribed spacer 2 exhibits a common secondary structure in Green algae and flowering plants. J. Mol. Evol. 44, 258–271. doi: 10.1007/PL00006143
Malfertheiner, L., Martínez-Pérez, C., Zhao, Z., Herndl, G. J., and Baltar, F. (2022). Phylogeny and metabolic potential of the candidate phylum SAR324. Biology 11:599. doi: 10.3390/biology11040599
Manton, I., and Parke, M. (1960). Further observations on small green flagellates with special reference to possible relatives of Chromulina pusilla butcher. J. Mar. Biol. Assoc. U. K. 39, 275–298. doi: 10.1017/S0025315400013321
Marchant, H. J., Buck, K. R., Garrison, D. L., and Thomsen, H. A. (1989). Mantoniella in Antarctic waters including the description of M. antarctica sp. nov. (Prasinophyceae). J. Phycol. 25, 167–174. doi: 10.1111/j.0022-3646.1989.00167.x
Marin, B., and Melkonian, M. (2010). Molecular phylogeny and classification of the Mamiellophyceae class. Nov. (Chlorophyta) based on sequence comparisons of the nuclear- and plastid-encoded rRNA operons. Protist 161, 304–336. doi: 10.1016/j.protis.2009.10.002
Mathieu-Rivet, E., Mati-Baouche, N., Walet-Balieu, M.-L., Lerouge, P., and Bardor, M. (2020). N- and O-glycosylation pathways in the microalgae polyphyletic group. Front. Plant Sci. 11:609993. doi: 10.3389/fpls.2020.609993
Mendes, F. K., Vanderpool, D., Fulton, B., and Hahn, M. W. (2021). CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518. doi: 10.1093/bioinformatics/btaa1022
Merget, B., Koetschan, C., Hackl, T., Förster, F., Dandekar, T., Müller, T., et al. (2012). The ITS2 database. J. Vis. Exp. JoVE 61:3806. doi: 10.3791/3806
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Monier, A., Worden, A. Z., and Richards, T. A. (2016). Phylogenetic diversity and biogeography of the Mamiellophyceae lineage of eukaryotic phytoplankton across the oceans. Environ. Microbiol. Rep. 8, 461–469. doi: 10.1111/1758-2229.12390
Moreau, H., Verhelst, B., Couloux, A., Derelle, E., Rombauts, S., Grimsley, N., et al. (2012). Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol. 13:R74. doi: 10.1186/gb-2012-13-8-r74
Nguyen, L.-T., Schmidt, H. A., Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Palenik, B., Grimwood, J., Aerts, A., Rouzé, P., Salamov, A., Putnam, N., et al. (2007). The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc. Natl. Acad. Sci. 104, 7705–7710. doi: 10.1073/pnas.0611046104
Piganeau, G., Eyre-Walker, A., Grimsley, N., and Moreau, H. (2011a). How and why DNA barcodes underestimate the diversity of microbial eukaryotes. PLoS One 6:e16342. doi: 10.1371/journal.pone.0016342
Piganeau, G., Grimsley, N., and Moreau, H. (2011b). Genome diversity in the smallest marine photosynthetic eukaryotes. Res. Microbiol. Genome Organ. Eukaryot. Microbes 162, 570–577. doi: 10.1016/j.resmic.2011.04.005
Raven, J. A., and Beardall, J. (2003). “Carbohydrate metabolism and respiration in algae” in Photosynthesis in algae. eds. A. W. D. Larkum, S. E. Douglas, and J. A. Raven (Dordrecht: Springer Netherlands).
Richards, E. J., and Ausubel, F. M. (1988). Isolation of a higher eukaryotic telomere from Arabidopsis thaliana. Cell 53, 127–136. doi: 10.1016/0092-8674(88)90494-1
Ronquist, F., Teslenko, M., Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Saulnier, D., De Decker, S., and Haffner, P. (2009). Real-time PCR assay for rapid detection and quantification of Vibrio aestuarianus in oyster and seawater: a useful tool for epidemiologic studies. J. Microbiol. Methods 77, 191–197. doi: 10.1016/j.mimet.2009.01.021
Schauer, R., and Kamerling, J. P. (2018). “Chapter One-Exploration of the sialic acid world,” in Advances in carbohydrate chemistry and biochemistry, ed. D. C. Baker (Academic Press Cambridge)
Schloerke, B., Cook, D., Larmarange, J., Briatte, F., Marbach, M., Thoen, E., et al. (2018). GGally: Extension to “ggplot2.”. Geneva: Zenodo.
Schultz, J., Maisel, S., Gerlach, D., Müller, T., and Wolf, M. (2005). A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA N. Y. N 11, 361–364. doi: 10.1261/rna.7204505
Seibel, P. N., Müller, T., Dandekar, T., Schultz, J., and Wolf, M. (2006). 4SALE – a tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinform. 7:498. doi: 10.1186/1471-2105-7-498
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinforma. Oxf. Engl. 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Simmons, M. P., Sebastian, S., Adam, M., Alexander, J. L., Valeria, J., Christopher, R. P., et al. (2016). Abundance and biogeography of Picoprasinophyte ecotypes and other phytoplankton in the eastern North Pacific Ocean. Appl. Environ. Microbiol. 82, 1693–1705. doi: 10.1128/AEM.02730-15
Simon, N., Foulon, E., Grulois, D., Six, C., Desdevises, Y., Latimier, M., et al. (2017). Revision of the Genus Micromonas Manton & Parke (Chlorophyta, Mamiellophyceae), of the type Species M. pusilla (butcher) Manton & Parke and of the Species M. Commoda van Baren, Bachy and Worden and description of two new species based on the genetic and phenotypic characterization of cultured isolates. Protist 168, 612–635. doi: 10.1016/j.protis.2017.09.002
Smit, A., Hubley, R., and Green, P. (2015). Repeat masker. Available at: http://www.repeatmasker.org (accessed October 3, 2023).
Stark, J. R., Cardon, Z. G., and Peredo, E. L. (2020). Extraction of high-quality, high-molecular-weight DNA depends heavily on cell homogenization methods in green microalgae. Appl. Plant Sci. 8:e11333. doi: 10.1002/aps3.11333
Subirana, L., Péquin, B., Michely, S., Escande, M.-L., Meilland, J., Derelle, E., et al. (2013). Morphology, genome plasticity, and phylogeny in the genus Ostreococcus reveal a cryptic species, O. mediterraneus sp. nov. (Mamiellales, Mamiellophyceae). Protist 164, 643–659. doi: 10.1016/j.protis.2013.06.002
Tatusov, R. L., Koonin, E. V., and Lipman, D. J. (1997). A genomic perspective on protein families. Science 278, 631–637. doi: 10.1126/science.278.5338.631
Tragin, M., and Vaulot, D. (2018). Green microalgae in marine coastal waters: the ocean sampling day (OSD) dataset. Sci. Rep. 8:14020. doi: 10.1038/s41598-018-32338-w
Tragin, M., and Vaulot, D. (2019). Novel diversity within marine Mamiellophyceae (Chlorophyta) unveiled by metabarcoding. Sci. Rep. 9:5190. doi: 10.1038/s41598-019-41680-6
van Baren, M. J., Bachy, C., Reistetter, E. N., Purvine, S. O., Grimwood, J., Sudek, S., et al. (2016). Evidence-based green algal genomics reveals marine diversity and ancestral characteristics of land plants. BMC Genomics 17:267. doi: 10.1186/s12864-016-2585-6
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. Plo S One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wang, F., Xie, Y., Wu, W., Sun, P., Wang, L., and Huang, B. (2019). Picoeukaryotic diversity and activity in the northwestern Pacific Ocean based on rDNA and rRNA high-throughput sequencing. Front. Microbiol. 9:3259. doi: 10.3389/fmicb.2018.03259
Wickham, H. (2006). An introduction to ggplot: an implementation of the grammar of graphics in R. Statistics. Available at: https://citeseerx.ist.psu.edu/document?repid=rep1&type=pdf&doi=7f3e2207d2ef8fc0cee74069879c8adf35303a91 (accessed October 3, 2023).
Worden, A. Z., Lee, J.-H., Mock, T., Rouzé, P., Simmons, M. P., Aerts, A. L., et al. (2009). Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 324, 268–272. doi: 10.1126/science.1167222
Wu, W., Huang, B., and Zhong, C. (2014). Photosynthetic picoeukaryote assemblages in the South China Sea from the Pearl River estuary to the SEATS station. Aquat. Microb. Ecol. 71, 271–284. doi: 10.3354/ame01681
Xu, Y., Wang, H., Sahu, S. K., Li, L., Liang, H., Günther, G., et al. (2022). Chromosome-level genome of Pedinomonas minor (Chlorophyta) unveils adaptations to abiotic stress in a rapidly fluctuating environment. New Phytol. 235, 1409–1425. doi: 10.1111/nph.18220
Xu, X., Yu, Z., Cheng, F., He, L., Cao, X., and Song, X. (2017). Molecular diversity and ecological characteristics of the eukaryotic phytoplankton community in the coastal waters of the Bohai Sea, China. Harmful Algae 61, 13–22. doi: 10.1016/j.hal.2016.11.005
Yau, S., Krasovec, M., Benites, L. F., Rombauts, S., Groussin, M., Vancaester, E., et al. (2020a). Virus-host coexistence in phytoplankton through the genomic lens. Sci. Adv. 6:eaay2587. doi: 10.1126/sciadv.aay2587
Yau, S., Santos, A. L., Eikrem, W., Ribeiro, C. G., Gourvil, P., Balzano, S., et al. (2020b). Mantoniella beaufortii and Mantoniella baffinensis sp. nov. (Mamiellales, Mamiellophyceae), two new green algal species from the high Arctic. J. Phycol. 56, 37–51. doi: 10.1111/jpy.12932
Yu, G., and Yu, M. G. (2018). Package “scatterpie.” Available at: cran.r-project.org/web/packages/scatterpie/ (accessed October 3, 2023).
Yung, C. C. M., Rey Redondo, E., Sanchez, F., Yau, S., and Piganeau, G. (2022). Diversity and evolution of Mamiellophyceae: early-diverging Phytoplanktonic Green algae containing many cosmopolitan species. J. Mar. Sci. Eng. 10:240. doi: 10.3390/jmse10020240
Zhou, X. (2022). Comparative genomics analysis 3: Specific node gene family enrichment analysis (non-model species GO/KEGG enrichment analysis). Available at: https://www.jianshu.com/p/6671f3189309 (accessed October 30, 2023).
Zimin, A. V., Marçais, G., Puiu, D., Roberts, M., Salzberg, S. L., and Yorke, J. A. (2013). The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677. doi: 10.1093/bioinformatics/btt476
Keywords: biogeography, genomics, Mamiellophyceae, Mantoniella tinhauana sp. nov., marine algae, metagenomics
Citation: Rey Redondo E, Xu Y and Yung CCM (2024) Genomic characterisation and ecological distribution of Mantoniella tinhauana: a novel Mamiellophycean green alga from the Western Pacific. Front. Microbiol. 15:1358574. doi: 10.3389/fmicb.2024.1358574
Edited by:
Jin Zhou, Tsinghua University, ChinaReviewed by:
Shengwei Hou, Southern University of Science and Technology, ChinaCopyright © 2024 Rey Redondo, Xu and Yung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charmaine Cheuk Man Yung, Y2NteXVuZ0B1c3QuaGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.