
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 18 March 2024
Sec. Microbial Symbioses
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1358137
This article is part of the Research TopicThe Regulation of Environmental Factors on the Microbial CooperationView all 13 articles
 Fan Tian1,2†Juncai Wang3†Fangjun Ding1,2Lianhui Wang1,2Yanbing Yang1,2Xinxiang Bai4Chengjiang Tan5
Fan Tian1,2†Juncai Wang3†Fangjun Ding1,2Lianhui Wang1,2Yanbing Yang1,2Xinxiang Bai4Chengjiang Tan5 Xiaofeng Liao3‡*
Xiaofeng Liao3‡*Introduction: Paphiopedilum barbigerum is currently the rarest and most endangered species of orchids in China and has significant ornamental value. The mature seeds of P. barbigerum are difficult to germinate owing to the absence of an endosperm and are highly dependent on mycorrhizal fungi for germination and subsequent development. However, little is known about the regulation mechanisms of symbiosis and symbiotic germination of P. barbigerum seeds.
Methods: Herein, transcriptomics and proteomics were used to explore the changes in the P. barbigerum seeds after inoculation with (FQXY019 treatment group) or without (control group) Epulorhiza sp. FQXY019 at 90 days after germination.
Results: Transcriptome sequencing revealed that a total of 10,961 differentially expressed genes (DEGs; 2,599 upregulated and 8,402 downregulated) were identified in the control and FQXY019 treatment groups. These DEGs were mainly involved in carbohydrate, fatty acid, and amino acid metabolism. Furthermore, the expression levels of candidate DEGs related to nodulin, Ca2+ signaling, and plant lectins were significantly affected in P. barbigerum in the FQXY019 treatment groups. Subsequently, tandem mass tag-based quantitative proteomics was performed to recognize the differentially expressed proteins (DEPs), and a total of 537 DEPs (220 upregulated and 317 downregulated) were identified that were enriched in processes including photosynthesis, photosynthesis-antenna proteins, and fatty acid biosynthesis and metabolism.
Discussion: This study provides novel insight on the mechanisms underlying the in vitro seed germination and protocorm development of P. barbigerum by using a compatible fungal symbiont and will benefit the reintroduction and mycorrhizal symbiotic germination of endangered orchids.
The Orchidaceae is one of the largest and most diverse families of flowering plants in the world, with >28,000 accepted species across 800 genera (Chao et al., 2014; Zhao et al., 2021). Orchids are considerably valuable from ecological, ornamental, medical, and evolutionary perspectives (Zhang et al., 2018; Yang et al., 2020). Paphiopedilum is one of the most primordial genera of Orchidaceae, which includes 107 species so far. Members of this genus have considerable horticultural value and development potential (Huang et al., 2016; Fang et al., 2021). The Paphiopedilum orchids are generally known as the Lady’s slipper orchid owing to their slipper-shaped pouch (Tsai et al., 2020). They are extremely popular and highly valued in the horticulture market because of their unique flowers that have abundant colors, shapes, and sizes and long floral lifespan (Tsai et al., 2020; Fang et al., 2021). Unfortunately, members of this genus are endangered with their population severely declining in recent decades owing to the native habitat loss, rampant smuggling, and over-collection (Fang et al., 2021). To conserve these endangered species, the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) placed all Paphiopedilum species on the conservation list in 2017 to ban illegal trade (Yang et al., 2020). Furthermore, the mature Paphiopedilum seeds are very small, have few nutrient reserves, and lack an endosperm and are consequently difficult to germinate under natural conditions, making them particularly vulnerable to extinction (Zhang et al., 2018; Mujica et al., 2021). Therefore, the Paphiopedilum seed germination requires investigation, which is important for appropriately utilizing and conserving species resources.
The traditional propagation of Paphiopedilum orchids using an axillary bud division from the mother plant is unproductive and time-consuming (Chen et al., 2004). Many Paphiopedilum seeds form a compatible and symbiotic relationship with mycorrhizal or endophytic fungi that facilitates their germination in conditions of their natural habitat (Xu et al., 2020; Li Y. Y. et al., 2021). During this process, regarded as symbiotic germination, fungi and seeds form a nonphotosynthetic spherical body named a protocorm, which completely relies on mycorrhizal fungi for nourishment (Fang et al., 2021). Therefore, symbiotic germination can be used as an ordinary and efficient approach for promoting the germination and the large-scale propagation of Paphiopedilum (Chen et al., 2004). Several studies have found that mycorrhizal fungi can provide carbon and nitrogen sources, amino acids, and other nutrients as well as metabolic regulatory substances such as hormones for the seed germination of Paphiopedilum plants, whereas the plants provide carbohydrates for the mycorrhizal fungi (Fochi et al., 2017; Mujica et al., 2021; Zhao et al., 2021). Furthermore, the existence of compatible mycorrhizal fungi in Paphiopedilum seedlings enhances the adaptability of plants to the environment to increase plant survival and growth rate (Zhang et al., 2018). However, Paphiopedilum seedlings do not simply form a symbiotic relationship with any fungus, and germination can only happen when an appropriate fungus is present (Pujasatria et al., 2020), which includes Tulasnellaceae, Ceratobasidiaceae, Serendipitaceae, Mycena, Helicogloea, Fusarium, or other fungi (Xu and Guo, 1989; Kottke et al., 2010; Sebastián et al., 2014; Jiang et al., 2019). Understanding the interaction between Paphiopedilum and mycorrhizal fungi is a significant for generative reproduction, augmenting existing populations, and maintaining species diversity in orchids (Yang et al., 2020). Currently, research on the interaction between mycorrhiza and Paphiopedilum plants mainly includes promoting seed germination, growth and development, nutrient absorption, improving stress resistance of plants and the symbiotic relationship (Gao et al., 2022; Tian et al., 2023; Xu et al., 2023; Yang H. et al., 2023; Yang J. X. et al., 2023), whereas the molecular regulation mechanism of the interaction underlying the symbiotic relationship need further investigation.
In recent decades, understanding of the symbiotic interaction mechanism between mycorrhizal fungi and orchids has entered a new era with the development of next-generation sequencing and multi-omics technologies (Chen et al., 2018, 2022; Ahmad et al., 2022a). Multi-omics technology (i.e., transcriptomics, proteomics, and metabolomics) has become an effective method of exploring the orchid-fungus relationship (Balilashaki et al., 2019). For example, Fochi et al. (2017) used transcriptomics analysis to identify two functional ammonium transporters and several amino acid transporters involved in nitrogen uptake and transfer to host plants by the orchid mycorrhiza fungus Tulasnella calospora. Ghirardo et al. (2020) applied untargeted metabolomics to report that many metabolites, especially lipid compounds, were significantly altered during the symbiotic relationship between the Mediterranean orchid Serapias vomeracea and the basidiomycete T. calospora. In addition, Chen et al. (2018) used proteomic analysis to identify 42 proteins associated with antioxidant pathways, protein metabolism, and energy metabolism in Phalaenopsis. Hence, the multi-omics techniques are regarded as a promising approach for exploring the molecular mechanisms of the symbiotic relationship between mycorrhizal fungi and orchids.
Herein, we investigated the germination of P. barbigerum, a rare Paphiopedilum native to southwest China with high ornamental value, in the presence of a mycorrhizal fungus, Epulorhiza sp. FQXY019, which we had previously isolated and which progressed seed germination of P. barbigerum (Tian et al., 2022). Based on this, we integrated transcriptomics analyses with proteomic techniques to investigate the genes and proteins differentially expressed in the seed germination of P. barbigerum with and without Epulorhiza sp. FQXY019 inoculation. Thus, this study aimed to (1) reveal the transcriptional changes in the major metabolic pathways, and (2) investigate the differences in the proteomic profiles of P. barbigerum inoculated and uninoculated Epulorhiza sp. FQXY019. We hypothesized that: FQXY019 would induce the different biological and metabolic pathways of the P. barbigerum seed germination, and activate numerous responsive genes and proteins involved in molecular pathways at transcriptomic and proteomic level. The results will provide a theoretical basis for further elucidating the molecular regulation mechanism of the symbiotic relationship between orchids and mycorrhizal fungi as well as for the propagation and conservation of orchids.
In January 2021, the mature and well-grown fruit capsules were collected from P. barbigerum plants in the greenhouse of the Institute of Biotechnology, Guizhou Academy of Forestry. Capsules were desiccated at room temperature and stored in a glass vessel at 4°C until sowing. Epulorhiza sp. FQXY019 had been isolated from the mycorrhizal roots of P. barbigerum in our previous study (Tian et al., 2022). First, a layer of cellophane was placed onto 9 cm Petri dishes containing 30 mL potato dextrose agar (PDA) medium (200 g potato, 10 g agar, 20 g glucose, and 1 L distilled water). Epulorhiza sp. FQXY019 was then inoculated onto the cellophane. After 2 weeks of culture, the healthy mycelium of the fungus filled the Petri dishes and was used for the symbiotic seed germination assay.
Capsules of P. barbigerum were surface sterilized in 70% ethanol for 30 s, and shook for 6 min in 0.1% mercuric chloride, before being washed five times with sterile distilled water (Tian et al., 2022). Subsequently, the fruit capsules were cut with a sterile scalpel and forceps, gently shaken, and approximately 100 seeds were evenly sown on Epulorhiza sp. FQXY019 mycelium for symbiotic germination testing as the FQXY019-treatment group. Symbiotic seed germination was obtained by placing surface-sterilized seeds directly on PDA culture medium without Epulorhiza sp. FQXY019 as the control group (CK). Each treatment had 30 plates, which represented three replicates. The average temperature of culture was 25/20°C (darkness/light), light intensity was 1,200 lux, and day/night photoperiod was 18/6 h (Meng et al., 2020; Tian et al., 2022). Symbiotic and asymbiotic plants were collected after culturing for 90 days. At this point, the seeds had germinated, and the seedlings had two or three leaves. Sampling (the whole seedling) was performed after 90 days of culture because we had previously found that the seeds of P. barbigerum inoculated with Epulorhiza sp. FQXY019 can further develop, and rooting occurred after 90 days of culture (Tian et al., 2022). The figures of plant growing with and without Epulorhiza sp. FQXY019 was showed in Supplementary Figure S1. Three biological replicates were collected for each experimental group. All samples were quickly flash-frozen in liquid nitrogen and stored at −80°C for further analysis.
Total RNA was extracted from approximately 200 mg of frozen P. barbigerum plant samples of each treatment using prechilled RNA extraction Kit (TIANGEN, No DP411, in China) following the manufacture’s protocols (Ahmad et al., 2022a). The extracted RNA concentration and purity were assessed using a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific), and the integrity of RNA was assessed using an Agilent Bioanalyzer 2,100 system (Agilent Technologies, USA) (Mumtaz et al., 2022). Three separate libraries were prepared from three biological replicates per treatment. An RNA integrity number > 8.0 was selected to construct the sequencing library. cDNA libraries were constructed using the TruSeq™ RNA sample prep kit (Illumina, San Diego, USA) following the manufacturing specification after mRNA was isolated from total RNA using oligo (dT) magnetic beads and broken randomly into numerous fragments. Subsequently, sequencing was performed using an NovaSeq 6,000 platform by Beijing Biomarker Technologies Co., Ltd. To obtain high-quality clean reads, the remaining adapters, ploy-N, and low-quality value raw reads were eliminated (Ahmad et al., 2022a). High-quality clean reads were further de novo assembled using Trinity software. The unigenes were annotated in accordance with the NR, GO, KEGG, KOG, Pfam, SwissProt, TrEMBL, and eggNOG databases. The raw data of transcriptomics were available in the NCBI under accession PRJNA868469.
Gene expression levels were assessed using the fragments per kilobase per transcript per million mapped reads (FPKM). To consider the correlation of gene expression levels between control and treatment group samples, Pearson’s correlation coefficient was evaluated. Screening standards for differentially expressed genes (DEGs) were |log2(fold change, FC)| > 1 and false discovery rates (FDR) < 0.01 (Li P. et al., 2021). DEGs were identified utilizing the DEseq R package. To deduce the supposed function of DEGs, we performed GO enrichment analysis and KEGG pathway analysis using the topGO R package and KOBAS software, respectively.
To confirmation the accuracy of RNA-seq data, 12 DEGs were randomly chosen for qRT-PCR validation. Primers were designed via Primer Premier version 5.0 and are listed in Supplementary Table S1. qRT-PCR was performed as reported by Li P. et al. (2021). The calculation method of 2−ΔΔCt was employed for the relative expression level of genes. Three biological replicates were measured per gene, and actin was used as a reference gene.
Plant material protein isolation and Tandem Mass Tag (TMT) label-based proteome analyzes were conducted by Beijing Biomarker Technologies Co., Ltd. The proteins of P. barbigerum were extracted as described by Hao et al. (2017) and Zhu et al. (2021). Briefly, frozen samples were ground into a fine powder under liquid nitrogen. Then, the powder was transferred into a centrifuge tube and sonicated on ice three times in lysis buffer of 8 M urea, 2 mM EDTA, 10 mM dithiothreitol (DTT), and 1% protease inhibitor cocktail. The sediment was pelleted by centrifugation at 12,000 × g for 20 min. The protein in the supernatant was deposited with 15% precooled TCA buffer for 2 h at −20°C. The supernatant was removed, and then residual sediment was rinsed with cold acetone. Finally, the protein was redissolved in 8 M urea/100 mM tetraethylammonium bromide buffer (pH 8.0). The amount of protein in each sample was measured via the Bradford method and examined by sodium dodecyl sulfate polyacrylamide gel electrophoresis.
The protein sample was reduced with 10 mM DTT for 1 h at 37°C followed by an alkylation reaction at room temperature for 45 min by incubation in 40 mM iodoacetamide without light. Subsequently, the protein solution was diluted with 100 mM TEAB to a urea concentration of <2 M. Trypsin was added at a ratio of 1:50 (enzyme: protein, w/w) for the first digestion overnight and then was secondly digested for 4 h at a ratio of 1:100. After digestion, the protein samples were peptide desalted using a Strata X C18 SPE column and vacuum dried. The peptide was reconstituted with 0.5 M TEAB in accordance with the manufacturer’s protocol for the TMT kit (Thermo-Scientific, United States). Briefly, one unit of TMT reagent was reconstituted in acetonitrile. The peptide mixtures were incubated at room temperature for 2 h, and merged, desalted, and dried by vacuum centrifugation (Grosjean et al., 2022).
The sample was fractionated and purified using high-pH reverse-phase HPLC via an Agilent Zorbax 300Extend-C18 column (250 × 4.6 mm, 5-mm particles). Peptides were divided into eighty fractions over 80 min using a 2–60% acetonitrile gradient in 10 mM ammonium bicarbonate (pH 10). Peptides were collected into 18 fractions and dried via vacuum centrifugation (Jian et al., 2020).
Peptide samples were redissolved in 0.1% formic acid (FA, buffer A) and then loaded onto an EASY-nLC 1,000 UPLC system (Thermo-Scientific, USA) equipped with an Agilent Zorbax 300Extend-C18 column as above. Peptides were eluted with a linear gradient from 7 to 26% solvent B (1% FA in 98% acetonitrile) for 22 min, 26 to 40% in 12 min and 40 to 80% in 3 min, then remaining at 80% for the last 3 min, at a flow rate of 400 nL/min. Target peptides were analyzed using mass spectrometry (MS/MS) with a Q Exactive™ Plus (Thermo-Scientific, USA) with a Nanospray Flex™ (NSI) source. The electrospray voltage applied was 2.4 kV. Full-scan MS spectra (m/z 400–1,500) were examined in an Orbitrap at a mass resolution of 60,000 at m/z 200. A data-dependent procedure was conducted with 15-s dynamic exclusion of one MS scan followed by 20 MS/MS scans. Automatic gain control (AGC) was set as 3e6. After the full scan, the top 40 intense precursor ions were chosen for MS2 analysis at a resolution of 17,500 at m/z 200. The MS2 activation type was higher-energy collisional dissociation, normalized collision energy was 30 eV; AGC was set as 5e4. The obtained MS/MS data were evaluated with the Maxquant search engine (v.1.5.2.8). Multiple p values were controlled to a FDR of 1%. Protein quantification analysis was conducted on at least two unique peptides, and protein confidence was set at a 95% confidence interval (Jian et al., 2020; Zhou et al., 2022).
DEPs were identified using the thresholds of |log2(fold change, FC)| > 1 and p-value <0.05 in three biological replicates. The relative expression of the studied proteins was utilized to perform hierarchical clustering analysis by Cluster 3.0. GO functional annotation of the proteome was derived from the UniProt-GOA database using the Blast2GO program. DEPs were divided into three categories: BP, CC, and MF based on GO terms. The KEGG database was used to categorize these screened protein species via Blastx/Blastp 2.2.24 software. Then, the pathway enrichment of GO and KEGG of the DEPs was conducted using the Fisher’ exact test, and pathways with a p-value <0.05 were considered significant (Peng et al., 2019; Yu et al., 2019).
Three independent replicates of P. barbigerum in the control (CK) and FQXY019 treatment groups were analyzed via RNA-seq to reveal the potential symbiosis mechanism. The number of clean reads generated from a total of six libraries (three repeats per treatment) ranged from 22,375,505 to 24,054,439, with a GC content >47.86%; the percentage of Q20, Q30. and mapped ratio value was more than 97.77, 93.77, and 73%, respectively (Supplementary Table S2). After assembling the high-quality reads, the total number of transcripts and unigenes were 175,386 and 100,136 with a mean length of 1071.96 and 800.22 bp and an N50 length of 1842 and 1,577 bp, respectively (Supplementary Table S3). Overall, these results illustrated the high quality of the RNA-Seq data and practicability for further analysis.
All 40,742 unigenes were annotated via eight public databases, and 39,850 (97.81%), 30,367 (74.53%), 23,334 (57.27%), 20,217 (49.62%), 24,422 (59.94%), 20,781 (51.01%), 36,768 (90.25%), and 30,161 (74.03%) unigenes were successfully annotated to known proteins in the National Centre for Biotechnology Information (NCBI) non-redundant protein sequences (NR), Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), eukaryotic Orthologous Groups (KOG), Protein family (Pfam), Swiss-prot protein Sequence (Swiss-Prot), TrEMBL, and Evolutionary Genealogy of Genes: Nonsupervised Orthologous Groups (eggNOG) databases, respectively (Supplementary Table S4). The homologous species distribution analysis showed that 29.49% of annotated unigenes were matched with Dendrobium catenatum, in the Orchidaceae family (Supplementary Figure S2). This result implied that some gene sequence results of D. catenatum could be used as reference in the further study of the molecular mechanism of the symbiotic germination of P. barbigerum seeds as P. barbigerum is a reference-free genome plant. In addition, since these two plants belong to different genera, many unknown functional genes may be present in P. barbigerum that are different from those in D. catenatum and need further exploration.
A correlation coefficient (R2) of biological samples between 0.8 and 1 is a strong correlation relationship (p < 0.05), and when R2 is <0.8, the correlation relationship between samples is low. In the case of the gene expression patterns in each treatment, the R2 among the three replicates was >0.9, implying a remarkably high correlation (Figure 1A). Meanwhile, principal component analysis (PCA) suggested that the expression of gene clusters in the control and inoculation treatments was distinguishable, indicating a distinct responsive effect of Epulorhiza sp. FQXY019 inoculation on the gene expression patterns (Figure 1B).
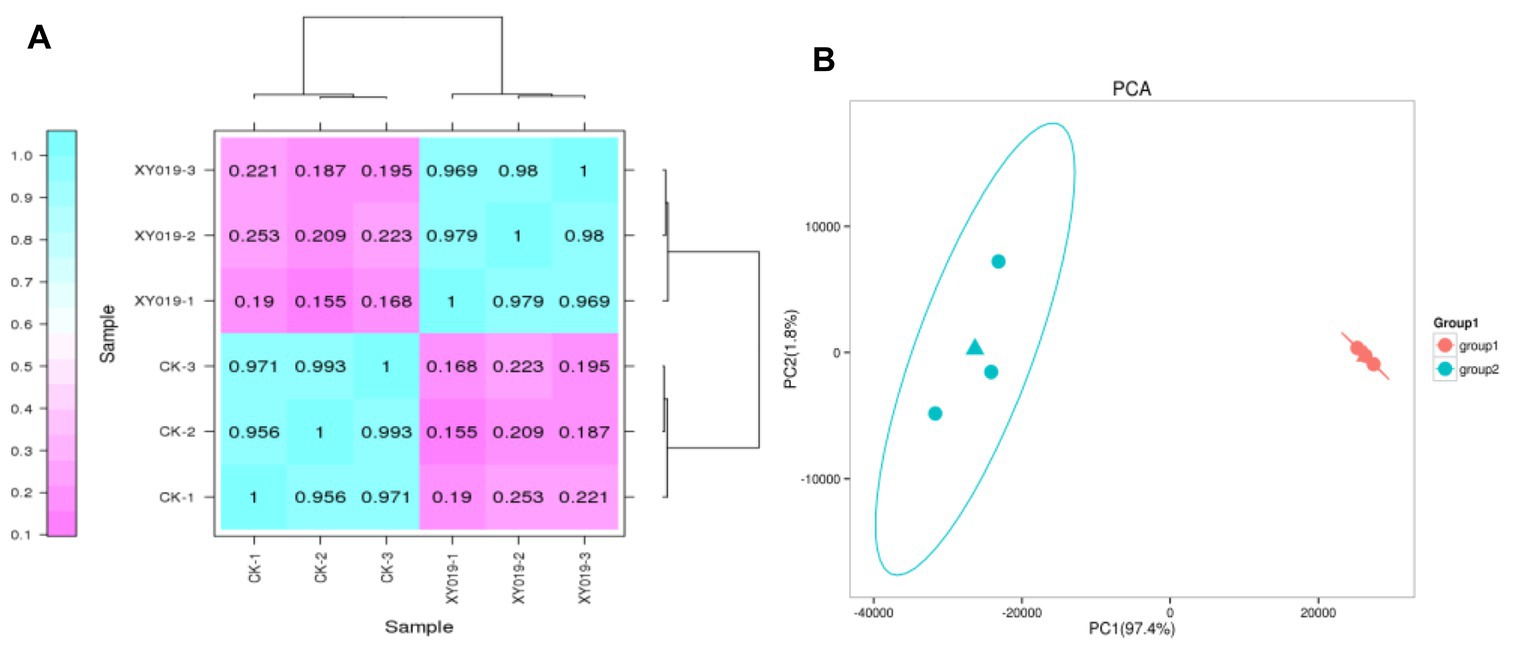
Figure 1. (A) Correlation analysis of the gene expression patterns in the FQXY019 treatment and CK groups. (B) Principal component analysis (PCA) of FPKM profiles in the FQXY019 treatment groups (group 1) and CK (group 2). The deeper the blue represents the stronger the positive correlation, and the deeper the purple the weaker the positive correlation (A). The blue and orange triangles represent the average coordinates of the three groups (B).
To identify DEGs in the two groups, we used a pairwise comparison. As shown in Figure 2, a total of 10,961 DEGs (2,599 upregulated and 8,402 downregulated) was identified in CK compared with expression in the FQXY019-treatment group (Supplementary Table S5). Subsequently, we annotated these DEGs using the GO database to investigate their functions. According to GO enrichment, a total of 6,828 DEGs were enriched to 57 GO terms, including 22, 20, and 15 terms in biological process (BP), cellular component (CC), and molecular function (MF), respectively (Figure 3A). Among them, the metabolic, cellular, and single-organism processes were the top three subcategories in BP; cell, cell part, and organelle were the top three subcategories in CC; and binding, catalytic activity, and transporter activity were the top three subcategories in MF (Figure 3A). Therefore, the DEGs between the two treatment groups were mainly involved in these processes, revealing that inoculation had an effect on these pathways.
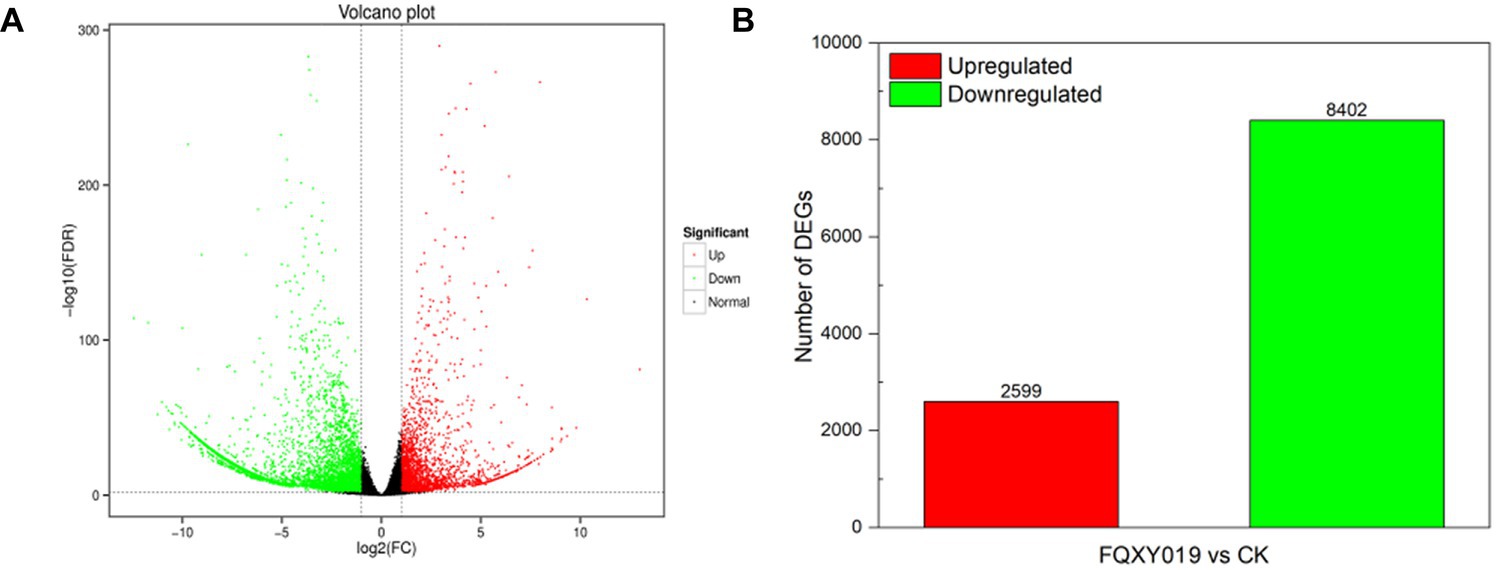
Figure 2. Differentially expressed genes (DEGs) for FQXY019 treatment groups vs. control (CK). (A) Volcano plot and (B) histogram; the number of up- and downregulated genes are represented by red and green, respectively.
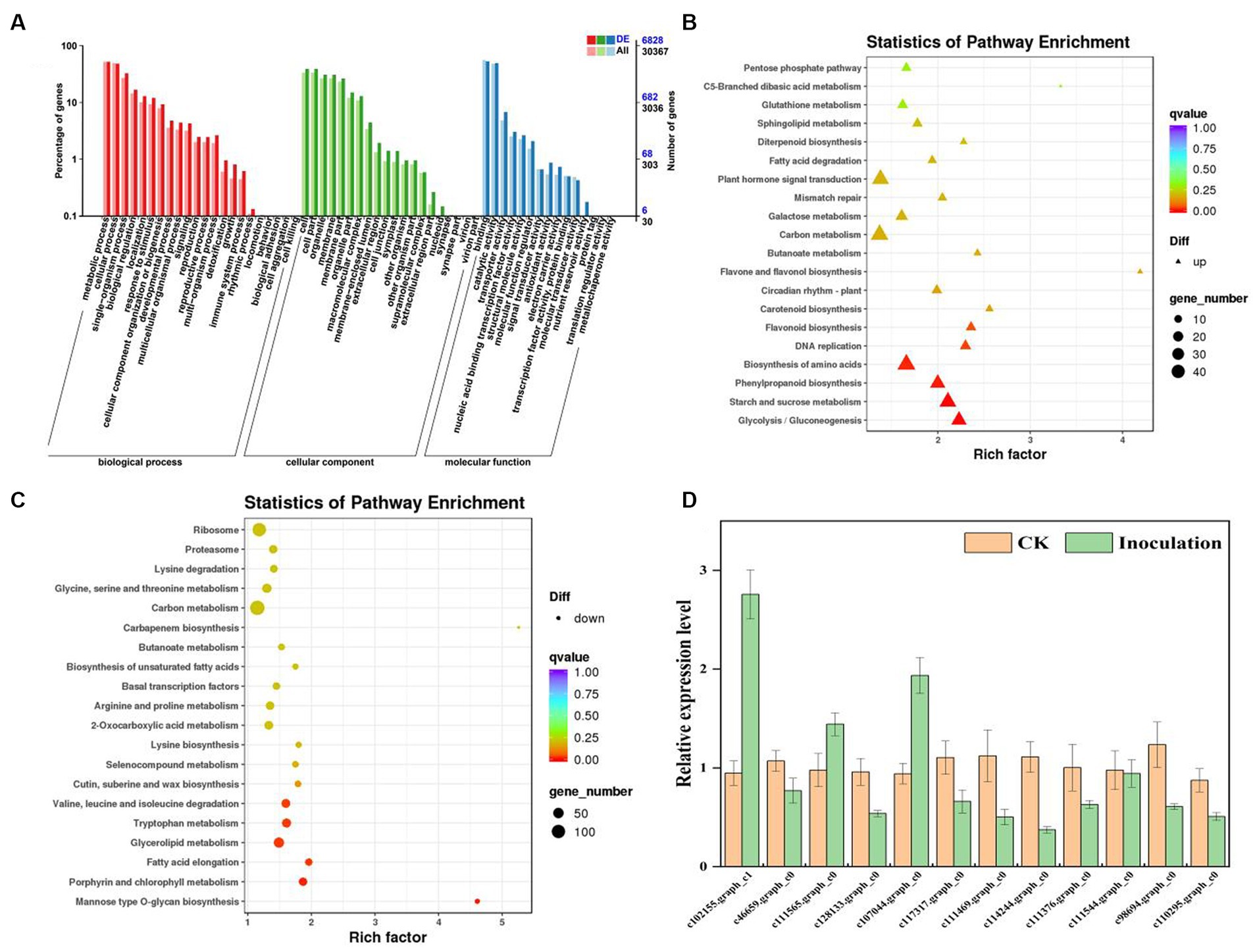
Figure 3. Graphical representation of significantly enriched pathways. (A) GO enrichment analysis of DEGs for FQXY019 treatment groups vs. CK. The bubble diagram shows the top 20 significant enriched pathways in the upregulated (B) and downregulated (C) DEGs for FQXY019 treatment groups vs. CK obtained via KEGG analysis. (D) qRT-PCR validation of DEG results. Values of qRT-PCR are mean ± SD (n = 3). Bars indicate SD. The detailed primer information is shown in Supplementary Table S1. The X-axis was the rich factor, which was the ratio of the DEG number to the total gene number in a certain pathway; the Y-axis represents the name of the pathway. The bubble size represents the number of DEGs involved. The bubble color indicates the enrichment degree of the pathway.
To reveal the biological functions and associated pathways of DEGs in Epulorhiza sp. FQXY019-induced symbiotic relationships, KEGG enrichment analysis was performed. Most DEGs were mainly annotated in metabolic pathways (Figures 3A–C; Supplementary Figure S3 and Supplementary Table S6), indicating that these pathways are important for symbiotic germination of P. barbigerum seeds. The top three most significant KEGG pathways were glycolysis/gluconeogenesis, starch and sucrose metabolism, and phenylpropanoid biosynthesis in upregulated DEGs in CK, compared with the FQXY019 treatment group (Figure 3B; Supplementary Figures S4–S6). The DEGs involved in glycolysis/gluconeogenesis, such as PFK, PFP, PDC, PGAM, etc. were significantly upregulated, but the DEGs (FBA, PDHA, DLD, FBP, etc.) were downregulated in the FQXY019 treatment. The DEGs (ISA, WAXY, E 2.4.1.14, etc) related to starch and sucrose metabolism were significantly upregulated and the DEGs (GYG1, SUS, INV, ENPP1, etc) were downregulated in the FQXY019 treatment. The genes (CCR, 4CL, CAD, COMT, etc.) of phenylpropanoid biosynthesis were significantly upregulated, and the genes (4CL, HCT, CCR, etc.) were significantly downregulated in the FQXY019 treatment relative to CK treatment (Supplementary Figures S4–S6). By contrast, the mannose type O-glycan biosynthesis, porphyrin and chlorophyl II metabolism, and fatty acid elongation pathways were ranked the top three significantly enriched pathways in downregulated DEGs in CK compared with that in the FQXY019 treatment group (Figure 3C; Supplementary Figures S7–S9). In addition, the largest number of upregulated DEGs was in amino acid biosynthesis, carbon metabolism, and plant hormone signal transduction, whereas the largest number of downregulated DEGs was in carbon metabolism and ribosome and amino acid biosynthesis. This suggests that the inoculation of FQXY019 affected the growth of seedlings by regulating metabolic pathways, including those of carbon and amino acid metabolism. In summary, these results imply that carbohydrate, fatty acid, and amino acid metabolism are the main physiological activity pathways used to maintain the stability of symbiotic relationships during the germination of P. barbigerum seeds.
To validate the reliability of the transcriptome results, we randomly selected 12 DEGs and examined them by qRT-PCR. According to the qRT-PCR analysis, the expression trends of 12 DEGs except in two DEGs (c46659.graph_c0 and c128133.graph_c0) were consistent with those obtained via transcriptome analysis (Figure 3D). The qRT-PCR results demonstrated that the transcriptome results were reliable and suitable for subsequent analysis.
To determine and identify up- or downregulated proteins in the P. barbigerum proteome inoculated with Epulorhiza sp. FQXY019 mycelium, we performed tandem mass tag (TMT) quantitative proteomic profiling of 90-day-old P. barbigerum seedlings in the CK and FQXY019 treatment groups, each with three biological replicates. After the combination of the two treatments of samples, 2,614 proteins were identified and quantified (Supplementary Table S10). Pairwise comparison of proteins with the criteria of a |log2(fold change, FC)| > 1 and a p-value <0.05 were screened as differentially expressed proteins (DEPs, Figure 4A). As shown in Figure 4B, 537 DEPs (220 upregulated and 317 downregulated) were detected in CK compared with the FQXY019 treatment group.
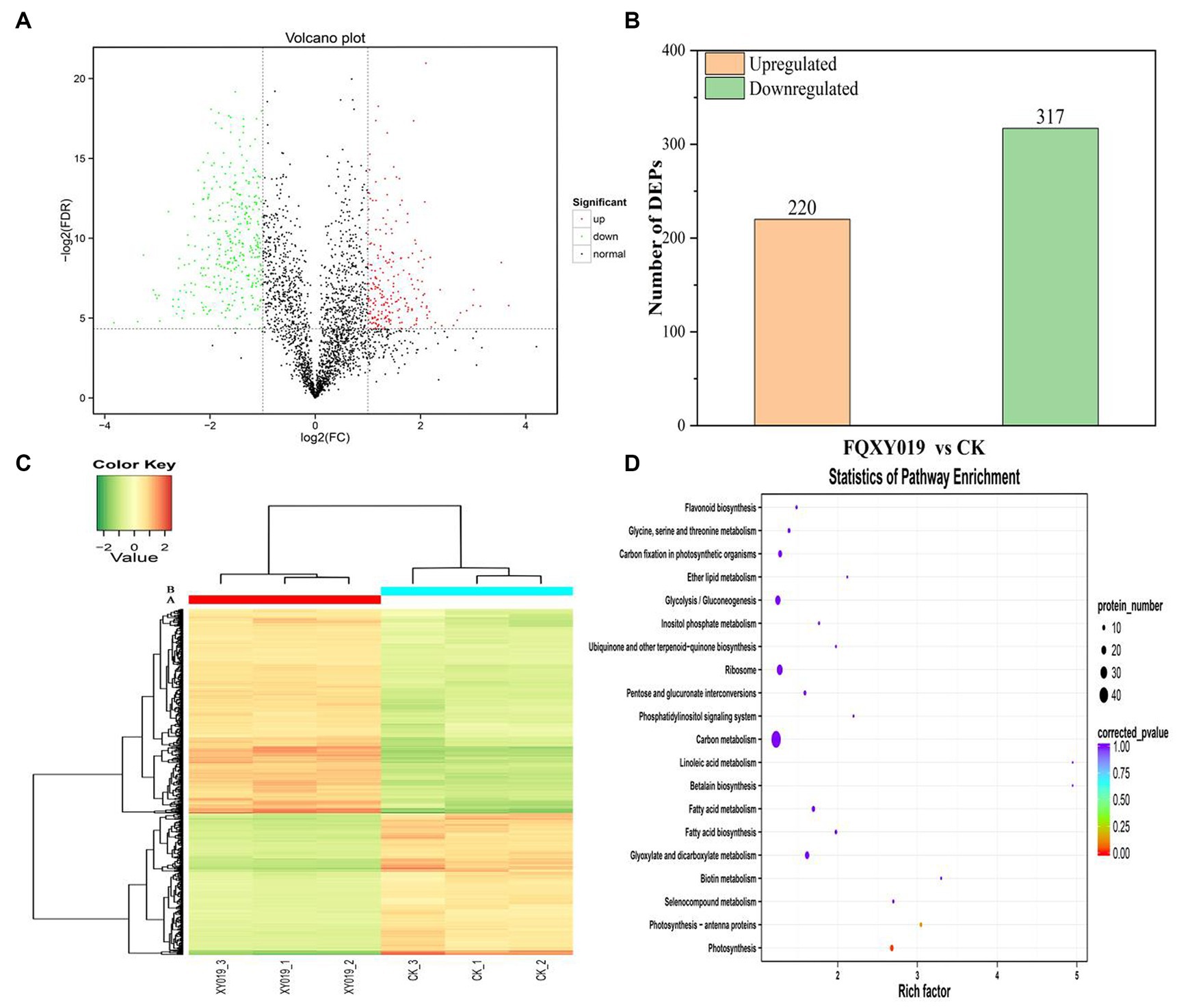
Figure 4. Differentially expressed proteins (DEPs) for FQXY019 treatment groups vs. control (CK). (A) Volcano plot and (B) histogram; the number of up- and downregulated genes are represented by red and green, respectively. (C) Hierarchical clustering analysis of the DEPs in different treatments. CK was the control treatment and FQXY019 was the inoculation treatment. Blue represents the CK, and red represents the FQXY019 treatment groups. (D) Bubble diagram showing the top 20 pathways with significant enrichment of DEPs for FQXY019 treatment groups and CK on KEGG pathway analysis. The X-axis was the rich factor, which was the ratio of the DEP number to the total protein number in a certain pathway; the Y-axis represents the name of the pathway. The bubble size represents the number of DEPs involved. The color of the circle represents p-value, which indicates the enrichment degree of the pathway.
We conducted a hierarchical clustering analysis of the DEPs in different treatments to explore the similarity between them. Each of the three replicates of the FQXY019 treatment groups and CK samples were clustered together independently and had high repeatability (Figure 4C). The protein abundance and the diversity of expression levels were markedly different between the FQXY019 treatment groups and the CK sample. PCA also showed similar results (Supplementary Figure S10).
To investigate the biological function of the DEPs, enrichment-based functional annotation was employed. GO functional annotation was performed on these DEPs, and 486 DEPs were successfully enriched into three categories including BP, CC, and MF. In the category of BP, the top three GO terms were metabolic process, cellular process, and single-organism process (Supplementary Figure S11a); the cell, cell part, and organelle were the largest subcategories in the CC category (Supplementary Figure S11b); and the top three GO terms were catalytic activity, binding, and structural molecule activity in MF analysis, respectively (Supplementary Figure S11c). Photosynthesis (BP), chloroplast thylakoid membrane (CC), and chlorophyl II binding (MF) were the most significantly enriched terms in these three processes (Supplementary Figure S11).
Proteins cannot accomplish their functions independently in an organism, and different proteins must coordinate with each other to achieve a series of biochemical reactions to conduct their biological functions. We therefore performed KEGG pathway enrichment analysis to help us understand how these DEPs were related to metabolism or signaling pathways and to reveal the biological functions and effect factors involved in the process. The DEPs were involved in 102 metabolic pathways, of which the top 20 pathways are shown in Figure 4D, including carbon metabolism, photosynthesis, amino acid biosynthesis, and fatty acid metabolism. Notably, the DEPs were primarily involved in metabolic pathways (Supplementary Figure S12); and the photosynthesis and photosynthesis-antenna proteins pathways were significantly enriched (Figures 4D, 5, 6). The DEPs (PsaA, PsaD, PsaK, PsaF, etc) related to photosynthesis I pathway and the DEPs (PsbA, PsbO, PsbQ, Psb27, etc) related to photosynthesis II pathway were significantly downregulated in the FQXY019 treatment compared with the CK treatment. The DEPs (Lhca1, Lhca3, Lhcb1, Lhcb2, etc.) involved in light-harvesting chlorophyl II protein complex pathway was significantly downregulated. These results indicating that symbiosis with Epulorhiza sp. FQXY019 could affect seed germination and seedling growth via changing the photosynthesis of P. barbigerum.
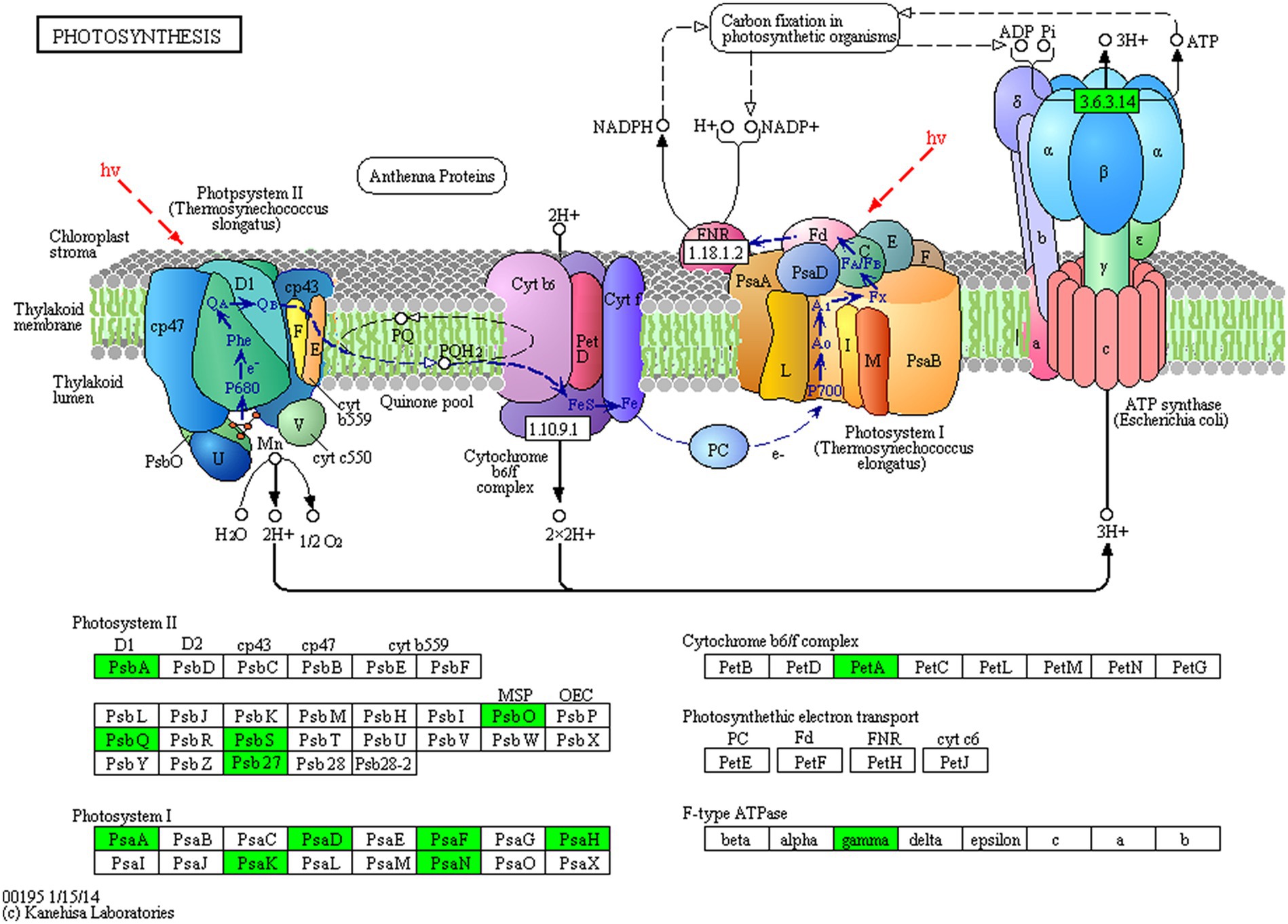
Figure 5. The expression levels of common DEPs in P. barbigerum involved in photosynthesis.
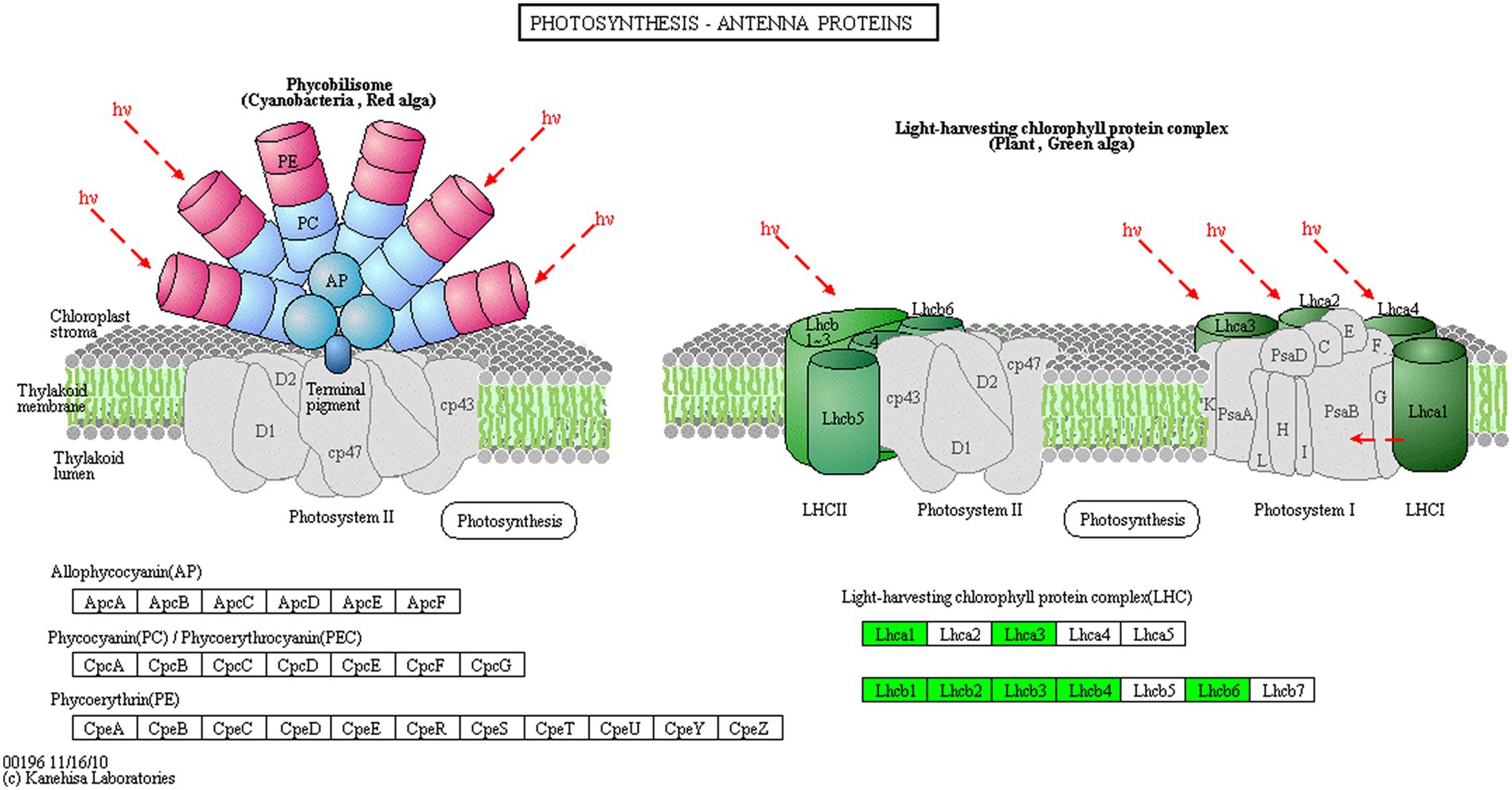
Figure 6. The expression levels of common DEPs in P. barbigerum related to photosynthesis-antenna proteins.
In this work, a combined transcriptomics, and proteomics analysis was performed to explore the symbiotic interaction of P. barbigerum inoculated and uninoculated Epulorhiza sp. FQXY019. Lots of DEGs were obtained using transcriptome sequencing during P. barbigerum seeds germination. The proteome changes, which were based on TMT labeling-based proteome quantification, also identified a large number of DEPs. Our study demonstrated that symbiosis induced substantial genes and proteins alterations, and regulate growth and development in P. barbigerum with FQXY019 (Figure 7). Several studies have proved that the beneficial relationship of carbon exchange in symbionts affects the nutritional balance between fungi and host plants (Li Y. Y. et al., 2021; Zhao et al., 2021). Fungi need to acquire carbohydrates from host plants, thereby regulating nutrient transport to play a dominant role in symbiosis (Vives-Peris et al., 2020). As they lack a mature endosperm, orchid seeds contain only a small amount of carbohydrates as a source of nutrients and need mycorrhizal fungi mycelium to obtain carbohydrates (Zhang et al., 2018; Zhao et al., 2021). However, to adapting to the environment and orchid metabolism, mycorrhizal fungi regulate their carbon and amino acid metabolism and improve their adaptability and survival competitiveness (Zhao et al., 2021).
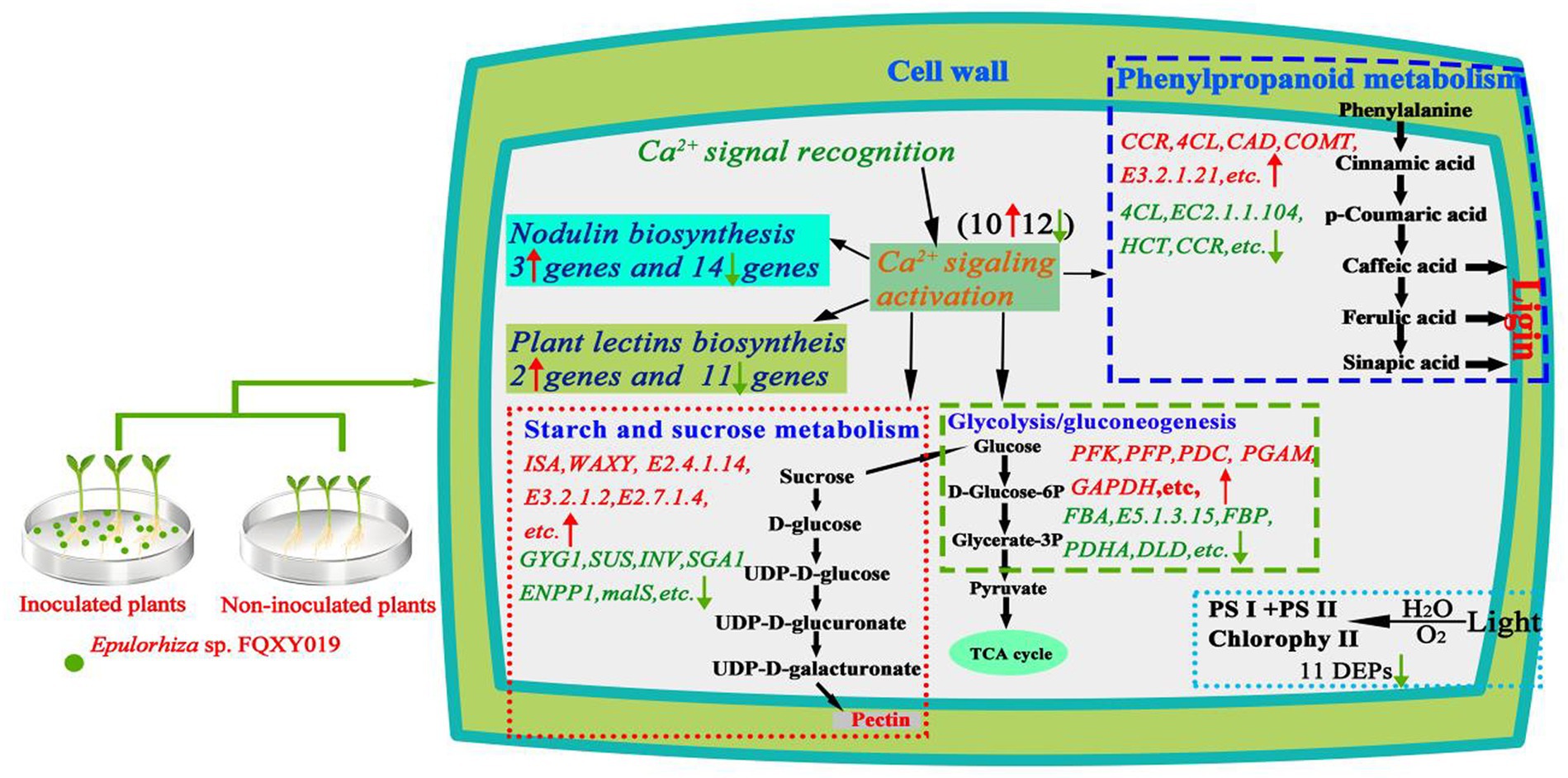
Figure 7. Schematic diagram of the potential mechanisms involved in the effect of Epulorhiza sp. FQXY019 symbiosis on the growth of P. barbigerum. Red and upward arrows indicate upregulated. Green and downward arrows indicate downregulated.
Nodulation genes are mainly involved in the formation and structural maintenance of nodules, protection of nitrogen fixation activity, and metabolism of nitrogen fixation products in nodule plants, especially in the establishment of the relationship between orchids and mycorrhizal fungi and the development of protocorm symbionts (Zhang et al., 2022). However, these genes not only play a role in nodulation but also in other developmental stages of plants. Several studies reported that nodulation genes and their encoded proteins are activated and actively expressed after arbuscular mycorrhizal fungi infection in plants (Wang et al., 2016; Ghirardo et al., 2020). These genes belong to the phytocyanin family of type I, and the expression products of this family are characterized by binding to type I copper ions and participate in the electron transfer process of the plant–microorganism interaction, plant cell growth, and development (Denance et al., 2014). We identified 17 genes that encoded proteins with a plastocyanin-like domain, which were predicted to be phytocyanin family genes, and one gene that encoded an early nodulation protein through screening the DEGs in CK compared with the FQXY019 treatment group (Supplementary Table S7). In a previous study, the effect of TaCLP1 (a gene encoding plastocyanin protein) on miRNA408 in wheat enhanced the resistance of wheat to stripe rust (Maunoury and Vaucheret, 2011). In addition, nodulin genes were found to be involved in the regulation of nodule formation in Arabidopsis, soybean, and other plants (Svistoonoff et al., 2010; Cheng et al., 2011; Gong et al., 2020). The induced expression of these genes indicated that inoculation could activate the expression of plastocyanin-like domain proteins, enhance the photosynthesis of seedlings, and accelerate seedling growth and development. These genes can also promote the formation of symbiotic nodules, which is beneficial to the uptake of nitrogen by the root system. In conclusion, the study of plant-mycorrhizal fungi interactions involving these genes still needs further study.
Ca2+ signaling plays a crucial role in regulating the growth and development of plants, such as plant carbon and nitrogen metabolism and ion water transmembrane transport, and the defense response of plants to abiotic environmental stress (Zhang et al., 2022). Ca ions are enriched around the nucleus of root hair cells, which is considered as the earliest cellular response of plants after the establishment of symbiotic relationship (Zhu, 2016). Plant cells have evolved a series of Ca2+ responsive receptors, including calmodulin-like protein (CLPs), calcium-binding protein (CBPs), calcium-dependent protein kinases (CDPKs), and calcineurin B-like proteins (CBLs), which can recognize and transmit Ca2+ signaling to regulate various cellular activities (De Bang et al., 2021). In this study, numerous genes related to Ca2+ signaling were screened, including three DEGs encoding CLPs, eight DEGs encoding CBPs, nine DEGs encoding CDPKs, and one DEG encoding a CBLa (Supplementary Table S8). The protein genes regulated by Ca2+ signals are known to be closely related to the formation of arbuscular mycorrhizal symbionts in rice, and the signals generated by these genes can regulate fungal colonization in plant cells (Campos-Soriano et al., 2011). The roots of Medicago truncatula activate Ca2+ signals and induce the expression of nodule genes to promote root nodule formation after recognizing mycorrhizal factors (such as nodulation factors) released by arbuscular mycorrhizal fungi (Kosuta et al., 2018). In addition, many genes involved in Ca2+ signaling have been found in the mycorrhizal symbionts of other orchids. For example, five DEGs encoding CDPKs were reported in the mycorrhizal symbionts of Epidendrum (Zhao et al., 2021), and five DEGs encoding CBPs and two DEGs encoding CDPKs were also found in the mycorrhizal symbionts of Cymbidium hybridum (Zhao et al., 2014). Therefore, our results indicated that after inoculation with Epulorhiza sp. FQXY019, P. barbigerum can induce the expression of Ca 2+ signaling-related genes, then transfer the signal from the upstream to the downstream gene expression in the root hair area, thus inducing the expression of orchid mycorrhizal symbiotic-related genes and the production of a variety of substances to identify fungi, digest mycelia, and establish an advantageous symbiotic relationship.
Plant lectins are a class of nonenzymatic proteins found in legumes that can agglutinate cells and precipitate monosaccharide or polysaccharide complexes (Kosuta et al., 2018; Ahmad et al., 2022b). Plant lectins play a crucial role in many signaling processes, for instance, signal transduction, plant defense, and immune response. Recent studies have reported that the specific recognition of plant symbiosis may be mediated by lectins, which can specifically bind carbohydrate complexes owing to their specific ability to bind monosaccharides and carbohydrate complexes, such as with mannitol-specific lectins (Houston et al., 2016; Andrade et al., 2019; Valadares et al., 2020). In this study, 13 DEGs were found to encode lectin-related proteins in the symbiotic mechanism of P. barbigerum between the FQXY019 treatment and CK group, six of which were downregulated DEGs that encoded mannose-specific lectin proteins (Supplementary Table S9). Our results were similar to previous studies. For example, significantly upregulated expression of mannose-specific lectin genes was found in several studies of orchid–fungi symbiotic relationships, such as between a Cymbidium hybrid and six different fungal strains (Reinhold-Hurek et al., 2015), Platanthera sikkimensis, T. calospora and Epidendrum secundum (Pujasatria et al., 2020). In addition, a mannose-specific lectin was reported to be involved in the establishment of the interaction with microorganisms during Gastrodia elata seed germination (Zeng et al., 2017). The induced expression of these genes indicates that they are one of the key factors for the establishment of symbiotic relationship between orchids and mycorrhizal fungi. Mycorrhizal fungi have faster growth and development than orchid plants and can quickly invade plant cells to establish a symbiotic relationship with plants (Dong et al., 2008; Xu et al., 2017). During this process, numerous plant lectin-related genes were induced and expressed, which played an important role in regulating and maintaining the interaction between orchids and mycorrhizal fungi.
Although most orchids can obtain carbon via photosynthesis, they still need fungi to provide nutrients. Zhang et al. (2020) found that Mycena increased the expression of photosynthesis-related proteins, such as antenna proteins, in Dendrobium officinale, thereby enhancing the photosynthetic efficiency of D. officinale. In a study on the symbiotic relationship between Oncidium sphacelatum and Ceratobasidium, the expression of photosynthesis-related proteins in the green protocorm of O. sphacelatum was found to be upregulated, including that of ribulose diphosphate carboxylase and chlorophyl a-apolipoprotein A1 of photosystem I, which affected their own photosynthesis. However, in this study, we observed that the expression of 13 DEPs related to photosynthesis was greatly downregulated in the CK in comparison with that in the FQXY019 treatment groups (Supplementary Table S11). In addition, eight DEPs related to photosynthesis antenna proteins were markedly downregulated in the CK in comparison with that in the FQXY019 treatment groups (Supplementary Table S12). These resulted indicate that the photosynthesis of P. barbigerum was gradually changed during the symbiotic development with Epulorhiza sp. FQXY019. Thus, this enables the energy requirement of seeding germination to be met with adjustment of the seed germination rate to quickly enable growth of seedlings of P. barbigerum.
During plant seed germination, starch in the endosperm is gradually degraded as the main source of ATP, which provides a precursor for the biosynthetic and metabolic reactions in embryos (Zhao et al., 2021). Nevertheless, orchid seeds are very small and nutrient storage is limited, and seed germination and subsequent developments of orchids in nature mainly depend on the colonization of mycorrhizal fungi (Li Y. Y et al., 2021). After the mycorrhizal fungi colonize the orchid seeds, the mycelium passes through the seed coat and produces amylase to hydrolyze starch in the embryo, which promotes the growth of the embryo protoplasm and absorbs nutrients from the surrounding environment to accelerate the further germination and growth of seeds (Pujasatria et al., 2020). In this study, transcriptomics showed that two pathways (starch and sucrose metabolism, and glycolysis/gluconeogenesis) related to energy metabolism were significantly enriched, involving a total of 72 DEGs, all of which were upregulated in the CK in comparison with the FQXY019 treatment groups (Supplementary Tables S13, S14). Interestingly, proteomics also showed that two pathways (pentose and glucuronate interconversions, and glycolysis/gluconeogenesis) related to energy metabolism were enriched and included a total of 31 DEPs, of which 20 DEPs were upregulated and 11 DEPs were downregulated in the CK in comparison with that in the FQXY019 treatment groups (Supplementary Tables S15, S16). These results demonstrated that the energy metabolism-related pathways in seeds of P. barbigerum rapidly changed after the seeds were infected by Epulorhiza sp. FQXY019, and the seeds germinated and formed seedlings. The energy metabolism pathways in seedlings also changed during the symbiotic development of seedlings and mycelium to meet the energy required for seedling growth, which is consistent with results from previous studies (Fochi et al., 2017).
Orchid seeds are among the smallest seeds in nature, and they are naturally rich in fatty acids (Chen et al., 2018). Fatty acids can generate intermediate products of sugar metabolism via glyoxylate and dicarboxylate metabolism and other metabolic processes and then convert them into carbohydrates via gluconeogenesis, which can provide sufficient energy for seed germination by glycolysis and pentose phosphate pathway (Ghirardo et al., 2020). Therefore, in this study, the DEGs and DEPs involved in the fatty acid metabolism in P. barbigerum were analyzed. The transcriptomics analysis showed that a total of 11 upregulated DEGs involved in fatty acid degradation, and 16 and 8 of the downregulated DEGs involved in fatty acid elongation and biosynthesis of unsaturated fatty acids, respectively, in the CK in comparison with that the FQXY019 treatment groups (Supplementary Table S17). A total of 13 DEPs (seven upregulated and six downregulated DEPs) were involved in fatty acid biosynthesis and metabolism in proteomics in the CK in comparison to the FQXY019 treatment groups, respectively (Supplementary Table S18). In addition, we found that two DEPs (one upregulated and one downregulated DEP) were related to linoleic acid metabolism in the CK in comparison with that in the FQXY019 treatment groups (Supplementary Table S18). These results indicate that inoculation with Epulorhiza sp. FQXY019 can affect the synthesis, decomposition, and metabolism of fatty acids in seeds of P. barbigerum by invading into seeds, so that the fatty acids stored in embryos can be more efficiently used to pass through the early stage of germination. Chen et al. (2022) found that the colonization by fungi could change the metabolic pattern and improve the nutrient utilization efficiency of D. officinale, especially when lipid metabolism was activated. In addition, genes encoding key enzymes involved in fatty acid metabolism, such as acetyl-CoA binding protein and β-oxidation polyfunctional protein genes, were significantly upregulated. Overall, our results indicated that the Epulorhiza sp. FQXY019 can induce the expression of genes and proteins related to fatty acid metabolism in P. barbigerum, and that the carbohydrates produced by this process can provide energy resources for seed germination and seedling growth.
This study demonstrated that inoculation of Epulorhiza sp. FQXY019 boosted and changed the transcriptomic and proteomic profile of P. barbigerum seed germination. Based on transcriptional evidence, numerous DEGs were identified in the CK and FQXY019 treatment groups, which are mainly associated with sugar, fatty acid, and amino acid metabolic pathway The expression levels of candidate DEGs related to nodulin, Ca2+ signaling, and plant lectins were significantly affected in P. barbigerum with FQXY019 treatment. Additionally, a total of 537 DEPs were identified in proteomic analysis and were involved in carbon metabolism, amino acid biosynthesis, and fatty acid metabolism. We identified several candidate DEPs related to photosynthesis, energy metabolism, and fatty acid metabolic pathways. Collectively, the identified genes and proteins will provide insight into the specific mechanism underlying the fungal inoculation of Paphiopedilum orchid seeds and their subsequent germination, and will act as a constructive reference resource to improve the genetic improvement of Paphiopedilum orchids.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
FT: Writing – original draft, Software, Investigation, Data curation. JW: Writing – original draft, Software, Methodology, Investigation, Data curation. FD: Writing – review & editing. LW: Writing – review & editing. YY: Writing – review & editing. XB: Writing – review & editing. CT: Writing – review & editing. XL: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the National key wildlife protection subsidy project: Paphiopedilum emersonii, P. barbigerum, and other Paphiopedilum plants rescue and protection project. The Scientific Search Projects of Guizhou Forestry Bureau [2019] 02; Innovative Talent Team Project of Guizhou Academy of Sciences (2019–07); The Project of Science & Technology of Guizhou Province [grant number: Qiankehezhicheng [2017]2524]; The Capacity Improvement Project of State Key Laboratory of Southwest Karst Mountain Biodiversity Conservation of National Forestry and Grassland Administration [2019–000411–05-01-001641]; Scientific Search Projects of Guizhou Forestry Bureau [2022] 36.
We are grateful to English language and grammar by editors of NES (www.nesediting.com) greatly improved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1358137/full#supplementary-material
Ahmad, S., Chen, J., Chen, G., Huang, J., Hao, Y., and Shi, X. (2022a). Transcriptional proposition for uniquely developed protocorm flowering in three orchid species: resources for innovative breeding. Front. Plant Sci. 13:942591. doi: 10.3389/fpls.2022.942591
Ahmad, S., Gao, J., Wei, Y., Lu, C., Zhu, G., and Yang, F. (2022b). The transcriptome profiling of flavonoids and bibenzyls reveals medicinal importance of rare orchid Arundina graminifolia. Front. Plant Sci. 13:1934. doi: 10.3389/fpls.2022.923000
Andrade, L. M., Peixoto-Junior, R. F., Ribeiro, R. V., Nóbile, P. M., Brito, M. S., and Marchiori, P. E. R. (2019). Biomass accumulation and cell wall structure of rice plants overexpressing a dirigent-jacalin of sugarcane (ShDJ) under varying conditions of water availability. Front. Plant Sci. 10:65. doi: 10.3389/fpls.2019.00065
Balilashaki, K., Zakizadeh, H., Olfati, J. A., Vahedi, M., Kumar, A., and Indracanti, M. (2019). Recent advances in Phalaenopsis orchid improvement using omics approaches. Plant Tissue Cult. Biotechnol. 29, 133–149. doi: 10.3329/ptcb.v29i1.41986
Campos-Soriano, L., Gómez-Ariza, J., Bonfante, P., and San, S. B. (2011). A rice calcium-dependent protein kinase is expressed in cortical root cells during the presymbiotic phase of the arbuscular mycorrhizal symbiosis. BMC Plant Biol. 11:90. doi: 10.1186/1471-2229-11-90
Chao, Y. T., Su, C. L., Jean, W. H., Chen, W. C., Chang, Y. C., and Shih, M. C. (2014). Identification and characterization of the microRNA transcriptome of a moth orchid Phalaenopsis aphrodite. Plant Mol. Biol. 84, 529–548. doi: 10.1007/s11103-013-0150-0
Chen, T. Y., Chen, J. T., and Chang, W. C. (2004). Plant regeneration through direct shoot bud formation from leaf cultures of Paphiopedilum orchids. Plant Cell Tissue Organ Cult. 76, 11–15. doi: 10.1023/A:1025858211320
Chen, J., Tang, Y., Kohler, A., Lebreton, A., Xing, Y., and Zhou, D. (2022). Comparative transcriptomics analysis of the symbiotic germination of D. officinale (Orchidaceae) with emphasis on plant cell wall modification and cell wall-degrading enzymes. Front. Plant Sci. 13:880600. doi: 10.3389/fpls.2022.880600
Chen, C., Zeng, L., and Ye, Q. (2018). Proteomic and biochemical changes during senescence of Phalaenopsis ‘red dragon’ petals. Int. J. Mol. Sci. 19:1317. doi: 10.3390/ijms19051317
Cheng, X. G., Wang, L., Wang, H., Yu, G. H., Bai, Y. L., and Liu, M. M. (2011). Specific expression of a novel nodulin gmN479 gene in the infected cells of soybean (glycine max) nodules. Agric. Sci. China 10, 1512–1524. doi: 10.1016/S1671-2927(11)60146-6
De Bang, T. C., Husted, S., Laursen, K. H., Persson, D. P., and Schjoerring, J. K. (2021). The molecular-physiological functions of mineral macronutrients and their consequences for deficiency symptoms in plants. New Phytol. 229, 2446–2469. doi: 10.1111/nph.17074
Denance, N., Szurek, B., and Noël, L. D. (2014). Emerging functions of nodulin-like proteins in non-nodulating plant species. Plant Cell Physiol. 55, 469–474. doi: 10.1093/pcp/pct198
Dong, F., Liu, H. X., Jin, H., and Luo, Y. B. (2008). Symbiosis between fungi and the hybrid cymbidium and its mycorrhizal microstructures. For. Stud. China 10, 41–44. doi: 10.1007/s11632-008-0002-1
Fang, L., Kong, X., Wen, Y., Li, J., Yin, Y., and Li, L. (2021). Characterization of embryo and protocorm development of Paphiopedilum spicerianum. Plant Physiol. Biochem. 167, 1024–1034. doi: 10.1016/j.plaphy.2021.09.001
Fochi, V., Chitarra, W., Kohler, A., Voyron, S., Singan, V. R., and Lindquist, E. A. (2017). Fungal and plant gene expression in the Tulasnella calospora-Serapias vomeracea symbiosis provides clues about nitrogen pathways in orchid mycorrhizas. New Phytol. 213, 365–379. doi: 10.1111/nph.14279
Gao, Y., Ji, J., Zhang, Y., Yang, N., and Zhang, M. (2022). Biochemical and transcriptomic analyses of the symbiotic interaction between Cremastra appendiculata and the mycorrhizal fungus Coprinellus disseminatus. BMC Plant Biol. 22:15. doi: 10.1186/s12870-021-03388-6
Ghirardo, A., Fochi, V., Lange, B., Witting, M., Schnitzler, J. P., Perotto, S., et al. (2020). Metabolomic adjustments in the orchid mycorrhizal fungus Tulasnella calospora during symbiosis with Serapias vomeracea. New Phytol. 228, 1939–1952. doi: 10.1111/nph.16812
Gong, Z., Xiong, L., Shi, H., Yang, S., Herrera-Estrella, L. R., and Xu, G. (2020). Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 63, 635–674. doi: 10.1007/s11427-020-1683-x
Grosjean, N., Le Jean, M., Armengaud, J., Schikora, A., Chalot, M., and Gross, E. M. (2022). Combined omics approaches reveal distinct responses between light and heavy rare earth elements in Saccharomyces cerevisiae. J. Hazard. Mater. 425:127830. doi: 10.1016/j.jhazmat.2021.127830
Hao, J., Guo, H., Shi, X., Wang, Y., Wan, Q., and Song, Y. B. (2017). Comparative proteomic analyses of two Taxus species (Taxus x media and Taxus mairei) reveals variations in the metabolisms associated with paclitaxel and other metabolites. Plant Cell Physiol. 58, 1878–1890. doi: 10.1093/pcp/pcx128
Houston, K., Tucker, M. R., Chowdhury, J., Shirley, N., and Little, A. (2016). The plant cell wall: a complex and dynamic structure as revealed by the responses of genes under stress conditions. Front. Plant Sci. 7:984. doi: 10.3389/fpls.2016.00984
Huang, J. Z., Lin, C. P., Cheng, T. C., Huang, Y. W., Tsai, Y. J., and Cheng, S. Y. (2016). The genome and transcriptome of Phalaenopsis yield insights into floral organ development and flowering regulation. PeerJ 4:e2017. doi: 10.7717/peerj.2017
Jian, M., Zhang, D., Wang, X., Wei, S., Zhao, Y., and Ding, Q. (2020). Differential expression pattern of the proteome in response to cadmium stress based on proteomics analysis of wheat roots. BMC Genomics 21:343. doi: 10.1186/s12864-020-6716-8
Jiang, J., Zhang, K., Cheng, S., Nie, Q., Zhou, S. X., and Chen, Q. (2019). Fusarium oxysporum KB-3 from Bletilla striata: an orchid mycorrhizal fungus. Mycorrhiza 29, 531–540. doi: 10.1007/s00572-019-00904-3
Kosuta, S., Chabaud, M., Lougnon, G., Gough, C., Dénarié, J., and Barker, D. G. (2018). A diffusible factor from arbuscular mycorrhizal fungi induces symbiosis-specific MtENOD 11 expression in roots of Medicago truncatula. Plant Physiol. 131, 952–962. doi: 10.1104/pp.011882
Kottke, I., Suárez, J. P., Herrera, P., Cruz, D., Bauer, R., and Haug, I. (2010). Atractiellomycetes belonging to the ‘rust’ lineage (Pucciniomycotina) form mycorrhizae with terrestrial and epiphytic Neotropical orchids. Proc. Biol. Sci. 277, 1289–1298. doi: 10.1098/rspb.2009.1884
Li, Y. Y., Boeraeve, M., Cho, Y. H., Jacquemyn, H., and Lee, Y. I. (2021). Mycorrhizal switching and the role of fungal abundance in seed germination in a fully mycoheterotrophic orchid, Gastrodia confusoides. Front. Plant Sci. 12:775290. doi: 10.3389/fpls.2021.775290
Li, P., Ruan, Z., Fei, Z., Yan, J., and Tang, G. (2021). Integrated transcriptome and metabolome analysis revealed that flavonoid biosynthesis may dominate the resistance of Zanthoxylum bungeanum against stem canker. J. Agric. Food Chem. 69, 6360–6378. doi: 10.1021/acs.jafc.1c00357
Maunoury, N., and Vaucheret, H. (2011). AGO1 and AGO2 act redundantly in miR408-mediated plantacyanin regulation. PLoS One 6:e28729. doi: 10.1371/journal.pone.0028729
Meng, X., Li, G., Gu, L., Sun, Y., Li, Z., and Liu, J. (2020). Comparative metabolomic and transcriptome analysis reveal distinct flavonoid biosynthesis regulation between petals of white and purple Phalaenopsis amabilis. J. Plant Growth Regul. 39, 823–840. doi: 10.1007/s00344-019-10025-y
Mujica, M. I., Cisternas, M., Claro, A., Simunovic, M., and Pérez, F. (2021). Nutrients and fungal identity affect the outcome of symbiotic germination in Bipinnula fimbriata (Orchidaceae). Symbiosis 83, 91–101. doi: 10.1007/s13199-020-00737-1
Mumtaz, M. A., Hao, Y., Mehmood, S., Shu, H., Zhou, Y., and Jin, W. (2022). Physiological and transcriptomic analysis provide molecular insight into 24-Epibrassinolide mediated Cr (VI)-toxicity tolerance in pepper plants. Environ. Pollut. 306:119375. doi: 10.1016/j.envpol.2022.119375
Peng, C., Chang, L., Yang, Q., Tong, Z., Wang, D., and Tan, Y. (2019). Comparative physiological and proteomic analyses of the chloroplasts in halophyte Sesuvium portulacastrum under differential salt conditions. J. Plant Physiol. 232, 141–150. doi: 10.1016/j.jplph.2018.10.028
Pujasatria, G. C., Miura, C., and Kaminaka, H. (2020). In vitro symbiotic germination: a revitalized heuristic approach for orchid species conservation. Plants 9:1742. doi: 10.3390/plants9121742
Reinhold-Hurek, B., Bünger, W., Burbano, C. S., Sabale, M., and Hurek, T. (2015). Roots shaping their microbiome: global hotspots for microbial activity. Annu. Rev. Phytopathol. 53, 403–424. doi: 10.1146/annurev-phyto-082712-102342
Sebastián, F., Vanesa, S., Eduardo, F., Graciela, T., and Silvana, S. (2014). Symbiotic seed germination and protocorm development of aa achalensis Schltr., a terrestrial orchid endemic from Argentina. Mycorrhiza 24, 35–43. doi: 10.1007/s00572-013-0510-2
Svistoonoff, S., Sy, M. O., Diagne, N., Barker, D. G., Bogusz, D., and Franche, C. (2010). Infection-specific activation of the Medicago truncatula Enod11 early nodulin gene promoter during actinorhizal root nodulation. Mol. Plant-Microbe Interact. 23, 740–747. doi: 10.1094/MPMI-23-6-0740
Tian, F., Liao, X. F., Wang, L. H., Bai, X. X., Yang, Y. B., and Luo, Z. Q. (2022). Isolation and identification of beneficial orchid mycorrhizal fungi in Paphiopedilum barbigerum (Orchidaceae). Plant Signal. Behav. 17:2005882. doi: 10.1080/15592324.2021.2005882
Tian, F., Wang, J. C., Bai, X. X., Yang, Y. B., Huang, L., and Liao, X. F. (2023). Symbiotic seed germination and seedling growth of mycorrhizal fungi in Paphiopedilum hirsutissimun (Lindl.Ex hook.) stein from China. Plant Signal. Behav. 18:2293405. doi: 10.1080/15592324.2023.2293405
Tsai, C. C., Liao, P. C., Ko, Y. Z., Chen, C. H., and Chiang, Y. C. (2020). Phylogeny and historical biogeography of Paphiopedilum Pfitzer (Orchidaceae) based on nuclear and plastid DNA. Front. Plant Sci. 11:126. doi: 10.3389/fpls.2020.00126
Valadares, R. B. S., Perotto, S., Lucheta, A. R., Santos, E. C., Oliveira, R. M., and Lambais, M. R. (2020). Proteomic and transcriptomic analyses indicate metabolic changes and reduced defense responses in mycorrhizal roots of Oeceoclades maculata (Orchidaceae) collected in nature. J. Fungi 6:148. doi: 10.3390/jof6030148
Vives-Peris, V., De Ollas, C., Gómez-Cadenas, A., and Pérez-Clemente, R. M. (2020). Root exudates: from plant to rhizosphere and beyond. Plant Cell Rep. 39, 3–17. doi: 10.1007/s00299-019-02447-5
Wang, Y., Xu, L., Tang, M., Jiang, H., Chen, W., and Zhang, W. (2016). Functional and integrative analysis of the proteomic profile of radish root under Pb exposure. Front. Plant Sci. 7:1871. doi: 10.3389/fpls.2016.01871
Xu, X., Fang, L., Li, L., Ma, G., Wu, K., and Zeng, S. (2020). Abscisic acid inhi bits asymbiotic germination of immature seeds of Paphiopedilum armeniacum. Int. J. Mol. Sci. 21:9561. doi: 10.3390/ijms21249561
Xu, J. T., and Guo, S. X. (1989). Fungus associated with nutrition of seed germination of Gastrodia elata-Mycena osmundicola Lange. Acta Mycol. Sin. 8, 221–226. doi: 10.13346/j.mycosystema.1989.03.011
Xu, L., Tian, J. N., Wang, T., and Li, L. B. (2017). Symbiosis established between orchid and Tulasnella spp. Fungi. J. Nucl. Agric. Sci. 31, 876–883. doi: 10.11869/j.issn.100-8551.2017.05.0876
Xu, Z. X., Zhu, X. M., Yin, H., Li, B., Chen, X. J., Fan, X. L., et al. (2023). Symbiosis between Dendrobium catenatum protocorms and Serendipita indica involves the plant hypoxia response pathway. Plant Physiol. 192, 2554–2568. doi: 10.1093/plphys/kiad198
Yang, J. X., Dierckxsens, N., Bai, M. Z., and Guo, Y. Y. (2023). Multichromosomal mitochondrial genome of Paphiopedilum micranthum: compact and fragmented genome, and rampant intracellular gene transfer. Int. J. Mol. Sci. 24:3976. doi: 10.3390/ijms24043976
Yang, H., Li, N. Q., and Gao, J. Y. (2023). A novel method to produce massive seedlings via symbiotic seed germination in orchids. Front. Plant Sci. 14:1114105. doi: 10.3389/fpls.2023.1114105
Yang, W. K., Li, T. Q., Wu, S. M., Finnegan, P. M., and Gao, J. Y. (2020). Ex situ seed baiting to isolate germination-enhancing fungi for assisted colonization in Paphiopedilum spicerianum, a critically endangered orchid in China. Glob. Ecol. Conserv. 23:e01147. doi: 10.1016/j.gecco.2020.e01147
Yu, R., Jiang, Q., Bu, S., and Shi, G. (2019). Comparative proteomics analysis of peanut roots reveals differential mechanisms of cadmium detoxification and translocation between two cultivars differing in cadmium accumulation. BMC Plant Biol. 19, 137–135. doi: 10.1186/s12870-019-1739-5
Zeng, X., Li, Y., Ling, H., Liu, S., Liu, M., and Chen, J. (2017). Transcriptomic analyses reveal clathrin-mediated endocytosis involved in symbiotic seed germination of Gastrodia elata. Bot. Stud. 58:31. doi: 10.1186/s40529-017-0185-7
Zhang, J. Y., Qin, J. L., and Liu, J. (2020). Transcriptomic analysis of Dendrobium officinale photosynthesis promoted by Mycena sp. Plant Physiol. J. 56, 1408–1418. doi: 10.13592/j.cnki.ppj.2020.0202
Zhang, S., Yang, Y., Li, J., Qin, J., Zhang, W., and Huang, W. (2018). Physiological diversity of orchids. Plant Divers. 40, 196–208. doi: 10.1016/j.pld.2018.06.003
Zhang, H., Zhu, J., Gong, Z., and Zhu, J. K. (2022). Abiotic stress responses in plants. Nat. Rev. Genet. 23, 104–119. doi: 10.1038/s41576-021-00413-0
Zhao, D. K., Selosse, M. A., Wu, L., Luo, Y., Shao, S. C., and Ruan, Y. L. (2021). Orchid reintroduction based on seed germination-promoting mycorrhizal fungi derived from protocorms or seedlings. Front. Plant Sci. 12:701152. doi: 10.3389/fpls.2021.701152
Zhao, X., Zhang, J., Chen, C., Yang, J., Zhu, H., Liu, M., et al. (2014). Deep sequencing-based comparative transcriptional profiles of Cymbidium hybridum roots in response to mycorrhizal and non-mycorrhizal beneficial fungi. BMC Genomics 15:747. doi: 10.1186/1471-2164-15-747
Zhou, Z., Lin, B., Tan, J., Hao, P., Hua, S., and Deng, Z. (2022). Tandem mass tag-based quantitative proteomics reveals implication of a late embryogenesis abundant protein (BnLEA57) in seed oil accumulation in Brassica napus L. Front. Plant Sci. 13:907244. doi: 10.3389/fpls.2022.907244
Zhu, J. K. (2016). Abiotic stress signaling and responses in plants. Cell 167, 313–324. doi: 10.1016/j.cell.2016.08.029
Zhu, Y., Qiu, W., He, X., Wu, L., Bi, D., and Deng, Z. (2021). Integrative analysis of transcriptome and proteome provides insights into adaptation to cadmium stress in sedum plumbizincicola. Ecotoxicol. Environ. Saf. 230:113149. doi: 10.1016/j.ecoenv.2021.113149
Keywords: Paphiopedilum barbigerum , mycorrhizal fungi, symbiotic germination, transcriptome, proteome
Citation: Tian F, Wang J, Ding F, Wang L, Yang Y, Bai X, Tan C and Liao X (2024) Comparative transcriptomics and proteomics analysis of the symbiotic germination of Paphiopedilum barbigerum with Epulorhiza sp. FQXY019. Front. Microbiol. 15:1358137. doi: 10.3389/fmicb.2024.1358137
Edited by:
Zhenlin Han, University of Hawaii at Manoa, United StatesReviewed by:
Taihe Xiang, Hangzhou Normal University, ChinaCopyright © 2024 Tian, Wang, Ding, Wang, Yang, Bai, Tan and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Liao, bHhmbnNkQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
‡ORCID: Xiaofeng Liao, https://orcid.org/0000-0002-0934-1798
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.