 Yuyang Shen
Yuyang Shen Chen Delai2,3
Chen Delai2,3 Taiguo Liu
Taiguo Liu Wanquan Chen
Wanquan Chen Haifeng Gao
Haifeng Gao Li Gao
Li Gao- 1Key Laboratory of Integrated Pest Management on Crop in Northwestern Oasis, Institute of Plant Protection, Xinjiang Academy of Agricultural Sciences, Ministry of P. R. China, Xinjiang, China
- 2State Key Laboratory for Biology of Plant Disease and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
- 3College of Plant Protection, Gansu Agricultural University, Lanzhou, China
Common bunt caused by Tilletia laevis Kühn is one of the most serious fungal diseases of wheat. The root–microbial associations play key roles in protecting plants against biotic and abiotic factors. Managing these associations offers a platform for improving the sustainability and efficiency of agriculture production. Here, by using high throughput sequencing, we aimed to identify the bacterial and fungal associations in wheat, alfalfa, and oat crops cultivated in different years in the Gansu province of China. Soil samples (0–6 cm below the surface) from infected wheat by T. laevis had significantly more bacterial and fungal richness than control samples as per the Chao1 analysis. We found some dominant fungi and bacterial phyla in infected wheat by T. laevis, such as Proteobacteria, Acidobacteria, Actinobacteria, Chloroflexi, Ascomycota, Basidiomycota, and Mortierello mycota. We also analyzed the chemical and enzymatic properties of soil samples after T. laevis inoculation. The total nitrogen, total kalium (TK), ammonium nitrogen, available kalium, organic carbon, invertase, phosphatase, and catalase were more in T. laevis-infected samples as compared to the control samples, while pH, total phosphorus, nitrate nitrogen, available phosphorus, and urease were more in control samples compared to T. laevis-infected samples. The results of this study will contribute to the control of wheat common bunt by candidate antagonistic microorganisms and adverse properties of soil.
1 Introduction
Tilletia laevis Kühn is a threatening pathogen of wheat crops, which causes huge damage worldwide, and mostly sporulation occurs in the plant ovary with host tissues in the kernel slowly replaced by masses of black teliospores (Nguyen et al., 2019). Losses in wheat crops reached 75–80% in many wheat-growing areas of the world (Qin et al., 2020). The relationship between plant pathogens and soil microbes can be either commensalistic, symbiotic, antagonistic, or parasitic (Ruby, 2008; Haegeman et al., 2009). For Tilletia, the characterization of the microbial communities in wheat tissues and rhizosphere soil (Din et al., 2021; Xu et al., 2021), characterization of rhizosphere microbial communities for disease incidence and optimized concentration of difenoconazole fungicide for controlling of wheat dwarf bunt (Jia et al., 2022), and microbiome signature of endophytes in wheat seed response to wheat dwarf bunt caused by Tilletia controversa Kühn were explored (Ren et al., 2020). The T. laevis, with a fishy smell, leads to the decreased quality and quantity of wheat crops (Lu et al., 2005). Some plants can alter soil biochemical properties (Hussain et al., 2011; Tang et al., 2015), and some plant pathogens can change rhizosphere microbial communities (Zhou and Wu, 2012; She et al., 2017) and alter the relative abundance of other soil-borne pathogens (Din et al., 2021). Soil microbial diversity is not only important for the soil life but also important for soil nutrient cycling (Berendsen et al., 2012). This microbial diversity plays an important role in the health of plants, increasing the soil fertility, and cycling of N, C, and many other nutrients (Berendsen et al., 2012; Miransari, 2013). Previous studies revealed that plant pathogens, such as T. laevis (Din et al., 2021), root know nematode (Zhou et al., 2019), and Erwinia spp. (She et al., 2017), changed rhizosphere microbial communities. Similarly Mendes et al. (2013) demonstrated that rhizosphere soil microbiome can alter the composition and structure of plant pathogenic and beneficial microorganisms (Mendes et al., 2013). Several studies have shown that soil microbial diversity is influenced by plant pathogens (Zhou et al., 2019; Din et al., 2021). Additionally, environmental factors, like pH, influence soil microbial diversity (Kim et al., 2016; Zhang et al., 2016). Therefore, it is very important to analyze the relationship between environmental factors and soil microbial diversity. However, limited studies have concerns about fungal and bacterial communities under different conditions with different plants after pathogen infection.
Wheat crops is a staple food crop in many countries of the world. Owing to its high amino acid contents, high protein, deliciousness, and use in many products, wheat is used as a food crop throughout the world (Chen et al., 2021). Alfalfa (Medicago sativa L.) is a Fabaceae perennial herb and is an important legume crop used for forage worldwide. Moreover, alfalfa is a key source of pollen and nectar throughout the world (Taha, 2015). Oats (Avena sativa L.) is an important crop for their high content of functional substances such as phytochemicals, dietary fibers, and several other substances with high nutritional value (Havrlentová et al., 2020). Because of the plant pathogens, the soil microbial community is influenced, which may increase or decrease the relative abundance of soil microorganisms (Din et al., 2021). Additionally, long-term continuous cropping alters the soil microbial community by increasing the relative abundance of soil-borne pathogens in the soil (Yang et al., 2012; Liu et al., 2014; Tang et al., 2015; She et al., 2017). Therefore, there is a need to investigate the effect of a plant pathogen on microbial diversity in different crops that have been growing continuously for a long period. It is known that plant species or soil environment influence the soil microbial diversity composition (Harrison and Bardgett, 2010; Huang et al., 2014). The plants adapt to biotic stresses by modifying the chemistry of their root exudates to assemble a health-promoting microbiome, such as the “cry for help” hypothesis, which provides a mechanistic explanation for previously described soil feedback responses to plant diseases, such as the development of disease-suppressive soils following continuous cultivation of take all-infected wheat (Rolfe et al., 2019). Din et al. (2021) revealed that the diversity and composition of the rhizosphere microbiome associated with wheat crops changed after T. laevis infection. However, issues associated with wheat, alfalfa, and oat crops are caused by alterations in the rhizosphere in response to diseases, the cultivation area of these crops has decreased sharply in recent years in Gansu province.
Here, to obtain an inclusive understanding of the rhizosphere soil microorganisms in wheat, alfalfa, and oat crops in Gansu province, China, after T. laevis infection, for comparatively exploring fungal and bacterial communities, we subjected fungal and bacterial communities from wheat, alfalfa, and oat cropping fields in Gansu province, China, to high-throughput sequencing, and we used redundancy analysis (RDA) to analyze relationships between soil microbial communities and soil properties with enzyme activities.
2 Results
2.1 Changes in physical and chemical properties of soil and its enzyme activities by Tilletia laevis
The basic chemical characteristics of soil from Gansu Province from the fields of wheat, alfalfa, and oat are listed in Table 1. The total nitrogen (TN), total phosphorus (TP), and total kalium (TK) of soil range from 0.46 to 1.28 g/kg, 0.66 to 0.94 g/kg, and 17.48 to 18.36 g/kg, respectively. Similarly, nitrate nitrogen (NO3(−)–N), ammonium nitrogen (NH4(+)–N), and available phosphorus (AP) ranged from 0.74 to 54.48 mg/kg, 0.47 to 2.52 mg/kg, and 6.79 to 22.44 mg/kg, respectively. The range of available kalium (AK), organic carbon (OC), moisture content (MC), and pH varied from 109.40 to 352.20 mg/kg, 4.53 to 11.18 g/kg, 0.77 to 15.30%, and 8.25 to 8.80, respectively (Table 1). The activities of various enzymes were investigated in topsoil and rhizosphere soil in various crops. The statistical analysis showed that invertase, phosphatase, urease, and catalase enzymes were significant in different treatments. The invertase was the highest in TFL2, with 46.29 (mg/g), and the lowest in CK1, with 9.19 (mg/g). The phosphatase and urease were the highest in TFL2 and CK4, with 3.29 (mg/g) and 2.01 (mg/g), respectively, and the lowest in CK and TFL1, with 1.32 (mg/g) and 0.14 (mg/g), respectively. Similarly, catalase was the highest in TFL2, with 1.53 (ml/g), and the lowest in CK5, with 1.57 mL/g (Table 2).
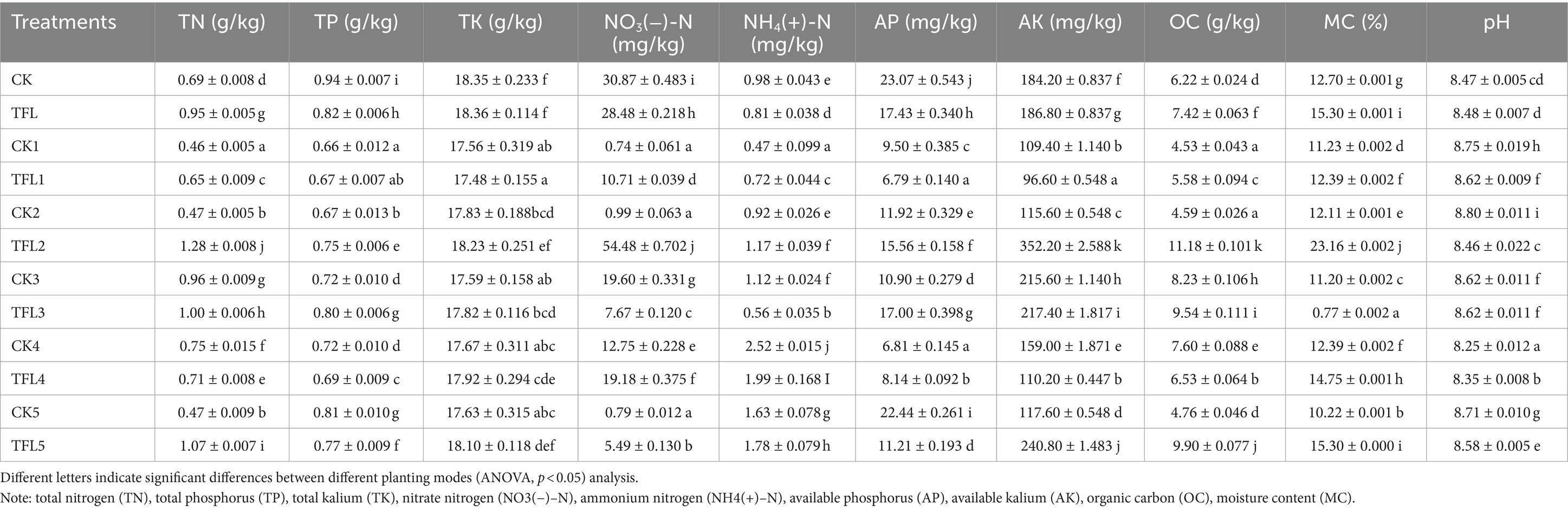
Table 1. Summary of soil physical and chemical properties under different planting modes.
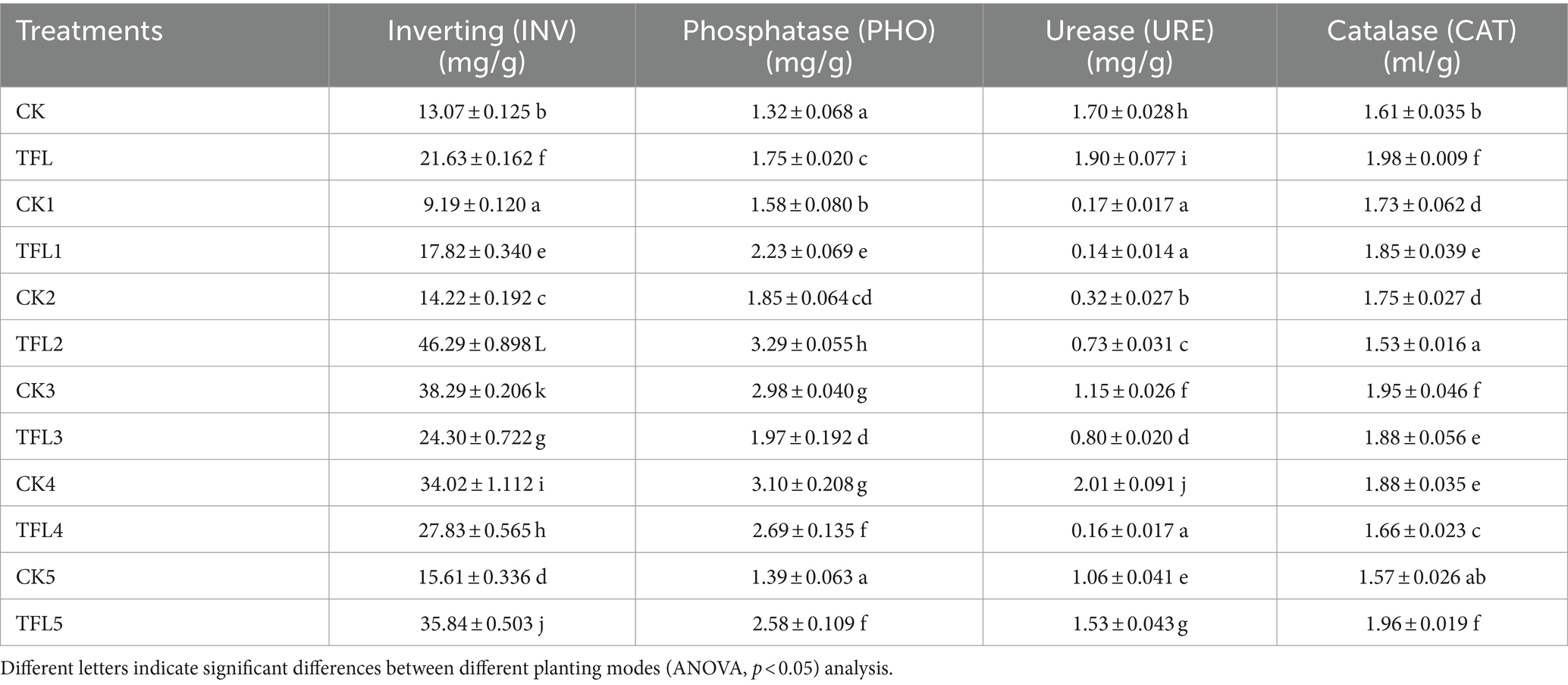
Table 2. Summary of soil enzyme activities under different planting patterns.
2.2 The influence of different types of soil on microbial diversity by Tilletia laevis
Across all (Garrett et al., 2018) rhizosphere soil samples, a total of 4,551,828 original bacterial sequences were obtained and 4,415,176 high-quality bacterial sequences were obtained from all samples. Similarly, 4,178,521 original fungal sequences were obtained, of which 3,900,304 were high-quality sequences. These bacterial and fungal sequences were on OTUs with 97% similarity levels. A total of 13,628 bacterial OTUs and 3,606 fungal OTUs were left after leveling (Supplementary Table S1).
2.3 Diversity and species richness of bacterial and fungal community by Tilletia laevis
Alpha diversity was analyzed based on the Chao1 and Shannon diversity indexes to assess the robustness of the dataset (Figure 1). The Chao1 index reflects species richness in samples, without considering the abundance of every species (Qiao et al., 2017). For bacteria, results showed that TFL2 and TFL3 soils have significantly higher species richness compared to CK1-5, TFL1, 4–5, TFL, and CK measured by Chao 1 index (Figure 1A). Additionally, for the Shannon diversity estimates, the CK5, TFL3 and TFL2 soils have significant higher diversity compared to TFL1, 4–5, CK1, 3–4, TFL, and CK (Figure 1B). For fungi, TFL1 and TFL rhizosphere soils have significant higher species richness than CK, CK1-5, and TFL2-5(Figure 1C). Additionally, for the Shannon diversity estimates, CK1 and TFL5 soils have significant higher diversity than CK, TFL, TFL1, 2–4, and CK2-5 (Figure 1D). We further conducted a comparison of the species diversity among different microbial communities. The principal coordinates analysis (PCoA) based on the Bray–Curtis distance between samples was visualized to analyze the differences in bacterial and fungal community diversity between groups. The samples of the same replicates clustered together indicated the level of significance. Additionally, samples formed distinct clusters, revealing that the largest source of variation was noted in the microbial community. The PCoA analysis bacterial OTUs showed the maximum variation of 14.22% (PC1) and 12.31% (PC2), as shown in Figure 2A and fungal OTUs showed the maximum variation of 15.44% (PC1) and 13.1% (PC2), as shown in Figure 2B.
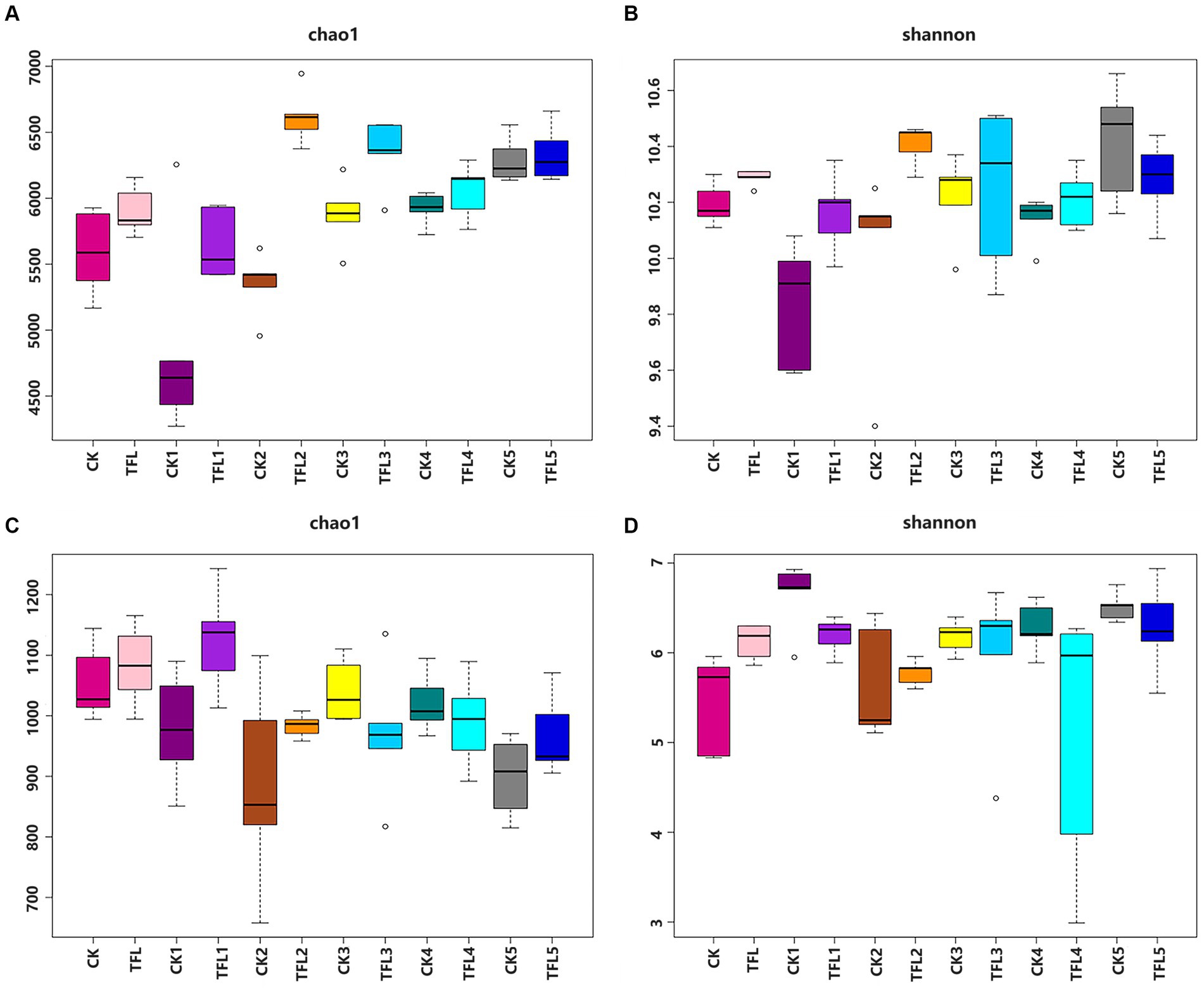
Figure 1. Chao1 and Shannon diversity analysis in the top layer and rhizosphere soil of wheat, alfalfa, and oat crops from pathogen trials inoculated with T. laevis. (A) Chao1 analysis of bacterial community. (B) Shannon analysis of bacterial community. (C) Chao1 analysis of fungal community. (D) Shannon analysis of fungal community.
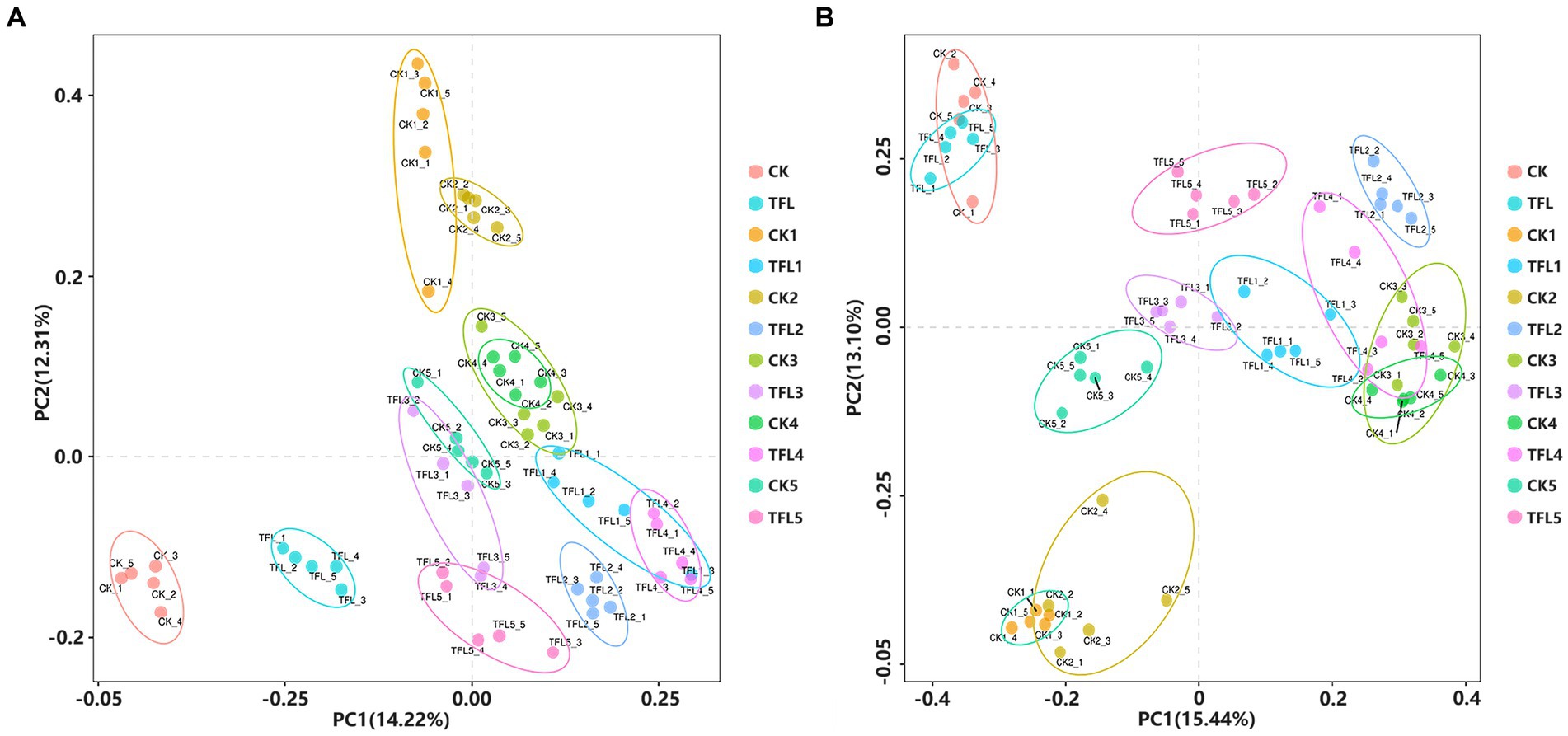
Figure 2. PCA of the OTUs detected major variations in the bacterial and fungal communities in three (wheat, alfalfa, and oat) crops. The OTUs differentiate based on the plant type and soil type. (A) PCAs analysis for bacterial community OTUs. (B) PCAs analysis for fungal community OTUs.
2.4 Dominant phyla and genera of bacterial and fungal communities
There were differences in the diversity indexes within the 12 samples analyzed demonstrating specific trends within different soil samples. The sequences that could not be classified into any known group are allocated as other and unidentified. The relative abundance of bacterial and fungal communities of T. laevis infected and control samples were different from each other. For bacteria, a total of 12 were distributed at the phylum level. Results showed that the phylum Proteobacteria, Acidobacteria, Actinobacteria, Chloroflexi, and Gemmatimonadetes were the dominant phyla in above samples than other phylum (Figure 3A). Similarly, for the fungus, the dominant phyla were Ascomycota, Basidiomycota, and Mortierellomycota, as compared to other phyla (Figure 3B).

Figure 3. The relative abundance of the dominant bacterial and fungal taxa in Gansu province in three (wheat, alfalfa, and oat) crops at the phylum and genus levels. (A) Relative abundance of bacterial community at the phylum level. (B) Relative abundance of fungal community at the phylum level. Sequences not classified into any known group were designated as “other”.
2.5 Correlation between microbial communities with soil properties and enzyme activities
In all samples, the bacterial and fungal OTUs were correlated with soil properties and enzyme activities using redundancy analysis (RDA). The RDA based on OTU reads, soil properties, and enzyme activities were carried out for the various soil samples in Gansu province, China. The relationship between bacterial communities and soil properties is illustrated in Figure 4A (RDA1 = 21.89%, RDA2 = 12.44%), the relationship between fungal communities and soil properties is illustrated in Figure 4B (RDA1 = 29.22%, RDA2 = 25.53%). Similarly, the relationship between bacterial communities and enzyme activity is illustrated in Figure 4C (RDA1 = 19.43%, RDA2 = 8.32%) and relationship between fungal communities and enzyme activity are illustrated in Figure 4D (RDA1 = 25.35% and RDA2 = 20.74%). The length of the arrow in the RDA plot indicates the degree of correlation among sample distribution, soil properties, and enzymatic activity. The results demonstrated that TP, AP, pH, OC, and TN showed the most significant correlation with bacterial community, while AP, NO3-H, and WC showed the least correlation with bacterial community structure in all soil samples (Figure 4A). Similarly, organic carbon (OC), TN, TP, AP, pH and NH4+-N showed the most significant correlation with fungal community, while NO3–N and moisture content (MC) revealed the least correlation with fungal community structure in all soil samples (Figure 4B). Additionally, bacterial community and enzyme activity analysis revealed that URE, INV, and PHO exhibited the most significant correlation in all samples, except CAT, which revealed the least correlation (Figure 4C). Moreover, PHO, INV, and URE enzymes showed the most significant correlation with fungal community structures in all samples (Figure 4D).
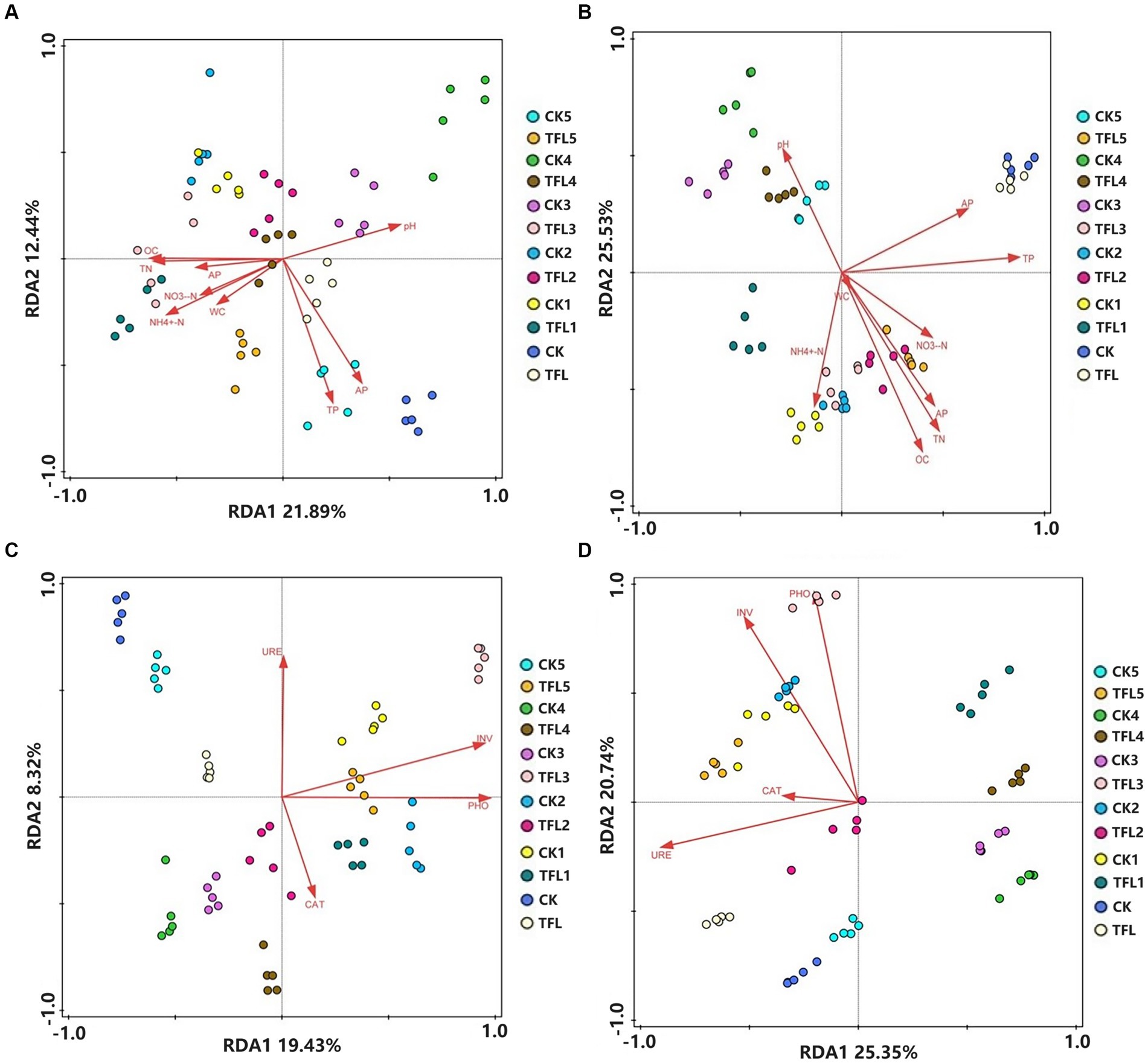
Figure 4. Redundancy analysis (RDA) based on bacterial and fungal OUT data with chemical properties and enzyme activity in three (wheat, alfalfa, and oat) crops after T. laevis infection. (A) The relationship between bacterial community and chemical properties of soil. (B) The relationship between fungal community and chemical properties of soil. (C) The relationship between bacterial community and enzyme activity. (D) The relationship between fungal community and enzyme activity.
3 Discussion
In this study, using high-throughput sequencing, we analyzed bacterial and fungal communities in wheat, alfalfa, and oat crop fields in Gansu Province, China. According to the α-diversity analysis, the overall diversity of bacterial and fungal community compositions differed among the soil samples. The Chao1 α-diversity and Shannon analysis revealed that the diversity of microbial communities is different in different crops (Figure 1). This may be due to different soil characters and crops in different periods. For bacteria, the Proteobacteria, Acidobacteria, Actinobacteria, Chloroflexi, Gemmatimonadetes, and Bacteroidetes, while for fungus, the Ascomycota, Basidiomycota, and Mortierellomycota were the dominant phyla (Figure 3), which was by the findings of previous studies (Zhou et al., 2019; Wang et al., 2020; Din et al., 2021). These phyla were also dominant in fields of soybean (Li et al., 2010), peanut (Li et al., 2014), and tobacco (She et al., 2017), as well as in T. laevis (Din et al., 2021) and root-knot nematode-infected (15)fields. The members of Proteobacteria play an important role in S, N, and C in soil (Nosheen et al., 2016). Previous studies revealed that there is greater abundance of Proteobacteria in fertile soil as compared to diseased soil (Wang et al., 2017). However, in our results, the percentage of Proteobacteria was the highest in TFL2 (alfalfa rhizosphere soil infected with T. laevis) from different crops after T. laevis infection. The Acidobacteria and Actinobacteria are key players in the suppression of fungal pathogen F. oxysporum (Trivedi et al., 2017). The Bacillus is a genus of Firmicutes, which has the role of controlling soil-borne pathogens and can stimulate plant growth activities as a beneficial microbe (Jos et al., 2008). For instance, Bacillus spp. inhibits R. solanacearum infection, which causes bacterial wilt (Guo et al., 2004; Tan et al., 2010; Muhae-ud-Din et al., 2018). Additionally, application of Bacillus spp. as a fertilizer can increase the soil microbial diversity (Huang et al., 2012). Therefore, Firmicutes are the best options to improve the soil microbial community and are influenced by soil-borne pathogens (Wang et al., 2020; Din et al., 2021). In our study, the relative abundance of fungal and bacterial rhizosphere microorganisms significantly changed in T. laevis-inoculated samples as compared to control samples with the increased abundance of Ascomycota, Basidiomycota, Proteobacteria, and Acidobacteria. These changes could be attributed to a change in the root exudation patterns in the presence of soil-borne pathogens, a higher prevalence of dead roots, and microbial competition (Jones et al., 2009; Berendsen et al., 2012; Zahar et al., 2014; Dudenhöffer et al., 2016; Gu et al., 2016). These rhizosphere soil microorganisms have a role in changing redox conditions, C flow, soil pH, and the production of rhizodeposits, including the release of root exudates of various natures (Hinsinger et al., 2003; Jones et al., 2009; Dennis et al., 2010). In our results, Ascomycota, Basidiomycota, and Mortierellomycota were the dominant phyla, which were consistent with the findings of previous studies (Xu et al., 2018; Din et al., 2021). The Basidiomycota and Ascomycota are important groups of fungi in most types of soils (Wallenstein et al., 2007; Unterseher et al., 2013), and species of these phyla are involved in crop cycling by degrading organic substances (Unterseher et al., 2013; Purahong et al., 2016). We observed significant changes in the relative abundance of Basidiomycota and Ascomycota in our samples, especially in CK-4, the relative abundance of Ascomycota was the highest compared to other soil samples.
The soil properties, including available N and soil pH, are influenced directly or indirectly by plant pathogens (Lazcano et al., 2021). Soil properties play an important role in plant nutrient acquisition and resistance to biotic and abiotic stresses (Eaton et al., 2012; Yu et al., 2022; Tiecher et al., 2023), such as adequate total nitrogen (TN) levels, vigorous plant growth, and higher yields. Phosphorus (TP) and total kalium deficiency can limit crop growth and yield, and low levels of NO3(−)-N can limit plant growth. NH4(+)-N can be influenced by soil pH and temperature, Adequate phosphorus (AP) levels are crucial for early root development and flowering. Available kalium (potassium) deficiency can increase susceptibility to diseases and stress. High organic carbon (OC) content, moisture content (MC), and pH can influence nutrient availability, microbial activity, and plant growth, excessive and low levels both will hinder nutrient uptake. Hence, we used RDA analysis for the relationship between environmental factors (including soil T, available K, soil pH, TN, and urease activity) and soil microbial composition. RDA results showed that environmental factors differentially affected the fungal and bacterial communities, which were proven by various previous studies (Zhang et al., 2005; DeAngelis et al., 2015; Zhou et al., 2017). Urease catalyzes the breakdown of urea into NH3 and CO2, which may be good for soil quality (Jezierska-Tys and Rutkowska, 2014). The plants and rhizosphere soil microorganisms release urease enzymes (Follmer, 2008).
The plant pathogens cause a decline in the urease activity, and positive correlations between soil micro-organisms and urease have been previously found (Lazcano et al., 2021).
Soil microbial communities were altered in response to pathogen infection, leading to changes in soil enzymatic activities and nutrient availability (Mendes et al., 2013). Pathogen-infected plants may exhibit altered nutrient uptake and cycling dynamics. For example, Phytophthora infestans infection in potato plants can lead to changes in phosphorus cycling and availability in the soil; pathogen infections can decrease crop yield globally, with significant variation depending on the pathogen and crop species (Garrett et al., 2018). Pathogen infections can alter the composition and function of soil microbial communities, which play crucial roles in nutrient cycling, disease suppression, and plant health. For example, Fusarium oxysporum infection in different common beans has been shown to reduce microbial diversity and alter soil bacterial community composition (Hollander, 2018). Similarly, in our results, the urease activity changed after T. laevis inoculation in different crops (Table 2). Previous studies revealed that N has a role in regulating the rhizosphere soil microbial community (Cleveland et al., 2007; Högberg et al., 2014), and urease activity increased by the N application from 247 to 433 mg/kg (Liang et al., 2016; Lei et al., 2018). Therefore, N provides a good means to increase the urease activity to increase the soil micro-biota. However, a high concentration of ammonia can reduce the activity of the urease enzyme (Piotrowska and Wilczewski, 2012). Additionally, TN has a major role in influencing the fungal and bacterial community (Wang et al., 2020).
In conclusion, according to the RDA analysis of rhizosphere microorganisms and environmental factors in Gansu province, a positive correlation was noted in the chemical properties and enzyme activity of rhizosphere and top-layer soil. We explored some dominant fungi and bacterial phyla in the rhizosphere and top soil in infected wheat by T. laevis, such as Proteobacteria, Acidobacteria, Actinobacteria, Chloroflexi, Ascomycota, Basidiomycota and Mortierellomycota, which were related to T. laevis, we may reduce the content of this may contribute to the control of T. laevis shortly, and we may isolate these to explore the interaction with T. laevis (Jin et al., 2023; Zhou et al., 2023). Even though some taxa belong to the same genus, they can have different functions in the control of different pathogens. Additionally, nitrogen, total kalium, ammonium nitrogen, available kalium, and organic carbon were increased after T. laevis infection, so, reducing these elements may also contribute to controlling wheat’s common bunt disease which is caused by T. laevis. Hope shortly, we can control the wheat’s common bunt disease with efficient and friendly microbiology and the elements mentioned above.
4 Materials and methods
4.1 Site description and sample collection
The experimental site was located in Gansu Province, 32°11′ - 42 °57″ N and 92 °13 ′-108 ° 46″ (E). The soil samples were collected from a depth of 6 cm with a stainless-steel cylindrical driller and immediately stored in a portable refrigerator at −20°C for further use. The samples were passed out from a 2 mm sieve to remove the debris and stored at −20°C for next use. We collected samples from topsoil and rhizosphere soil from five plants and pooled them into one sample. A total of 12 soil samples from wheat, alfalfa, and oat crops were collected and stored in plastic bags and shifted on ice to the laboratory. One-half of each soil sample was stored at −20°C for biochemical and biological analyses, and the remaining were used for chemical analysis. Every sample was investigated in triplicate. Detailed information about samples is illustrated in Table 3. The T. laevis culture was collected from the Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China. With the teliospores from infected wheat tassels and the concentration of T. laevis, infectious hyphae were adjusted to 106 cfu/mL with an OD600 of 0.15. Five inoculations of T. laevis infectious hyphae were inoculated into the root zone of all the above-mentioned crop varieties, with three biological replicates as described (Din et al., 2021), and three sets of each variety were used as controls.
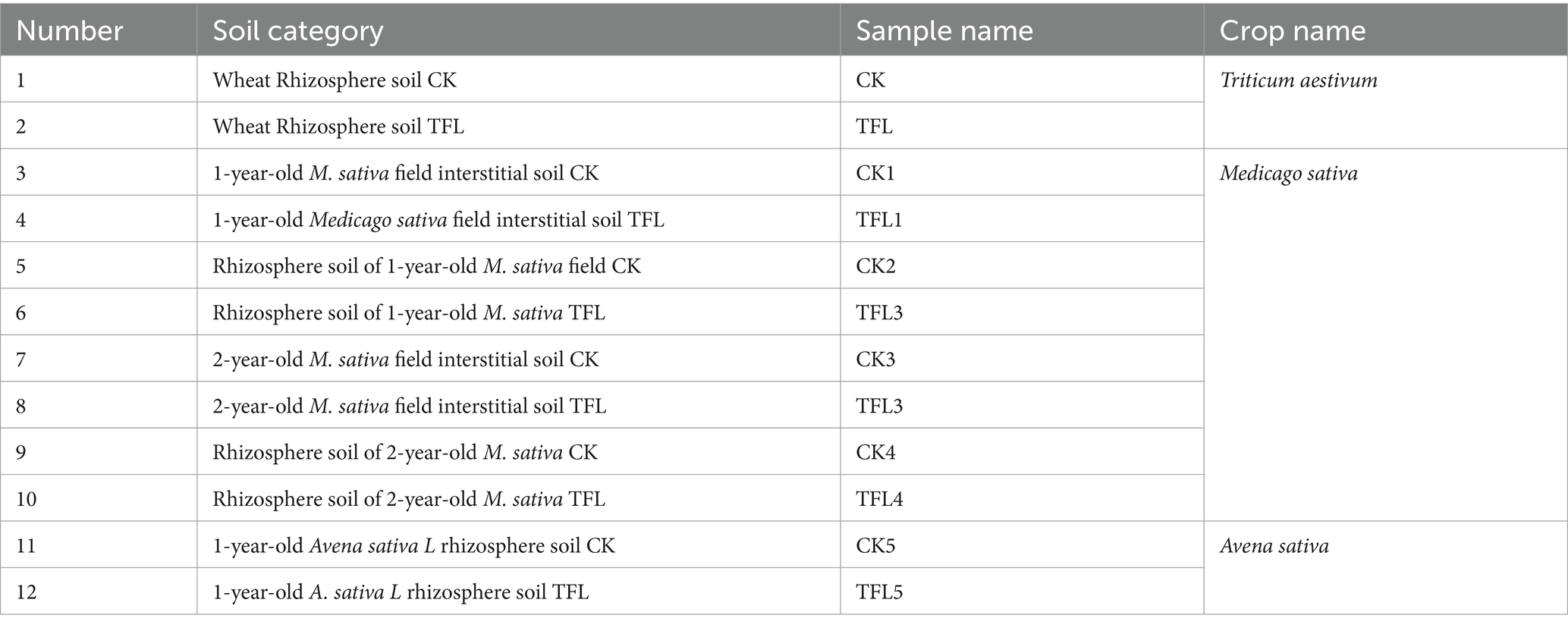
Table 3. Samples information.
4.2 Analysis of soil basic properties and enzymatic properties
Soil basic properties, including TP, AP, pH, NO3(−)-N, NH4(+)-N, OC, and TN were analyzed by using redundancy analysis (RDA) with CANOCO 4.5 (Biometrics, Wageningen, The Netherlands). These basic properties of soil were analyzed by following the method of previous reports (Xu et al., 2018; Wang et al., 2020). The sodium phenate and sodium hypochlorite colorimetric methods were used to determine soil urease and other enzyme activities (Vlek et al., 1980).
4.3 DNA extraction and PCR amplification
DNA extraction was performed from 5 gm of each homogenized soil sample as previously described (DeSantis et al., 2005) and purified using the PowerSoil® DNA isolation kit (MO BIO, Carlsbad, CA, United States), according to the manufacturer’s instructions. DNA concentration was quantified on a NanoDrop spectrophotometer (Thermo Scientific). The primer sequences for T. laevis were ITS1F (5- CTTGGTCATTTAGAGGAAGTAA −3) and ITS2 (5- TGCGTTCTTCATCGATGC -3). PCR amplification was performed by using 25 μL mixture, including 12.5 μL KAPA 2G robust hot start ready mix, 1 μL forward primer (5 μM), 1 μL reverse primer (5 μM), 5 μL DNA (30 ng), and 5.5 μL ddH2O. Following an initial denaturation at 95°C for 5 min, PCR was cycled 28 times at 95°C for 45 s, 55°C for 50 s, and a final extension at 72°C for 10 min. PCR products were purified using the AMPure XP kit (Beckman Coulter, Life Sciences).
4.4 High-throughput sequencing and data analysis
Deep sequencing was performed on MiSeq platform allergens Technology Inc. (Biotechnology, Beijing). After the run, image analysis, base calling, and error estimation were performed using Illumina analysis pipeline version 2.6. The samples were sequenced based on the following bases: (1) the sequence with precise primers and bar codes; (2) quality score ˃20; and (3) the sequences >230 bp in length. The data analysis was done by following the method of published procedures (Wang et al., 2020; Din et al., 2021). Additionally, visualization of beta-diversity information was achieved via ordination plotting with non-metric multidimensional scaling (NMDS) (Tian et al., 2018).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
YS: Data curation, Writing – original draft. CD: Data curation, Writing – original draft. TL: Formal analysis, Writing – review & editing. WC: Formal analysis, Writing – review & editing. HG: Formal analysis, Writing – review & editing. LG: Conceptualization, Data curation, Funding acquisition, Investigation, Resources, Supervision, Writing – original draft, Writing – review & editing. GL: Formal analysis, data curation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Xinjiang Major Science and Technology projects (Research, development, and demonstration of key technologies for the green control of major pests on special and superiority crops in Xinjiang, 2023A02009). LG was supported by Xinjiang Uygur Autonomous Region’s first batch of “2 + 5” key talent plan. We thanked Ghulam Muhae-Ud-Din and Han Weng for helping on editing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1343946/full#supplementary-material
References
Berendsen, R. L., Pieterse, C. M., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Chen, D., Muhae-Ud-din, G., Liu, T., Chen, W., Liu, C., and Gao, L. (2021). Wheat varietal response to Tilletia controversa J. G. Kühn using qrt-pcr and laser confocal microscopy. Genes 12:425. doi: 10.3390/genes12030425
Cleveland, C. C., Nemergut, D. R., Schmidt, S. K., and Townsend, A. R. (2007). Increases in soil respiration following labile carbon additions linked to rapid shifts in soil microbial community composition. Biogeochemistry 82, 229–240. doi: 10.1007/s10533-006-9065-z
DeAngelis, K. M., Pold, G., TopçuoÄŸlu, B. Ã.¼. D., van Diepen, L. T. A., Varney, R. M., Blanchard, J. L., et al. (2015). Long-term forest soil warming alters microbial communities in temperate forest soils. Front. Microbiol. 6:104. doi: 10.3389/fmicb.2015.00104
Dennis, P. G., Miller, A. J., and Hirsch, P. R. (2010). Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol. Ecol. 72, 313–327. doi: 10.1111/j.1574-6941.2010.00860.x
DeSantis, T. Z., Stone, C. E., Murray, S. R., Moberg, J. P., and Andersen, G. L. (2005). Rapid quantification and taxonomic classification of environmental DNA from both prokaryotic and eukaryotic origins using a microarray. FEMS Microbiol. Lett. 245, 271–278. doi: 10.1016/j.femsle.2005.03.016
Din, G. M. U., Du, Z., Zhang, H., Zhao, S., Liu, T., Chen, W., et al. (2021). Effects of Tilletia foetida on microbial communities in the rhizosphere soil of wheat seeds coated with different concentrations of Jianzhuang. Microb. Ecol. 82, 736–745. doi: 10.1007/s00248-021-01696-w
Dudenhöffer, J. H., Scheu, S., and Jousset, A. (2016). Systemic enrichment of antifungal traits in the rhizosphere microbiome after pathogen attack. J. Ecol. 104, 1566–1575. doi: 10.1111/1365-2745.12626
Eaton, W., Roed, M., and ChassotO, B. D. (2012). Differences in soil moisture, nutrients and the microbial community between forests on the upper Pacific and Caribbean slopes at Monteverde, cordillera de Tilaran: implications for responses to climate change. Trop. Ecol. 53, 235–240. doi: 10.1016/j.tpb.2012.01.004
Follmer, C. (2008). Insights into the role and structure of plant ureases. Phytochemistry 69, 18–28. doi: 10.1016/j.phytochem.2007.06.034
Garrett, K. A., Alcalá-Briseño, R. I., Andersen, K. F., Buddenhagen, C. E., Choudhury, R. A., Fulton, J. C., et al. (2018). Network analysis: a systems framework to address grand challenges in plant pathology. Annu. Rev. Phytopathol. 56, 559–580. doi: 10.1146/annurev-phyto-080516-035326
Gu, Y., Wei, Z., Wang, X., Friman, V. P., Huang, J., Wang, X., et al. (2016). Pathogen invasion indirectly changes the composition of soil microbiome via shifts in root exudation profile. Biol. Fertil. Soils 52, 997–1005. doi: 10.1007/s00374-016-1136-2
Guo, J. H., Qi, H. Y., Guo, Y. H., Ge, H. L., Gong, L. Y., Zhang, L. X., et al. (2004). Biocontrol of tomato wilt by plant growth-promoting rhizobacteria. Biol. Control 29, 66–72. doi: 10.1016/S1049-9644(03)00124-5
Haegeman, A., Vanholme, B., Jacob, J., Vandekerckhove, T. T. M., Claeys, M., Borgonie, G., et al. (2009). An endosymbiotic bacterium in a plant-parasitic nematode: member of a new Wolbachia supergroup. Int. J. Parasitol. 39, 1045–1054. doi: 10.1016/j.ijpara.2009.01.006
Harrison, K. A., and Bardgett, R. D. (2010). Influence of plant species and soil conditions on plant-soil feedback in mixed grassland communities. J. Ecol. 98, 384–395. doi: 10.1111/j.1365-2745.2009.01614.x
Havrlentová, M., Gregusová, V., Šliková, S., Nemeček, P., Hudcovicová, M., and Kuzmová, D. (2020). Relationship between the content of β -D-glucans and infection with Fusarium pathogens in oat. Plan. Theory 9:1776. doi: 10.3390/plants9121776
Hinsinger, P., Plassard, C., Tang, C., and Jaillard, B. (2003). Origins of root-mediated pH changes in the rhizosphere and their responses to environmental constraints: a review. Plant Soil 248, 43–59. doi: 10.1023/A:1022371130939
Högberg, M. N., Yarwood, S. A., and Myrold, D. D. (2014). Fungal but not bacterial soil communities recover after termination of decadal nitrogen additions to boreal forest. Soil Biol. Biochem. 72, 35–43. doi: 10.1016/j.soilbio.2014.01.014
Hollander, M. (2018). Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 12, 212–224. doi: 10.1038/ismej.2017.158
Huang, X., Chaparro, J. M., Reardon, K. F., Zhang, R., Shen, Q., and Vivanco, J. M. (2014). Rhizosphere interactions: root exudates, microbes, and microbial communities. Botany 92, 267–275. doi: 10.1139/cjb-2013-0225
Huang, X., Zhang, N., Yong, X., Yang, X., and Shen, Q. (2012). Biocontrol of Rhizoctonia solani damping-off disease in cucumber with Bacillus pumilus SQR-N43. Microbiol. Res. 167, 135–143. doi: 10.1016/j.micres.2011.06.002
Hussain, Q., Liu, Y., Zhang, A., Pan, G., Li, L., Zhang, X., et al. (2011). Variation of bacterial and fungal community structures in the rhizosphere of hybrid and standard rice cultivars and linkage to CO 2 flux. FEMS Microbiol. Ecol. 78, 116–128. doi: 10.1111/j.1574-6941.2011.01128.x
Jezierska-Tys, S., and Rutkowska, A. (2014). Zmiany chemiczne oraz enzymatyczne gleby poddanej działaniu glufosynatu amonowego. J. Elem. 19, 129–141.
Jia, H. Y., Muhae-Ud-Din, G., Zhang, H., Zong, Q. Q., Zhao, S. F., Guo, Q. Y., et al. (2022). Characterization of rhizosphere microbial communities for disease incidence and optimized concentration of Difenoconazole fungicide for controlling of wheat dwarf bunt. Front. Virol. 13:863176. doi: 10.3389/fmicb.2022.853176
Jin, X., Rahman, M. K., Ma, C. L., Zheng, X. Q., Wu, F. Z., and Zhou, X. G. (2023). Silicon modification improves biochar’s ability to mitigate cadmium toxicity in tomato by enhancing root colonization of plant -benificial bacteria. Ecotoxicol. Environ. Saf. 249:114407. doi: 10.1016/j.ecoenv.2022.114407
Jones, D. L., Nguyen, C., and Finlay, R. D. (2009). Carbon flow in the rhizosphere: carbon trading at the soil-root interface. Plant Soil 321, 5–33. doi: 10.1007/s11104-009-9925-0
Jos, M., Raaijmakers, T. C. P., Christian Steinberg, C. A., and Moënne-Loccoz, Y. (2008). The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 7, 2008–2010. doi: 10.1007/s11104-008-9568-6
Kim, J. M., Roh, A. S., Choi, S. C., Kim, E. J., Choi, M. T., Ahn, B. K., et al. (2016). Soil pH and electrical conductivity are key edaphic factors shaping bacterial communities of greenhouse soils in Korea. J. Microbiol. 54, 838–845. doi: 10.1007/s12275-016-6526-5
Lazcano, C., Boyd, E., Holmes, G., Hewavitharana, S., Pasulka, A., and Ivors, K. (2021). The rhizosphere microbiome plays a role in the resistance to soil-borne pathogens and nutrient uptake of strawberry cultivars under field conditions. Sci. Rep. 11:3188. doi: 10.1038/s41598-021-82768-2
Lei, T., Gu, Q., Guo, X., Ma, J., Zhang, Y., and Sun, X. (2018). Urease activity and urea hydrolysis rate under coupling effects of moisture content, temperature, and nitrogen application rate. Int. J. Agric. Biol. Eng. 11, 132–138. doi: 10.25165/j.ijabe.20181102.3784
Li, X., Ding, C. F., Zhang, T. L., and Wang, X. X. (2014). Fungal pathogen accumulation at the expense of plant-beneficial fungi as a consequence of consecutive peanut monoculturing. Soil Biol. Biochem. 72, 11–18. doi: 10.1016/j.soilbio.2014.01.019
Li, C., Li, X., Kong, W., Wu, Y., and Wang, J. (2010). Effect of monoculture soybean on soil microbial community in the Northeast China. Plant Soil 330, 423–433. doi: 10.1007/s11104-009-0216-6
Liang, Y., Li, F., Nong, M., Luo, H., and Zhang, J. (2016). Microbial activity in paddy soil and water-use efficiency of rice as affected by irrigation method and nitrogen level. Commun. Soil Sci. Plant Anal. 47, 19–31. doi: 10.1080/00103624.2015.1104336
Liu, X., Zhang, J., Gu, T., Zhang, W., Shen, Q., Yin, S., et al. (2014). Microbial community diversities and taxa abundances in soils along a seven-year gradient of potato monoculture using high throughput pyrosequencing approach. PLoS One 9:e86610. doi: 10.1371/journal.pone.0086610
Lu, Z. X., Gaudet, D. A., Frick, M., Puchalski, B., Genswein, B., and Laroche, A. (2005). Identification and characterization of genes differentially expressed in the resistance reaction in wheat infected with Tilletia tritici, the common bunt pathogen. J. Biochem. Mol. Biol. 38, 420–431. doi: 10.5483/bmbrep.2005.38.4.420
Mendes, R., Garbeva, P., and Raaijmakers, J. M. (2013). The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 37, 634–663. doi: 10.1111/1574-6976.12028
Miransari, M. (2013). Soil microbes and the availability of soil nutrients. Acta Physiol. Plant. 35, 3075–3084. doi: 10.1007/s11738-013-1338-2
Muhae-ud-Din, G., Moosa, A., Ghummen, U. F., Jabran, M., Abbas, A., Naveed, M., et al. (2018). Host status of commonly planted ornamentals to Meloidogyne incognita and management through endophytic bacteria. Pak. J. Zool. 50, 1393–1402. doi: 10.17582/journal.pjz/2018.50.4.1393.1402
Nguyen, H. D. T., Sultana, T., Kesanakurti, P., and Hambleton, S. (2019). Genome sequencing and comparison of five Tilletia species to identify candidate genes for the detection of regulated species infecting wheat. IMA Fungus 10:10. doi: 10.1186/s43008-019-0011-9
Nosheen, A., Bano, A., Yasmin, H., Keyani, R., Habib, R., Shah, S. T. A., et al. (2016). Protein quantity and quality of safflower seed improved by NP fertilizer and rhizobacteria (Azospirillum and Azotobacter spp.). Front. Plant Sci. 7:104. doi: 10.3389/fpls.2016.00104
Piotrowska, A., and Wilczewski, E. (2012). Effects of catch crops cultivated for green manure and mineral nitrogen fertilization on soil enzyme activities and chemical properties. Geoderma 189-190, 72–80. doi: 10.1016/j.geoderma.2012.04.018
Purahong, W., Wubet, T., Lentendu, G., Schloter, M., Pecyna, M. J., Kapturska, D., et al. (2016). Life in leaf litter: novel insights into community dynamics of bacteria and fungi during litter decomposition. Mol. Ecol. 25, 4059–4074. doi: 10.1111/mec.13739
Qiao, Q., Wang, F., Zhang, J., Chen, Y., Zhang, C., Liu, G., et al. (2017). The variation in the rhizosphere microbiome of cotton with soil type, genotype and developmental stage. Sci. Rep. 7:3940. doi: 10.1038/s41598-017-04213-7
Qin, C. C., Xu, T. S., Liu, T. G., Chen, W. Q., and Li, G. (2020). First report of wheat common bunt caused by Tilletia laevis in Henan province, China. Plant Dis. 79:212. doi: 10.1094/PDIS-05-20-1121-PDN
Ren, Z. Y., Liu, J. J., Muhae-Ud-Din, G., Zhang, H., Du, Z. Z., Chen, W. Q., et al. (2020). Transcriptome analysis of wheat spikes in response to Tilletia controversa Kühn which cause wheat dwarf bunt. Sci. Rep. 10:21567. doi: 10.1038/s41598-020-78628-0
Rolfe, S. A., Griffiths, J., and Ton, J. (2019). Crying out for help with root exudates: adaptive mechanisms by which stressed plants assemble health-promoting soil microbiomes. Curr. Opin. Microbiol. 49, 73–82. doi: 10.1016/j.mib.2019.10.003
Ruby, E. G. (2008). Symbiotic conversations are revealed under genetic interrogation. Nat. Rev. Microbiol. 6, 752–762. doi: 10.1038/nrmicro1958
She, S., Niu, J., Zhang, C., Xiao, Y., Chen, W., Dai, L., et al. (2017). Significant relationship between soil bacterial community structure and incidence of bacterial wilt disease under continuous cropping system. Arch. Microbiol. 199, 267–275. doi: 10.1007/s00203-016-1301-x
Taha, E. A. (2015). A study on nectar and pollen sources for honeybee, Apis mellifera L. In Al-Ahsa Saudi Arabia. J. Entomol. Zool. Stud. 3, 272–277. https://www.entomoljournal.com/archives/2015/vol3issue3/PartD/3-3-91.pdf
Tan, S., Jiang, Y., Song, S., Huang, J., Ling, N., Xu, Y., et al. (2010). Characterization of an antimicrobial material from a newly isolated Bacillus amyloliquefaciens from mangrove for biocontrol of Capsicum bacterial wilt. Biol. Control 54, 359–365. doi: 10.1016/j.biocontrol.2010.06.015
Tang, J., Xue, Z., Daroch, M., and Ma, J. (2015). Impact of continuous Salvia miltiorrhiza cropping on rhizosphere actinomycetes and fungi communities. Ann. Microbiol. 65, 1267–1275. doi: 10.1007/s13213-014-0964-2
Tian, J., He, N., Hale, L., Niu, S., Yu, G., Liu, Y., et al. (2018). Soil organic matter availability and climate drive latitudinal patterns in bacterial diversity from tropical to cold temperate forests. Funct. Ecol. 32, 61–70. doi: 10.1111/1365-2435.12952
Tiecher, T., Fontoura, S. M. V., Ambrosini, V. G., Araújo, E. A., Alves, L. A., Bayer, C., et al. (2023). Soil phosphorus forms and fertilizer use efficiency are affected by tillage and soil acidity management. Geoderma 435:116495. doi: 10.1016/j.geoderma.2023.116495
Trivedi, P., Delgado-Baquerizo, M., Trivedi, C., Hamonts, K., Anderson, I. C., and Singh, B. K. (2017). Keystone microbial taxa regulate the invasion of a fungal pathogen in agro-ecosystems. Soil Biol. Biochem. 111, 10–14. doi: 10.1016/j.soilbio.2017.03.013
Unterseher, M., Peršoh, D., and Schnittler, M. (2013). Leaf-inhabiting endophytic fungi of European beech (Fagus sylvatica L.) co-occur in leaf litter but are rare on decaying wood of the same host. Fungal Divers. 60, 43–54. doi: 10.1007/s13225-013-0222-0
Vlek, P. L. G., Stumpe, J. M., and Byrnes, B. H. (1980). Urease activity and inhibition in flooded soil systems. Fertil. Res. 1, 191–202. doi: 10.1007/BF01053131
Wallenstein, M. D., McMahon, S., and Schimel, J. (2007). Bacterial and fungal community structure in Arctic tundra tussock and shrub soils. FEMS Microbiol. Ecol. 59, 428–435. doi: 10.1111/j.1574-6941.2006.00260.x
Wang, Y., Xu, X., Liu, T., Wang, H., Yang, Y., Chen, X., et al. (2020). Analysis of bacterial and fungal communities in continuous-cropping ramie (Boehmeria nivea L. Gaud) fields in different areas in China. Sci. Rep. 10:3264. doi: 10.1038/s41598-020-58608-0
Wang, R., Zhang, H., Sun, L., Qi, G., Chen, S., and Zhao, X. (2017). Microbial community composition is related to soil biological and chemical properties and bacterial wilt outbreak. Sci. Rep. 7:343. doi: 10.1038/s41598-017-00472-6
Xu, T. S., Jiang, W. L., Qin, D. D., Liu, T. G., Zhang, J. M., Chen, W. Q., et al. (2021). Characterization of the microbial communities in wheat tissues and rhizosphere soil caused by dwarf bunt of wheat. Sci. Rep. 11:5773. doi: 10.1038/s41598-021-85281-8
Xu, J., Zhang, Y., Zhang, P., Trivedi, P., Riera, N., Wang, Y., et al. (2018). The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 9:4894. doi: 10.1038/s41467-018-07343-2
Yang, J., Ruegger, P. M., McKenry, M. V., Becker, J. O., and Borneman, J. (2012). Correlations between root-associated microorganisms and peach replant disease symptoms in a California soil. PLoS One 7:e46420. doi: 10.1371/journal.pone.0046420
Yu, Y., Liu, T. H., Liu, L. X., Chen, Y., Tang, J., Peng, W. H., et al. (2022). Application of the mushroom volatile 1-octen-3-ol to suppress a norel disease caused by Paecilomyces penicillatus. Environ. Biotechnol. 106, 4787–4799. doi: 10.1007/s00253-022-12038-2
Zahar, H., Santaella, F., Heulin, C., and Achouak, W. (2014). Root exudates mediated interactions belowground. Soil Biol. Biochem. 77, 69–80. doi: 10.1016/j.soilbio.2014.06.017
Zhang, W., Parker, K. M., Luo, Y., Wan, S., Wallace, L. L., and Hu, S. (2005). Soil microbial responses to experimental warming and clipping in a tallgrass prairie. Glob. Chang. Biol. 11, 266–277. doi: 10.1111/j.1365-2486.2005.00902.x
Zhang, T., Wang, N. F., Liu, H. Y., Zhang, Y. Q., and Yu, L. Y. (2016). Soil pH is a key determinant of soil fungal community composition in the Ny-Ålesund region, Svalbard (high Arctic). Front. Microbiol. 7:227. doi: 10.3389/fmicb.2016.00227
Zhou, D., Feng, H., Schuelke, T., De Santiago, A., Zhang, Q., Zhang, J., et al. (2019). Rhizosphere microbiomes from root knot nematode non-infested plants suppress nematode infection. Microb. Ecol. 78, 470–481. doi: 10.1007/s00248-019-01319-5
Zhou, W. P., Shen, W. J., Li, Y. E., and Hui, D. F. (2017). Interactive effects of temperature and moisture on composition of the soil microbial community. Eur. J. Soil Sci. 68, 909–918. doi: 10.1111/ejss.12488
Zhou, X., and Wu, F. (2012). Dynamics of the diversity of fungal and Fusarium communities during continuous cropping of cucumber in the greenhouse. FEMS Microbiol. Ecol. 80, 469–478. doi: 10.1111/j.1574-6941.2012.01312.x
Keywords: fungal community, bacterial community, Tilletia laevis , RNA sequencing, soil properties, enzyme activities
Citation: Shen Y, Delai C, Liu T, Chen W, Li G, Gao H and Gao L (2024) Analysis of microbial communities in wheat, alfalfa, and oat crops after Tilletia laevis Kühn infection. Front. Microbiol. 15:1343946. doi: 10.3389/fmicb.2024.1343946
Edited by:
Francisca Suárez-Estrella, University of Almeria, SpainReviewed by:
Sakineh Abbasi, Institut National de recherche pour L’agriculture, L’alimentation et L’environnement, FranceHao Tan, Sichuan Academy of Agricultural Sciences, China
Xingang Zhou, Northeast Agricultural University, China
Copyright © 2024 Shen, Delai, Liu, Chen, Li, Gao and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haifeng Gao, Z2hmMjAwNDQ2NjZAMTYzLmNvbQ==; Guangkuo Li, MTQ0ODgzMjc2NEBxcS5jb20=; Li Gao, eGlhb2dhb3N4QGhvdG1haWwuY29t