
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 02 February 2024
Sec. Infectious Agents and Disease
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1334268
This article is part of the Research TopicConference Research Topic: 9th Symposium on Antimicrobial Resistance in Animals and the Environment (ARAE 2023)View all 21 articles
 Flavia Zendri1
Flavia Zendri1 Vanessa Schmidt2
Vanessa Schmidt2 Norman Mauder3
Norman Mauder3 Anette Loeffler4
Anette Loeffler4 Rosanne Ellen Jepson4
Rosanne Ellen Jepson4 Cajsa Isgren5
Cajsa Isgren5 Gina Pinchbeck6Sam Haldenby7
Gina Pinchbeck6Sam Haldenby7 Dorina Timofte1*
Dorina Timofte1*Introduction: The emergence of multi-drug resistant (MDR) pathogens linked to healthcare-associated infections (HCAIs) is an increasing concern in modern veterinary practice. Thus, rapid bacterial typing for real-time tracking of MDR hospital dissemination is still much needed to inform best infection control practices in a clinically relevant timeframe. To this end, the IR Biotyper using Fourier-Transform InfraRed (FTIR) spectroscopy has the potential to provide fast cluster analysis of potentially related organisms with substantial cost and turnaround time benefits.
Materials and methods: A collection of MDR bacterial isolates (n = 199, comprising 92 Klebsiella pneumoniae and 107 Pseudomonas aeruginosa) obtained from companion animal (i.e., dogs, cats and horses) clinical investigations, faecal and environmental screening from four veterinary facilities between 2012 and 2019 was analysed retrospectively by FTIR spectroscopy. Its performance was compared against MLST extracted from whole genomes of a subset of clustering isolates (proportionally to cluster size) for investigation of potential nosocomial transmission between patients and the surrounding hospital environments.
Results: Concordance between the FTIR and MLST types was overall high for K. pneumoniae (Adjusted Rand Index [ARI] of 0.958) and poor for P. aeruginosa (ARI of 0.313). FTIR K. pneumoniae clusters (n = 7) accurately segregated into their respective veterinary facility with evidence of intra-hospital spread of K. pneumoniae between patients and environmental surfaces. Notably, K. pneumoniae ST147 intensely circulated at one Small Animal Hospital ICU. Conversely, Pseudomonas aeruginosa FTIR clusters (n = 18) commonly contained isolates of diversified hospital source and heterogeneous genetic background (as also genetically related isolates spread across different clusters); nonetheless, dissemination of some clones, such as P. aeruginosa ST2644 in the equine hospital, was apparent. Importantly, FTIR clustering of clinical, colonisation and/or environmental isolates sharing genomically similar backgrounds was seen for both MDR organisms, highlighting likely cross-contamination events that led to clonal dissemination within settings.
Conclusion: FTIR spectroscopy has high discriminatory power for hospital epidemiological surveillance of veterinary K. pneumoniae and could provide sufficient information to support early detection of clonal dissemination, facilitating implementation of appropriate infection control measures. Further work and careful optimisation need to be carried out to improve its performance for typing of P. aeruginosa veterinary isolates.
Healthcare-associated infections (HCAIs) and the emergence of multi-drug resistant (MDR) nosocomial pathogens in companion animal (dogs, cats and horses) medicine have been increasingly reported in the last two decades, where outbreaks are often associated with, and complicated by, the antimicrobial resistant and zoonotic nature of the pathogens involved (Murphy et al., 2010; Steneroden et al., 2010; Wieler et al., 2011; Walther et al., 2014, 2018; Soza-Ossandón et al., 2020). Advancements in modern veterinary practice, including the progress in patient management, medical procedures and the development of hospital facilities, have given rise to favourable conditions for harbouring veterinary nosocomial MDR opportunistic pathogens, as seen in human hospitals. Despite this, the frequency and nature of HCAIs in veterinary hospitals are not known and the progress made in the field of veterinary infection control (IC) has been slow compared to human medicine (Weese, 2011; Walther et al., 2017). Nowadays, IC is an essential component in the operation of all veterinary hospitals delivering high-quality care; risks associated with lack of biosecurity and infection control programs include outbreaks of HCAIs in hospitalised patients and zoonotic infections of hospital personnel and animal owners, leading to increased morbidity, mortality, medical costs and welfare issues in both animals and people. Screening hospital surfaces in conjunction with cases of clinical infection is advisable as contaminated hospital environments may be an important source of infections in hospitalised human (Weber et al., 2013; Nutman et al., 2016) and animal patients (Weese et al., 2004; Loeffler et al., 2005; Hoet et al., 2011). Furthermore, MDR pathogens can persist in the hospital environment for long periods of time, providing continuous exposure for colonisation and subsequent infection in hospitalised patients (Kramer et al., 2006; Bortolami et al., 2017; Keck et al., 2020).
Rapid typing of bacterial isolates is valuable for outbreak management and hospital surveillance of MDR pathogens by revealing their clonal relationships and possible routes of transmission and by linking clinical isolates to environmental reservoirs (Foxman et al., 2005). These results can guide the implementation of focused interventions to tackle and prevent HCAIs. However, the quick detection of pathogen cross-transmissions in healthcare settings remains challenging despite the broad choice of typing methods available. Molecular typing methods relying on genomic fingerprinting, such as Pulsed-Field Gel Electrophoresis (PFGE), Multi-Locus Sequence Typing (MLST) and Whole Genome Sequencing (WGS) are widely used in hospital epidemiological surveillance and outbreak investigation to discern and track patterns of infection and transmission sources (Boccia et al., 2015; Timofte et al., 2016). However, while molecular tools are the gold standard for diagnostic accuracy and discriminatory power, they are mainly used in retrospective epidemiological studies, thereby lacking clinical applicability. Hence, a quick and reliable typing method detecting pathogen cross-transmissions in the clinical microbiology laboratory is still needed.
Fourier-Transform InfraRed (FTIR) spectroscopy is a phenotypic method generating highly-specific metabolic fingerprint-like signatures (from nucleic acids, proteins, carbohydrates and lipids) that are widely used to differentiate, identify and classify a range of microbial species and strains (Yang et al., 2020). The principle of this technique is that the absorption of the infrared (IR) radiation by bacterial cells causes excitation of the different molecules; different cell components absorb radiation at different wavelengths, originating characteristic spectral peaks that present as a specific fingerprint-like pattern (Novais et al., 2019). The potential of this technique as a quick (results can be available as early as 2–3 h from culture harvest), inexpensive, and high-throughput tool for bacterial typing is widely accepted, which makes it an attractive alternative to current gold standard typing methods for routine diagnostic applications (Finlayson et al., 2019).
Clinical research exploring the performance of FTIR in human hospitals indicated this could be a promising tool for infection control purposes, particularly for fast typing of gram-negative organisms associated with HCAIs’ outbreaks (Dinkelacker et al., 2018; Martak et al., 2019; Vogt et al., 2019; Rakovitsky et al., 2020). In our previous work exploring rapid bacterial typing of veterinary hospital-associated MDR bacteria, we reported unexpectedly high circulation of MDR gram-negative organisms belonging to the ESKAPE group of pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.) within the intensive care units (ICUs) of two veterinary hospitals (one equine and one small animal; Zendri et al., 2023). Here, we consider the integration of this technology into infection control programmes by assessing its value in strain typing of Klebsiella pneumoniae and Pseudomonas aeruginosa from clinical cases, colonised inpatients and clinical environments in veterinary clinics/hospitals.
Sixty-seven isolates were selected from a previous pilot study (PS) which evaluated the epidemiology of multidrug resistant gram-negative (MDR-GN) bacteria in the ICUs of two veterinary hospitals, one small animal (SAH1) and one equine (EH), at the University of Liverpool (UoL), United Kingdom (Zendri et al., 2023), where n = 31 K. pneumoniae (Table 1) and n = 36 P. aeruginosa (Table 2) isolated across both hospitals, were selected for FTIR testing.
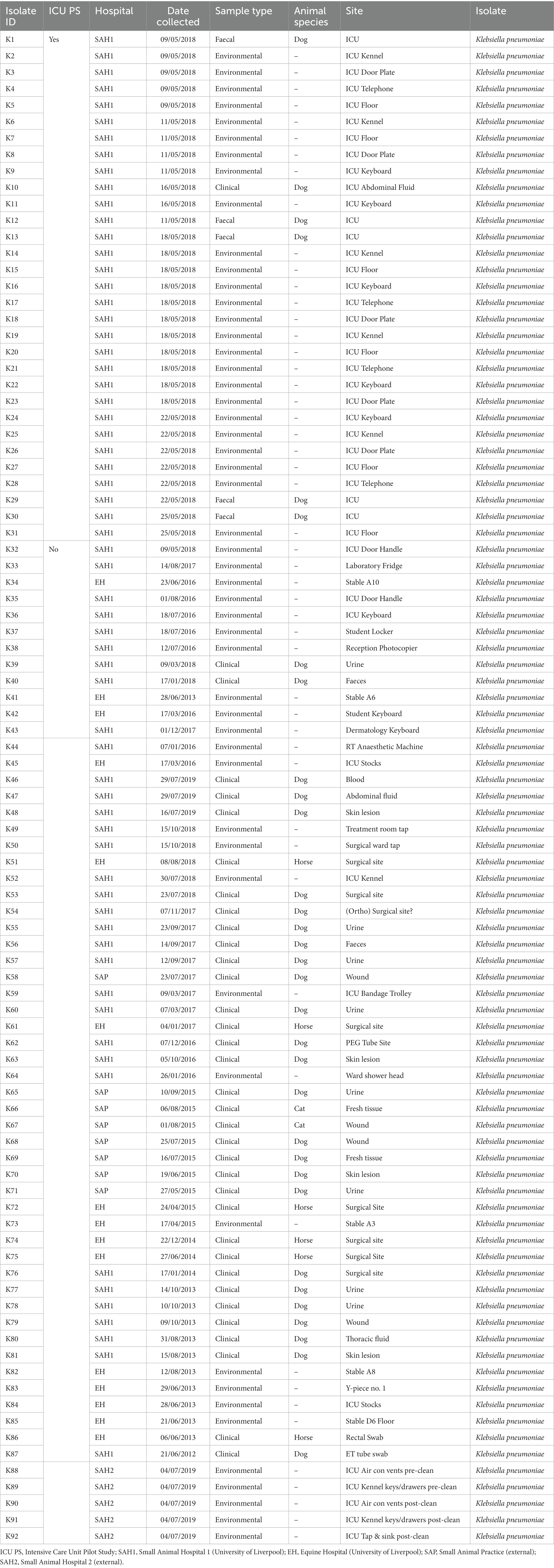
Table 1. Sample type and origin of clinical, faecal and environmental MDR Klebsiella pneumoniae spp. pneumoniae (n = 92) from four companion animal veterinary facilities in the UK (2012–2019).
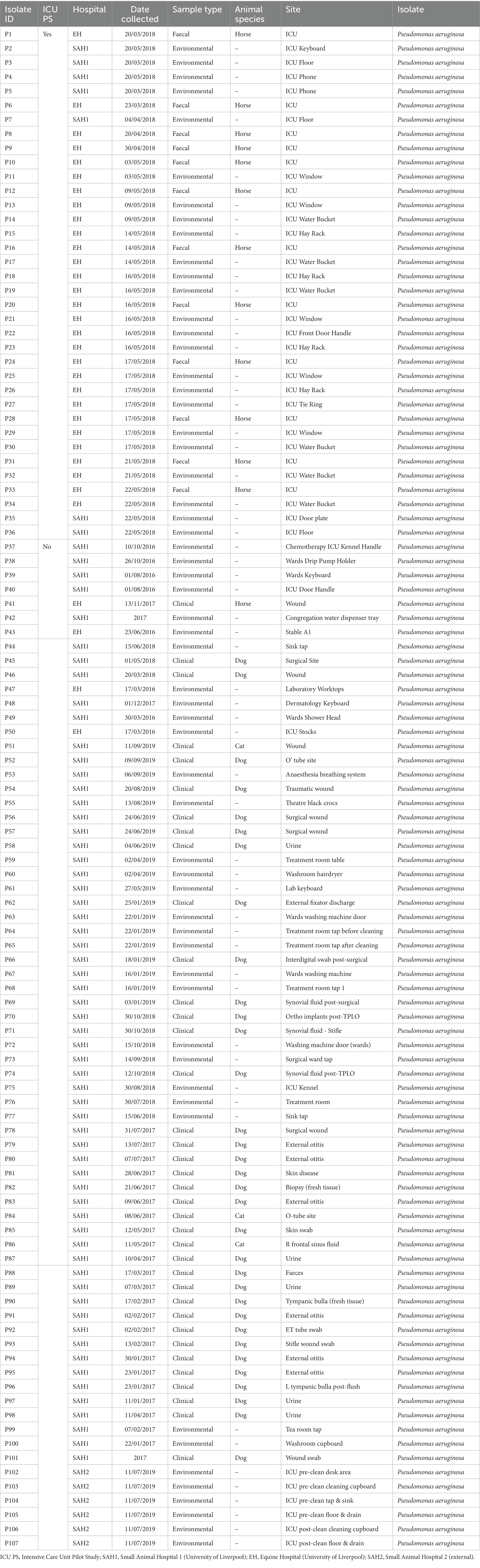
Table 2. Sample type and origin of clinical, faecal and environmental MDR Pseudomonas aeruginosa (n = 107) from three companion animal veterinary facilities in the UK (2016–2019).
In addition, MDR K. pneumoniae (n = 48) and P. aeruginosa (n = 65) isolated through routine diagnostics (RD) of clinical (CL) and environmental (ENV) specimens from the same UoL hospitals, were included in the analysis. These isolates covered a broader range of patients and hospital areas than the ICUs, and were collected from 2012 to 2019 for K. pneumoniae (Table 1) and from 2016 to 2019 for P. aeruginosa (Table 2). Furthermore, several isolates (n = 13 K. pneumoniae and n = 6 P. aeruginosa) received in the microbiology laboratory from two external small animal facilities (one hospital [SAH2] and one practice [SAP]) for environmental surveillance (n = 11) or for routine clinical diagnostics (n = 8) were also included, resulting in n = 199 bacterial isolates analysed overall with FTIR spectroscopy, whose summary is presented in Table 3.
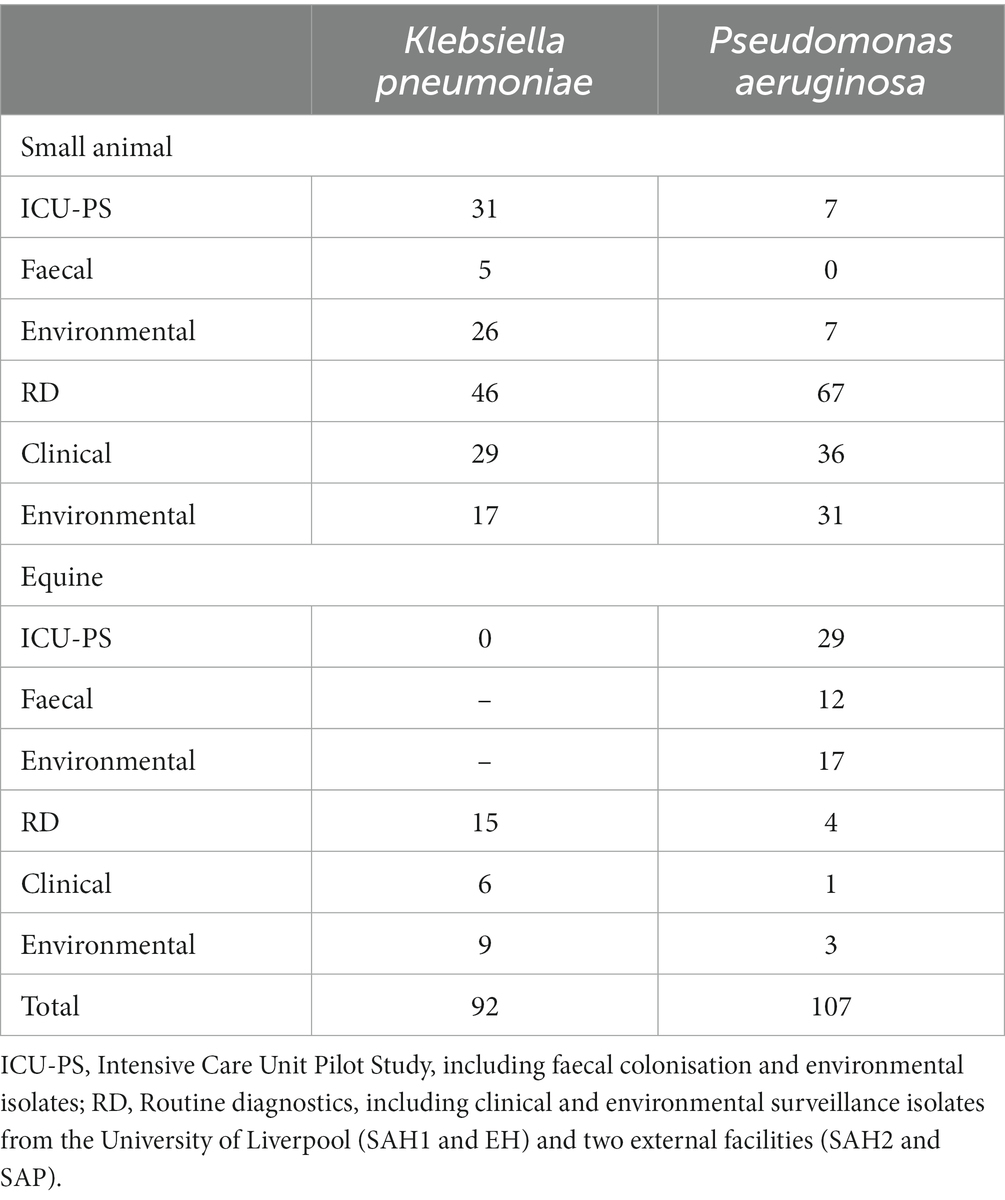
Table 3. Summary of Klebsiella pneumoniae and Pseudomonas aeruginosa isolates (n = 199) from veterinary settings (small animal and equine) analysed by Fourier-transform Infrared (FTIR) spectroscopy.
Clinical and environmental specimens processing was performed as previously described (Zendri et al., 2023). In brief, clinical isolates were obtained through routine microbiology diagnostics, processed in accordance with local laboratory protocols for culture and antimicrobial susceptibility testing (AST) using the CLSI methodology and interpretative criteria (Clinical and Laboratory Standards Institute (CLSI), 2018). All isolates were identified using MALDI-TOF MS (MALDI Biotyper 4.1.100 Software, Bruker Daltonics, Bremen, Germany) with a score > 2.0. Hospital environmental bacterial isolates were obtained from routine environmental surveillance samples, collected aseptically by veterinary nurses using sterile gauzes placed individually in Buffered Peptone Water (BPW) and enriched overnight at 37°C aerobically. To screen for MDR isolates, a small BPW inoculum in which faeces and ENV samples were placed, was plated onto eosin methylene blue agar (EMBA; Thermo Scientific) containing 1 mg/mL of cefotaxime (Sigma-Aldrich Ltd., United Kingdom) and incubated for 24 h at 37°C aerobically. EMBA negative morphotypes identified as Klebsiella pneumoniae and Pseudomonas aeruginosa using MALDI-TOF MS were included in the downstream analysis. All environmental surveillance and clinical MDR isolates were continuously stored at-80°C in the local strain collection; inclusion of these isolates in the study was based on phenotypic resistance to extended-spectrum cephalosporins and MDR status upon broth microdilution and/or disc diffusion antimicrobial susceptibility testing (AST) performed at the time of diagnostics.
A total of n = 199 MDR bacterial isolates, of which n = 92 K. pneumoniae and n = 107 P. aeruginosa, from all hospitals/clinics were typed using FTIR spectroscopy (IR Biotyper, Bruker Daltonics, Bremen, Germany) to determine their clonal relatedness. Bacterial isolates were grown on 5% sheep Blood Agar under strictly controlled temperature, time and humidity conditions (22 h incubation at 37°C under aerobic atmosphere) and harvested in Eppendorf tubes to create bacterial suspensions in equal parts of 70% ethanol and molecular grade water before spotting the target plate. Tests were run with three technical replicates in each of three independent experiments with two bacterial standards for internal quality control included in each run. Spectra were acquired in transmission mode using the OPUS v7.5 software (Bruker Optics GmbH). Spectra were then pre-processed (calculated 2nd derivative, cut to 1,300–800 cm-1, vector-normalised) and further analysed with the IR Biotyper Client Software v1.5 (Bruker Daltonics GmbH). Dendrograms expressing Hierarchical Cluster Analysis (HCA) were generated by the IR Biotyper Client Software v1.5 using the Euclidean distance and average linkage clustering methods using Cut-Off Values (COVs) to define clusters of 0.246 and of 0.287 for P. aeruginosa and K. pneumoniae, respectively. An optimisation procedure was conducted beforehand to determine optimal COVs for these organisms that were specific to our local laboratory conditions. The isolates used for species-specific optimisation of the COVs included epidemiologically related and non-related (e.g., reference strains) small animal K. pneumoniae and P. aeruginosa from external sources. The COVs were established according to manufacturer’s recommendations on three repetitive experiments each using five technical and/or biological replicates of K. pneumoniae (n = 8) and P. aeruginosa (n = 9) isolates, previously typed by MLST or WGS. The automatically generated COVs were adjusted on the dendograms based on prior knowledge of the genetic relatedness of the K. pneumoniae and P. aeruginosa subsets of isolates used for the optimisation process.
To evaluate the performance of FTIR spectroscopy against a gold standard typing method, a subset of isolates demonstrating clonal relatedness was further characterised by WGS. Fifty-four P. aeruginosa and 27 K. pneumoniae isolates were selected, covering most FTIR clusters in a proportional measure to the cluster size. Fragment libraries (NEBNext Ultra II FS Kit; ∼300 base pair inserts) were created from purified genomic DNA (Qiagen QIAmp DNA mini kit) and sequenced using a 2 × 150 base pair paired-end protocol (Illumina NovaSeq SP; CGR, University of Liverpool). De novo assembly was performed using SPAdes 3.16.0 (Bankevich et al., 2012) and genome annotation via Prokka 1.14.5 (Seemann, 2014). MLST profiles and allele sequences were obtained.1 All allele sequences were aligned to assemblies using Bowtie2 version 2.3.5.1 (Langmead and Salzberg, 2012) in sensitive mode. The allele that aligned best for each locus was selected and the sequence type was determined by comparing perfectly detected alleles against the database profiles. Sequence types were used to infer eBURST groups, using goeBURST,2 where group members shared at least 2 ST locus alleles. In silico identification of virulence and AMR genes was performed using the Virulence Factor Database (VFDB; Chen et al., 2005) and the Comprehensive Antibiotic Resistance Database (CARD; McArthur et al., 2013), respectively. Incompatibility (Inc) group plasmids were identified in K. pneumoniae via PlasmidFinder (Carattoli et al., 2014). Using assemblies as input, pangenome reconstruction and concatenated core gene multiple alignment was carried using Panaroo (Tonkin-Hill et al., 2020). Subsequently, the multiple alignment data was used as input to IQ-TREE using the GTR model. The resulting tree was imported to the Interactive Tree of Life (iTOL; Letunic and Bork, 2021). Genomic sequences are deposited on the European Nucleotide Archive (ENA) under the project accession: PRJEB70897.
The clusters defined by MLST extrapolated from genomic sequences were compared with those obtained by FTIR biotyping using the service3 by calculating the Adjusted Rand index (AR) and Adjusted Wallace coefficient (AW) with 95% confidence (Carriço et al., 2006). AR compares partitions without consideration of the reference method and evaluates the congruence between two typing methods. In comparison, AW provides inter-cluster distances and evaluates the directional agreement between typing methods (Carriço et al., 2006; Pinto et al., 2008; Severiano et al., 2011a,b). In essence, AR and AW equal to 1 indicate a perfect correlation between the two typing methods. The optimal cut-offs for each species were defined by maximising AR between MLST and FTIR results.
After FTIR spectra acquisition, dendrograms were built for each species. FTIR analysis identified several clusters for both organisms tested, suggesting clonal relatedness of the isolates within individual clusters. Overall, n = 25 FTIR clusters were identified spanning across both pathogens; the number of isolates within each cluster varied, ranging from two to 43, with an average cluster size of 6.8 isolates. Fourteen of 25 clusters (56%) contained a mixture of environmental and clinical and/or faecal carriage isolates in various combinations, typically several environmental isolates linked to fewer clinical/carriage ones. Wound and skin isolates were the most common clinical specimen type represented within mixed clusters. Environmental isolates only, collected from different hospitals’ sites, also accounted for some of the clusters observed. Several isolates collected from the ICUs contributed to cluster formation, either with or without association to isolates collected from the broader hospital patients and departments. Details are provided in the following paragraphs for each organism.
Seven K. pneumoniae clusters were defined overall totalling 83/92 isolates (Figure 1A); of these, the largest (Cluster 167) contained 43 small animal isolates including environmental, faecal carriage and clinical isolates collected over a 19-month period (March 2017–October 2018); this cluster included isolates predominantly obtained from the SAH1 ICU during the PS (31/43) with fewer non-ICU isolates also found in this cluster. All isolates sequenced from this large cluster (n = 11) belonged to ST147. The ICU K. pneumoniae isolates from Cluster 167, were identified on numerous ICU surfaces and canine faecal carriage samples within a relatively short time-frame (31 isolates over 16 days). Contaminated environmental sites included ICU telephone receiver, computer keyboards, door handles, equipment trolley, floor and patients’ kennels. One clinical K. pneumoniae was cultured from a case of hospital-acquired septic peritonitis in a dog enrolled in the ICU study that was previously found to be a faecal carrier of the same strain. Sixteen isolates accounted for the second largest K. pneumoniae FTIR cluster (Cluster 168), comprising clinical and environmental isolates collected from the wider SAH1 environments over a 6-year period (from 2012 to 2018). Clinical isolates included in this group consisted of skin and wound isolates (n = 2 sequenced, ST11) but also sterile fluid ones (thoracic fluid and cystocentesis urine) alongside environmental isolates from clinical (e.g., anaesthetic machine and ICU surfaces; n = 2 sequenced, ST11) and non-clinical (e.g., reception photocopier, lockers) hospital areas.

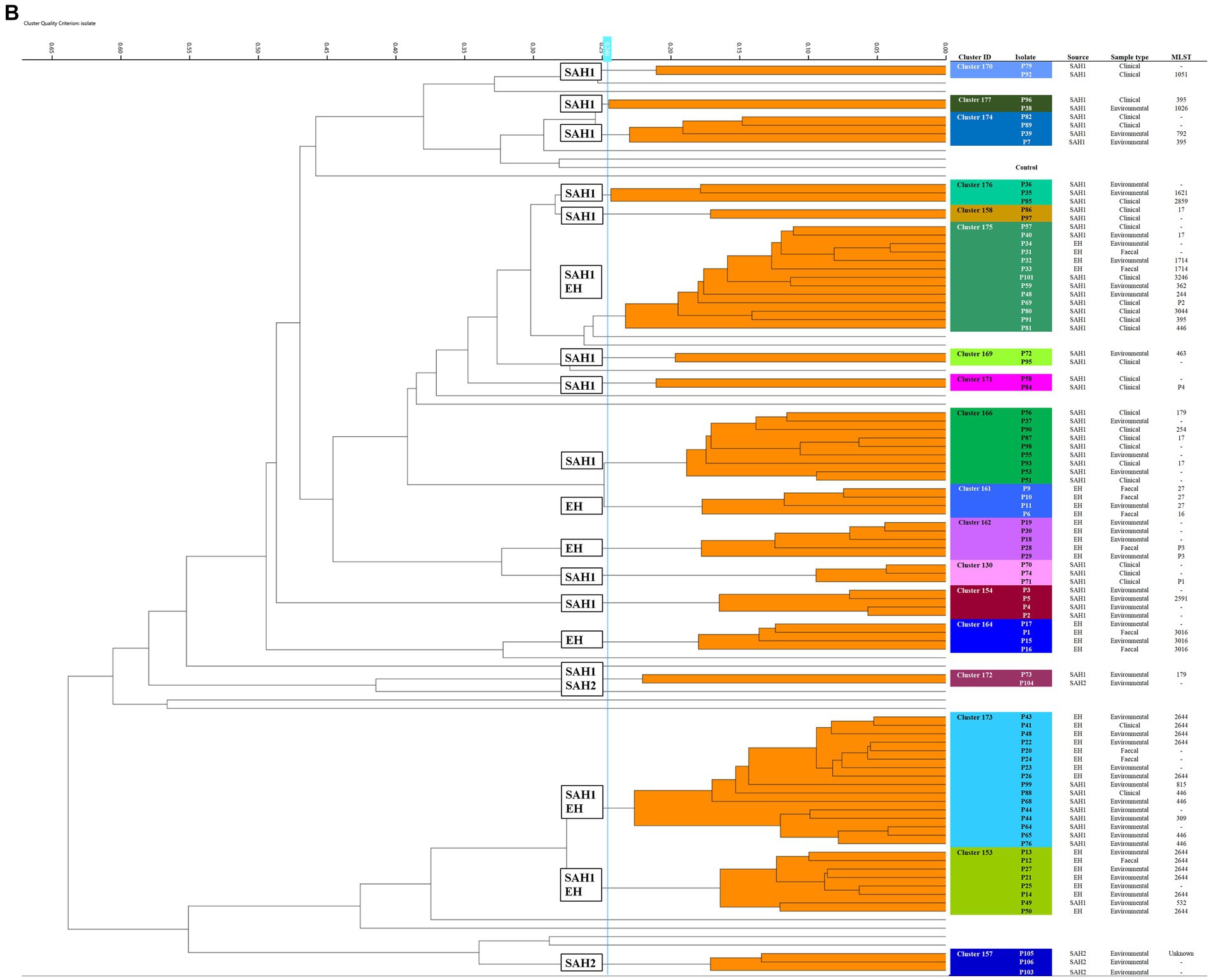
Figure 1. Fourier-transform Infrared (FTIR) biotyping of Klebsiella pneumoniae subsp. pneumoniae (A) and Pseudomonas aeruginosa (B) veterinary isolates. Dendograms were obtained by Euclidean average spectra analysis of K. pneumoniae (n = 92) and P. aeruginosa (n = 107) from clinical, faecal and environmental surveillance specimens collected in small animal and equine veterinary facilities. Analysis was based on three technical replicates of each isolate and included one control strain per each organism; the blue line represents the adjusted cut-off values (COVs) of 0.287 and 0.246 for K. pneumoniae and P. aeruginosa, respectively. Clusters comprising two or more isolates are shaded in orange. Isolate data is shown for clusters only. SAH1: Small Animal Hospital 1 (University of Liverpool); EH, Equine Hospital (University of Liverpool); SAP, Small Animal Practice (external); SAH2, Small Animal Hospital 2 (external).
Of note, all K. pneumoniae from external small animal settings accurately clustered with isolates from their respective hospital source (SAP for Cluster 153 and SAH2 for Cluster 165).
Two small clusters (of 5 and 4 isolates; Clusters 161 and 163) of environmental K. pneumoniae (e.g., from stables, ICU) in association with post-surgical infection isolates were identified in the EH; these belonged to ST307 and ST789, respectively.
Importantly, clonal dissemination of K. pneumoniae was identified in all veterinary settings; two clones were circulating in each one of the UoL hospitals (ST147 and ST11 in the SAH1 and ST307 and ST789 in the EH). In addition, single clones were identified in the two external facilities included in the study, i.e., Cluster 165 composed of environmental isolates from the SAH2 ICU (ST307) and Cluster 153 of clinical isolates originating from the SAP (ST336).
Of the 18 P. aeruginosa FTIR clusters, the two largest contained 16 (Cluster 173) and 13 (Cluster 175) mixed EH and SAH1 isolates collected over two distinct 3-year periods (Figure 1B). Cluster 173 comprised P. aeruginosa isolated from a horse faecal samples and multiple ICU environmental sites surrounding the patient (n = 2 sequenced, ST2644), alongside few other clinical and environmental isolates from the broader EH one to 2 years prior (n = 3 sequenced, ST2644). Small animal P. aeruginosa in Cluster 173 were mostly water-borne environmental isolates (mostly typed to ST446, but also ST309 and ST815) cultured from diverse sink taps within the SAH1 (e.g., treatment room taps, wards shower head and tea room tap) over a 3-year interval. Cluster 175 comprised P. aeruginosa isolates linked to a single ICU PS horse (n = 2 sequenced, ST1714) and the remainder from the SAH1. In this cluster, small animal isolates were mostly clinical P. aeruginosa from surgical wounds and otitis cases cultured between 2017 and 2019 (n = 8 sequenced, multiple STs). Small ICU clusters (4–6 isolates) of PS isolates (faecal carriage and environmental) linking 2–3 patients over short periods (from 2 days to 7 weeks) were established in the EH (Clusters 161, 162, 164 and 153) but not in the SAH1 (Cluster 154 was linked to a single patient). Some other clusters (2–8 isolates; Clusters 170, 174, 176, 177, 158, 169, 171, 166 and 130) comprised various combinations of environmental and/or clinical isolates within the SAH1; P. aeruginosa isolated from a range of clinical specimens featured in these clusters, including both normally contaminated (wound, skin and ear) and sterile (cystocentesis urine and synovial fluid) body sites.
In total, 81 (n = 81) isolates underwent WGS (at least one isolate per cluster and up to nine) and MLST, eBURST and other genotypic data were extracted from whole genomes. MLST, eBURST groups and AMR genes are summarised in Table 4 (full characterisation including virulence genes and plasmid types is found in Supplementary Table S1). Concordance between the FTIR and WGS findings was overall high for K. pneumoniae and poor for P. aeruginosa using the established COVs. For K. pneumoniae, both typing methods, FTIR and WGS, achieved the same level of discriminatory power, dividing the isolates into seven clusters each. The Simpson’s diversity index (SDI; Simpson, 1949; Hunter and Gaston, 1988) for both was 0.789 and 0.781, respectively. FTIR and WGS typing showed very high agreement, resulting in an adjusted Rand index (ARI; Carriço et al., 2006) of 0.958. The adjusted Wallace index (AWI; Severiano et al., 2011b) for FTIR predicting WGS types was 0.983, while in the reverse direction it was 0.934. For P. aeruginosa, the congruence of typing results was low. First, WGS was able to distinguish 28 different types (SDI 0.941) while FTIR could only distinguish 18 clusters (SDI 0.243). This was also reflected in quite asymmetrical AWIs, where FTIR predicted WGS types with 0.243, but WGS predicted FTIR clusters with 0.437. Overall congruence was 0.313 using the ARI.
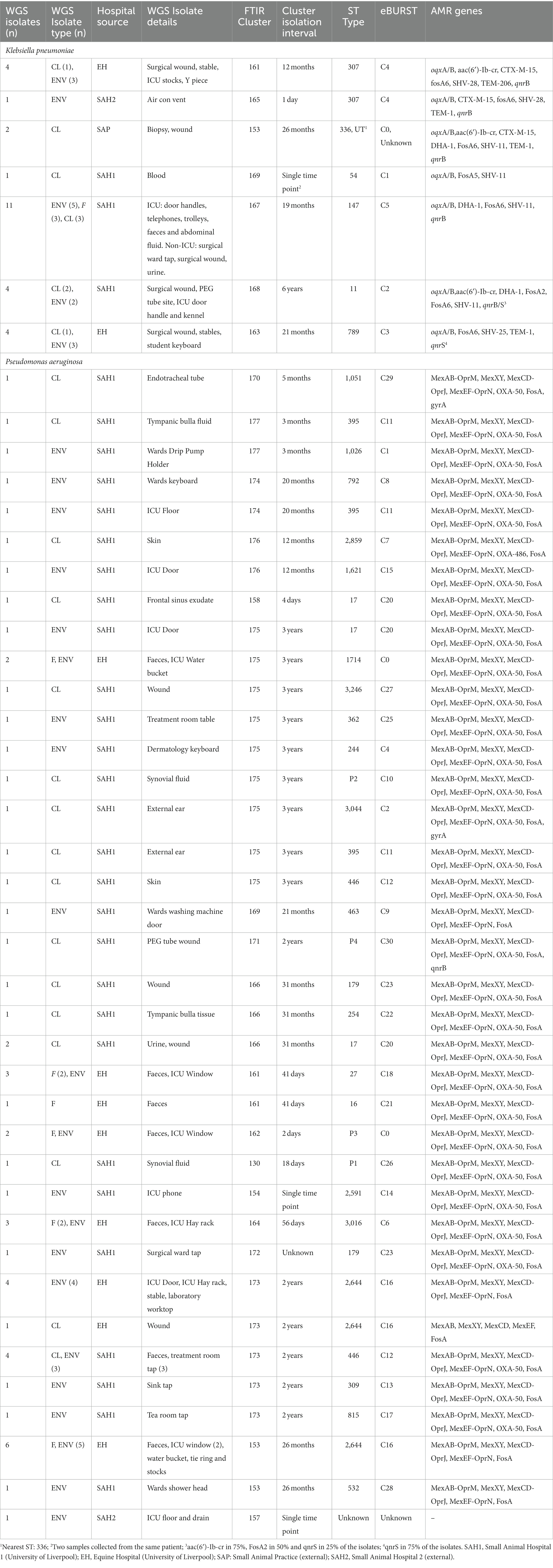
Table 4. Comparison of Fourier-Transform InfraRed (FTIR) Spectroscopy and Multi-Locus Sequence Typing (MLST) results extracted from Whole-Genome Sequences (WGS).
Twenty-seven K. pneumoniae across the seven FTIR clusters had WGS data available. Results indicate high level agreement on the identification of related isolates between the spectroscopic and the genomic methods (Figures 1A, 2A); all sequenced K. pneumoniae isolates clustered by FTIR spectroscopy belonged to the same or closest ST (one isolate in Cluster 153 was of unknown ST type; however, the nearest ST match aligned with its cluster’s ST) with six STs identified overall. Notably, ST307 was detected in two adjacent Clusters, 161 and 165; the former gathering isolates from the EH while the latter from the SAH2, supporting the FTIR Biotyper ability to discriminate between isolates of the same sequence type. One important finding was the association of ST147 harbouring plasmid-encoded quinolone resistance genes qnrB and oqxA/B, SHV-11 and DHA-1 beta-lactamase types with Cluster 167, the largest FTIR cluster of K. pneumoniae intensely circulating in the SAH1 ICU which included the hospital-acquired infection case in a dog. ST11 was associated to K. pneumoniae in Cluster 168, linking isolates from the wider SAH1 patients and areas, and additionally carried the fluoroquinolone acetylating aminoglycoside-(6)-N-acetyltransferase (aac[6′]-Ib-cr) gene. CTX-M-15-producing K. pneumoniae ST307 was identified in the former of two EH clusters (Cluster 161) and qnrS-positive ST789 in the latter (Cluster 163). Beyond beta-lactamase resistance genes present in all isolates, multidrug efflux pump genes oqxA/B were detected in 100% (27/27) of sequenced K. pneumoniae, qnr and aac(6′)-Ib-cr genes mediating resistance to fluoroquinolones were identified in 81.8% (25/27) and 29.6% (8/27) of the sequenced K. pneumoniae isolates, respectively, while CTX-M-15 type enzyme in 22% (6/27) of isolates. IncR type plasmid was common among ST147 and ST11 while IncFIB and IncFII plasmids were both found among ST307 and ST336. Concordance between AMR genes (Clusters 168 and 163) and Inc. types (Clusters 161, 167, 168 and 163) among sequenced isolates was not absolute within some clusters (Table 4), as some but not all (generally ≥50%) isolates carried several certain determinants, such as qnrS and IncFIA in 3/4 of Cluster 163 isolates.
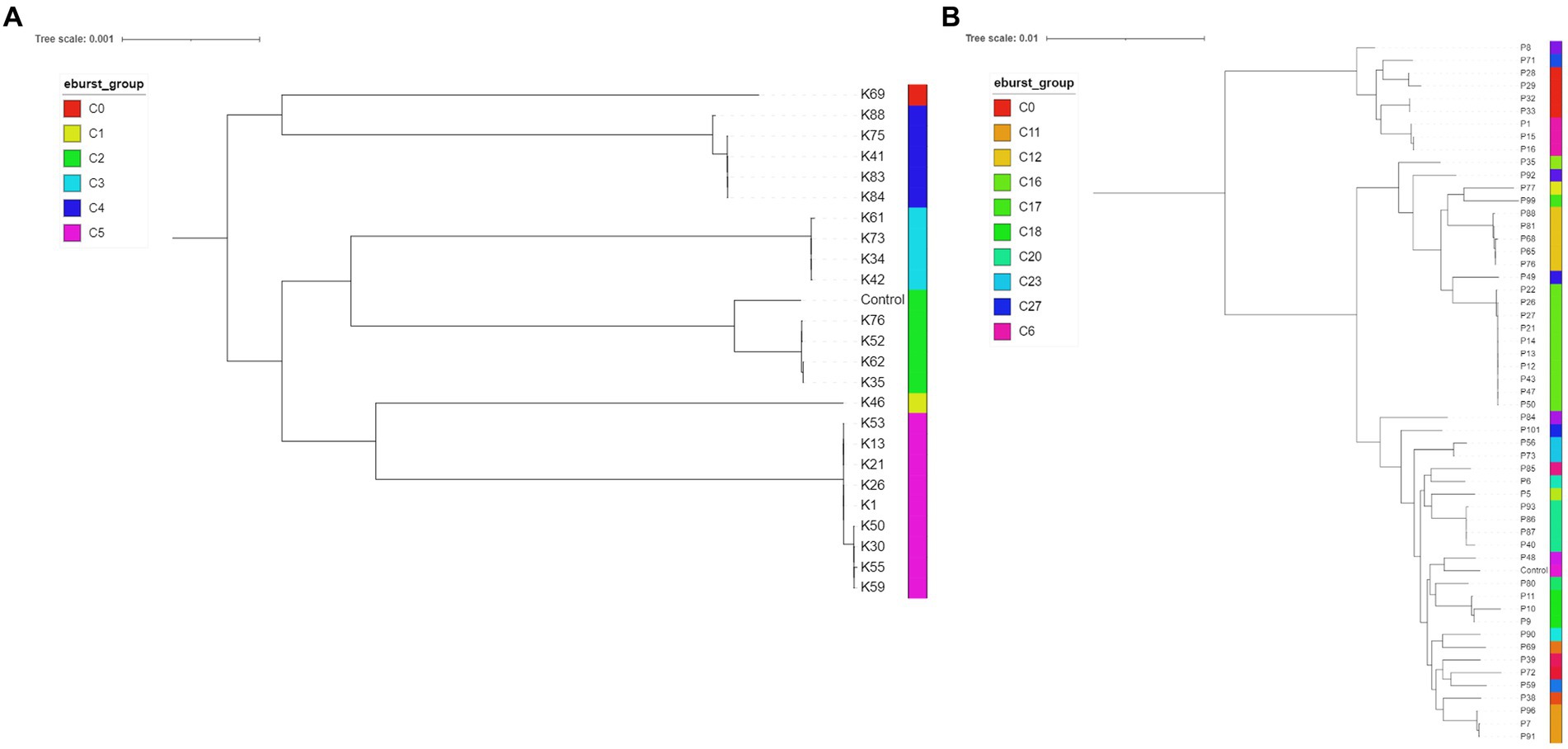
Figure 2. Midpoint rooted phylogenetic trees of Klebsiella pneumoniae (A, n = 24; isolates K10, K32 and K67 had incomplete core genome assembly, therefore they are not included in the below figure) and Pseudomonas aeruginosa (B, n = 53; P105 had incomplete genome assembly) isolates. The eBURST groups derived from MLST profiles extracted from the assembled genome sequences are shown. Klebsiella pneumoniae HS12286 and Pseudomonas aeruginosa PAO1 strains were used as internal controls.
Fifty-four P. aeruginosa spanning over 18 FTIR clusters underwent WGS; in contrast to K. pneumoniae, the accuracy of FTIR clusters was generally poor when compared to WGS (Figures 1B, 2B). Heterogeneity of MLST types was observed in the vast majority of P. aeruginosa clusters where more than one isolate was sequenced, with up to nine STs detected in Cluster 175 (where 10 of 13 were sequenced). Multiple STs were also identified in Clusters 173 (n = 4 STs over 11 sequenced isolates) and 153 (n = 2 STs over 7 sequenced isolates), others combining SAH1 and EH isolates as Cluster 175. In all these mixed clusters, the equine isolates typically accounted for a single ST; notably, the majority of ST2644 isolates from Clusters 173 and 153 were isolated from EH ICU patients (faecal carriage) and surroundings during the pilot study over a short time. In contrast, the SAH1 isolates from any of the mixed clusters were of discordant ST (when more than one small animal isolate was found), apart from ST446 in Cluster 173 (here two thirds of sequenced small animal isolates belonged to this ST). Cluster 162 and 164 (n = 2 and 3 isolates sequenced, respectively), both comprising EH ICU pilot study isolates, were the only examples of genomically highly similar isolates appropriately clustered by the FTIR. SAH1 only clusters where more than one isolate was sequenced displayed substantial within-cluster ST heterogeneity, such as Clusters 166, 174 and 176. In addition to genomically divergent isolates inadequately clustered together by the FTIR, examples of genotypically highly similar isolates inappropriately not clustered were also seen, e.g., ST2644, ST395, ST17 and ST446 isolates from the same facility are seen across more than one cluster (usually 2 or 3). Finally, P. aeruginosa STs associated to clusters where single isolates were sequenced differed from one another and from STs in larger clusters, except for ST17. Overall, 28 STs were identified with ST2644, ST446, ST17, ST395, ST27 and ST3016 being most represented (Table 4). Significantly less variability was observed among P. aeruginosa with regards to AMR determinants. Sequenced isolates harboured a large arsenal of efflux pumps, particularly of the Mex (A/B, C/D, E/F, X/Y, M/J, and X/Y types amongst others) and Opr (OprJ, M and N) families and 73.7% contained OXA-50 (42/57) beta-lactamase; gyrA mutation and qnrB gene conferring fluoroquinolone resistance were detected in 5.3% (3/57) and 1.8% (1/57) of isolates, respectively.
To the best of the authors’ knowledge, this is the first study to evaluate the FTIR potential for bacterial strain typing in a clinically-relevant timeframe within veterinary settings. We assessed the discriminatory power of FTIR spectroscopy by typing two ESKAPE MDR gram-negative bacterial species with potential implication in HCAIs, namely K. pneumoniae and P. aeruginosa, isolated from cases of clinical infection in hospitalised or vet-visiting companion animals (i.e., dogs, cats and horses), as well as faecal colonisation and environmental sites across veterinary clinics/hospitals. The IR Biotyper could determine clonal relatedness of MDR gram-negative isolates circulating in veterinary hospitals within 2–3 h from fresh culture harvest, making it an attractive tool for rapid strain typing of organisms associated with HCAIs. This technique has the potential to be integrated into the routine diagnostic workflow of a clinical microbiology laboratory for “real-time” monitoring of MDR pathogens’ transmission events to support prompt and targeted infection control measures during hospital outbreaks.
In our study, we found that the discriminatory power of FTIR spectroscopy was high for K. pneumoniae veterinary isolates using a COV of 0.287, which demonstrated overall concordance (ARI of 0.958) with WGS results for the fraction of isolates that underwent the gold standard technique. This is in agreement with previous studies evaluating the IR Biotyper performance on K. pneumoniae-associated human hospital outbreaks (Rakovitsky et al., 2020; Rodrigues et al., 2020; Silva et al., 2020; Hu et al., 2021). In one study assessing the IR Biotyper performance on environmental isolates of K. pneumoniae and other gram-negative bacilli from nine human hospitals, however, the instrument displayed limited sensitivity to cluster isolates compared with WGS (Aranega Bou et al., 2023); this contrasts with our findings where both environmental and clinical/carriage K. pneumoniae were adequately clustered according to WGS data. In addition, some authors evaluated the performance of the FTIR as a first-line typing tool for the identification of ESBL-producing Klebsiella pneumoniae outbreaks in the hospital setting in comparison with conventional epidemiology (CE) investigations and found both AR and AW were significantly higher for FTIR clustering than CE clustering (Wang-Wang et al., 2022). Importantly, the majority of K. pneumoniae isolates circulating in veterinary hospitals in our study were consistent with critically important human lineages and they segregated into well-defined hospital source clusters. Clonally related K. pneumoniae ST147 formed the largest SAH1 cluster comprising 72% of isolates collected from the ICU over 16 days. This lineage was identified simultaneously in carriage (faeces), environmental (patients kennels, ICU door handles, keyboards and phone) and one clinical infection case (septic peritonitis) in an ICU hospitalised dog. K. pneumoniae ST147 is an emerging high-risk human clone with worldwide distribution (Navon-Venezia et al., 2017) and the potential to become a major public health threat due to its ability to cause serious infections and association with AMR, including pan-resistance. This successful clone consists of multiple clades/clusters, often encoding for ESBLs and carbapenemases such as CTX-M-15, KPC-2 and NDM-1 amongst others (Peirano et al., 2020). To date, K. pneumoniae ST147 has been occasionally described in companion and exotic pets’ clinical specimens (Ovejero et al., 2017; Marques et al., 2019; Davies et al., 2022) without association to carbapenem resistance. Similar considerations may be formulated for K. pneumoniae ST11 identified in the SAH1 and ST307 and ST789 isolated from the EH; these epidemic human-associated clones (except for ST789) have already been described in companion animals more extensively than ST147 (Harada et al., 2016; Loncaric et al., 2020; Garcia-Fierro et al., 2022), with reports of OXA-48-producing ST11 strains causing animal infection in some European countries (Pulss et al., 2018). In contrast, K. pneumoniae ST15 broadly reported among dogs, cats and horses (Haenni et al., 2012; Stolle et al., 2013; Ewers et al., 2014; Maeyama et al., 2018), was not detected in the present study. Altogether, these findings point towards the possibility of clonal transmission of K. pneumoniae within all veterinary facilities investigated, as outlined by the FTIR association of clusters (and STs) with their hospital sources and, for isolates within some clusters, by the short sample collection window (e.g., the SAH1 ICU isolates in Cluster 167). Furthermore, the tight clustering of clinical isolates from the SAP (Cluster 153) on one hand, and of the environmental isolates from the SAH2 (Cluster 165), also points towards cross-transmission within these facilities.
Compared to K. pneumoniae, FTIR-based clustering of P. aeruginosa veterinary isolates was much less congruent with WGS results; its ability to correctly detect and cluster isolates of a given ST using a COV of 0.246 was poor, as reflected by an ARI of 0.313, with multiple examples of genomically related isolates spread across different clusters and genomically divergent isolates incorrectly clustered together by the IR Biotyper. Nevertheless, comparable diagnostic resolution to that of current reference methods has been previously reported for this organism by other authors (Martak et al., 2019; Hu et al., 2023). In these studies analysing clinical P. aeruginosa isolates from human hospital outbreaks, optimal FTIR spectroscopy COVs varied for this organism; one publication reports the optimal range 0.184 to 0.374 (AR, 0.936; Martak et al., 2019) while the other determined a more stringent COV range of 0.132 to 0.173 (AR, 0.711; Hu et al., 2023). Both studies employed Mueller-Hinton (MH) agar medium to passage P. aeruginosa isolates undergoing FTIR Biotyping, and Hu et al., 2023 demonstrated that this resulted in better discriminatory power than 5% Blood Agar medium used in our study, which could have had a negative effect on our results for veterinary P. aeruginosa isolates. It may be that our FTIR validation process to optimise the culture scheme and COV determination, which constitutes the main and, perhaps, only complication of applying FTIR to routine laboratory settings, should be revisited and improved for typing veterinary P. aeruginosa. No correlation was found between FTIR spectroscopy and molecular methods in another study for P. aeruginosa strain classification (Oho et al., 2021). These authors analysed clinical P. aeruginosa from multiple institutions and concluded that differences in the measurement principles may account for classification mismatches of P. aeruginosa organisms; WGS data could help understanding the genomic variations between isolates that modify carbohydrate composition and therefore impact the FTIR spectrum. As this was a retrospective analysis of veterinary hospital isolates collected over several years, it is plausible that genomic changes which could be reflected in the carbohydrate composition of the bacterial cell wall may have occurred over time, and this may have affected Pseudomonas aeruginosa more than Klebsiella pneumoniae in light of its larger genome. The genetic background and population structure of P. aeruginosa isolates in the present study was greatly diversified; nonetheless a clear association was found between ST2644 isolates and the EH, particularly from the ICU (including colonisation, environmental and clinical isolates). This clone has been previously described in MDR virulent P. aeruginosa isolates from small animal hospitals in Japan (Hayashi et al., 2021) and it is herein reported in horses for the first time. Other potential high-risk clones identified in this study included ST395 and ST244, both associated to small animal clinical or environmental isolates. The former ST has been described among carbapenemase-negative carbapenem-nonsusceptible P. aeruginosa small animal clinical isolates in France (Haenni et al., 2017) whereas the latter is a global epidemic human clone often expressing acquired carbapenem-resistance genes and MDR/XDR profiles (Chen et al., 2014; del Barrio-Tofiño et al., 2020; Pérez-vázquez et al., 2020) that has also been reported in veterinary infections (Haenni et al., 2015) and hospital environments (Soonthornsit et al., 2023). Finally, P. aeruginosa isolated across multiple sink taps across the SAH1 belonged to ST446, a recently identified high-risk clone associated with multidrug resistance (Pincus et al., 2020).
The reasons behind numerous FTIR clusters containing isolates collected at broad time intervals (≥3 years), as herein seen for both organisms, warrants more focused prospective studies. For example, it would be useful to understand whether this is determined by the persistence of MDR-GNs in the hospital environments, their re-introduction or by the highly clonal nature of some isolates within certain bacterial species. Furthermore, larger prospective studies would be beneficial to understand and measure the impact of FTIR real-time hospital surveillance to infection prevention and control. Finally, it is important to bear in mind that, in our study, we compared the concordance of FTIR spectroscopy and WGS-based cluster analyses where only a subset of FTIR clustering isolates underwent the gold standard method (few isolates for each defined cluster, proportional to cluster size) which represents the main limitation of this work.
In conclusion, we found that FTIR spectroscopy accurately clustered veterinary K. pneumoniae isolates belonging to the same clone but may be less accurate in discriminating P. aeruginosa isolates. The resolution of this method was high for K. pneumoniae for both clinical/colonisation and environmental hospital isolates, suggesting that FTIR spectroscopy alone could provide sufficient information to support early and appropriate infection control measures for hospital outbreak and epidemiological surveillance of K. pneumoniae. The rapid turnaround time combined with its ease of performance, low cost per sample and lack of requirement for specialised training, bring considerable advantages over the current molecular reference methods (i.e., PFGE, MLST and WGS) for implementation into the routine workflow of a veterinary microbiology laboratory conducting infection control work. Here, we have shown that FTIR spectroscopy has the potential to become a valuable tool for rapid identification of Klebsiella pneumoniae (and potentially other pathogens) transmission events between patients and the veterinary clinical environment, therefore providing real-time surveillance information to aid infection prevention in veterinary settings.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: ENA—PRJEB70897.
The animal studies were approved by University of Liverpool Veterinary Research Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
FZ: Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. VS: Resources, Writing – review & editing. NM: Data curation, Formal analysis, Resources, Writing – review & editing. AL: Resources, Writing – review & editing. RJ: Resources, Writing – review & editing. CI: Resources, Writing – review & editing. GP: Funding acquisition, Writing – review & editing. SH: Data curation, Formal analysis, Resources, Writing – review & editing. DT: Conceptualization, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Whole-genome sequencing of Pseudomonas aeruginosa and Klebsiella pneumoniae equine isolates was supported by the Horse Trust (Registered Charity 585 No: 231748).
The authors would like to thank the Veterinary Microbiology Diagnostic Laboratory at the University of Copenhagen for kindly providing fully characterised bacterial isolates used for the technical optimisation of the FTIR spectroscopy cut-offs. We warmly thank the hospitals’ staff for helping with the environmental sample collection, the Veterinary Microbiology Diagnostic Laboratory (VMDL) at the University of Liverpool for sample processing and the Centre for Genomic Research (CGR) at the University of Liverpool for performing WGS and bioinformatics.
NM is employed at Bruker Daltonics GmbH. CI was employed at the School of Veterinary Science, Philip Leverhulme Equine Hospital, University of Liverpool at the time of commencing the study and moved to Western Counties Equine Hospital Ltd where is currently employed.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1334268/full#supplementary-material
Aranega Bou, P., Cornbill, C., Rodger, G., Bird, M., Moore, G., Roohi, A., et al. (2023). Evaluation of Fourier transform infrared spectroscopy (IR biotyper) as a complement to whole genome sequencing (WGS) to characterise Enterobacter cloacae, Citrobacter freundii and Klebsiella pneumoniae isolates recovered from hospital sinks. Fortschr. Med. 4. doi: 10.1101/2023.04.24.23289028
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Boccia, S., Pasquarella, C., Colotto, M., Barchitta, M., Quattrocchi, A., and Agodi, A. (2015). Molecular epidemiology tools in the management of healthcare-associated infections: towards the definition of recommendations. Epidemiol. Prev. 39, 21–26.
Bortolami, A., Williams, N. J., McGowan, C. M., Kelly, P. G., Archer, D. C., Corrò, M., et al. (2017). Environmental surveillance identifies multiple introductions of MRSA CC398 in an equine veterinary hospital in the UK, 2011–2016. Sci. Rep. 7:5499. doi: 10.1038/s41598-017-05559-8
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Carriço, J. A., Silva-Costa, C., Melo-Cristino, J., Pinto, F. R., De Lencastre, H., Almeida, J. S., et al. (2006). Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J. Clin. Microbiol. 44, 2524–2532. doi: 10.1128/jcm.02536-05
Chen, Y., Sun, M., Wang, M., Lu, Y., and Yan, Z. (2014). Dissemination of IMP-6-producing Pseudomonas aeruginosa ST244 in multiple cities in China. Eur. J. Clin. Microbiol. Infect. Dis. 33, 1181–1187. doi: 10.1007/s10096-014-2063-5
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Clinical and Laboratory Standards Institute (CLSI). (2018). Performance standards for antimicrobial disk and dilution susceptibility tests for bacteria isolated from animals. 4th edition, VET08. Wayne, PA.
Davies, Y. M., Cunha, M. P. V., Dropa, M., Lincopan, N., Gomes, V. T. M., Moreno, L. Z., et al. (2022). Pandemic clones of CTX-M-15 producing Klebsiella Pneumoniae ST15, ST147, and ST307 in companion parrots. Microorganisms 10:1412. doi: 10.3390/microorganisms10071412
del Barrio-Tofiño, E., López-Causapé, C., and Oliver, A. (2020). Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update. Int. J. Antimicrob. Agents 56:106196. doi: 10.1016/j.ijantimicag.2020.106196
Dinkelacker, A. G., Vogt, S., Oberhettinger, P., Mauder, N., Rau, J., Kostrzewa, M., et al. (2018). Typing and species identification of clinical Klebsiella isolates by Fourier transform infrared spectroscopy and matrix-assisted laser desorption ionization–time of flight mass spectrometry. J. Clin. Microbiol. 56, e00843–e00818. doi: 10.1128/jcm.00843-18
Ewers, C., Stamm, I., Pfeifer, Y., Wieler, L. H., Kopp, P. A., Schønning, K., et al. (2014). Clonal spread of highly successful ST15-CTX-M-15 Klebsiella pneumoniae in companion animals and horses. J. Antimicrob. Chemother. 69, 2676–2680. doi: 10.1093/jac/dku217
Finlayson, D., Rinaldi, C., and Baker, M. J. (2019). Is infrared spectroscopy ready for the clinic? Anal. Chem. 91, 12117–12128. doi: 10.1021/acs.analchem.9b02280
Foxman, B., Zhang, L., Koopman, J. S., Manning, S. D., and Marrs, C. F. (2005). Choosing an appropriate bacterial typing technique for epidemiologic studies. Epidemiologic perspectives & innovations 2, 1–8. doi: 10.1186/1742-5573-2-10
Garcia-Fierro, R., Drapeau, A., Dazas, M., Saras, E., Rodrigues, C., Brisse, S., et al. (2022). Comparative phylogenomics of ESBL-, AmpC-and carbapenemase-producing Klebsiella pneumoniae originating from companion animals and humans. J. Antimicrob. Chemother. 77, 1263–1271. doi: 10.1093/jac/dkac041
Haenni, M., Bour, M., Châtre, P., Madec, J.-Y., Plésiat, P., and Jeannot, K. (2017). Resistance of animal strains of Pseudomonas aeruginosa to carbapenems. Front. Microbiol. 8:1847. doi: 10.3389/fmicb.2017.01847
Haenni, M., Hocquet, D., Ponsin, C., Cholley, P., Guyeux, C., Madec, J.-Y., et al. (2015). Population structure and antimicrobial susceptibility of Pseudomonas aeruginosa from animal infections in France. BMC Vet. Res. 11, 9–5. doi: 10.1186/s12917-015-0324-x
Haenni, M., Ponsin, C., Métayer, V., Médaille, C., and Madec, J.-Y. (2012). Veterinary hospital-acquired infections in pets with a ciprofloxacin-resistant CTX-M-15-producing Klebsiella pneumoniae ST15 clone. J. Antimicrob. Chemother. 67, 770–771. doi: 10.1093/jac/dkr527
Harada, K., Shimizu, T., Mukai, Y., Kuwajima, K., Sato, T., Usui, M., et al. (2016). Phenotypic and molecular characterization of antimicrobial resistance in Klebsiella spp. isolates from companion animals in Japan: clonal dissemination of multidrug-resistant extended-spectrum β-lactamase-producing Klebsiella pneumoniae. Front. Microbiol. 7:1021. doi: 10.3389/fmicb.2016.01021
Hayashi, W., Izumi, K., Yoshida, S., Takizawa, S., Sakaguchi, K., Iyori, K., et al. (2021). Antimicrobial resistance and type III secretion system virulotypes of Pseudomonas aeruginosa isolates from dogs and cats in primary veterinary hospitals in Japan: identification of the international high-risk clone sequence type 235. Microbiol. Spectrum 9, e00408–e00421. doi: 10.1128/Spectrum.00408-21
Hoet, A. E., Johnson, A., Nava-Hoet, R. C., Bateman, S., Hillier, A., Dyce, J., et al. (2011). Environmental methicillin-resistant Staphylococcus aureus in a veterinary teaching hospital during a nonoutbreak period. Vector-Borne and Zoonotic Dis. 11, 609–615. doi: 10.1089/vbz.2010.0181
Hu, Y., Zhou, H., Lu, J., Sun, Q., Liu, C., Zeng, Y., et al. (2021). Evaluation of the IR biotyper for Klebsiella pneumoniae typing and its potentials in hospital hygiene management. Microb. Biotechnol. 14, 1343–1352. doi: 10.1111/1751-7915.13709
Hu, Y., Zhu, K., Jin, D., Shen, W., Liu, C., Zhou, H., et al. (2023). Evaluation of IR biotyper for carbapenem-resistant Pseudomonas aeruginosa typing and its application potential for the investigation of nosocomial infection. Front. Microbiol. 14:1068872. doi: 10.3389/fmicb.2023.1068872
Hunter, P. R., and Gaston, M. A. (1988). Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol. 26, 2465–2466. doi: 10.1128/jcm.26.11.2465-2466.1988
Keck, N., Dunie-merigot, A., Dazas, M., Hirchaud, E., Laurence, S., Gervais, B., et al. (2020). Long-lasting nosocomial persistence of chlorhexidine-resistant Serratia marcescens in a veterinary hospital. Vet. Microbiol. 245:108686. doi: 10.1016/j.vetmic.2020.108686
Kramer, A., Schwebke, I., and Kampf, G. (2006). How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect. Dis. 6, 1–8. doi: 10.1186/1471-2334-6-130
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Loeffler, A., Boag, A. K., Sung, J., Lindsay, J. A., Guardabassi, L., Dalsgaard, A., et al. (2005). Prevalence of methicillin-resistant Staphylococcus aureus among staff and pets in a small animal referral hospital in the UK. J. Antimicrob. Chemother. 56, 692–697. doi: 10.1093/jac/dki312
Loncaric, I., Cabal Rosel, A., Szostak, M. P., Licka, T., Allerberger, F., Ruppitsch, W., et al. (2020). Broad-spectrum cephalosporin-resistant Klebsiella spp. isolated from diseased horses in Austria. Animals 10:332. doi: 10.3390/ani10020332
Maeyama, Y., Taniguchi, Y., Hayashi, W., Ohsaki, Y., Osaka, S., Koide, S., et al. (2018). Prevalence of ESBL/AmpC genes and specific clones among the third-generation cephalosporin-resistant Enterobacteriaceae from canine and feline clinical specimens in Japan. Vet. Microbiol. 216, 183–189. doi: 10.1016/j.vetmic.2018.02.020
Marques, C., Menezes, J., Belas, A., Aboim, C., Cavaco-Silva, P., Trigueiro, G., et al. (2019). Klebsiella pneumoniae causing urinary tract infections in companion animals and humans: population structure, antimicrobial resistance and virulence genes. J. Antimicrob. Chemother. 74, 594–602. doi: 10.1093/jac/dky499
Martak, D., Valot, B., Sauget, M., Cholley, P., Thouverez, M., Bertrand, X., et al. (2019). Fourier-transform infrared spectroscopy can quickly type gram-negative bacilli responsible for hospital outbreaks. Front. Microbiol. 10:1440. doi: 10.3389/fmicb.2019.01440
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Murphy, C. P., Reid-Smith, R. J., Boerlin, P., Weese, J. S., Prescott, J. F., Janecko, N., et al. (2010). Escherichia coli and selected veterinary and zoonotic pathogens isolated from environmental sites in companion animal veterinary hospitals in southern Ontario. Can. Vet. J. 51, 963–972.
Navon-Venezia, S., Kondratyeva, K., and Carattoli, A. (2017). Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol. Rev. 41, 252–275. doi: 10.1093/femsre/fux013
Novais, Â., Freitas, A. R., Rodrigues, C., and Peixe, L. (2019). Fourier transform infrared spectroscopy: unlocking fundamentals and prospects for bacterial strain typing. Eur. J. Clin. Microbiol. Infect. Dis. 38, 427–448. doi: 10.1007/s10096-018-3431-3
Nutman, A., Lerner, A., Schwartz, D., and Carmeli, Y. (2016). Evaluation of carriage and environmental contamination by carbapenem-resistant Acinetobacter baumannii. Clin. Microbiol. Infect. 22, 949.e5–949.e7. doi: 10.1016/j.cmi.2016.08.020
Oho, M., Nagasawa, Z., Funashima, Y., Ueda, O., Watamabe, S., Cui, L., et al. (2021). Correlation of Strain Classification with IR Biotyper and Molecular Epidemiological Method of Pseudomonas aeruginosa. Rinsho Biseibutsu Jinsoku Shindan Kenkyukai shi= JARMAM: J. Assoc. Rapid Method Auto. Microbiol. 31, 29–40.
Ovejero, C. M., Escudero, J. A., Thomas-Lopez, D., Hoefer, A., Moyano, G., Montero, N., et al. (2017). Highly tigecycline-resistant Klebsiella pneumoniae sequence type 11 (ST11) and ST147 isolates from companion animals. Antimicrob. Agents Chemother. 61:e02640-16. doi: 10.1128/aac.02640-16
Peirano, G., Chen, L., Kreiswirth, B. N., and Pitout, J. D. D. (2020). Emerging antimicrobial-resistant high-risk Klebsiella pneumoniae clones ST307 and ST147. Antimicrob. Agents Chemother. 64:e01148–20. doi: 10.1128/AAC.01148-20
Pérez-vázquez, M., Sola-campoy, P. J., Zurita, Á. M., Ávila, A., Gómez-bertomeu, F., Solís, S., et al. (2020). Carbapenemase-producing Pseudomonas aeruginosa in Spain: interregional dissemination of the high-risk clones ST175 and ST244 carrying blaVIM-2, blaVIM-1, blaIMP-8, blaVIM-20 and blaKPC-2. Int. J. Antimicrob. Agents 56:106026. doi: 10.1016/j.ijantimicag.2020.106026
Pincus, N. B., Bachta, K. E., Ozer, E. A., Allen, J. P., Pura, O. N., Qi, C., et al. (2020). Long-term persistence of an extensively drug-resistant subclade of globally distributed Pseudomonas aeruginosa clonal complex 446 in an academic medical center. Clin. Infect. Dis. 71, 1524–1531. doi: 10.1093/cid/ciz973
Pinto, F. R., Melo-Cristino, J., and Ramirez, M. (2008). A confidence interval for the Wallace coefficient of concordance and its application to microbial typing methods. PLoS One 3:e3696. doi: 10.1371/journal.pone.0003696
Pulss, S., Stolle, I., Stamm, I., Leidner, U., Heydel, C., Semmler, T., et al. (2018). Multispecies and clonal dissemination of OXA-48 carbapenemase in Enterobacteriaceae from companion animals in Germany, 2009—2016. Front. Microbiol. 9:1265. doi: 10.3389/fmicb.2018.01265
Rakovitsky, N., Frenk, S., Kon, H., Schwartz, D., Temkin, E., Solter, E., et al. (2020). FT-IR spectroscopy is a new option for outbreak investigation: a retrospective analysis of an ESBL-producing Klebsiella pneumoniae outbreak in a neonatal intensive care unit. J. Clin. Microbiol. 58, e00098–e00020. doi: 10.1128/JCM.00098-20
Rodrigues, C., Sousa, C., Lopes, J. A., Novais, Â., and Peixe, L. (2020). A front line on Klebsiella pneumoniae capsular polysaccharide knowledge: Fourier transform infrared spectroscopy as an accurate and fast typing tool. Msystems 5, e00386–e00319. doi: 10.1128/msystems.00386-19
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Severiano, A., Carriço, J. A., Robinson, D. A., Ramirez, M., and Pinto, F. R. (2011a). Evaluation of jackknife and bootstrap for defining confidence intervals for pairwise agreement measures. PLoS One 6:e19539. doi: 10.1371/journal.pone.0019539
Severiano, A., Pinto, F. R., Ramirez, M., and Carriço, J. A. (2011b). Adjusted Wallace coefficient as a measure of congruence between typing methods. J. Clin. Microbiol. 49, 3997–4000. doi: 10.1128/jcm.00624-11
Silva, L., Rodrigues, C., Lira, A., Leão, M., Mota, M., Lopes, P., et al. (2020). Fourier transform infrared (FT-IR) spectroscopy typing: a real-time analysis of an outbreak by carbapenem-resistant Klebsiella pneumoniae. Eur. J. Clin. Microbiol. Infect. Dis. 39, 2471–2475. doi: 10.1007/s10096-020-03956-y
Soonthornsit, J., Pimwaraluck, K., Kongmuang, N., Pratya, P., and Phumthanakorn, N. (2023). Molecular epidemiology of antimicrobial-resistant Pseudomonas aeruginosa in a veterinary teaching hospital environment. Vet. Res. Commun. 47, 73–86. doi: 10.1007/s11259-022-09929-0
Soza-Ossandón, P., Rivera, D., Tardone, R., Riquelme-Neira, R., García, P., Hamilton-West, C., et al. (2020). Widespread environmental presence of multidrug-resistant Salmonella in an equine veterinary hospital that received local and international horses. Front. Vet. Sci. 7:346. doi: 10.3389/fvets.2020.00346
Steneroden, K., van Metre, D. C., Jackson, C., and Morley, P. S. (2010). Detection and control of a nosocomial outbreak caused by Salmonella Newport at a large animal hospital. J. Vet. Intern. Med. 24, 606–616. doi: 10.1111/j.1939-1676.2010.0484.x
Stolle, I., Prenger-Berninghoff, E., Stamm, I., Scheufen, S., Hassdenteufel, E., Guenther, S., et al. (2013). Emergence of OXA-48 carbapenemase-producing Escherichia coli and Klebsiella pneumoniae in dogs. J. Antimicrob. Chemother. 68, 2802–2808. doi: 10.1093/jac/dkt259
Timofte, D., Maciuca, I. E., Williams, N. J., Wattret, A., and Schmidt, V. (2016). Veterinary hospital dissemination of CTX-M-15 extended-Spectrum Beta-lactamase–producing Escherichia coli ST410 in the United Kingdom. Microb. Drug Resist. 22, 609–615. doi: 10.1089/mdr.2016.0036
Tonkin-Hill, G., MacAlasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J. A., et al. (2020). Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21:180. doi: 10.1186/s13059-020-02090-4
Vogt, S., Löffler, K., Dinkelacker, A. G., Bader, B., Autenrieth, I., Peter, S. M., et al. (2019). Fourier-transform infrared (FTIR) spectroscopy for typing of clinical Enterobacter cloacae complex isolates. Front. Microbiol. 10:2582. doi: 10.3389/fmicb.2019.02582
Walther, B., Klein, K.-S., Barton, A.-K., Semmler, T., Huber, C., Wolf, S. A., et al. (2018). Extended-spectrum beta-lactamase (ESBL)-producing Escherichia coli and Acinetobacter baumannii among horses entering a veterinary teaching hospital: the contemporary “trojan horse”. PLoS One 13:e0191873. doi: 10.1371/journal.pone.0191873
Walther, B., Lübke-Becker, A., Stamm, I., Gehlen, H., Barton, A. K., Janssen, T., et al. (2014). Suspected nosocomial infections with multi-drug resistant E. coli, including extended-spectrum beta-lactamase (ESBL)-producing strains, in an equine clinic. Berl. Munch. Tierarztl. Wochenschr. 127, 421–427. doi: 10.2376/0005-9366-127-421
Walther, B., Tedin, K., and Lübke-Becker, A. (2017). Multidrug-resistant opportunistic pathogens challenging veterinary infection control. Vet. Microbiol. 200, 71–78. doi: 10.1016/j.vetmic.2016.05.017
Wang-Wang, J. H., Bordoy, A. E., Martró, E., Quesada, M. D., Pérez-Vázquez, M., Guerrero-Murillo, M., et al. (2022). Evaluation of Fourier transform infrared spectroscopy as a first-line typing tool for the identification of extended-Spectrum β-lactamase-producing Klebsiella pneumoniae outbreaks in the hospital setting. Front. Microbiol. 13:897161. doi: 10.3389/fmicb.2022.897161
Weber, D. J., Anderson, D., and Rutala, W. A. (2013). The role of the surface environment in healthcare-associated infections. Curr. Opin. Infect. Dis. 26, 338–344. doi: 10.1097/QCO.0b013e3283630f04
Weese, J. S. (2011). Infection control in veterinary practice; the time is now. J. Small Anim. Pract. 52, 507–508. doi: 10.1111/j.1748-5827.2011.01140.x
Weese, J. S., DaCosta, T., Button, L., Goth, K., Ethier, M., and Boehnke, K. (2004). Isolation of methicillin-resistant Staphylococcus aureus from the environment in a veterinary teaching hospital. J. Vet. Intern. Med. 18, 468–470. doi: 10.1111/j.1939-1676.2004.tb02568.x
Wieler, L. H., Ewers, C., Guenther, S., Walther, B., and Lübke-Becker, A. (2011). Methicillin-resistant staphylococci (MRS) and extended-spectrum beta-lactamases (ESBL)-producing Enterobacteriaceae in companion animals: nosocomial infections as one reason for the rising prevalence of these potential zoonotic pathogens in clinical samples. Int. J. Med. Microbiol. 301, 635–641. doi: 10.1016/j.ijmm.2011.09.009
Yang, H., Wu, F., Xu, F., Tang, K., Ding, C., Shi, H., et al. (2020). Bacterial typing and identification based on Fourier transform infrared spectroscopy. Protocol Exchange. doi: 10.21203/rs.2.23337/v1
Keywords: veterinary, infection control, Fourier-transform infrared (FTIR) spectroscopy, veterinary settings, companion animals, Klebsiella pneumoniae, Pseudomonas aeruginosa
Citation: Zendri F, Schmidt V, Mauder N, Loeffler A, Jepson RE, Isgren C, Pinchbeck G, Haldenby S and Timofte D (2024) Rapid typing of Klebsiella pneumoniae and Pseudomonas aeruginosa by Fourier-transform Infrared spectroscopy informs infection control in veterinary settings. Front. Microbiol. 15:1334268. doi: 10.3389/fmicb.2024.1334268
Edited by:
Sebastien Olivier Leclercq, INRA Centre Val de Loire, FranceReviewed by:
Simone Ambretti, IRCCS University Hospital of Bologna Sant Orsola Polyclinic, ItalyCopyright © 2024 Zendri, Schmidt, Mauder, Loeffler, Jepson, Isgren, Pinchbeck, Haldenby and Timofte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dorina Timofte, ZC50aW1vZnRlQGxpdi5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.