
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 24 January 2024
Sec. Microbial Physiology and Metabolism
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1328321
 Jiaqi Mao†Min Zhang†Wenjuan DaiChenghao FuZhanzhan WangXiuwen WangQingshou YaoLinghui Kong*
Jiaqi Mao†Min Zhang†Wenjuan DaiChenghao FuZhanzhan WangXiuwen WangQingshou YaoLinghui Kong* Jiayang Qin*
Jiayang Qin*The available resources of Streptomyces represent a valuable repository of bioactive natural products that warrant exploration. Streptomyces albulus is primarily utilized in the industrial synthesis of ε-poly-L-lysine (ε-PL). In this study, the NADP-dependent glyceraldehyde 3-phosphate dehydrogenase (GapN) from Streptococcus mutans was heterologously expressed in S. albulus CICC11022, leading to elevated intracellular NADPH levels and reduced NADH and ATP concentrations. The resulting perturbation of S. albulus metabolism was comprehensively analyzed using transcriptomic and metabolomic methodologies. A decrease in production of ε-PL was observed. The expression of gapN significantly impacted on 23 gene clusters responsible for the biosynthesis of secondary metabolites. A comprehensive analysis revealed a total of 21 metabolites exhibiting elevated levels both intracellularly and extracellularly in the gapN expressing strain compared to those in the control strain. These findings underscore the potential of S. albulus to generate diverse bioactive natural products, thus offering valuable insights for the utilization of known Streptomyces resources through genetic manipulation.
Natural products (NPs) generated from microorganisms and their derivatives are important sources of bioactive substances such as antibiotics, pesticides, antiparasites, anti-infectives, and antitumor drugs (Al-shaibani et al., 2021; Jose et al., 2021; Reddy et al., 2021). Among all the microorganisms, Actinomycetes contribute significantly to the production of bioactive compounds, including well-known secondary metabolites (SMs), such as streptomycin, vancomycin, erythromycin, rifamycin and tetracycline (Kong et al., 2019; Al-shaibani et al., 2021; Jose et al., 2021; Reddy et al., 2021). To obtain new bioactive compounds, scientists have strived to screen Actinomycetes, especially Streptomyces, from a variety of environments, including marine and extreme environments (Donald et al., 2022). Another resource that should not be overlooked is the known Streptomyces. The number of Streptomyces genomes that have been sequenced is continuously increasing, and a large number of secondary metabolite biosynthesis gene clusters (SM-BGCs) have been discovered (Belknap et al., 2020; Lee et al., 2020). However, the majority of these BGCs, are silent when tested under standard laboratory conditions (Liu et al., 2021). Therefore, numerous strategies, including heterologous expression (Kang and Kim, 2021), overexpression in native host (Li H. et al., 2022), promoter engineering (Liu X. F. et al., 2022), transcriptional regulation engineering (Li et al., 2019; Martínez-Burgo et al., 2019) and ribosome engineering (Zhang et al., 2019; Li et al., 2021) have been developed to activate these BGCs in NP discovery studies (Beck et al., 2021; Liu et al., 2021). However, since most of these studies focused on one or more metabolites, the full potential of Streptomyces has rarely been studied.
Streptomyces albulus is a species of Streptomyces whose main SM is ε-poly-L-lysine (ε-PL); thus, it has become an industrial production strain for ε-PL (Wang et al., 2021; Li S. et al., 2022). ε-PL is an L-lysine homopolymer with broad-spectrum antibacterial activity and thus has a wide range of uses in the food, medical, chemical, and other industries (Wang et al., 2021). S. albulus is also capable of synthesizing other bioactive substances, such as wuyiencin (Liu B. et al., 2022; Yang et al., 2022), salinomycin (Zhang et al., 2019), tetramycin A and B (Yamanaka et al., 2020), and toyocamycin (Liu B. et al., 2022). In our previous study, we sequenced the genome of S. albulus CICC11022 and subsequently predicted 37 SM-BGCs on its chromosome (Lian et al., 2022). These results indicate that the metabolism of S. albulus CICC11022 is complicated, and the potential of this strain to produce bioactive compounds needs to be further clarified.
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is a glycolytic enzyme that catalyze the conversion of glyceraldehyde 3-phosphate into 1,3-bisphosphoglycerate. This reaction is usually accompanied by the reduction of NAD+ to produce NADH. However, another type of GAPDH, GapN (EC 1.2.1.9), is found in nature. GapN is found in both photosynthetic organisms (Mateos and Serrano, 1992) and some Gram-positive bacteria, such as Streptococcus mutans (Boyd et al., 1995). GapN catalyzes the non-reversible oxidation of glyceraldehyde 3-phosphate to 3-phosphoglycerate along with NADPH formation. Since four molecules of NADPH are required for the biosynthesis of one molecule of L-lysine in Corynebacterium glutamicum, the expression of gapN can alleviate the dependence of cells on the pentose phosphate pathway and thus increase the production of L-lysine (Bommareddy et al., 2014; Takeno et al., 2016; Wu et al., 2019).
The biosynthesis of ε-PL in S. albulus involves the diaminopimelate pathway, as in C. glutamicum and uses L-lysine as a precursor (Lian et al., 2022). The impact of gapN expression on ε-PL production in S. albulus has yet to be determined, despite its favorable effect on L-lysine production in C. glutamicum. In the present study, gapN from Streptococcus mutans was heterologously expressed in S. albulus CICC11022. The effects of gapN expression on the metabolism of S. albulus CICC11022 were determined using transcriptomic and metabolomic approaches. The potential of S. albulus CICC11022 to synthesize various NPs was subsequently emphasized.
The strains, plasmids and primers used for strain construction are listed in Supplementary Table 1. The host bacterium and vector used to express gapN were S. albulus CICC11022 and pSET152, respectively (Wang et al., 2020). Spores of S. albulus strains were cultured on MS solid medium containing 20 g/L mannitol, 20 g/L soybean powder, and 20 g/L agar powder. The seed culture was prepared using M3G medium composed of 50 g/L glucose, 10 g/L (NH4)2SO4, 5 g/L yeast extract, 0.5 g/L MgSO4⋅7H2O, 0.8 g/L K2HPO4, 1.36 g/L KH2PO4, 0.03 g/L FeSO4⋅7H2O, and 0.04 g/L ZnSO4⋅7H2O at an initial pH of 6.8. The fermentation medium for S. albulus was the M3G medium supplemented with 5 g/L sodium citrate. Cell growth and SM production ability were compared by initially culturing S. albulus strains on the MS solid medium for 5–6 days at 30°C. Afterward, the spores were collected, inoculated into 50 mL of M3G medium, and cultured at 30°C and 220 rpm for 48 h. Finally, 5 mL of the seed culture was inoculated into 50 mL of the fermentation medium and cultured at 30°C and 220 rpm for 168 h. Cells of S. albulus Q-152 and Q-gapN were harvested after 48 h of fermentation for RNA sequencing, as was the quantification of NAD(P)H and ATP. Samples for metabolomics analysis were collected after 7 days of fermentation. E. coli strains were aerobically cultured at 37°C in Luria–Bertani (LB) medium, which contained 10 g/L tryptone, 5 g/L yeast extract, and 10 g/L sodium chloride. When needed, antibiotics were used at the following concentrations: 50–80 μg/mL apramycin, 25–50 μg/mL chloramphenicol, 40–50 μg/mL kanamycin, and 25 μg/mL nalidixic acid.
The SP43 promoter, the SR41 ribosome-binding site (RBS) (Bai et al., 2015), and the codon-optimized gapN gene (accession number: OR257568) from Streptococcus mutans GS-5 were chemically synthesized (Supplementary Figure 1). The primers gapN-F and gapN-R were subsequently used to amplify the SP43-SR40-gapN DNA fragment. SP43-SR40-gapN was digested using XbaI and BamHI, ligated to pSET152 and subsequently transformed into E. coli Trans5α. After screening and verification, the recombinant expression plasmid was named pSET152-gapN, which was subsequently transferred into E. coli ET12567/pUZ8002 and then into S. albulus CICC11022 via intergeneric conjugation in accordance with a previously reported method (Lian et al., 2022). The obtained gapN-expressing strain, S. albulus Q-gapN, harbored pSET152-gapN at the attB site of its chromosome. Moreover, a control strain, S. albulus Q-152, which harbors pSET152, was also constructed.
The total RNA of S. albulus Q-152 (control) and Q-gapN (gapN) was isolated after 48 h of fermentation. Three biological replicates were prepared for each group. An Illumina HiSeq 2000 platform (Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China) was used for RNA sequencing. The sequencing data were deposited in the NCBI Sequence Read Archive under accession numbers SRR20727629 (control) and SRR20727628 (gapN). The online platform Majorbio Cloud Platform1 was used for conducting Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses, facilitating functional annotation, classification, and enrichment analysis of the genes (Ren et al., 2022). Additionally, the genome sequence of S. albulus CICC11022 (BioProject accession number: PRJNA859656) served as a reference. Genes that exhibited twofold or greater changes [fold change (FC) ≥ 2 or ≤ -2; FDR < 0.05] between samples were defined as differentially expressed genes (DEGs). Functional enrichment analyses were performed as previously reported (Lian et al., 2022).
The gapN gene expression levels in S. albulus Q-gapN and Q-152 were compared via quantitative real-time PCR (qRT-PCR) using the primers RT-gapN-F and RT-gapN-R (Supplementary Table 3). qRT-PCR was also used to verify the RNA sequencing data. The genes and primers used are shown in Supplementary Table 2. All samples were taken after 48 h of fermentation. The PCR experiments were performed as reported previously (Lian et al., 2022).
The effect of gapN expression on the intracellular and extracellular metabolites of S. albulus was investigated using an untargeted metabolomic method. The samples were divided into four groups, representing two strains. The intracellular and extracellular metabolite samples of the gapN-expressing strain were labeled gapN and gapN_s, while the intracellular and extracellular metabolite samples of the control strain Q-152 were labeled control and control_s, respectively. Cells and cell culture supernatants were taken after 7 days of fermentation. Six biological replicates were prepared for each group. Liquid chromatography-mass spectrometry (LC-MS)-based metabolomic detection was conducted following a previously reported methodology (Lian et al., 2022). The LC-MS sample preparation procedure was conducted as follows: A precisely weighed 50 mg of cell or supernatant sample was placed into a 2 mL centrifuge tube, followed by the addition of a ground bead with a diameter of 6 mm and 400 μL of methanol and acetonitrile in equal proportions. Subsequently, the samples were subjected to grinding using a frozen tissue grinder for 6 min at −10°C and 50 Hz, followed by ultrasonic extraction for 30 min at 5°C and 40 kHz. Afterward, the samples were frozen at −20°C for 30 min and subsequently centrifuged at 13,000 × g and 4°C for 15 min. The next step involved redissolving the samples by adding 120 μL of solution composed of acetonitrile and water in equal proportions. The mixture was then vortexed for 30 s and subjected to an additional ultrasonic extraction for five more minutes. Following another round of centrifugation, the resulting supernatant was collected and utilized in LC-MS analysis. The UHPLC-Q Exactive HF-X system, equipped with a Q-Exactive quadrupole-Orbitrap mass spectrometer and a heated electrospray ionization (ESI) source (Thermo Fisher Scientific, Waltham, MA, USA), was employed as the analytical instrument for metabolic profiling analysis in both ESI-positive and ESI-negative ion modes. The column used was ACQUITY UPLC HSS T3 (100 mm × 2.1 mm i.d., 1.8 μm; Waters, Milford, USA). Mobile phase A consisted of 95% water and 5% acetonitrile (with 0.1% formic acid), while mobile phase B consisted of 47.5% acetonitrile, 47.5% isopropanol, and 5% water (containing 0.1% formic acid). The injection volume was 3 μL, and the column temperature was 40°C. Quality control samples were prepared by combining equal volumes of extracts from all samples. LC-MS raw data were acquired and processed using ProgenesisQI software (Waters Corporation, Milford, USA). Subsequently, the software was used to search for characteristic peaks and identify metabolites by matching MS and tandem mass spectra (MS/MS) information with the metabolic databases under an MS mass error threshold of less than 10 ppm. The databases utilized included the HMDB,2 the Metlin database,3 and a proprietary database developed by Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China. Differentially abundant metabolites with variable importance plot (VIP) values > 1 and P ≤ 0.05 were screened.
NADPH and NADH concentrations were measured using an NADP+/NADPH Assay Kit (Beyotime, China) and an NAD+/NADH Assay Kit (Beyotime, China), respectively, based on the WST-8 method (Chamchoy et al., 2019). The ATP concentration was determined using a firefly luciferase-based ATP Assay Kit (Beyotime, China) (Lundin, 2000). An Enhanced BCA Protein Assay Kit (Beyotime, China) was used to determine the protein concentrations of the cells used for NADPH, NADH, and ATP determination. The NADPH, NADH, and ATP concentrations per microgram of protein were subsequently calculated.
Cell growth and glucose concentration were measured by using a spectrophotometer and an SBA-40E biosensor analyzer, respectively (Wang et al., 2020). The concentration of ε-PL was measured as described previously (Lian et al., 2022). The concentrations of anisomycin were determined using a previously described method (Shen et al., 2019). GraphPad Prism 8.3 (GraphPad Software, USA) was used to plot the results and for statistical analysis.
Initially, a heterologous gapN gene expression strain was constructed. A schematic diagram of the expression vector pSET152-gapN is shown in Supplementary Figure 2A. The gapN expression levels in S. albulus Q-gapN and Q-152 were compared via qRT-PCR (Supplementary Figure 2B). These results indicate that the gapN gene heterologously expressed strain was successfully constructed. It was subsequently used in cofactor concentration detection, fermentation product analysis, and omics investigations.
The intracellular NADPH, NADH and ATP concentrations were subsequently determined to study the effect of heterologous gapN expression. As shown in Figure 1, the NADPH concentration in S. albulus Q-gapN was 1.74 ± 0.02 μM/μg protein, which was 28.0% greater than the 1.35 ± 0.05 μM/μg protein concentration in the control strain. Moreover, the NADH concentration in S. albulus Q-gapN was only 0.88 ± 0.13 μM/μg protein, which was 24.0% lower than the 1.34 ± 0.05 μM/μg protein concentration in the control strain. The total NADP+ (NADP+ + NAPDH) concentrations in S. albulus Q-gapN and Q-152 were 5.22 ± 0.16 and 3.73 ± 0.22 μM/μg protein, respectively. Moreover, the total NAD+ (NAD+ + NADH) concentrations in S. albulus Q-gapN and Q-152 were 5.36 ± 0.03 and 8.48 ± 0.41 μM/μg protein, respectively. These results suggest that the heterologous expression of gapN in S. albulus indeed perturbs the intracellular concentration balance between NADPH and NADH. We further examined the effect of this perturbation on the intracellular ATP content. The ATP concentration in S. albulus Q-gapN was 2.98 ± 0.63 μM/μg protein, which was 52.1% lower than the 6.22 ± 1.32 μM/μg protein concentration in the control strain (Figure 1).
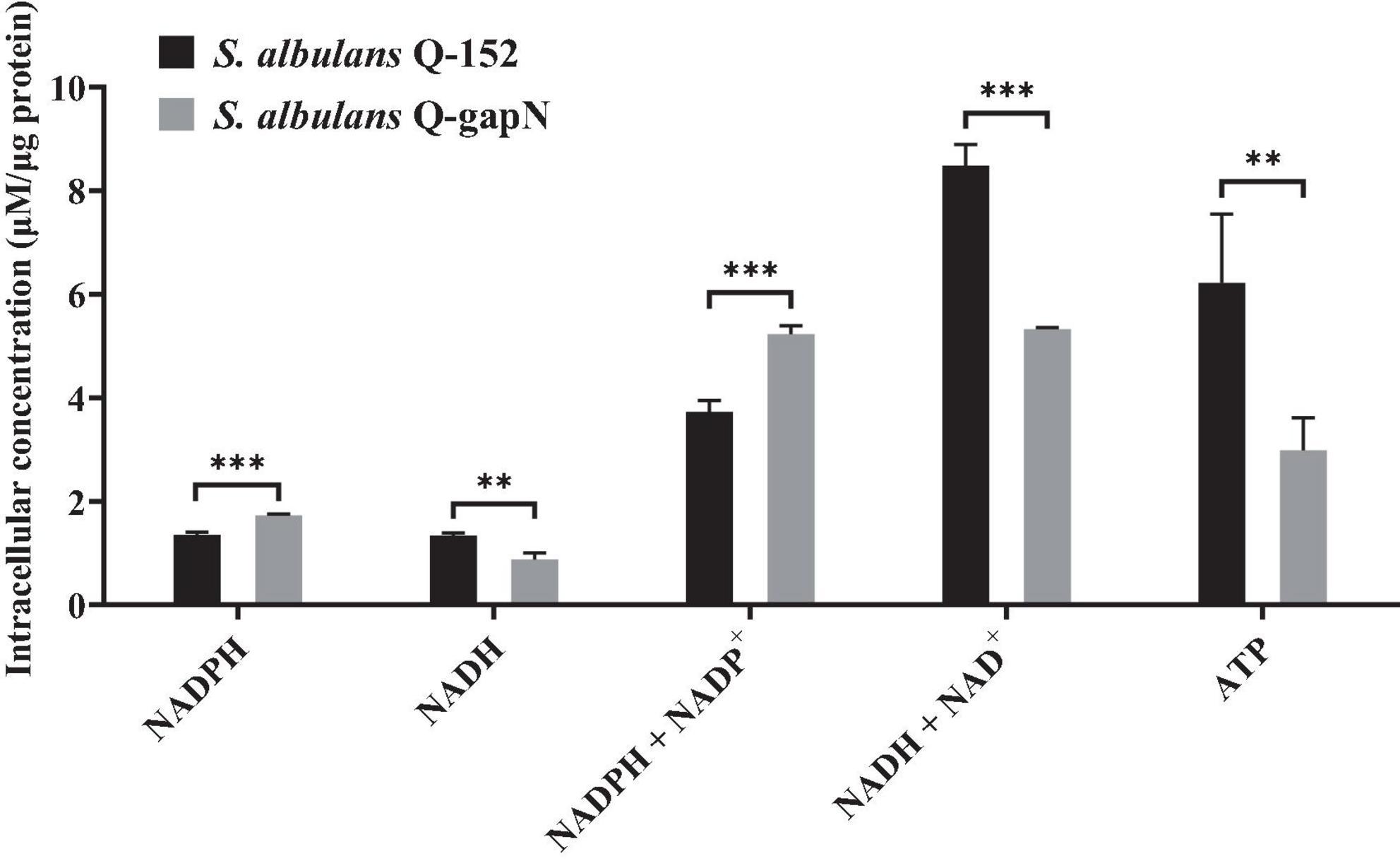
Figure 1. Effect of gapN expression on intracellular NADPH, NADH and ATP concentrations. **P < 0.01; ***P < 0.001.
The cell growth, residual glucose, and ε-PL production of S. albulus Q-gapN and Q-152 were subsequently compared; the results are shown in Figure 2. For the first 24 h, the cell growth and ε-PL production of both strains were similar. Afterward, significant differences were observed in the growth and production of the two strains. By 72 h, the maximum OD values of S. albulus Q-gapN and Q-152 were 13.13 and 17.33, respectively. The ε-PL concentrations produced by the Q-gapN and Q-152 strains were 0.35 and 1.23 g/L, respectively. Although the final ε-PL yields were quite different, the glucose consumption of the two strains was very similar. These results indicate that the heterologous expression of gapN resulted in slower strain growth and reduced ε-PL production.
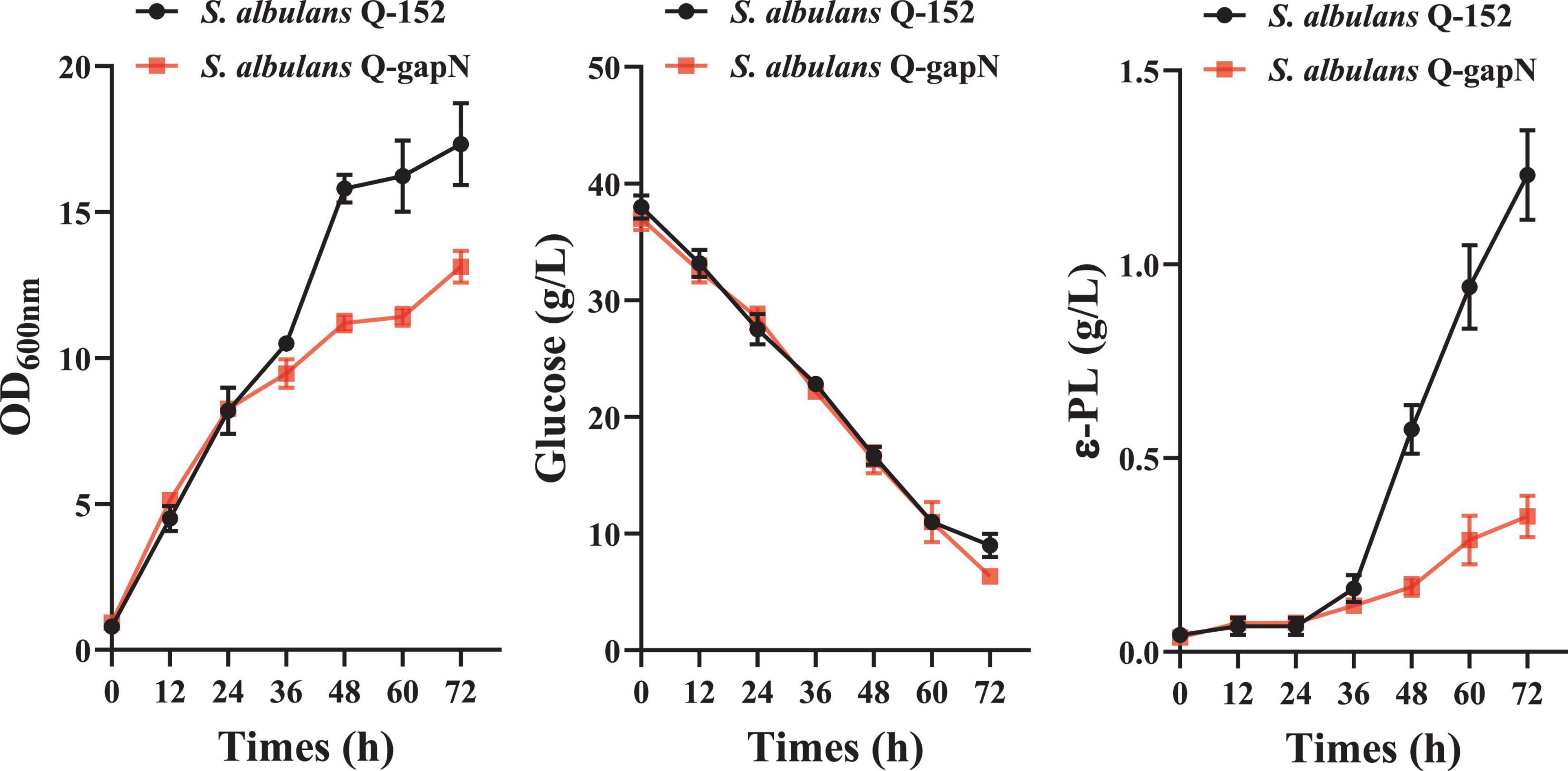
Figure 2. Effects of heterologous gapN expression on cell growth, glucose consumption, and ε-PL production.
A volcano plot of the significant DEGs between S. albulus Q-gapN (gapN) and Q-152 (control) is shown in Supplementary Figure 3A. The heterologous expression of gapN in S. albulus CICC11022 resulted in 2,935 DEGs, of which 1,318 and 1,617 genes were significantly up- and downregulated, respectively. Moreover, 76 and 208 sRNAs were found to be significantly up- and downregulated, respectively, by gapN expression. The accuracy of the transcriptome results was verified by qRT-PCR (Supplementary Table 3). The distribution of DEGs across various KEGG pathways (Ren et al., 2022) is presented in Figure 3A. Amino acid metabolism was the primary KEGG pathway associated with the highest number of DEGs, followed by carbohydrate metabolism. Noteworthy DEGs within the KEGG pathway are listed in Figure 3B, while additional information can be found in Supplementary Table 4. KEGG enrichment analyses of all the DEGs revealed that the most significantly regulated pathways in the gapN expressing strain were “steroid degradation” (KEGG: ko00984), “beta-alanine metabolism” (KEGG: ko00410), “beta-lactam resistance” (KEGG: ko01501), “C5-branched dibasic acid metabolism” (KEGG: ko00660), and “xylene degradation” (KEGG: ko00622) (P < 0.05) (Supplementary Figure 3B). GO enrichment analyses of all the DEGs revealed that the most significantly regulated GO terms in the gapN expressing strain were “iron ion binding” (GO:0005506), “monooxygenase activity” (GO:0004497), “heme binding” (GO:0020037), “tetrapyrrole binding” (GO:0046906), and “modified amino acid binding” (GO:0072341), among others (FDR < 0.001) (Supplementary Figure 3C).
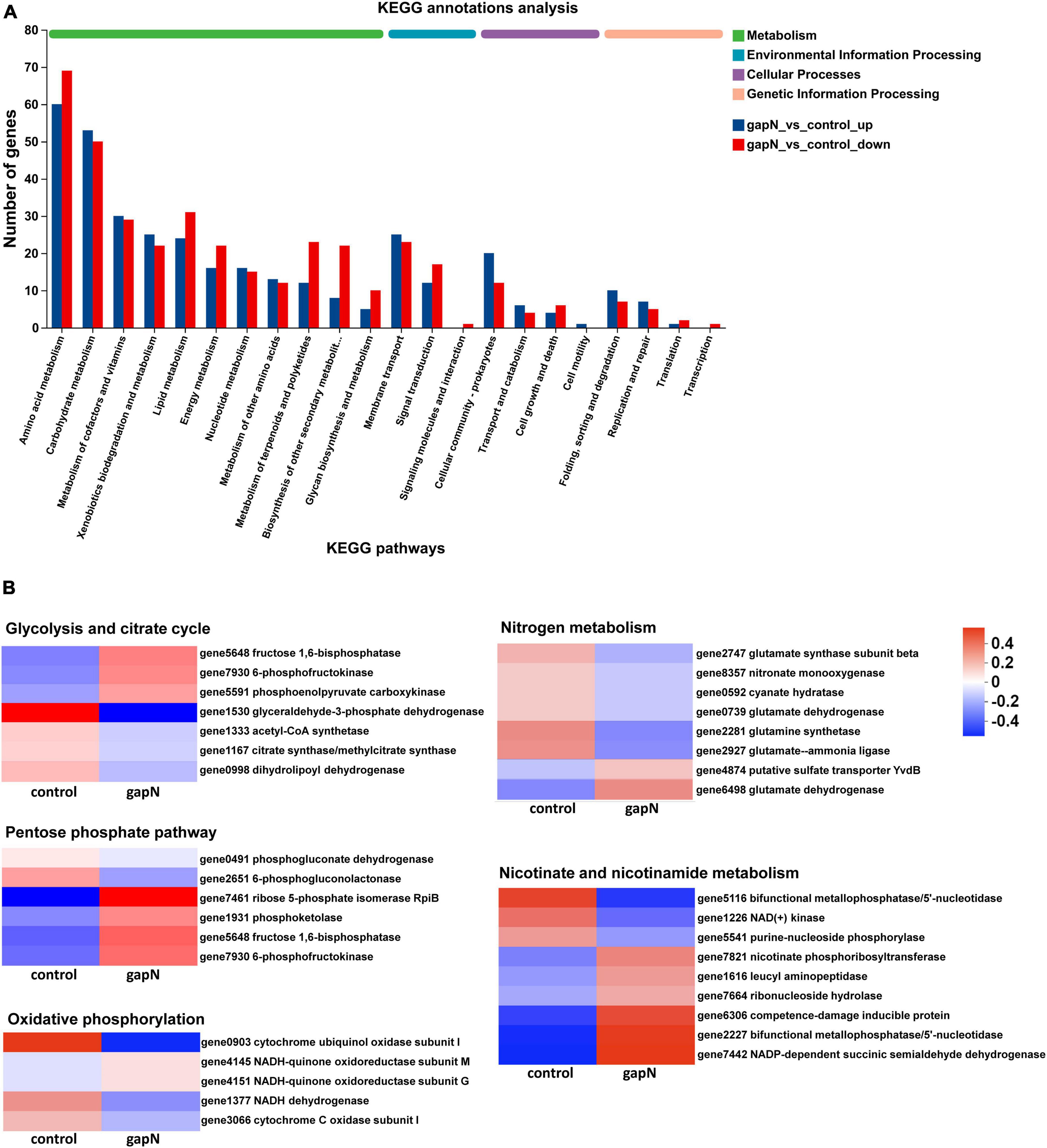
Figure 3. KEGG annotation analysis of the DEGs. (A) Classification of DEGs with KEGG annotation results. (B) Noteworthy DEGs within specific KEGG pathways.
Liquid chromatography-mass spectrometry-based metabolomic approaches were used to study the effects of gapN expression on the intracellular and extracellular metabolites of S. albulus. A total of 10,494 positive ion peaks and 8,700 negative ion peaks were detected in all four groups. Partial least squares-discriminant analysis (PLS-DA) and Venn plot analysis demonstrated global metabolic changes in the four groups (Supplementary Figures 4A–D).
After data preprocessing and comparative analysis, 970 differential intracellular metabolites were identified between gapN and the control (Figures 4A, B), whereas 1,149 differential extracellular metabolites were identified between gapN_s and control_s (Figures 4A, C). KEGG enrichment analysis based on the differential intracellular metabolites revealed 12 enriched pathways (corrected p value < 0.05), and the top 5 pathways were “tryptophan metabolism” (map00380), “biosynthesis of plant secondary metabolites” (map01060), “purine metabolism” (map00230), “phenylpropanoid biosynthesis” (map00940), and “biosynthesis of alkaloids derived from histidine and purine” (map01065) (Figure 4D). KEGG enrichment analysis based on the differential extracellular metabolites revealed 9 enriched pathways (corrected p value < 0.05), and the top 5 pathways were “biosynthesis of alkaloids derived from histidine and purine” (map01065), “purine metabolism” (map00230), “tryptophan metabolism” (map00380), “biosynthesis of plant secondary metabolites” (map01060) and “steroid hormone biosynthesis” (map00140) (Figure 4E). The pathways that which were enriched both intracellularly and extracellularly included “biosynthesis of plant secondary metabolites,” “tryptophan metabolism,” “purine metabolism,” “biosynthesis of alkaloids derived from histidine and purine,” and “beta-alanine metabolism.”
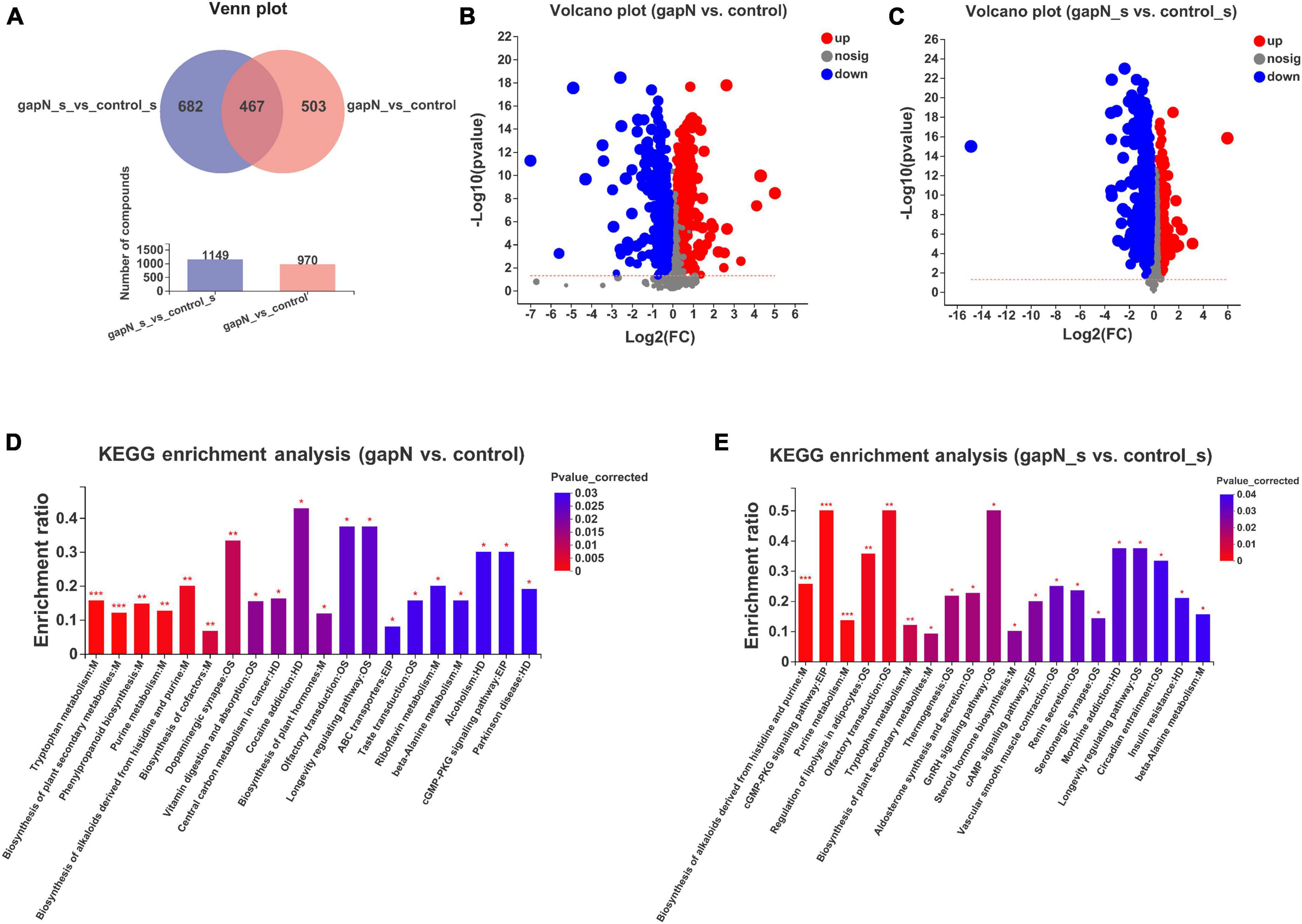
Figure 4. Intracellular and extracellular metabolomic comparisons of the gapN-expressing strain and the control strain. (A) Venn plot of the differential intracellular and extracellular metabolites. (B) Volcano plot of the differential intracellular metabolites. (C) Volcano plot of the differential extracellular metabolites. (D) KEGG enrichment analysis based on intracellular metabolites. (E) KEGG enrichment analysis based on extracellular metabolites. *P < 0.05; **P < 0.01; ***P < 0.001.
Transcriptomics analysis was primarily employed to elucidate the impact of gapN expression on ε-PL metabolism; hence, samples were chosen at the 48-h mark. Metabolomics, on the other hand, was primarily utilized to discern alterations in SMs of S. albulus influenced by gapN expression; thus, samples were selected after 7 days. Despite the disparate sampling timeframes, certain correlations were observed between the data generated in these two experiments. The network diagrams of differential intracellular metabolites and differential genes identified via KEGG enrichment analysis are presented in Supplementary Figures 5A, B, respectively. The analysis revealed enrichment of multiple metabolic pathways in both the transcriptomic and metabolomic data. Notably, “beta-alanine metabolism” metabolic pathway exhibited enrichment in both studies, indicating the influence of gapN expression on the metabolism and biosynthesis of amino acids. Given the significance of amino acids as crucial precursors for SM biosynthesis, several metabolic pathways associated with SM biosynthesis were enriched in the metabolomic studies. The metabolic perturbation of S. albulus due to gapN expression was visualized using the interactive Pathways Explorer v3 (iPath 3).4 This visualization was based on KEGG annotations of the transcriptome and intracellular metabolism results, as shown in Supplementary Figure 5C.
The expression of gapN in S. albulus resulted in decreased ε-PL production. The effects of gapN expression on the ε-PL biosynthesis pathway were then further analyzed via transcriptomic and metabolomic analyses. As shown in Figure 5, the expression of phosphoenolpyruvate carboxylase (ppc) and phosphoenolpyruvate carboxykinase (pckA) was significantly downregulated and upregulated, respectively, by gapN expression. These results indicate that the conversion equilibrium between oxaloacetate (OAA) and phosphoenolpyruvate was disrupted, and the latter was favored. Isocitrate lyase (aceA) and malate synthase (aceB) were significantly downregulated, indicating that the glyoxylate shunt was negatively affected by gapN expression. Interestingly, the intracellular concentration of L-aspartate in the gapN-expressing strain greatly increased. Moreover, the expression levels of genes encoding the L-ectoine and hydroxyectoine synthesis pathway (ectA, ectB, ectC, and ectD) and the gene encoding pyruvate oxidase (poxB) were significantly upregulated by gapN expression. These two pathways serve as competitive metabolic pathways for ε-PL biosynthesis. L-Aspartate 4-semialdehyde plays a crucial role as an intermediate metabolite in both the biosynthetic pathways of L-lysine and L-ectoine, as depicted in Figure 5. These two metabolic pathways compete for the utilization of this specific compound. Furthermore, there was a significant increase in the extracellular concentration of L-ectoine (Figure 6B), confirming the competitive relationship between the two pathways. Pyruvate oxidase catalyzes the conversion of pyruvate to acetate, leading to a limited availability of acetyl-CoA. The insufficiency of acetyl-CoA may impact the availability of oxaloacetate, a crucial initial metabolite for L-lysine biosynthesis. Additionally, there was a significant downregulation of 6-phosphogluconolactonase (pgl), leading to a reduced intracellular accumulation of D-ribose-5P. This result may be due to the reduced dependence of the strain on the pentose phosphate pathway as a result of the increased intracellular NADPH concentrations. Moreover, 2,3,4,5-tetrahydropyridine-2,6-dicarboxylate N-succinyltransferase (dapD) and diaminopimelate decarboxylase (lysA) were downregulated, whereas the ε-PL degrading enzyme (pld) was upregulated.
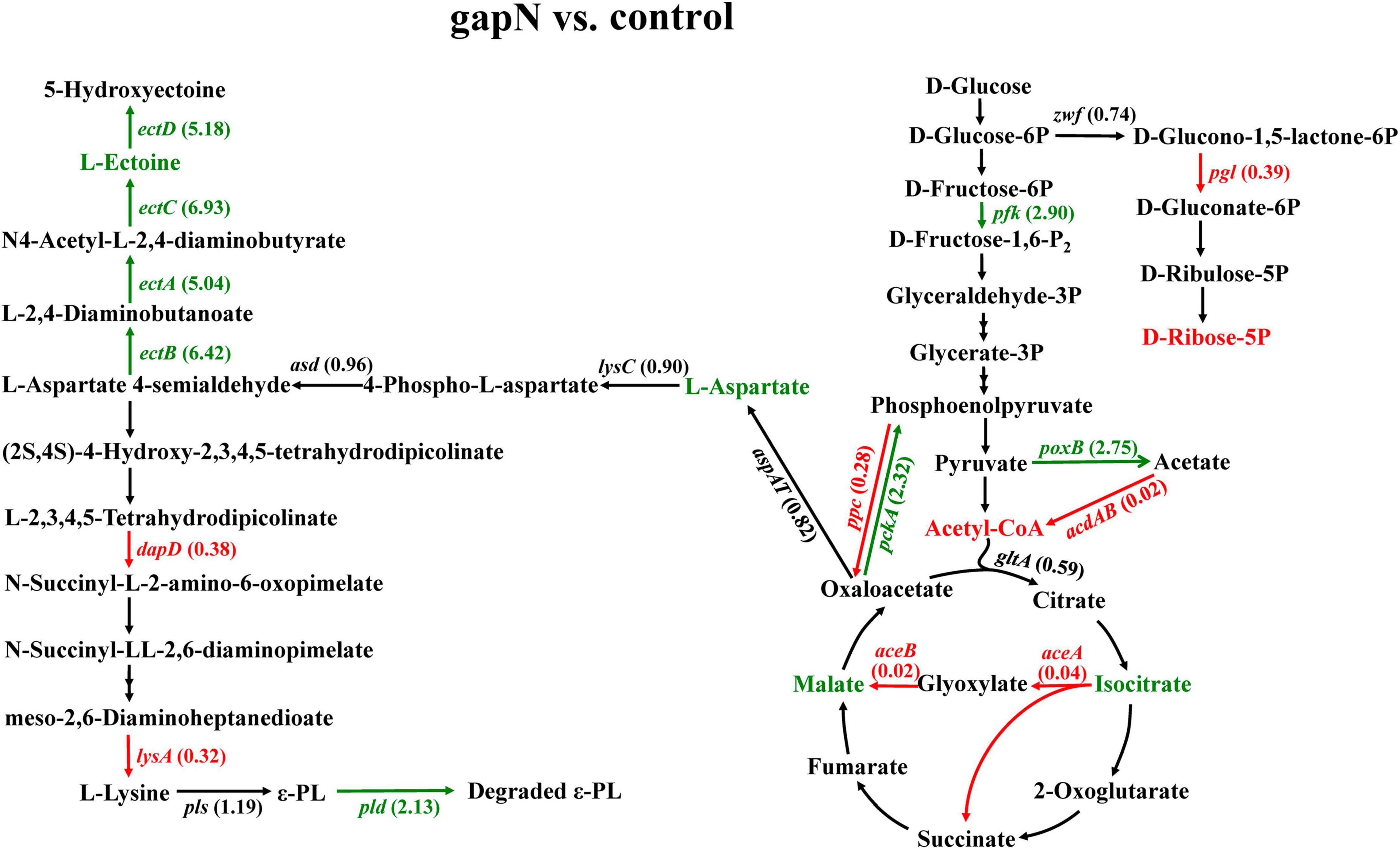
Figure 5. Effects of gapN expression on the ε-PL biosynthesis pathway. The green lines, arrows, and gene names represent genes whose expression was significantly upregulated at each step, whereas the red lines, arrows and gene names represent genes whose expression was significantly downregulated. The numbers that follow the gene names are the FCs in gene expression. The green and red metabolite names correspond to the upregulated and downregulated metabolites detected by metabolomics, respectively. zwf, glucose-6-phosphate dehydrogenase; pgl, 6-phosphogluconolactonase; pfk, 6-phosphofructokinase; ppc, phosphoenolpyruvate carboxylase; pckA, phosphoenolpyruvate carboxykinase; poxB, pyruvate oxidase; acdAB, acetate-CoA ligase (ADP-forming); gltA, citrate synthase; aceA, isocitrate lyase; aceB, malate synthase A; lysC, aspartate kinase; asd, aspartate-semialdehyde dehydrogenase; ectA, L-2,4-diaminobutyrate acetyltransferase; ectB, diaminobutyrate-2-oxoglutarate transaminase; ectC, L-ectoine synthase; ectD, ectoine hydroxylase; dapA, 4-hydroxytetrahydrodipicolinate synthase; dapD, 2,3,4,5-tetrahydropyridine-2,6-dicarboxylate N-succinyltransferase; argD, acetylornithine aminotransferase; lysA, diaminopimelate decarboxylase; pls, polylysine synthetase; and pld, ε-PL degrading enzyme.
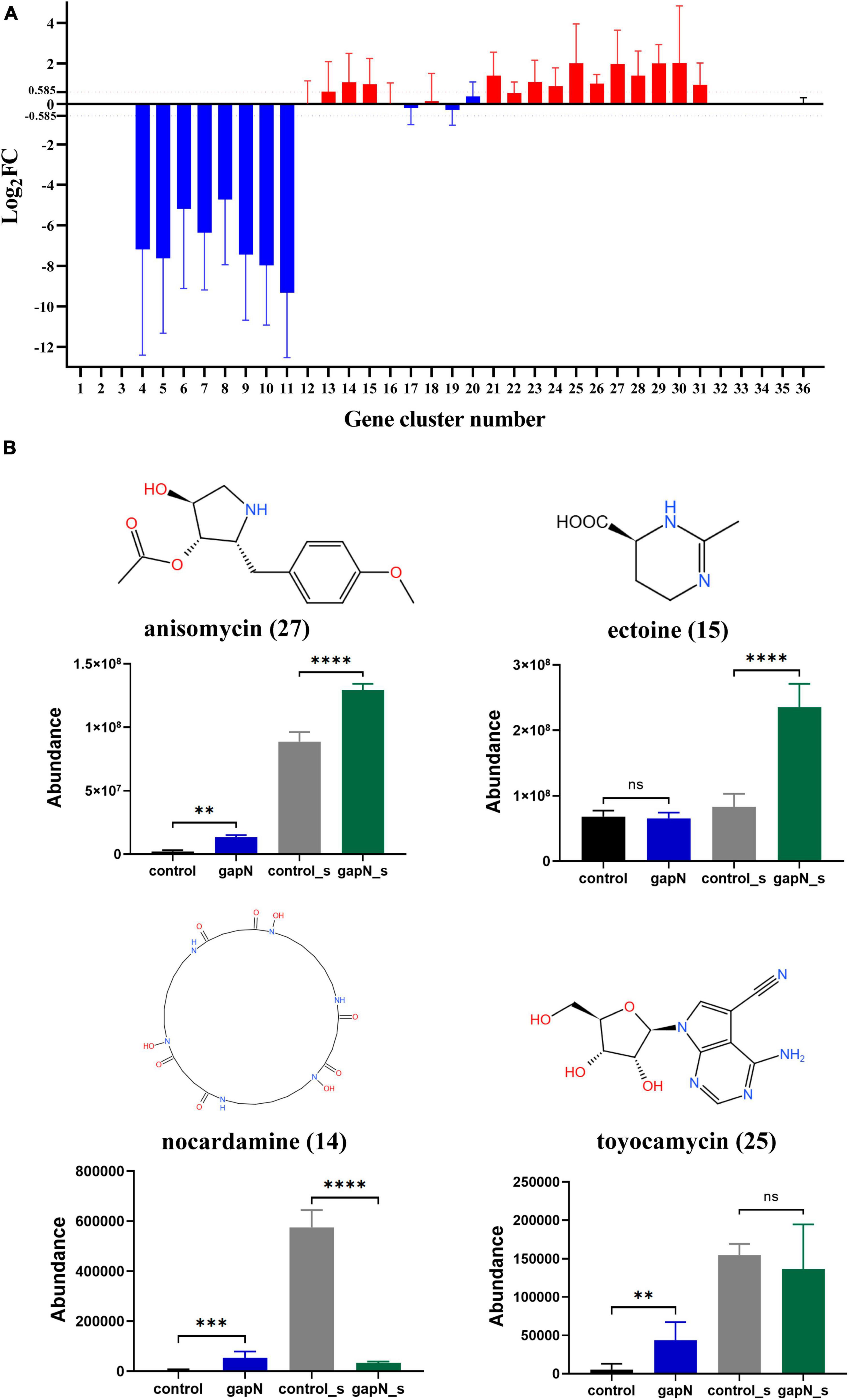
Figure 6. Effects of gapN expression on SM-BGC expression and SM biosynthesis in S. albulus CICC11022. (A) Regulation of the 36 SM-BGCs by gapN expression. The numbers on the x-axis correspond to the SM-BGC numbers. (B) The predicted secondary metabolites of the SM-BGCs that were identified in the metabolomic data. The numbers in parentheses after the metabolite names are the SM-BGC numbers. **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
To summarize, the alterations in gene expression levels and metabolites resulting from gapN expression suggest a potential correlation between the decline in ε-PL production in S. albulus and a decrease in intracellular L-lysine availability, an increase in ε-PL degradation, and, notably, a reduction in the intracellular ATP concentration.
A total of 36 SM-BGCs in the S. albulus CICC11022 genome were predicted by antiSMASH 7.1.0. These SM-BGCs were numbered 1 to 36 according to their location (Supplementary Figure 6; Supplementary Table 5). To determine the effect of gapN expression on each SM-BGC, the average Log2FC values for all genes in each gene cluster were calculated. SM-BGCs with an average | log2FC| > 0.585 (FC > 1.5) were considered to be significantly affected by gapN expression. As shown in Figure 6A, SM-BGCs 1–3 and 32–36 were almost completely silent with or without gapN expression, indicating that they cannot be activated by gapN expression. Heterologous gapN expression resulted in the almost complete silencing of SM-BGCs 4–11. SM-BGCs 13–15, 21, and 23–31 were significantly upregulated by gapN expression. In summary, a total of 28 SM-BGCs were affected by gapN expression, 13 and 8 of which were significantly up- and downregulated, respectively.
The metabolomic data for the predicted SMs of the 21 SM-BGCs affected by gapN expression were then carefully checked. Four metabolites were identified, namely, anisomycin, ectoine, nocardamine, and toyocamycin. The intracellular amounts of anisomycin, toyocamycin, and nocardamine and the extracellular amounts of ectoine in the gapN-expressing strain were significantly greater than those in the control strain (Figure 6B). These results are consistent with the changes in the expression levels of the corresponding gene clusters (Figure 6A).
To determine the metabolites that were genuinely influenced by gapN expression, we further identified 21 metabolites that exhibited upregulation both intracellularly and extracellularly (Supplementary Table 6). The expression profiles and VIP values of these metabolites are illustrated in Figure 7. Notably, among the metabolites displaying the two highest VIP values both intracellularly and extracellularly was 6-aminopenicillanic acid.
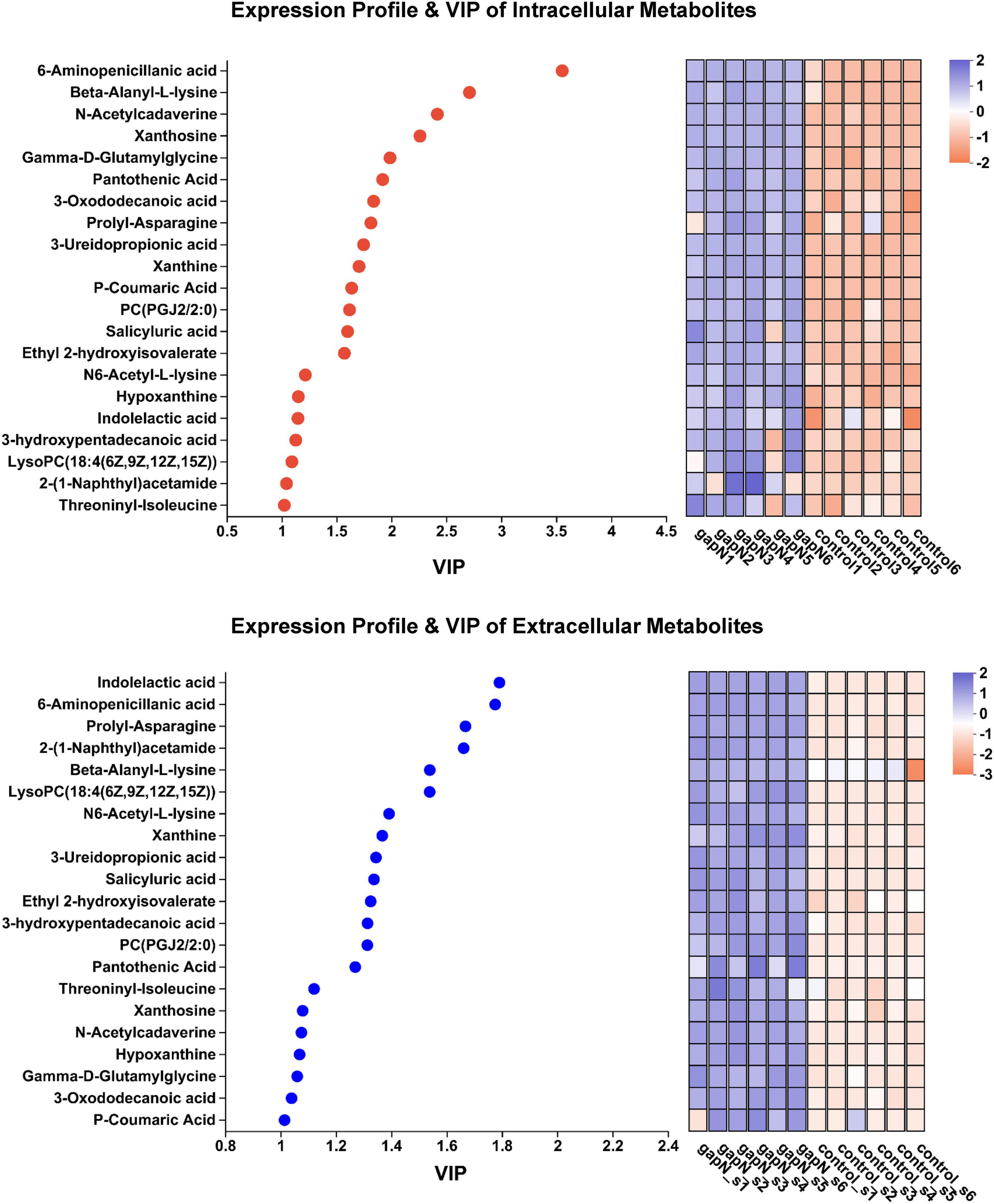
Figure 7. Expression profiles and VIP analysis of the metabolites that were upregulated both intracellularly and extracellularly.
In the present study, the expression of gapN was found to have a detrimental effect on the production of ε-PL. One notable distinction between the production of L-lysine and ε-PL is the requirement of ATP for polymerization. The findings of this study indicate that gapN expression leads to an increase in the intracellular NADPH concentration and a decrease in the NADH concentration, consequently resulting in a decrease in the intracellular ATP concentration. These alterations in the balance of intracellular cofactors ultimately lead to metabolic shunts in the biosynthesis pathway of ε-PL. The reliance of cells on the pentose phosphate pathway was significantly decreased due to the downregulation of the 6-phosphogluconolactonase gene and the decrease in D-ribose-5P levels in the gapN expressing strain (Figure 5). The activation of the L-ectoine biosynthetic pathway was observed as a consequence of gapN expression, leading to the upregulation of the ectABCD gene cluster (Figure 5) and subsequent augmentation of L-ectoine synthesis (Figure 6B). In our previous investigation, it was discovered that the activation of the L-ectoine biosynthetic pathway occurs upon deletion of the gene encoding polylysine synthetase (Lian et al., 2022). Consequently, the L-ectoine biosynthetic pathway serves as a competitive metabolic bypass for the ε-PL biosynthetic pathway in S. albulus CICC11022. These findings suggest that while gapN expression can mitigate the reliance of the L-lysine synthesis pathway on the pentose phosphate pathway, the substantial reduction in ATP supply resulting from gapN expression significantly hampers ε-PL production.
The heterologous expression of gapN had an unexpected and interesting positive impact on the biosynthesis of bioactive SMs in S. albulus. A total of 21 SM-BGCs were significantly affected by gapN expression, including three gene clusters associated with antibiotic production, namely, anisomycin, nocardamine, and toyocamycin (Supplementary Table 5; Supplementary Figure 6). Anisomycin is a pyrrolidine alkaloid antibiotic that has notable antifungal, antigenic, antiviral, and antitumor properties (Ye et al., 2019; Quintana et al., 2020). Nocardamine, is a cyclic hydroxamic acid siderophore that has been shown to demonstrate antitumor and antimalarial effects (Mahmud et al., 2022). Finally, toyocamycin, is an adenosine analog that has antitumor and antifungal effects (Pandey et al., 2022; Song et al., 2022). The aforementioned three antibiotics were also identified through metabolomics analysis, and their intracellular levels were positively influenced by gapN expression (Figure 6B). Additionally, after 7 days, the concentrations of anisomycin in the fermentation broth of both the gapN-expressing strain and the control strain were found to be 155.66 mg/L and 86.24 mg/L, respectively. These findings are consistent with the expected alterations in metabolome profiles. Given the well-established BGCs associated with these antibiotics, it is anticipated that overexpression of their BGCs will further enhance their production. Additionally, 21 metabolites were increased in the gapN-expressing strain both intracellularly and extracellularly (Supplementary Table 6). The application of VIP analysis demonstrated that 6-aminopenicillanic acid was among the top two metabolites upregulated (Figure 7). 6-Aminopenicillanic acid, the core β-lactam compound of penicillins, is an important active pharmaceutical intermediate that can be used as the main starting block for the preparation of numerous semisynthetic penicillins (Sawant et al., 2020). With respect to the genome of S. albulus CICC11022, we identified a total of 31 penicillin-related genes, including two genes (gene 2388 and gene 3892) encoding penicillin acylases. These finding suggest the potential involvement of these genes in the biosynthesis pathway of 6-aminopenicillanic acid.
In summary, the expression of gapN in S. albulus significantly increased SM synthesis. This phenomenon could be attributed to an ATP deficit, which is known to trigger the activation of oxidative metabolism to reestablish the energetic balance. This activation is essential for ATP generation concomitant with oxidative stress (Esnault et al., 2017; Lejeune et al., 2022; Apel et al., 2023), and potential detrimental effects should be mitigated by various processes. These processes include the production of specific metabolites with antioxidant properties (Virolle, 2020). Research in the literature has indicated that pantothenic acid (Revuelta et al., 2016), p-coumaric acid (Boz, 2015; Boo, 2019), 3-ureidopropionic acid (Noguer et al., 2014) and indolelactic acid (Negatu et al., 2020) indeed exhibit antioxidant properties. Furthermore, the downregulation of poxB is expected to lead to a decrease in acetyl-CoA generation and thus to reduced feeding and activity in the TCA cycle. Similarly, the downregulation of ppc and upregulation of pckA are predicted to lead to a depletion of oxaloacetate from the TCA cycle. Both processes are thought to contribute to the reduction in the activity of the TCA cycle and thus to limit the generation of NADH, whose reoxidation by the respiratory chain is the origin of the generation of ROS/NOS responsible for oxidative stress. Since our present findings point to a significant concern regarding oxidative stress in the gapN-expressing strain, we examined the expression of 12 genes classified as involved in the oxidative stress response. Among them, 4 exhibited upregulated expression in the gapN-expressing strain (Supplementary Figure 7), including the redox-sensitive transcriptional activator SoxR, a peptide-methionine (R)-S-oxide reductase and two catalases, whereas two genes encoding catalase-peroxidase and hydroperoxide resistance protein were significantly downregulated in the gapN-expressing strain.
This study provided a comprehensive view of the metabolic perturbations in S. albulus induced by heterologous expression of gapN. Moreover, this approach unexpectedly led to the upregulation of the biosynthesis of valuable bioactive SMs. Consequently, this research can serve as a valuable reference for the utilization of genetic engineering techniques to exploit the high metabolic richness of the numerous Streptomyces species present on earth for the discovery of novel bioactive SMs whose biosynthesis is directed by the cryptic pathways present in the genomes of these actinobacteria.
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/, SRR20727629 (control) and SRR20727628 (gapN).
JM: Formal analysis, Investigation, Methodology, Writing—original draft. MZ: Investigation, Methodology, Writing—original draft. WD: Investigation, Writing—original draft. CF: Methodology, Writing—original draft. ZW: Investigation, Writing—original draft. XW: Resources, Visualization, Writing—review and editing. QY: Funding acquisition, Supervision, Writing—review and editing. LK: Funding acquisition, Supervision, Writing—review and editing. JQ: Conceptualization, Writing—review and editing, Funding acquisition, Resources, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (32370094), the Natural Science Foundation of Shandong Province (ZR2021MC134 and ZR2022QC004), and the Shandong Provincial Youth Innovation Team Education Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1328321/full#supplementary-material
Al-shaibani, M. M., Radin Mohamed, R. M. S., Sidik, N. M., Enshasy, H. A. E., Al-Gheethi, A., Noman, E., et al. (2021). Biodiversity of secondary metabolites compounds isolated from phylum Actinobacteria and its therapeutic applications. Molecules 26:4504. doi: 10.3390/molecules26154504
Apel, C., Levasseur, M., Lejeune, C., Korch, S. B., Guérard, F., David, M., et al. (2023). Metabolic adjustments in response to ATP spilling by the small DX protein in a strain. Front. Cell Dev. Biol. 11:1129009. doi: 10.3389/fcell.2023.1129009
Bai, C., Zhang, Y., Zhao, X., Hu, Y., Xiang, S., Miao, J., et al. (2015). Exploiting a precise design of universal synthetic modular regulatory elements to unlock the microbial natural products in Streptomyces. Proc. Natl. Acad. Sci. U.S.A. 112, 12181–12186. doi: 10.1073/pnas.151102711
Beck, C., Blin, K., Gren, T., Jiang, X., Mohite, O. S., Palazzotto, E., et al. (2021). “Metabolic engineering of filamentous Actinomycetes,” in Metabolic engineering, eds J. Nielsen, G. Stephanopoulos, and S. Lee (Hoboken, NJ: Wiley), 653–688. doi: 10.1002/9783527823468.ch17
Belknap, K. C., Park, C. J., Barth, B. M., and Andam, C. P. (2020). Genome mining of biosynthetic and chemotherapeutic gene clusters in Streptomyces bacteria. Sci. Rep. 10:2003. doi: 10.1038/s41598-020-58904-9
Bommareddy, R. R., Chen, Z., Rappert, S., and Zeng, A.-P. (2014). A de novo NADPH generation pathway for improving lysine production of Corynebacterium glutamicum by rational design of the coenzyme specificity of glyceraldehyde 3-phosphate dehydrogenase. Metab. Eng. 25, 30–37. doi: 10.1016/j.ymben.2014.06.005
Boo, Y. C. (2019). p-Coumaric acid as an active ingredient in cosmetics: A review focusing on its antimelanogenic effects. Antioxidants 8:275. doi: 10.3390/antiox8080275
Boyd, D. A., Cvitkovitch, D. G., and Hamilton, I. R. (1995). Sequence, expression, and function of the gene for the nonphosphorylating, NADP-dependent glyceraldehyde-3-phosphate dehydrogenase of Streptococcus mutans. J. Bacteriol. 177, 2622–2627. doi: 10.1128/jb.177.10.2622-2627.1995
Boz, H. (2015). p-Coumaric acid in cereals: Presence, antioxidant and antimicrobial effects. Int. J. Food Sci. Technol. 50, 2323–2328. doi: 10.1111/ijfs.12898
Chamchoy, K., Pakotiprapha, D., Pumirat, P., Leartsakulpanich, U., and Boonyuen, U. (2019). Application of WST-8 based colorimetric NAD(P)H detection for quantitative dehydrogenase assays. BMC Biochemy. 20:4. doi: 10.1186/s12858-019-0108-1
Donald, L., Pipite, A., Subramani, R., Owen, J., Keyzers, R. A., and Taufa, T. (2022). Streptomyces: Still the biggest producer of new natural secondary metabolites, a current perspective. Microbiol. Res. 13, 418–465. doi: 10.3390/microbiolres13030031
Esnault, C., Dulermo, T., Smirnov, A., Askora, A., David, M., Deniset-Besseau, A., et al. (2017). Strong antibiotic production is correlated with highly active oxidative metabolism in Streptomyces coelicolor M145. Sci. Rep. 7:200. doi: 10.1038/s41598-017-00259-9
Jose, P. A., Maharshi, A., and Jha, B. (2021). Actinobacteria in natural products research: Progress and prospects. Microbiol. Res. 246:126708. doi: 10.1016/j.micres.2021.126708
Kang, H.-S., and Kim, E.-S. (2021). Recent advances in heterologous expression of natural product biosynthetic gene clusters in Streptomyces hosts. Curr. Opin. Biotechnol. 69, 118–127. doi: 10.1016/j.copbio.2020.12.016
Kong, D., Wang, X., Nie, J., and Niu, G. (2019). Regulation of antibiotic production by signaling molecules in Streptomyces. Front. Microbiol. 10:2927. doi: 10.3389/fmicb.2019.02927
Lee, N., Hwang, S., Kim, J., Cho, S., Palsson, B., and Cho, B.-K. (2020). Mini review: Genome mining approaches for the identification of secondary metabolite biosynthetic gene clusters in Streptomyces. Comput. Struct. Biotechnol. J. 18, 1548–1556. doi: 10.1016/j.csbj.2020.06.024
Lejeune, C., Sago, L., Cornu, D., Redeker, V., and Virolle, M.-J. (2022). A proteomic analysis indicates that oxidative stress is the common feature triggering antibiotic production in Streptomyces coelicolor and in the pptA mutant of Streptomyces lividans. Front. Microbiol. 12:813993. doi: 10.3389/fmicb.2021.813993
Li, D., Tian, Y., Liu, X., Wang, W., Li, Y., Tan, H., et al. (2021). Reconstitution of a mini-gene cluster combined with ribosome engineering led to effective enhancement of salinomycin production in Streptomyces albus. Microb. Biotechnol. 14, 2356–2368. doi: 10.1111/1751-7915.13686
Li, H., Gao, W., Cui, Y., Pan, Y., and Liu, G. (2022). Remarkable enhancement of bleomycin production through precise amplification of its biosynthetic gene cluster in Streptomyces verticillus. Sci. China Life Sci. 65, 1248–1256. doi: 10.1007/s11427-021-1998-8
Li, S., Mao, Y., Zhang, L., Wang, M., Meng, J., Liu, X., et al. (2022). Recent advances in microbial ε-poly-L-lysine fermentation and its diverse applications. Biotechnol. Biofuels Bioprod. 15:65. doi: 10.1186/s13068-022-02166-2
Li, Z.-Y., Bu, Q.-T., Wang, J., Liu, Y., Chen, X.-A., Mao, X.-M., et al. (2019). Activation of anthrachamycin biosynthesis in Streptomyces chattanoogensis L10 by site-directed mutagenesis of rpoB. J. Zhejiang Univ. Sci. B 20, 983–994. doi: 10.1631/jzus.B1900344
Lian, C., Zhang, M., Mao, J., Liu, Y., Wang, X., Kong, L., et al. (2022). Transcriptomic and metabolomic analyses for providing insights into the influence of polylysine synthetase on the metabolism of Streptomyces albulus. Microb. Cell Fact. 21:224. doi: 10.1186/s12934-022-01953-8
Liu, B., Wei, Q., Yang, M., Shi, L., Zhang, K., and Ge, B. (2022). Effect of toyF on wuyiencin and toyocamycin production by Streptomyces albulus CK-15. World J. Microbiol. Biotechnol. 38:65. doi: 10.1007/s11274-022-03234-3
Liu, X.-F., Wang, J.-X., Chen, X.-A., Liu, Y., and Li, Y.-Q. (2022). Activation and characterization of lanthomicins A–C by promoter engineering in Streptomyces chattanoogensis L10. Front. Microbiol. 13:902990. doi: 10.3389/fmicb.2022.902990
Liu, Z., Zhao, Y., Huang, C., and Luo, Y. (2021). Recent advances in silent gene cluster activation in Streptomyces. Front. Bioeng. Biotechnol. 9:632230. doi: 10.3389/fbioe.2021.632230
Lundin, A. (2000). Use of firefly luciferase in atp-related assays of biomass, enzymes, and metabolites. Methods Enzymol. 305, 346–370. doi: 10.1016/S0076-6879(00)05499-9
Mahmud, F., Lai, N. S., How, S. E., Gansau, J. A., Mustaffa, K. M. F., Leow, C. H., et al. (2022). Bioactivities and mode of actions of dibutyl phthalates and nocardamine from Streptomyces sp. H11809. Molecules 27:2292. doi: 10.3390/molecules27072292
Martínez-Burgo, Y., Santos-Aberturas, J., Rodríguez-García, A., Barreales, E. G., Tormo, J. R., Truman, A. W., et al. (2019). Activation of secondary metabolite gene clusters in Streptomyces clavuligerus by the PimM regulator of Streptomyces natalensis. Front. Microbiol. 10:580. doi: 10.3389/fmicb.2019.00580
Mateos, M. I., and Serrano, A. (1992). Occurrence of phosphorylating and non-phosphorylating NADP+-dependent glyceraldehyde-3-phosphate dehydrogenases in photosynthetic organisms. Plant Sci. 84, 163–170. doi: 10.1016/0168-9452(92)90130-E
Negatu, D. A., Gengenbacher, M., Dartois, V., and Dick, T. (2020). Indole propionic acid, an unusual antibiotic produced by the gut microbiota, with anti-inflammatory and antioxidant properties. Front. Microbiol. 11:575586. doi: 10.3389/fmicb.2020.575586
Noguer, M. A., Cerezo, A. B., Moyá, M. L., Troncoso, A. M., and García-Parrilla, M. C. (2014). Synergism effect between phenolic metabolites and endogenous antioxidants in terms of antioxidant activity. Adv. Chem. Eng. Sci. 4, 258–265. doi: 10.4236/aces.2014.42029
Pandey, S., Djibo, R., Darracq, A., Calendo, G., Zhang, H., Henry, R. A., et al. (2022). Selective CDK9 inhibition by natural compound toyocamycin in cancer cells. Cancers 14:3340. doi: 10.3390/cancers14143340
Quintana, V. M., Selisko, B., Brunetti, J. E., Eydoux, C., Guillemot, J. C., Canard, B., et al. (2020). Antiviral activity of the natural alkaloid anisomycin against dengue and Zika viruses. Antivir. Res. 176:104749. doi: 10.1016/j.antiviral.2020.104749
Reddy, S., Sinha, A., and Osborne, W. J. (2021). “Chapter 1 - Microbial secondary metabolites: Recent developments and technological challenges,” in Volatiles and metabolites of microbes, eds A. Kumar, J. Singh, and J. Samuel (Cambridge, MA: Academic Press), 1–22. doi: 10.1016/B978-0-12-824523-1.00007-9
Ren, Y., Yu, G., Shi, C., Liu, L., Guo, Q., Han, C., et al. (2022). Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta 1:e12. doi: 10.1002/imt2.12
Revuelta, J. L., Buey, R. M., Ledesma-Amaro, R., and Vandamme, E. J. (2016). Microbial biotechnology for the synthesis of (pro)vitamins, biopigments and antioxidants: Challenges and opportunities. Microb. Biotechnol. 9, 564–567. doi: 10.1111/1751-7915.12379
Sawant, A. M., Sunder, A. V., Vamkudoth, K. R., Ramasamy, S., and Pundle, A. (2020). Process development for 6-aminopenicillanic Acid production using lentikats-encapsulated Escherichia coli cells expressing penicillin V acylase. ACS Omega 5, 28972–28976. doi: 10.1021/acsomega.0c02813
Shen, J., Kong, L., Li, Y., Zheng, X., Wang, Q., Yang, W., et al. (2019). A LuxR family transcriptional regulator AniF promotes the production of anisomycin and its derivatives in Streptomyces hygrospinosus var. Beijingensis. Synth. Syst. Biotechnol. 4, 40–48. doi: 10.1016/j.synbio.2018.12.004
Song, Y., Zhang, Z., Zhang, X., Yao, J., Yu, X., and Shentu, X. (2022). Genome shuffling mutant of Streptomyces diastatochromogenes for substantial improvement of toyocamycin production. Fermentation 8:535. doi: 10.3390/fermentation8100535
Takeno, S., Hori, K., Ohtani, S., Mimura, A., Mitsuhashi, S., and Ikeda, M. (2016). L-Lysine production independent of the oxidative pentose phosphate pathway by Corynebacterium glutamicum with the Streptococcus mutans gapN gene. Metab. Eng. 37, 1–10. doi: 10.1016/j.ymben.2016.03.007
Virolle, M.-J. (2020). A challenging view: Antibiotics play a role in the regulation of the energetic metabolism of the producing bacteria. Antibiotics 9:83. doi: 10.3390/antibiotics9020083
Wang, A., Tian, W., Cheng, L., Xu, Y., Wang, X., Qin, J., et al. (2020). Enhanced ε-poly-L-lysine production by the synergistic effect of ε-poly-L-lysine synthetase overexpression and citrate in Streptomyces albulus. Front. Bioeng. Biotechnol. 8:288. doi: 10.3389/fbioe.2020.00288
Wang, L., Zhang, C., Zhang, J., Rao, Z., Xu, X., Mao, Z., et al. (2021). Epsilon-poly-L-lysine: Recent advances in biomanufacturing and applications. Front. Bioeng. Biotechnol. 9:748976. doi: 10.3389/fbioe.2021.748976
Wu, W., Zhang, Y., Liu, D., and Chen, Z. (2019). Efficient mining of natural NADH-utilizing dehydrogenases enables systematic cofactor engineering of lysine synthesis pathway of Corynebacterium glutamicum. Metab. Eng. 52, 77–86. doi: 10.1016/j.ymben.2018.11.006
Yamanaka, K., Hamano, Y., and Oikawa, T. (2020). Enhancement of metabolic flux toward ε-poly-l-lysine biosynthesis by targeted inactivation of concomitant polyene macrolide biosynthesis in Streptomyces albulus. J. Biosci. Bioeng. 129, 558–564. doi: 10.1016/j.jbiosc.2019.12.002
Yang, M., Zhang, W., Lv, Z., Shi, L., Zhang, K., and Ge, B. (2022). Evaluation of the inhibitory effects of wuyiencin, a secondary metabolite of Streptomyces albulus CK-15, against sclerotinia sclerotiorum in vitro. Plant Dis. 106, 156–164. doi: 10.1094/PDIS-05-21-0987-RE
Ye, W., Ni, Z., Yicheng, S., Pan, H., Huang, Y., Xiong, Y., et al. (2019). Anisomycin inhibits angiogenesis in ovarian cancer by attenuating the molecular sponge effect of the lncRNA-Meg3/miR-421/PDGFRA axis. Int. J. Oncol. 55, 1296–1312. doi: 10.3892/ijo.2019.4887
Keywords: Streptomyces albulus, metabolic perturbation, NADP-dependent glyceraldehyde 3-phosphate dehydrogenase, transcriptome and metabolome, ε-poly-L-lysine, bioactive natural products
Citation: Mao J, Zhang M, Dai W, Fu C, Wang Z, Wang X, Yao Q, Kong L and Qin J (2024) Metabolic perturbation of Streptomyces albulus by introducing NADP-dependent glyceraldehyde 3-phosphate dehydrogenase. Front. Microbiol. 15:1328321. doi: 10.3389/fmicb.2024.1328321
Received: 26 October 2023; Accepted: 08 January 2024;
Published: 24 January 2024.
Edited by:
Yuriy Rebets, Explogen LLC., UkraineReviewed by:
Marie-Joelle Virolle, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2024 Mao, Zhang, Dai, Fu, Wang, Wang, Yao, Kong and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linghui Kong, a29uZ2xpbmdodWlAYnptYy5lZHUuY24=; Jiayang Qin, cWluanlAYnptYy5lZHUuY24=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.