 Sangam Kandel1
Sangam Kandel1 Susanna L. Hartzell2Ashton K. Ingold2Grace A. Turner2
Susanna L. Hartzell2Ashton K. Ingold2Grace A. Turner2 Joshua L. Kennedy2,3,4
Joshua L. Kennedy2,3,4 David W. Ussery1*
David W. Ussery1*- 1Department of Biomedical Informatics, University of Arkansas for Medical Sciences, Little Rock, AR, United States
- 2Arkansas Children's Research Institute, Little Rock, AR, United States
- 3Department of Pediatrics, University of Arkansas for Medical Sciences, Little Rock, AR, United States
- 4Department of Internal Medicine, University of Arkansas for Medical Sciences, Little Rock, AR, United States
Introduction: Whole Genome Sequencing (WGS) of the SARS-CoV-2 virus is crucial in the surveillance of the COVID-19 pandemic. Several primer schemes have been developed to sequence nearly all of the ~30,000 nucleotide SARS-CoV-2 genome, using a multiplex PCR approach to amplify cDNA copies of the viral genomic RNA. Midnight primers and ARTIC V4.1 primers are the most popular primer schemes that can amplify segments of SARS-CoV-2 (400 bp and 1200 bp, respectively) tiled across the viral RNA genome. Mutations within primer binding sites and primer-primer interactions can result in amplicon dropouts and coverage bias, yielding low-quality genomes with ‘Ns’ inserted in the missing amplicon regions, causing inaccurate lineage assignments, and making it challenging to monitor lineage-specific mutations in Variants of Concern (VoCs).
Methods: In this study we used a set of seven long-range PCR primer pairs to sequence clinical isolates of SARS-CoV-2 on Oxford Nanopore sequencer. These long-range primers generate seven amplicons approximately 4500 bp that covered whole genome of SARS-CoV-2. One of these regions includes the full-length S-gene by using a set of flanking primers. We also evaluated the performance of these long-range primers with Midnight primers by sequencing 94 clinical isolates in a Nanopore flow cell.
Results and discussion: Using a small set of long-range primers to sequence SARS-CoV-2 genomes reduces the possibility of amplicon dropout and coverage bias. The key finding of this study is that long range primers can be used in single-molecule sequencing of RNA viruses in surveillance of emerging variants. We also show that by designing primers flanking the S-gene, we can obtain reliable identification of SARS-CoV-2 variants.
1 Introduction
Whole Genome Sequencing (WGS) is widely used for the surveillance of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), the causative agent of the pandemic disease COVID-19 (Huang et al., 2020; Wu et al., 2020; Zhou et al., 2020). At the time of writing (August, 2023), there are more than 15.8 million genomes available in the GISAID database1 and more than 8.1 million genomes in GenBank.2 Sequencing SARS-CoV-2 genomes is crucial in tracking viral mutations that can affect viral transmission (Kupferschmidt and Wadman, 2021; Brito et al., 2022; Escalera et al., 2022; Carabelli et al., 2023), disease pathogenesis (Bakhshandeh et al., 2021), vaccine efficacy (Hoffmann et al., 2021; Madhi et al., 2021; Chatterjee et al., 2023), and virulence (Issa et al., 2020; Carabelli et al., 2023). A variety of methods, including metagenomic sequencing, hybridization capture, direct RNA sequencing, and target enrichment using multiplex PCR have been used for sequencing SARS-CoV-2 (Carbo et al., 2020; Charre et al., 2020; Deng et al., 2020; Wu et al., 2020; Xiao et al., 2020; Butler et al., 2021; Liu et al., 2021; Rehn et al., 2021; Gerber et al., 2022; Vacca et al., 2022). Most of the target enrichment methods require reverse transcription to generate a double-stranded cDNA copy of the genomic RNA (gRNA) and then utilize this cDNA as a template for DNA sequencing, using multiplex primers to cover the whole genome of SARS-CoV-2 (Grubaugh et al., 2019).
Target enrichment using PCR amplicons and subsequent Oxford Nanopore Sequencing is extremely popular and relatively inexpensive (~$10 per sample), with a quick turnaround time (~24 h from sample to GenBank file). Target enrichment using publicly available ARTIC Network PCR primers (Quick 2020; Tyson et al., 2020), Entebbe primers (1.5 kb-2Kb) (Cotten et al., 2021), MRL primers (1.5 kb-2.5 kb) (Arana et al., 2022), and Midnight Primers (Freed et al., 2020) are used to sequence SARS-CoV-2 with Oxford Nanopore flow cells. Among these primer schemes, ARTIC primers and Midnight primers are the most used to sequence clinical isolates of SARS-CoV-2 (Table 1; Figure 1). ARTIC primers V4 includes 98 primer pairs, each amplifying ~400 bp fragments along the viral genome, which can be sequenced on either Illumina or Oxford Nanopore platforms. The ‘Midnight primers’ have 29 primer pairs that generate amplicons with a targeted size of 1,200 base pairs, taking advantage of the longer read lengths of third-generation sequencing, including Oxford Nanopore flow cells. Generation of full-length high-quality consensus sequences depends upon the quality and quantity of the viral load in clinical samples, as well as the mutations occurring within the primer binding regions of the viral genome (Davis et al., 2021; Liu et al., 2021; Kuchinski et al., 2022). Amplicon dropouts and coverage bias at different amplicon regions have been observed with the sequencing protocols based on ARTIC (Itokawa et al., 2020; Kuchinski et al., 2022) as well as Midnight primers (Bei et al., 2022; Kuchinski et al., 2022). Mutations within the primer binding site can prevent primer-annealing and result in ‘dropout’ or loss of that amplicon, leading to incomplete genome sequences (Sanderson and Barrett, 2021; Bei et al., 2022). Furthermore, primer-primer interactions could result in amplification bias of interacting amplicons (Itokawa et al., 2020), resulting in coverage bias and affecting the identification of mutations in the viral genome that are key in the nomenclature of emerging variants.
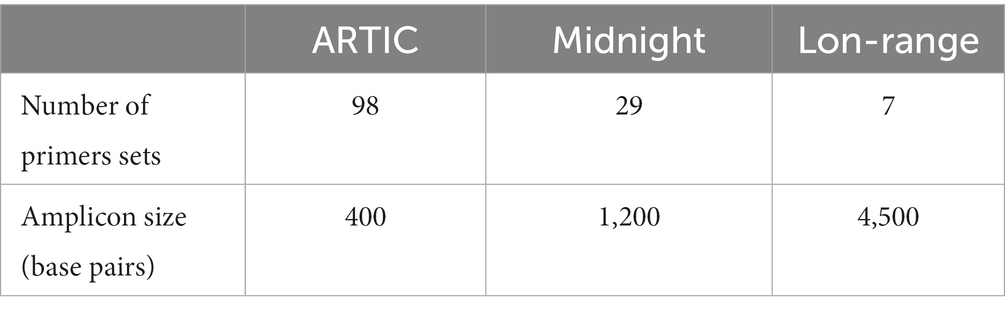
Table 1. Comparison of ARTIC, Midnight, and Long-range primers used to sequence SARS-CoV-2 clinical isolates.
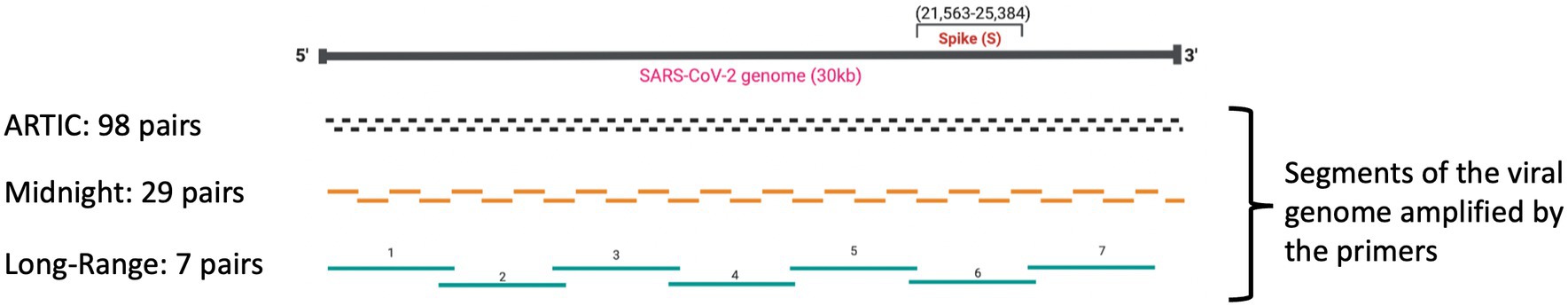
Figure 1. Comparison of ARTIC, Midnight, and Long-Range PCR primers.
The variants of SARS-CoV-2 are determined by a combination of several mutations that occur mainly within the Spike gene. For example, in the Alpha variant (B.1.1.7), there are 14 critical lineage-defining mutations within the S gene (Galloway et al., 2021). Similarly, Omicron subvariant B.1.1.529 has 60 mutations within the viral genome, including 15 key mutations within the receptor binding domain (He et al., 2021). The characteristic mutation within the S gene for the Alpha variant B.1.1.7 (Meng et al., 2021; Clark et al., 2022) and the Omicron variants B.1.1.529, BA.1, BA.1.1 (Clark et al., 2022) is the deletion of two amino acids at positions 69 and 70 (del H69/V70)3. This deletion inhibits the PCR amplification of the S-gene (S-Gene Target Failure, or SGTF) in diagnostic PCR assays such as the ThermoFisher TaqPath™ COVID-19 Combo Kit RT-PCR (Davies et al., 2021; Clark et al., 2022) that targets the N, ORF1ab, and S gene regions. This deletion (del H69/V70) results in a false-negative result for the S-gene targeted diagnostic test. SGTF became a proxy for early detection of Alpha and Omicron B.1.1.529 variants (Galloway et al., 2021). In addition, a mutation at position 27,807 (Cytosine substituted to Thymine) within amplicon 28, also a primer annealing site (Primer 28_LEFT, pool B of Midnight primer) (Supplementary Figure 1), caused a common dropout in the Delta variant genome when using Midnight Primers (Kuchinski et al., 2022). Spiking Primer pool B with a custom primer designed by substituting Cytosine with Thymine base not only corrected the dropout but also increased the coverage at this region (Constantinides et al., 2022). Furthermore, the genome sequences of two BA.2 Omicron variants from Arkansas (GenBank Accession: OM863926, ON831693) sequenced using Midnight Primers in Oxford Nanopore GridION have a complete dropout at amplicon region 21 (20,677-21,562). The Omicron and the Alpha variant waves taught us that tests and primers designed toward regions within the S gene could result in false-negative tests because this gene encodes a surface protein, subjecting it to varying selectional pressures (Julenius and Pedersen, 2006). Variations can lead to problems that are troublesome in deciding the public health interventions needed to control the transmission and spread of COVID-19 disease.
Multiplex primers used to sequence SARS-CoV-2 viral isolates must be targeted to bind regions that are conserved with little variance to avoid dropout failures secondary to the primers not binding. Long-range PCR primers targeting the amplification of 4,500 bp can prevent the ‘S-gene dropouts’, as the primer binding sites flanking the S-gene region are located within highly conserved regions on either side of the S gene. The S gene is approximately 3,822 base pairs long and stretches between the nucleotide position 21,563 to 25,384 along the viral genome. Therefore, these long-range PCR primers can generate amplicons around 4,500 bp that will cover the entire S gene, making the chances of amplicon dropout within the S-gene minimal. We have previously demonstrated whole-genome cDNA sequences from Mumps genomes using long-range PCR yielding fragments of ~5,000 bp in length from buccal samples (Alkam et al., 2019). In addition to our work, long-range semi-nested PCR have been used to sequence Ebola virus (Seifert et al., 2018), Middle East respiratory syndrome coronavirus (MERS-CoV) (Seifert et al., 2021), Hendra virus (HeV), Nipah virus (NiV) and Cedar virus (CedPV) (Yinda et al., 2020) on an Oxford Nanopore MinION sequencer. More recently, long-range primers were used to sequence clinical isolates of Monkeypox virus (MPXV) generating amplicons around 5,000 base pairs (Isabel et al., 2023) to sequence the much larger DNA viral genome of approximately 200,000 bp. We have previously identified conserved regions withing the SARS-CoV-2 genome, including regions that flank the S gene (Wassenaar et al., 2022). In this study, we designed long-range PCR primers to target these conserved areas flanking the S gene and to sequence SARS-CoV-2 isolates. Our objective was to improve the quality of the sequences generated and minimize the amplicon dropouts, as the designed primers are outside the highly variable regions.
2 Methods
2.1 Primer design
A total of 7,046 Omicron sub-variant genomes (BA.2, BA.3, BA.4, BF.5, BA.5.1, BA.5.2.1, BA.5.2) were downloaded from GISAID on 12 August, 2022. Pangolin v4.0.6 (O'Toole et al., 2021) was used to assign lineages to the genomes, and any ‘unclassified’ genomes were removed. Genome sequences that were 100% identical were then filtered out to avoid redundancy, and genome sequences having gaps of 5Ns or more in their sequences were removed that resulted in a set of 1,205 high-quality genomes that were used for multiple sequence alignment using MAFT (Katoh et al., 2019). MSA Viewer4 was used to visualize the alignment, and consensus sequences were downloaded from MSA Viewer. PrimalScheme (Quick et al., 2017) was used to generate primer schemes using the consensus genome generated from the alignment of 1,205 high-quality genomes, including different sub-variants of Omicron. Primers were designed using the PrimalScheme tool using the command line (Table 2):
primalscheme multiplex <fasta-file> -a 4500 –o <path-to-output> -n <primers_name> -t 30 -p -g
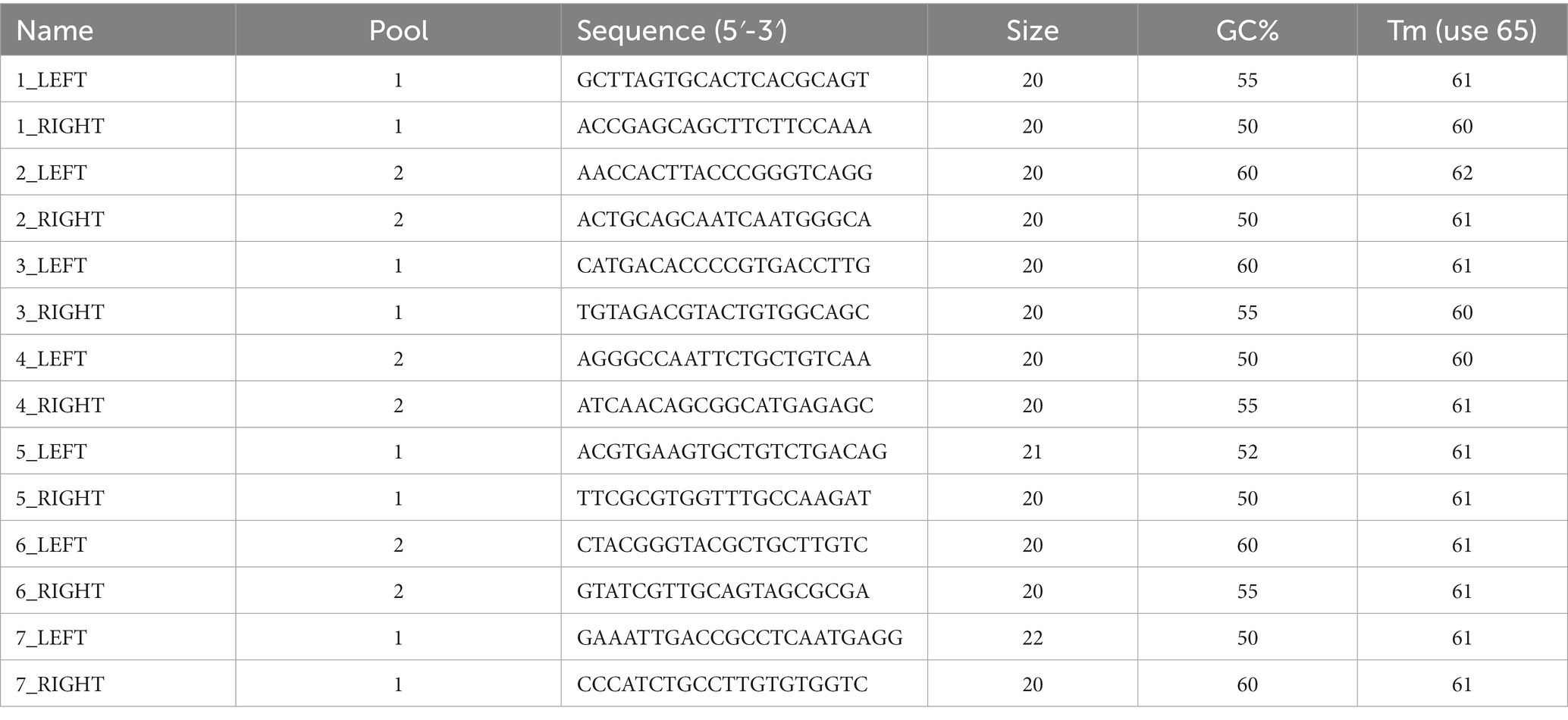
Table 2. List of 7 primer pairs designed using PrimalScheme.
Primers were ordered from Integrated DNA Technology (IDT) (Coralville, IA) in lab-ready form. Individual primers in each pool were mixed and resuspended to a final concentration of 100 μM. Each primer was normalized to 3 nmol during synthesis. Primers were diluted in Nuclease-free water (Sigma) to use in a final concentration of 10 μM.
High-quality genomes were downloaded from GenBank, and a consensus sequence was generated using the most recent dominant variants of SARS-CoV-2 from GenBank collected between December 2022 and March 2023. Quality filtering was done to include only those genomes that did not contain any non-ATCGN bases and those that did not have any ‘N’s in the genome sequence. The consensus sequence from this set of genomes was used to manually design the alternative primers, including amplicons 5, 6, and 7.
2.2 Primer analysis
MFEprimer (Wang et al., 2019) was used to predict the various quality metrics of the primer scheme designed using PrimalScheme. This method predicted that forward primer for amplicon region 5 and forward primer for amplicon region 7 could form self-dimers. Self-dimers could prevent primer annealing to the template and hence prevent the amplification of the targets resulting in the drop of amplicon. However, the experimental results suggested a high coverage at all amplicon regions. Based on our experimental results, we can conclude that primers 5 and 7 worked well.
2.3 Detection and quantification of SARS-CoV-2 viral mRNA
All the samples used in this study were collected at Arkansas Children’s Hospital and the University of Arkansas for Medical Sciences as routine surveillance between (November 2022 and Jan 2023). Nasal swab samples were collected in a 3 mL M4RT transport media (Remel, San Diego, CA). Samples were tested for the SARS-CoV-2 using the Aptima® SARS-CoV-2 (Panther® System, Hologic, San Diego, CA) nucleic acid amplification assay. Positive samples were stored frozen at −80°C until they could be further processed.
2.4 RNA extraction, library preparation, and whole genome sequencing
A summary of the sequencing protocol is described in Figure 2. Two hundred fifty microliters of viral transport media from clinical nasal swabs were used for viral RNA extraction using the MagMax Viral/Pathogen Nucleic Isolation Kit (Applied Biosystems) on the Kingfisher Flex automated instrument (Thermofisher). Viral RNA was reverse transcribed to generate cDNA using LunaScript RT SuperMix (NEB #E3010) as described (Freed et al., 2020). Each reverse transcription reaction contained 8 μL template RNA and 2 μL LunaScript RT SuperMix (NEB #E3010). The reaction condition for reverse transcription was: 25°C for 10 min, followed by 50°C for 10 min and 85°C for 5 min. Subsequent cDNA amplification and sequencing were done using a modified sequencing protocol (Quick 2020; Table 3). In brief, viral cDNA was used in the tiling PCR method to amplify the SARS-CoV-2 viral genome using long-range PCR primers in 2 reaction pools. These primers generate PCR amplicons of around 4,500 bp size. Pool A consisted of the primers specific to amplicon regions 1, 3, 5, and 7, whereas Pool B consisted of the primers specific to amplicon regions 2, 4, and 6. A 25 μL PCR reaction mixture contained 2.5 μL template cDNA, 8.9 μL RNase-free water, 1.1 μL Primer pool A or Primer pool B (10 μM), 12.5 μL Q5 Hot Start HF 2x Master Mix (NEB # M0494X). The PCR conditions used were: 98°C for 30 s (Initial denaturation), 40 cycles of: 98°C for 10 s (Denaturation), 65°C for 30 s followed by 72°C for 5 min (Annealing and extension), and a final extension of 72°C for 5 min. Pool 1 and Pool 2 amplicons were pooled together, and 7.5 μL of each sample were barcoded using 2.5 μL of rapid barcodes available with the kit SQK-RBK004 (ONT). Barcoded samples were pooled together and cleaned using 0.8 X AMPure beads (Beckman Coulter, USA) to retain larger DNA fragments. The sequencing library was prepared using sequencing kit SQK-RBK004 (ONT), loaded onto a MinION flow cell (ONT), and sequenced for 28 h using a Minion R9.4.1 flow cell on GridION with the MinKNOW application.
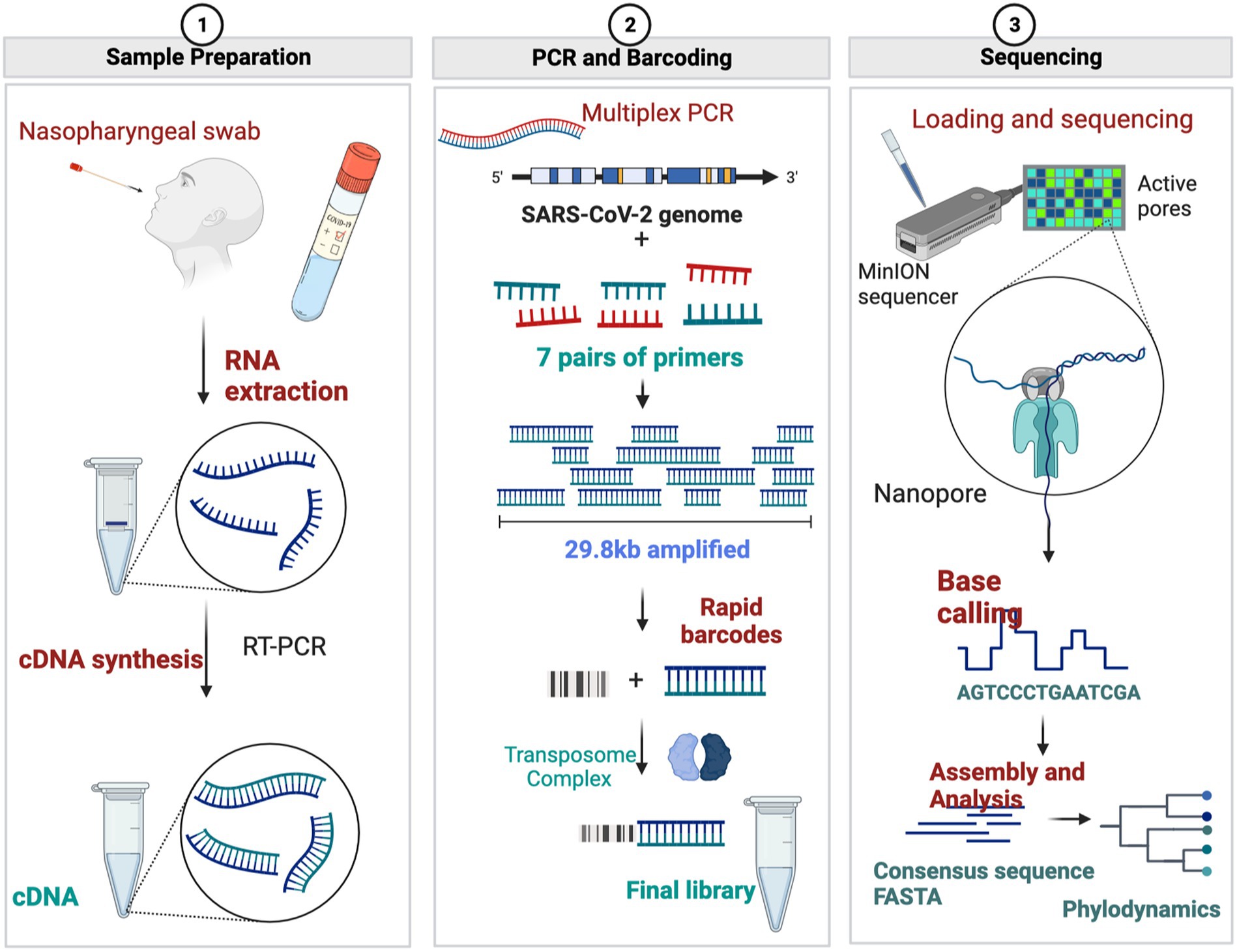
Figure 2. Diagrammatic representation of Oxford Nanopore Sequencing of SARS-CoV-2 using long-range PCR primers. (Figures made using BioRender.com).
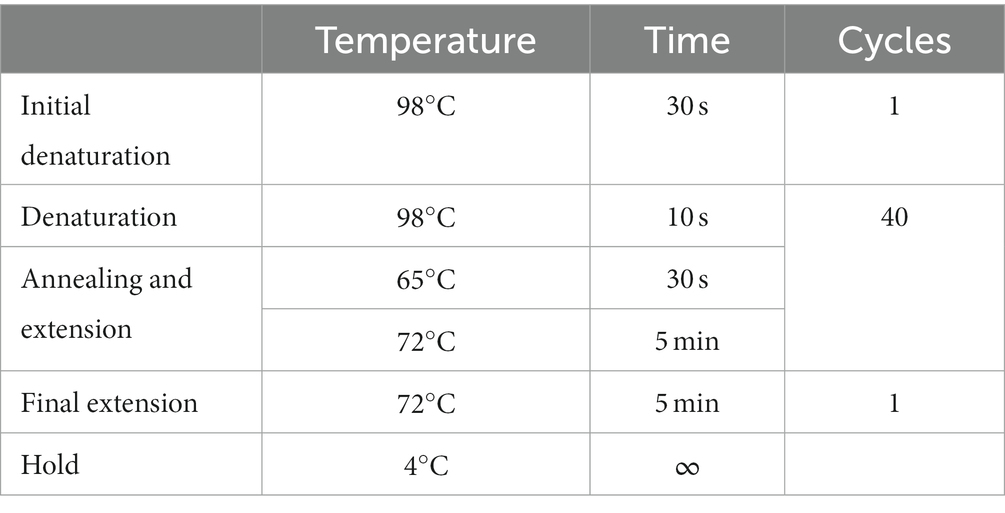
Table 3. Optimized PCR conditions for cDNA amplification to sequence SARS-CoV-2 clinical isolates.
2.5 Bioinformatics analysis
Basecalling and demultiplexing the sequencing reads in FAST5 format was done in real-time using Guppy v5.0.7 (Wick et al., 2019) with a high-accuracy model. A minimum quality score of 9 was used to remove low-quality bases. Demultiplexed FASTQ files were processed using the ARTIC Network Bioinformatics pipeline5. Sequencing reads were quality filtered using artic gupplyplex method, and reference-based genome assembly was done using medaka from the artic minion method of the ARTIC bioinformatics pipeline. ONTdeCIPHER (Cherif et al., 2022) was used for generating visualization plots for genome coverage at different amplicon regions. The consensus sequence was generated by mapping to NC_045512.2 as a reference. Read depth was calculated using samtools depth (Li et al., 2009). Pangolin v4.0.6 was used to assign lineages to the genomes sequenced (O'Toole et al., 2021). Nextclade (Aksamentov et al., 2021) was used for assigning lineage as well as visualization and comparison of mutations within the viral genome.
3 Results
Long-range primers were used to sequence a set of four samples, with various cycle threshold (CT) values, on an Oxford Nanopore GridION machine. A lower cycle threshold is associated with higher levels of the virus in the sample, requiring fewer amplification cycles for detection. These samples, identified as: V05476, V06110, V05450, and V06106, and had CT values of 11.6, 14.3, 15.1, and 18.3, respectively. A total of 4.8 million reads were generated from the four samples with an N50 of 2,640 bases after 28 h of sequencing. The mean read coverage was approximately the same (7,529, 7,646, 7,673, and 7,725, respectively) for the four samples (Table 4). All the samples had high genome coverage (>98%), and each was assigned the BA.5 variant of Omicron. The number of reads mapped to each amplicon position is summarized in Figure 3 and Table 5. Out of seven amplicons, amplicon 4 had the highest number of reads mapped to the reference.
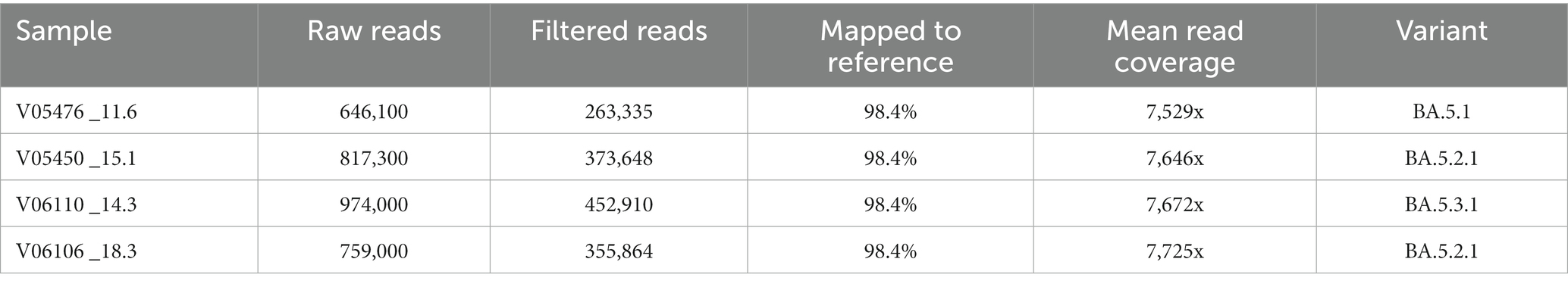
Table 4. Sequencing summary of four samples showing different quality metrics.
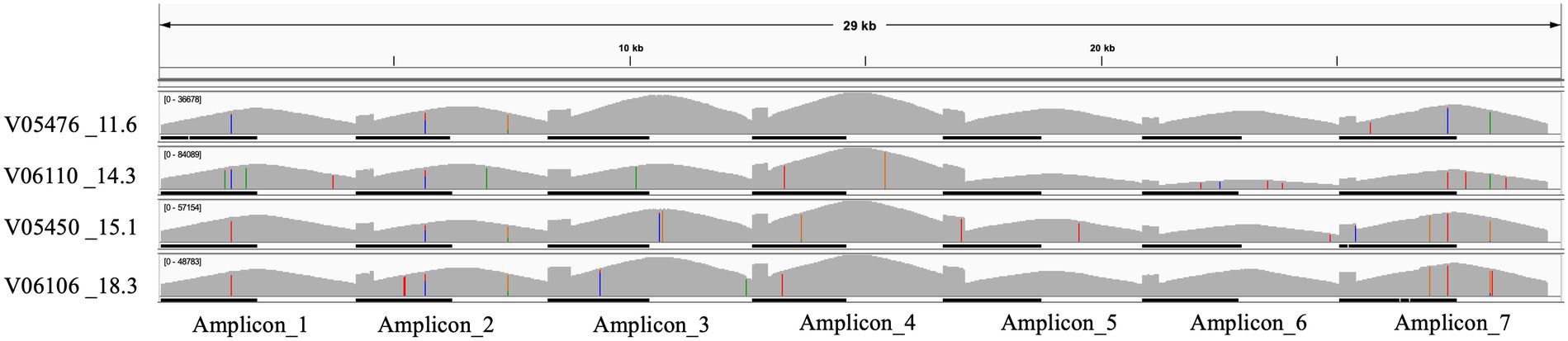
Figure 3. IGV plot showing seven different amplicons mapped to the SARS-CoV-2 reference genome for four samples with low CT values.
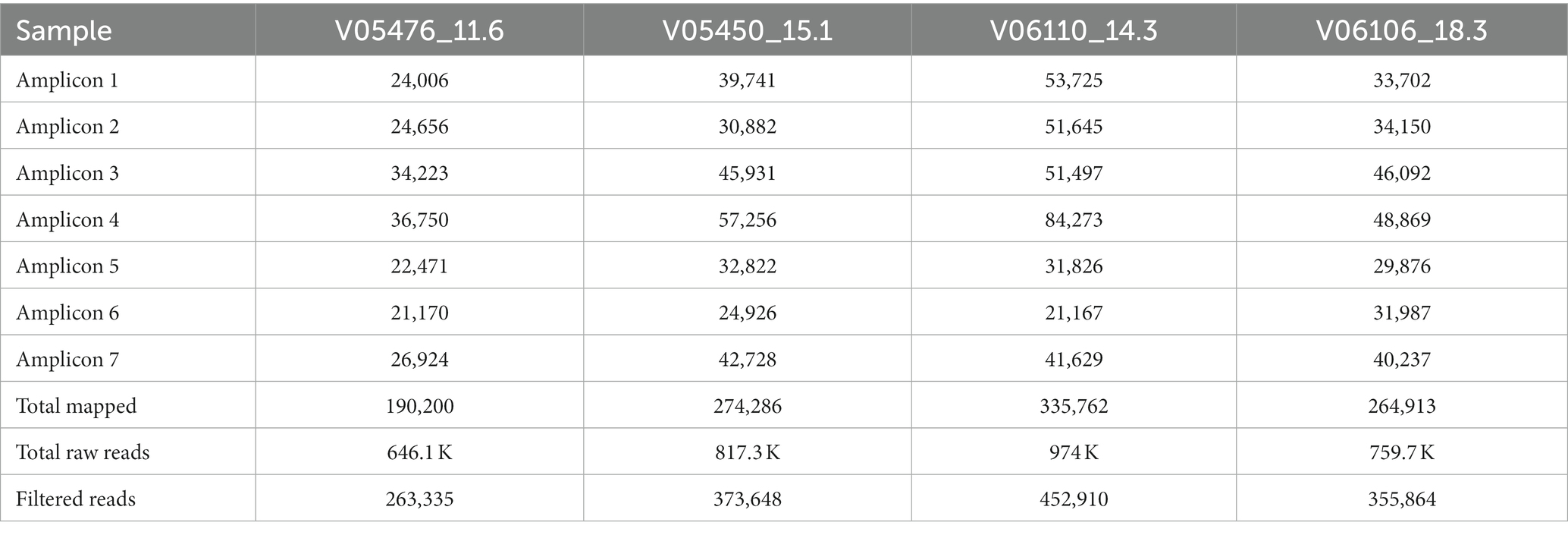
Table 5. Total number of raw reads, filtered reads, and reads that mapped to the reference genome at seven amplicon regions.
A 96-well plate containing samples with different CT values spanning from 11 to 16 (n = 19), 17 to 20 (n = 14), 21 to 25 (n = 15), 26 to 30 (n = 15), 31 to 35 (n = 15), and 36 to 42 (n = 16) were sequenced using long-range and Midnight primers for comparison, as shown in Figure 3. With the long-range primers, 100% of the samples with CT values 11 to 16 passed quality, whereas 95% of samples within the range of this CT value passed quality when sequenced with midnight primers. Long-range primers performed similarly to the midnight primers for sequencing samples with CT values between 17 and 20 (Long-range: 73% and Midnight: 88% passing quality). For samples with CT values of 21–25, 47% passed quality with Midnight primers, whereas only 33% passed quality with long-range primers. With midnight primers, only two samples passed quality with CT values greater than 26. The long-range and the midnight primers generated no quality sequences in those samples with CT values greater than 26 (Figure 4 and Table 6). A heatmap of coverage for each amplicon regions is shown in the Supplementary Figures 2, 3.
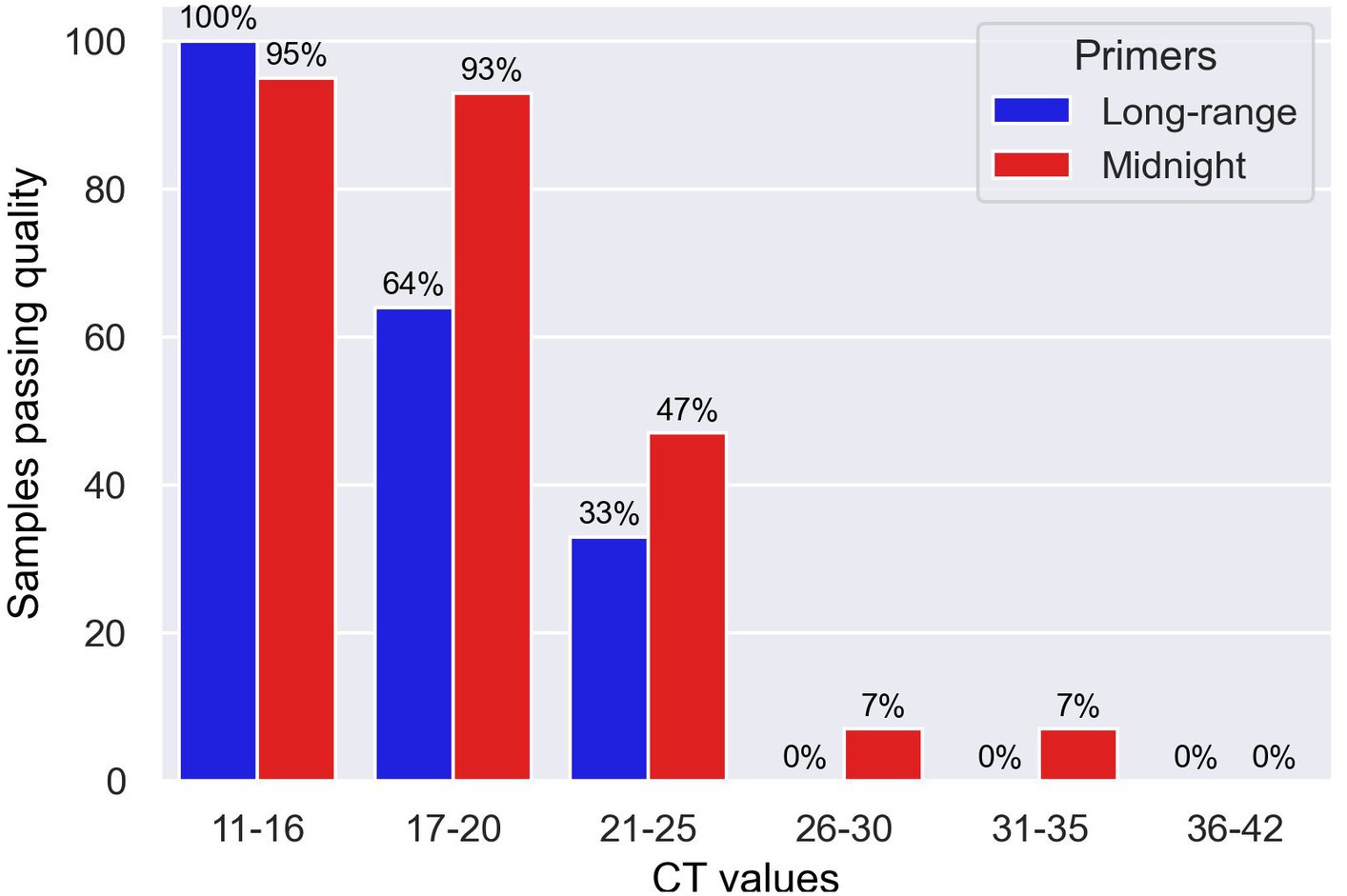
Figure 4. Bar chart showing samples sequenced with Midnight and Long-range primers with different CT values that passed quality.

Table 6. Comparison of total samples passing quality standards by CT values.
Although the samples from a 96-plex sequencing run that passed quality were accurately assigned to a lineage, we have found, in some cases, there was low coverage of some regions. Some of the amplicon regions such as 5, 6 and 7 had low coverage for some samples. We updated the primers specific to amplicon regions 5, 6 and 7 using the reference genomes of SARS-CoV-2 that were the most dominant during the time of writing this manuscript (Dec, 2022 to March, 2023). The updated primers were: 5-LEFT: 5ʹ-GTG ACT GGA CAA ATG CTG GTG A-3ʹ,5-RIGHT: 5’-CAT GAC ATA ACC ATC TAT TTG TTC GC-3ʹ, 6-LEFT: 5ʹ-GGT TCC GTG GCT ATA AAG ATA ACA G-3ʹ, 6-RIGHT: 5ʹ-AGG CTT GTA TCG GTA TCG TTG C-3ʹ, 7-LEFT: 5ʹ- GCC ATG GTA CAT TTG GCT AGG-3ʹ, 7-RIGHT: 5’-GCT CTT CCA TAT AGG CAG CTC-3ʹ. These alternative primers generated high-quality genomes with a lineage assigned to the consensus sequence of the genome (Supplementary Table 1). As the virus continues to mutate, it will likely be necessary to adjust the primers to maintain optimal coverage for all regions.
4 Discussion
We have developed and evaluated novel long-range primers to sequence SARS-CoV-2 clinical isolates using Oxford Nanopore sequencing. These novel primers can amplify regions approximately 4,500 base pairs. Using our primer set, the entire S-gene was sequenced using a single primer set. We compared the performance of long-range primers with midnight primers and found that long-range primers work as good as the midnight primers regarding the quality of genome sequences and coverage. This finding depends upon the amount of viral RNA in the sample. We decided to focus on comparison of the Midnight primers with the long-range PCR primers, and to exclude ARTIC primers, since much has already been published and discussed about the S-gene knockouts in the many short regions amplified by the ARTIC primers (Radhakrishnan et al., 2021; Carattini et al., 2023). Based on the results shown in Table 6, we can conclude that the optimal CT value for the long-range primers is 16 or lower, where we can consistently get 100% coverage. We can conclude that Midnight primers and long-range primers have better performance with CT values less than 20. Midnight primers are better than Long-range primers, however, long-range primers are as good as midnight primers with the advantage of having longer reads and the ability to minimize amplicon dropouts.
It is true that long-range primers are not better than midnight primers in generating high quality consensus sequences from clinical isolates of SARS-CoV-2 with very high CT values. However, long-range primers work as good as midnight primers for samples with CT values less than 20 as shown in Table 6 or Figure 4, Midnight primers generated 88% whereas long-range primers generate 73% of good quality genomes for GenBank. There are two advantages of long-range primers: first, this can solve the problems of amplicon dropouts due to mutations within primer binding site, and second, using single-molecule sequencing, these primers can be used to sequence across the entire S-gene region, and to assign lineages for population surveillance. Furthermore, the main objective of genomic surveillance during COVID-19 pandemic is to sequence as many genomes as possible with rapid and faster turnaround time.
We used 7,000 reference genomes from GISAID to generate a consensus sequence to design these long-range primers. Genome coverage is improved when primer schemes are created using multiple reference genome sequences compared to those designed using a single reference genome (Bei et al., 2022). ARTIC v3 and Midnight-1200 primers were designed using just one reference genome of SARS-CoV-2. In contrast, other primer schemes, such as the updated ARTIC (ARTIC v4.1), VarSkip Short v2, and VarSkip Long primers, were designed using multiple reference genomes. Long-range PCR primers can minimize the amplicon dropout due to mutations within the primer binding site (Bei et al., 2022).
After the ARTIC protocol was made public on January 22, 2020, these primers were adopted globally to sequence millions of SARS-CoV-2 genomes. After the introduction, there have been several improvements and updates to these primers to resolve dropouts and improve sequencing coverage (Grubaugh et al., 2019; Tyson et al., 2020; Davis et al., 2021). In addition to ARTIC primers, midnight primers that are extremely popular for sequencing SARS-CoV-2 clinical isolates using Nanopore sequencing were also updated to resolve amplicon dropouts and coverage bias along different regions of the viral genome (Constantinides et al., 2022). Several studies have been conducted to compare different sequencing protocols, using multiplex PCR primers to increase the genome coverage, improve the sequencing reading quality, eliminate amplicon dropouts, and improve coverage bias at different amplicon regions (Bei et al., 2022; Constantinides et al., 2022; Lambisia et al., 2022). As the virus mutates and spreads throughout communities, the primers and protocols need to be updated to avoid amplicon dropouts and avoid coverage bias. Because the S-gene is roughly 3,821 base pairs long, amplifying the entire S-gene requires multiple primer pairs using short-range primer pairs that are currently popular. Therefore, if any mutation occurs within the primer binding regions within S-gene, a significant fraction of S-gene could be dropped from final consensus sequence.
Long-range primers to sequence SARS-CoV-2 have previously not been reported, apart from a few primer schemes amplifying regions up to 2,500 base pairs (Arana et al., 2022). Because the S-gene is approximately 3,821 base pairs, amplifying the entire S-gene requires more than one primer. Therefore, mutations within S-gene could result in dropout within S-gene. As an alternative to this problem, leveraging the long-read sequencing available with Oxford Nanopore flow cells, we have developed long-range primers, which sequence the entire S-gene using just one primer pair, thereby eliminating the possibility of amplicon dropout due to mutations within S-gene.
The accuracy of PCR reactions for the long-range primers depends upon many factors, including the specificity of the primers, experimental conditions such as number of PCR cycles, cycle parameters, and environmental conditions such as Mg2+ concentrations, and which DNA polymerases are used for amplification. For the purposes of the experiments outlined here, ‘accuracy’ is important in terms of obtaining full length cDNA sequences for the segments of interest. In principle, we could quantitate the transcripts using something like droplet digital PCR (ddPCR; Hindson et al., 2013; Kojabad et al., 2021). However, the main purpose of this current work is to reduce the number of primers used, and to verify that we are indeed getting full length sequences of the regions of interest (which is verified by sequence alignment).
A limitation of this approach is that a mutation within the primer binding sites can result in a drop out of that entire region, leading to a more significant gap in the consensus sequence that significantly affects the quality of the genome sequence. However, since the primer sites were designed using conserved regions, we anticipate that this will continue to work, although, as necessary, it is easy to update the primers for novel strains. Another limitation is associated with viral load in the sample. We have found that although these long-range primers can amplify larger segments of the viral genome, these primers are not well suited to sequence samples with higher CT values (greater than 25).
Single-read sequencing technologies could quantitate viral genome fragmentation. For example, the viral genome is sheared into smaller pieces, most of the sequence reads would be short (and little amplification would occur, since this would be essentially linear extension of only one strand). We have designed primers in well conserved regions, that will hopefully have few mutations. One of the advantages of long-range primers is that fewer primer sites are necessary, and also more flexibility is given with respect to the placement of the primers, allowing for better optimization of primer binding sites within conserved regions.
Although WHO lifted the global health emergency due to a significant reduction in positive cases, we are entering into a new phase of COVID-19 as 1 out of 10 people have long-haul COVID (Thaweethai et al., 2023). Looking back to historical epidemics due to coronavirus and the evolutionary relatedness of the SARS-CoV-2 with previous outbreaks of SARS and MERS, future pandemics are inevitable. COVID-19 is still circulating as local outbreaks continue. The long-range PCR method outlined here can help with surveillance of community infections through wastewater monitoring. With single reads over the entire S-gene region, it is possible to quantitate variant diversity within a sample. This will allow monitoring of emerging variants as well as keeping track of known variants of concern.
Table 7. GenBank accession number of the samples used to validate this study’s long-range primers.
In conclusion, in this study we have shown the applications of long-range primers to sequence SARS-CoV-2 mainly in the context of surveillance to address the issues of amplicon dropouts as observed by using primers that amplify short regions. While the long-range primers might be affected by the quality of the viral genetic material, we have shown that we can sequence the most important region of the virus using just one primer set (flanking the S-gene) which would help to determine the emerging variants.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material. The datasets analyzed in this study are deposited in NCBI, accession numbers: OP576060, OQ079743, OQ079740, OQ938311, OQ938315, OQ938316 (Table 7).
Author contributions
SK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. SH: Methodology, Writing – review & editing. AI: Methodology, Writing – review & editing. GT: Methodology, Writing – review & editing. JK: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. DU: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Center for Translational Pediatric Research (NIH/NIGMS P20GM121293) at Arkansas Children’s Hospital, the Translational Research Institute (NIH/NCATS UL1TR003107) at the University of Arkansas for Medical Sciences, an NSF award (no. OIA-1946391), and funding from the Arkansas Children’s Research Institute and the Arkansas Research Alliance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1272972/full#supplementary-material
Footnotes
2. ^https://www.ncbi.nlm.nih.gov/sars-cov-2/
4. ^https://www.ncbi.nlm.nih.gov/projects/msaviewer/
5. ^https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html
References
Aksamentov, I., Roemer, C., Hodcroft, E. B., and Neher, R. A. (2021). Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 6:3773. doi: 10.21105/joss.03773
Alkam, D., Jenjaroenpun, P., Wongsurawat, T., Udaondo, Z., Patumcharoenpol, P., Robeson, M., et al. (2019). Genomic characterization of mumps viruses from a large-scale mumps outbreak in Arkansas, 2016. Infect. Genet. Evol. 75:103965. doi: 10.1016/j.meegid.2019.103965
Arana, C., Liang, C., Brock, M., Zhang, B., Zhou, J., Chen, L., et al. (2022). A short plus Long-amplicon based sequencing approach improves genomic coverage and variant detection in the SARS-CoV-2 genome. PLoS One 17:e0261014. doi: 10.1371/journal.pone.0261014
Bakhshandeh, B., Jahanafrooz, Z., Abbasi, A., Goli, M. B., Sadeghi, M., Mottaqi, M. S., et al. (2021). Mutations in SARS-CoV-2; consequences in structure, function, and pathogenicity of the virus. Microb. Pathog. 154:104831. doi: 10.1016/j.micpath.2021.104831
Bei, Y., Pinet, K., Vrtis, K. B., Borgaro, J. G., Sun, L., Campbell, M., et al. (2022). Overcoming variant mutation-related impacts on viral sequencing and detection methodologies. Front. Med. 9:989913. doi: 10.3389/fmed.2022.989913
Brito, A. F., Semenova, E., Dudas, G., Hassler, G. W., Kalinich, C. C., Kraemer, M. U. G., et al. (2022). "global disparities in SARS-CoV-2 genomic surveillance." nature. Communications 13:7003. doi: 10.1038/s41467-022-33713-y
Butler, D., Mozsary, C., Meydan, C., Foox, J., Rosiene, J., Shaiber, A., et al. (2021). Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat. Commun. 12:1660. doi: 10.1038/s41467-021-21361-7
Carabelli, A. M., Peacock, T. P., Thorne, L. G., Harvey, W. T., and Hughes, J. (2023). SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat. Rev. Microbiol. 21, 162–177. doi: 10.1038/s41579-022-00841-7
Carattini, Y. L., Griswold, A., Williams, S., Valiathan, R., Zhou, Y., Shukla, B., et al. (2023). Combined use of RT-qPCR and NGS for identification and surveillance of SARS-CoV-2 variants of concern in residual clinical laboratory samples in Miami-Dade County, Florida. Viruses 15:593. doi: 10.3390/v15030593
Carbo, E. C., Sidorov, I. A., Zevenhoven-Dobbe, J. C., Snijder, E. J., Claas, E. C., Laros, J. F. J., et al. (2020). Coronavirus discovery by metagenomic sequencing: a tool for pandemic preparedness. J. Clin. Virol. 131:104594. doi: 10.1016/j.jcv.2020.104594
Charre, C., Ginevra, C., Sabatier, M., Regue, H., Destras, G., Brun, S., et al. (2020). Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol 6:veaa075. doi: 10.1093/ve/veaa075
Chatterjee, S., Bhattacharya, M., Nag, S., Dhama, K., and Chakraborty, C. (2023). A detailed overview of SARS-CoV-2 omicron: its sub-variants, mutations and pathophysiology, clinical characteristics, immunological landscape, immune escape, and therapies. Viruses 15:167. doi: 10.3390/v15010167
Cherif, E., Thiam, F. S., Salma, M., Rivera-Ingraham, G., Justy, F., Deremarque, T., et al. (2022). ONTdeCIPHER: an amplicon-based Nanopore sequencing pipeline for tracking pathogen variants. Bioinformatics 38, 2033–2035. doi: 10.1093/bioinformatics/btac043
Clark, C., Schrecker, J., Hardison, M., and Taitel, M. S. (2022). Validation of reduced S-gene target performance and failure for rapid surveillance of SARS-CoV-2 variants. PLoS One 17:e0275150. doi: 10.1371/journal.pone.0275150
Constantinides, B., Webster, H., Gentry, J., Bastable, J., Dunn, L., and Oakley, S. (2022). Rapid turnaround multiplex sequencing of SARS-CoV-2: comparing tiling amplicon protocol performance. medRxiv 12:21268461. doi: 10.1101/2021.12.28.21268461
Cotten, M., Bugembe, D. L., Kaleebu, P., and Phan, M. V. T. (2021). Alternate primers for whole-genome SARS-CoV-2 sequencing. Virus Evol. 7:veab006. doi: 10.1093/ve/veab006
Davies, N. G., Jarvis, C. I., John Edmunds, W., Jewell, N. P., Diaz-Ordaz, K., and Keogh, R. H. (2021). Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 593, 270–274. doi: 10.1038/s41586-021-03426-1
Davis, J. J., Wesley Long, S., Christensen, P. A., Olsen, R. J., Olson, R., Shukla, M., et al. (2021). Analysis of the ARTIC version 3 and version 4 SARS-CoV-2 primers and their impact on the detection of the G142D amino acid substitution in the spike protein. Microbiol. Spectr. 9, e01803–e01821. doi: 10.1128/Spectrum.01803-21
Deng, X., Achari, A., Federman, S., Guixia, Y., Somasekar, S., Bártolo, I., et al. (2020). Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 5, 443–454. doi: 10.1038/s41564-019-0637-9
Escalera, A., Gonzalez-Reiche, A. S., Aslam, S., Mena, I., Laporte, M., Pearl, R. L., et al. (2022). Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe 30, 373–387.e7. doi: 10.1016/j.chom.2022.01.006
Freed, N. E., Vlková, M., Faisal, M. B., and Silander, O. K. (2020). Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 Bp tiled amplicons and Oxford Nanopore rapid barcoding. Biol. Methods Protoc. 5:bpaa014. doi: 10.1093/biomethods/bpaa014
Galloway, S. E., Paul, P., MacCannell, D. R., Johansson, M. A., Brooks, J. T., MacNeil, A., et al. (2021). Emergence of SARS-CoV-2 B.1.1.7 lineage United States, December 29, 2020–January 12, 2021. Morb. Mortal. Wkly Rep. 70, 95–99. doi: 10.15585/mmwr.mm7003e2
Gerber, Z., Daviaud, C., Delafoy, D., Sandron, F., Alidjinou, E. K., Mercier, J., et al. (2022). A comparison of high-throughput SARS-CoV-2 sequencing methods from nasopharyngeal samples. Sci. Rep. 12:12561. doi: 10.1038/s41598-022-16549-w
Grubaugh, N. D., Gangavarapu, K., Quick, J., Matteson, N. L., De Jesus, J. G., Main, B. J., et al. (2019). An amplicon-based sequencing framework for accurately measuring Intrahost virus diversity using PrimalSeq and IVar. Genome Biol. 20:8. doi: 10.1186/s13059-018-1618-7
He, X., Hong, W., Pan, X., Guangwen, L., and Wei, X. (2021). SARS-CoV-2 omicron variant: characteristics and prevention. MedComm. 2, 838–845. doi: 10.1002/mco2.110
Hindson, C. M., Chevillet, J. R., Briggs, H. A., Gallichotte, E. N., Ruf, I. K., Hindson, B. J., et al. (2013). Absolute quantification by droplet digital PCR versus Analog real-time PCR. Nat. Methods 10, 1003–1005. doi: 10.1038/nmeth.2633
Hoffmann, M., Arora, P., Groß, R., Seidel, A., Hörnich, B. F., Hahn, A. S., et al. (2021). SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cells 184, 2384–2393.e12. doi: 10.1016/j.cell.2021.03.036
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Yi, H., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/S0140-6736(20)30183-5
Isabel, S., Eshaghi, A., Duvvuri, V. R., Gubbay, J. B., Cronin, K., Li, A., et al. (2023). Targeted amplification-based whole genome sequencing of Monkeypox virusin clinical specimens. Microbiol. Spectr. :e0297923. doi: 10.1128/spectrum.02979-23
Issa, E., Merhi, G., Panossian, B., Salloum, T., and Tokajian, S. (2020). SARS-CoV-2 and ORF3a: nonsynonymous mutations, functional domains, and viral pathogenesis. MSystems 5, e00266–e00220. doi: 10.1128/mSystems.00266-20
Itokawa, K., Sekizuka, T., Hashino, M., Tanaka, R., and Kuroda, M. (2020). Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS One 15:e0239403. doi: 10.1371/journal.pone.0239403
Julenius, K., and Pedersen, A. G. (2006). Protein evolution is faster outside the cell. Mol. Biol. Evol. 23, 2039–2048. doi: 10.1093/molbev/msl081
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kojabad, A. A., Farzanehpour, M., Galeh, H. E. G., Dorostkar, R., Jafarpour, A., Bolandian, M., et al. (2021). Droplet digital PCR of viral DNA/RNA, current Progress, challenges, and future perspectives. J. Med. Virol. 93, 4182–4197. doi: 10.1002/jmv.26846
Kuchinski, K. S., Nguyen, J., Lee, T. D., Hickman, R., Jassem, A. N., Hoang, L. M. N., et al. (2022). Mutations in emerging variant of concern lineages disrupt genomic sequencing of SARS-CoV-2 clinical specimens. Int. J. Infect. Dis. 114, 51–54. doi: 10.1016/j.ijid.2021.10.050
Kupferschmidt, K., and Wadman, M. (2021). Delta variant triggers new phase in the pandemic. Science 372, 1375–1376. doi: 10.1126/science.372.6549.1375
Lambisia, A. W., Mohammed, K. S., Makori, T. O., Ndwiga, L., Mburu, M. W., Morobe, J. M., et al. (2022). Optimization of the SARS-CoV-2 ARTIC network V4 primers and whole genome sequencing protocol. Front. Med. 9:836728. doi: 10.3389/fmed.2022.836728
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). Goncalo Abecasis, Richard Durbin, and 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, T., Chen, Z., Chen, W., Chen, X., Hosseini, M., Yang, Z., et al. (2021). A benchmarking study of SARS-CoV-2 whole-genome sequencing protocols using COVID-19 patient samples. IScience 24:102892. doi: 10.1016/j.isci.2021.102892
Madhi, S. A., Baillie, V., Cutland, C. L., Voysey, M., Koen, A. L., Fairlie, L., et al. (2021). Efficacy of the ChAdOx1 NCoV-19 Covid-19 vaccine against the B.1.351 variant. N. Engl. J. Med. 384, 1885–1898. doi: 10.1056/NEJMoa2102214
Meng, B., Kemp, S. A., Papa, G., Datir, R., Ferreira, I. A. T. M., Marelli, S., et al. (2021). Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the alpha variant B.1.1.7. Cell Rep. 35:109292. doi: 10.1016/j.celrep.2021.109292
O'Toole, Á., Scher, E., Underwood, A., Jackson, B., Hill, V., McCrone, J. T., et al. (2021). "assignment of epidemiological lineages in an emerging pandemic using the pangolin tool." virus. Evolution 7:veab064. doi: 10.1093/ve/veab064
Quick, Josh. (2020). NCoV-2019 Sequencing Protocol. Available at:https://www.protocols.io/view/ncov-2019-sequencing-protocol-bbmuik6w.
Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith, A. D., Gangavarapu, K., et al. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276. doi: 10.1038/nprot.2017.066
Radhakrishnan, C., Divakar, M. K., Jain, A., Viswanathan, P., Bhoyar, R. C., Jolly, B., et al. (2021). Initial insights into the Genetic Epidemiology of SARS-CoV-2 isolates from Kerala suggest local spread from limited introductions. Front. Genet. 12:630542. doi: 10.3389/fgene.2021.630542
Rehn, A., Braun, P., Knüpfer, M., Wölfel, R., Antwerpen, M. H., and Walter, M. C. (2021). Catching SARS-CoV-2 by sequence hybridization: a comparative analysis. MSystems 6:e0039221. doi: 10.1128/mSystems.00392-21
Sanderson, T., and Barrett, J. C. (2021). Variation at spike position 142 in SARS-CoV-2 Delta genomes is a technical Artifact caused by dropout of a sequencing amplicon. Wellcome Open Res. 6:305. doi: 10.12688/wellcomeopenres.17295.1
Seifert, S. N., Schulz, J. E., Jeremiah Matson, M., Bushmaker, T., Marzi, A., and Munster, V. J. (2018). Long-range polymerase chain reaction method for sequencing the Ebola virus genome from ecological and clinical samples. J. Infect. Dis. 218, S301–S304. doi: 10.1093/infdis/jiy290
Seifert, S. N., Schulz, J. E., Ricklefs, S., Letko, M., Yabba, E., Hijazeen, Z. S., et al. (2021). Limited Genetic diversity detected in Middle East respiratory syndrome-related coronavirus variants circulating in dromedary camels in Jordan. Viruses 13:592. doi: 10.3390/v13040592
Thaweethai, T., Jolley, S. E., Karlson, E. W., Levitan, E. B., Levy, B., McComsey, G. A., et al. (2023). Development of a definition of Postacute sequelae of SARS-CoV-2 infection. JAMA 329, 1934–1946. doi: 10.1001/jama.2023.8823
Tyson, J. R., James, P., Stoddart, D., Sparks, N., Wickenhagen, A., Hall, G., et al. (2020). Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 4:2020.09.04.283077. doi: 10.1101/2020.09.04.283077
Vacca, D., Fiannaca, A., Tramuto, F., Cancila, V., La Paglia, L., and Mazzucco, W. (2022). Direct RNA Nanopore sequencing of SARS-CoV-2 extracted from critical material from swabs. Life 12:69. doi: 10.3390/life12010069
Wang, K., Li, H., Yue, X., Shao, Q., Yi, J., Wang, R., et al. (2019). MFEprimer-3.0: quality control for PCR primers. Nucleic Acids Res. 47, W610–W613. doi: 10.1093/nar/gkz351
Wassenaar, T. M., Wanchai, V., Buzard, G., and Ussery, D. W. (2022). The first three waves of the Covid-19 pandemic hint at a limited Genetic repertoire for SARS-CoV-2. FEMS Microbiol. Rev. 46:fuac003. doi: 10.1093/femsre/fuac003
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of neural network Basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20:129. doi: 10.1186/s13059-019-1727-y
Wu, F., Zhao, S., Bin, Y., Chen, Y.-M., Wang, W., Song, Z.-G., et al. (2020). A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269. doi: 10.1038/s41586-020-2008-3
Xiao, M., Liu, X., Ji, J., Li, M., Li, J., Yang, L., et al. (2020). Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 12:57. doi: 10.1186/s13073-020-00751-4
Yinda, C. K., Seifert, S. N., Macmenamin, P., van Doremalen, N., Kim, L., Bushmaker, T., et al. (2020). A novel field-deployable method for sequencing and analyses of Henipavirus genomes from complex samples on the MinION platform. J. Infect. Dis. 221, S383–S388. doi: 10.1093/infdis/jiz576
Keywords: SARS-CoV-2, surveillance, nanopore, long-range primers, sequencing, genomic epidemiology
Citation: Kandel S, Hartzell SL, Ingold AK, Turner GA, Kennedy JL and Ussery DW (2024) Genomic surveillance of SARS-CoV-2 using long-range PCR primers. Front. Microbiol. 15:1272972. doi: 10.3389/fmicb.2024.1272972
Edited by:
Pragya Dhruv Yadav, ICMR-National Institute of Virology, IndiaReviewed by:
Mohamad S. Hakim, Gadjah Mada University, IndonesiaCongyue Peng, Clemson University, United States
Copyright © 2024 Kandel, Hartzell, Ingold, Turner, Kennedy and Ussery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David W. Ussery, RFdVc3NlcnlAdWFtcy5lZHU=