 Carolin J. Schiffer
Carolin J. Schiffer Christian Grätz2
Christian Grätz2 Matthias A. Ehrmann
Matthias A. Ehrmann- 1Chair of Technical Microbiology, TUM School of Life Sciences, Technical University of Munich, Freising, Germany
- 2Chair of Animal Physiology and Immunology, TUM School of Life Sciences, Technical University of Munich, Freising, Germany
Restriction modification (RM) systems are known to provide a strong barrier to the exchange of DNA between and within bacterial species. Likewise, DNA methylation is known to have an important function in bacterial epigenetics regulating essential pathways such as DNA replication and the phase variable expression of prokaryotic phenotypes. To date, research on staphylococcal DNA methylation focused mainly on the two species Staphylococcus aureus and S. epidermidis. Less is known about other members of the genus such as S. xylosus, a coagulase-negative commensal of mammalian skin. The species is commonly used as starter organism in food fermentations but is also increasingly considered to have an as yet elusive function in bovine mastitis infections. We analyzed the methylomes of 14 S. xylosus strains using single-molecular, real-time (SMRT) sequencing. Subsequent in silico sequence analysis allowed identification of the RM systems and assignment of the respective enzymes to the discovered modification patterns. Hereby the presence of type I, II, III and IV RM systems in varying numbers and combinations among the different strains was revealed, clearly distinguishing the species from what is known for other members of the genus so far. In addition, the study characterizes a newly discovered type I RM system, encoded by S. xylosus but also by a variety of other staphylococcal species, with a hitherto unknown gene arrangement that involves two specificity units instead of one (hsdRSMS). Expression of different versions of the operon in E. coli showed proper base modification only when genes encoding both hsdS subunits were present. This study provides new insights into the general understanding of the versatility and function of RM systems as well as the distribution and variations in the genus Staphylococcus.
1. Introduction
Staphylococcus xylosus is a Gram-positive, coagulase-negative commensal of mammalian skin with high biotechnological value, as it is commonly used as starter organisms in food fermentations (Ravyts et al., 2012; Leroy et al., 2017). However, over the past years, studies have associated S. xylosus with infections, i.e., bovine mastitis infections, as well (Supré et al., 2011; Condas et al., 2017). In addition, a growing number of studies is addressing the potential of coagulase-negative staphylococci (CoNS) to serve as reservoir for virulence-associated factors (Otto, 2013; Rossi et al., 2017; Heilmann et al., 2019; Marincola et al., 2021). Considering that spread of virulence and acquisition of antibiotic resistance genes are emerging topics nowadays (Karkman et al., 2018; Heilmann et al., 2019; Lindsay, 2019), it is of great concern that such genes can fuel pathogen evolution of highly pathogenic species such as S. aureus when transferred via horizontal gene transfer (HGT). Therefore, it is important to understand the probability and extent to which HGT occurs as well as the natural barriers that bacteria possess against HGT. One way, in which bacteria protect themselves from the uptake of exogenous, foreign DNA is by restriction modification (RM) systems. Active RM systems have been shown to be one of the major factors preventing inter- and intraspecies HGT (Tock and Dryden, 2005; Lindsay, 2014; Atack et al., 2018). Thereby, they shape bacterial genome evolution and impact host adaption mechanisms (Kobayashi, 2001; Park et al., 2022). The basic principle of distinguishing between foreign and own DNA is the site-specific modification of the individual DNA by methyltransferases combined with the expression of effective restriction endonucleases that recognize and cleave any unmodified, foreign DNA (Wilson and Murray, 1991; Sadykov, 2016).
Four major types of bacterial restriction (modification) systems (type I - IV) have been described to date. They are distinguished based on their enzymatic subunits, mechanism of action, DNA specificity/sequence recognition motifs as well as co-factor requirements and reaction conditions (Wilson and Murray, 1991; Loenen et al., 2014; De Ste Croix et al., 2017; Oliveira and Fang, 2021).
Type I systems are heterooligomeric complexes composed of three subunits, a methyltransferase (hsdM), modifying the host DNA by adding a methyl group in a specific sequence, a restriction endonuclease cleaving non-modified DNA (hsdR) and a specificity unit (hsdS) determining the recognition sequence of the system (Murray, 2000; Loenen et al., 2014; Gao et al., 2020). Hereby, hsdM and hsdS are usually transcribed from a common promoter, while hsdR is under the control of its own promoter (Murray, 2000). HsdS-HsdM complexes are active in methylation within the recognition sequence while HsdS-HsdM-HsdR complexes are active in restriction at unspecified sites on the unmodified DNA (Gao et al., 2020). Their cleavage sites are generally far from the methylation sites. How these sites are determined has not yet been fully clarified (Ishikawa et al., 2009).
Currently, type I RM systems are subdivided into five families (IA – IE) based on sequence homology and genetic complementation (Titheradge et al., 2001; Cooper et al., 2017). While hsdM and hsdR are very conserved within one family, with sequence similarity values reported between 70 - 90%, hsdS consists of two highly variable regions (Murray, 2000; Chin et al., 2004; Monk et al., 2015; Cooper et al., 2017). These variable regions encode the target recognition domains (TRDs) of HsdS, each of them specifying one half of the bipartite recognition motif (Murray, 2000; Loenen et al., 2014; Costa et al., 2017). The motif comprises two specific 3-4 bp long sequences, separated by a 5-8 bp long, non-specific, spacer sequence (Loenen et al., 2014; Costa et al., 2017; Atack et al., 2018). Since individual TRDs can shuffle between different HsdS proteins through various rearrangements between hsdS alleles, an extensive variety of target recognition motifs exists (Furuta et al., 2011; Loenen et al., 2014; Cooper et al., 2017; Atack et al., 2020).
In contrast to the polycistronic organization of type I systems, type II systems mostly include two separate enzymes: a methyltransferase, that targets a specific base in a specific sequence and a restriction endonuclease that cleaves DNA either at a defined site within or by the recognition sequence. Recognition motifs of type II systems are usually 4-8 bp in length and palindromic (Pingoud et al., 2014). A particular representative of the class are type IIG systems, in which the restriction enzyme and methyltransferase are transcribed from a single gene to form a contiguous polypeptide (Pingoud et al., 2014).
Type III systems are heterooligomeric complexes consisting of a methyltransferase that also determines sequence specificity (mod) and an endonuclease (res), that carries out restriction cleavage near an unmethylated recognition sequence when bound to the Mod subunit (Rao et al., 2014). Type III systems usually recognize short (5-6 bp), asymmetric motifs and have been reported to occur only rarely in staphylococci such as S. aureus (Sadykov, 2016; Lee et al., 2019).
Type IV systems are anti-methylation systems and only composed of one to two endonucleases. They distinguish themselves from type I to III systems as they are not associated with a respective methyltransferase. Hereby, type IV restriction enzymes solely cleave DNA harboring a particular type of methylation with a loose sequence specificity (Loenen and Raleigh, 2014). They are further thought to defend genomes against competing genome methylation systems by cell suicide (Fukuda et al., 2008).
RM systems are also relevant in respect to the genetic manipulability of cloning hosts. Particularly wildtype strains often possess strong barriers to incoming, exogenous DNA and are therefore much harder to transform compared to laboratory strains. An approach to overcome the hosts RM systems is plasmid artificial modification (PAM). PAM mimics the host target strains methylation profile by pre-passaging plasmid DNA through modified E. coli strains and has been shown to increase the transformation efficiency of many bacteria (Deng et al., 2000; Suzuki and Yasui, 2011; Monk et al., 2015).
The fact that methylation patterns not only serve as barrier to invading DNA but also play an important role in the context of epigenetics, should not go unmentioned here. There is a growing number of studies addressing the influence of DNA methylation mediated by the methyltransferases of RM systems on the regulation of important cellular mechanisms including replication control, the expression of virulence factors and phenotypes such as biofilm formation and host colonization as well as the phase-variable expression of genes which in turn enables cells to switch flexibly between different physiological states (Furuta et al., 2014; Atack et al., 2018, 2020; Nye et al., 2020; Yano et al., 2020; Oliveira and Fang, 2021).
The methylome of more well-known members of the genus Staphylococcus such as S. aureus and S. epidermidis has been extensively characterized in the past (Sadykov, 2016; Lee et al., 2019). Only little is known about the less well studied species such as S. xylosus, for which the presence of RM systems has only been named sporadically in a few studies (Harrison et al., 2013) but has never been characterized in detail. In this study we determined the methylome, thus all methyl-modified DNA sequences in selected S. xylosus strains using single molecule real-time (SMRT) sequencing in order to obtain more information about methylation patterns and the presence of RM systems within the species.
2. Materials and methods
2.1. Bacterial strains, growth, reagents
All bacterial strains, oligonucleotides and plasmids used in this study are listed in Supplementary Table 1. Escherichia coli and Staphylococcus sp. were routinely cultured at 37°C, 200 rpm in Lysogeny Broth (LB, tryptone 10 g/l, yeast extract 5 g/l, NaCl 5 g/l) and Trypticase soy broth (TSB, casein peptone 15 g/L, soy peptone 15 g/L, yeast extract 3 g/L), respectively, unless required and stated otherwise. For the respective agar plates, the liquid media were solidified with 1.5% agar (Carl Roth). All antibiotics were purchased from Carl Roth and used at the following concentrations: ampicillin (100 μg/ml), kanamycin (20 μg/ml). Oligonucleotides were obtained from Eurofins Genomics, Germany. Restriction enzymes, Gibson assembly mix, T4 DNA ligase as well as PCR components (Q5 high fidelity PCR kit) were obtained from New England Biolabs (NEB). For plasmid isolation, DNA gel extraction and PCR product purification, the NEB Monarch Plasmid Miniprep, DNA gel extraction and PCR and DNA Cleanup kits were used, respectively.
2.2. Transformation
Transformation of E. coli strains was performed by washing E. coli cells electrocompetent using standard protocols (Wood, 1983). Basically, 100 ml of overnight culture was harvested during mid-exponential phase (OD600 0.5-0.7), placed on ice for 10 min and centrifuged at 5,000 x g, 4°C for 10 min. The supernatant was poured off and the pellet was resuspended in 100 ml of 10% glycerol. Centrifugation and resuspension steps were repeated twice more with decelerating volumes of resuspension buffer and cells were finally resuspended with 500 μl of 10% glycerol. Transformation of E. coli cells by electroporation was performed in a 0.1 cm cuvette (Gene pulser MicroPulser cuvette) at 1.8 kV (MicroPulser electroporator, Bio-Rad Laboratories).
2.3. Expression of type I and type II modification enzymes in E. coli
Staphylococcus xylosus methyltransferases were heterologously expressed in E. coli. The respective genes were integrated into E. coli strain K12 DC10B at site-specific locations (attB sites) of the chromosome, in a single cloning and chromosomal integration step (St-Pierre et al., 2013). We hoped that the expression of modification genes from the chromosome rather than multicopy plasmid, would result in less metabolic burden for the cell, a stable expression and subsequent complete modification (Englaender et al., 2017). The applied method is based on bacteriophage integrases mediating site-specific insertions of the genes of interest into prokaryotic chromosomes (attB sites). Within this study, the integrases of coliphages λ (pOSIP-KL) and 186 (pOSIP-KO) were used. The type II methyltransferase of S. xylosus TMW 2.1324 was amplified using primers PN25_MT_F and RS_MT_R at first, followed by a subsequent PCR reaction complemented with the dimerized oligonucleotides of promoter PN25 and primers PN25_MT_F and RS_MT_R. The promoter-gene construct was excised from an agarose gel, purified, and ligated into the linearized (SacI/PstI) vector pOSIP-KL. The different variants of type I systems of TMW 2.1023 and TMW 2.1324 (hsdSMS/hsdMS/hsdMStr) were ligated into vector pOSIP-KO (KpnI/SphI) in the same way, using primer pairs PN25_hsdSMS_F/PN25_hsdMS_F and RS_hsdS_R/RS_hsdS_tr_R at first, followed by overamplification with RS_PN25_F and RS_hsdS_R/RS_hsdS_tr_R, respectively. Integration of the pBla-MTase construct was performed by amplifying the promoter from plasmid pE-Flp using primers vec_pBla_1F and Bla_Mtase_1R and the methyltransferase of TMW 2.1324 using Bla_Mtase_2F and Mtase_186_2R with subsequent Gibson assembly of all PCR products into the linearized vector pOSIP-KO (KpnI/PstI).
Assembled vectors were transformed into E. coli by electroporation, and FLP-mediated excision of the backbone was achieved by transforming cells with plasmid pE-FLP. Integration, screening for successful transformants, excision and final screening for successful integrants were performed according to the step-by-step protocol provided by Cui and Shearwin (2017).
2.4. Real-time quantitative reverse transcription PCR
For RT-qPCR experiments, RNA was isolated from the E. coli CM strains at first in three biological replicates each. Therefore, 3 ml of liquid culture were harvested during early exponential phase and RNA was extracted using the Monarch® Total RNA Miniprep Kit (NEB) according to the manufacturer’s instructions. As recommended in the NEB protocol, two DNase I digestions were performed on all samples to remove residual gDNA, namely a one-column treatment as well as an in-tube treatment after purification. In-tube digestion was performed by incubating 1.35 μg of RNA at 25°C for 5 min with 2.73 Kunitz units DNAse I and 0.1x RDD buffer from the RNase-Free DNase Set (QIAgen) in a total volume of 40 μl. DNAse I was subsequently inactivated by adding 5 μl of a 25 mM EDTA solution and incubation at 75°C for 5 min. Complementary DNA (cDNA) was generated from 180 ng DNAse-treated RNA from each sample using the QuantiTect Reverse Transcription Kit (QIAgen) according to the manufacturer’s protocol. Additionally, a no reverse transcriptase (NRT) was generated for each sample. After reverse transcription, the cDNA was diluted 1:12 with nuclease-free water (Omega Bio-tek). qPCR was performed on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.) using 6 ng of cDNA, the Luna Universal qPCR Master Mix (NEB) and the primer pairs shown in Supplementary Table 2. Cycling parameters were set to 95°C (1 min), 40 cycles of 95°C (15 s) and 60°C (30 s with plate read on SYBR channel) each, and a melt curve from 60 to 95°C with an increment of 0.5°C per 5 s and plate read on SYBR channel after each increment.
2.5. SMRT sequencing
Single molecule real-time (SMRT) sequencing was performed to identify modified bases of S. xylosus and genetically modified E. coli strains (Clark et al., 2012). DNA isolation was performed using the E.Z.N.A. Bacterial DNA-kit (Omega Bio-tek) according to the manufacturers instruction, except that lysostaphin (0.5 mg/ml) was included into the lysis buffer of the staphylococcal samples to weaken the cell wall. Library construction and sequencing (PacBio RS II) of S. xylosus followed the protocol described by Schiffer et al. (2019). E. coli sequencing was performed on a PacBio Sequel instrument (SMRT cell 1M), partly at the functional genomics center Zurich (ETH Zürich), and partly at the research unit for environmental genomics Munich (Helmholtz Zentrum München). Therefore, the Sequel® Binding Kit 3.0 (Pacific Biosciences of California) was used and the libraries were size-selected to around 6 to 7 kb. SMRT Analysis version 7.0 (Pacific Biosciences) was used for assembly (HGAP4), base modification and motif analysis of S. xylosus, SMRT Link version 10.1 for assembly, base modification and motif analysis of E. coli. For S. xylosus the assembled genomes were used as their own reference, while for E. coli, the assembly of strain DH10B available on NCBI (NC_010473) was used as a reference.
2.6. Bioinformatic analysis and data availability
Sequence alignments were made using CLC main workbench 8.1.41 with the built-in Clustal Omega plugin and subsequent construction of pairwise comparison matrices and phylogenetic trees (neighbor-joining). Blasting against two databases [NCBI’s conserved domain database (Marchler-Bauer et al., 2015) and the restriction enzyme database REBASE (Roberts and Macelis, 2001)] was used to confirm the affiliation of the identified enzymes to one of the restriction modification families, to identify enzymatic domains and to search for RM systems with the same DNA target sequence. The Blast Diagnostic Gene finder tool (BADGE) was used for comparative genomics in order to match the corresponding RM genes and modification patterns (Behr et al., 2016). The online available NCBI blastn and blastp tools were used to search for RM components besides the ones already annotated. The protein fold recognition server PHYRE2 (Kelley et al., 2015) helped in predicting secondary structure conformation of the identified polypeptides. In a previous study, a full proteome dataset was generated for S. xylosus TMW 2.1023 and TMW 2.1523 (Schiffer et al., 2021), which was taken into account in this study to verify the expression of single genes (Supplementary Table 3). The dataset is available under the identifier PXD029728 at the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2019). All S. xylosus genome sequences are deposited at GenBank under the accession numbers provided in Supplementary Table 1. For in silico analysis, additionally available genomes on the NCBI server that were sequenced using PacBio technology were included into the analysis. The respective accession numbers are also included in Supplementary Table 1. The assembled sequences of the E. coli CM strains as well as the base modification analysis outputs were submitted to GenBank, too.
3. Results
3.1. Analyzing the methylome of S. xylosus
In order to better understand the presence of active RM systems in S. xylosus, we determined the DNA methylation profile of selected S. xylosus strains using PacBio SMRT sequencing technology and further explored the occurrence of restriction modification systems by detailed bioinformatic analysis of the genomes. Hereby, we were able to assign the respective modification and restriction enzymes to the identified methylated DNA sequences with a high degree of certainty as mostly not more than one respective open reading frame was available for choice. Table 1 provides an overview of identified RM systems and the corresponding modification patterns. Supplementary Table 4 displays the full base modification output of the sequenced strains. Six other PacBio sequenced strains of S. xylosus listed on REBASE were included into the overview to provide a more comprehensive picture of the prevalence of RM systems within the species.
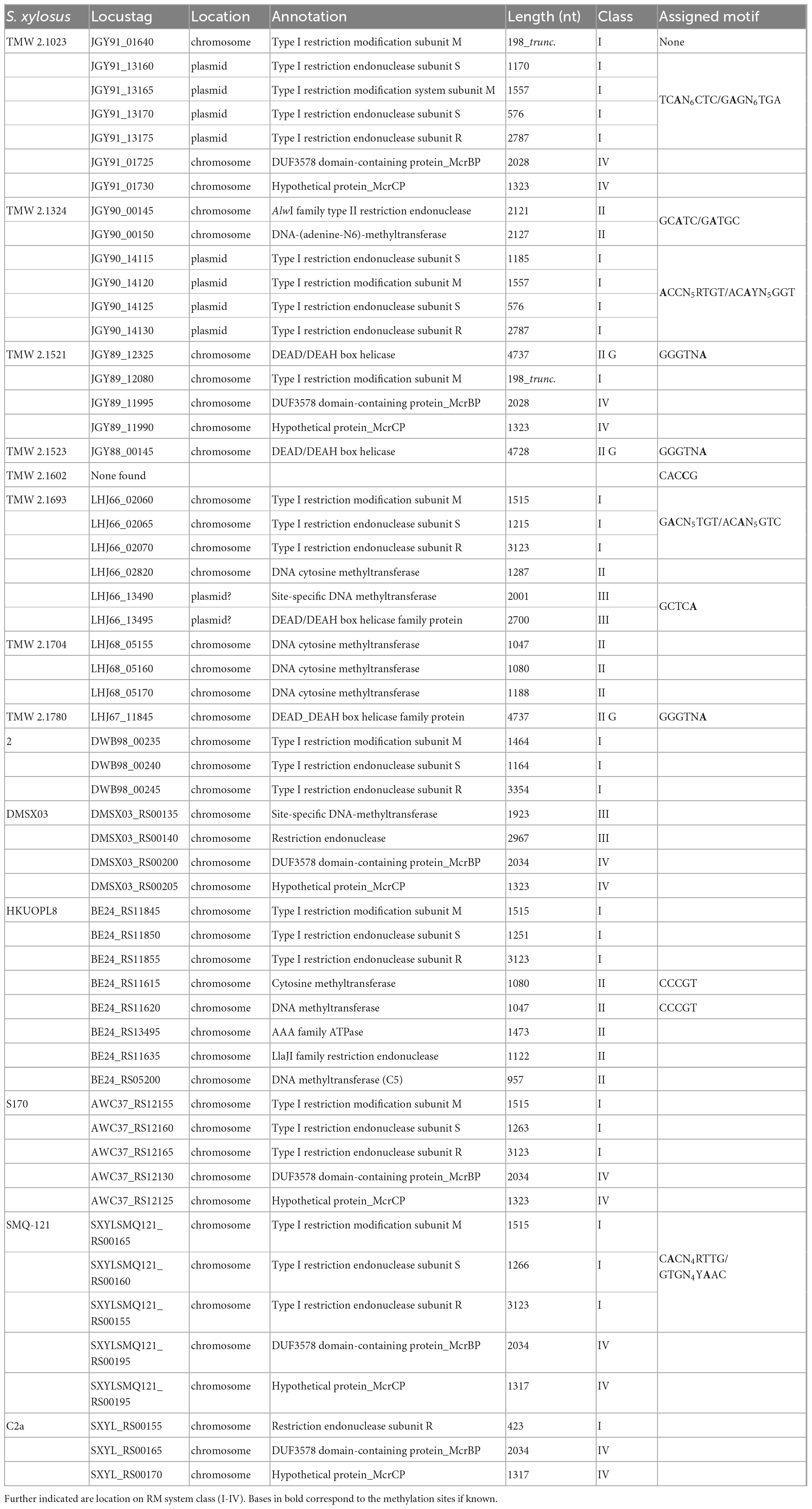
Table 1. Overview of present restriction modification systems in selected S. xylosus strains and the respective base modification motifs derived from SMRT sequencing.
In terms of type I RM systems, out of the 14 strains analyzed, seven carry a complete type I RM system in their genome (presence of hsdM, hsdS, and hsdR). None of the S. xylosus strains harbors more than one type I RM system, nor any orphan hsdS genes. Notably we found two different types of operons, five strains harbor a chromosomally encoded type I system organized as a contiguous three-gene (hsdMSR) operon while two strains (TMW 2.1023 and 2.1324) carry a plasmid encoded four-gene (hsdRSMS) operon.
Despite type I base modifications, other motifs such as GCATC, a common type II motif with more than 600 hits on REBASE, present across a wide range of species such as Mycoplasma bovis, Mannheimia haemolytica and Streptococcus pneuomoniae were also identified in strains such as TMW 2.1324. Interestingly, three S. xylosus strains (TMW 2.1521, 2.1523 and 2.1780) possess a type IIG system, which comprises a single enzyme, mediating methyltransferase as well as endonuclease activity. The detected type IIG systems are all associated with the same modification pattern (GGGTNA) and gene sequence analysis did not reveal any frameshifts in the gene sequences. Furthermore, data derived from whole proteome analysis (Schiffer et al., 2021) confirmed the expression of a functional type IIG enzyme of the expected amino acid sequence in TMW 2.1523 (Supplementary Table 3).
Blasting of methyltransferase genes against the REBASE database also revealed the presence of type III systems in strains TMW 2.1693 (LHJ66_13490-95) and DMSX03 (DMSX03_RS00135-40). The only strain, for which no respective methyltransferase could be assigned to the determined modification pattern is TMW 2.1602. According to its kinetic signature during PacBio sequencing, the strain modifies the motif CACCG, which could be a type II or type III motif. Nevertheless, using comparative genomics, no strain-specific methyltransferases or endonucleases were identifiable for this strain, nor did the online available Restriction-ModificationFinder (Roer et al., 2016) identify any RM systems in the genome of TMW 2.1602. The motif is not listed on REBASE either, therefore no further conclusions on this strain’s methylation can be drawn at the time. Genome analysis additionally revealed, that some S. xylosus strains, namely TMW 2.1693, TMW 2.1704, HKUOPL8 and S04010, encode cytosine methyltransferases, probably mediating 5-methylcytosine (m5C) modification. However, checking whether these enzymes are active or which motifs they modify is difficult since it is challenging to use SMRT sequencing technology to distinguish m5C from cytosine (Clark et al., 2012; Loenen et al., 2014).
Lastly, endonucleases belonging to the type IV RM family were identified in strains TMW 2.1023, TMW 2.1521, DMSX03, S170, SMQ-121 and C2a (Table 1). The identified type IV systems encompass two endonucleases which are encoded in tandem with overlapping reading frames on the chromosome. The type IV system includes a DUF3578 domain and resembles, in its sequence structure, the well-characterized, two subunits containing McrBC 5-methylcytosine restriction system of Escherichia coli K-12.
3.2. In silico analysis of type I RM systems reveals a new operon structure
Type I RM systems were identified in seven out of the 14 analyzed S. xylosus strains, making their presence within the species non-ubiquitous. While a common gene order of the hsd operon (hsdRSM/hsdMSR) is chromosomally encoded in strains TMW 2.1693, 2, HKUOPL8, S170 and SMQ-121, an unusual gene arrangement (hsdRSMS) was found on the plasmids harbored by TMW 2.1023 and TMW 2.1324, with two genes of different lengths, both annotated as hsdS surrounding the methyltransferase (hsdM). To confirm that none of the hsdS subunits is truncated, the proteomic dataset obtained from a previous study was consulted again, confirming the expression of both hsdS subunits in TMW 2.1023 (Supplementary Table 3). The first hsdS (hsdS_short) subunit of the system is 191 aa in length and the second one (hsdS_long) around 390 - 400 aa. Furthermore, the 3′ end of hsdM overlaps by 8 bp the 5′ end of the second hsdS_long subunit. Because of the organization of the ORFs directly to one another (hsdS-hsdM-hsdS), with the hsdM-hsdS 8 bp overlap and a conserved Shine-Dalgarno binding site preceding each ORF, it can be assumed that the genes are co-transcribed under the control of a single promoter in both S. xylosus strains. We also note that putative promoter sequences (canonical consensus σ70 -35/-10) are present in front of hsdR and hsdS_short. Polycistronic gene organization facilitates enhanced regulatory control through translational coupling between genes of related functional partners to control subunit stoichiometry and was previously described for type I restriction systems (Deng et al., 2000; Roberts et al., 2012). Interestingly, hsdRSMS systems are part of a large plasmid in both S. xylosus strains. Blasting the individual genes of the operon reveals that the system is located on at least eleven further staphylococcal plasmids (hsdRSMSPL) as well as it was found that some staphylococcal species also carry the four gene operon on their chromosome (hsdRSMSCHRM). Yet, it appears as if hsdRSMSCHRM is mostly encoded on mobile genetic elements (MGEs) on the chromosome, often being part of staphylococcal cassette chromosome (SCC) genomic islands. Also, recombinases are frequently encoded just a few genes upstream or downstream of the operon. Table 2 lists all strains encoding the four gene hsdRSMS operon on a plasmid as well as a selection of strains that have the operon encoded on their chromosome.
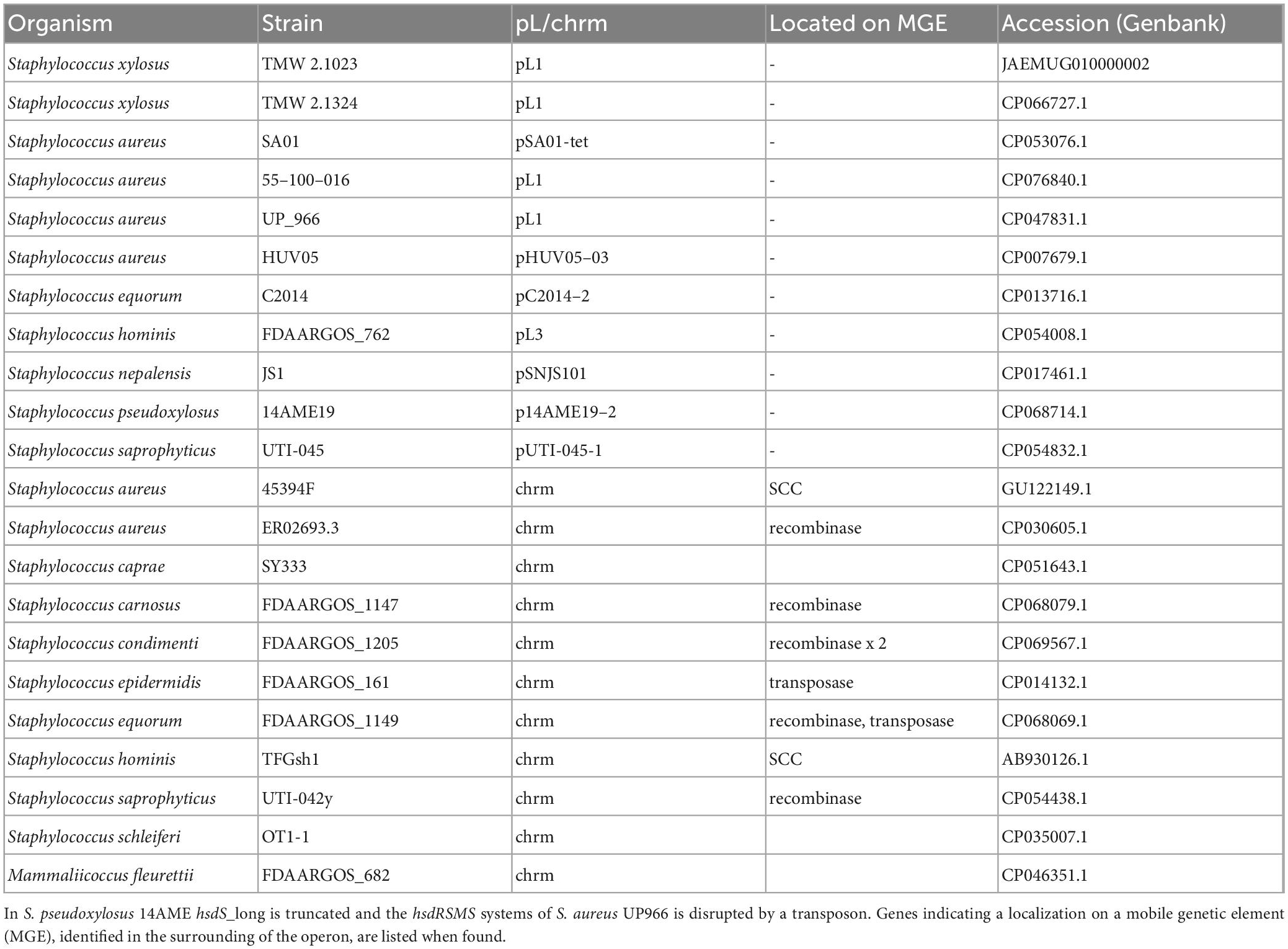
Table 2. Overview of organisms harboring the hsdRSMS systeme either on a plasmid (pL) or on the chromosome (chrm).
Alignments and gene topology analysis was performed to classify all discovered S. xylosus type I RM systems (three and four gene operons) into one of the five existing type I families (A-E). Percent identity and distant values for alignments of hsdR and hsdM with the reference genes of the respective families are provided in the comparison matrix of Supplementary Figure 1. Hereby, hsdM and hsdR of the S. xylosus three-gene hsdMSR operons are closest to the reference genes of family type ID RM systems (StySBLI) sharing 50% (hsdM) and 40% (hsdR) percent sequence identity, respectively. An exception is S. xylosus strain 2, which cannot clearly be categorized as it carries a hsdMSR system with identity values below 30% to any of the reference genes. Methyltransferases (hsdM) and endonucleases (hsdR) of the hsdRSMS four gene operon display as little as 7% identity to family IB (M.EcoAI) and ID (M.StySBLI) and a maximum of 48% (hsdM) and 40% (hsdR) identity to the type IC reference genes (EcoR124I). Interestingly, intraspecies percent identity values of hsdM genes, namely hsdM of S. xylosus hsdRSMS operons and hsdM of S. xylosus hsdMSR operons were all below 10%, substantiating the hypothesis that the hsdRSMS operon represents a differentiated system. The phylogenetic trees provided in Figure 1 (hsdM) and Supplementary Figure 2 (hsdR) further reveal that neither hsdR nor hsdM of the hsdRSMS operon cluster with any of the type I family reference genes. In contrast, the phylogenetic distance of the three-gene operon (hsdMSR) of the other S. xylosus strains to type ID systems is smaller and they group together. Interestingly, the trees even distinguish between the two types of hsdRSMS systems found, with hsdM and hsdR of hsdRSMSPL systems showing a distinct phylogenetic distance to hsdRSMSCHRM systems. The only exception here is S. equorum FDAARGOS_1149, that carries a hsdRSMSCHRM system clustering together with the hsdRSMSPL systems. Alignments of each gene of hsdRSMSPL separately revealed that hsdM, hsdR and hsdS_short are well conserved along the entire sequence (Figure 2A). Alignments of hsdS_long resulted in a typical conservation plot often seen for hsdS subunits, with three conserved regions (N-, C -terminal, central) flanking two variable regions, each dedicated as one TRD (Figures 2B, C). No tetra amino acid repeats as previously described for type IC hsdS subunits (Adamczyk-Popławska et al., 2003) could be identified in the central conserved region. We did identify two short repeating stretches in the central region, though (2x LEEQK), as well as part of the central sequence is repeated in the N- and C- terminal conserved regions, respectively, (Figure 2C). Important to mention is that long and short hsdS subunits of one hsdRSMS operon don’t share any common, homologous regions. Further, the repeats found in the long hsdS subunits do not exist in the short ones. Taking secondary structure into consideration a typical protein fold was predicted by PHYRE (Kelley et al., 2015) for hsdS_long with the two TRDs connected by alpha helices/coiled coil structures in an antiparallel order (Figure 2D), whereas for hsdS_short, a strikingly similar structure to a halfsize hsdS subunit is predicted. Referring to the NCBI Conserved Domain Database (Marchler-Bauer et al., 2015), hsdS_long subunits consist of two TRDs and hsdS_short of one. However, while this result is consistent for hsdS_long genes as they comprise two variable regions flanked by conserved regions, it is less clear for hsdS_short as the entire sequence is conserved, not harboring any variable parts. Furthermore, when blasting the individual TRDs of hsdS_long, it yields hits on other hsdS subunits, emphasizing the dynamic, interallelic recombination of single TRDs between hsdS subunits. However, a contrary result is obtained when hsdS_short is blasted against the NCBI database. According to the results obtained, hsdS_short seems to never occur as part of a hsdS_long subunit. This fact clearly votes for hsdS_short not being a halfsize or truncated hsdS subunit but rather an individual gene with a specific function, not flipping and recombining with hsdS_long subunits. One last noteworthy fact is, that it has been reported previously that hsdS genes, even if they are not part of the same family, share high homology (> 50%) among their variable regions determining the TRDs, if they recognize the same nucleotide motif (Murray, 2000). According to REBASE, the type I system of numerous E. coli strains (e.g., NCTC9029) as well as Anaerobiospirillum thomasii NCTC12467 recognize the same motif as S. xylosus TMW 2.1023 as well as certain S. aureus strains (AUS0325, WBG8366, MRSA - AMRF 6, MRSA - AMRF 4, ER09113.3) and TMW 2.1324 share a type I system recognizing the same target DNA sequence. When aligning the TRDs accordingly, percent identity values of 69% (N-TRD) and 63% (C-TRD), respectively, were obtained for TMW 2.1324 and the HsdS subunits of the S. aureus strains, compared to 20% amino acid sequence identity when aligning the TMW 2.1023 TRDs with HsdS of the S. aureus strains. In contrast, the TRDs of TMW 2.1023 did not show any significant similarity to neither the HsdS subunits of A. thomasii NCTC12467 nor E. coli NCTC9029, despite recognizing the same sequence motif (percent identity values around 21%).
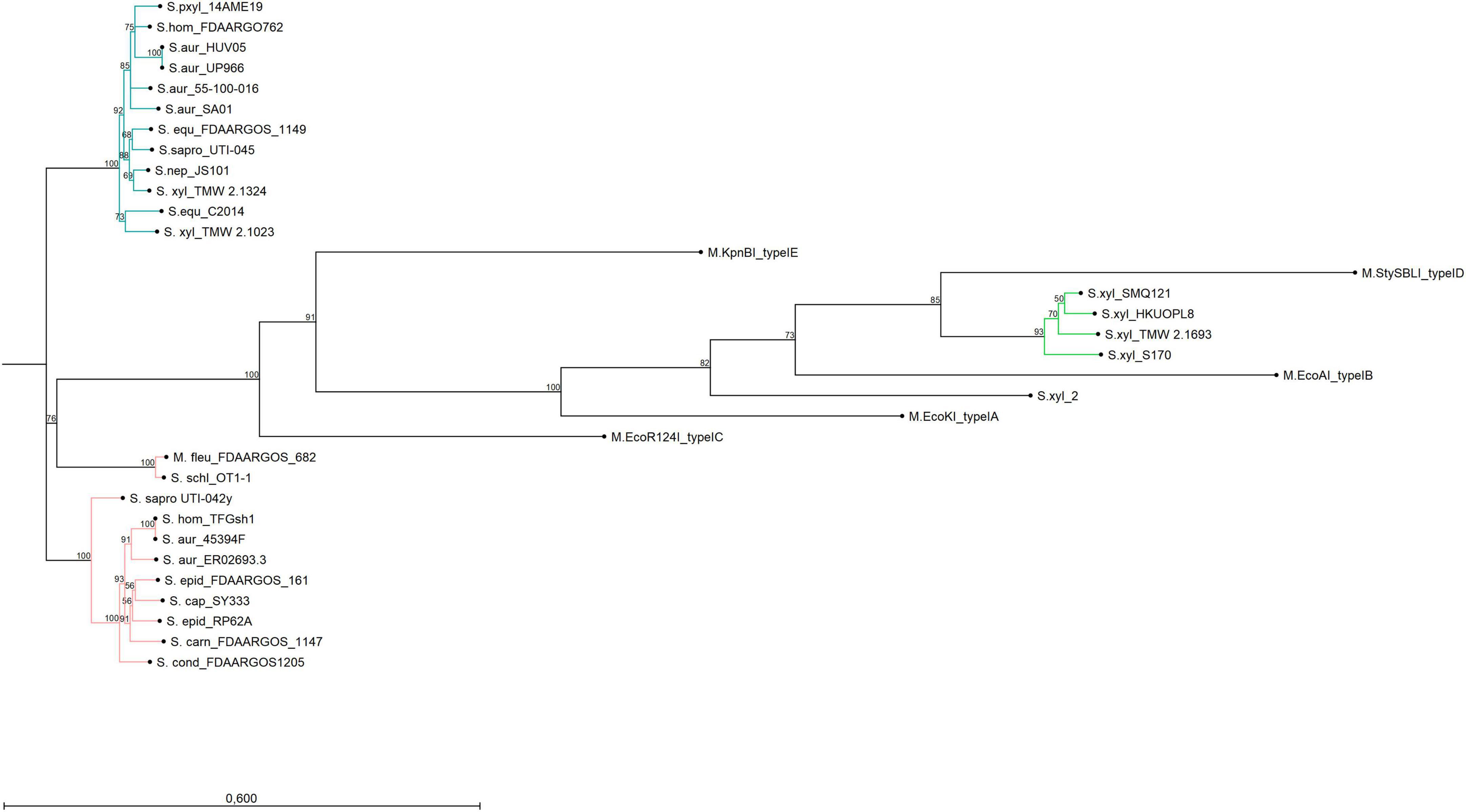
Figure 1. Neighbor-joining tree displaying the phylogenetic topology of hsdM from type I RM systems of different bacterial organisms and strains. The turquoise group represents hsdM genes of hsdRSMSPL systems, the green group belongs to S. xylosus chromosomal hsdMSR systems and the group in rose encompasses hsdM genes of hsdRSMSCHRM systems. The only outlier is hsdM of S. equorum FDAARGOS_1149 which is chromosomally encoded but clusters with the plasmid-based group. Reference genes of type I systems (A-E) were included into the Figure. The bar indicates 60% sequence divergence.
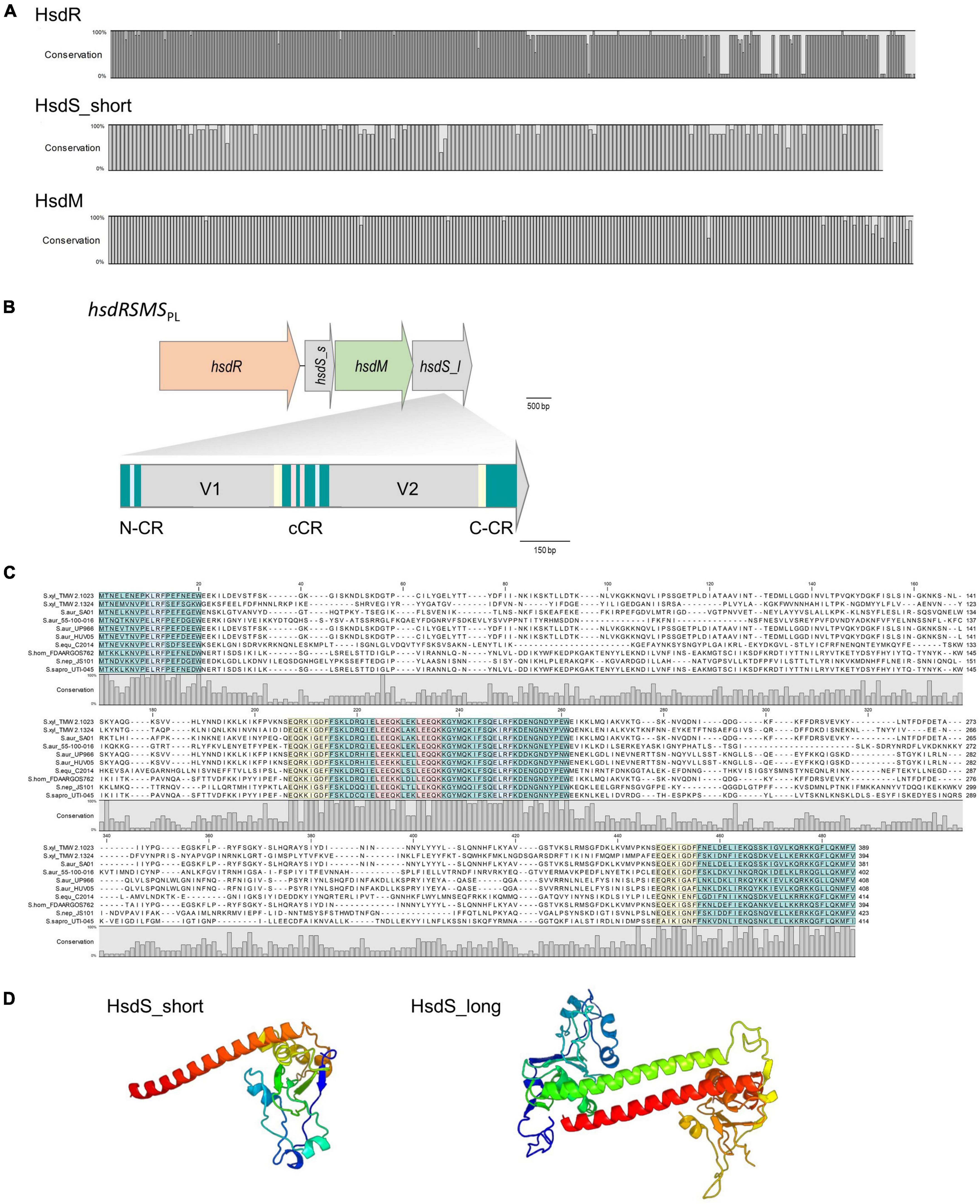
Figure 2. (A) Conservation plots based on amino acid alignments of HsdR, HsdM and HsdS_short derived from hsdRSMSPL systems (11 sequences each were aligned, full alignments are shown in Supplementary Figures 4, 5). (B) Gene arrangement of the four genes containing hsdRSMS operon. hsdS_s = hsdS_short, hsdS_l = hsdS_long. HsdS_long is composed of two variable regions (V1 and V2) as well as an N-terminal (N-CR), C-terminal (C-CR) and central conserved region (cCR). All conserved regions are marked in turquoise. Other repeating sequences are marked in red, blue and yellow, respectively. Note the frameshift at the junction between coding regions: in hsdRSMS, hsdM overlaps hsdS_long by 8 bp (C). HsdS_long subunit alignment on amino acid level of all hsdRSMSPL systems listed in Table 2. Conservation plot shows the low conservation in the variable region as well as the repeating sections (blue, yellow, red) of the conserved (turquoise) regions. (D) Protein fold prediction based on PHYRE for HsdS_short and HsdS_long exemplarily for strain TMW 2.1023. The coiled coil region (red/green) displays the conserved region connecting the two TRDs which are colored in blue and yellow to orange (99.9% modeling confidence).
3.3. Expression of modification systems in E. coli
In order to confirm the specificity of selected methyltransferases and to characterize the function of the newly detected hsdRSMS system in more detail, methyltransferases and specificity units were heterologously expressed in E. coli. As expression host functioned E. coli DC10B, a dcm - negative K12 derivate, unable to methylate cytosine. Modification enzymes were integrated into and expressed from the chromosome as the expression on a plasmid has previously been associated with instability and inadequate base modification (Lee et al., 2019). To determine the most suitable promoter, which provides a complete methylation of the target DNA but does not pose a too heavy burden for the cell, the less complex type II system of TMW 2.1324 was used as a test system. Therefore, the corresponding type II methyltransferase gene (motif GATGC/GCATC) was integrated into the E. coli chromosome transcribed from two different constitutive promoters, the β-lactamase promoter Pbla as well as the T5 coliphage promoter PN25. Subsequent sequencing and base modification analysis revealed that only 42-76% of the existing motifs were modified when Pbla was used (Table 3, E. coli CM2) compared to 99.7% modification when the gene is under the control of PN25 (E. coli CM93). The difference in methylation propensity is also clear when digesting isolated plasmid DNA from these two strains with SfaNI. SfaNI recognizes the same motif as the type II system of TMW 2.1324, thus, proper modification by the respective methyltransferase should protect the plasmid from restriction. While a complete restriction digest was visible on the gel when plasmid of E. coli DC10B was used, an incomplete digest was detectable for plasmids isolated from E. coli CM2 (Pbla) and no digestion was visible when plasmid was isolated from E. coli CM93 (PN25) (Supplementary Figure 3).
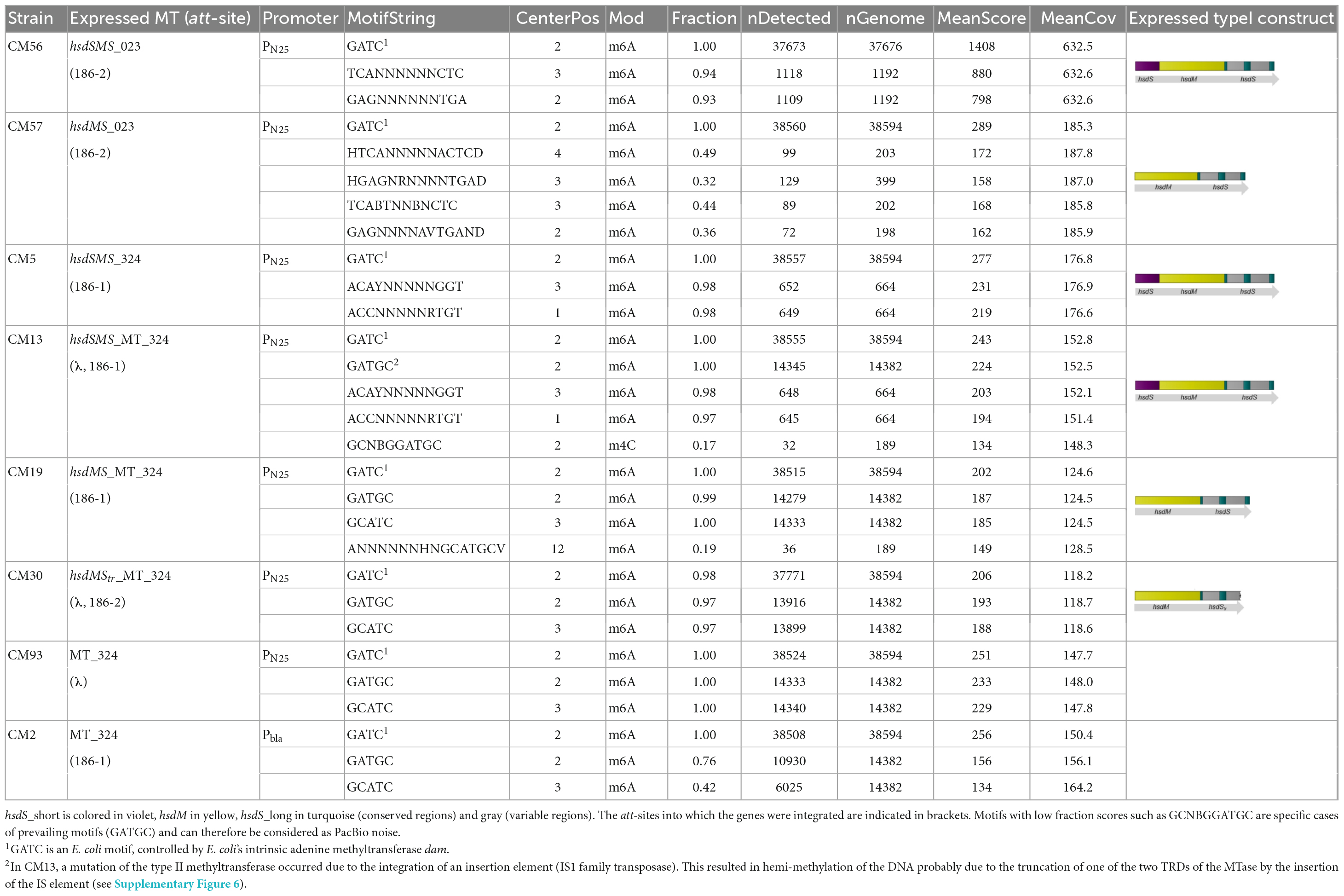
Table 3. Base modification analysis of heterologously expressed S. xylosus methyltransferases in E. coli using different gene combinations and promoters.
Expressing from promoter PN25, modification genes of the newly detected four gene type I operons were integrated into the chromosome of E. coli, namely the full hsdSMS system of S. xylosus strains TMW 2.1023 (E. coli_CM56) and 2.1324 (E. coli CM5 and CM13), respectively, as well as hsdMS only, neglecting hsdS_short (E. coli CM57 and CM19). Results are listed in Table 3. In both cases, the expected motif was only properly methylated when the full hsdSMS operon was expressed in E. coli (CM56, CM13/CM5). On the contrary, if hsdS_short was missing, random motifs and/or modified motifs with a changed specificity and low fraction scores appeared (CM19, CM57). At this point, it should also be mentioned that attempts to complement hsdS_short in CM19 and CM57 to restore the full methylation specificity of the type I system remained unsuccessful. Expression of hsdS_short under the control of PN25 from the common E. coli expression vector pET3a resulted in toxic effects on the cell. Only cells in which IS elements had inserted into the construct or the plasmid had recombined so that expression of the protein no longer occurred, were able to survive. We could not fully elucidate the exact cause of the toxic effect of hsdS_short in this study, but the data reconfirm how profoundly an artificial methylation pattern can interfere with the regulation of essential processes in the cell.
To confirm that the observed differences in methylation are not caused by differences in gene expression, RT-qPCR was employed to quantify transcription levels. The data has been normalized to three housekeeping reference genes (cysG, hcaT, and recA) to be able to compare inter-strain mRNA transcription levels and to control for errors between samples. The relative gene expression between the different E. coli CM strains (fold change) was calculated from the mean Cq values between all biological replicates using the 2 ΔΔCq method. The normalized mean quantification values clearly showed that all heterologously expressed constructs (hsdSMS, hsdMS) were transcribed in the respective CM strains in similar amounts (Supplementary Table 5). Precisely, the expression levels of hsdM and hsdS of the hsdMS constructs in comparison with the expression levels of the corresponding hsdSMS constructs are within a range of 0.36-fold to a maximum of 1.23-fold expression for hsdM and between 0.52 and 1.06 for the respective hsdS genes (Supplementary Table 6). Due to the small differences in expression levels, we conclude that the differences in methylation are not due to a reduced expression of the genes. Yet, it should be mentioned at this point that no data on translation are available, as we did not determine the proteins in this case.
The function of hsdS_long and the influence of the presence of hsdS_short was further tested in another experiment. For type IC RM systems, deletion of one half of the specificity unit (hsdS) does not impair the function of the system, it only results in a change of the TRM to a symmetric, palindrome specificity (Abadjieva et al., 1993; MacWilliams and Bickle, 1996). Attempts to reproduce these results by expressing only the N-terminal part (TRD1 and central conserved region) of TMW 2.1324 hsdS_long in E. coli (CM30) in the absence of hsdS_short resulted in no modification patterns at all.
4. Discussion
In this study we describe the prevalence of RM systems in Staphylococcus xylosus. We found, that S. xylosus harbors a variety of RM systems that are distinctly different from those of other well-studied staphylococci, such as S. aureus and S. epidermidis. Among the most prevailing differences is the presence of at most one type I system in S. xylosus, which is always arranged in a contiguous operon. On the contrary, up to three functioning type I systems per isolate have been reported for S. epidermidis strains, yet at the same time around 38% of S. epidermidis genomes were found to contain no functional type I RM system at all (Lee et al., 2019). Studies on S. aureus report hsdR to be spatially distant on the chromosome from hsdMS and that members of the species harbor at least one and up to three functional systems per strain (Monk et al., 2015; Lee et al., 2019). Furthermore, type IIG systems have been reported as inactive in S. aureus and type III systems as rarely present (Jones et al., 2015; Sadykov, 2016). In S. xylosus on the contrary, both systems appear to be active and more common as proven in this study by methylome, bioinformatic and proteomic analysis. In terms of type IV systems, the S. aureus subsp. aureus USA300 type IV system SauUSI has been deeply studied, consisting of one endonuclease polypeptide recognizing and cleaving the cytosine modified motif SCNGS (Xu et al., 2011). Again, S. xylosus differs here, as all identified type IV systems appear to consist of two subunits rather than one single restriction endonuclease.
Special emphasis of this work was laid on type I RM systems, which we could identify in seven out of the 14 investigated S. xylosus strains. Among the type I-positive S. xylosus isolates, we found two different types of hsd-operons. Firstly, chromosomally encoded three-gene hsdMSR operons, resembling in their gene and sequence structure other type I systems described for staphylococci and also other Gram-positive bacteria before (Lee et al., 2019; Reva et al., 2019; Finn et al., 2021). Namely, they are arranged in an operon like structure, in the order of transcription, including the three typical genes, hsdR, hsdM, hsdS. Secondly, we identified a hitherto undescribed variant of type I systems, hsdRSMS. The operon shares some common features with other staphylococcal type I systems such as the localization on mobile genetic elements (MGEs) of the chromosome and on plasmids (Lee et al., 2019) as well as the usual gene arrangement with hsdSMS all being transcribed from a shared promoter and hsdR being associated with its own promoter (Murray, 2000). Yet, in contrast to other type I systems, hsdRSMS requires two specificity units for proper and stable base modification, a long and a short subunit. While hsdS_long resembles known specificity units in its composition consisting of variable regions (TRDs) flanked by conserved regions, for hsdS_short such typical structural features are not evident, as it is lacking any variable regions and is highly conserved among different strains methylating different motifs. This makes it unlikely that hsdS_short is involved in target sequence recognition. Likewise, there is no indication for hsdS_short being a remnant, truncated half-size hsdS polypeptide. Fragmented hsdS genes have been reported for other type IC systems [e.g., NgoAV, EcoDXXI, EcoR124I (Abadjieva et al., 1993; MacWilliams and Bickle, 1996; Piekarowicz et al., 2001)] with the C-terminal domain coding for the long hsdS peptide usually missing, resulting in palindromic recognition motifs. Our data showed that hsdS_short does not exist as part of a long hsdS subunit though. It is functionally expressed as well as it contributes to specific base modification of non-palindromic motifs. Upon methylation of DNA, type I methyltransferases usually form a M2S trimer, whereas for restriction a pentamer consisting of either R2M2S1 or R1M2S1 is formed (Gao et al., 2020). One could speculate that hsdS_short might have a stabilizing role in these complexes, somehow promoting binding of hsdS_long to hsdM since missing hsdS_short resulted in DNA target motifs with a modified specificity and low modification scores. Further studies are needed to determine the exact role of hsdS_short during complex assembly of the newly discovered type I RM system.
Classification of type I RM systems into one of the five existing families is based on sequence similarity values of hsdR and hsdM genes, as they are usually well conserved. However, clear cutoff values have not been determined so far and values specified in the literature vary strongly. Yet in trying to find consent, one could conclude that hsdM and hsdR share usually over 70% sequence similarity when they are members of the same family and < 30% when they are part of different families (Murray, 2000; Titheradge et al., 2001; Chin et al., 2004; Cooper et al., 2017; Gao et al., 2020). HsdR and hsdM of the hsdRSMS system share highest percent identity values with the reference gene of type IC systems (EcoR124I), namely 40 and 48%, respectively. Thereby, they are just at the interface between classifying them into the type IC family or establishing a new family for them. Voting for classifying them into the family of type IC systems is their occurrence on plasmids and MGEs which is characteristic for many RM systems especially members of the type IC family (Kobayashi, 2001; Youell and Firman, 2008; Loenen et al., 2014). Moreover, according to Gao et al. (2020), HsdM of type IC families is composed of three domains, namely a N-terminal (aa 11 – 190), a catalytic (aa 198 – 473) and a C-terminal (aa 481 – 510) domain. HsdM of HsdRSMSPL systems displays 33% protein sequence identity to the N-terminal, 44% to the catalytic and 13-23% to the C-terminal domain of type IC M.EcoR124I (data not shown). Thus, even though both methyltransferases are arranged into a similar domain structure, single domains are not reaching sequence identity values over 44%. Therefore, voting against grouping the new operon into the family of type IC systems is not just the overall comparatively low sequence homology (∼40%) of hsdR and hsdM with the respective type I reference genes but also that hsdS_long is lacking some important structural and functional characteristics. Most importantly, the long subunit is not able to function independently without the presence of hsdS_short. Additionally, type IC hsdS subunits usually harbor characteristic tandem tetra amino acid repeats [e.g., TAEL, LEAT, SEAL or TSEL (Adamczyk-Popławska et al., 2003)] in their central conserved region. These repeats define among others, the spacer length between the two specificity elements of the recognition motif, with two and three repeats correlating with a 6-7 bp spacer, respectively, (Abadjieva et al., 1993; Adamczyk-Popławska et al., 2003). No such tetra amino acid repeats were identified in the central conserved region of hsdS from the hsdRSMS system, though we did find two short repeating amino acid stretches in the central conserved region (2x LEEQK). However, they are separated by 3 non-specific amino acids, thus not arranged in tandem and they do not seem to influence spacer length, as both hsdS_long subunits investigated in this study harbor two of such repeats but the TRDs of the TMW 2.1023 motif are divided by a 6 bp spacer compared to a 5 bp spacer in the motif of TMW 2.1324.
By methylome analysis of S. xylosus, this study provided new insights into the diversity of RM systems encoded by the genus Staphylococcus. The study further revealed the presence of a new variant of type I RM system that seems to require two specificity units for specific and thorough DNA methylation. Interestingly, the occurrence of this variant is not restricted to S. xylosus, as it was found to be present in other staphylococcal species as well. Additional approaches such as subunit complementation tests or antibody cross reactivity assays could further define the family affiliation in the future. All in all, the results obtained from this study present another piece in the mosaic of the diversity of methylation systems in bacteria.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in this article/Supplementary material.
Author contributions
CS: conceptualization, data curation, investigation, methodology, software, visualization, and writing—original draft. CG: investigation, methodology, and software. MP: supervision and methodology. RV: funding acquisition, supervision, and writing—review and editing. ME: conceptualization, investigation, methodology, supervision, and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the German Federal Ministry for Economic Affairs and Energy via the German Federation of Industrial Research Associations (AiF), project 19690N.
Acknowledgments
We thank Dr. Barbara Fösel from Helmholtz Zentrum Münich as well as Dr. Weihong Qi, Dr. Natalia Zajac, and Simon Grüter from ETH Zurich for their support of sequencing and bioinformatic analysis of the CMx E. coli strains.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.946189/full#supplementary-material
Footnotes
References
Abadjieva, A., Patel, J., Webb, M., Zinkevich, V., and Firman, K. (1993). A deletion mutant of the type IC restriction endonuclease EcoR1241 expressing a novel DNA specificity. Nucleic Acids Res. 21, 4435–4443. doi: 10.1093/nar/21.19.4435
Adamczyk-Popławska, M., Kondrzycka, A., Urbanek, K., and Piekarowicz, A. (2003). Tetra-amino-acid tandem repeats are involved in HsdS complementation in type IC restriction-modification systems. Microbiology 149, 3311–3319. doi: 10.1099/mic.0.26497-0
Atack, J. M., Guo, C., Yang, L., Zhou, Y., and Jennings, M. P. (2020). DNA sequence repeats identify numerous Type I restriction-modification systems that are potential epigenetic regulators controlling phase-variable regulons; phasevarions. FASEB J. 34, 1038–1051. doi: 10.1096/fj.201901536RR
Atack, J. M., Tan, A., Bakaletz, L. O., Jennings, M. P., and Seib, K. L. (2018). Phasevarions of bacterial pathogens: Methylomics sheds new light on old enemies. Trends Microbiol. 26, 715–726. doi: 10.1016/j.tim.2018.01.008
Behr, J., Geissler, A. J., Schmid, J., Zehe, A., and Vogel, R. F. (2016). The identification of novel diagnostic marker genes for the detection of beer spoiling Pediococcus damnosus strains using the BlAst diagnostic gene findEr. PLoS One 11:e0152747. doi: 10.1371/journal.pone.0152747
Chin, V., Valinluck, V., Magaki, S., and Ryu, J. (2004). KpnBI is the prototype of a new family (IE) of bacterial type I restriction-modification system. Nucleic Acids Res. 32:e138. doi: 10.1093/nar/gnh134
Clark, T. A., Murray, I. A., Morgan, R. D., Kislyuk, A. O., Spittle, K. E., Boitano, M., et al. (2012). Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 40:e29. doi: 10.1093/nar/gkr1146
Condas, L. A. Z., de Buck, J., Nobrega, D. B., Carson, D. A., Roy, J.-P., Keefe, G. P., et al. (2017). Distribution of non-aureus staphylococci species in udder quarters with low and high somatic cell count, and clinical mastitis. J. Dairy Sci. 100, 5613–5627. doi: 10.3168/jds.2016-12479
Cooper, L. P., Roberts, G. A., White, J. H., Luyten, Y. A., Bower, E. K. M., Morgan, R. D., et al. (2017). DNA target recognition domains in the Type I restriction and modification systems of Staphylococcus aureus. Nucleic Acids Res. 45, 3395–3406. doi: 10.1093/nar/gkx067
Costa, S. K., Donegan, N. P., Corvaglia, A.-R., François, P., and Cheung, A. L. (2017). Bypassing the restriction system to improve transformation of Staphylococcus epidermidis. J. Bacteriol. 199:e00271–17. doi: 10.1128/JB.00271-17
Cui, L., and Shearwin, K. E. (2017). Clonetegration using OSIP plasmids: One-step DNA Assembly and site-specific genomic integration in bacteria. Methods Mol. Biol. 1472, 139–155. doi: 10.1007/978-1-4939-6343-0_11
De Ste Croix, M., Vacca, I., Kwun, M. J., Ralph, J. D., Bentley, S. D., Haigh, R., et al. (2017). Phase-variable methylation and epigenetic regulation by type I restriction-modification systems. FEMS Microbiol. Rev. 41, S3–S15. doi: 10.1093/femsre/fux025
Deng, Y. M., Liu, C. Q., and Dunn, N. W. (2000). LldI, a plasmid-encoded type I restriction and modification system in Lactococcus lactis. DNA Seq. 11, 239–245. doi: 10.3109/10425170009033237
Englaender, J. A., Jones, J. A., Cress, B. F., Kuhlman, T. E., Linhardt, R. J., and Koffas, M. A. G. (2017). Effect of genomic integration location on heterologous protein expression and metabolic engineering in E. coli. ACS Synth. Biol. 6, 710–720. doi: 10.1021/acssynbio.6b00350
Finn, M. B., Ramsey, K. M., Tolliver, H. J., Dove, S. L., and Wessels, M. R. (2021). Improved transformation efficiency of group A Streptococcus by inactivation of a type I restriction modification system. PLoS One 16:e0248201. doi: 10.1371/journal.pone.0248201
Fukuda, E., Kaminska, K. H., Bujnicki, J. M., and Kobayashi, I. (2008). Cell death upon epigenetic genome methylation: A novel function of methyl-specific deoxyribonucleases. Genome Biol. 9:R163. doi: 10.1186/gb-2008-9-11-r163
Furuta, Y., Kawai, M., Uchiyama, I., and Kobayashi, I. (2011). Domain movement within a gene: A novel evolutionary mechanism for protein diversification. PLoS One 6:e18819. doi: 10.1371/journal.pone.0018819
Furuta, Y., Namba-Fukuyo, H., Shibata, T. F., Nishiyama, T., Shigenobu, S., Suzuki, Y., et al. (2014). Methylome diversification through changes in DNA methyltransferase sequence specificity. PLoS Genet. 10:e1004272. doi: 10.1371/journal.pgen.1004272
Gao, Y., Cao, D., Zhu, J., Feng, H., Luo, X., Liu, S., et al. (2020). Structural insights into assembly, operation and inhibition of a type I restriction-modification system. Nat. Microbiol. 5, 1107–1118. doi: 10.1038/s41564-020-0731-z
Harrison, E. M., Paterson, G. K., Holden, M. T. G., Morgan, F. J. E., Larsen, A. R., Petersen, A., et al. (2013). A Staphylococcus xylosus isolate with a new mecC allotype. Antimicrob. Agents Chemother. 57, 1524–1528. doi: 10.1128/AAC.01882-12
Heilmann, C., Ziebuhr, W., and Becker, K. (2019). Are coagulase-negative staphylococci virulent? Clin. Microbiol. Infect. 25, 1071–1080. doi: 10.1016/j.cmi.2018.11.012
Ishikawa, K., Handa, N., and Kobayashi, I. (2009). Cleavage of a model DNA replication fork by a Type I restriction endonuclease. Nucleic Acids Res. 37, 3531–3544. doi: 10.1093/nar/gkp214
Jones, M. J., Donegan, N. P., Mikheyeva, I. V., and Cheung, A. L. (2015). Improving transformation of Staphylococcus aureus belonging to the CC1, CC5 and CC8 clonal complexes. PLoS One 10:e0119487. doi: 10.1371/journal.pone.0119487
Karkman, A., Do, T. T., Walsh, F., and Virta, M. P. J. (2018). Antibiotic-resistance genes in waste water. Trends Microbiol. 26, 220–228. doi: 10.1016/j.tim.2017.09.005
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kobayashi, I. (2001). Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29, 3742–3756. doi: 10.1093/nar/29.18.3742
Lee, J. Y. H., Carter, G. P., Pidot, S. J., Guérillot, R., Seemann, T., da Silva, A. G., et al. (2019). Mining the methylome reveals extensive diversity in Staphylococcus epidermidis restriction modification. MBio 10:e02451–19. doi: 10.1128/mBio.02451-19
Leroy, S., Vermassen, A., Ras, G., and Talon, R. (2017). Insight into the genome of Staphylococcus xylosus, a ubiquitous species well adapted to meat products. Microorganisms 5:52. doi: 10.3390/microorganisms5030052
Lindsay, J. A. (2014). Staphylococcus aureus genomics and the impact of horizontal gene transfer. Int. J. Med. Microbiol. 304, 103–109. doi: 10.1016/j.ijmm.2013.11.010
Lindsay, J. A. (2019). Staphylococci: Evolving Genomes. Microbiol. Spectr. 7, 485–498. doi: 10.1128/microbiolspec.GPP3-0071-2019
Loenen, W. A. M., and Raleigh, E. A. (2014). The other face of restriction: Modification-dependent enzymes. Nucleic Acids Res. 42, 56–69. doi: 10.1093/nar/gkt747
Loenen, W. A. M., Dryden, D. T. F., Raleigh, E. A., and Wilson, G. G. (2014). Type I restriction enzymes and their relatives. Nucleic Acids Res. 42, 20–44. doi: 10.1093/nar/gkt847
MacWilliams, M. P., and Bickle, T. A. (1996). Generation of new DNA binding specificity by truncation of the type IC EcoDXXI hsdS gene. EMBO J. 15, 4775–4783.
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Marincola, G., Liong, O., Schoen, C., Abouelfetouh, A., Hamdy, A., Wencker, F. D. R., et al. (2021). Antimicrobial resistance profiles of coagulase-negative staphylococci in community-based healthy individuals in Germany. Front. Public Health 9:684456. doi: 10.3389/fpubh.2021.684456
Monk, I. R., Tree, J. J., Howden, B. P., Stinear, T. P., and Foster, T. J. (2015). Complete bypass of restriction systems for major Staphylococcus aureus Lineages. MBio 6:e00308–15. doi: 10.1128/mBio.00308-15
Murray, N. E. (2000). Type I restriction systems: Sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev. 64, 412–434. doi: 10.1128/MMBR.64.2.412-434.2000
Nye, T. M., Fernandez, N. L., and Simmons, L. A. (2020). A positive perspective on DNA methylation: Regulatory functions of DNA methylation outside of host defense in Gram-positive bacteria. Crit. Rev. Biochem. Mol. Biol. 55, 576–591. doi: 10.1080/10409238.2020.1828257
Oliveira, P. H., and Fang, G. (2021). Conserved DNA Methyltransferases: A window into fundamental mechanisms of epigenetic regulation in bacteria. Trends Microbiol. 29, 28–40. doi: 10.1016/j.tim.2020.04.007
Otto, M. (2013). Coagulase-negative staphylococci as reservoirs of genes facilitating MRSA infection: Staphylococcal commensal species such as Staphylococcus epidermidis are being recognized as important sources of genes promoting MRSA colonization and virulence. Bioessays 35, 4–11. doi: 10.1002/bies.201200112
Park, S., Jung, D., O’Brien, B., Ruffini, J., Dussault, F., Dube-Duquette, A., et al. (2022). Comparative genomic analysis of Staphylococcus aureus isolates associated with either bovine intramammary infections or human infections demonstrates the importance of restriction-modification systems in host adaptation. Microb. Genom. 8:000779. doi: 10.1099/mgen.0.000779
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Piekarowicz, A., Kłyz, A., Kwiatek, A., and Stein, D. C. (2001). Analysis of type I restriction modification systems in the Neisseriaceae: Genetic organization and properties of the gene products. Mol. Microbiol. 41, 1199–1210. doi: 10.1046/j.1365-2958.2001.02587.x
Pingoud, A., Wilson, G. G., and Wende, W. (2014). Type II restriction endonucleases–a historical perspective and more. Nucleic Acids Res. 42, 7489–7527. doi: 10.1093/nar/gku447
Rao, D. N., Dryden, D. T. F., and Bheemanaik, S. (2014). Type III restriction-modification enzymes: A historical perspective. Nucleic Acids Res. 42, 45–55. doi: 10.1093/nar/gkt616
Ravyts, F., Vuyst, L. D., and Leroy, F. (2012). Bacterial diversity and functionalities in food fermentations. Eng. Life Sci. 12, 356–367. doi: 10.1002/elsc.201100119
Reva, O. N., Swanevelder, D. Z. H., Mwita, L. A., Mwakilili, A. D., Muzondiwa, D., Joubert, M., et al. (2019). Genetic, epigenetic and phenotypic diversity of four Bacillus velezensis strains used for plant protection or as probiotics. Front. Microbiol. 10:2610. doi: 10.3389/fmicb.2019.02610
Roberts, G. A., Chen, K., Cooper, L. P., White, J. H., Blakely, G. W., and Dryden, D. T. F. (2012). Removal of a frameshift between the hsdM and hsdS genes of the EcoKI Type IA DNA restriction and modification system produces a new type of system and links the different families of Type I systems. Nucleic Acids Res. 40, 10916–10924. doi: 10.1093/nar/gks876
Roberts, R. J., and Macelis, D. (2001). REBASE–restriction enzymes and methylases. Nucleic Acids Res. 29, 268–269. doi: 10.1093/nar/29.1.268
Roer, L., Hendriksen, R. S., Leekitcharoenphon, P., Lukjancenko, O., Kaas, R. S., Hasman, H., et al. (2016). Is the evolution of Salmonella enterica subsp. enterica linked to restriction-modification systems? mSystems 1:e00009–16. doi: 10.1128/mSystems.00009-16
Rossi, C. C., Souza-Silva, T., Araújo-Alves, A. V., and Giambiagi-Demarval, M. (2017). CRISPR-cas systems features and the gene-reservoir role of coagulase-negative staphylococci. Front. Microbiol. 8:1545. doi: 10.3389/fmicb.2017.01545
Sadykov, M. R. (2016). Restriction-modification systems as a barrier for genetic manipulation of Staphylococcus aureus. Methods Mol. Biol. 1373, 9–23. doi: 10.1007/7651_2014_180
Schiffer, C. J., Abele, M., Ehrmann, M. A., and Vogel, R. F. (2021). Bap-independent biofilm formation in Staphylococcus xylosus. Microorganisms 9:2610. doi: 10.3390/microorganisms9122610
Schiffer, C., Hilgarth, M., Ehrmann, M., and Vogel, R. F. (2019). Bap and cell surface hydrophobicity are important factors in Staphylococcus xylosus biofilm formation. Front. Microbiol. 10:1387. doi: 10.3389/fmicb.2019.01387
St-Pierre, F., Cui, L., Priest, D. G., Endy, D., Dodd, I. B., and Shearwin, K. E. (2013). One-step cloning and chromosomal integration of DNA. ACS Synth. Biol. 2, 537–541. doi: 10.1021/sb400021j
Supré, K., Haesebrouck, F., Zadoks, R. N., Vaneechoutte, M., Piepers, S., and de Vliegher, S. (2011). Some coagulase-negative Staphylococcus species affect udder health more than others. J. Dairy Sci. 94, 2329–2340. doi: 10.3168/jds.2010-3741
Suzuki, T., and Yasui, K. (2011). “Plasmid artificial modification: A novel method for efficient DNA transfer into bacteria,” in Strain engineering: Methods and protocols, ed. J. A. Williams (Totowa, NJ: Humana Press), 309–326.
Titheradge, A. J., King, J., Ryu, J., and Murray, N. E. (2001). Families of restriction enzymes: An analysis prompted by molecular and genetic data for type ID restriction and modification systems. Nucleic Acids Res. 29, 4195–4205. doi: 10.1093/nar/29.20.4195
Tock, M. R., and Dryden, D. T. F. (2005). The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 8, 466–472. doi: 10.1016/j.mib.2005.06.003
Wilson, G. G., and Murray, N. E. (1991). Restriction and modification systems. Annu. Rev. Genet. 25, 585–627. doi: 10.1146/annurev.ge.25.120191.003101
Wood, E. J. (1983). Molecular cloning. A laboratory manual. Biochem. Educ. 11:82. doi: 10.1016/0307-4412(83)90068-7
Xu, S.-Y., Corvaglia, A. R., Chan, S.-H., Zheng, Y., and Linder, P. (2011). A type IV modification-dependent restriction enzyme SauUSI from Staphylococcus aureus subsp. aureus USA300. Nucleic Acids Res. 39, 5597–5610. doi: 10.1093/nar/gkr098
Yano, H., Alam, M. Z., Rimbara, E., Shibata, T. F., Fukuyo, M., Furuta, Y., et al. (2020). Networking and specificity-changing DNA Methyltransferases in Helicobacter pylori. Front. Microbiol. 11:1628. doi: 10.3389/fmicb.2020.01628
Keywords: Staphylococcus xylosus, restriction modification systems, methylome, epigenetics, DNA methylation, methyltransferases (MTases)
Citation: Schiffer CJ, Grätz C, Pfaffl MW, Vogel RF and Ehrmann MA (2023) Characterization of the Staphylococcus xylosus methylome reveals a new variant of type I restriction modification system in staphylococci. Front. Microbiol. 14:946189. doi: 10.3389/fmicb.2023.946189
Received: 17 May 2022; Accepted: 13 February 2023;
Published: 08 March 2023.
Edited by:
Feng Gao, Tianjin University, ChinaCopyright © 2023 Schiffer, Grätz, Pfaffl, Vogel and Ehrmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolin J. Schiffer, Y2Fyb2xpbi5zY2hpZmZlckB0dW0uZGU=
†Present Address: Carolin J. Schiffer and Matthias A. Ehrmann, Chair of Microbiology, TUM School of Life Sciences, Technical University of Munich, Freising, Germany