
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 09 November 2023
Sec. Virology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1293846
This article is part of the Research TopicThe Role of Viruses in Marine EnvironmentsView all 9 articles
 Yuanfang Liu1†
Yuanfang Liu1† Xue Meng1†
Xue Meng1† Hongrui Zheng1†
Hongrui Zheng1† Lanlan Cai2Shuzhen Wei3
Lanlan Cai2Shuzhen Wei3 Minglu He4Jiale He5Yue Hao5Chang Ge5
Minglu He4Jiale He5Yue Hao5Chang Ge5 Jihua Liu1
Jihua Liu1 Feng Chen6
Feng Chen6 Yongle Xu1*
Yongle Xu1*Cyanophages affect the abundance, diversity, metabolism, and evolution of picocyanobacteria in marine ecosystems. Here we report an estuarine Synechococcus phage, S-CREM2, which represents a novel viral genus and leads to the establishment of a new T4-like cyanophage clade named cluster C. S-CREM2 possesses the longest tail (~418 nm) among isolated cyanomyoviruses and encodes six tail-related proteins that are exclusively homologous to those predicted in the cluster C cyanophages. Furthermore, S-CREM2 may carry three regulatory proteins in the virion, which may play a crucial role in optimizing the host intracellular environment for viral replication at the initial stage of infection. The cluster C cyanophages lack auxiliary metabolic genes (AMGs) that are commonly found in cyanophages of the T4-like clusters A and B and encode unique AMGs like an S-type phycobilin lyase gene. A variation in the composition of tRNA and cis-regulatory RNA genes was observed between the marine and freshwater phage strains in cluster C, reflecting their different modes of coping with hosts and habitats. The cluster C cyanophages are widespread in estuarine and coastal regions and exhibit equivalent or even higher relative abundance compared to those of clusters A and B cyanophages in certain estuarine regions. The isolation of cyanophage S-CREM2 provides new insights into the phage–host interactions mediated by both newly discovered AMGs and virion-associated proteins and emphasizes the ecological significance of cluster C cyanophages in estuarine environments.
Cyanophages, which infect cyanobacteria, are an important component of marine viruses and play a crucial role in marine ecosystems by influencing the population dynamics, community structure, metabolism, and evolution of cyanobacteria (Mann and Clokie, 2012). Cyanophages isolated from marine ecosystems can be classified into three morphological groups: cyanomyoviruses, cyanopodoviruses, and cyanosiphoviruses (Sullivan et al., 2005, 2009, 2010). Among the three groups of cyanophages, cyanomyoviruses are the most frequently isolated, with the T4-like as the dominant member (Wang et al., 2023). It is estimated that T4-like cyanomyoviruses are the most abundant and widely distributed cyanophages in marine environments (Mann and Clokie, 2012; Huang et al., 2015). About 106 T4-like cyanophage genomes have been sequenced (Jiang et al., 2020; Wang et al., 2023) and are found possessing a collection of core genes associated with DNA replication and structure forming (Sullivan et al., 2010). Marine T4-like cyanophage isolates were previously classified into two lineages, namely clusters A and B, based on phylogenetic analysis of the core genes (Ignacio-Espinoza and Sullivan, 2012). The freshwater cyanophage S-CRM01 exhibits high homology with the marine T4-like cyanophage isolates but is divergent from the previously identified clusters A and B in terms of phylogenetic lineage, which sheds new light on the phylogeny of T4-like cyanophages (Dreher et al., 2011). As more cyanophage isolates are discovered, the classification of T4-like cyanophages necessitates further refinement.
The T4-like cyanophages usually encode auxiliary metabolic genes (AMGs), which play important roles in regulating host metabolisms to increase the phage fitness (Sullivan et al., 2010). The AMG composition varies greatly among T4-like cyanophages. A large number of T4-like cyanophages, such as P-SSM2, S-SCSM1, and S-SZBM1 (Sullivan et al., 2005; Rong et al., 2022; Wang et al., 2022), encode a variety of AMGs, including those involved in photosynthesis (psbA, psbD, hli, petE, petF, ptoX, ho1, pcy, pebS, and cpeT), carbon metabolism (talC, gnd, zwf, cp12, and MPI), phosphorus acquisition (pstS and phoA), vitamin B12 synthesis (cobS, cobO, and cobA), and cell wall synthesis and modification (GDP-mannose glycosyl hydrolase, GDP-L-fucose synthase, and GDP-mannose-3,5-epimerase) (Sullivan et al., 2010; Wang et al., 2022). Furthermore, multiple copies of certain genes have been observed in the T4-like cyanophage genomes. For example, three hlis were found in the P-SSM2 genome (Sullivan et al., 2010), 24 2OG-Fe(II) oxygenase family genes are predicted in the S-SCSM1 genome (Wang et al., 2022). Additionally, some AMGs are prevalent in the T4-like cyanophage genomes. Over 90% of T4-like cyanophages encode psbA, hli, phoH, mazG, cobS, and hsp20, while more than 60% of T4-like cyanophages encode psbD, talC, petE, cpeT, and cp12 (Jiang et al., 2020). However, some T4-like cyanophages encode a limited number of AMGs. S-CRM01 only contains seven AMGs that are commonly found in T4-like cyanophages. S-H34, S-N03, and S-B68 even lack the most commonly observed photosynthesis-related genes, psbA and hli, in T4-like cyanophages (Jiang et al., 2020). Different AMG profiles in cyanophage genomes can have diverse influences on host metabolisms. An illustrative instance involves cyanophages S-RSM4 and S-PM2. S-RSM4 has multiple genes related to carbon metabolism (talC, gnd, zwf, and cp12) and stops the host’s carbon fixation reaction 2 h earlier than S-PM2, which lacks these carbon metabolism-related genes, resulting in much less carbon fixation in the S-RSM4-infected cells (Puxty et al., 2016).
tRNA genes are widely distributed in the T4-like cyanophage genomes, with numbers ranging from 0 to 33 (Enav et al., 2012). Enav et al. propose that phages carry tRNAs to optimize the codon usage discrepancy between phages and hosts, enabling phage cross-infectivity of hosts with divergent G + C contents (Enav et al., 2012). In addition, cyanophage tRNA genes are predicted to facilitate the expression of specific AMGs (Enav et al., 2012; Xu et al., 2018). Cyanophage genomes also contain non-coding RNA (ncRNA) genes with regulatory functions. Currently, T4-like cyanophages have been found to contain several cis-regulatory RNAs, including glnA, manA, PhotoRC-II, and wcaG, which are thought to play a regulatory role in important host bioprocesses, including photosynthesis, nitrogen metabolism, and exopolysaccharide production (Weinberg et al., 2010; Wang et al., 2022; Zheng et al., 2023).
Generally, virions (i.e., an infectious virus particle) consist of structural proteins. However, certain viral proteomes have been found to encapsulate non-structural proteins related to host metabolism regulations. The most well-known phage non-structural protein is the RNA polymerase in N4 podoviruses (Falco et al., 1980; Kazmierczak et al., 2002). Virion-associated protein kinases (VAPKs) have been identified in various virus families except for dsRNA and ssDNA viruses. Especially in animal and plant viruses, VAPKs are prevalent and play crucial roles in multiple stages of the viral life cycle, including infection, uncoating, transcription, and replication (Hui, 2002). Furthermore, VAPKs have also been detected within the virion of cyanophages. For instance, putative serine/therine protein kinases have been identified in the S-CRM01 and S-TIM5 virions (Dreher et al., 2011; Sabehi et al., 2012). APH, ChoK, and Rio2 kinases were detected in the virions of a non-T4 cyanomyovirus, S-CBWM1, and are predicted to be involved in host bioprocesses like antibiotic resistance, protein binding to phospholipids and choline, and ribosome biogenesis (Xu et al., 2018). In addition, nicotinamide/nicotinate monomucleotide adenylytransferase (NMNAT)-like proteins are found in the virion proteomes of cyanomyoviruses, S-CBWM1, S-SZBM1, and S-SCSM1, which are thought to be involved in NAD+ synthesis during infection (Xu et al., 2018; Rong et al., 2022; Wang et al., 2022). These regulatory proteins are thought to create an optimal environment for viral replication upon entry into the host cell.
The discovery of novel cyanophage isolates is always enhancing our comprehension of viral genetic diversity, evolution, phage–host interactions, and potential ecological functions. Here, we characterized a new T4-like cyanophage, S-CREM2, which was isolated from the Changjiang River Estuary. S-CREM2 represents a new viral genus and possesses the longest tail ever found in cyanomyoviruses. The identification of S-CREM2 promoted the establishment of a novel T4-like cyanophage lineage, referred to as cluster C, which is as prevalent as the previously identified clusters A and B in the estuarine environment. The isolation and characterization of S-CREM2 provide new insights into phage–host interactions and the ecological distribution of the newly established cluster C cyanophages.
Synechococcus sp. CRE1902 was isolated from the surface water of Changjiang River Estuary (31.52°N, 122.64°E) in July 2019, and used as a host organism for cyanophage isolation. Synechococcus sp. CRE1902 was grown in seawater-based SN medium with a salinity of 25‰ (SN25) (Waterbury and Willey, 1988), and incubated at a temperature of 22°C under a constant cool-white light intensity of 20 μE m−2 s−1. Cyanophage S-CREM2 was obtained from the surface seawater sample collected in the Changjiang River Estuary (31.31°N, 122.49°E) in July 2019. The viral seawater used for cyanophage isolation was prepared by a 0.22-μm filtration to remove bacterial cells and subsequently stored in the dark at 4°C until further use. Cyanophages were first enriched by adding 20 μl of the above viral seawater to 180 μl of exponentially growing Synechococcus sp. CRE1902 cultures (optical density at 750 nm (OD750) = 0.5) in a 96-well microtiter plate. After the lysis of Synechococcus cells, the lysates were collected and centrifuged at 10,000 × g, 4°C for 10 min. The supernatants were filtered through 0.22-μm-pore-size sterile syringe filters (Millipore, Millex®-G, USA) and subsequently used for phage purification. Phage purification was performed using the plaque assay method (Suttle and Chen, 1992) and repeated three times.
Nine Synechococcus strains were used for the host range detection of S-CREM2, which included five estuarine strains, CB0101, CRE1901, CRE1902, CBW1107, CBW1101, and four oceanic strains, CC9311, WH8102, WH7803, WH7805. About 20 μl of S-CREM2 suspensions were added to 180 μl exponentially growing Synechococcus cultures in 96-well microtiter plates in triplicate, while the controls received 20 μl of SN25 medium. All plates were incubated under the same condition as described above and observed daily for cell lysis.
To amplify S-CREM2 phage, phage suspensions were added into 2 L of exponentially growing Synechococcus sp. CRE1902 cultures (OD750 = 0.5) at a multiplicity of infection of 0.01. The resulting lysates were treated with DNase І and RNase A both at a concentration of 2 μg mL−1 at room temperature for 1 h. Subsequently, the NaCl concentration of the lysates was adjusted to 1 M, and the lysates were ice-bathed for 30 min (Xu et al., 2015). The treated lysates were then centrifuged at 10,000 × g, 4°C for 20 min. The resulting supernatants were filtered through 0.45-μm-pore-size polycarbonate membrane filters to remove cell debris. Phage particles in the supernatants were concentrated using 10% (w/v) polyethylene glycol 8,000 at 4°C for 24 h and then precipitated by centrifugation at 12,000 × g for 1 h. The resulting S-CREM2 pellet was resuspended in TM buffer (20 mM Tris-Cl and 10 mM MgSO4) and subjected to CsCl-gradient centrifugation (200,000 × g at 4°C for 6 h) using a SW 41Ti rotor (Beckman Optima L-100XP, Beckman Coulter, CA, USA). The visible phage band was extracted and underwent a 30-kDa centrifugal ultrafiltration to remove CsCl from the phage suspension.
Ten microliters of the CsCl-purified phage suspension were absorbed onto a 200-mesh carbon-coated copper film for 1 min. Subsequently, they were negatively stained with 2% (w/v) uranyl acetate for 30 s. The excess dye was gently removed using filter paper, and the staining process was repeated. After drying for 30 min, the prepared sample was observed using a Tecnai G2 Spirit BioTwin transmission electron microscope (FEI Tecnai G2 F20, Thermo Fisher Scientific, Waltham, MA, USA). The Xplore3D image transmission system (USA) was utilized to capture high-quality images of the phage particles.
Phage particles were first treated with a cocktail buffer containing proteinase K (100 mg mL−1), SDS (10%, wt/vol), and EDTA (0.5 M). Subsequently, phage DNA was extracted using the phenol-chloroform method as previously described (Chen et al., 2006). A whole-genome shotgun strategy was used to construct the PE150 library. The obtained raw data were subjected to quality filtering, trimming, and de novo assembly using IDBA v1.1.3 (Peng et al., 2012) and megahit v1.2.9 (Li et al., 2016). Any remaining gaps in the cyanophage genome were closed using pilon v1.24 and bcftools v1.17 (Narasimhan et al., 2016). The complete genome sequence has been submitted to the GenBank database under accession no. OR473000.
The putative open reading frames (ORFs) of S-CREM2 were predicted using the GeneMarkS1 (Besemer and Borodovsky, 2005), the RAST server2 (Brettin et al., 2015), and the MetaGene Annotator3 (Noguchi et al., 2008). Translated ORFs were annotated by combining the results of homolog search against the NCBI non-redundant (NR) database, conserved domain prediction, and remote homolog search using the HHpred server4 (Söding et al., 2005). ORF homolog search against the NR database was conducted using BLASTP with an e-value cutoff of <10−5 and a bit core of >40 (Pruitt et al., 2007). Conserved domains within ORFs were predicted by searching against the NCBI Conserved Domain Database (CDD) (Marchler-Bauer et al., 2011), with an e-value cutoff <10−3, a bit score of >40, and a coverage of >40%. For ORFs without predicted conserved domains, HHpred search against PDB_mmCIF70_18_Jun, UniProt-SwissProt-viral70_3_NOV_2021, SCOPe70_2.08 structural/domain databases was conducted, with a probability cutoff of >90%, to supplement the ORF annotation. tRNA genes in the S-CREM2 genome were identified using tRNAscan-SE (Chan and Lowe, 2019). Other ncRNA genes were predicted by searching against the Rfam database5 (Yao et al., 2007). Comparative genomic analyses of cluster C cyanophages were conducted and visualized by using Easyfig v2.2.3 (Sullivan et al., 2011).
Phylogenetic analyses of the phycobilin lyase and CP12 genes were conducted using the MEGA 7.0 software package (Kumar et al., 2016). The phycobilin lyase phylogenetic tree was constructed based on amino acid sequences, while the CP12 phylogenetic trees utilized nucleotide sequences. The maximum-likelihood method with the Jones-Taylor-Thornton (JTT) model and the neighbor-joining method with the p-distance model were both used in the phylogenetic tree construction with 1,000 bootstrap replicates. Phylogenomic analyses of S-CREM2 and 40 T4-like cyanophages were performed based on the amino acid sequences of 31 core genes. The core genes were identified among the 41 cyanophages using OrthoFinder v2.5.2 (Emms and Kelly, 2015), aligned using MAFFT v7.52 (Katoh et al., 2009), and edited using TrimAI v22.9.0 (Capella-Gutiérrez et al., 2009). The phylogenomic tree was constructed by RAxML v8.2.12 (Stamatakis, 2014) using the maximum-likelihood method with the PROTGAMMAJTT model (bootstrap replicates = 100). Five cyanophage representatives in clusters A, B, and C were selected to analyze the intergenomic similarity by VIRIDIC6 (Moraru et al., 2020). All five phages in cluster C, S-CREM2, S-CRM01, S-B68, S-H34, and S-N03, were used in the analysis. Cyanophages S-PM2, S-RSM4, S-SM2, S-SSM7, and P-HM1 were chosen to represent cluster A, while S-ShM2, Syn10, S-RIM8, S-IOM18, and S-RIM2 were selected to act for cluster B.
The CsCl-purified phage suspensions were used for the virion protein determination. Virion proteins were digested using the FASP methods procedure described by Wiśniewski et al. (2009). The resulting tryptic peptides were analyzed using a Q Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), coupled to an Easy nLC 1,000 system (Thermo Fisher Scientific) (Michalski et al., 2011). Generated mass spectra were searched against the S-CREM2 genome by using the Mascot2.2 software (Matrix Science, London, UK) to retrieve the data.
To estimate the relative abundances and distributional patterns of T4-like cyanophage clusters A, B, and C, fragment recruitment was performed using virome datasets from both marine and freshwater environments. Five representative cyanophages in each cluster used in the intergenomic similarity analysis were selected, and core genes shared among clusters A, B, and C were used for the recruitment analyses. Viromes used in this study include Global Ocean Virome 2.0 (GOV 2.0) (Gregory et al., 2019), Delmarva Estuarine Virome (DEV) (Sun et al., 2021), and Pearl River Estuary Virome (PREV) (Xu et al., 2022; Supplementary Table S1). The GOV 2.0 datasets were downloaded from the iMicrobe website7. The DEV datasets were obtained from the NCBI SRA database8. The PREV was sourced from the National Omics Data Encyclopedia9. Core gene homolog recruitment was conducted using BLASTN, with specific thresholds: an e-value of <1e−5, a bit score of >40, a nucleotide identity of >95%, an alignment length of >90 bp, and a coverage of >40% (Mizuno et al., 2016; Martinez-Hernandez et al., 2017). The relative abundances of T4-like cyanophage clusters A, B, and C were normalized by the total recruited nucleotides (kb) per kilobase of core genes per gigabase of metagenome (KPKG) (Martinez-Hernandez et al., 2017).
Cyanophage S-CREM2 and its host, Synechococcus sp. CRE1902 which is a member of Synechococcus subcluster 5.1 clade VI, were both isolated from the surface seawater of the Changjiang River Estuary in July 2019. Transmission electron microscopy observation reveals that S-CREM2 is a myovirus, possessing an isometric capsid (~96 nm in diameter) and an extraordinarily long contractile tail (~418 nm in length) (Figure 1; Supplementary Figure S1). Cyanomyovirus isolates rarely have tails longer than 200 nm (Sullivan et al., 2005; Clokie et al., 2008; Dreher et al., 2011; Sabehi et al., 2012; Xu et al., 2018; Rong et al., 2022; Wang et al., 2022; Zheng et al., 2023). S-CREM2 has the longest tail among the isolated cyanomyoviruses, even the isolated myoviruses, discovered so far. In contrast to the strong cross-infectivity of most cyanomyovirus isolates (Sullivan et al., 2003, 2008; Wang et al., 2022), S-CREM2 exhibits a narrow host range (Table 1). Among the nine Synechococcus strains examined in this study, S-CREM2 exclusively infected its original host, failing to cross-infect any other strains, even those in the same phylogenetic clade as the host. Previous studies have also emphasized the limited host range of two cyanomyoviruses, S-CREM1 and S-SZBM1 (Rong et al., 2022; Zheng et al., 2023), which are also isolated from estuarine or coastal environments. More cyanophage isolation would facilitate a better understanding of the picocyanobacteria–phage interactions occurring in the eutrophic marine environment.

Figure 1. Transmission electron microscopy image of Synechococcus phage S-CREM2.
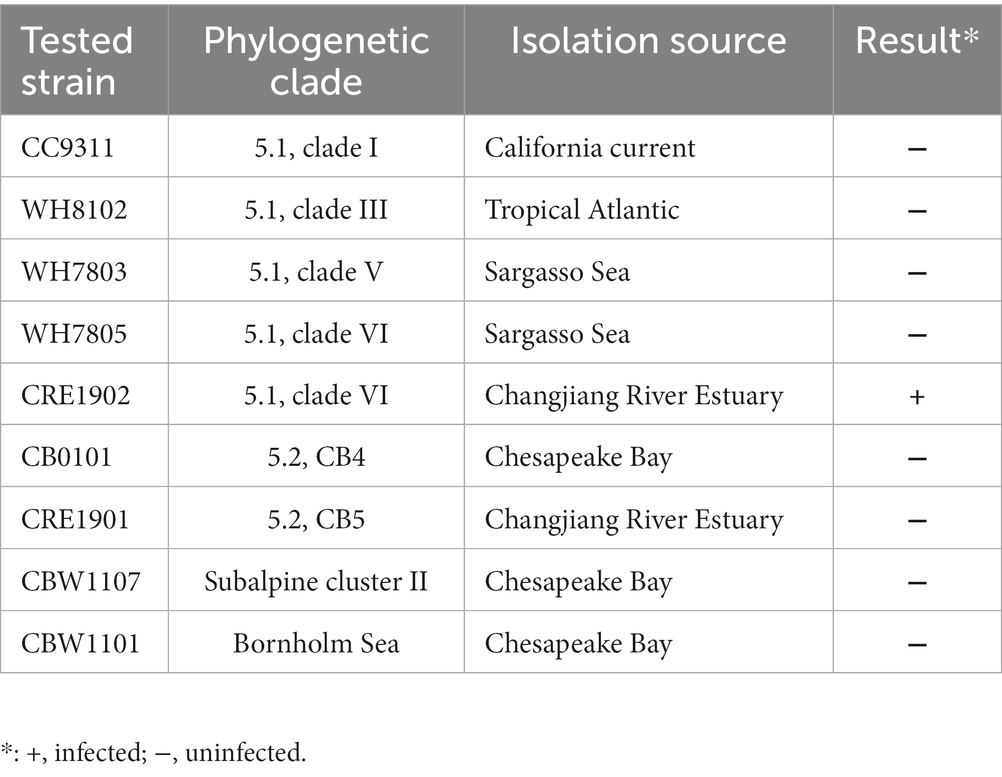
Table 1. Host range of S-CREM2.
The genome of S-CREM2 was assembled into a circularly permuted, double-stranded DNA molecule with a length of 174,876 bp and a G + C content of 47.92%. A total of 219 ORFs, two tRNA genes, and a cis-regulatory RNA gene were predicted in the S-CREM2 genome (Figure 2; Supplementary Table S2). Of the 219 ORFs, 92 were annotated with predicted functions and categorized into four categories, structural formation (32 ORFs), DNA replication and metabolism (29 ORFs), regulation (27 ORFs), and lysis (4 ORFs) (Figure 2; Supplementary Tables S2, S3). The remaining 127 ORFs had unknown functions, with 49 ORFs having no matches in the NR database. A total of 159 ORFs of S-CREM2 showed homology with those of T4-like cyanophages that infect Prochlorococcus and Synechococcus, which indicates that S-CREM2 is a member of the T4-like cyanophages (Supplementary Table S2).
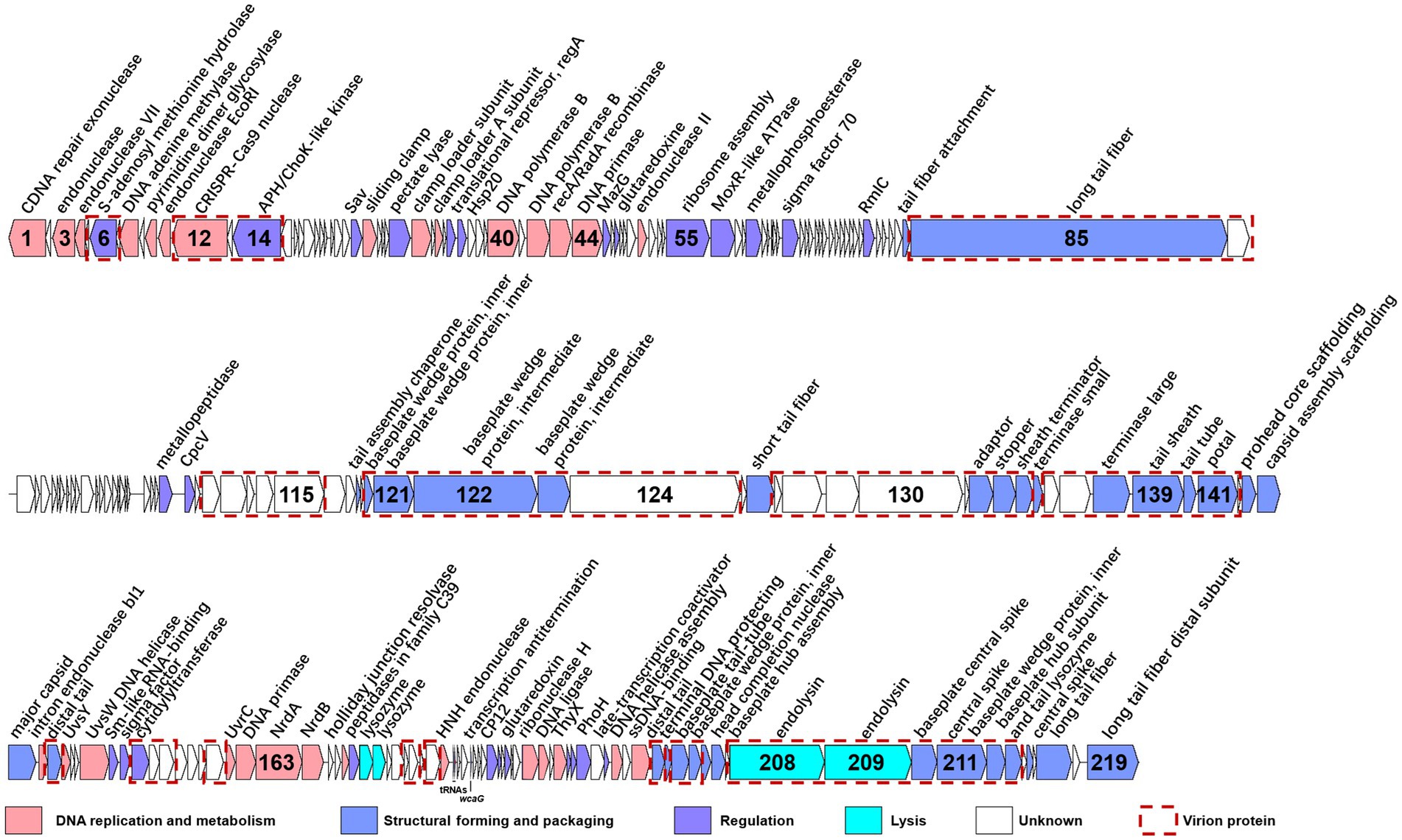
Figure 2. Genome organization of S-CREM2. ORFs with different functions are indicated by different colored arrows, and red dotted boxes represent virion proteins identified by mass spectrometry analysis. The number inside the arrow indicates the ORF number. ncRNA genes are labeled below the ORF bars.
To investigate the phylogenetic relationship between S-CREM2 and other T4-like cyanophages, a set of 31 core genes were identified in S-CREM2 (Supplementary Table S4) and 40 referenced T4-like cyanophages. Phylogenomic analysis based on these 31 core genes revealed that S-CREM2 clustered with Synechococcus phages S-B68, S-N03, S-H34, and S-CRM01 and formed a discrete clade, which is divergent from the well-characterized clusters A and B proposed by Ignacio-Espinoza and Sullivan (2012) (Figure 3). The new clade encompassing S-CREM2, S-B68, S-N03, S-H34, and S-CRM01 were named as cluster C, in which the marine phage strains S-CREM2, S-B68, S-N03, and S-H34 exhibit closer phylogenetic relationship with each other and are relatively distant from the freshwater strain S-CRM01. In addition, the G + C contents of marine strains in cluster C (47.9–51.7%) are much higher than that of the freshwater strain S-CRM01 (39.7%) (Table 2) and those of cluster A (37.8–43%) and B (36.7–42.2%) (Jiang et al., 2020). A total of 150 ORFs in S-CREM2 showed homology with the T4-like cluster C cyanophages and 46 of them were exclusively homologous to cluster C. Within cluster C, S-CREM2 shares the largest number of homologous genes (144) with S-H34. In addition, S-CREM2 has 137, 135, and 78 ORFs homologous with S-N03, S-B68, and S-CRM01, respectively (Supplementary Figure S2; Supplementary Table S2).
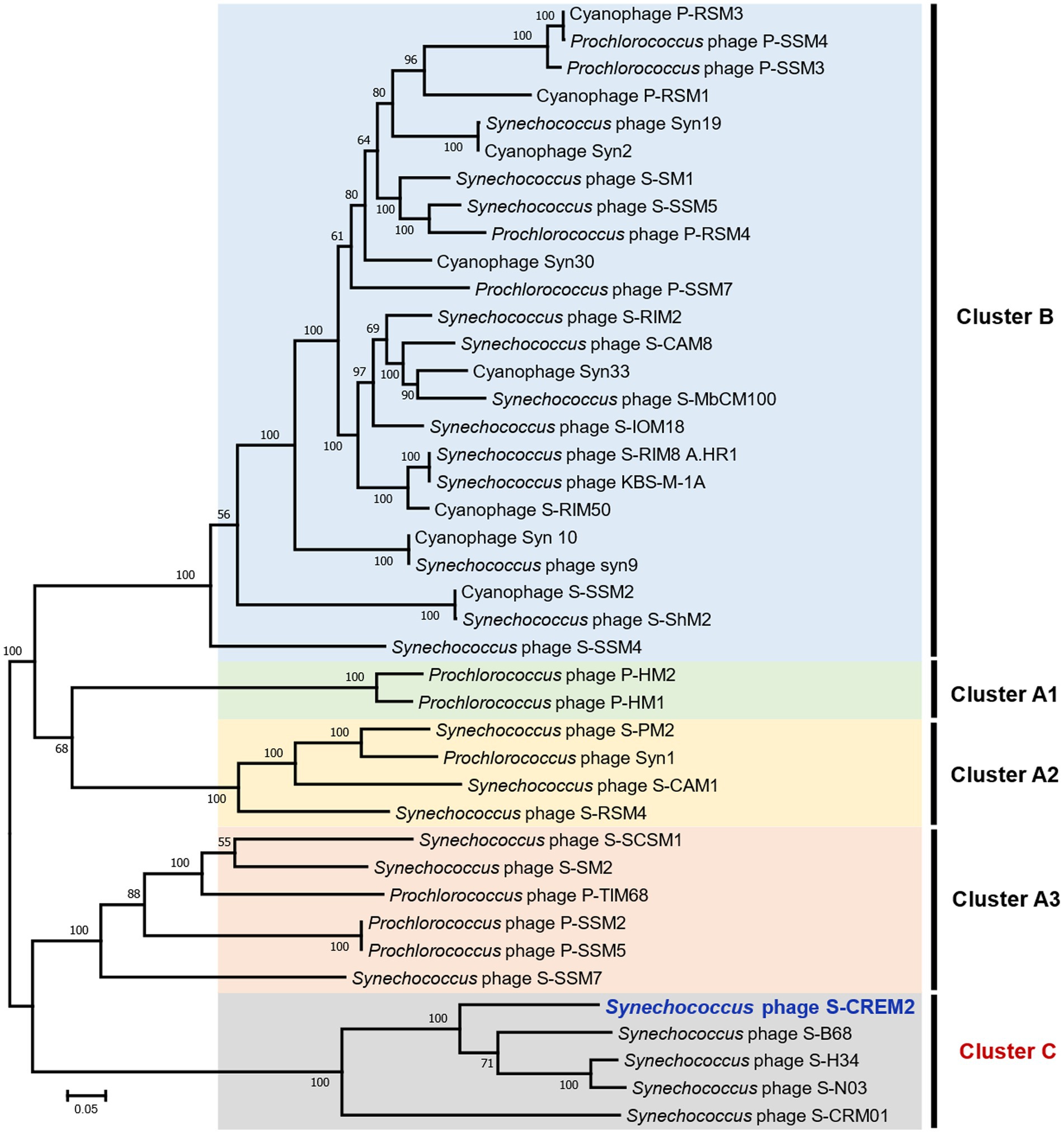
Figure 3. The maximum-likelihood phylogenomic tree based on the 31 core genes among S-CREM2 and 40 T4-like cyanophages. Bootstrap values are calculated based on 100 replicates. The 31 core genes contain 11 DNA replication-related genes, 13 structure-related genes, four AMGs, and three hypothetical genes.
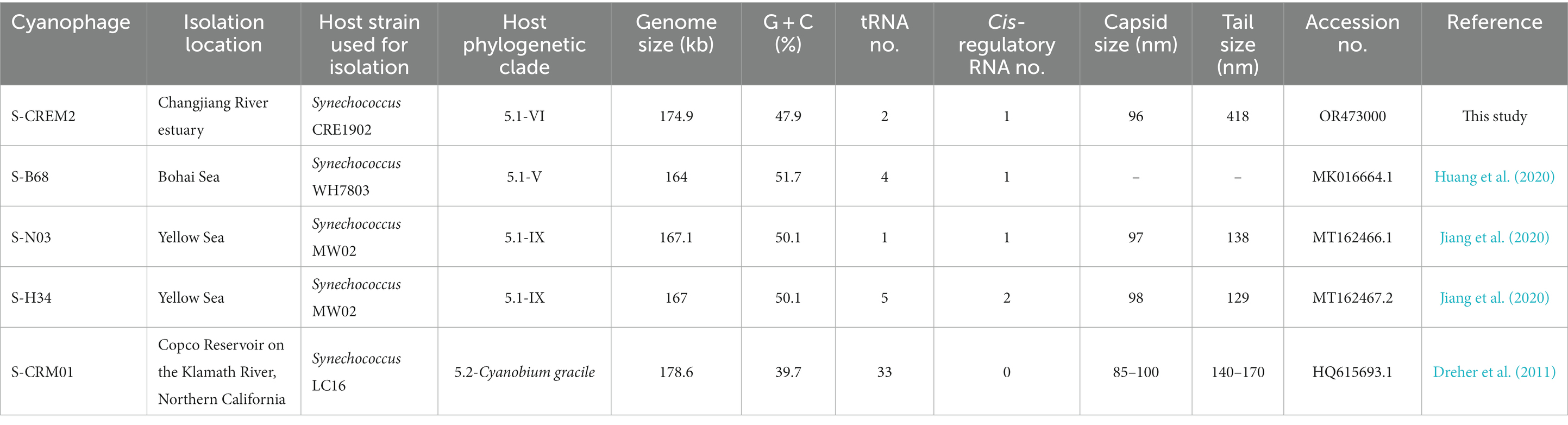
Table 2. Genomic and morphology features of the T4-like cyanophages in cluster C.
Five cyanophages in each T4-like cluster were selected as representatives to calculate nucleotide-based intergenomic similarities. The nucleotide similarities between cluster C cyanophages and representatives in clusters A and B (4.4–6.8%) are much lower than those within cluster C (11.4–68%) (Supplementary Figure S3). S-CREM2 showed nucleotide similarities of 11.4–32.8% with the other four cyanophages in cluster C, with the highest similarity observed with S-B68 and the lowest with S-CRM01. Following the genus-level classification criteria in phage taxonomy, the nucleotide similarity less than 50% of the whole genome is indicative of different genera (Adriaenssens and Brister, 2017). Thus, we propose classifying S-CREM2 as a representative of a new viral genus.
A total of 29 ORFs encode genes related to DNA replication and metabolism in the S-CREM2 genome, including DNA polymerase, helicase, primase, ligase, various endonucleases, and enzymes involved in nucleotide metabolism and DNA damage repair. Nucleotide metabolism genes encoded in the S-CREM2 genome include nrdA (ORF163), nrdB (ORF164), thyX (ORF192), and DNA adenine methylase gene (ORF8). Specifically, nrdA, nrdB, and thyX can provide DNA monomers for viral replication (Myllykallio et al., 2002; Gon et al., 2006; Koehn and Kohen, 2010). DNA adenine methylase, which is involved in the process of nucleotide methylation, plays an important role in enhancing DNA stability (Miller et al., 2003). Among the DNA damage repair genes, the product of ORF10 (putative pyrimidine dimer DNA glycosylase) may function as a base-cutting repair protein, thereby reducing the occurrence of pyrimidine dimer formation caused by UV damage (Grafstrom et al., 1982; Walker et al., 2006), while the S-CREM2 putative UvsY (ORF1) and CDNA repair exonuclease SbcCD ATPase subunit (ORF148) may be able to remove mutated bases and nucleotide fragments (Wilson and Murray, 1991). The expression of these genes may be crucial in maintaining accurate transcriptional translation when the virus or host is subjected to external damage (Kemp and Hu, 2017). Additionally, a CRISPR-Cas9 nuclease (ORF12) gene is predicted in the S-CREM2 genome. Bacterial CRISPR-Cas9 nuclease is associated with chromosome rearrangement and genotoxicity, and it functions as a component of the adaptive immune system, which serves to defend against viral infection by degrading DNA originating from invading viruses or other foreign sources (Cui et al., 2020). The S-CREM2 CRISPR-Cas9 nuclease gene may be acquired from cyanobacterial hosts through horizontal gene transfer.
Among the 29 DNA replication and metabolism ORFs predicted in the S-CREM2 genome, 27 ORFs are homologous to those predicted in other T4-like cluster C cyanophages, with the highest amino acid identity for each gene ranging from 32.7 to 94.6% and averaged at 73.1%. Additionally, 28 out of the 29 DNA replication and metabolism ORFs showed homology with genes predicted in T4-like clusters A and B cyanophages, with the highest amino acid identity of each gene ranging from 29.3 to 81.4% and averaged at 54.6% (Supplementary Figure S4A).
A total of 32 ORFs were predicted to encode structural proteins in the S-CREM2 genome, including terminase large subunit, terminase small subunit, portal protein, adaptor, stopper, sheath terminator, capsid-related proteins, and tail-related proteins (Figure 2; Supplementary Tables S2, S3), 24 of which were detected in the virion proteome by mass spectrometry analysis (Table 3). Of the 32 structural ORFs, 31 show homology to ORFs predicted in other cluster C cyanophages, with the highest amino acid identity for each gene ranging from 30.6 to 92.9% and averaged at 64.6% (Supplementary Figure S4B). The S-CREM2 ORF219, encoding a long tail fiber distal subunit, shows no homology with any cyanophages, but is homologous to genes predicted in heterotrophic bacteria, other bacteriophages, and Ostreococcus lucimarinus viruses, with amino acid identities ranging from 26.5 to 67% (Supplementary Table S2). Of the 32 S-CREM2 structure-related ORFs, 25 are homologous with those predicted in the T4-like cluster A and B cyanophages, with the highest amino acid identity of each gene ranging from 26.8 to 70.5% and averaged at 46.9% (Supplementary Figure S4B). It is worth noting that the S-CREM2 ORFs involved in structure formation exhibit a lower degree of conservation compared to ORFs related to DNA replication and metabolism (Supplementary Figure S4). Notably, six tail-related ORFs of S-CREM2 are exclusively homologous to genes predicted in the T4-like cluster C cyanophages. By comparing the S-CREM2 structural proteins involved in the virion formation with those of the T4 phage, the overall architecture of the S-CREM2 virion was predicted (Figure 4). Twenty-three proteins were mapped to the virion structure, with 21 detected in the virion proteome (Figure 4). Most of the structural proteins of S-CREM2 highly resemble those of the T4 phage. However, the adopter and the long tail fiber are different from those of the T4 phage and show homology with those of Escherichia phage vB_EcoP_SU10 and Escherichia phage K1F, respectively (Supplementary Table S5).
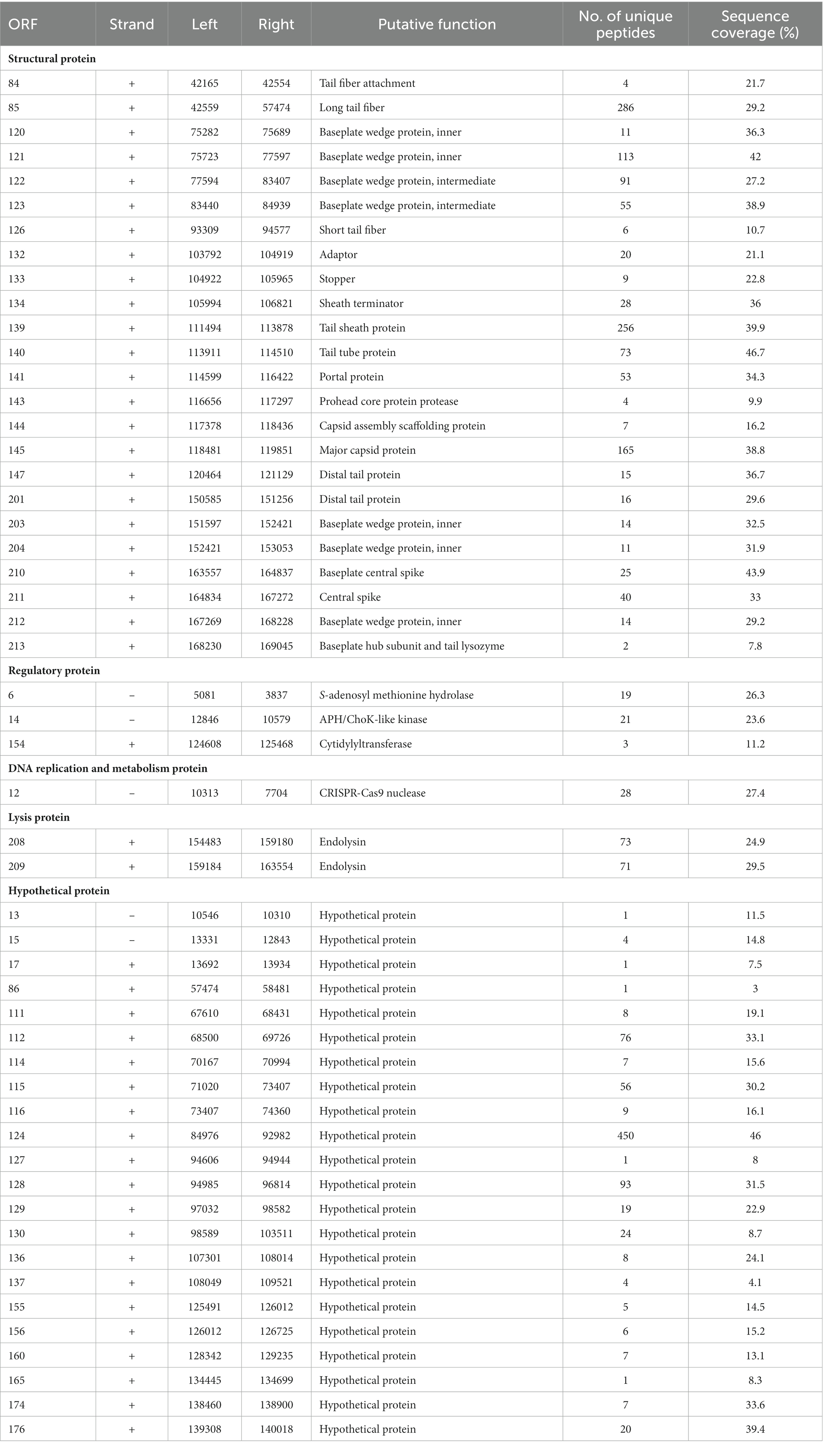
Table 3. The virion proteome of S-CREM2.
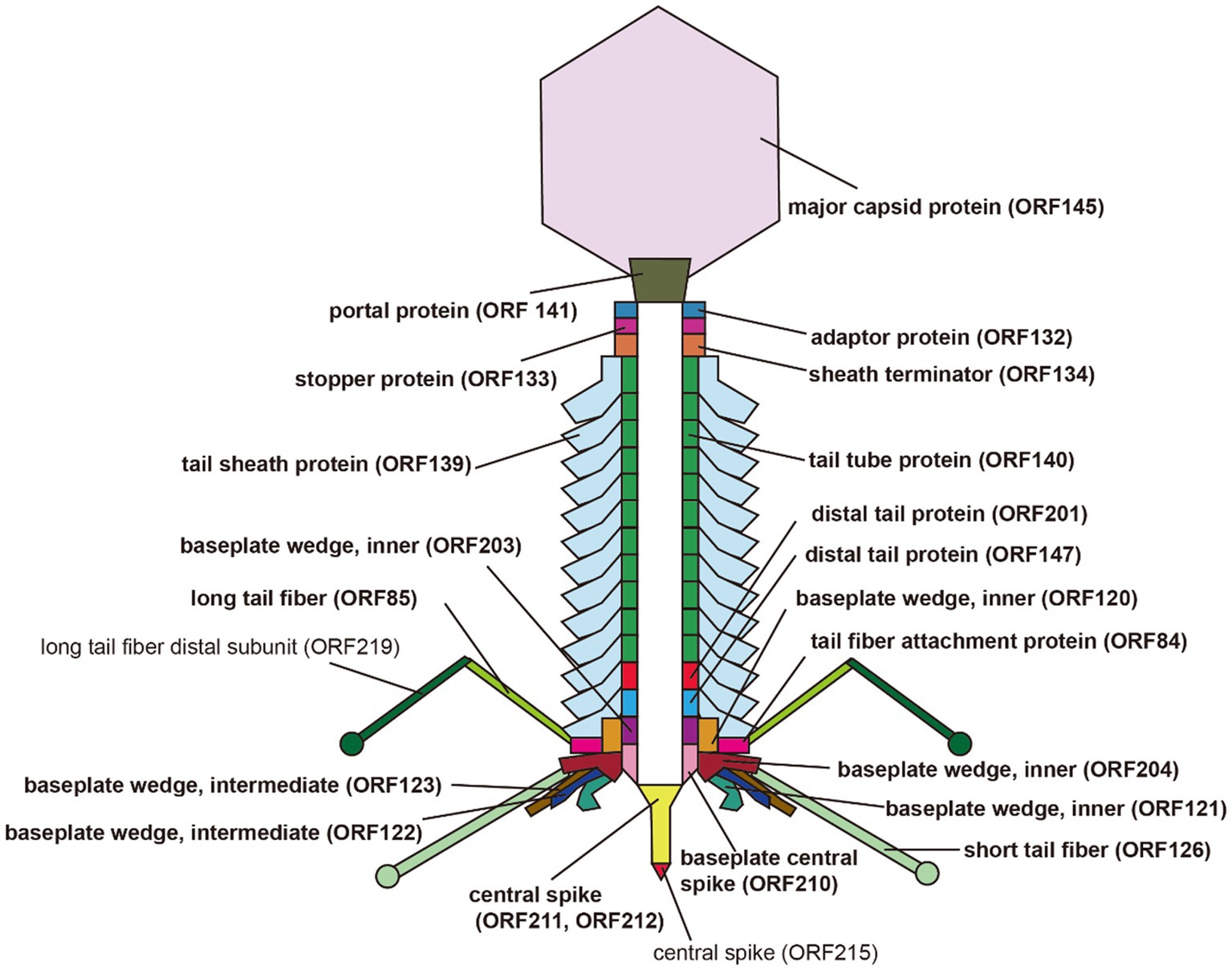
Figure 4. Predicted architecture of the S-CREM2 virion. Bold fonts indicate structural proteins detected by mass spectrometry analysis.
A total of 52 S-CREM2-encoded proteins were detected in the virion proteome by mass spectrometry analysis, including 24 structural proteins, six non-structural proteins, and 22 proteins with unknown function (Figure 2; Table 3). Notably, among the 22 protein genes with unknown function, 13 are located in the genome region (ORF84–145) primarily associated with structural genes and may also encode structural proteins, indicating distinctive proteins contributing to the formation of the unique virion, which has an extraordinarily long tail. The six non-structural proteins include S-adenosyl methionine (SAM) hydrolase (ORF6), APH/ChoK-like kinase (ORF14), cytidylyltransferase (ORF154), CRISPR-Cas9 nuclease (ORF12), and two endolysins (ORF208, 209) and may be encapsulated within the capsid as virion-associated proteins.
SAM hydrolase is essential for the degradation of S-adenosine methionine (SAM) (Jerlström Hultqvist et al., 2018). SAM serves as a crucial methyl donor for methyltransferases that function on nucleic acids, proteins, and lipids in bacteria cells (Loenen, 2006). As a defense mechanism, bacteria employ SAM to differentiate their own DNA from that of foreign invaders (Wilson and Murray, 1991). It is reported that the phage-encoded SAM hydrolase can degrade SAM, switching off the bacterial defense (Jerlström et al., 2018; Guo et al., 2021). The entry of the phage-encoded SAM hydrolase into the host cell upon infection may protect the phage genomic DNA from attacks of the host restriction-modification systems. VAPKs are common in enveloped viruses infecting animals and plants but are rarely discovered in phages (Hui, 2002). Recently, protein kinase-like proteins have been continuously detected in the cyanophage virion proteomes (Dreher et al., 2011; Sabehi et al., 2012; Xu et al., 2018) and are speculated to regulate host bioprocesses by phosphorylating specific substrates like serine, threonine, or tyrosine residues of proteins, aminoglycosides, and choline. The putative APH/ChoK-like kinase detected in the S-CREM2 virions is homologous and shares an amino acid identity of 32% with the putative protein kinase detected in the S-CRM01 virions. In prokaryotes, APHs phosphorylate and inactivate aminoglycoside antibiotics (Wright and Thompson, 1999), ChoKs facilitate the formation of phosphorylcholine and play an important role in phosphorylcholine-associated lipopolysaccharide modifications on cell surface and cell stress (Thomsen et al., 2003). The S-CREM2 APH/ChoK-like kinase may influence antibiotic resistance and the stress tolerance of the host cells (Wright and Thompson, 1999; Thomsen et al., 2003). Cytidyltransferase is a homolog of NMNAT. Previous studies have speculated that phage-encoded NMNAT may regulate host metabolism by affecting NAD+ levels in the cell and promote the production of phage progeny (Raffaelli et al., 1997, 1999, 2001; Wang et al., 2022). The frequent detection of protein or small molecule kinases and cytidyltransferases in the virion proteomes of cyanophages suggests that these proteins may be carried by the virion and able to enter the host cells to create an optimized intracellular environment that fosters phage replication at the initial stage of phage infection. However, it is also possible that these phage-encoded regulatory proteins are highly expressed during phage infection and were not separated from virions in the CsCl purification. Further efforts are needed to verify their presence in the phage virions.
In contrast to the numerous and diverse AMGs identified in T4-like cyanophages of clusters A and B, only a limited number of AMGs were predicted in the T4-like cluster C cyanophages (Figure 5). Only the freshwater strain, S-CRM01, contains the six most commonly found AMGs, psbA, hli, phoH, mazG, cobS, and hsp20, in T4-like cyanophages of clusters A and B (Ignacio-Espinoza and Sullivan, 2012; Jiang et al., 2020). While, four marine strains in cluster C, S-CREM2, S-H34, S-N03, and S-B68, only encode phoH, mazG, and hsp20 (Figure 5).
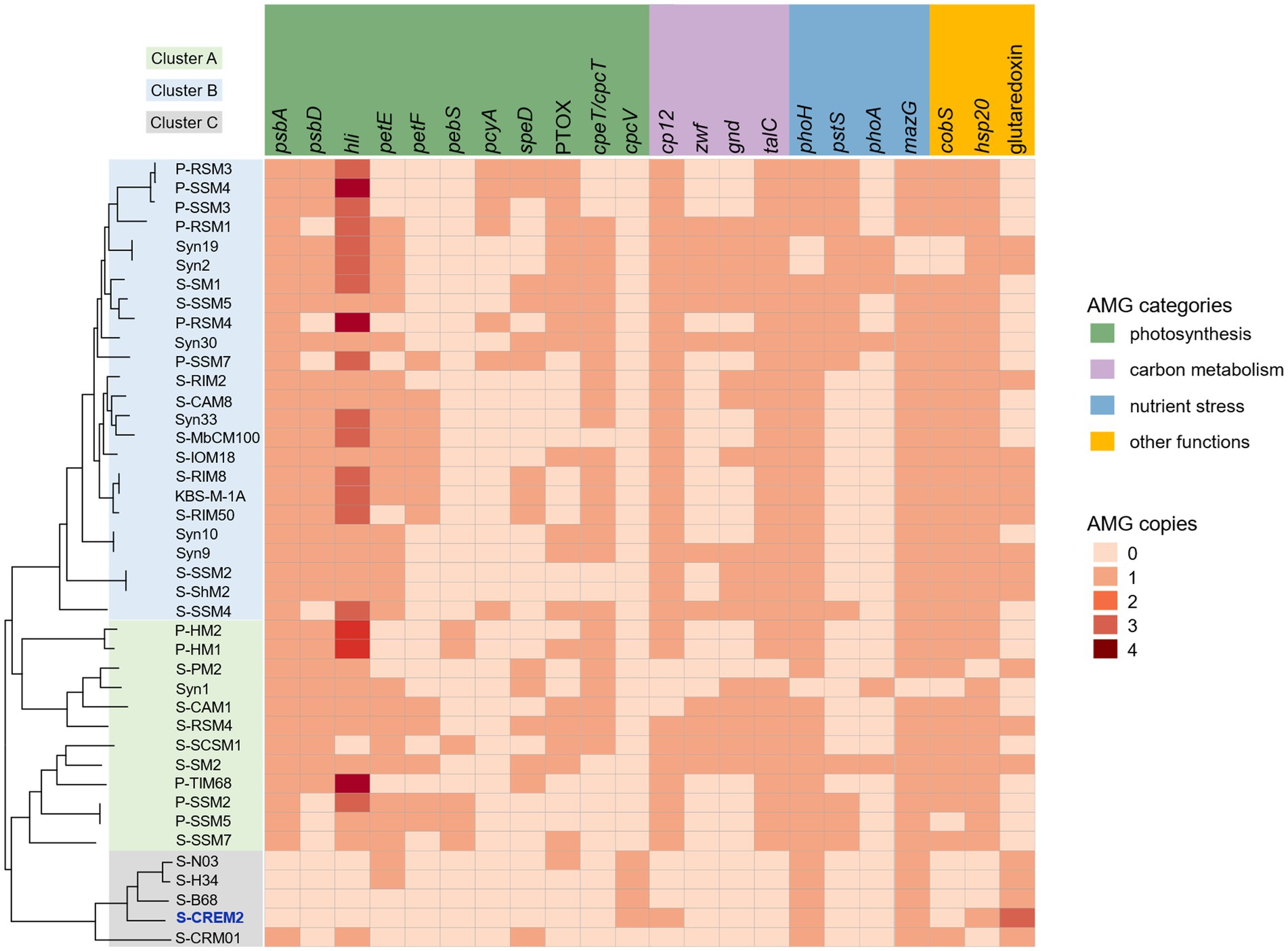
Figure 5. Comparative analysis of the AMGs in different T4-like cyanomyovirus clusters. Phylogenomic tree of S-CREM2 and 40 T4-like cyanophage based on 31 core genes. Colored boxes on the left signify T4-like cyanomyovirus clusters.
Tough lacking the commonly found photosynthesis genes psbA and hli, four marine cyanophage strains in cluster C encode an S-type phycobilin lyase gene, cpcV (Figure 5). Phycobilin lyases catalyze the covalent ligation between phycobilin chromophores and phycobiliproteins at specific binding sites, facilitating the synthesis of phycobilisome (Bretaudeau et al., 2013). The expression of phage cpcVs may assist the light absorption in infected host cells, providing energy for phage replication (Six et al., 2007; Xu et al., 2018). Phycobilin lyase genes are common in T4-like cyanophages of clusters A and B. However, all of the phycobilin lyase genes in clusters A and B are T-type, cpeT or cpcT. The cpcV is only found in T4-like cluster C cyanophages and S-CBWM1 (Xu et al., 2018). Phylogenetic analysis revealed that cpcT and cpeT of cyanophages in clusters A and B grouped into a stable branch with those of picocyanobacteria. The cpcV of cluster C cyanophages formed an individual clade with those of S-CBWM1 and a putative prophage of Synechococcus sp. SYN20, but did not cluster with any host-derived cpcV (Figure 6A). The discovery of more cpcV homologs from both cyanophage and cyanobacteria would facilitate the illustration of the evolutionary source and trajectory of the cyanophage cpcVs in future studies.
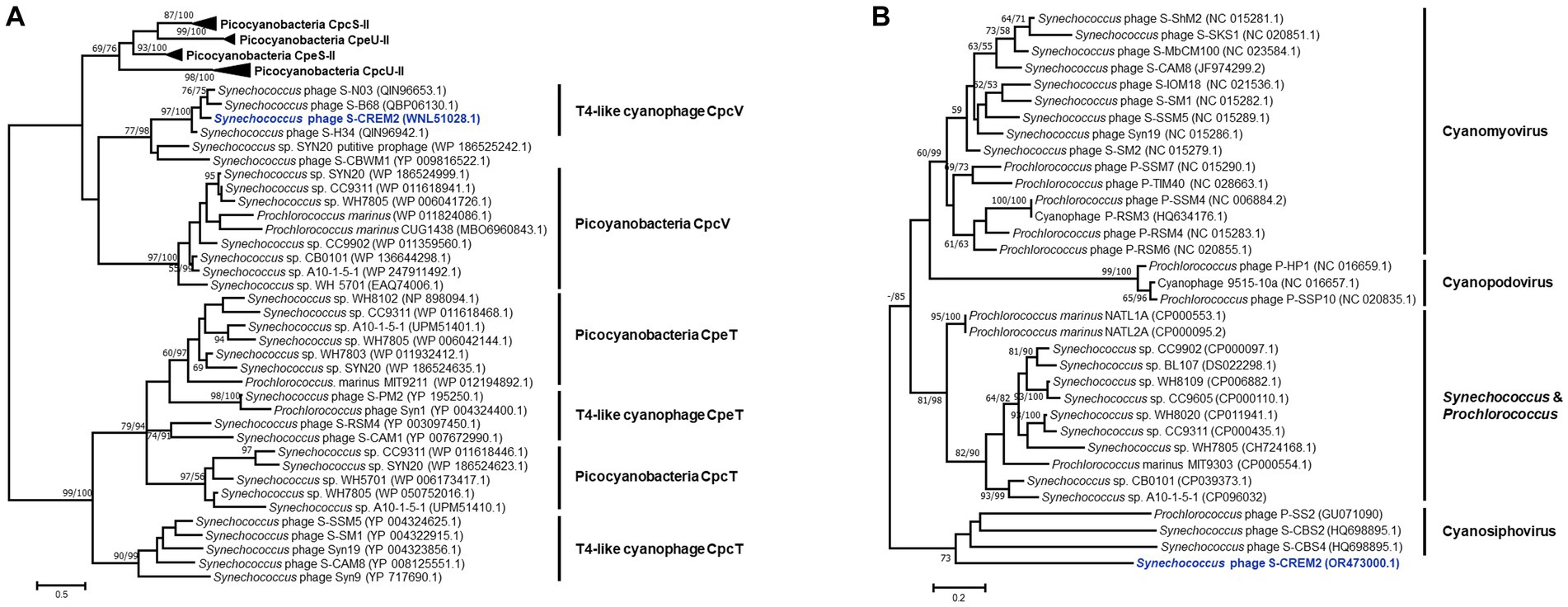
Figure 6. Maximum-likelihood phylogenetic trees of phycobilin lyase genes (A) and cp12s (B) from picocyanobacteria and cyanophages. The phylogenetic analyses of the phycobilin lyase genes were performed based on the amino acid sequences, while the cp12 trees were constructed using nucleotide sequences. Numbers near each branch node represent the bootstrap values (maximum-likelihood/neighbor-joining, ML/NJ) of ≥50%. The bootstrap replicates = 1,000.
In addition, the S-CREM2 genome carries a cp12 that is related to carbon metabolism. As a Calvin cycle inhibitor, the phage-encoded CP12 was proposed to redirect the host carbon flow from the Calvin cycle to the pentose phosphate pathway, resulting in ATP, NADPH, and pentose accumulation that are favorable for phage dNTP biosynthesis (Thompson et al., 2011). The phylogeny of cp12 revealed that cp12 homologs from marine T4-like cyanophages of clusters A and B and cyanopodoviruses both grouped with those of marine picocyanobacteria, indicating that cyanophages may acquire cp12 from their hosts. However, the S-CREM2 cp12 clustered with those of three cyanosiphoviruses and formed a very deep branch (Figure 6B), which suggests that the S-CREM2 cp12 evolves from a different origin or has experienced a divergent evolutionary trajectory from those of T4-like cyanophages in clusters A and B.
Three ncRNA genes were identified in the S-CREM2 genome, including two tRNA genes and a cis-regulatory RNA gene (wcaG) (Figure 2). The numbers of tRNA genes vary greatly among cyanophages in T4-like cluster C (Table 2). The marine phages in cluster C contain no more than five tRNA genes, while the freshwater strain, S-CRM01, contains 33 tRNA genes which cover all 20-amino-acid specificities. Notably, the G + C content of S-CRM01 (39.7%) is much lower than those of the marine phage strains in cluster C (47.9–51.7%). While, the G + C contents of their hosts are the opposite. The G + C contents of Synechococcus CRE1902 and WH7803, which are hosts of S-CREM2 and S-B68, are 57.4 and 60.2%. The G + C content of the S-CRM01 host, Synechococcus LC16, is not available. Synechococcus LC16 is a member of the Cyanobium gracile cluster. Since the G + C contents of cyanobacteria in the same phylogenetic clade are usually similar, the G + C content of Synechococcus LC16 can be estimated from that of the type strain in the Cyanobium gracile cluster, Synechococcus PCC6307, which is 68.5% and much higher than those of Synechococcus CRE1902 and WH7803. It is speculated that phages carry tRNAs to overcome the codon usage difference from its hosts (Enav et al., 2012). The large difference in tRNA number between the S-CRM01 and the marine phages in cluster C can be illustrated by the larger discrepancy of G + C contents between S-CRM01 and its host than those between the marine phage strains and their hosts. A cis-regulatory RNA gene, wcaG, was also predicted in genomes of the other three marine phages of cluster C. S-H34 also contains an extra glnA. However, no cis-regulatory elements were found in the S-CRM01 genome (Table 2). In prokaryotes, the cis-regulatory RNA wcaG acts as a regulator of exopolysaccharide production-related genes, glnA regulates gene expressions related to nitrogen metabolism (Weinberg et al., 2010). Phage cis-regulatory RNAs may also play similar roles in altering host metabolisms during infection. Different compositions of tRNA and cis-regulatory RNA genes between the marine and freshwater phages in cluster C may reflect their different modes of coping with their hosts and habitats.
The distribution and relative abundance of the T4-like cluster C cyanophages in the marine environment were investigated by metagenomic fragment recruitment analyses and compared with those of T4-like cluster A and B cyanophages. Among the 120 viromes employed for recruitment analyses, the cluster C-like cyanophages were detected in 29 viromes retrieved from various ecosystems, including temperate and subtropic estuaries, diverse coastal regions, and open oceans in both tropic and polar regions (Figure 7A). The five cluster C-like cyanophages are widespread in estuarine and coastal regions. Specially, the S-CREM2-like cyanophages are more abundant in coastal environments, the S-H34, S-N03, and S-B68-like cyanophages are more prevalent in estuarine environments. Whereas, ORF homologs of the freshwater strain, S-CRM01, are rarely detected in marine ecosystems (Figure 7B). However, the residence of these five cluster C-like cyanophages in the open sea is quite limited. Only four out of 18 open sea viromes used in this study exhibit the presence of cluster C-like cyanophages (Figure 7). The distributional pattern of the cluster C-like cyanophages is congruent with those of their hosts, Synechococcus subcluster 5.1 clade V, VI, and IX (Table 2), which also thrive in the estuarine and coastal environments but are rarely observed in the open sea (Xia et al., 2015; Sohm et al., 2016). The cluster A and B-like cyanophages are prevalent across various marine ecosystems. Despite consistently lower relative abundance compared to specific cluster A and B-like members in various marine ecosystems, specific members of cluster C-like cyanophages exhibit comparable or even higher relative abundances in certain estuarine regions (Figure 7B). This suggests that cluster C-like cyanophages play important ecological roles in the estuarine environment, which has been previously overlooked due to a lack of awareness regarding their existence.
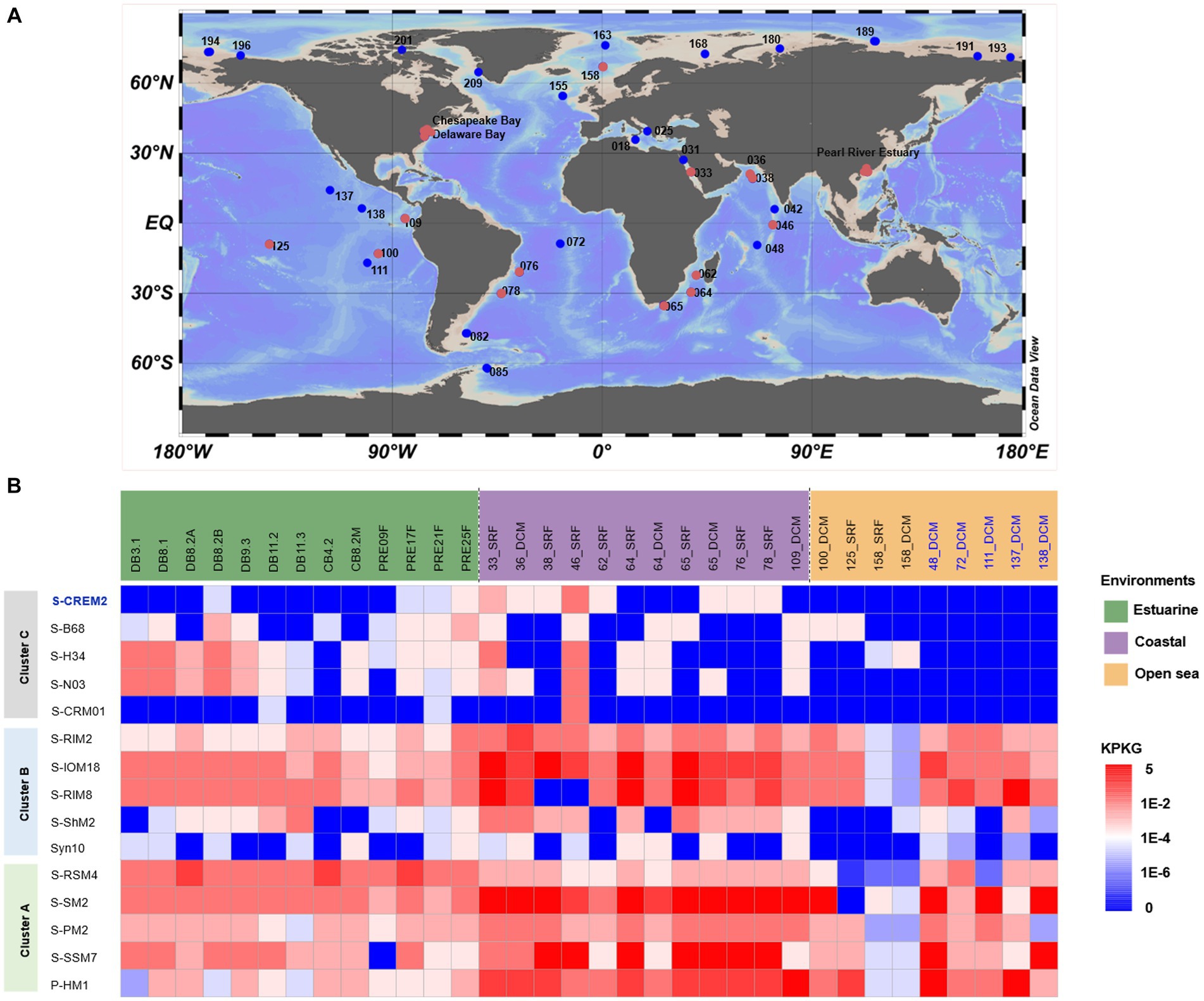
Figure 7. Comparison of environmental distribution of T4-like clusters A, B, and C-like cyanomyoviruses. (A) Location of publicly available viriomes used for the distributional analyses of the T4-like cyanophages. Red and blue dots indicate the presence and absence of the cluster C-like cyanophages; (B) The relative abundances of five representative strains of cyanophages in each cluster in metagenome databases. The results for 29 viromes containing cluster C-like cyanophages were shown, while those for additional five open sea viromes were also exhibited to better demonstrate the distributional pattern of cyanophages in clusters A and B in the open ocean. Relative abundance was normalized by KPKG.
Cyanophage S-CREM2 represents a new viral genus. The discovery of S-CREM2 refreshes our knowledge of the tail length of cyanomyoviruses and leads to the establishment of a new T4-like cyanophage clade, cluster C. Much less and unique AMGs and various virion-associated regulatory proteins of S-CREM2 may drive different phage–host interactions from those of clusters A and B cyanophages. The T4-like cluster C cyanophages are widespread in the estuarine and coastal environment. Specific members of this cluster may play important roles in certain estuarine ecosystems due to their equivalent or even higher relative abundance compared to cyanophages of clusters A and B. The isolation of S-CREM2 and establishment of the T4-like cluster C cyanophages provide new insights into the phage diversity, evolution, and phage–host interactions in the marine environment.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
YL: Data curation, Formal analysis, Visualization, Writing – original draft. XM: Formal analysis, Methodology, Writing – original draft. HZ: Data curation, Methodology, Writing – review & editing. LC: Writing – review & editing. SW: Writing – original draft, Visualization. MH: Writing – original draft, Visualization. JH: Writing – original draft, Visualization. YH: Writing – original draft, Visualization. CG: Writing – original draft, Visualization. JL: Supervision, Writing – review & editing. FC: Funding acquisition, Supervision, Writing – review & editing. YX: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Joint Funds of the National Natural Science Foundation of China (U1906216), the National Key Research and Development Program of China (2018YFA0605800), and the National Natural Science Foundation of China (41706161).
We greatly appreciated Xiaoju Li and Haiyan Sui from Shandong University Core Facilities for Life and Environmental Sciences for their help in the transmission electron microscopy observation. We thank Zefeng Zhang for the help in data analyses.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at:https://www.frontiersin.org/articles/10.3389/fmicb.2023.1293846/full#supplementary-material
1. ^http://exon.gatech.edu/GeneMark/
3. ^http://metagene.nig.ac.jp/
4. ^https://toolkit.tuebingen.mpg.de/tools/hhpred
6. ^http://rhea.icbm.uni-oldenburg.de/VIRIDIC/
Adriaenssens, E. M., and Brister, J. R. (2017). How to name and classify your phage: an informal guide. Viruses 9:70. doi: 10.3390/v9040070
Besemer, J., and Borodovsky, M. (2005). GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33, W451–W454. doi: 10.1093/nar/gki487
Bretaudeau, A., Coste, F., Humily, F., Garczarek, L., le Corguillé, G., Six, C., et al. (2013). CyanoLyase: a database of phycobilin lyase sequences, motifs and functions. Nucleic Acids Res. 41, D396–D401. doi: 10.1093/nar/gks1091
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chan, P. P., and Lowe, T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chen, F., Wang, K., Stewart, J., and Belas, R. (2006). Induction of multiple prophages from a marine bacterium: a genomic approach. Appl. Environ. Microbiol. 72, 4995–5001. doi: 10.1128/AEM.00056-06
Clokie, M. R. J., Thalassinos, K., Boulanger, P., Slade, S. E., Stoilova-McPhie, S., Cane, M., et al. (2008). A proteomic approach to the identification of the major virion structural proteins of the marine cyanomyovirus S-PM2. Microbiology 154, 1775–1782. doi: 10.1099/mic.0.2007/016261-0
Cui, Y. R., Wang, S. J., Chen, J., Li, J., Chen, W., Wang, S., et al. (2020). Allosteric inhibition of CRISPR-Cas9 by bacteriophage-derived peptides. Genome Biol. 21:51. doi: 10.1186/s13059-020-01956-x
Dreher, T. W., Brown, N., Bozarth, C. S., Schwartz, A. D., Riscoe, E., Thrash, C., et al. (2011). A freshwater cyanophage whose genome indicates close relationships to photosynthetic marine cyanomyophages. Environ. Microbiol. 13, 1858–1874. doi: 10.1111/j.1462-2920.2011.02502.x
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Enav, H., Béjà, O., and Mandel-Gutfreund, Y. (2012). Cyanophage tRNAs may have a role in cross-infectivity of oceanic Prochlorococcus and Synechococcus hosts. ISME J. 6, 619–628. doi: 10.1038/ismej.2011.146
Falco, S. C., Zehring, W., and Rothman-Denes, L. B. (1980). DNA-dependent RNA polymerase from bacteriophage N4 virions. Purification and characterization. J. Biol. Chem. 255, 4339–4347. doi: 10.1016/S0021-9258(19)85670-3
Gon, S., Faulkner, M. J., and Beckwith, J. (2006). In vivo requirement for glutaredoxins and thioredoxins in the reduction of the ribonucleotide reductases of Escherichia coli. Antioxid. Redox Signal. 8, 735–742. doi: 10.1089/ars.2006.8.735
Grafstrom, R. H., Park, L., and Grossman, L. (1982). Enzymatic repair of pyrimidine dimer-containing DNA. A 50 dimer DNA glycosylase: 30-apyrimidinic endonuclease mechanism from Micrococcus luteus. J. Biol. Chem. 257, 13465–13474. doi: 10.1016/S0021-9258(18)33472-0
Gregory, A. C., Zayed, A. A., Conceição-Neto, N., Temperton, B., Bolduc, B., Alberti, A., et al. (2019). Marine DNA viral macro- and microdiversity from pole to pole. Cells 177, 1109–1123.e14. doi: 10.1016/j.cell.2019.03.040
Guo, X., Söderholm, A., Kanchugal, P. S., Isaksen, G. V., Warsi, O., Eckhard, U., et al. (2021). Structure and mechanism of a phage-encoded SAM lyase revises catalytic function of enzyme family. eLife 10:e61818. doi: 10.7554/eLife.61818
Huang, L., Liu, Q., Liu, X., Wang, Q., Zhao, Q., Wang, M., et al. (2020). Isolation and complete genome sequence of a novel cyanophage S-B68. Curr. Microbiol. 77, 2385–2390. doi: 10.1007/s00284-020-02045-9
Huang, S., Zhang, S., Jiao, N., and Chen, F. (2015). Marine cyanophages demonstrate biogeographic patterns throughout the global ocean. Appl. Environ. Microbiol. 81, 441–452. doi: 10.1128/aem.02483-14
Hui, E. K. (2002). Virion-associated protein kinases. Cell. Mol. Life Sci. 59, 920–931. doi: 10.1007/s00018-002-8479-6
Ignacio-Espinoza, J. C., and Sullivan, M. B. (2012). Phylogenomics of T4 cyanophages: lateral gene transfer in the 'core' and origins of host genes. Environ. Microbiol. 14, 2113–2126. doi: 10.1111/j.1462-2920.2012.02704.x
Jerlström Hultqvist, J., Warsi, O., Söderholm, A., Knopp, M., Eckhard, U., Vorontsov, E., et al. (2018). A bacteriophage enzyme induces bacterial metabolic perturbation that confers a novel promiscuous function. Nat. Ecol. Evol. 2, 1321–1330. doi: 10.1038/s41559-018-0568-5
Jiang, T., Guo, C., Wang, M., Wang, M., Zhang, X., Liu, Y., et al. (2020). Genome analysis of two novel Synechococcus phages that lack common auxiliary metabolic genes: possible reasons and ecological insights by comparative analysis of cyanomyoviruses. Viruses 12:800. doi: 10.3390/v12080800
Katoh, K., Asimenos, G., and Toh, H. (2009). Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 537, 39–64. doi: 10.1007/978-1-59745-251-9_3
Kazmierczak, K. M., Davydova, E. K., Mustaev, A. A., and Rothman-Denes, L. B. (2002). The phage N4 virion RNA polymerase catalytic domain is related to single-subunit RNA polymerases. EMBO J. 21, 5815–5823. doi: 10.1093/emboj/cdf584
Kemp, M. G., and Hu, J. (2017). Postexcision events in human nucleotide excision repair. Photochem. Photobiol. 93, 178–191. doi: 10.1111/php.12641
Koehn, E. M., and Kohen, A. (2010). Flavin-dependent thymidylate synthase: a novel pathway towards thymine. Arch. Biochem. Biophys. 493, 96–102. doi: 10.1016/j.abb.2009.07.016
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, D., Luo, R., Liu, C. M., Leung, C. M., Ting, H. F., Sadakane, K., et al. (2016). MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi: 10.1016/j.ymeth.2016.02.020
Loenen, W. A. (2006). S-adenosylmethionine: jack of all trades and master of everything? Biochem. Soc. Trans. 34, 330–333. doi: 10.1042/BST20060330
Mann, N. H., and Clokie, M. R. J. (2012). “Cyanophages, p 535–557” in Ecology of cyanobacteria II: Their diversity in space and time. ed. B. A. Whitton (Netherlands: Springer, Dordrecht), 535–557.
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229. doi: 10.1093/nar/gkq1189
Martinez-Hernandez, F., Fornas, O., Lluesma Gomez, M., Bolduc, B., de la Cruz Peña, M. J., Martínez, J. M., et al. (2017). Single-virus genomics reveals hidden cosmopolitan and abundant viruses. Nat. Commun. 8:15892. doi: 10.1038/ncomms15892
Michalski, A., Damoc, E., Hauschild, J. P., Lange, O., Wieghaus, A., Makarov, A., et al. (2011). Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 10:M111.011015. doi: 10.1074/mcp.M111.011015
Miller, E. S., Kutter, E., Mosig, G., Arisaka, F., Kunisawa, T., and Rüger, W. (2003). Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 67, 86–156. doi: 10.1128/MMBR.67.1.86-156.2003
Mizuno, C. M., Ghai, R., Saghaï, A., López-García, P., and Rodriguez-Valera, F. (2016). Genomes of abundant and widespread viruses from the deep ocean. MBio 7, e00805–e00816. doi: 10.1128/mBio.00805-16
Moraru, C., Varsani, A., and Kropinski, A. M. (2020). VIRIDIC-A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 12:1268. doi: 10.3390/v12111268
Myllykallio, H., Lipowski, G., Leduc, D., Filee, J., Forterre, P., and Liebl, U. (2002). An alternative flavin-dependent mechanism for thymidylate synthesis. Science 297, 105–107. doi: 10.1126/science.1072113
Narasimhan, V., Danecek, P., Scally, A., Xue, Y., Tyler-Smith, C., and Durbin, R. (2016). BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–1751. doi: 10.1093/bioinformatics/btw044
Noguchi, H., Taniguchi, T., and Itoh, T. (2008). MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 15, 387–396. doi: 10.1093/dnares/dsn027
Peng, Y., Leung, H. C., Yiu, S. M., and Chin, F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Pruitt, K. D., Tatusova, T., and Maglott, D. R. (2007). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65. doi: 10.1093/nar/gkl842
Puxty, R. J., Millard, A. D., Evans, D. J., and Scanlan, D. J. (2016). Viruses inhibit CO2 fixation in the most abundant phototrophs on earth. Curr. Biol. 26, 1585–1589. doi: 10.1016/j.cub.2016.04.036
Raffaelli, N., Emanuelli, M., Pisani, F. M., Amici, A., Lorenzi, T., Ruggueru, S., et al. (1999). Identification of the archaeal NMN adenylytransferase gene. Mol. Cell. Biochem. 193, 99–102. doi: 10.1023/A:1006968328186
Raffaelli, N., Pisani, F. M., Lorenzi, T., Emanuelli, M., Amici, A., Ruggieri, S., et al. (1997). Characterization of nicotinamide mononucleotide adenylyltransferase from thermophilic archaea. J. Bacteriol. 179, 7718–7723. doi: 10.1128/jb.179.24.7718-7723.1997
Raffaelli, N., Pisani, F. M., Lorenzi, T., Emanuelli, M., Amici, A., Ruggieri, S., et al. (2001). Nicotinamide-mononucleotide adenylyltransferase from Methanococcus jannaschii. Methods Enzymol. 331, 292–298. doi: 10.1016/s0076-6879(01)31066-2
Rong, C., Zhou, K., Li, S., Xiao, K., Xu, Y., Zhang, R., et al. (2022). Isolation and characterization of a novel cyanophage encoding multiple auxiliary metabolic genes. Viruses 14:887. doi: 10.3390/v14050887
Sabehi, G., Shaulov, L., Silver, D. H., Yanai, I., Harel, A., and Lindell, D. (2012). A novel lineage of myoviruses infecting cyanobacteria is widespread in the oceans. Proc. Natl. Acad. Sci. U. S. A. 109, 2037–2042. doi: 10.1073/pnas.1115467109
Six, C., Thomas, J. C., Garczarek, L., Ostrowski, M., Dufresne, A., Blot, N., et al. (2007). Diversity and evolution of phycobilisomes in marine Synechococcus spp.: a comparative genomics study. Genome Biol. 8:R259. doi: 10.1186/gb-2007-8-12-r259
Söding, J., Biegert, A., and Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248. doi: 10.1093/nar/gki408
Sohm, J. A., Ahlgren, N. A., Thomson, Z. J., Williams, C., Moffett, J. W., Saito, M. A., et al. (2016). Co-occurring Synechococcus ecotypes occupy four major oceanic regimes defined by temperature, macronutrients and iron. ISME J. 10, 333–345. doi: 10.1038/ismej.2015.115
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sullivan, M. B., Coleman, M. L., Quinlivan, V., Rosenkrantz, J. E., DeFrancesco, A. S., Tan, G., et al. (2008). Portal protein diversity and phage ecology. Environ. Microbiol. 10, 2810–2823. doi: 10.1111/j.1462-2920.2008.01702.x
Sullivan, M. B., Coleman, M. L., Weigele, P., Rohwer, F., and Chisholm, S. W. (2005). Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 3:e144. doi: 10.1371/journal.pbio.0030144
Sullivan, M. B., Huang, K. H., Ignacio-Espinoza, J. C., Berlin, A. M., Kelly, L., Weigele, P. R., et al. (2010). Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 12, 3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x
Sullivan, M. B., Krastins, B., Hughes, J. L., Kelly, L., Chase, M., Sarracino, D., et al. (2009). The genome and structural proteome of an ocean siphovirus: a new window into the cyanobacterial 'mobilome'. Environ. Microbiol. 11, 2935–2951. doi: 10.1111/j.1462-2920.2009.02081.x
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Sullivan, M. B., Waterbury, J. B., and Chisholm, S. W. (2003). Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature 424, 1047–1051. doi: 10.1038/nature01929
Sun, M., Zhan, Y., Marsan, D., Páez-Espino, D., Cai, L., and Chen, F. (2021). Uncultivated viral populations dominate estuarine viromes on the spatiotemporal scale. mSystems. 6:e01020. doi: 10.1128/mSystems.01020-20
Suttle, C. A., and Chen, F. (1992). Mechanisms and rates of decay of marine viruses in seawater. Appl. Environ. Microbiol. 58, 3721–3729. doi: 10.1128/aem.58.11.3721-3729.1992
Thompson, L. R., Zeng, Q., Kelly, L., Huang, K. H., Singer, A. U., Stubbe, J. A., et al. (2011). Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. U. S. A. 108, E757–E764. doi: 10.1073/pnas.1102164108
Thomsen, L. E., Chadfield, M. S., Bispham, J., Wallis, T. S., Olsen, J. E., and Ingmer, H. (2003). Reduced amounts of LPS affect both stress tolerance and virulence of Salmonella enterica serovar Dublin. FEMS Microbiol. Lett. 228, 225–231. doi: 10.1016/S0378-1097(03)00762-6
Walker, R. K., Mccullough, A. K., and Lloyd, R. S. (2006). Uncoupling of nucleotide flipping and DNA bending by the T4 pyrimidine dimer DNA glycosylase. Biochemistry 45, 14192–14200. doi: 10.1021/bi060802s
Wang, Q., Cai, L., Zhang, R., Wei, S., Li, F., Liu, Y., et al. (2022). A unique set of auxiliary metabolic genes found in an isolated cyanophage sheds new light on marine phage-host interactions. Microbiol. Spectr. 10:e0236722. doi: 10.1128/spectrum.02367-22
Wang, Y., Ferrinho, S., Connaris, H., and Goss, R. J. M. (2023). The impact of viral infection on the chemistries of the earth’s most abundant photosynthesizes: metabolically talented aquatic cyanobacteria. Biomol. Ther. 13:1218. doi: 10.3390/biom13081218
Waterbury, J. B., and Willey, J. M. (1988). “Isolation and growth of marine planktonic cyanobacteria” in Methods in enzymology—cyanobacteria. eds. L. Packer and A. N. Glazer, vol. 167 (San Diego, CA: Academic Press), 100–105.
Weinberg, Z., Wang, J. X., Bogue, J., Yang, J., Corbino, K., Moy, R. H., et al. (2010). Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol. 11:R31. doi: 10.1186/gb-2010-11-3-r31
Wilson, G. G., and Murray, N. E. (1991). Restriction and modification systems. Annu. Rev. Genet. 25, 585–627. doi: 10.1146/annurev.ge.25.120191.003101
Wiśniewski, J. R., Zougman, A., Nagaraj, N., and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Wright, G. D., and Thompson, P. R. (1999). Aminoglycoside phosphotransferases: proteins, structure, and mechanism. Front. Biosci. 4, D9–D21. doi: 10.2741/wright
Xia, X., Vidyarathna, N. K., Palenik, B., Lee, P., and Liu, H. (2015). Comparison of the seasonal variations of Synechococcus assemblage structures in estuarine waters and coastal waters of Hong Kong. Appl. Environ. Microbiol. 81, 7644–7655. doi: 10.1128/AEM.01895-15
Xu, B., Li, F., Cai, L., Zhang, R., Fan, L., and Zhang, C. (2022). A holistic genome dataset of bacteria, archaea and viruses of the Pearl River estuary. Sci Data. 9:49. doi: 10.1038/s41597-022-01153-4
Xu, Y., Zhang, R., and Jiao, N. (2015). Complete genome sequence of Paracoccus marcusii phage vB_PmaS-R3 isolated from the South China Sea. Stand. Genomic Sci. 10:94. doi: 10.1186/s40793-015-0089-7
Xu, Y., Zhang, R., Wang, N., Cai, L., Tong, Y., Sun, Q., et al. (2018). Novel phage-host interactions and evolution as revealed by a cyanomyovirus isolated from an estuarine environment. Environ. Microbiol. 20, 2974–2989. doi: 10.1111/1462-2920.14326
Yao, Z., Barrick, J., Weinberg, Z., Neph, S., Breaker, R., Tompa, M., et al. (2007). A computational pipeline for high- throughput discovery of cis-regulatory noncoding RNA in prokaryotes. PLoS Comput. Biol. 3:e126. doi: 10.1371/journal.pcbi.0030126
Keywords: T4-like cyanophages, cluster C, longest tail, auxiliary metabolic gene, virion-associated protein, ecological distribution
Citation: Liu Y, Meng X, Zheng H, Cai L, Wei S, He M, He J, Hao Y, Ge C, Liu J, Chen F and Xu Y (2023) A novel long-tailed myovirus represents a new T4-like cyanophage cluster. Front. Microbiol. 14:1293846. doi: 10.3389/fmicb.2023.1293846
Edited by:
Xiaobo Zhang, Zhejiang University, ChinaReviewed by:
Fang Qin, Xuzhou Medical University, ChinaCopyright © 2023 Liu, Meng, Zheng, Cai, Wei, He, He, Hao, Ge, Liu, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongle Xu, eHV5b25nbGVAc2R1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.