
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 23 November 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1288286
 Mengcheng You1,2†Qiuyun Zhao2†Yuansheng Liu1,2Wenhao Zhang1,2Zhewei Shen1Zhenxing Ren3*
Mengcheng You1,2†Qiuyun Zhao2†Yuansheng Liu1,2Wenhao Zhang1,2Zhewei Shen1Zhenxing Ren3* Chenggang Xu2*
Chenggang Xu2*Mesophilic, anaerobic, and cellulolytic Ruminiclostridium-type bacterial species can secrete an extracellular, multi-enzyme machinery cellulosome, which efficiently degrades cellulose. In this study, we first reported the complete genome of Ruminiclostridium papyrosolvens DSM2782, a single circular 5,027,861-bp chromosome with 37.1% G + C content, and compared it with other Ruminiclostridium-type species. Pan-genome analysis showed that Ruminiclostridium-type species share a large number of core genes to conserve basic functions, although they have a high level of intraspecific genetic diversity. Especially, KEGG mapping revealed that Ruminiclostridium-type species mainly use ABC transporters regulated by two-component systems (TCSs) to absorb extracellular sugars but not phosphotransferase systems (PTSs) that are employed by solventogenic clostridia, such as Clostridium acetobutylicum. Furthermore, we performed comparative analyses of the species-specific repertoire of CAZymes for each of the Ruminiclostridium-type species. The high similarity of their cohesins suggests a common ancestor and potential cross-species recognition. Additionally, both differences between the C-terminal cohesins and other cohesins of scaffoldins and between the dockerins linking with cellulases and other catalytic domains indicate a preference for the location of cellulosomal catalytic subunits at scaffoldins. The information gained in this study may be utilized directly or developed further by genetic engineering and optimizing enzyme systems or cell factories for enhanced biotechnological biomass deconstruction and biofuel production.
Lignocellulose is observed plentifully in nature and is obtained globally, considering it a fascinating source of feedstock for bio-based energy and chemicals. In nature, the direct hydrolysis of lignocellulose is carried out exclusively by microorganisms. Cellulolytic clostridia, such as Clostridium (Hungateiclostridium) thermocellum (Mazzoli and Olson, 2020), Clostridium (Thermoclostridium) stercorarium (Poehlein et al., 2013), Clostridium (Ruminiclostridium) cellulolyticum (Desvaux, 2005), and Clostridium cellulovorans (Tamaru et al., 2010, 2011), which are ubiquitous in cellulosic anaerobic environments, represent a major paradigm for efficient biological degradation of cellulosic biomass (Demain et al., 2005; Ransom-Jones et al., 2012). Many of these anaerobes digest cellulose via a cell surface-attached extracellular enzymatic complex called the cellulosome, where primarily catalytic components (including glycoside hydrolases, carbohydrate esterases, and polysaccharide lyases) are integrated onto a non-catalytic macromolecular scaffoldin subunit (Bayer et al., 2004, 2008). The scaffoldins bear modules called cohesin that interact with their modular counterparts, called dockerins, usually conjugated to enzymatic subunits or other scaffoldins. In addition, the scaffoldin may contain a carbohydrate-binding module (CBM) that guides the complex and its intricate set of component enzymes to the surface of the cellulosic substrate (Guillen et al., 2010; Hyeon et al., 2013).
To distinguish them from non-cellulolytic clostridia, these cellulolytic clostridia from Ludwig et al.’s (2019) clostridial cluster III (Garrity et al., 2010; Galperin et al., (2012) were first placed in the new genus “Ruminiclostridium” proposed by Yutin et al. (2012), Garrity et al. (2010), Galperin et al. (2012). Whereas the members of clostridial cluster III were further distributed into four new sublineages based on the phylogenetic analysis of 16S rRNA gene sequences made by Zhang X. et al. (2018), including three new genera, Thermoclostridium, Hungateiclostridium, and Ruminiclostridium. Members of Ruminiclostridium were mesophilic, spore-forming, cellulosome-producing, and cellulose-degrading bacteria, including R. cellobioparum, R. cellulolyticum, R. hungatei, R. josui, R. papyrosolvens, R. termitidis, and R. sufflavum. Their genome size ranged from 4.1 to 6.4 Mb, and the G + C content varied from 36 to 42 mol%. It is noteworthy that they harbor the cip-cel gene cluster, which encodes major cellulosomal components that are essential for cellulose degradation. Up to now, 10 whole-genome sequences of Ruminiclostridium-type species have been published in NCBI, which allowed us to perform a detailed analysis of the architecture, putative regulation, and evolution of the cellulolytic machinery of mesophilic cellulosome-producing clostridia.
In this study, we first sequenced and completed the genome of R. papyrosolvens DSM2782 to understand the mechanism of lignocellulose degradation in mesophilic and cellulolytic clostridia. We further compared it with other nine Ruminiclostridium-type species and C. cellulovorans 743B, which is involved in mesophilic cellulosome-producing species. Their evolutionary information, genomic diversity, cellulose degradation profiles, and cellulosomal structures were explored using comparative genome analysis. The results of comparative genomics among multiple Ruminiclostridium-type species offer new insights into genome evolution involving lignocellulose degradation.
The genome and protein sequences of Clostridium acetobutylicum ATCC824, Clostridium cellulovorans 743B, Ruminiclostridium sp. BNL1100, Ruminiclostridium cellobioparum DSM1351, Ruminiclostridium termitidis CT1112, Ruminiclostridium cellulolyticum H10, Ruminiclostridium herbifermentans MA18, Ruminiclostridium hungatei DSM14427, Ruminiclostridium josui JCM17888, Ruminiclostridium papyrosolvens C7, Ruminiclostridium sufflavum DSM19573, and Ruminiclostridium papyrosolvens DSM2782 strains (Supplementary Table S1) were downloaded from the NCBI database1 for comparative analysis.
Ruminiclostridium papyrosolvens DSM2782 was cultured anaerobically at 35°C in 250-ml flasks with a 100-ml working volume of GS-2 liquid medium (Johnson et al., 1981; Cui et al., 2012) (K2HPO4 2.9 g/L, KH2PO4 1.5 g/L, urea 2.1 g/L, resazurin 1.0 mg/L, yeast extract 6.0 g/L, cysteine–HCl 0.5 g/L, MOPS 10.0 g/L, and trisodium citrate 3.0 g/L, pH 7.4) supplemented with 3.0 g/L of cellobiose (Yuanye Biotechnology, Shanghai, China). The medium for cultivation was depleted of oxygen in an anaerobic chamber (COY, United States) using resazurin (0.0005% g/L) as the indicator, and then sterilized at 121°C for 20 min. The genomic DNA of R. papyrosolvens DSM2782 was extracted using (Illumina, United States) and Template Prep Kit 1.0 (PacBio, United States). The quantity and purity of extracted DNA were determined using a Hou et al. (2021) (Thermo Scientific, United States). The integrity of genomic DNA was further checked by agarose gel electrophoresis to evaluate its quality. DNA was stored at −20°C until use.
The raw sequence data generated from Illumina and PacBio sequencing was utilized for bioinformatics investigation; the whole-genome sequence was assembled using both Illumina and PacBio quality reads. For quality trimming, a value data statistic was used, from which the low-value information could be eliminated to form clean reads (Supplementary Table S2). The reads were then assembled into contigs by the Unicycler (Wick et al., 2017). The final step was completed and finished manually, generating a whole genome with seamless chromosomes. After genome annotation and genes prediction by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) server (Tatusova et al., 2016), further bioinformatics analysis was performed.
The complete genomes of R. papyrosolvens DSM2782 were compared with those of other Ruminiclostridium species, C. acetobutylicum ATCC824, and C. cellulovorans 743B using the BLAST Ring Image Generator (BRIG) (Alikhan et al., 2011) to determine the overall sequence similarity between the strains. Each circular genomic map was drawn using the genome of one reference strain based on a local BLAST+ with standard parameters (50% lower and 70% upper cutoff for identity and an E-value of 1e−5). The ring color gradients correspond to varying degrees of the identity of BLAST matches (Altschul et al., 1990). Circular genomic maps also include information on GC skew and GC content, and their evolutionary relationship was inferred by FastTree (Price et al., 2009) using the representative genomes in the Genome Taxonomy Database (GTDB) (Parks et al., 2022) as references. The phylogenetic tree was visualized using the Interactive Tree of Life (iTOL) (Letunic and Bork, 2021).
All the protein sequences were calculated usingOrthoFinder2 software with the DIAMOND method to identify homologous groups of protein families in the pan-genome (Emms and Kelly, 2019; Buchfink et al., 2021). The core genome families represented the genes or proteins shared by all 10 Ruminiclostridium species. The necessary genome families comprised the genes or proteins shared by at least two strains but not by all 10 Ruminiclostridium species. The remaining genes or proteins occurring only in one Ruminiclostridium were clustered into unique genome families. To gain more information on the functional characteristics of Ruminiclostridium-type species, all the homologous protein sequences were annotated to COG2 by using BLASTp (Altschul et al., 1997); a search was performed against the COG database with an E-value cutoff of 1 × 10−5. The top of the annotation results was selected as the best annotation for homologous families, and then it was assigned to functional categories.
Identification of genes encoding ABC transporter systems and PTSs in 12 Clostridia species was performed using BlastKOALA3 (Kanehisa et al., 2016b). Additionally, genes encoding TCSs were searched using HMMER 3.0 with TCS characteristic domains from the Pfam database4 as references. The sequences were manually sorted to remove the redundancy, and the remaining proteins were considered as identified TCS proteins.
Genes encoding carbohydrate-active enzymes (CAZymes) in the genomes of Ruminiclostridium species were predicted based on dbCAN2 (Zhang H. et al., 2018) and classified into families of glycoside hydrolases (GH), carbohydrate esterases (CE), polysaccharide lyases (PL), carbohydrate-binding modules (CBMs), auxiliary activities (AAs), and S-layer homology (SLH) by running a hmmscan of HMMER with an E-value cutoff of 1 × 10−15. The Easyfig software (Sullivan et al., 2011) was applied to reveal the homology of the cellulosome gene cluster relationships. Furthermore, cohesin and dockerin modules were predicted using Pfam-supported families as queries. Phylogenetic trees of cohesin and dockerin modules were built with MEGAX software (Kumar et al., 2018).
In this study, the genome of R. papyrosolvens DSM2782 was sequenced and completed by next-generation sequencing (NGS) and third-generation PacBio single-molecule sequencing technology, consisting of a single circular 5,027,861 bp chromosomewith a G + C content of 37.1%. In total, 4,274 coding DNA sequences (CDSs) were predicted, along with 24 rRNA and 62 tRNA genes (GenBank Accession Number CP119677.1). However, 40 SNPs and 200 InDels were identified by comparison with the previous version of the draft genome (GenBank Accession Number ACXX00000000.2) (Hemme et al., 2010). The majority (35) of SNPs were observed in the coding regions, of which 13 variants are nonsense mutations. Almost half of the InDels (95) occurred in polynucleotide regions, which corrected 27 pseudogenes predicted in the previous version (Supplementary Table S3). Thus, the resequencing results revealed that the genome of R. papyrosolvens DSM2782 in our laboratory has mutated compared with the original strain.
Furthermore, the complete genome of R. papyrosolvens DSM2782 was compared with that of other Ruminiclostridium-type species (R. papyrosolvens C7, R. sp. BNL1100, R. josui JCM17888, R. cellulolyticum H10, R. termitidis CT1112, R. cellobioparum DSM1351, R. hungatei DSM14427, R. sufflavum DSM19573, and R. herbifermentans MA18) and C. cellulovorans 743B using BLAST Ring Image Generator (BRIG) (Figure 1A). It indicated that most regions in the test strains show an identity higher than 70% of the alignment reference genome of R. papyrosolvens DSM2782. There is a position with higher GC content in the region ranging from 2,500 to 2,600 kbp, which harbors several genes encoding SDR family NAD(P)-dependent oxidoreductases. Meanwhile, several gaps highlighting the missing regions are visible at positions of 700–800, 1800–1900, and 2000–2,100 kbp, where genes encoding the ABC transporter system and different types of domain-containing proteins are enriched (Figure 1A). Finally, based on the BRIG analysis, the result represents a snapshot of the genetic diversity and close relatedness of the bacteria which belong to the Clostridia group.
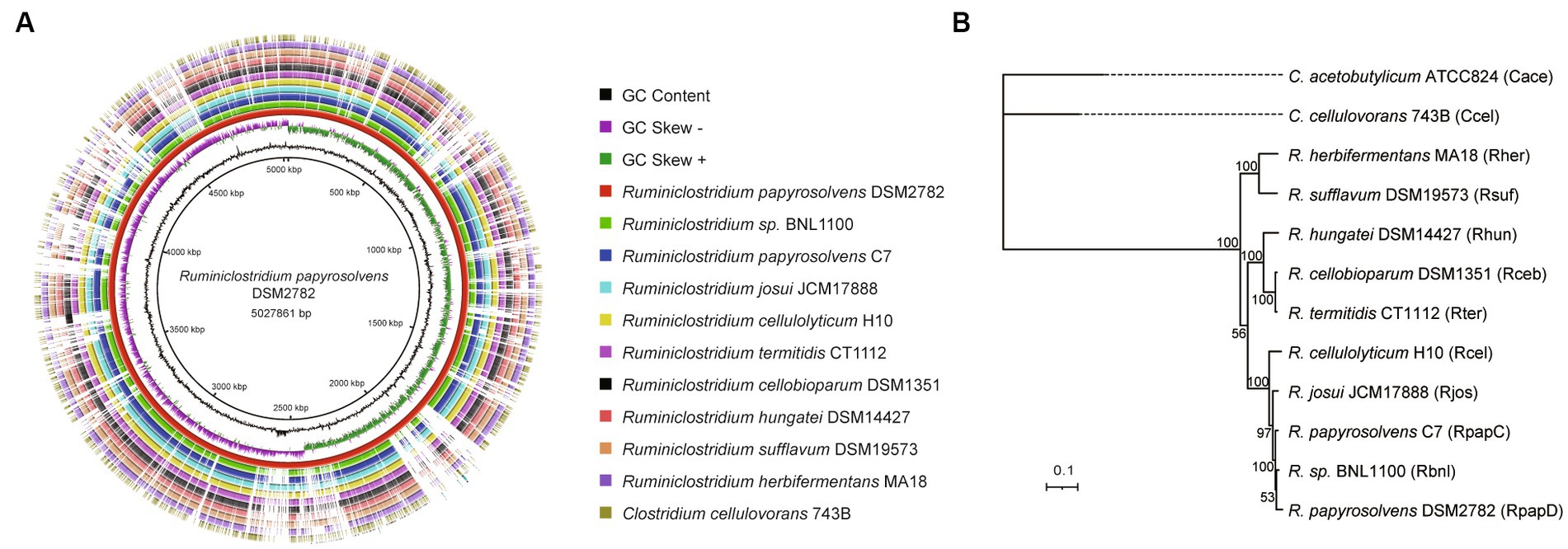
Figure 1. Whole-genome comparisons of Ruminiclostridium-type species. (A) Whole-genome comparisons, from outer to inner ring: C. cellulovorans 743B, R. herbifermentans MA18, R. sufflavum DSM19573, R. hungatei DSM14427, R. cellobioparum DSM1351, R. termitidis CT1112, R. cellulolyticum H10, R. josui JCM17888, R. papyrosolvens C7, R. sp. BNL1100; reference genome: R. papyrosolvens DSM2782. The color intensity in each ring represents the BLAST match identity. (B) Phylogenetic tree of mesophilic cellulolytic clostridia using the representative genomes in the Genome Taxonomy Database (GTDB) as references. Bootstrap values on nodes are indicated by >50. Bar 0.1 represents the nucleotide substitutions per position.
Moreover, a phylogenetic analysis of Ruminiclostridium-type species was performed by GTDB-Tk (Chaumeil et al., 2022) based on whole-genome sequences compared with C. cellulovorans 743B and C. acetobutylicum ATCC824, which are mesophilic cellulosome-producing clostridia but not belong to Ruminiclostridium-type species (Figure 1B). It showed that Ruminiclostridium species are phylogenetically distant from the Clostridium species of C. cellulovorans 743B and C. acetobutylicum ATCC824 and can be regarded as three clades supported by high bootstrap BP values. It was uncovered that R. papyrosolvens DSM2782 is closely related to Ruminiclostridium species BNL1100, R. papyrosolvens C7, R. josui JCM17888, and R. cellulolyticum H10 and belonged to the same clade, which had the farthest relationship with clostridia species.
To capture the entire genomic diversity of these mesophilic and cellulolytic Ruminiclostridium species, we performed a pan-genome analysis (Tettelin et al., 2008; Lee et al., 2021). It showed that the total genomes of 10 Ruminiclostridium-type species included 41,055 proteins with 8,414 orthologous gene families, of which 1,582 orthologous gene families were shared by all 10 analyzed genomes regarded as the core genome (Figure 2A). The necessary genome sharing by at least two species and the unique genome found in only one strain included 4,497 and 2,335 gene families, respectively. Among these mesophilic and cellulolytic Ruminiclostridium-type species, R. hungatei DSM14427 possessed more unique genes, with 349 genes (Figure 2A). The variable genes account for approximately 81.20% of the whole pan-genome, signifying the high level of genetic diversity in the members of mesophilic and cellulolytic Ruminiclostridium-type species.
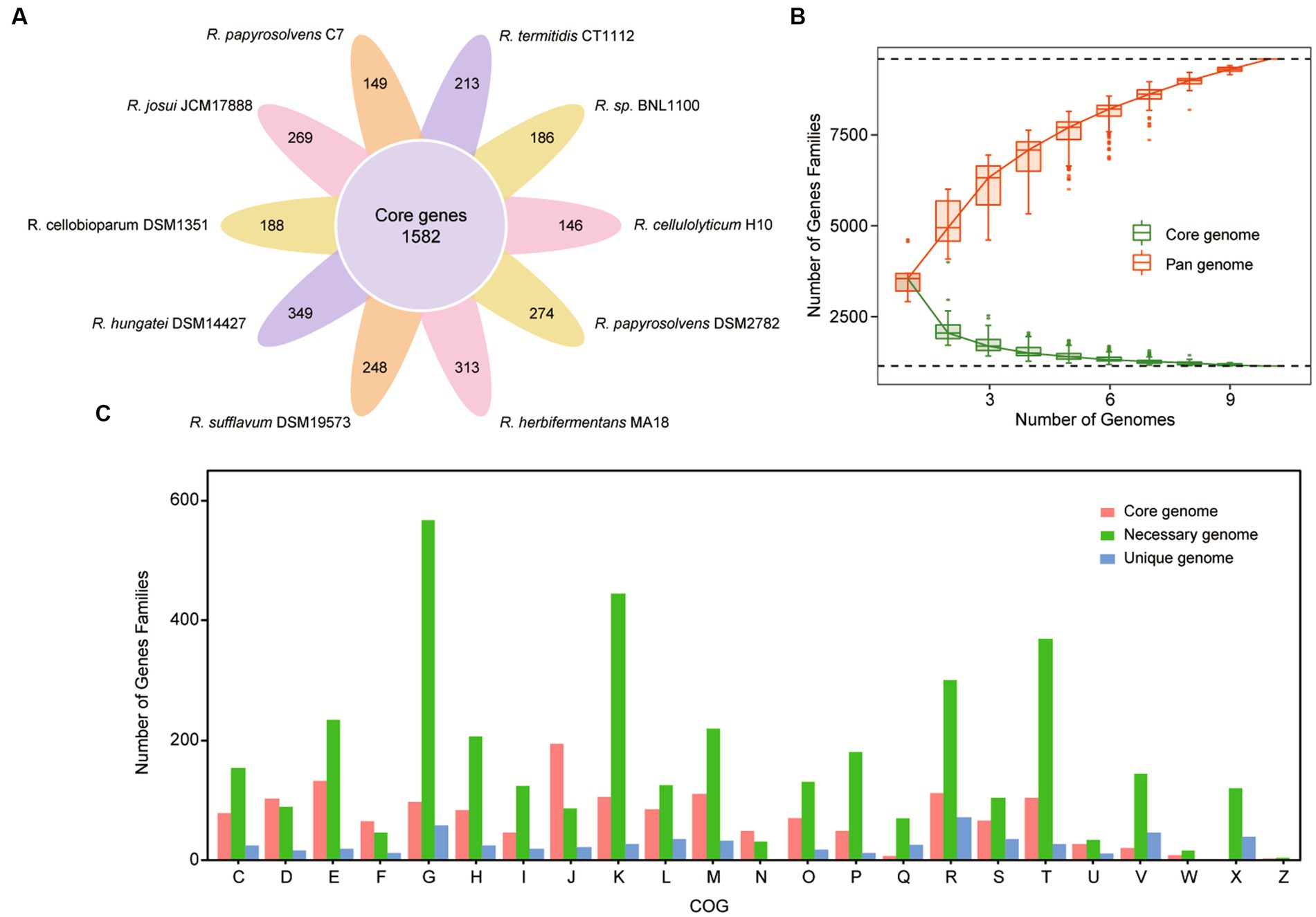
Figure 2. Pan-genome structure and gene functional distribution of Ruminiclostridium-type species. (A) Core and unique gene families in each Ruminiclostridium species. The number of core genomes shared by all species is in the center (1,582). (B) The size of the pan-genome (red) and core genome (green) shared by different Ruminiclostridium species. (C) Distribution of the Clusters of Orthologous Groups (COG) categories in Ruminiclostridium core, necessary, and unique genes (C, energy production and conversion; D, Cell cycle control, cell division, chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure and biogenesis; K, transcription; L, replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; V, defense mechanisms; W, extracellular structures; X, mobilome: prophages, transposons; Z, cytoskeleton).
Furthermore, the curves of the core genome and pan-genome size of these genomes with the increase in the number of genomes showed that the pan-genome size increased almost exponentially with the number of genomes, while the core genome size was being narrowed (Figure 2B). When the number of added genomes reached 10, the size of the pan-genome still increased. According to Figure 2B, the red line represented pan-genome, it’s increased. The measured size of the pan-genome was well fitted with a power law function (y = Axb, where A is 3648.8 and b is 0.3785), suggesting that the pan-genome might still be influenced by the inclusion of new genome sequences. The complete pan-genome of the Ruminiclostridium genus is likely to be substantially larger than that estimated by these 10 genomes. Thus, although the mesophilic and cellulolytic Ruminiclostridium species have a high level of intraspecific genetic diversity, their core genes have a stronger tendency to conserve basic functions.
Moreover, to clarify the functional characteristics of the Ruminiclostridium species genome, an analysis of the clusters of orthologous groups (COGs) was performed (Galperin et al., 2015). The functional categories of the genes were assigned to core, necessary, and unique classes, and the results showed that the gene families in the Ruminiclostridium core genome were enriched for genes involved in “translation, ribosomal structure, and biogenesis” (Figure 2C). The overall proportion of genes involved in “translation, ribosomal structure, and biogenesis” in the core genome was 11.9% (194/1,620), whereas that in the necessary and unique genomes was 2.28% (87/3,807) and 3.75% (22/586), respectively. Therefore, the COG analysis results highlighted that more core genes perform fundamental housekeeping functions than necessary and unique genes.
We further predicted and compared the transporters, including phosphoenolpyruvate-dependent phosphotransferase systems (PTSs), ATP-binding cassette (ABC) transporters, and TCSs, among these cellulolytic clostridia (Dassa and Bouige, 2001; Tian et al., 2017; Cheng et al., 2021), which the cellulolytic bacteria usually use two-component systems (TCSs) to sense extracellular sugars and regulate the expression of transporters and CAZymes (Joseph et al., 2002; Xu et al., 2013). It showed that the number of ABC transporters in each genome was similar to that of TCSs but was more than that of PTSs. Meanwhile, the number of ABC transporters and TCSs appeared to be positively related to the size of genomes. For example, the number of ABC transporters and TCSs in R. cellobioparum and R. termitidis was more than twice as much as that of other clostridia, and their genome size was more than 6 Mb, surpassing that of other clostridia (Figure 3A, Supplementary Table S4).
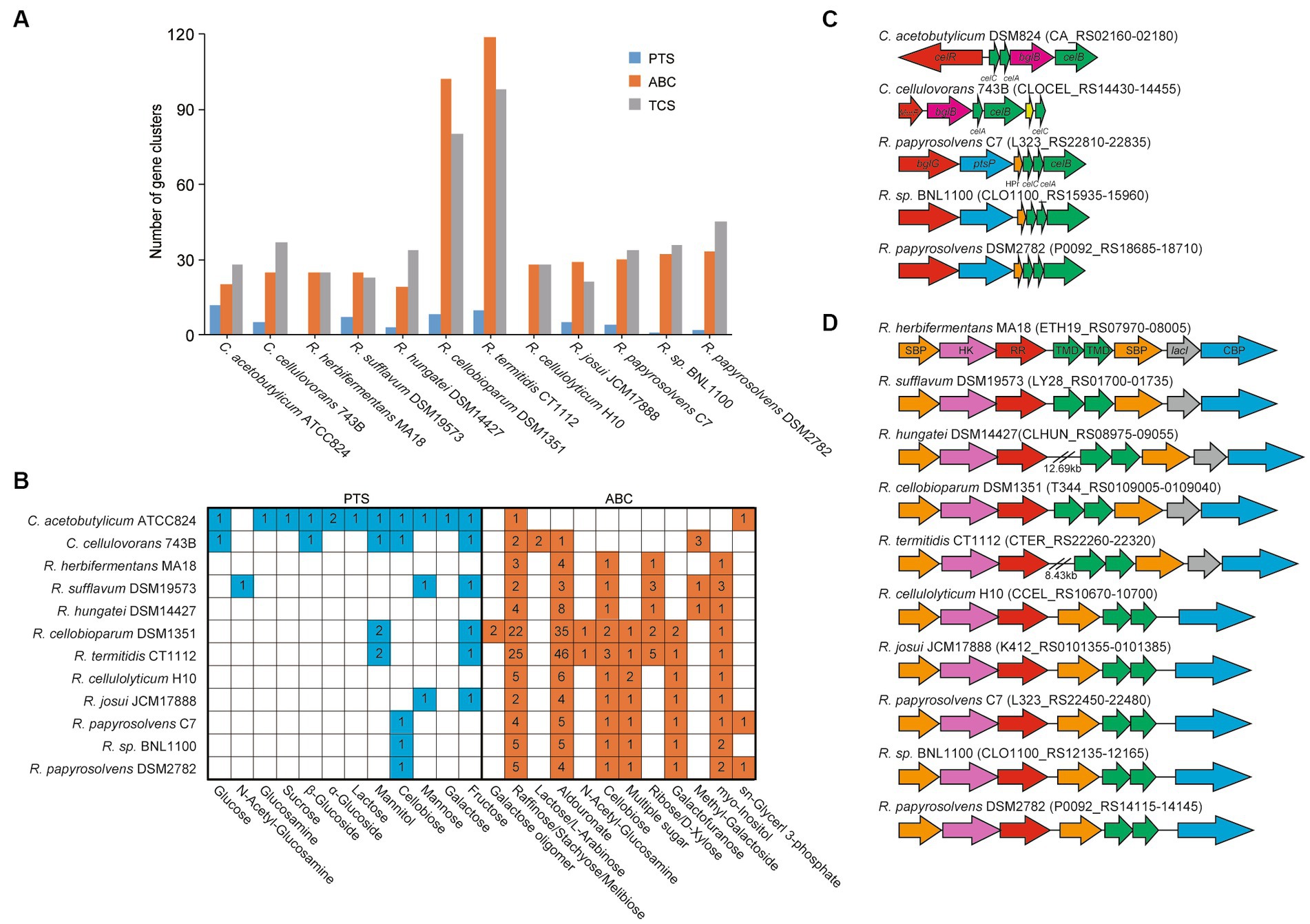
Figure 3. ABC transport systems and PTSs in Ruminiclostridium-type species compared with C. acetobutylicum and C. cellulovorans. (A) Comparsion of the number of gene clusters encoding ABC transporters, PTSs, and TCSs. (B) The number of ABC transporters and PTSs involved in the transportation of various sugars. (C) The gene clusters encoding PTSs and (D) ABC transport systems for cellobiose transportation. Soulte-binding protein (SBP), histidine kinase (HK), response regulator (RR), transmembrane domain (TMD), and cellobiose phosphorylase (CBP).
Furthermore, the transportation of various sugars in Ruminiclostridium-type species predicted by the KEGG database (Kanehisa et al., 2016a) was compared with that of C. acetobutylicum and C. cellulovorans. It was indicated that the number of ABC transporters for sugars in Ruminiclostridium-type species was much higher than in PTSs (Voigt et al., 2014), confirming that Ruminiclostridium species mainly employ ABC transporters to absorb extracellular sugars (Xu et al., 2013; Fosses et al., 2017). Specifically, R. cellulolyticum and R. herbifermentans harbor no PTSs for sugars. However, it is the exact opposite of C. acetobutylicum, in which there are 12 PTSs and only 2 ABC transporters for sugars. As for C. cellulovorans, it has 5 PTSs and 8 ABC transporters for the importation of sugars (Figure 3B, Supplementary Table S5), suggesting that PTSs are as important as ABC transporters for the importation of sugars in C. cellulovorans.
Moreover, the function of ABC and PTS transporters encoded in genomes was first annotated based on the KEGG database (map02010 for ABC transporters and map02060 for PTSs). The putative transporters involved in sugars in Ruminiclostridium-type species were compared with those of C. acetobutylicum and C. cellulovorans. C. acetobutylicum and C. cellulovorans harbor a cellobiose PTS (Figures 3B,C), while all Ruminiclostridium-type species harbor an orthologous cellobiose ABC transporter that is regulated by its upstream TCS (Fosses et al., 2017) (Figures 3B,D). However, in addition to the ABC transporter, R. papyrosolvens C7 and DSM2782, and Ruminiclostridium sp. BNL1100, the farthest relationship between Ruminiclostridium and C. acetobutylicum also evolves an orthologous cellobiose PTS that is not homologous with that of C. acetobutylicum and C. cellulovorans, which is potentially regulated by its upstream BglG-type transcriptional regulator (Figures 3B,C) (Tangney and Mitchell, 2007).
To understand the complex functions of carbohydrate degradation, carbohydrate-active enzymes (CAZymes) and cellulosomal subunits were predicted and compared among Ruminiclostridium-type species. R. papyrosolvens DSM2782 harbors 203 putative CAZymes, including one auxiliary activity (AA), 56 carbohydrate-binding modules (CBMs), 23 carbohydrate esterases (CEs), 111 glycoside hydrolases (GHs), and three polysaccharide lyases (PLs). It also has 73 putative cellulosomal subunits which contain cohesin (3) and dockerin domains (70). The number of CAZymes in R. papyrosolvens DSM2782 is similar to that of R. herbifermentans MA18, R. cellulolyticum H10, R. josui JCM17888, R. papyrosolvens C7, and Ruminiclostridium sp. BNL1100, which is higher than R. sufflavum DSM19573 and R. hungatei DSM14427 but much lower than R. cellobioparum DSM1351 and R. termitidis CT1112. However, R. cellobioparum DSM1351 and R. termitidis CT1112 harbor the largest number of CAZymes (298 and 322) but feature the least portfolio of cellulosomal subunits (12 and 11) (Figure 4A, Supplementary Table S6).
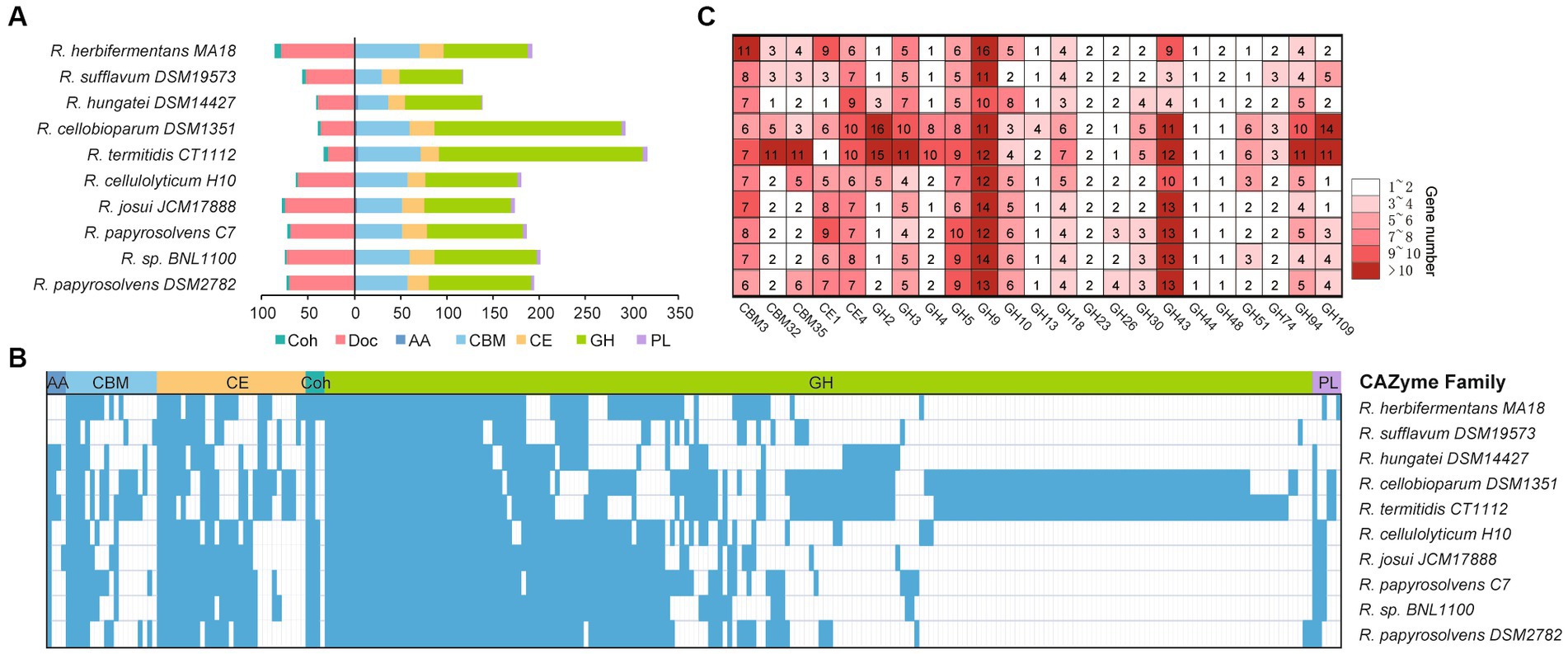
Figure 4. Statistical analysis of CAZymes modules in Ruminiclostridium-type species. (A) The number of CAZymes is denoted for each genome of a Ruminiclostridium-type species. (B) Presence (color)/absence (blank) pattern of CAZyme orthologous families in each Ruminiclostridium-type species. Glycoside hydrolases (GHs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), carbohydrate-binding modules (CBMs), cohesins (Coh), dokerins (Doc). (C) A detailed count of the major CAZyme families.
Phylogenetic analysis of CAZymes revealed that these 10 Ruminiclostridium species included 252 orthologous CAZyme families, with 42 orthologous families shared by all strains. In particular, most of the CAZymes (210) in R. papyrosolvens DSM2782, R. cellulolyticum H10, R. josui JCM17888, R. papyrosolvens C7, and Ruminiclostridium sp. BNL1100 were orthologous (38.6, 34.3, 35.2, 42.4, and 39.5% of all CAZymes, respectively), while both R. cellobioparum DSM1351 and R. termitidis CT1112 shared the most orthologous CAZyme families (Figure 4B). This was consistent with the evolutionary relationship of Ruminiclostridium species determined by genome alignment (Figure 1B). Furthermore, we counted the number of each CAZyme family to determine the major enzymes for lignocellulose degradation in Ruminiclostridium-type species. It was revealed that CAZymes belonging to 3 CBM families, 2 CE families, and 18 GH families are shared in all Ruminiclostridium-type species, such as CBM3, GH9, GH43, GH5, GH94 and CE4 families with the largest number (Figure 4C), suggesting that these CAZyme families are critical to cellulose degradation.
For profiling the cellulosomal system of each genome, we focused on its two gene clusters, cip-cel and xyl-doc (Maamar et al., 2006; Xu et al., 2013), and the organization of the cellulosomal protein modules. We first performed the homology analysis for the cip-cel (Figure 5A) and xyl-doc (Figure 5B) gene clusters by using Easyfig. This revealed that both gene clusters have high similarity among various Ruminiclostridium-type species. However, the similarities of cip-cel gene clusters between R. herbifermentans MA18 and R. sufflavum DSM19573 and between R. termitidis CT1112 and R. cellobioparum DSM1351 were higher than those of others, which were consistent with their phylogenetic tree of genomes (Figure 1B).
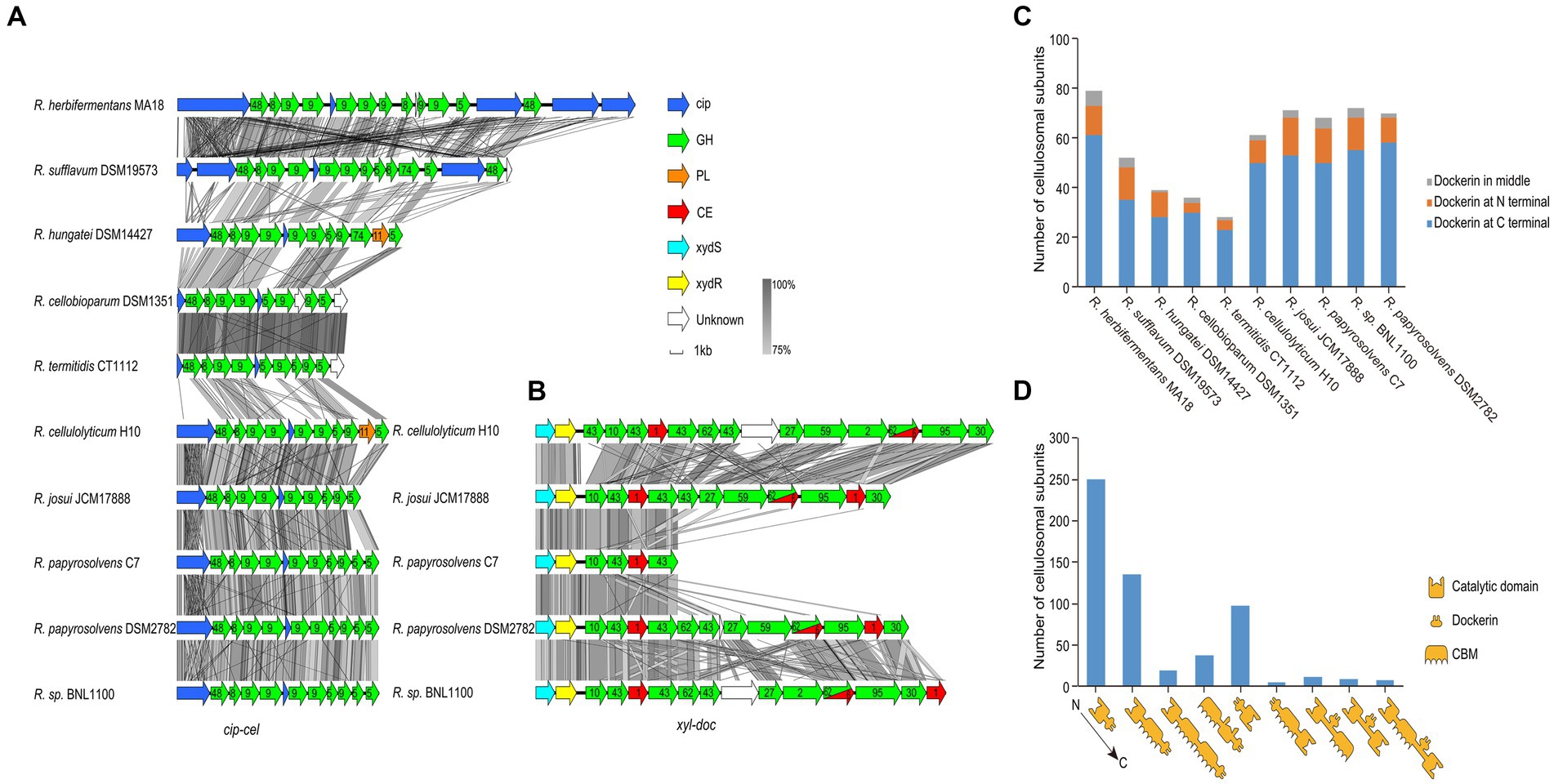
Figure 5. Gene clusters and architectures of catalytic subunits of cellulosomes. (A,B) Linear comparisons of the cip-cel and xyl-doc gene clusters from mesophilic Ruminiclostridium-type species. The major scaffoldin gene is represented by cip, and CAZymes are denoted by their family numbers. (C) Number of cellulosomal subunits with different locations of dockerin domains for each species. (D) Statistical analysis of catalytic subunits of cellulosomes with various architectures.
The cip-cel operon exists in all the examined species and is critical for cellulose degradation. The major scaffoldin gene, termed cip (Tomazetto et al., 2016), is the first gene, followed by 10 to 16 genes coding for cellulolytic enzymes from the GH 48, GH9, and GH5 families (Morag et al., 1991; Zverlov et al., 2005; Ravachol et al., 2014). In between the genes of the cluster lies a conserved gene, termed orfX, which codes for a cohesin-containing protein (Figure 5A) (Doi and Kosugi, 2004). The overall gene organization of the cluster is comparable to that of all species, suggesting that the cellulosomes of the mesophilic Ruminiclostridium species originated from a common ancestor. Nevertheless, we still observed two patterns of gene architecture among the different bacteria. R. herbifermentans MA18 and R. sufflavum DSM19573 harbor more scaffoldin genes in the cip-cel cluster than other Ruminiclostridium-type species. Both R. herbifermentans and R. sufflavum have another copy of the scaffoldin and GH48 genes downstream of the general cip-cel cluster (Figure 5A).
In addition to the cip-cel operon, another gene cluster encoding exclusively secreted dockerin-containing hemicellulases, termed xyl-doc, was found in four Ruminiclostridium species, which had the farthest relationship with species in clostridia, suggesting that xyl-doc evolved later than cip-cel. In the upstream of this gene cluster, there are two genes encoding the two-component system (TCS) (Figure 5B).
Furthermore, we analyzed the module organization of the cellulosomal catalytic subunits harboring dockerin domains (576) from all Ruminiclostridium-type species. These subunits include three modules: the catalytic domain (CD), CBM, and dockerin (Doc). The majority of dockerins (76.9%) were found at the C-terminal of catalytic subunits, followed by the N-terminal (18.1%) and the middle (5.0%) (Figure 5C). Specifically, the modular structures of catalytic subunits harboring C-terminal dockerins mainly included CD-Doc (250), CD-CBM-Doc (135), CD-CBM-CBM-Doc (19), and CBM-CD-Doc (38), while those of N-terminal dockerins are Doc-CD (98) and Doc-CBM-CD (5). Subunits harboring the middle dockerins contain three modular structures: CD-Doc-CBM (11), CD-Doc-CD (9), and CD-CBM-Doc-CD (8) (Figure 5D). Thus, the modular structures of catalytic subunits mainly have CD-Doc, CD-CBM-Doc, and Doc-CD, which account for 83.9% of the total.
Ruminiclostridium-type species usually harbor a large cellulosomal scaffoldin, but R. herbifermentans and R. sufflavum, respectively, have four and three scaffoldins. The cellulosomal scaffoldins of Ruminiclostridium-type species mainly include cohesin domains interacting with dockerin domains of enzymes, CBM3s binding to cellulose, and CBMX2 binding to cellulose and bacterial cell walls (Poole et al., 1992; Mosbah et al., 2000). The CBM domains are always located at the N-terminal of scaffoldins, except for those encoded by LY28_RS19570. They are followed by cohesin domains ranging in number from two 2 to 14, some of which are separated by CBM X2 (Figure 6A). To determine the difference between cohesins from various Ruminiclostridium-type species, the phylogenetic relationship of cohesins was analyzed. A total of 105 cohesins were classified into six groups. It was found that the cohesins between R. herbifermentans MA18 and R. sufflavum DSM19573 are closely related and are mainly clustered into Groups I, III, and V, while the cohesins from the other eight Ruminiclostridium-type species were mainly clustered into Group IV. However, the cohesins located at the C terminal of scaffoldins were separately clustered into Group II (Figure 6B). Thus, it was suggested that cohesins have interspecific similarity and intraspecific dissimilarity among Ruminiclostridium-type species, and the intraspecific dissimilarity is related to the location of scaffoldin.
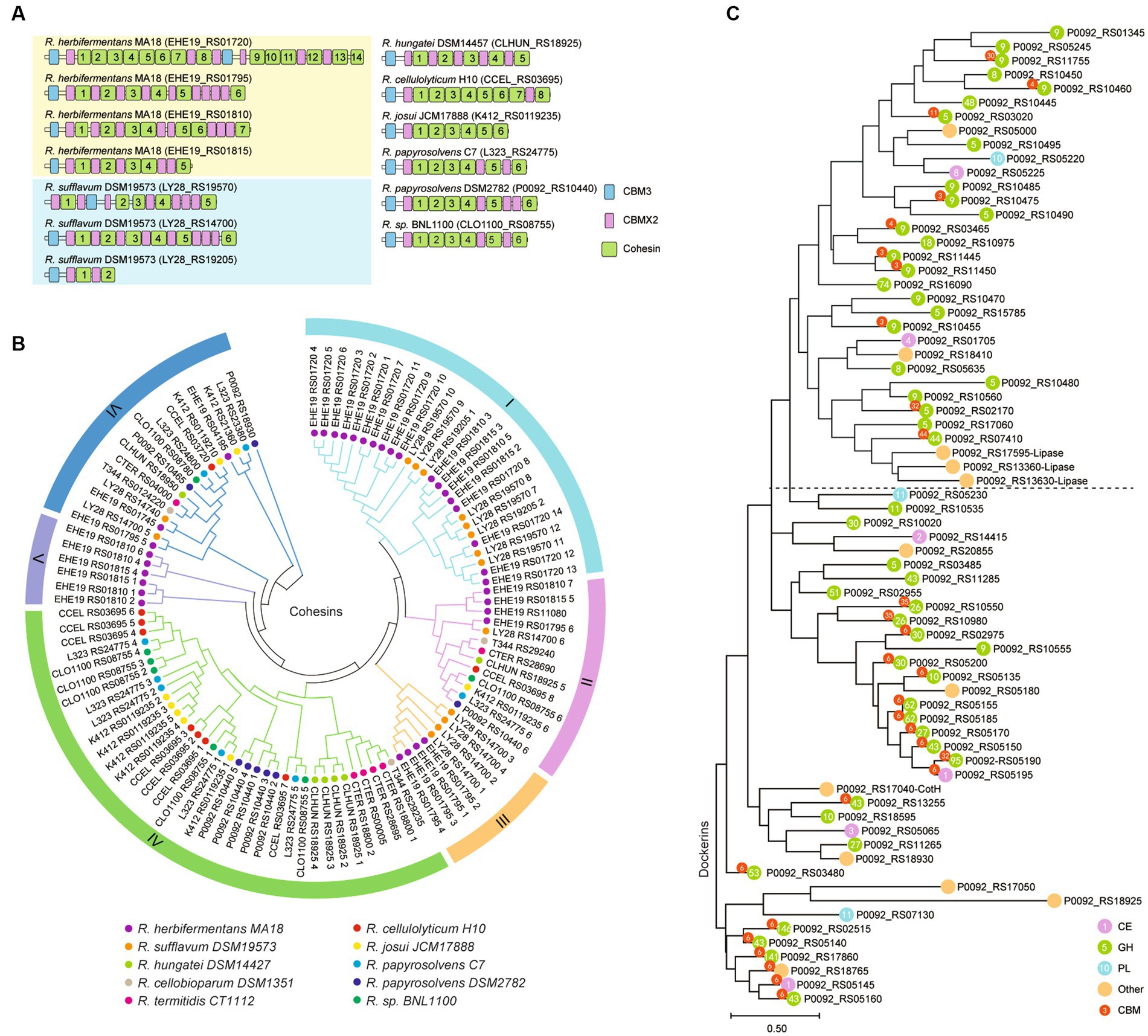
Figure 6. Phylogenetic relationship of dockerin and cohesin modules. (A) Modular and domain architectures of the primary scaffoldins of Ruminiclostridium-type species. (B) Phylogenetic analysis of cohesin domains from 10 Ruminiclostridium-type species using MEGAX. Six major branches are labeled with different colors. (C) Phylogenetic tree of all dockerins derived from catalytic subunits of the cellulosome in R. papyrosolvens DSM2782. The tree was built through 100 bootstraps using a maximum composite likelihood approach based on neighbor-joining algorithms. Bar 0.50 represents the nucleotide substitutions per position.
In addition, we used the dockerins of R. papyrosolvens DSM2782 as an example to analyze the link between dockerins and the catalytic domains they belong to. Phylogenetic analysis of dockerins of R. papyrosolvens DSM2782 showed that the dockerins fused with CBM3 and GH5, GH8, GH9, and GH48 families and others involved in cellulose degradation were clustered at the same clade in the evolutionary tree. The dockerins derived from cellulosomal subunits encoded by the cip-cel operon also belong to this clade. However, the other dockerins linked with CBM6 and GH10, GH26, GH30, and GH43 families, including enzymes encoded by the xyl-doc gene cluster and others involved in hemicellulose degradation, were mainly grouped into three clades (Figure 6C). It is suggested that the dockerin domain may co-evolve with its linking catalytic domain. Thus, the location of cellulosomal subunits in scaffoldin may not be random, but rather preferential due to the sequence dissimilarity of cohesins and dockerins, as indicated by the scaffoldin locations and the function of the fused catalytic domains.
The ability to grow on and ferment lignocellulose into valuable end products, such as ethanol, makes Ruminiclostridium-type species attractive and a potential candidate for biofuel production via consolidated bioprocessing. Understanding the composition and structure of CAZymes and cellulosomes associated with lignocellulose degradation is important for improving our understanding of cellulolytic physiology and identifying engineering targets for improving biomass biofuel production.
Ruminiclostridium papyrosolvens is one of the most highly evolved species among Ruminiclostridium-type species (Zou et al., 2018). We have developed a suitable genetic manipulation system in R. papyrosolvens (Ren et al., 2019; Wang et al., 2023) and analyzed its secretome in our previous studies, suggesting that it is a potential chassis cell for producing cellulosic biofuels, but further research is limited due to its incomplete genetic information. Thus, the genome of R. papyrosolvens DSM2782 was sequenced, completed, and compared with other mesophilic and cellulolytic clostridia in this study. It was revealed that R. papyrosolvens DSM2782 is closely related to Ruminiclostridium sp. BNL1100, R. papyrosolvens C7, R. josui JCM17888, and R. cellulolyticum H10. They are similar with respect to the composition of CAZymes, the transportation of sugars derived from lignocelluloses, and the gene clusters encoding the cellulosome.
Although C. cellulovorans is also an anaerobic, mesophilic, and cellulolytic Clostridium harboring the cip-cel cluster, it is phylogenetically distant from Ruminiclostridium-type species, suggesting that C. cellulovorans evolved in a different direction from Ruminiclostridium. For example, C. cellulovorans employs the PTS for the uptake of cellobiose and cellodextrin that derive from cellulose, while Ruminiclostridium-type species mainly transport them through ABC transporters, suggesting that PTSs are as important as ABC transporters for the importation of sugars in C. cellulovorans. Thus, Ruminiclostridium-type species, C. acetobutylicum, and C. cellulovorans evolve different strategies to import sugars (Servinsky et al., 2010; Wu et al., 2022). It seems that clostridia evolve more and more ABC transporters to import extracellular sugars as the ability of lignocellulose degradation increases. Additionally, the cip-cel cluster in C. cellulovorans does not contain a GH8 gene that is conserved in all Ruminiclostridium-type species and is shorter than that of Ruminiclostridium-type species (Tamaru et al., 2011).
Ruminiclostridium cellobioparum and R. termitidis, respectively, isolated from the rumen of cattle and the gut of termites (Lal et al., 2013; Mukherjee et al., 2017), have a close phylogenetic relationship. They have the largest genome size (6.13 and 6.42 Mb) among the Ruminiclostridium species considered in this study, harboring significantly higher CAZymes than other Ruminiclostridium strains. Especially, GH families are spread across more than 50 families in these two strains, including many characteristic/unique GH families, such as GH2, GH3, GH4, and GH51 hemicellulases, GH94 phosphorylases, and GH109 N-acetylhexosaminidase, highlighting the potential ability to produce a wide variety of enzymes needed to breakdown different types of complex biomass components. Considering the open state of the Ruminiclostridium pan-genome, the results of the COG enrichment analysis of Ruminiclostridium necessary genes and unique genes, especially “carbohydrate transport and metabolism” (G), “transcription” (K), “signal transduction mechanisms” (T), “general function prediction only” (R), and “cell wall/membrane/envelope biogenesis” (M), were consistent with the perspective that larger genomes tend to accumulate functions to enable organisms to achieve a higher degree of ecological diversification. However, R. cellobioparum and R. termitidis possess the least number of cellulosomal subunits among Ruminiclostridium species, which may limit their cellulose degradation capacity, suggesting that there may be abundant non-cellulosic polysaccharide in their living surroundings–animal guts.
Intriguingly, R. herbifermentans and R. sufflavum, phylogenetically distant from other Ruminiclostridium-type species, harbor more scaffoldin genes in the cip-cel cluster than other Ruminiclostridium-type species. Both R. herbifermentans and R. sufflavum have another copy of the scaffoldin and GH48 genes downstream of the cip-cel cluster. In addition, R. herbifermentans still has two scaffoldin genes following the copy of the GH 48 gene, while R. sufflavum has another shorter cip gene upstream of the cip-cel cluster. However, all these encoded scaffoldins in the cip-cel clusters are the primary scaffoldins incorporating enzymes, unlike the multiple scaffoldin gene clusters in H. thermocellum, which also encode the anchoring scaffoldins that contain type-II cohesins for attachment to the cell surface of the corresponding number of primary scaffoldins (Dassa et al., 2012). Thus, the multiple scaffoldin genes in the cip-cel cluster in R. herbifermentans and R. sufflavum may be the result of a gene duplication event.
The phylogenetic relationship of cohesins showed interspecific similarity among Ruminiclostridium-type species, indicating a general cross-species interaction between their scaffoldins and enzyme subunits in nature, and would imply their conservation in the same ecological niche. However, their putative conserved recognition residues are different from those of the complex cellulosomes from A. thermocellus, B. cellulosolvens, R. clariflavum, and R. flavefaciens (Dassa et al., 2017), suggesting that the cohesins of Ruminiclostridium-type species cannot exhibit cross-type recognition of the dockerins of complex cellulosomes, but the dockerin of A. thermocellus XynB is an exception, which is recognized by the cohesin from R. cellulolyticum (Haimovitz et al., 2008). On the other hand, the phylogenetic relationship between cohesins and dockerins showed that the cohesins located at the C-terminal of scaffoldins were significantly different from cohesins at other locations of scaffoldins, and dockerins linking with cellulases were clustered into a distinct clade. This may suggest the location preferences of catalytic subunits in scaffoldins for the best synergistic effect. It has been proven in the minimal cellulosome of Clostridium saccharoperbutylacetonicum, in which scaffoldin has only two cohesins (named Coh1 and Coh2). These two cohesins exhibited remarkably different binding patterns. Coh1 presented varied affinities toward the dockerin-containing enzymes, whereas Coh2 was much less selective and exhibited higher affinity toward all dockerins (Levi Hevroni et al., 2020).
Hydrolysis of cellulosic substrates is a major biotechnological challenge. Reconstitution of the biological principle of native cellulosomes may provide a basis for improved cellulolytic activity. In this study, we first sequenced and completed the genome of R. papyrosolvens DSM2782, which represented a single circular chromosome (5,027,861 bp, 37.1% G + C content), containing 4,407 coding DNA sequences and 90 RNA-coding genes. Comparative genome analysis showed that Ruminiclostridium-type species share a large number of core genes to conserve basic functions, such as the transcription of extracellular sugars and degradation of lignocelluloses, although they have a high level of intraspecific genetic diversity. However, some variations in the number and organization of genes encoding CAZymes and cellulosomes were found. Our analyses, described here, contribute to the understanding of the variety and similarity of genomes in Ruminiclostridium-type species involved in lignocellulose degradation. They should help in the design and construction of cellular systems for the robust and green conversion of lignocellulose into valuable products.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
MY: Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft. QZ: Formal analysis, Investigation, Methodology, Software, Visualization, Writing - original draft. YL: Formal analysis, Writing – review & editing. WZ: Writing – review & editing, Data curation, Software. ZS: Writing – review & editing, Formal analysis, Visualization. ZR: Writing – review & editing, Investigation, Methodology, Validation, Writing – original draft. CX: Visualization, Writing – review & editing, Methodology, Formal analysis, Conceptualization, Funding acquisition, Investigation, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants 32070045 and 32170053 from the National Natural Science Foundation of China and 2022LFR065 from the Science Development Foundation of Zhejiang A&F University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1288286/full#supplementary-material
1. ^https://www.ncbi.nlm.nih.gov/
2. ^https://ftp.ncbi.nih.gov/pub/COG/COG2020/data/
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). Blast ring image generator (Brig): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped Blast and psi-Blast: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bayer, E. A., Belaich, J. P., Shoham, Y., and Lamed, R. (2004). The Cellulosomes: multienzyme machines for degradation of plant Cell Wall polysaccharides. Annu. Rev. Microbiol. 58, 521–554. doi: 10.1146/annurev.micro.57.030502.091022
Bayer, E. A., Lamed, R., White, B. A., and Flint, H. J. (2008). From Cellulosomes to Cellulosomics. Chem. Rec. 8, 364–377. doi: 10.1002/tcr.20160
Buchfink, B., Reuter, K., and Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using Diamond. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2022). Gtdb-Tk V2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316. doi: 10.1093/bioinformatics/btac672
Cheng, C., Han, X., Xu, J., Sun, J., Li, K., Han, Y., et al. (2021). Yjbh mediates the oxidative stress response and infection by regulating Spxa1 and the phosphoenolpyruvate-carbohydrate phosphotransferase system (pts) in Listeria Monocytogenes. Gut Microbes 13, 1–19. doi: 10.1080/19490976.2021.1884517
Cui, G. Z., Hong, W., Zhang, J., Li, W. L., Feng, Y., Liu, Y. J., et al. (2012). Targeted gene engineering in Clostridium Cellulolyticum H10 without methylation. J. Microbiol. Methods 89, 201–208. doi: 10.1016/j.mimet.2012.02.015
Dassa, B., Borovok, I., Lamed, R., Henrissat, B., Coutinho, P., Hemme, C. L., et al. (2012). Genome-wide analysis of Acetivibrio Cellulolyticus provides a blueprint of an elaborate Cellulosome system. BMC Genomics 13:210. doi: 10.1186/1471-2164-13-210
Dassa, B., Borovok, I., Lombard, V., Henrissat, B., Lamed, R., Bayer, E. A., et al. (2017). Pan-Cellulosomics of mesophilic Clostridia: variations on a theme. Microorganisms 5:74. doi: 10.3390/microorganisms5040074
Dassa, E., and Bouige, P. (2001). The Abc of Abcs: a phylogenetic and functional classification of Abc systems in living organisms. Res. Microbiol. 152, 211–229. doi: 10.1016/S0923-2508(01)01194-9
Demain, A. L., Newcomb, M., and Wu, J. H. D. (2005). Cellulase, clostridia, and ethanol. Microbiol. Mol. Biol. Rev. 69, 124–154. doi: 10.1128/MMBR.69.1.124-154.2005
Desvaux, M. (2005). Clostridium Cellulolyticum: model organism of mesophilic cellulolytic Clostridia. FEMS Microbiol. Rev. 29, 741–764. doi: 10.1016/j.femsre.2004.11.003
Doi, R. H., and Kosugi, A. (2004). Cellulosomes: plant-Cell-Wall-degrading enzyme complexes. Nat. Rev. Microbiol. 2, 541–551. doi: 10.1038/nrmicro925
Emms, D. M., and Kelly, S. (2019). Orthofinder: phylogenetic Orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Fosses, A., Mate, M., Franche, N., Liu, N., Denis, Y., Borne, R., et al. (2017). A seven-gene cluster in Ruminiclostridium Cellulolyticum is essential for signalization, uptake and catabolism of the degradation products of cellulose hydrolysis. Biotechnol. Biofuels 10:250. doi: 10.1186/s13068-017-0933-7
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the cog database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Galperin, M. Y., Mekhedov, S. L., Puigbo, P., Smirnov, S., Wolf, Y. I., and Rigden, D. J. (2012). Genomic determinants of sporulation in Bacilli and Clostridia: towards the minimal set of sporulation-specific genes. Environ. Microbiol. 14, 2870–2890. doi: 10.1111/j.1462-2920.2012.02841.x
Garrity, G., Vos, P., Jones, D., Kreig, N., and Whitman, W. B. (2010). Bergey’s manual of systematic bacteriology. Springer, 3, 1–17.
Guillen, D., Sanchez, S., and Rodriguez-Sanoja, R. (2010). Carbohydrate-binding domains: multiplicity of biological roles. Appl. Microbiol. Biotechnol. 85, 1241–1249. doi: 10.1007/s00253-009-2331-y
Haimovitz, R., Barak, Y., Morag, E., Voronov-Goldman, M., Shoham, Y., Lamed, R., et al. (2008). Cohesin-Dockerin microarray: diverse specificities between two complementary families of interacting protein modules. Proteomics 8, 968–979. doi: 10.1002/pmic.200700486
Hemme, C. L., Mouttaki, H., Lee, Y. J., Zhang, G., Goodwin, L., Lucas, S., et al. (2010). Sequencing of multiple Clostridial genomes related to biomass conversion and biofuel production. J. Bacteriol. 192, 6494–6496. doi: 10.1128/JB.01064-10
Hou, N., Wang, Q., Sun, Y., Li, X., Song, Q., Jiang, X., et al. (2021). A novel biodemulsifier of Bacillus mojavensis XH1 - Oxalate decarboxylase with the potential for demulsification of oilfield emulsion. J Hazard Mater, 407:124737.
Hyeon, J. E., Jeon, S. D., and Han, S. O. (2013). Cellulosome-based, Clostridium-derived multi-functional enzyme complexes for advanced biotechnology tool development: advances and applications. Biotechnol. Adv. 31, 936–944. doi: 10.1016/j.biotechadv.2013.03.009
Johnson, E. A., Madia, A., and Demain, A. L. (1981). Chemically defined minimal medium for growth of the anaerobic cellulolytic thermophile Clostridium Thermocellum. Appl. Environ. Microbiol. 41, 1060–1062. doi: 10.1128/aem.41.4.1060-1062.1981
Joseph, P., Fichant, G., Quentin, Y., and Denizot, F. (2002). Regulatory relationship of two-component and Abc transport systems and clustering of their genes in the Bacillus/Clostridium group, suggest a functional link between them. J. Mol. Microbiol. Biotechnol. 4, 503–513.
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016a). Kegg as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kanehisa, M., Sato, Y., and Morishima, K. (2016b). Blastkoala and Ghostkoala: Kegg tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). Mega X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lal, S., Ramachandran, U., Zhang, X., Munir, R., Sparling, R., and Levin, D. B. (2013). Draft genome sequence of the cellulolytic, mesophilic, anaerobic bacterium Clostridium Termitidis strain Ct1112 (Dsm 5398). Genome Announc. 1:e00281-13. doi: 10.1128/genomeA.00281-13
Lee, H. H., Park, J., Jung, H., and Seo, Y. S. (2021). Pan-genome analysis reveals host-specific functional divergences in Burkholderia Gladioli. Microorganisms 9:1123. doi: 10.3390/microorganisms9061123
Letunic, I., and Bork, P. (2021). Interactive tree of life (Itol) V5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Levi Hevroni, B., Morais, S., Ben-David, Y., Morag, E., and Bayer, E. A. (2020). Minimalistic Cellulosome of the Butanologenic bacterium Clostridium Saccharoperbutylacetonicum. MBio 11:e00443-20. doi: 10.1128/mBio.00443-20
Ludwig, W., Schileifer, K., Whitman, W., Vos, P., Garrity, G., Jones, D., et al. (2019). Bergey’s Manual of Systematic Bacteriology. Springer, Volume 3:1–8.
Maamar, H., Abdou, L., Boileau, C., Valette, O., and Tardif, C. (2006). Transcriptional analysis of the Cip-Cel gene cluster from Clostridium Cellulolyticum. J. Bacteriol. 188, 2614–2624. doi: 10.1128/JB.188.7.2614-2624.2006
Mazzoli, R., and Olson, D. G. (2020). Clostridium Thermocellum: a microbial platform for high-value chemical production from lignocellulose. Adv. Appl. Microbiol. 113, 111–161. doi: 10.1016/bs.aambs.2020.07.004
Morag, E., Halevy, I., Bayer, E. A., and Lamed, R. (1991). Isolation and properties of a major Cellobiohydrolase from the Cellulosome of Clostridium Thermocellum. J. Bacteriol. 173, 4155–4162. doi: 10.1128/jb.173.13.4155-4162.1991
Mosbah, A., Belaich, A., Bornet, O., Belaich, J. P., Henrissat, B., and Darbon, H. (2000). Solution structure of the module X2 1 of unknown function of the Cellulosomal scaffolding protein Cipc of Clostridium Cellulolyticum. J. Mol. Biol. 304, 201–217. doi: 10.1006/jmbi.2000.4192
Mukherjee, S., Seshadri, R., Varghese, N. J., Eloe-Fadrosh, E. A., Meier-Kolthoff, J. P., Goker, M., et al. (2017). 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat. Biotechnol. 35, 676–683. doi: 10.1038/nbt.3886
Parks, D. H., Chuvochina, M., Rinke, C., Mussig, A. J., Chaumeil, P. A., and Hugenholtz, P. (2022). Gtdb: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794. doi: 10.1093/nar/gkab776
Poehlein, A., Zverlov, V. V., Daniel, R., Schwarz, W. H., and Liebl, W. (2013). Complete genome sequence of Clostridium Stercorarium Subsp. Stercorarium strain Dsm 8532, a thermophilic degrader of plant Cell Wall fibers. Genome Announc. 1:E0007313. doi: 10.1128/genomeA.00073-13
Poole, D. M., Morag, E., Lamed, R., Bayer, E. A., Hazlewood, G. P., and Gilbert, H. J. (1992). Identification of the cellulose-binding domain of the Cellulosome subunit S1 from Clostridium Thermocellum Ys. FEMS Microbiol. Lett. 78, 181–186. doi: 10.1016/0378-1097(92)90022-g
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Ransom-Jones, E., Jones, D. L., Mccarthy, A. J., and Mcdonald, J. E. (2012). The Fibrobacteres: an important phylum of cellulose-degrading Bacteria. Microb. Ecol. 63, 267–281. doi: 10.1007/s00248-011-9998-1
Ravachol, J., Borne, R., Tardif, C., De Philip, P., and Fierobe, H. P. (2014). Characterization of all Family-9 glycoside hydrolases synthesized by the Cellulosome-producing bacterium Clostridium Cellulolyticum. J. Biol. Chem. 289, 7335–7348. doi: 10.1074/jbc.M113.545046
Ren, Z., You, W., Wu, S., Poetsch, A., and Xu, C. (2019). Secretomic analyses of Ruminiclostridium Papyrosolvens reveal its enzymatic basis for lignocellulose degradation. Biotechnol. Biofuels 12:183. doi: 10.1186/s13068-019-1522-8
Servinsky, M. D., Kiel, J. T., Dupuy, N. F., and Sund, C. J. (2010). Transcriptional analysis of differential carbohydrate utilization by Clostridium Acetobutylicum. Microbiology 156, 3478–3491. doi: 10.1099/mic.0.037085-0
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Tamaru, Y., Miyake, H., Kuroda, K., Nakanishi, A., Kawade, Y., Yamamoto, K., et al. (2010). Genome sequence of the Cellulosome-producing mesophilic organism Clostridium Cellulovorans 743b. J. Bacteriol. 192, 901–902. doi: 10.1128/JB.01450-09
Tamaru, Y., Miyake, H., Kuroda, K., Nakanishi, A., Matsushima, C., Doi, R. H., et al. (2011). Comparison of the mesophilic Cellulosome-producing Clostridium Cellulovorans genome with other Cellulosome-related Clostridial genomes. Microb. Biotechnol. 4, 64–73. doi: 10.1111/j.1751-7915.2010.00210.x
Tangney, M., and Mitchell, W. J. (2007). Characterisation of a glucose phosphotransferase system in Clostridium Acetobutylicum Atcc 824. Appl. Microbiol. Biotechnol. 74, 398–405. doi: 10.1007/s00253-006-0679-9
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). Ncbi prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial Pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Tian, L., Song, T., He, R., Zeng, Y., Xie, W., Wu, Q., et al. (2017). Genome-wide analysis of Atp-binding cassette (Abc) transporters in the Sweetpotato whitefly, Bemisia Tabaci. BMC Genomics 18:330. doi: 10.1186/s12864-017-3706-6
Tomazetto, G., Hahnke, S., Koeck, D. E., Wibberg, D., Maus, I., Puhler, A., et al. (2016). Complete genome analysis of Clostridium Bornimense strain M2/40(T): a new Acidogenic Clostridium species isolated from a mesophilic two-phase laboratory-scale biogas reactor. J. Biotechnol. 232, 38–49. doi: 10.1016/j.jbiotec.2015.08.001
Voigt, C., Bahl, H., and Fischer, R. J. (2014). Identification of pts(Fru) as the major fructose uptake system of Clostridium Acetobutylicum. Appl. Microbiol. Biotechnol. 98, 7161–7172. doi: 10.1007/s00253-014-5809-1
Wang, D., You, M., Qiu, Z., Li, P., Qiao, M., and Xu, C. (2023). Development of an efficient Clostron system for gene disruption in Ruminiclostridium Papyrosolvens. Appl. Microbiol. Biotechnol. 107, 1801–1812. doi: 10.1007/s00253-023-12427-1
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:E1005595. doi: 10.1371/journal.pcbi.1005595
Wu, S., You, M., Wang, N., Ren, Z., and Xu, C. (2022). Internal transcription terminators control stoichiometry of Abc transporters in cellulolytic Clostridia. Microbiol. Spectr. 10:E0165621. doi: 10.1128/spectrum.01656-21
Xu, C., Huang, R., Teng, L., Wang, D., Hemme, C. L., Borovok, I., et al. (2013). Structure and regulation of the cellulose Degradome in Clostridium Cellulolyticum. Biotechnol. Biofuels 6:73. doi: 10.1186/1754-6834-6-73
Yutin, N., Puigbo, P., Koonin, E. V., and Wolf, Y. I. (2012). Phylogenomics of prokaryotic ribosomal proteins. PLoS One, 7, e36972.
Zhang, X., Tu, B., Dai, L. R., Lawson, P. A., Zheng, Z. Z., Liu, L. Y., et al. (2018). Petroclostridium Xylanilyticum gen. Nov., Sp. Nov., a Xylan-degrading bacterium isolated from an oilfield, and reclassification of Clostridial cluster iii members into four novel genera in a new Hungateiclostridiaceae fam. nov. Int. J. Syst. Evol. Microbiol. 68, 3197–3211. doi: 10.1099/ijsem.0.002966
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). Dbcan2: a Meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zou, X., Ren, Z., Wang, N., Cheng, Y., Jiang, Y., Wang, Y., et al. (2018). Function analysis of 5'-Utr of the Cellulosomal Xyl-doc cluster in Clostridium Papyrosolvens. Biotechnol. Biofuels 11:43. doi: 10.1186/s13068-018-1040-0
Keywords: Ruminiclostridium papyrosolvens, comparative genomics, pan-genome, CAZymes, cellulosome
Citation: You M, Zhao Q, Liu Y, Zhang W, Shen Z, Ren Z and Xu C (2023) Insights into lignocellulose degradation: comparative genomics of anaerobic and cellulolytic Ruminiclostridium-type species. Front. Microbiol. 14:1288286. doi: 10.3389/fmicb.2023.1288286
Edited by:
Mamoru Yamada, Yamaguchi University, JapanReviewed by:
Grzegorz Janusz, Maria Curie-Skłodowska University, PolandCopyright © 2023 You, Zhao, Liu, Zhang, Shen, Ren and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenxing Ren, enhyZW5Ac3h1LmVkdS5jbg==; Chenggang Xu, eHVjZ0B6YWZ1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.