 Arista Fourie
Arista Fourie Stephanus N. Venter
Stephanus N. Venter Bernard Slippers
Bernard Slippers Gerda Fourie
Gerda Fourie
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 30 November 2023
Sec. Microbial Symbioses
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1284397
Stink bug species (Pentatomoidea superfamily) have developed an interdependence with obligate bacterial gut symbionts in specialized midgut crypts (M4 sub-region). Species of the Enterobacteriaceae family (predominantly Pantoea) are vertically transferred to their offspring and provide nutrients that cannot be obtained from plant sap food sources. However, the bacteria in the other gut compartments of stink bugs have rarely been investigated. The two-spotted stink bug, Bathycoelia distincta, is a serious pest of macadamias in South Africa. Nothing is currently known regarding its gut microbiome or how symbionts are transferred between insect generations. In this study, the consistency of B. distincta gut bacteria across geographic locations and life stages was determined with 16S rRNA metabarcoding, considering both the M4 and other gut compartments. A novel Pantoea species was found to be the primary M4 gut symbiont and is vertically transferred to the offspring. The other gut compartments had a low bacterial diversity and genera varied between stink bug populations but a Sodalis species was prominent in all populations. Sequence data of the M4 compartment were used to produce high-quality metagenome-assembled genomes (MAGs) for the Pantoea and Sodalis species. Functional analyses suggested a similar role in nutrient provision for the host, yet also unique metabolites produced by each species. The Sodalis sp. also had additional traits, such as secretion systems, that likely allowed it to establish itself in the host. The Pantoea species was described as Pantoea bathycoeliae sp. nov based on the rules of the SeqCode.
Symbiotic associations between insects and bacteria have been established over long evolutionary time scales, and many of these associations are beneficial to the insect host (Cornwallis et al., 2023). Associations with gut symbionts have been investigated in numerous insects such as bees, termites, tsetse flies, beetles, aphids, and stink bugs (Engel and Moran, 2013; Gupta and Nair, 2020). These symbionts can contribute to nutrient provision and nutrient processing (Hansen and Moran, 2011; Moran, 2015), improving host immunity (Koch and Schmid-Hempel, 2011), overcoming plant host defense responses (Medina et al., 2018), protection against parasites (Oliver et al., 2009), or insecticide resistance (Kikuchi et al., 2012a).
One prominent long-standing insect-bacterial symbiosis is the gut symbionts of stink bugs (Duron and Noël, 2016; Hosokawa et al., 2016b). Stink bugs form part of the Pentatomoidea superfamily in the hemiptera order, consisting of more than 8,000 species (Rider et al., 2018). These insects have prominent associations with conserved extracellular symbionts that are maintained in midgut crypts, located in a subsection of their midgut (M4 region) (Salem et al., 2015; Hosokawa et al., 2016b). Usually, a single bacterial strain is maintained in high densities in this section. Physiological studies in a closely related family (Alydidae) suggest a constriction between the anterior part of the gut (M1–M3) and the M4 posterior section, allowing the obligate symbiont to enter the constricted section while food is bypassed from the M3 section to hemolymph and Malpighian tubules and then to the hind gut for excretion (Ohbayashi et al., 2015). In other species, once insects reach adulthood, the crypts become constricted and separated from the gut lumen and food can then again pass through this section (Oishi et al., 2019).
The obligate gut symbiont is generally transferred vertically from the mother to the offspring, through different methods in different species. Species in the Pentatomidae family perform an egg smearing on the surface of the eggs during oviposition (Calizotti and Panizzi, 2014), whereas the Urostylididae secrete a thick jelly that serves as a food source for the nymphs (Kaiwa et al., 2014). The Parastrachiidae secrete a thick mucus on the eggs just before hatching (Hosokawa et al., 2012), and the Plataspidae deposit small symbiont capsules along with the eggs (Hosokawa et al., 2005). The instars acquire the symbiont from the egg surface (or the symbiont capsules) after hatching and it is then maintained throughout their life cycle.
Many of the M4 gut symbionts are essential for the viability of their stink bug hosts. Experimental removal of the symbionts from Nezara viridula and Plautia splendens resulted in the mortality of the offspring (Tada et al., 2011; Hayashi et al., 2015). In other species, the effects were less severe but ranged from slower developmental rates to a reduction in body size or change in body color (Prado and Almeida, 2009; Kikuchi et al., 2012b; Hosokawa et al., 2013). In Halyomorpha halys, the detrimental effects, such as reduced fertility and viability, were only visible in the second generation (Taylor et al., 2014). Genome annotations and comparisons of numerous stink bug symbionts suggest that most of these species serve as nutritional symbionts, primarily contributing to the synthesis of essential amino acids and other vitamins and co-factors (Otero-Bravo et al., 2018; Kashkouli et al., 2021; Otero-Bravo and Sabree, 2021), but a role in uric acid recycling has also been suggested (Hosokawa et al., 2010). As stink bugs are xylem/phloem sap feeders, they obtain limited nutrients from their diet and additional amino acid and vitamin supplements are, therefore, essential (Douglas, 2006; Serteyn et al., 2020).
In the Pentatomoidea superfamily, each stink bug species shows a strong conservation of a specific bacterial species as an obligate symbiont. These bacterial species all form part of the Enterobacteriaceae family of Gammaproteobacteria, but different genera are prominent in the different Pentatomoidea families. For example, the largest stink bug family, Pentatomidae, predominantly contains Pantoea or Erwinia species (Duron and Noël, 2016), whereas all Acanthosomatidae contain one unique species, Candidatus Rosenkranzia clausaccus (Kikuchi et al., 2009), and the Parastrachiidae family maintains the species Candidatus Benitsuchiphilus tojoi (Mondal et al., 2020).
Limited studies have, however, focused on the bacterial species present in the other midgut compartments in stink bugs. A study of N. viridula identified two bacterial species in the M1–M3 section of the midgut (Klebsiella pneumoniae and Enterococcus faecalis), based on 16S PCR amplification and cloning (Hirose et al., 2006). Later studies identified both transient species, such as Bacillus spp., Micrococcus spp., Streptomyces spp., and Staphylococcus spp., and non-transient species including Yokenella spp. and Enterococcus spp (Medina et al., 2018). In Antestiopsis thunbergia, 16S rRNA PCR amplification and cloning from the midgut and ovaries identified Sodalis, Spiroplasma, and Rickettsia species throughout the gut and in other body tissues (Matsuura et al., 2014). In Acrosternum arabicum, Gammaproteobacteria and other taxa were detected in the M1 gut section (although less than in the M4 section) (Kashkouli et al., 2021). As most studies have focused on the M4 section, bacterial species in the rest of the digestive tract in stink bugs have likely been missed.
The two-spotted stink bug, Bathycoelia distincta (Hemiptera: Pentatomidae), is native to South Africa and is a serious pest in agricultural industries. It is one of the main contributors to nut losses in the macadamia industry in South Africa (est. 15.3 million USD annually) (Taylor et al., 2018), which is of great concern since South Africa is one of the largest exporters of this crop (SAMAC (South African Macadamia Association), 2021, Loss Factor Benchmark). The stink bug invasions in the orchards are predominantly controlled with insecticides, which is damaging to the environment and could lead to the development of insecticide resistance (Schoeman, 2019). The development of an integrated pest management approach, for example, targeting essential gut symbionts could improve control measures and reduce the use of insecticides (Gonella et al., 2020).
Nothing is currently known about the gut symbionts of B. distincta whether it also maintains an obligate symbiont in the M4 midgut section or how symbionts are transferred to the offspring. Additional facultative symbionts might also be hosted in other compartments of the stink bug gut. This study aimed to identify and compare the M4 midgut symbionts of B. distincta with those of other members of the Pentatomidae family, using a metabarcoding approach. In addition, the bacteria in the other gut compartments were also identified. To identify conserved symbionts, different geographic locations were included. To determine the potential transmission route of the symbionts and the microbes that are vertically transmitted, different developmental stages were considered. The functional roles of the primary symbionts in the host were predicted using metagenomic sequence data.
A lab-reared population of B. distincta is being maintained at the Forestry and Agricultural Biotechnology Institute Biological Control Centre of the University of Pretoria, South Africa. This population was originally collected from an orchard in the Limpopo province (Limpopo 2) and has been maintained for nearly 4 years, at the time of the experiment. Rearing conditions entailed a constant temperature of 25°C–27°C and a 16:8 h day/night cycle. Insects were maintained in plastic containers with sufficient aeration and fed on Zea mays (maize) kernels on the cob.
Adult insects were collected from three orchards in the Limpopo province, namely Limpopo 1, Limpopo 2, and Limpopo 3, and from two orchards in Mpumalanga, namely Mpumalanga 1 and Mpumalanga 2, to determine if bacterial gut symbionts are conserved across geographic locations (Table 1). Insects were collected from knockdown sprays performed during routine scouting surveys. Different blocks were selected each week and 10 random trees were targeted, including both edges and trees deeper in the blocks. Collected insects were frozen at −20°C and stored in these conditions until dissections were performed. Between 7 and 11 insects were obtained from each of the orchards and from the lab-reared population for comparison of gut symbiont communities. Egg packages and different developmental stages were also collected from the lab-reared population to determine which bacterial species are likely vertically transmitted.
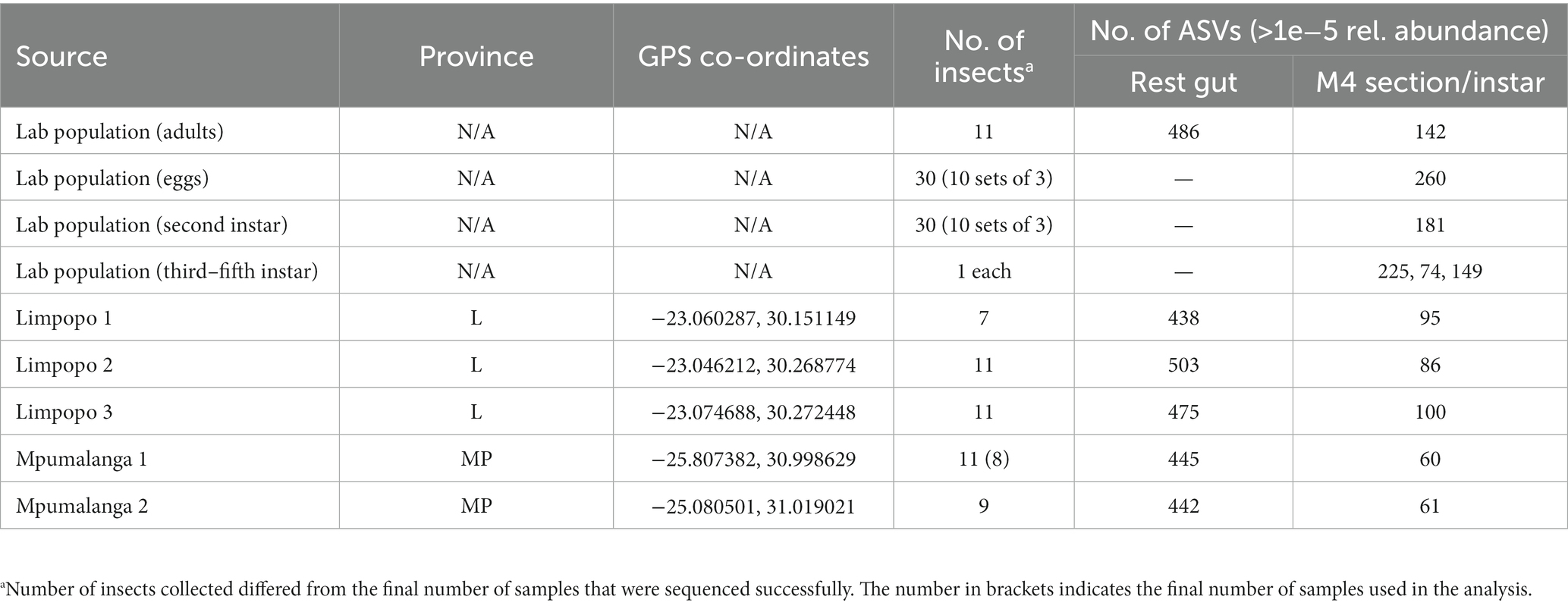
Table 1. Insect collections from the lab population, Limpopo (L) and Mpumalanga (MP) used for metabarcoding analyses and the number of ASVs observed in each.
Adult insects were dissected under a dissection microscope to obtain the gut. Insects were first surface sterilized for 2 min each in 100% ethanol, then 2 × 70% ethanol, and rinsed in sterile water. The dorsal part of the insect was removed after which it was pinned onto filtered wax (Paraplast, Sigma-Aldrich, South Africa) in a Petri dish and dissected under phosphate-buffered saline solution with fine forceps. The M4 section of the midgut was separated from the rest, and both parts were rinsed in 70% ethanol and Sabax water and then stored at −20°C for DNA extractions (Figure 1). Due to the small size, all the M4 sections per orchard were combined and a bulk extraction was performed per orchard, but the rest of the gut was extracted individually for each insect. Single representatives of the other life stages (third, fourth, and fifth instar) were also included to track the dominant bacteria throughout the developmental stages. A similar dissection procedure was followed for these insects, but the gut was extracted as a whole. For the eggs and second instars, several samples were pooled to obtain 10 extractions of each stage. These pools either consisted of three egg packages each or three second instars each. Eggs were not surface sterilized, and second instars were sterilized by removing the head and legs and rinsing them in a similar manner as the adult insects.
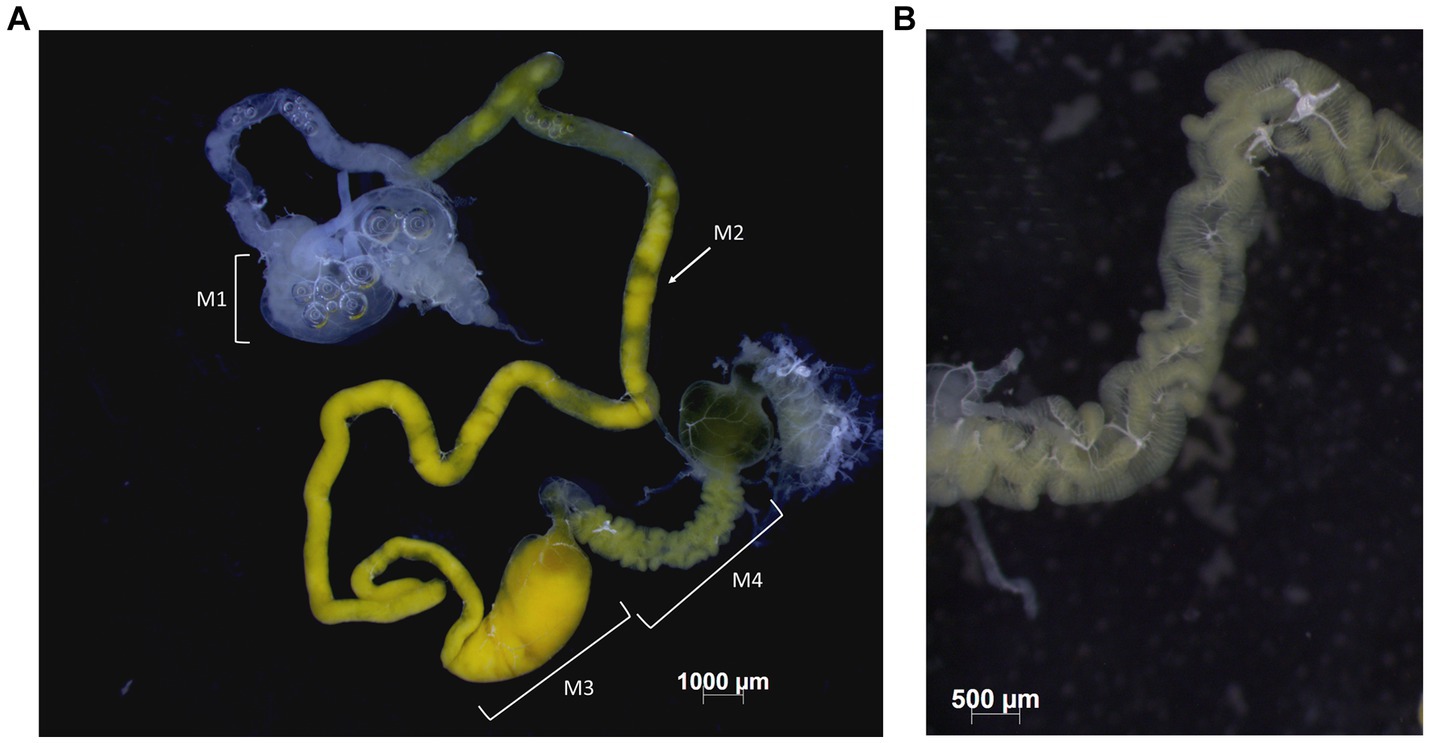
Figure 1. Midgut of B. distincta. (A) The entire midgut, indicating the different gut sections, from M1 to M4. (B) A close-up of the M4 midgut section where the midgut crypts, which contain a high abundance of the obligate symbiont, are clearly visible.
DNA extractions for Illumina MiSeq 16S rRNA barcoding were performed for all samples (Table 1), using the QIAamp PowerFecal DNA Kit, and following the manufacturer’s instructions (Qiagen, Johannesburg, South Africa). DNA purity and concentrations were determined using a Thermo Scientific Nanodrop ND_100 spectrophotometer (Wilmington, DE, United States).
A 450 bp fragment of the V3/V4 section of the 16S ribosomal RNA region was amplified in all DNA samples using the Bakt_341F and Bakt_805R primer set (Klindworth et al., 2013), with Illumina adapter sequences added to the ends (forward: 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG and reverse: 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG). PCR reactions were performed in triplicate per sample and then pooled before sequencing. PCR reactions were conducted in 25 μL reactions, consisting of ±50 ng DNA, 0.4 U Aq90 High fidelity DNA polymerase (Ampliqon, Denmark) and associated master mix, 0.5 μL of each primer (10 μM), 1–2 μL MgCl2 (25 mM stock), and Sabax water. The PCR conditions included 95°C for 3 min, 35 cycles of 95°C for 20 s, 51°C–54°C (ranged for optimization) for 30 s and 72°C for 30 s, and a final extension step at 72°C for 5 min. To determine PCR success, amplicons were analyzed on a 2% agarose gel via gel electrophoresis and visualized under UV light. Amplicons were purified with Agencourt AMPure XP beads (Beckman Coulter, CA, United States). Further library preparation included the addition of barcode sequences after which samples were combined for multiplex sequencing. Paired-end sequence data were generated using Illumina MiSeq technology. Library preparation and sequencing were performed at the Bioinformatics division, Agricultural Research Council, Pretoria, South Africa.
Illumina MiSeq metabarcoding sequence reads were processed by removing primer sequences using Cutadapt (Martin, 2011). Reads were further filtered, trimmed, and denoised using DADA2 (Callahan et al., 2016) in R v. 4.0.3 (R Core Team, 2020), with parameters maxN = 0, maxEE = c(3,5), and truncQ = 2. Reads were then merged and chimeras were removed with DADA2 to obtain amplicon sequence variants (ASVs). Taxonomy was assigned using the SILVA database v.138,1 and ASVs assigned as chloroplast sequences were removed from the dataset. ASVs represented by very low numbers of reads (average relative abundance <1e−5) were removed from the dataset with phyloseq (McMurdie and Holmes, 2013) using R v. 4.0.3 (R Core Team, 2020) in RStudio (RStudio Team, 2019).
The ASV data were separated into two datasets for downstream analysis. The first dataset contained the M4 samples from all populations and the early developmental stages (eggs and second instars) from the lab-reared population (M4_LifeStages dataset). The second dataset contained the rest of the gut, excluding the M4, from all populations (Rest_gut dataset).
The abundance of different bacterial genera was investigated in the M4_LifeStages and the Rest_gut dataset of each population, based on ASVs with a relative abundance above 1e−5. ASVs were clustered according to genus classification using the Microbiome package (Lahti and Shetty, 2017). Bacteria were considered as core genera if they were present in more than 75% of the individuals with a relative abundance >1e−5 in all samples. For summarizing and visualizing the most abundant genera, all genera with a relative abundance >1% were used to construct a bar graph with ggplot2, for both datasets (Wickham, 2016). A bubble plot was also constructed for the Rest_gut dataset, based on relative abundance >1e−5, to illustrate the genera present in each individual using the ggplot2 package (McMurdie and Holmes, 2013). All analyses were done with RStudio (RStudio Team, 2019).
In the M4 gut section, a single bacterial species was expected, but various ASVs were obtained with the metabarcoding analysis. BLASTn analysis of the ASVs on the NCBI GenBank database (Sayers et al., 2019) either had similar matches to Erwinia tasmaniensis and Pantoea septica or matched to Pantoea agglomerans. The 16S rRNA gene region cannot properly differentiate between all Pantoea and Erwinia species (Brady et al., 2008); thus, a multilocus sequence typing (MLST) was used to determine the proper classification of the symbiont as an initial step. A DNA sample from the M4 section of a lab-reared insect, for which its 16S rRNA gene sequence was identical to the most abundant ASV, was used. PCR amplification was performed for three additional bacterial housekeeping genes (atpD, rpoB, gyrB) that can differentiate between Pantoea and Erwinia (Brady et al., 2008). The PCR reaction consisted of 50 ng DNA, 5 μL 5 × MyTaq™ buffer, 0.2 μL of 1 U MyTaq™ DNA polymerase (Bioline Ltd., United Kingdom), 0.5 μL of each primer (10 mM), and sterile Sabax water up to a total volume of 25 μL. PCR cycler and Sanger sequencing conditions were identical to Brady et al. (2008).
The consensus sequences obtained for each of the three gene regions were aligned with all Pantoea species for which sequence data for these genes were available in GenBank, as well symbionts in other stinkbugs classified as Pantoea sp. (Duron and Noël, 2016). Kluyvera ascorbata (strain ATCC 33433) and Yokenella regensburgei (strain ATCC 49455) were included as outgroups. Alignments were performed in MEGA v. 5 (Tamura et al., 2011), using Muscle. Sequences were concatenated and a phylogenetic tree was constructed, based on a maximum likelihood analysis, using the GTRGAMMA setting and 1,000 bootstrap replications in RaxML v. 8.2.11 (Stamatakis, 2014).
To determine the phylogenetic placement and clustering of the 10 most abundant Pantoea ASVs (metabarcoding data) from the M4 gut sections and the different life stages, a reduced phylogenetic tree was constructed, including the 16S rRNA sequences of closely related Pantoea species and stink bug symbionts. The symbiont of a related Bathycoelia species, Bathycoelia indica, was specifically included to test for any relatedness between their symbionts. Pseudomonas syringae (strain ICMP 3023) was used as an outgroup for the tree. Phylogenetic relationships were determined, as described above. This is the only step where the third–fifth instar ASVs were included in analyses since these samples were represented by single individuals.
The other prominent species, observed in the Rest_gut dataset of all geographic locations and in the different developmental stages, was a Sodalis species. To use the full-length 16S rRNA of this species for more accurate phylogenetic placement, the sequence was obtained from the metagenome-assembled genome (MAG, see section 2.7) and the five most abundant Sodalis ASVs were aligned to this sequence to determine similarity. Each ASV differed in only one nucleotide (thus 99.77% similarity) from the metagenomic 16S rRNA sequence, and hence, this sequence was used to represent the phylogenetic placement of the Sodalis species. Additional Sodalis isolates collected from other insects, including various stink bug species, were included (available on NCBI, GenBank) (Sayers et al., 2019) and alignments and phylogenetic analyses were performed similar to the methods described above.
The richness and evenness (alpha diversity) of the bacterial communities present in the rest of the gut of each individual were determined using Shannon’s diversity index (richness and evenness with more weight on richness), Simpson’s index (richness and evenness with more weight on evenness) and Chao1 diversity index (richness). The absolute read counts for each ASV were used, excluding ASVs with less than <1e−5 relative abundance. Significant differences in the alpha diversity of the different populations were tested with Kruskal–Wallis, followed by a Wilcoxon rank sum test to identify the populations that differed significantly. The Benjamini and Hochberg method was used to adjust p-values for multiple comparisons.
To determine if any of the populations had distinct microbial communities, a β-diversity analysis was performed with the Rest_gut data, using the relative abundance of the ASVs (abundance >1e−5). Distances were calculated based on Jaccard distance (which does not consider the abundance of different ASVs) and Bray–Curtis dissimilarity (which considers composition and abundance of ASVs). The homogeneity of data dispersion in each group was confirmed, using betadisper, followed by a permutational multivariate analysis of variance (permanova) using adonis in the vegan package (Oksanen et al., 2020). To determine the populations that were significantly different from one another, a pairwise comparison was performed using the pairwiseAdonis package and p-values were adjusted with the Benjamini and Hochberg method (Martinez, 2020). In addition, the distances were used to construct an NMDS ordination plot to visualize the clustering of individuals from all populations. All analyses were performed with R v. 4.0.3 (R Core Team, 2020) in RStudio (RStudio Team, 2019).
Given the potentially unique species identified through the MLST approach described above, DNA extractions for metagenome sequencing were performed for the M4 section. The M4 gut section of three female insects from the lab-reared population were combined. DNA was extracted using the Macherey-Nagel NucleoSpin Tissue kit (Düren, Germany) and an RNase A treatment was included in the protocol. DNA purity was determined with a Thermo Scientific Nanodrop ND_100 spectrophotometer (Wilmington, DE, United States), and concentrations were determined with a Qubit® 2.0 Fluorometer (ThermoFisher Scientific). The DNA that passed the purity filters of 260/280 ratio >1.7 and minimum concentration >20 ng/μL was sent for Illumina TruSeq PCR-free sequencing at Macrogen, Seoul, Republic of Korea. The DNA libraries were constructed using the TruSeq Nano DNA kit (Illumina, CA, United States). DNA was randomly fragmented, 5′ and 3′ adapters were ligated, and a 350 bp insert size was used for paired-end sequencing.
The sequence reads obtained from Illumina TruSeq were quality checked using fastqc (Andrews, 2010) and trimmed using Trimmomatic v. 0.38 (Bolger et al., 2014), based on a phred quality score above 30, minimum length 100 bp, using a sliding window of 4:30 and an average read quality of 20. The trimmed reads were analyzed with fastqc again to ensure all adapters were removed. To remove host-contaminating DNA, the reads were mapped to an in-house available draft genome sequence of the B. distincta host, using bowtie2 (Langmead and Salzberg, 2012). All unmapped reads were collected for metagenome assembly.
Metagenomic reads were assembled using the MetaSpades mode in Spades v. 3.15.3 (Nurk et al., 2017). Quality filtered reads were mapped back to the assembly, using bowtie2 (Langmead and Salzberg, 2012), to determine the assembly success rate and for contig binning. Contigs were grouped into bins using MaxBin 2.0 (Wu et al., 2015) and MetaBAT 2 (Kang et al., 2019), respectively, after which bins were dereplicated using MiGA (Rodriguez-R et al., 2018). Phylogenetic identity, genome completeness, and contamination of the bins were determined with the MiGA webserver2. Two bins with high genome completeness were obtained and were identified as Pantoea and Sodalis, respectively. To identify any contaminant contigs in the bins (MAGs), taxonomic classification of contigs was performed using the Contig Annotation Tool (CAT) (von Meijenfeldt et al., 2019). Genome completeness of the MAGs was confirmed with BUSCO (Manni et al., 2021), using the core Pseudomonodota (previous Proteobacteria) protein dataset as a reference. The closest related genomes in the Genome Taxonomy Database (GTDB) were identified for both genomes, using GTDB-Tk-v1.7.0 (Chaumeil et al., 2019) on the Kbase server (Arkin et al., 2018).
Gene prediction and functional annotation were performed for the Pantoea and Sodalis MAGs, using Prokka (Seemann, 2014), including the –compliant and –rfam options and providing the appropriate genus as a reference for annotation. Pseudogenes were predicted with Pseudofinder (Syberg-Olsen et al., 2022), which compares proteins both to a non-redundant protein database and to its closest reference genome to identify genes distinctly smaller or larger than its average ortholog, containing frameshifts, early stop codons, or no stop codons as parameters for pseudogene prediction. The genomes Pantoea septica (GCF_902386985.1) and Sodalis glossinidius (GCF_000010085.1) were used as reference genomes for each species, respectively, and additional parameters that were modified included --length_pseudo 0.5, --shared_hits 0.7, --intergenic_length 100, --intergenic_threshold 0.9, and --hitcap 30. Specific functional groups, such as carbohydrate-active enzymes (CAZymes) and peptidases, were further summarized using dbCAN2 (Zhang et al., 2018) (only considering features detected by both HMMER and Diamond) and BLASTp to the MEROPS database (E-value <1e−6, >40% ID, >40% query coverage) (Rawlings et al., 2013), respectively. KEGG functional annotations were added to the proteins, using KofamKOALA (Aramaki et al., 2019), and metabolic pathways were identified with KEGG Mapper (Kanehisa and Sato, 2020). Secondary metabolite gene clusters were predicted with antiSMASH (Blin et al., 2021), and secretion systems were predicted with MacSyFinder (Abby et al., 2014).
The MAG of the primary symbiont, Pantoea sp., was further investigated to determine its relatedness to other Pantoea species, both free-living and other stink bug symbionts. Publicly available genomes for all well-defined free-living Pantoea species, as well as symbionts from other Pentatomidae stink bugs, were obtained from NCBI (Supplementary Table S1). The average nucleotide identity (ANI) between the Pantoea symbiont and other Pantoea genomes was determined using MASH (Ondov et al., 2016).
The similarity in gene content between the genomes was determined by identifying shared and unique orthologs, using OrthoFinder v2.2.6 (Emms and Kelly, 2019) with standard parameters. This could identify any genes unique to the Pantoea symbiont. The OrthoFinder output was also used to identify the core set of single-copy genes that were used for phylogenomic tree reconstruction. The Pseudomonas syringae strain BIM B-268 (GCF_016694755.2) was used as an outgroup to root the phylogenetic tree and was thus included in the OrthoFinder analysis of single-copy core genes. A total of 328 single-copy genes were identified, and each was individually aligned with MAFFT v7.407 (Katoh and Standley, 2013); poorly aligned regions were removed with trimAl (Capella-Gutiérrez et al., 2009), after which all genes were concatenated with FASconCAT-G (Kück and Longo, 2014). A phylogenomic tree was constructed with RaxML v8.2.12 (Stamatakis, 2014) with the concatenated gene alignment, as well as partition data for each gene as generated by FASconCAT-G, using the GAMMA model of rate heterogeneity. One thousand bootstrap replicates were performed to determine branch support values.
The Illumina MiSeq data obtained for the metabarcoding ranged from 56,000 to 190,000 raw paired reads per sample, with an average of 121,000 reads. After quality filtering and chimera removal, the reads ranged from 166 to 140,000, with an average of 45,500 read pairs per sample. Three of the samples had very few reads retained after filtering and chimera removal (all three from Mpumalanga 1), and these were excluded from further analyses.
A total of 1,447 ASVs (excluding chloroplast sequences) were obtained in the full dataset from the DADA2 analyses. After the removal of ASVs with a relative abundance <1e−5, 331 of these ASVs were retained in the M4_LifeStages dataset and 603 in the Rest_gut dataset. Of these filtered ASVs, individuals each contained between 61 and 239 ASVs (avg. 119) in the M4_LifeStages dataset (Supplementary Table S2) and between 20–364 ASVs (avg. 168) in the Rest_gut dataset (Supplementary Table S3), respectively.
The bacteria present at the different developmental stages of the lab-reared population and those in the M4 gut section of all populations were compared to identify genera maintained from birth and conserved across locations. Sodalis, Serratia, Siccibacter, and Pantoea were present in the different life stages, of which only Sodalis and Pantoea were consistent in all stages (Figure 2). For example, the most abundant Serratia ASV (ASV7) was only present in the eggs, 2nd and 5th instar with a high abundance in the 2nd instar. The M4 section predominantly contained one Pantoea ASV (ASV1), but an additional Pantoea strain/species was present in the Limpopo 3 population (Figure 2). Additional ASVs, classified as Pantoea, were 99.7%–99.9% similar to ASV1 (ASV4, ASV9, ASV10, ASV13, ASV18). The alternative Pantoea strain was represented by four ASVs (ASV16, ASV26, ASV28, ASV45) that were predominantly present in the Limpopo 3 population. These ASVs were also 99.8% similar to one another but 98.5% similar to ASV1, suggesting a distinct strain.

Figure 2. Relative abundance of the most prominent bacterial genera (>1%) present in the M4 midgut section of stink bugs from different locations as well as in the early developmental stages of the lab-reared population. The samples were bulked and represent a number of individuals (8–11).
Both Sodalis and Pantoea were present on the eggs and throughout the life stages, suggesting a vertical transfer of these bacteria from parents to offspring. Newly hatched instars also displayed a probing behavior on the egg surfaces after hatching (Supplementary material S1), where they likely obtained the bacteria. However, Sodalis could also have been transferred transovarially as it is known to be an endocellular symbiont.
In the Rest_gut dataset, the Enterobacterales order (Pseudomonodota phylum—previously known as the Proteobacteria) was most abundant, including the genera Sodalis, Pantoea, Yokenella, and Serratia (Figure 3). Less abundant ASVs (yet still >1%) were assigned to the Bacillota phylum (previously Firmicutes), including the genera Aerococcus, Bacillus, Granulicatella, Leuconostoc, Lactococcus, Oceanobacillus, Rummeliibacillus, Staphylococcus, Streptococcus, and Veilonella and other gammaproteobacteria of the genera Acinetobacer, Buttiauxella, Enterobacter, Haemophilus, and Pseudomonas. All genera observed in the populations (RA >1e−5) are represented in Supplementary Figure S1.
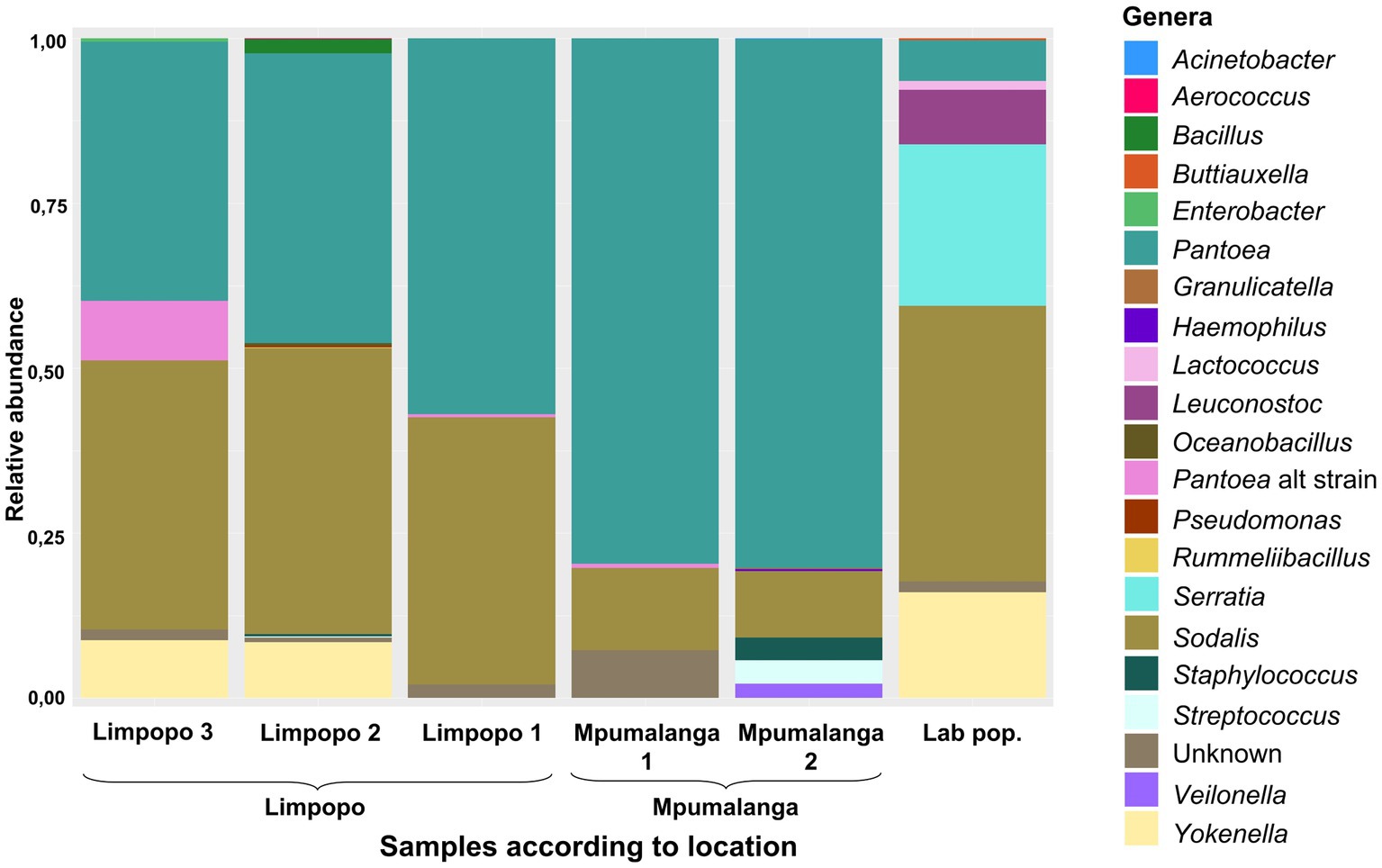
Figure 3. Relative abundance of the most prominent bacterial genera (>1%) present in the rest of the gut (excluding the M4 region), according to geographic location. Each location is represented by 7–11 samples.
The core genera (present in >75% of the samples and RA >1e−5) were Pantoea, Serratia, and Sodalis, while others were in lower abundance (or only abundant in some populations) such as Atlantibacter, Buttiauxella, Leuconostoc, Siccibacter, Staphylococcus, and Streptococcus. Sodalis was primarily represented by five abundant ASVs, all with 99.7% similarity. The species was present in all populations (although at variable RA in individuals) and was in all individuals of the lab and Limpopo populations but only in 50% of the individuals of the Mpumalanga populations (Supplementary Figure S1). Pantoea, however, was present in all individuals, which confirms this as the most conserved symbiont. This species was represented by 10 abundant ASVs. ASV4, ASV9, and ASV15 were all 99.8% similar to ASV1. Although in low abundance, ASV4 and ASV15 were present in most individuals, while ASV9 was abundant in some but absent from many individuals. The additional strain/species of Pantoea (such as ASV16) was observed in the Limpopo 3 population although this was predominantly in one individual with very low abundances in other individuals (Figure 3; Supplementary Figure S1). Serratia occurred in low abundance in most individuals but was more abundant in some individuals in the lab population. Sequence similarity between ASVs of Serratia were all above 99.2%.
In the phylogenetic tree of the combined gene regions (atpD, rpoB, and gyrB), the conserved B. distincta symbiont grouped closest to the free-living Pantoea species, P. septica, and was also closely related to “P. latae” and the gut symbionts of an Acrosternum stink bug species and the Plautia stali symbiont B (Figure 4). This phylogenetic tree supports that the B. distincta Pantoea symbiont is a novel species and is from here on referred to as Pantoea bathycoeliae, for ease of reference. A formal description of the species follows at the end of this section based on the MAG obtained from the metagenomic data of the M4 gut section.
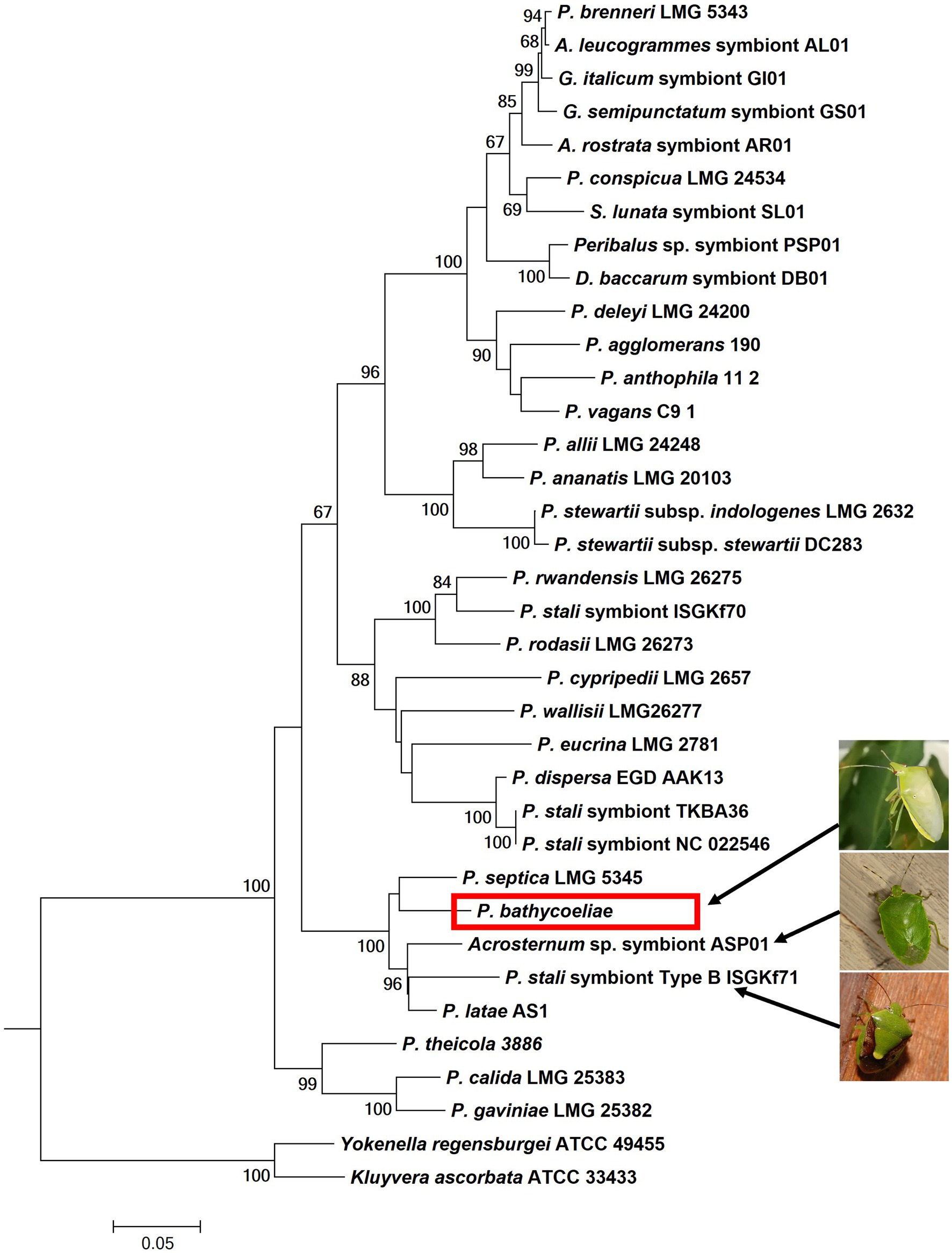
Figure 4. Phylogenetic placement of the essential gut symbiont of B. distincta (red block), based on the combined sequence of the atpD, rpoB, and gyrB genes. This tree represents the most likely tree, based on maximum likelihood analyses, and branch support based on 1,000 bootstrap replications is indicated on the branches. This illustrates the most closely related free-living species is Pantoea septica and the closest stink bug symbionts are those from Acrosternum sp. and Plautia stali (indicated by the arrows and pictures).
The most abundant Pantoea ASVs, identified in the 16S rRNA metabarcodes, could be classified in a 16S rRNA phylogenetic tree (Figure 5). Six of the ASVs grouped close to Pantoea septica and the symbionts from four stink bug species. These six ASVs were 99.6%–99.9% similar to one another and are likely variations introduced due to minor sequence errors, or could be different strains of the same species. The additional Pantoea strain/variant that was present in the M4 section and rest of the gut in the Limpopo 3 samples (represented by four ASVs) was clearly a distinct species from P. bathycoeliae and grouped with Pantoea vagans and Pantoea brenneri. The Pantoea symbiont from the closely related stink bug species, B. indica, did not show any close relation to the B. distincta symbiont.
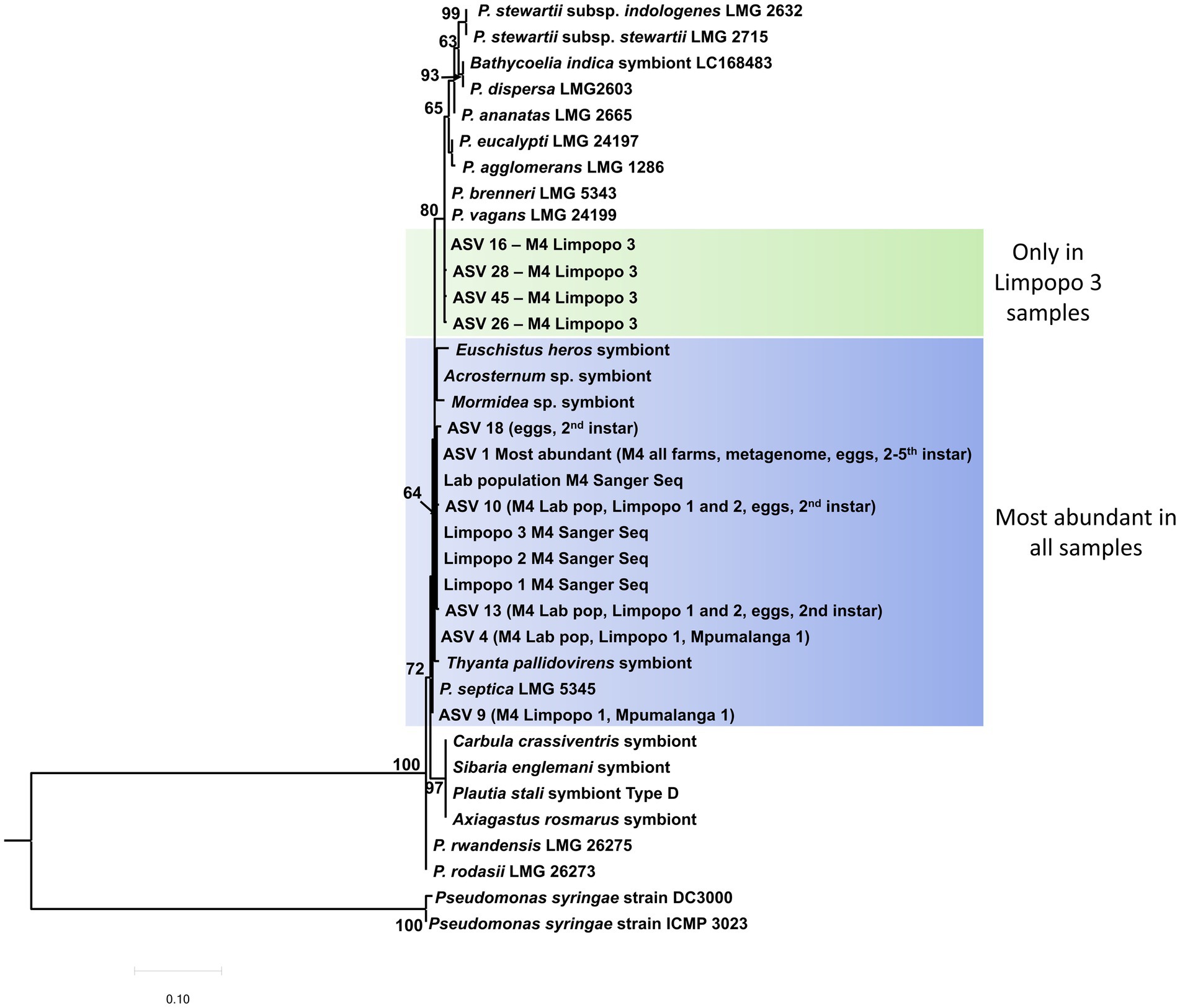
Figure 5. Maximum likelihood tree of the 10 most abundant Pantoea ASVs (16S rRNA metabarcoding) present in the M4 gut sections from all locations and the different developmental stages. The most abundant ASV (ASV1) was present in the M4 section from all locations and in all developmental stages (eggs, second–fifth instar). An additional strain/species was present in the Limpopo 3 samples, which grouped with P. vagans and P. brenneri. Branch support values >60% are indicated at the branches, based on 1,000 bootstrap replicates. Pseudomonas syringae was selected as outgroup to root the tree.
In the 16S rRNA phylogenetic tree of the Sodalis symbiont, most of the strains could not be properly differentiated by this gene region or had low branch support values (Figure 6). The Sodalis isolate from B. distincta was nearly identical to a strain previously collected from Antestiopsis thunbergia (Hemiptera: Pentatomidae), a pest of coffee plants in Africa. This is likely the same bacterial species occurring in different stink bug species but the availability of a full genome would provide more clarification.
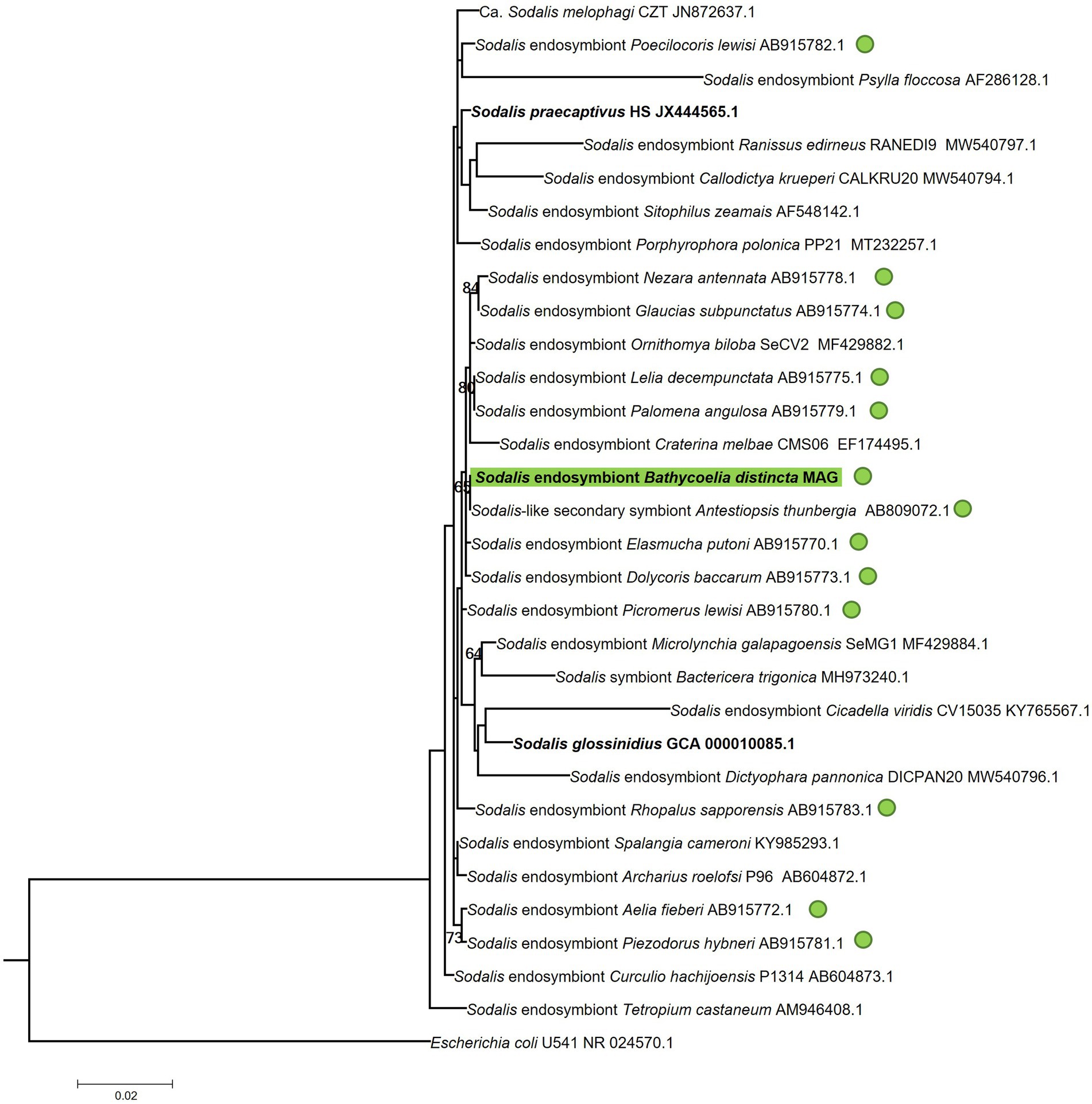
Figure 6. Maximum likelihood tree of the 16S rRNA gene region of the Sodalis symbiont identified in the B. distincta gut in relation to Sodalis strains identified in other stink bugs and other insects. The B. distincta symbiont is highlighted in green, and all other stink bug species are indicated with green dots. Escherichia coli was selected as outgroup to root the tree. Branch support values >60% are indicated, based on 1,000 bootstrap replicates.
The alpha diversity of each population (Rest_gut dataset) was compared to determine if any populations differ in diversity. The Mpumalanga populations had a significantly lower diversity compared to each of the Limpopo populations and the lab population, based on multiple diversity indices (Chao1, Shannon and Simpson; Pairwise Wilcoxon Rank Sum Tests; Supplementary Figure S2). Interestingly, the lab population had higher Shannon’s and Simpson’s diversity indices than its original source population, Limpopo 2, but they were not more diverse than the other Limpopo populations.
There was a significant difference in community composition between the populations, based on both Jaccard (R2 = 0.17552) and Bray–Curtis distances (R2 = 0.1859, p < 0.05). This was mainly attributed to the difference between the lab population and the Mpumalanga populations (pairwise comparison p = 0.015, Supplementary Table S4). Overall, the Mpumalanga populations (Mpumalanga 1 and Mpumalanga 2) were relatively distinct from the lab and Limpopo populations, as seen in the NMDS plot (Supplementary Figure S3). This is likely due to the lower species diversity in these populations.
Nearly 207.3 million reads were retained from Illumina TruSeq sequencing of the metagenome, after quality filtering and removal of host reads. The contig assembly, with MetaSpades, resulted in 354,262 contigs with an N50 value of 2,687 bp. The assembly rate, based on the percentage of reads that mapped back to the assembly, was 88.14%. The contig binning resulted in four bins of which two where high-quality bins with low contamination (<2%) and high completeness scores (>95%). Bin1 was classified as a Pantoea species and bin2 as a Sodalis species based on CheckM analysis. Comparison to the GTDB did not find any closely related species to the Pantoea genome, and the closest match to the Sodalis genome (98.7% ANI) was Sodalis praecaptivus. Alignment of the 16S rRNA sequences from the two metagenome-assembled genomes (MAGs) to the ASVs from the metabarcoding data confirmed these species to be identical to the most abundant Pantoea (P. bathycoeliae) and Sodalis ASVs (ASV1 and ASV2, respectively).
The Pantoea bathycoeliae MAG had a 100% completeness and 1.9% contamination score, based on CheckM, and a 97% completeness based on BUSCO analysis. The genome consisted of 127 contigs with a total length of 3.16 Mbp, an N50 of 70.5 Kbp, and a GC content of 54.85%. The genome coverage had an average sequence depth of 3,333. The number of predicted genes was 4,180, including 4,058 protein-coding genes, 48 tRNA, and 71 other RNA transcripts. The Pseudofinder analysis joined 460 ORFs as fragmented pseudogenes, resulting in a final prediction of 1,747 pseudogenes and 2,128 intact protein-coding genes.
The Sodalis sp. MAG was 95.3% complete with 0.9% contamination (CheckM) and a 98.4% BUSCO completeness score. The genome consisted of 453 contigs with a total length of 3.57 Mbp, an N50 of 15.6 Kbp, and a 56.7% GC content. The genome coverage had an average sequence depth of 686. A total of 3,822 genes were predicted, including 3,688 protein-coding genes, 45 tRNA genes, and 88 other RNA transcripts. The Pseudofinder analysis predicted the joining of 94 ORFs as fragmented pseudogenes, resulting in a final set of 753 pseudogenes and 2,916 intact protein-coding genes. The genome assembly JAUHTO000000000 is available under the GenBank BioProject PRJNA987703 and BioSample SAMN36080331.
The metabolic pathway modules predicted in the genomes of the P. bathycoeliae and Sodalis sp. MAGs indicated a high similarity in their metabolic potential, with primary metabolic modules (>80% complete) related to carbon fixation, carbohydrate metabolism, nucleotide metabolism, ATP synthesis, and biosynthesis of amino acids, vitamins, and other co-factors (Figure 7A). Complete or near-complete (one enzyme missing) modules were present for 15 and 13 amino acids in the P. bathycoeliae and Sodalis sp. MAGs, respectively. All nine essential amino acids had complete pathway modules in P. bathycoeliae. In the Sodalis sp., the leucine, methionine, and histidine modules had one enzyme missing (LeuA, MetA/X, and HisD/HIS4) (Figure 7B) of which the LeuA and HisD genes were predicted pseudogenes, suggesting they are no longer functional. In addition, the Sodalis sp. had a complete pathway for proline, while some genes were not functional in P. bathycoeliae. P. bathycoeliae and Sodalis each had complete (or near-complete) modules for 13 vitamins and co-factors. The vitamins and co-factors were highly similar between the two species: P. bathycoeliae uniquely encoded for pantothenate and molybdenum metabolism and Sodalis sp. for menaquinone and thiamine.
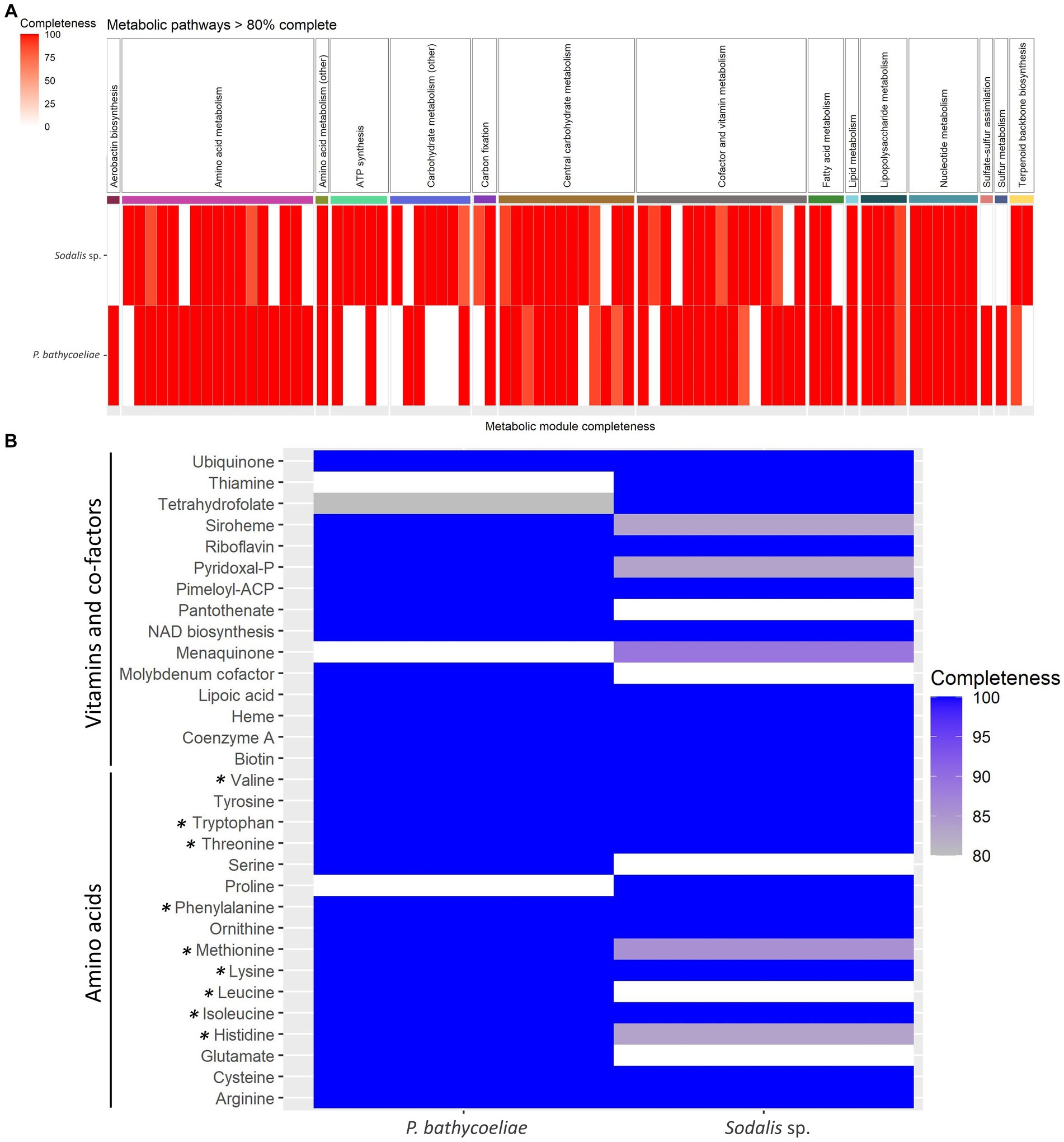
Figure 7. (A) All metabolic models detected in the genomes of the Pantoea and Sodalis symbiont of B. distincta with >80% completeness. (B) Detailed summary of amino acids, vitamins, and co-factors modules. All essential amino acids are labeled with an asterisk.
Additional metabolic modules of importance, that were present in both species, included lipopolysaccharides, and the terpenoid C5 isoprenoid biosynthesis, while only P. bathycoeliae had a complete pathway for aerobactin production (iron chelating agent) and for sulfur metabolism and sulfate–sulfur assimilation. Sulfur assimilation, through sulfate reduction, contributes to the incorporation of sulfur into methionine and cysteine amino acids (Hansen and Moran, 2014).
Pantoea bathycoeliae encoded for 30 carbohydrate-active enzymes (CAZymes), involved in the synthesis or breakdown of carbohydrates. These primarily consisted of glycoside hydrolases and glycosyl transferases (Supplementary Table S5A). A total of 78 peptidases and four inhibitors were encoded, of which metallopeptidases and serine peptidases were most abundant (Supplementary Table S5B). Of particular interest were families with high abundance in the genome, including C26 (gamma-glutamyl hydrolase), M20A (glutamate carboxypeptidase), and M23B (beta-lytic metallopeptidase). The Sodalis sp. encoded for 44 CAZymes and had more GH and GT families than P. bathycoeliae. The highest abundance was six peptidoglycan lyases GH23, and it had more glycosyltransferases (GT2, GT4, GT9) involved in the breakdown of sugars and lipopolysaccharides. The species encoded for 116 peptidases and four inhibitors but encoded for more cysteine peptidases than P. bathycoeliae and had less proteins of the M20A family but more of the M38 (isoaspartyl dipeptidase) and S33 (prolyl aminopeptidase) families.
Only one known, complete secondary metabolite cluster was identified using antiSMASH, a terpenoid biosynthetic gene cluster encoding for carotenoid synthesis in the P. bathycoeliae genome. The cluster had 78% similarity to a carotenoid cluster in a related Erwiniaceae species, Kalamiella piersonii, and 50% similarity to clusters in Pantoea agglomerans strains. All the genes from this pathway matched the carotenoid pathway of Pantoea ananatis on the Minimum Information about a Biosynthetic Gene cluster (MiBIG) database. It should be noted that two of the P. bathycoeliae genes were predicted to be pseudogenes, including an alcohol dehydrogenase and one of the two LysR transcriptional regulators, although the other LysR regulator was still functional. In the Sodalis sp., a potential acyl homoserine lactone gene cluster was detected, but because the cluster was located at the end of a contig the complete gene cluster could not be confirmed and will need further investigation. The genes present had 68% similarity to the gene cluster in Sodalis praecaptivus.
Bacterial secretion systems were also investigated in both symbionts. Pantoea bathycoeliae encoded for one Type 5 secretion system (T5aSS) but was predicted as a potential pseudogene due to a 40% shorter gene than its orthologs. T5aSSs consist of a single protein that also encodes for the secretion product. InterPro analyses classified the secreted domain as a pectin lyase (IPR012332), which can be considered a virulence factor. When compared to the other Pantoea symbiont genomes, orthologs were present only in the Mormidea symbiont, Plautia stali symbionts A, C, and F, and in the free-living species P. dispersa and P. vagans. The Sodalis symbiont encoded for a Type 1 secretion system (T1SS), three Type 3 secretion system (T3SS), and one T5cSS system. The T1SS can secrete a range of substrates, involved in biofilm formation, toxins, iron uptake, proteases, or lipases. The T5cSS encodes for adhesin proteins that play a role in cell attachment, and the T3SS can assist with host immune suppression and establishment in the host. The one T3SS gene cluster contained three predicted pseudogenes, and only the other two T3SSs have likely remained functional.
Pantoea bathycoeliae had the highest genome similarity (ANI) to “P. latae” and P. septica (87% similarity). The phylogenomic analysis of 328 single-copy genes confirmed “P. latae” and P. septica as the closest related species, and the closest stink bug symbionts were Candidatus P. persica (from Acrosternum arabicum) and the Plautia stali symbiont B (Figure 8). This confirms that P. bathycoeliae is a novel Pantoea species. The genome size of the species is 1 Mb smaller than those of the closest free-living Pantoea species but 310–730 kb larger than the aforementioned Pantoea symbionts associated with other stink bugs. Nonetheless, the symbiotic species, Candidatus P. persica and Plautia stali symbiont B, had a highly similar number of intact protein-coding genes compared with P. bathycoeliae and had a pseudogene/gene ratio of 35%–45% (Supplementary Table S1).
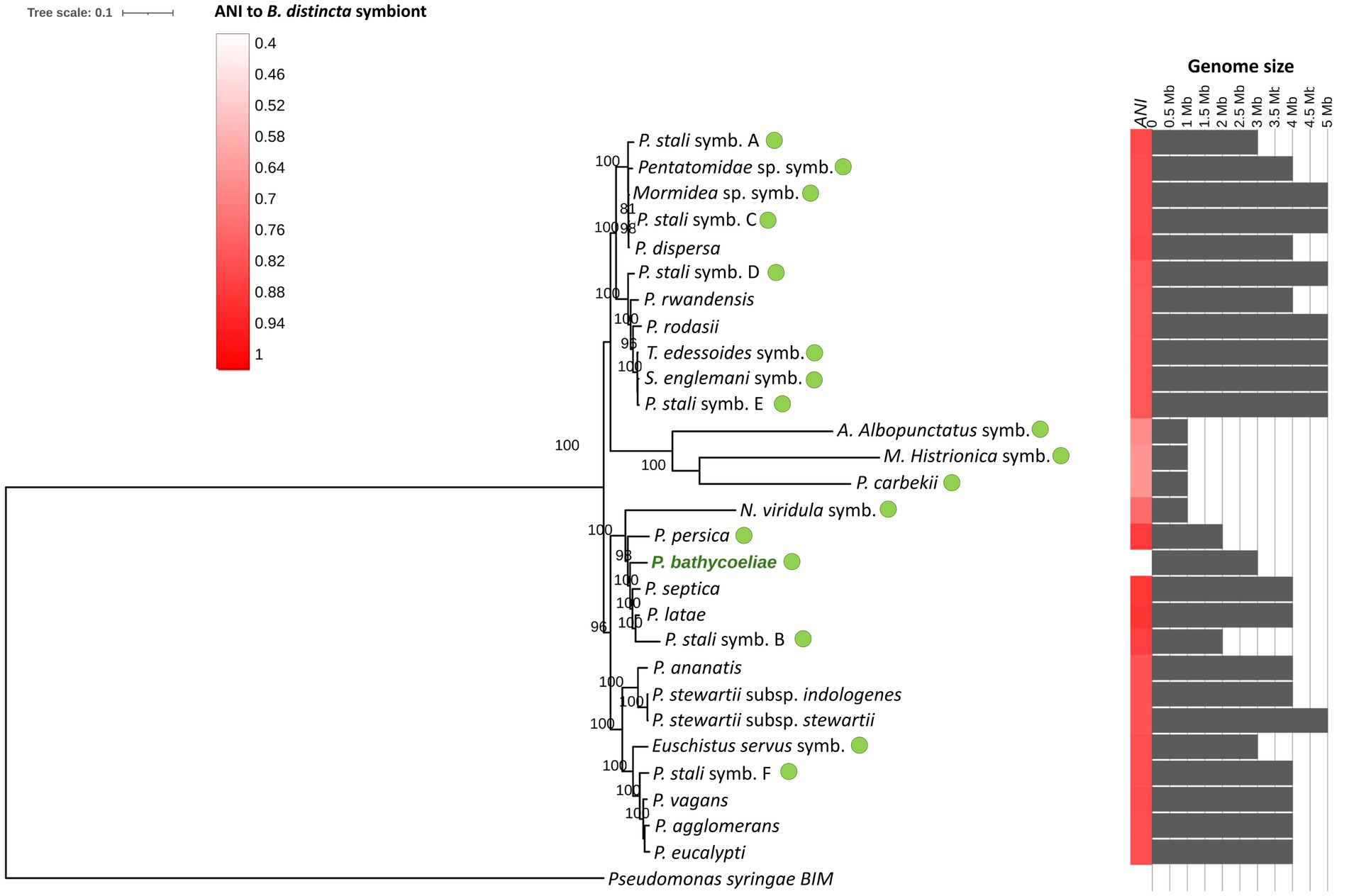
Figure 8. Maximum likelihood phylogenomic tree of the B. distincta symbiont and related Pantoea species and other stink bug gut symbionts (green dots), based on 358 single-copy orthologous genes (GenBank numbers of genomes are presented in Supplementary Table S1). The heatmap on the right indicates the average nucleotide identity (ANI) between the Pantoea symbiont and other genomes and the gray bar chart indicates genome sizes (Mb). Branch support values, based on 1,000 bootstrap replicates, are indicated on the branches.
As comprehensive genome comparisons were recently reported for the Pantoea gut symbionts of stink bugs (Kashkouli et al., 2021; Otero-Bravo and Sabree, 2021), only the main genome structure and any unique genes of P. bathycoeliae are reported in this study. The shared and unique orthologs among the Pantoea genomes were determined with OrthoFinder. A total of 311 genes were unique to P. bathycoeliae. Although many unique genes were identified, these were not linked to distinct functions in this species. The unique proteins were assigned to KEGG functional categories associated with carbohydrate metabolism, energy metabolism, nucleotide metabolism, amino acid metabolism, glycan biosynthesis and metabolism, genetic information processing, membrane transport, signal transduction, and cellular processes such as quorum sensing and biofilm formation.
Pantoea bathycoeliae (ba.ti.coe.liae L. fem. pl. suff. Bathycoeliae, from the Bathycoelia distincta stink bug species, as this bacterium was obtained from the gut of this insect).
A MAG representing this species was obtained from metagenomic sequencing of the M4 gut region of B. distincta. The majority of stink bug symbionts within the Pantoea genus are obligate symbionts and cannot be cultured in vitro. Despite various attempts P. bathycoeliae could not be isolated on nutrient agar. Phylogenetic inference of species in the Pantoea genus robustly supports this species as distinct in the genus and the delineation of the species is supported by a low average nucleotide identity (ANI), below 88%, with the closest related species. The genome JAUHTP000000000, available under GenBank BioProject PRJNA987703 and BioSample SAMN36078333, is the designated nomenclatural type for the species. Extracted DNA from the B. distincta gut, which predominantly consists of this species, is stored at the Forestry and Agricultural Biotechnology Institute, South Africa.
The gut of the two-spotted stink bug, Bathycoelia distincta, has a clear compartmentalization, as seen in other Pentatomidae, with an obligate Pantoea symbiont in the M4 section and various secondary symbionts in the rest of the gut. The M4 symbiont was confirmed as a highly conserved and vertically transmitted species, based on its presence on the egg surfaces and all developmental life stages and in adults across geographic locations. Multilocus genotyping and phylogenomics confirmed this to be a distinct species, described in this study as Pantoea bathycoeliae. Most stink bug studies have only focused on the bacteria in the M4 gut section, but, in the current study, we highlight the facultative as well as core genera present in the rest of the gut. This also allowed the identification of a prominent secondary symbiont, Sodalis sp., with high functional overlap with P. bathycoeliae.
Pantoea bathycoeliae is considered a highly conserved and primary symbiont of B. distincta. The same Pantoea strain was present in the M4 gut section of all B. distincta stink bugs collected from five orchards in two provinces and the lab-reared population. In other stinkbug species, that were also collected from different locations, a single symbiont species was conserved in all populations (Duron and Noël, 2016; Hosokawa et al., 2016b), or minor haplotype differences were observed (Otero-Bravo and Sabree, 2018). The metabarcoding sequence approach identified 10 abundant Pantoea ASV sequences in B. distincta, of which six were classified as the P. bathycoeliae symbiont species, with minor sequence variations. Some of these variants likely relate to different strains of the species or different copies of the 16S rRNA gene in a single individual. However, some could also be the result of sequence errors, a caveat of Illumina sequence technologies.
A different species to the primary M4 midgut symbiont has rarely been observed in Pentatomidae (Hosokawa et al., 2016a, 2019). A species distinct from P. bathycoeliae was identified in this study (predominantly in the Limpopo 3 population) that was highly similar to P. brenneri and P. vagans, which are free-living Pantoea species. P. brenneri has been isolated from humans, while P. vagans was isolated from plant sources (Brady et al., 2010), making the latter a more likely source of the M4 symbiont. One prominent example of symbiont replacement is in Plautia stali, which were collected from different climatic regions in Japan (Hosokawa et al., 2016a). Six lineages of Enterobacteriaceae M4 symbionts (lineages A–F) were predominantly separated based on geographic location. Some of these lineages were free-living environmental strains and others obligate symbionts. In B. distincta, only one insect had a high abundance of the alternative Pantoea strain. This might suggest a symbiont replacement event in the Limpopo province, but follow-up collections are needed to confirm the prominence of the alternative species.
Pantoea bathycoeliae was present on the eggs and in the second instar nymphs, suggesting the instars acquired the symbiont by probing the egg surface. This probing behavior was observed in the current study and has also been recorded in other Pentatomidae species (Calizotti and Panizzi, 2014; Koga et al., 2022). In other stinkbug species, the M4 symbiont has been confirmed on the egg surface by methods such as egg surface sterilization, microscopy, and 16S rRNA sequencing (Prado et al., 2006; Kenyon et al., 2015; Kashkouli et al., 2020). A recent metabarcoding study of N. viridula egg microbiomes also detected a Pantoea symbiont on the egg surfaces but a Sodalis sp. was also present (Geerinck et al., 2022). Similarly, a Sodalis sp. was also present on the eggs of B. distincta; however, the vertical transfer of this additional symbiont is a new observation from our study. Whether this transfer is also through egg smearing or via the ovaries needs to be confirmed since Sodalis spp. can be transmitted transovarially (Kaiwa et al., 2010).
The phylogenetic and phylogenomic analyses from this study suggest that P. bathycoeliae is a novel species. The species was, therefore, named following the rules of the SeqCode (Whitman et al., 2022). The phylogenetic placement of the species suggests that it is closest to two free-living Pantoea species (P. septica and “P. latae”). Pantoea septica has predominantly been isolated from humans (Brady et al., 2010; Nadarasah and Stavrinides, 2014) with a single report from olive oil (Pizzolante et al., 2018), while “P. latae” was obtained from the rhizosphere of a cycad (Zamia floridana) but has rarely been reported since. The plant-associated species would be a more plausible ancestor of the B. distincta symbiont, but the closest relative to this species is more likely still unsampled.
The genome of P. bathycoeliae was smaller (3.16 Mb) than those of most free-living Pantoea species (4.33 Mb–5.68 Mb) yet is larger than the genomes of the closely related obligate stink bug symbionts (Candidatus P. persica—2.85 Mb and Plautia stali symb B—2.43 Mb). Nonetheless, both P. bathycoeliae and Candidatus P. persica encoded for a similar number of ORFs and had a similar proportion of predicted pseudogenes (43%–46%). Genome reduction is a common trend in obligate stink bug symbionts as a result of their dependence on the host for energy and resources. A gradual adaptation has been hypothesized where larger genomes contain many pseudogenes (up to 40%) that are purged over time, while small genomes contain few pseudogenes since most have already been lost from the genome (Hosokawa et al., 2016a; Kashkouli et al., 2021; Otero-Bravo and Sabree, 2021). Pantoea bathycoeliae is also in this intermediate stage of genome reduction since many protein-coding genes were predicted, but 43% are potentially non-functional. It should be noted that different approaches to pseudogene identification were used in the related studies and thus direct comparisons should be made with caution.
Despite a large number of predicted pseudogenes in P. bathycoeliae, the functional roles predicted for the intact genes were highly similar to those of other stink bug symbionts. Metabolic pathways predominantly encoded for amino acids, vitamins, and co-factors. The species can produce more amino acids than some of the other stink bug symbionts, including all nine essential amino acids, whereas branched-chain amino acids (leucine, isoleucine, and valine) were incomplete in many other species (Kenyon et al., 2015; Otero-Bravo and Sabree, 2021). Pantoea bathycoeliae also encoded many of the same vitamins and co-factors as other stink bug symbionts, such as NAD, riboflavin, pyridoxal-phosphate, coenzyme A, and heme. Similar to other obligate symbionts (Kenyon et al., 2015; Otero-Bravo et al., 2018; Kashkouli et al., 2021; Otero-Bravo and Sabree, 2021), this species thus serves an important beneficial role in nutrient supplementation for B. distincta.
In addition to amino acid and vitamin provision, P. bathycoeliae likely produces a carotenoid metabolite. Carotenoids contribute to important functions in insects such as pigmentation, antioxidants, precursor of hormones, or the production of vitamin A (Heath et al., 2013). These compounds are often obtained from plant food sources but can also be produced by microbes. For example, Benitsuchiphilus tojoi, the symbiont of the parastrachiid stink bug Parastrachia japonensis has a carotenoid biosynthesis pathway (Mondal et al., 2020). Carotenoid production has also been confirmed in other free-living Pantoea species, such as β-carotene in P. septica (Pizzolante et al., 2018), and carotenoid biosynthetic gene clusters are encoded in P. agglomerans, P. stewartii, and P. ananatis (Kautsar et al., 2019). Biochemical assays (HPLC studies) in four stink bug species, including Halyomorpha halys and Nezara antenata, identified four carotenoid derivates (β-carotene, β-cryptoxanthin, lutein, and zeaxanthin) in these species (Maoka et al., 2021). These were suggested to have a plant origin but production by bacterial symbionts cannot be excluded. It is thus possible that P. bathycoeliae produces a carotenoid derivative, advantageous to its host, but functional studies are required to confirm this.
The microbiome composition in the rest of the B. distincta gut (excluding the M4 section) was not highly diverse, as seen in the low alpha diversities of the populations. A similar low alpha diversity was observed in the M1–M3 gut sections of N. viridula, with many transient microbes and few core microbiota (Medina et al., 2018). This is also a general phenomenon in insects such as bees, aphids, and herbivorous Hemipterans (Gauthier et al., 2015; Kwong and Moran, 2016; Li et al., 2022). Interestingly, the lab-reared population did not have a lower alpha diversity than the orchard-collected samples, suggesting the lab-rearing did not reduce gut microbe diversity. In addition, many of the genera present in the native starter population of the lab population (Limpopo 2 farm) were still present in their guts, indicating the long-term maintenance of gut microbes over many generations.
The two populations from Mpumalanga had a lower gut microbiome diversity than the other populations and little variation between the orchards. This high similarity could be due to similar pest management practices since the two orchards are managed by the same company. For example, a specific pesticide regime could influence the bacterial composition and diversity of the populations. Pesticide treatments in honey bee populations have shown modifications of the gut microbial community by some neurotoxic insecticides (Rouzé et al., 2019; Zhu et al., 2020), while others had no effect on the microbiome (Raymann et al., 2018; Wintermantel et al., 2018). Due to limited sampling sites in the two different provinces, other confounding factors such as climatic differences between the provinces cannot be excluded.
Only three core genera were detected in the rest of the gut in all populations (>75% of individuals), Pantoea, Sodalis, and Serratia. Some genera were more region-specific such as Yokenella and the alternative Pantoea strain that were predominantly in the Limpopo and lab populations, while Acinetobacter and others were only in Mpumalanga. This suggests that many of the bacterial species are transient with no strong selection and could even have been obtained from food sources. Interestingly, Yokenella was one of the few non-transient genera in N. viridula and might thus be a more common gut microbe of stink bugs (Medina et al., 2018). Other transient genera detected in N. viridula, which were also present in P. bathycoeliae, were Bacillus and Staphylococcus.
Serratia species were present in most of the B. distincta individuals but were only abundant in some individuals of the lab population. Some Serratia species can be beneficial to their host, such as S. symbiotica in aphids, that provides additional vitamins or protection against parasites (Oliver et al., 2014; Renoz et al., 2022). Yet, Serratia marcescens is considered an insect pathogen (Slatten and Larson, 1967). This pathogen was detected in N. viridula midguts and was more abundant in lab populations compared to wild samples collected from soya fields (Medina et al., 2022). Further sequencing would be required to confirm the species present in B. distincta.
The most prominent secondary gut symbiont was Sodalis as it was present in all populations and highly abundant in some individuals. This species could not be clearly distinguished, phylogenetically, from other Sodalis stink bug symbionts and likely the same species can infect various stink bug species. It was most closely related to the Sodalis strain collected from the stink bug species, Antestiopsis thunbergia, where it was present in the gut and other tissues of about half of the individuals investigated (Matsuura et al., 2014). Sodalis symbionts were first reported in tsetse flies but have since been observed in weevils, psyllids, aphids, mealybugs, and a diversity of stink bugs (Hosokawa et al., 2015; Ghosh et al., 2020). However, in most stink bug species, Sodalis infections are rare (<5%), except for the Urostylididae family which shows very high infection rates (60%–100%) (Kaiwa et al., 2010, 2014; Hosokawa et al., 2015). The high prevalence of Sodalis in B. distincta is thus a rare phenomenon and suggests a strong selection for the symbiont in this stink bug species.
Despite the presence of both species in the M4 and the other gut compartments, Sodalis sp. cannot outcompete P. bathycoeliae in the M4 section, whereas in the rest of the gut, the Sodalis sp. is sometimes in much higher proportions. This might be attributed to a physical constriction in the M4 compartment, as well as physical traits of the bacterium (such as motility), as seen in Rhiptortus pedestris and its Burkholderia symbiont (Ohbayashi et al., 2015). With competitive infection assays other bacteria could also colonize the M4 region but when co-inoculated with the Burkholderia symbiont, the latter outcompeted them (Itoh et al., 2019). The location of Sodalis could also have an impact since it can be localized intracellularly in gut cells (Ghosh et al., 2020) and in this manner occupy a different niche.
The Sodalis sp. has some traits that might allow it to compete with other bacteria, in the rest of the gut. It encodes for a Type 5c secretion system and three copies of the type 3 secretion system. The former plays a role in cellular adhesion, which might be advantageous for the bacterium to attach and colonize the host gut (Łyskowski et al., 2011). T3SSs have generally been linked to infection and establishment advantages for bacteria in insect hosts (Dale and Moran, 2006). More specifically, Sodalis glossinidius encodes for two T3SSs that have been suggested to contribute to its invasion and establishment in tsetse flies (Toh et al., 2006). Likely these nanoweapons contribute to the establishment of Sodalis sp. in B. distincta, yet in the M4 compartment, other unknown factors prevent this bacterium from establishing itself.
The maintenance of both P. bathycoeliae and the Sodalis sp. in the gut is likely due to their highly similar metabolic capacities and some unique metabolites that can complement each other. Both species encode for most known amino acids and many vitamins but P. bathycoeliae has more complete pathways for at least three amino acids. Conversely, only the Sodalis sp. produces proline, thiamine, and menaquinone. Thiamine production was conserved in the Parastrachia japonensis symbiont and is likely important for stinkbugs (Mondal et al., 2020). Although the two species had similar CAZymes and peptidases, the unique families and higher numbers of specific families in each likely contribute to different capacities of nutrient metabolism of importance to the host. The recruitment and conservation of an additional symbiont has been observed in various aphid species, where the primary symbiont lost some metabolic capacities that could be complemented by another facultative symbiont, which eventually invades the symbiotic organ (Renoz et al., 2022; Manzano-Marín et al., 2023). It could be that the Pantoea and Sodalis species in B. distincta are also in this process of symbiont complementation and establishment of a second important gut symbiont.
In this study, we identified a unique Pantoea species maintained in the M4 midgut section of B. distincta. This is in correspondence with other Pentatomidae, all of which maintain a unique Pantoea species. An additional vertically transferred secondary symbiont, Sodalis sp., was prominent in the rest of the gut. Other than these two abundant symbionts, most bacteria in the rest of the gut were likely obtained from the environment and were more variable. High-quality MAGs were produced for both core symbionts and large metabolic overlaps were observed in their genomes. Sodalis is likely maintained in B. distincta outside the midgut crypts for additional beneficial functions not provided by P. bathycoeliae. This study contributed to our understanding of the composition of B. distincta microbial symbionts, which is essential for the effective control of these pests. This information could contribute in future to the development of alternative biocontrol measures such as eliminating essential symbionts or performing symbiont replacements, as has been explored in other stink bug species (Sassera et al., 2013; Taylor et al., 2017; Gonella et al., 2022).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, BioProject PRJNA972719 and PRJNA987703.
The manuscript presents research on animals that do not require ethical approval for their study.
AF: Conceptualization, Data curation, Formal analysis, Investigation, Project administration, Writing – original draft, Writing – review & editing. SV: Data curation, Methodology, Writing – original draft, Writing – review & editing. BS: Conceptualization, Writing – original draft, Writing – review & editing. GF: Conceptualization, Funding acquisition, Methodology, Resources, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was made possible by the Forestry and Agricultural Biotechnology Institute at the University of Pretoria, which provided the facilities and resources for this research, and the South African Macadamia Association (SAMAC), which provided a postdoctoral research grant and additional research funds for this project.
The stink bugs collected from macadamia orchards were all provided by local macadamia growers, and the authors thank each of the growers who contributed to the sample collections. The authors also thank Mancha Mabaso, Kinosha Moodley, and researchers from the Kostas Lab who provided guidance on the data analysis in the project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1284397/full#supplementary-material
Abby, S. S., Néron, B., Ménager, H., Touchon, M., and Rocha, E. P. C. (2014). MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS One 9:e110726. doi: 10.1371/journal.pone.0110726
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom
Aramaki, T., Blanc-Mathieu, R., Endo, H., Ohkubo, K., Kanehisa, M., Goto, S., et al. (2019). KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252. doi: 10.1093/bioinformatics/btz859
Arkin, A. P., Cottingham, R. W., Henry, C. S., Harris, N. L., Stevens, R. L., Maslov, S., et al. (2018). KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 36, 566–569. doi: 10.1038/nbt.4163
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van Wezel, G. P., Medema, M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi: 10.1093/nar/gkab335
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brady, C., Cleenwerck, I., Venter, S., Vancanneyt, M., Swings, J., and Coutinho, T. (2008). Phylogeny and identification of Pantoea species associated with plants, humans and the natural environment based on multilocus sequence analysis (MLSA). Syst. Appl. Microbiol. 31, 447–460. doi: 10.1016/j.syapm.2008.09.004
Brady, C. L., Cleenwerck, I., Venter, S. N., Engelbeen, K., De Vos, P., and Coutinho, T. A. (2010). Emended description of the genus Pantoea, description of four species from human clinical samples, Pantoea septica sp. nov., Pantoea eucrina sp. nov., Pantoea brenneri sp. nov. and Pantoea conspicua sp. nov., and transfer of Pectobacterium cypripedii (Hori 1911) Brenner et al. 1973 emend. Hauben et al. 1998 to the genus as Pantoea cypripedii comb. nov. Int. J. Syst. Evol. Microbiol. 60, 2430–2440. doi: 10.1099/ijs.0.017301-0
Calizotti, G. S., and Panizzi, A. R. (2014). Behavior of first instar nymphs of Edessa meditabunda (F.) (Hemiptera: Pentatomidae) on the egg mass. Fla. Entomol. 97, 277–280. doi: 10.1653/024.097.0139
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Cornwallis, C. K., Van’t Padje, A., Ellers, J., Klein, M., Jackson, R., Kiers, E. T., et al. (2023). Symbioses shape feeding niches and diversification across insects. Nat. Ecol. Evol. 7, 1022–1044. doi: 10.1038/s41559-023-02058-0
Dale, C., and Moran, N. A. (2006). Molecular interactions between bacterial symbionts and their hosts. Cells 126, 453–465. doi: 10.1016/j.cell.2006.07.014
Douglas, A. E. (2006). Phloem-sap feeding by animals: problems and solutions. J. Exp. Bot. 57, 747–754. doi: 10.1093/jxb/erj067
Duron, O., and Noël, V. (2016). A wide diversity of Pantoea lineages are engaged in mutualistic symbiosis and cospeciation processes with stinkbugs. Environ. Microbiol. Rep. 8, 715–727. doi: 10.1111/1758-2229.12432
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Engel, P., and Moran, N. A. (2013). The gut microbiota of insects—diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Gauthier, J.-P., Outreman, Y., Mieuzet, L., and Simon, J.-C. (2015). Bacterial communities associated with host-adapted populations of pea aphids revealed by deep sequencing of 16S ribosomal DNA. PLoS One 10:e0120664. doi: 10.1371/journal.pone.0120664
Geerinck, M. W. J., Van Hee, S., Gloder, G., Crauwels, S., Colazza, S., Jacquemyn, H., et al. (2022). Diversity and composition of the microbiome associated with eggs of the southern green stinkbug, Nezara viridula (Hemiptera: Pentatomidae). MicrobiologyOpen 11:e1337. doi: 10.1002/mbo3.1337
Ghosh, S., Sela, N., Kontsedalov, S., Lebedev, G., Haines, L. R., and Ghanim, M. (2020). An intranuclear Sodalis-like symbiont and Spiroplasma Coinfect the carrot psyllid, Bactericera trigonica (Hemiptera, Psylloidea). Microorganisms 8:692. doi: 10.3390/microorganisms8050692
Gonella, E., Orrù, B., and Alma, A. (2022). Symbiotic control of Halyomorpha halys using a microbial biopesticide. Entomol. Gen. 42, 901–909. doi: 10.1127/entomologia/2022/1707
Gonella, E., Orrù, B., Marasco, R., Daffonchio, D., and Alma, A. (2020). Disruption of host-symbiont associations for the symbiotic control and management of pentatomid agricultural pests—a review. Front. Microbiol. 11:547031. doi: 10.3389/fmicb.2020.547031
Gupta, A., and Nair, S. (2020). Dynamics of insect-microbiome interaction influence host and microbial symbiont. Front. Microbiol. 11:1357. doi: 10.3389/fmicb.2020.01357
Hansen, A. K., and Moran, N. A. (2011). Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc. Natl. Acad. Sci. U.S.A. 108, 2849–2854. doi: 10.1073/pnas.1013465108
Hansen, A. K., and Moran, N. A. (2014). The impact of microbial symbionts on host plant utilization by herbivorous insects. Mol. Ecol. 23, 1473–1496. doi: 10.1111/mec.12421
Hayashi, T., Hosokawa, T., Meng, X.-Y., Koga, R., and Fukatsu, T. (2015). Female-specific specialization of a posterior end region of the midgut symbiotic organ in Plautia splendens and allied stinkbugs. Appl. Environ. Microbiol. 81, 2603–2611. doi: 10.1128/aem.04057-14
Heath, J. J., Cipollini, D. F., and Stireman Iii, J. O. (2013). The role of carotenoids and their derivatives in mediating interactions between insects and their environment. Arthropod Plant Interact. 7, 1–20. doi: 10.1007/s11829-012-9239-7
Hirose, E., Panizzi, A. R., De Souza, J. T., Cattelan, A. J., and Aldrich, J. R. (2006). Bacteria in the gut of southern green stink bug (Heteroptera: Pentatomidae). Ann. Entomol. Soc. Am. 99, 91–95. doi: 10.1603/0013-8746(2006)099[0091:Bitgos]2.0.Co;2
Hosokawa, T., Hironaka, M., Inadomi, K., Mukai, H., Nikoh, N., and Fukatsu, T. (2013). Diverse strategies for vertical symbiont transmission among subsocial stinkbugs. PLoS One 8:e65081. doi: 10.1371/journal.pone.0065081
Hosokawa, T., Hironaka, M., Mukai, H., Inadomi, K., Suzuki, N., and Fukatsu, T. (2012). Mothers never miss the moment: a fine-tuned mechanism for vertical symbiont transmission in a subsocial insect. Anim. Behav. 83, 293–300. doi: 10.1016/j.anbehav.2011.11.006
Hosokawa, T., Imanishi, M., Koga, R., and Fukatsu, T. (2019). Diversity and evolution of bacterial symbionts in the gut symbiotic organ of jewel stinkbugs (Hemiptera: Scutelleridae). Appl. Entomol. Zool. 54, 359–367. doi: 10.1007/s13355-019-00630-4
Hosokawa, T., Ishii, Y., Nikoh, N., Fujie, M., Satoh, N., and Fukatsu, T. (2016a). Obligate bacterial mutualists evolving from environmental bacteria in natural insect populations. Nat. Microbiol. 1:15011. doi: 10.1038/nmicrobiol.2015.11
Hosokawa, T., Kaiwa, N., Matsuura, Y., Kikuchi, Y., and Fukatsu, T. (2015). Infection prevalence of Sodalis symbionts among stinkbugs. Zool. Lett. 1:5. doi: 10.1186/s40851-014-0009-5
Hosokawa, T., Kikuchi, Y., Meng, X. Y., and Fukatsu, T. (2005). The making of symbiont capsule in the plataspid stinkbug Megacopta punctatissima. FEMS Microbiol. Ecol. 54, 471–477. doi: 10.1016/j.femsec.2005.06.002
Hosokawa, T., Kikuchi, Y., Nikoh, N., Meng, X.-Y., Hironaka, M., and Fukatsu, T. (2010). Phylogenetic position and peculiar genetic traits of a midgut bacterial symbiont of the stinkbug Parastrachia japonensis. Appl. Environ. Microbiol. 76, 4130–4135. doi: 10.1128/aem.00616-10
Hosokawa, T., Matsuura, Y., Kikuchi, Y., and Fukatsu, T. (2016b). Recurrent evolution of gut symbiotic bacteria in pentatomid stinkbugs. Zool. Lett. 2:24. doi: 10.1186/s40851-016-0061-4
Itoh, H., Jang, S., Takeshita, K., Ohbayashi, T., Ohnishi, N., Meng, X.-Y., et al. (2019). Host-symbiont specificity determined by microbe-microbe competition in an insect gut. Proc. Natl. Acad. Sci. U.S.A. 116, 22673–22682. doi: 10.1073/pnas.1912397116
Kaiwa, N., Hosokawa, T., Kikuchi, Y., Nikoh, N., Meng, X. Y., Kimura, N., et al. (2010). Primary gut symbiont and secondary, Sodalis-allied symbiont of the scutellerid stinkbug Cantao ocellatus. Appl. Environ. Microbiol. 76, 3486–3494. doi: 10.1128/aem.00421-10
Kaiwa, N., Hosokawa, T., Nikoh, N., Tanahashi, M., Moriyama, M., Meng, X.-Y., et al. (2014). Symbiont-supplemented maternal investment underpinning host’s ecological adaptation. Curr. Biol. 24, 2465–2470. doi: 10.1016/j.cub.2014.08.065
Kanehisa, M., and Sato, Y. (2020). KEGG mapper for inferring cellular functions from protein sequences. Protein Sci. 29, 28–35. doi: 10.1002/pro.3711
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, –e7359. doi: 10.7717/peerj.7359
Kashkouli, M., Castelli, M., Floriano, A. M., Bandi, C., Epis, S., Fathipour, Y., et al. (2021). Characterization of a novel Pantoea symbiont allows inference of a pattern of convergent genome reduction in bacteria associated with Pentatomidae. Environ. Microbiol. 23, 36–50. doi: 10.1111/1462-2920.15169
Kashkouli, M., Fathipour, Y., and Mehrabadi, M. (2020). Habitat visualization, acquisition features and necessity of the gammaproteobacterial symbiont of pistachio stink bug, Acrosternum heegeri (Hem.: Pentatomidae). Bull. Entomol. Res. 110, 22–33. doi: 10.1017/S0007485319000245
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kautsar, S. A., Blin, K., Shaw, S., Navarro-Muñoz, J. C., Terlouw, B. R., van der Hooft, J. J. J., et al. (2019). MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 48, D454–D458. doi: 10.1093/nar/gkz882
Kenyon, L. J., Meulia, T., and Sabree, Z. L. (2015). Habitat visualization and genomic analysis of “Candidatus Pantoea carbekii,” the primary symbiont of the brown marmorated stink bug. Genome Biol. Evol. 7, 620–635. doi: 10.1093/gbe/evv006
Kikuchi, Y., Hayatsu, M., Hosokawa, T., Nagayama, A., Tago, K., and Fukatsu, T. (2012a). Symbiont-mediated insecticide resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 8618–8622. doi: 10.1073/pnas.1200231109
Kikuchi, Y., Hosokawa, T., Nikoh, N., and Fukatsu, T. (2012b). Gut symbiotic bacteria in the cabbage bugs Eurydema rugosa and Eurydema dominulus (Heteroptera: Pentatomidae). Appl. Entomol. Zool. 47, 1–8. doi: 10.1007/s13355-011-0081-7
Kikuchi, Y., Hosokawa, T., Nikoh, N., Meng, X.-Y., Kamagata, Y., and Fukatsu, T. (2009). Host-symbiont co-speciation and reductive genome evolution in gut symbiotic bacteria of acanthosomatid stinkbugs. BMC Biol. 7:2. doi: 10.1186/1741-7007-7-2
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Koch, H., and Schmid-Hempel, P. (2011). Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc. Natl. Acad. Sci. U.S.A. 108, 19288–19292. doi: 10.1073/pnas.1110474108
Koga, R., Moriyama, M., Onodera-Tanifuji, N., Ishii, Y., Takai, H., Mizutani, M., et al. (2022). Single mutation makes Escherichia coli an insect mutualist. Nat. Microbiol. 7, 1141–1150. doi: 10.1038/s41564-022-01179-9
Kück, P., and Longo, G. C. (2014). FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 11:81. doi: 10.1186/s12983-014-0081-x
Kwong, W. K., and Moran, N. A. (2016). Gut microbial communities of social bees. Nat. Rev. Microbiol. 14, 374–384. doi: 10.1038/nrmicro.2016.43
Lahti, L., and Shetty, S. (2017). Tools for microbiome analysis in R. Available at: http://microbiome.github.com/microbiome
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, G., Sun, J., Meng, Y., Yang, C., Chen, Z., Wu, Y., et al. (2022). The impact of environmental habitats and diets on the gut microbiota diversity of true bugs (Hemiptera: Heteroptera). Biology 11:1039. doi: 10.3390/biology11071039
Łyskowski, A., Leo, J. C., and Goldman, A. (2011). “Structure and biology of trimeric autotransporter adhesins” in Bacterial adhesion: chemistry, biology and physics. eds. D. Linke and A. Goldman (Dordrecht: Springer), 143–158.
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., and Zdobnov, E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654. doi: 10.1093/molbev/msab199
Manzano-Marín, A., D’acier, A. C., Clamens, A.-L., Cruaud, C., Barbe, V., and Jousselin, E. (2023). Co-obligate symbioses have repeatedly evolved across aphids, but partner identity and nutritional contributions vary across lineages. Peer Commun. J. 3:e46. doi: 10.1101/2022.08.28.505559
Maoka, T., Kawase, N., Hironaka, M., and Nishida, R. (2021). Carotenoids of hemipteran insects, from the perspective of chemo-systematic and chemical ecological studies. Biochem. Syst. Ecol. 95:104241. doi: 10.1016/j.bse.2021.104241
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Martinez, A. P. (2020). pairwiseAdonis: Pairwise multilevel comparison using adonis. R package version 0.4
Matsuura, Y., Hosokawa, T., Serracin, M., Tulgetske, G. M., Miller, T. A., and Fukatsu, T. (2014). Bacterial symbionts of a devastating coffee plant pest, the stinkbug Antestiopsis thunbergii (Hemiptera: Pentatomidae). Appl. Environ. Microbiol. 80, 3769–3775. doi: 10.1128/aem.00554-14
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Medina, V., Rosso, B. E., Soria, M., Gutkind, G. O., Pagano, E. A., and Zavala, J. A. (2022). Feeding on soybean crops changed gut bacteria diversity of the southern green stinkbug (Nezara viridula) and reduced negative effects of some associated bacteria. Pest Manag. Sci. 78, 4608–4617. doi: 10.1002/ps.7080
Medina, V., Sardoy, P. M., Soria, M., Vay, C. A., Gutkind, G. O., and Zavala, J. A. (2018). Characterized non-transient microbiota from stinkbug (Nezara viridula) midgut deactivates soybean chemical defenses. PLoS One 13:e0200161. doi: 10.1371/journal.pone.0200161
Mondal, S. I., Akter, A., Koga, R., Hosokawa, T., Dayi, M., Murase, K., et al. (2020). Reduced genome of the gut symbiotic bacterium “Candidatus Benitsuchiphilus tojoi” provides insight into its possible roles in ecology and adaptation of the host insect. Front. Microbiol. 11:840. doi: 10.3389/fmicb.2020.00840
Moran, N. A. (2015). Genomics of the honey bee microbiome. Curr. Opin. Insect Sci. 10, 22–28. doi: 10.1016/j.cois.2015.04.003
Nadarasah, G., and Stavrinides, J. (2014). Quantitative evaluation of the host-colonizing capabilities of the enteric bacterium Pantoea using plant and insect hosts. Microbiology 160, 602–615. doi: 10.1099/mic.0.073452-0
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Ohbayashi, T., Takeshita, K., Kitagawa, W., Nikoh, N., Koga, R., Meng, X.-Y., et al. (2015). Insect’s intestinal organ for symbiont sorting. Proc. Natl. Acad. Sci. U.S.A. 112, E5179–E5188. doi: 10.1073/pnas.1511454112
Oishi, S., Moriyama, M., Koga, R., and Fukatsu, T. (2019). Morphogenesis and development of midgut symbiotic organ of the stinkbug Plautia stali (Hemiptera: Pentatomidae). Zool. Lett. 5:16. doi: 10.1186/s40851-019-0134-2
Oksanen, J., Blanchet, F. G., Friendly, M., Roeland Kindt, P., Legendre, D. M., Minchin, P. R., et al. (2020). vegan: Community Ecology Package. R package version 2.5-7. Available at: https://CRAN.R-project.org/package=vegan
Oliver, K. M., Degnan, P. H., Hunter, M. S., and Moran, N. A. (2009). Bacteriophages encode factors required for protection in a symbiotic mutualism. Science 325, 992–994. doi: 10.1126/science.1174463
Oliver, K. M., Smith, A. H., and Russell, J. A. (2014). Defensive symbiosis in the real world—advancing ecological studies of heritable, protective bacteria in aphids and beyond. Funct. Ecol. 28, 341–355. doi: 10.1111/1365-2435.12133
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Otero-Bravo, A., Goffredi, S., and Sabree, Z. L. (2018). Cladogenesis and genomic streamlining in extracellular endosymbionts of tropical stink bugs. Genome Biol. Evol. 10, 680–693. doi: 10.1093/gbe/evy033
Otero-Bravo, A., and Sabree, Z. L. (2018). Comparing the utility of host and primary endosymbiont loci for predicting global invasive insect genetic structuring and migration patterns. Biol. Control 116, 10–16. doi: 10.1016/j.biocontrol.2017.04.003
Otero-Bravo, A., and Sabree, Z. L. (2021). Multiple concurrent and convergent stages of genome reduction in bacterial symbionts across a stink bug family. Sci. Rep. 11:7731. doi: 10.1038/s41598-021-86574-8
Pizzolante, G., Durante, M., Rizzo, D., Di Salvo, M., Tredici, S. M., Tufariello, M., et al. (2018). Characterization of two Pantoea strains isolated from extra-virgin olive oil. AMB Express 8:113. doi: 10.1186/s13568-018-0642-z
Prado, S. S., and Almeida, R. P. P. (2009). Role of symbiotic gut bacteria in the development of Acrosternum hilare and Murgantia histrionica. Entomol. Exp. Appl. 132, 21–29. doi: 10.1111/j.1570-7458.2009.00863.x
Prado, S. S., Rubinoff, D., and Almeida, R. P. P. (2006). Vertical transmission of a pentatomid caeca-associated symbiont. Ann. Entomol. Soc. Am. 99, 577–585. doi: 10.1603/0013-8746(2006)99[577:vtoapc]2.0.co;2
R Core Team (2020). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available at: https://www.R-project.org/
Rawlings, N. D., Waller, M., Barrett, A. J., and Bateman, A. (2013). MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 42, D503–D509. doi: 10.1093/nar/gkt953
Raymann, K., Motta, E. V. S., Girard, C., Riddington, I. M., Dinser, J. A., and Moran, N. A. (2018). Imidacloprid decreases honey bee survival rates but does not affect the gut microbiome. Appl. Environ. Microbiol. 84, e00545–e00518. doi: 10.1128/AEM.00545-18
Renoz, F., Ambroise, J., Bearzatto, B., Fakhour, S., Parisot, N., Ribeiro Lopes, M., et al. (2022). The di-symbiotic systems in the aphids Sipha maydis and Periphyllus lyropictus provide a contrasting picture of recent co-obligate nutritional endosymbiosis in aphids. Microorganisms 10:1360. doi: 10.3390/microorganisms10071360
Rider, D. A., Schwertner, C. F., Vilímová, J., Rédei, D., Kment, P., and Thomas, D. B. (2018). “Chapter 2: higher systematics of the Pentatomoidea” in Invasive stink bugs and related species (Pentatomoidea): biology, higher systematics, semiochemistry, and management. ed. J. E. McPherson (Boca Raton, FL: CRC Press), 27–141.
Rodriguez-R, L. M., Gunturu, S., Harvey, W. T., Rosselló-Mora, R., Tiedje, J. M., Cole, J. R., et al. (2018). The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288. doi: 10.1093/nar/gky467
Rouzé, R., Moné, A., Delbac, F., Belzunces, L., and Blot, N. (2019). The honeybee gut microbiota is altered after chronic exposure to different families of insecticides and infection by Nosema ceranae. Microbes Environ. 34, 226–233. doi: 10.1264/jsme2.ME18169
RStudio Team (2019). RStudio: integrated development for R. RStudio, Inc., Boston, MA: Available at: http://www.rstudio.com/
Salem, H., Florez, L., Gerardo, N., and Kaltenpoth, M. (2015). An out-of-body experience: the extracellular dimension for the transmission of mutualistic bacteria in insects. Proc. R. Soc. B 282:20142957. doi: 10.1098/rspb.2014.2957
SAMAC (South African Macadamia Association) (2021). Industry statistics on the South African Macadamia industry 2021. Available at: https://www.samac.org.za/industry-statistics/
Sassera, D., Epis, S., Pajoro, M., and Bandi, C. (2013). Microbial symbiosis and the control of vector-borne pathogens in tsetse flies, human lice, and triatomine bugs. Pathog. Glob. Health 107, 285–292. doi: 10.1179/2047773213Y.0000000109
Sayers, E. W., Cavanaugh, M., Clark, K., Ostell, J., Pruitt, K. D., and Karsch-Mizrachi, I. (2019). GenBank. Nucleic Acids Res. 48, D84–D86. doi: 10.1093/nar/gkz956
Schoeman, P. S. (2019). Stink bug resistance—causes and action plans. (SAMAC pest management technical information sheets). Available at: https://www.samac.org.za/
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Serteyn, L., Ponnet, L., Backus, E. A., and Francis, F. (2020). Characterization of electropenetrography waveforms for the invasive heteropteran pest, Halyomorpha halys, on Vicia faba leaves. Arthropod Plant Interact. 14, 113–126. doi: 10.1007/s11829-019-09722-y
Slatten, B. H., and Larson, A. D. (1967). Mechanism of pathogenicity of Serratia marcescens: I. Virulence for the adult boll weevil. J. Invertebr. Pathol. 9, 78–81. doi: 10.1016/0022-2011(67)90046-8
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Syberg-Olsen, M. J., Garber, A. I., Keeling, P. J., McCutcheon, J. P., and Husnik, F. (2022). Pseudofinder: detection of pseudogenes in prokaryotic genomes. Mol. Biol. Evol. 39:msac153. doi: 10.1093/molbev/msac153
Tada, A., Kikuchi, Y., Hosokawa, T., Musolin, D. L., Fujisaki, K., and Fukatsu, T. (2011). Obligate association with gut bacterial symbiont in Japanese populations of the southern green stinkbug Nezara viridula (Heteroptera: Pentatomidae). Appl. Entomol. Zool. 46, 483–488. doi: 10.1007/s13355-011-0066-6
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Taylor, C., Johnson, V., and Dively, G. (2017). Assessing the use of antimicrobials to sterilize brown marmorated stink bug egg masses and prevent symbiont acquisition. J. Pest. Sci. 90, 1287–1294. doi: 10.1007/s10340-016-0814-z
Taylor, C. M., Coffey, P. L., DeLay, B. D., and Dively, G. P. (2014). The importance of gut symbionts in the development of the brown marmorated stink bug, Halyomorpha halys (Stål). PLoS One 9, –e90312. doi: 10.1371/journal.pone.0090312
Taylor, P. J., Grass, I., Alberts, A. J., Joubert, E., and Tscharntke, T. (2018). Economic value of bat predation services—a review and new estimates from macadamia orchards. Ecosyst. Serv. 30, 372–381. doi: 10.1016/j.ecoser.2017.11.015
Toh, H., Weiss, B. L., Perkin, S. A. H., Yamashita, A., Oshima, K., Hattori, M., et al. (2006). Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res. 16, 149–156. doi: 10.1101/gr.4106106
von Meijenfeldt, F. A. B., Arkhipova, K., Cambuy, D. D., Coutinho, F. H., and Dutilh, B. E. (2019). Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 20:217. doi: 10.1186/s13059-019-1817-x
Whitman, W. B., Chuvochina, M., Hedlund, B. P., Hugenholtz, P., Konstantinidis, K. T., Murray, A., et al. (2022). Development of the SeqCode: a proposed nomenclatural code for uncultivated prokaryotes with DNA sequences as type. Syst. Appl. Microbiol. 45:126305. doi: 10.1016/j.syapm.2022.126305
Wintermantel, D., Locke, B., Andersson, G. K. S., Semberg, E., Forsgren, E., Osterman, J., et al. (2018). Field-level clothianidin exposure affects bumblebees but generally not their pathogens. Nat. Commun. 9:5446. doi: 10.1038/s41467-018-07914-3
Wu, Y.-W., Simmons, B. A., and Singer, S. W. (2015). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Keywords: gut microbiome, stink bug, symbionts, Pantoea, Sodalis, gastric caeca
Citation: Fourie A, Venter SN, Slippers B and Fourie G (2023) Pantoea bathycoeliae sp. nov and Sodalis sp. are core gut microbiome symbionts of the two-spotted stink bug. Front. Microbiol. 14:1284397. doi: 10.3389/fmicb.2023.1284397
Edited by:
Ryuichi Koga, National Institute of Advanced Industrial Science and Technology (AIST), JapanReviewed by:
Yudai Nishide, National Agriculture and Food Research Organization (NARO), JapanCopyright © 2023 Fourie, Venter, Slippers and Fourie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerda Fourie, Z2VyZGExLmZvdXJpZUBmYWJpLnVwLmFjLnph
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.