
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 15 November 2023
Sec. Biology of Archaea
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1281628
This article is part of the Research TopicMolecular Biology of Archaea - 2022View all 19 articles
 Evgenii Protasov1‡
Evgenii Protasov1‡ James O. Nonoh1†‡
James O. Nonoh1†‡ Joana M. Kästle Silva1‡
Joana M. Kästle Silva1‡ Undine S. Mies1
Undine S. Mies1 Vincent Hervé1
Vincent Hervé1 Carsten Dietrich1Kristina Lang1Lena Mikulski1Katja Platt1
Carsten Dietrich1Kristina Lang1Lena Mikulski1Katja Platt1 Anja Poehlein2Tim Köhler-Ramm1Edouard Miambi3
Anja Poehlein2Tim Köhler-Ramm1Edouard Miambi3 Hamadi I. Boga1Christopher Feldewert1
Hamadi I. Boga1Christopher Feldewert1 David K. Ngugi1Rudy Plarre4David Sillam-Dussès5
David K. Ngugi1Rudy Plarre4David Sillam-Dussès5 Jan Šobotník6
Jan Šobotník6 Rolf Daniel2
Rolf Daniel2 Andreas Brune1*
Andreas Brune1*Methane emission by terrestrial invertebrates is restricted to millipedes, termites, cockroaches, and scarab beetles. The arthropod-associated archaea known to date belong to the orders Methanobacteriales, Methanomassiliicoccales, Methanomicrobiales, and Methanosarcinales, and in a few cases also to non-methanogenic Nitrososphaerales and Bathyarchaeales. However, all major host groups are severely undersampled, and the taxonomy of existing lineages is not well developed. Full-length 16S rRNA gene sequences and genomes of arthropod-associated archaea are scarce, reference databases lack resolution, and the names of many taxa are either not validly published or under-classified and require revision. Here, we investigated the diversity of archaea in a wide range of methane-emitting arthropods, combining phylogenomic analysis of isolates and metagenome-assembled genomes (MAGs) with amplicon sequencing of full-length 16S rRNA genes. Our results allowed us to describe numerous new species in hitherto undescribed taxa among the orders Methanobacteriales (Methanacia, Methanarmilla, Methanobaculum, Methanobinarius, Methanocatella, Methanoflexus, Methanorudis, and Methanovirga, all gen. nova), Methanomicrobiales (Methanofilum and Methanorbis, both gen. nova), Methanosarcinales (Methanofrustulum and Methanolapillus, both gen. nova), Methanomassiliicoccales (Methanomethylophilaceae fam. nov., Methanarcanum, Methanogranum, Methanomethylophilus, Methanomicula, Methanoplasma, Methanoprimaticola, all gen. nova), and the new family Bathycorpusculaceae (Bathycorpusculum gen. nov.). Reclassification of amplicon libraries from this and previous studies using this new taxonomic framework revealed that arthropods harbor only CO2 and methyl-reducing hydrogenotrophic methanogens. Numerous genus-level lineages appear to be present exclusively in arthropods, suggesting long evolutionary trajectories with their termite, cockroach, and millipede hosts, and a radiation into various microhabitats and ecological niches provided by their digestive tracts (e.g., hindgut compartments, gut wall, or anaerobic protists). The distribution patterns among the different host groups are often complex, indicating a mixed mode of transmission and a parallel evolution of invertebrate and vertebrate-associated lineages.
Methanogenic archaea play an important role in the fermentative breakdown of organic matter (Müller et al., 2018). They are common constituents of the intestinal microbiota of both invertebrate and vertebrate animals, where they thrive on the products of bacterial fermentations, namely molecular hydrogen, formate, methanol, and methylamines (Brune, 2019; Chibani et al., 2022).
Methane emission by termites was documented half a century ago by the seminal work of Breznak and coworkers (Brune, 2019). Although the phenomenon attracted attention because of its implications for the global methane budget, methane emissions from termites are dwarfed by those from ruminants and wetlands. Subsequent surveys of other invertebrates revealed that methanogenesis is restricted to only a few distinct groups of terrestrial arthropods, namely millipedes, termites and cockroaches (Blattodea), and scarab beetles (Hackstein and Stumm, 1994; Hackstein and van Alen, 2018; Brune, 2019).
Methanogens in arthropod guts are typically restricted to specific hindgut compartments, where they are localized on the cuticular lining, attached to filamentous bacteria on the hindgut wall, and associated with anaerobic protists (ciliates in cockroaches and millipedes; flagellates in all termite families except Termitidae or higher termites) (e.g., Leadbetter and Breznak, 1996; Sprenger et al., 2000). They consist almost exclusively of uncultured representatives, which have been identified in 16S rRNA-based surveys as members of the orders Methanobacteriales (phylum Methanobacteriota), Methanomicrobiales and Methanosarcinales (both phylum “Halobacteriota”), and Methanomassiliicoccales (phylum “Thermoplasmatota”); only a few species of the genera Methanobrevibacter and Methanimicrococcus have been isolated in pure culture (see reviews by Brune, 2018, 2019; Hackstein and van Alen, 2018). Some studies also identified non-methanogenic Bathyarchaeales and Nitrososphaerales (both phylum Thermoproteota) (e.g., Friedrich et al., 2001; Loh et al., 2021).
Despite these efforts, all major host groups are severely undersampled, and the diversity of methanogens in arthropods remains poorly resolved. The reference databases lack resolution because both full-length 16S rRNA gene sequences and genomes of arthropod-associated archaea are scarce. Also, the taxonomy of existing lineages is not well developed, and the names of many taxa are provisional and not validly published, while other taxa are under-classified and require revision (Rinke et al., 2021).
To address these issues, we conducted a phylogenomic analysis of all archaeal genomes from arthropods using the taxonomic framework of the Genome Taxonomy Database (Parks et al., 2021), including a large number of metagenome-assembled genomes (MAGs) from termite guts (85 MAGs from 34 termite species) and diverse, so far undescribed, isolates obtained in our laboratory from cockroaches and millipedes. In parallel, we prepared full-length 16S rRNA gene libraries from more than 70 species of methane-emitting arthropods and incorporated them into the alignment of the SILVA database (version 138), together with full-length sequences from the termite gut metagenomes and unpublished clone libraries from our laboratory. Based on this comprehensive reference database, we reconstructed phylogenetic trees for all archaeal lineages in arthropod guts. In order to revise the taxonomy of the respective lineages, including a number of provisional Candidatus taxa from the literature, we then linked the lineages in the respective phylogenies via the 16S rRNA genes in the genomes, which allowed us to describe new species and higher taxa under the Code of Nomenclature of Prokaryotes Described from Sequence Data (SeqCode) (Hedlund et al., 2022; Whitman et al., 2022). Finally, we reclassified the archaeal 16S rRNA-gene libraries from arthropods guts from this study and selected datasets from the literature to provide an overview of the distribution of archaeal lineages across all host groups at the genus level.
High-throughput sequencing of long-read amplicon libraries of archaeal 16S rRNA genes from the intestinal tract of cockroaches, termites, and millipedes (47 species, Supplementary Tables 1, 4) and hitherto unpublished clone libraries of archaeal 16S rRNA genes from termites and millipedes (15 species, Supplementary Tables 1, 5) substantially expanded literature information on archaeal diversity in methane-emitting arthropods (18 species, Supplementary Table 5). Phylogenetic analysis revealed that the archaeal communities consist mostly of methanogenic archaea of the orders Methanobacteriales, Methanomicrobiales, Methanosarcinales, and Methanomassiliicoccales. In a few species of soil-feeding termites and litter-feeding millipedes, the archaeal communities comprised also non-methanogenic Bathyarchaeales and Nitrososphaerales.
Phylogenomic analysis of 85 archaeal MAGs from gut metagenomes of 34 termite species and 9 genomes of methanogens isolated from 5 millipede and 3 cockroach species revealed that almost all genus-level lineages occurring in arthropods were represented by one or more high-quality genomes (Figure 1 and Supplementary Table 2). Since the relative evolutionary divergence (RED) of several genera considerably exceeded the average values of other genus-level lineages in the respective phyla (Parks et al., 2018; Rinke et al., 2021), we harmonized the taxonomic ranks of the respective lineages by introducing additional genus-level taxa (Figure 2).
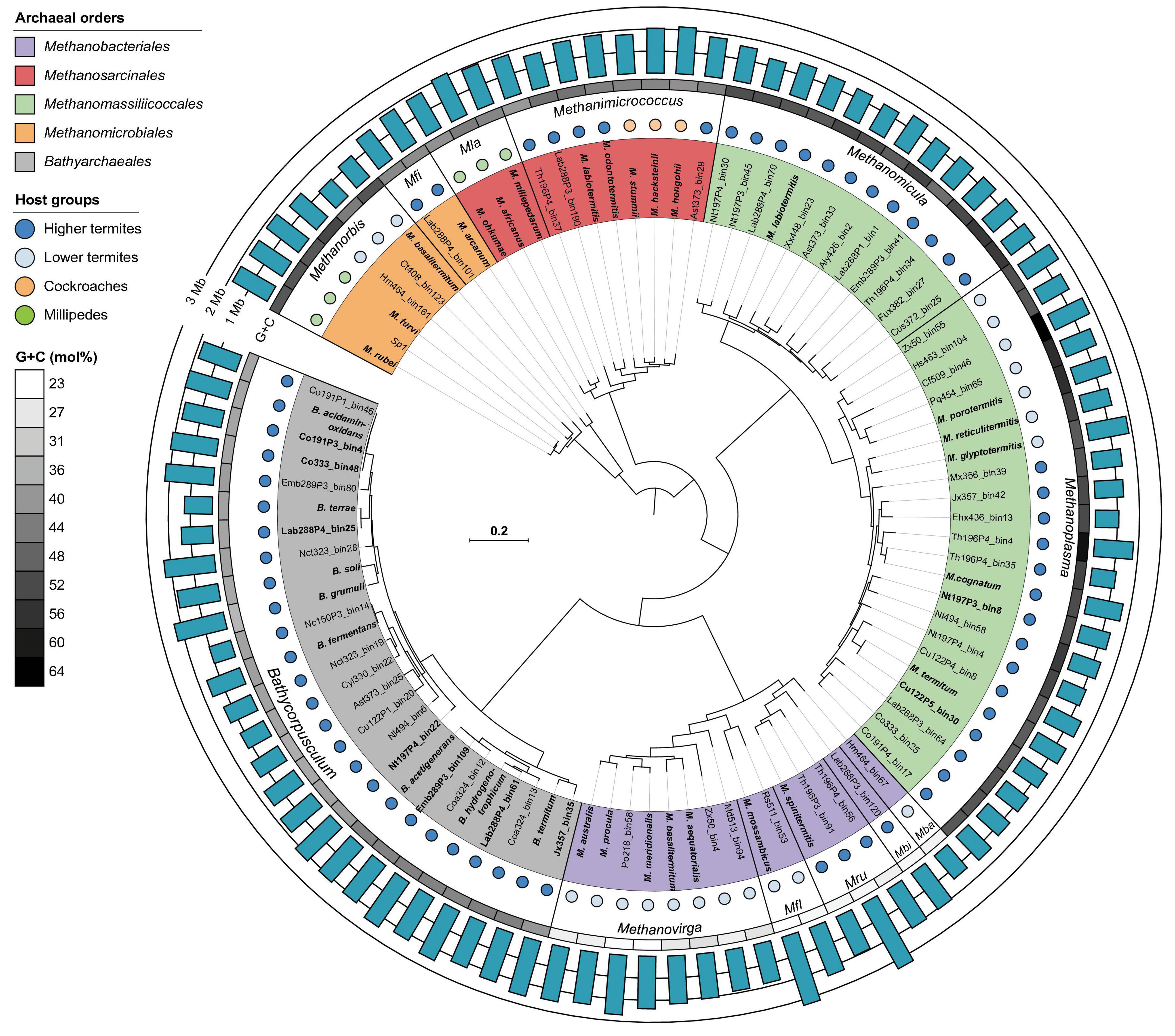
Figure 1. Phylogenomic analysis of archaeal genomes obtained from lower and higher termites, cockroaches, and millipedes. The tree is based on a concatenated alignment of 53 markers and was reconstructed with IQ-TREE under the LG + F + I + G4 model of evolution. High-quality genomes are in bold. For genome accession numbers and other details, see Supplementary Table 2. Mfi, Methanofilum; Mla, Methanolapillus; Mba, Methanobaculum; Mbi, Methanobinarius; Mru, Methanorudis; Mfl, Methanoflexus.
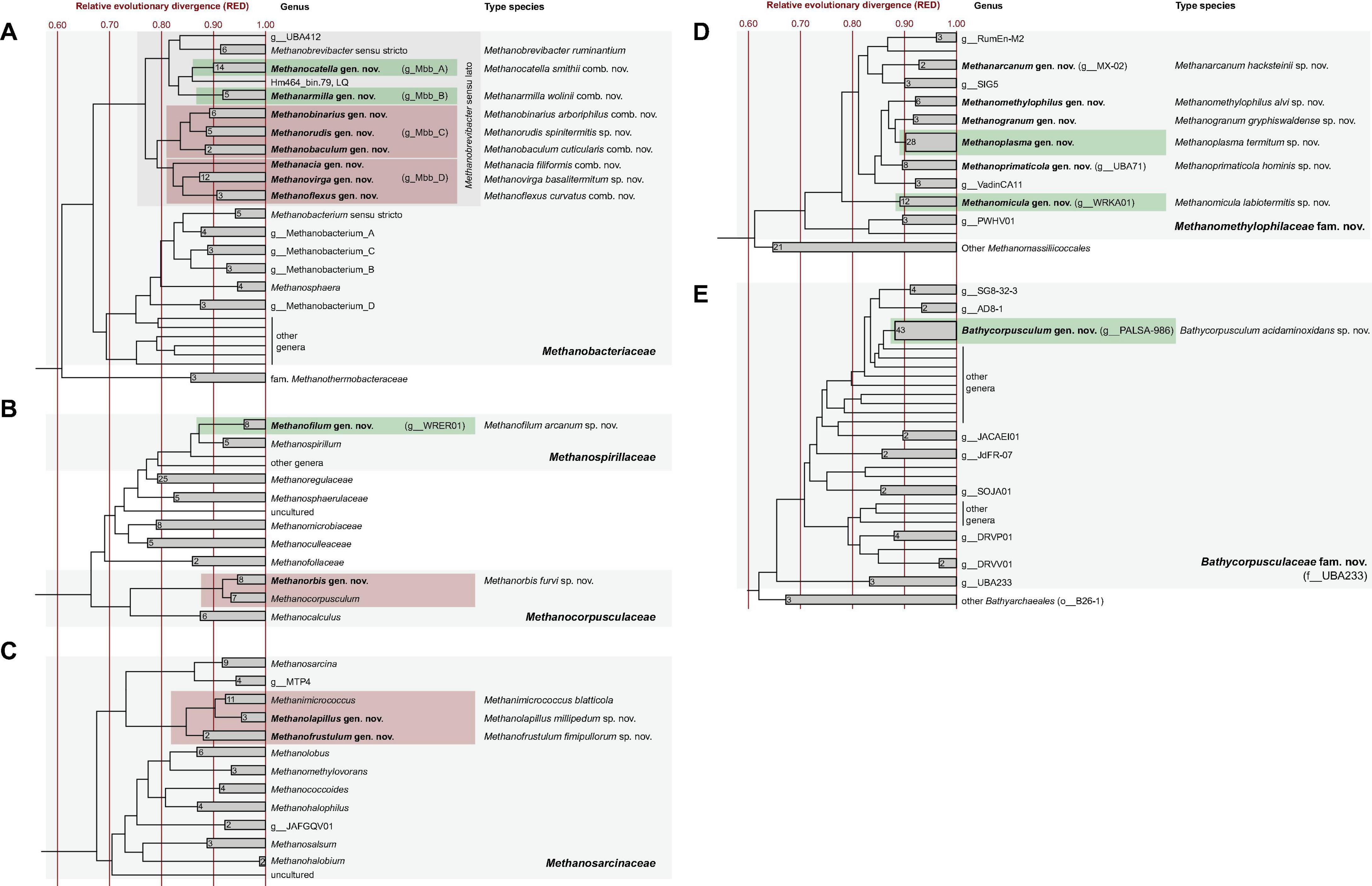
Figure 2. Rank-normalized phylogenies of the archaeal families that harbor isolates or MAGs from arthropod guts (A–E). Taxa with representatives from arthropod guts are highlighted in color; green shading indicates genera that are recognized also in GTDB, red shading indicates those that are expanded in the present study. Newly proposed genera are in bold; the corresponding type species are given. Provisional names in the GTDB taxonomy are in parentheses. The maximum-likelihood tree is based on an alignment of 53 marker genes and was normalized using relative evolutionary divergence (RED) values determined with PhyloRank. The number of genomes in the collapsed clades is indicated. For an expanded version of the original tree, including genome accession numbers, see Supplementary Figure 1.
Using the 16S rRNA genes from the genomic datasets, it was possible to link most clades in the 16S rRNA-based trees to this new taxonomic framework (Figures 3–7). In each order except Nitrososphaerales, the sequences from arthropod guts typically formed one or more lineages that comprised only members of a particular host group, often without cultured representatives. Most lineages had representatives with sequenced genomes of sufficient quality to serve as nomenclatural type for the description of new species under SeqCode (see section “Taxonomy”).
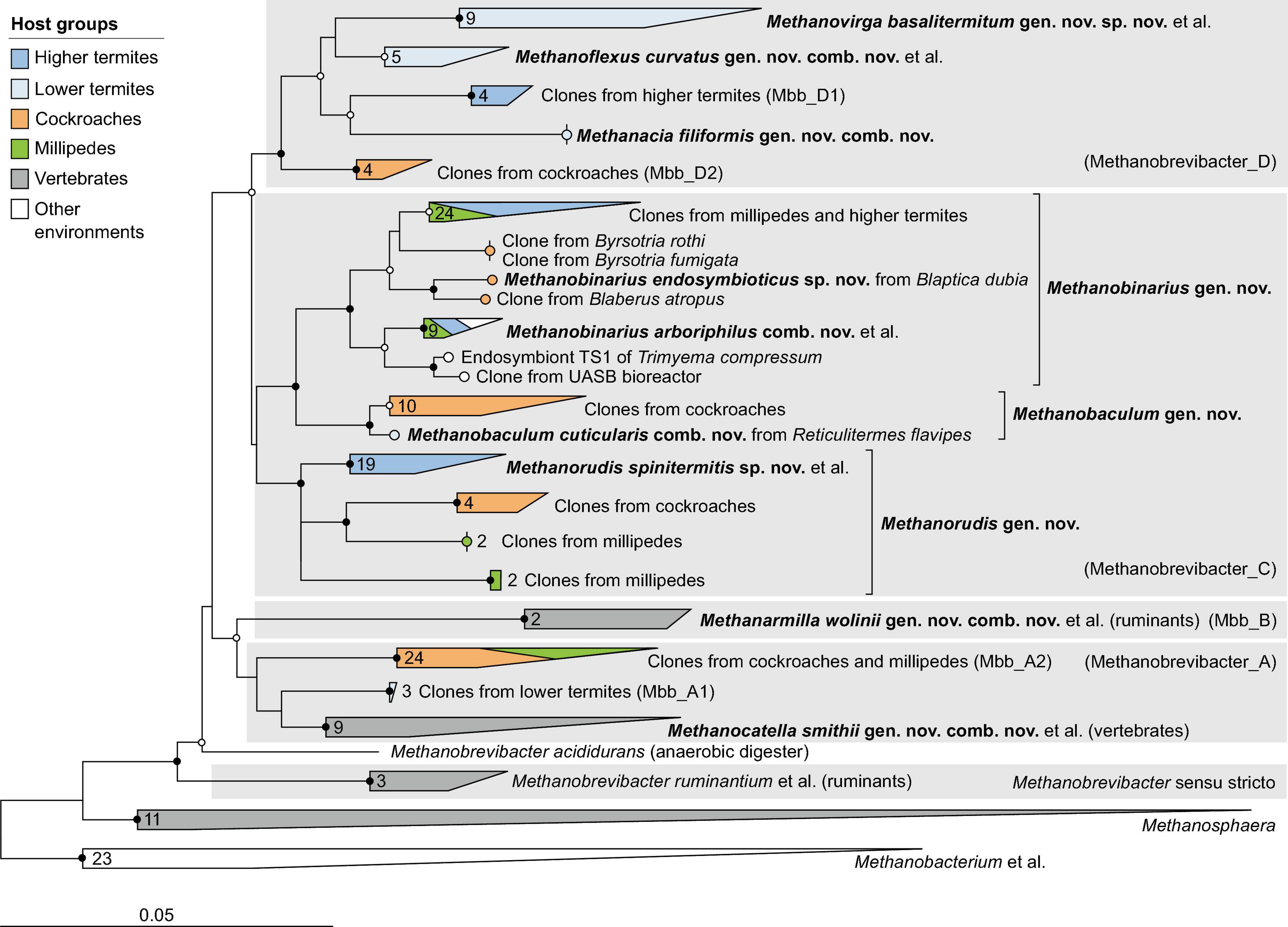
Figure 3. Phylogenetic tree of Methanobacteriaceae, illustrating the position of the sequences from arthropods obtained in this and previous studies. Other Methanobacteriales were used as outgroup. The maximum-likelihood tree is based on a curated alignment of near-full-length 16S rRNA genes (>1,400 sites) and was generated using IQ-TREE under the GTR + I + G4 model of evolution. Bullets on internal nodes indicate SH-aLRT/ultrafast bootstrap support ( , both ≥95/99%; ○, both ≥80/95%; 1,000 replicates each). The scale bar indicates the number of substitutions per site. Color coding indicates host groups. Newly described taxa and their type species are in bold; provisional names in the GTDB taxonomy in parentheses. The number of sequences in the collapsed clades is indicated. A fully expanded version of the tree, including accession numbers, is included in Supplementary Figure 3.
, both ≥95/99%; ○, both ≥80/95%; 1,000 replicates each). The scale bar indicates the number of substitutions per site. Color coding indicates host groups. Newly described taxa and their type species are in bold; provisional names in the GTDB taxonomy in parentheses. The number of sequences in the collapsed clades is indicated. A fully expanded version of the tree, including accession numbers, is included in Supplementary Figure 3.
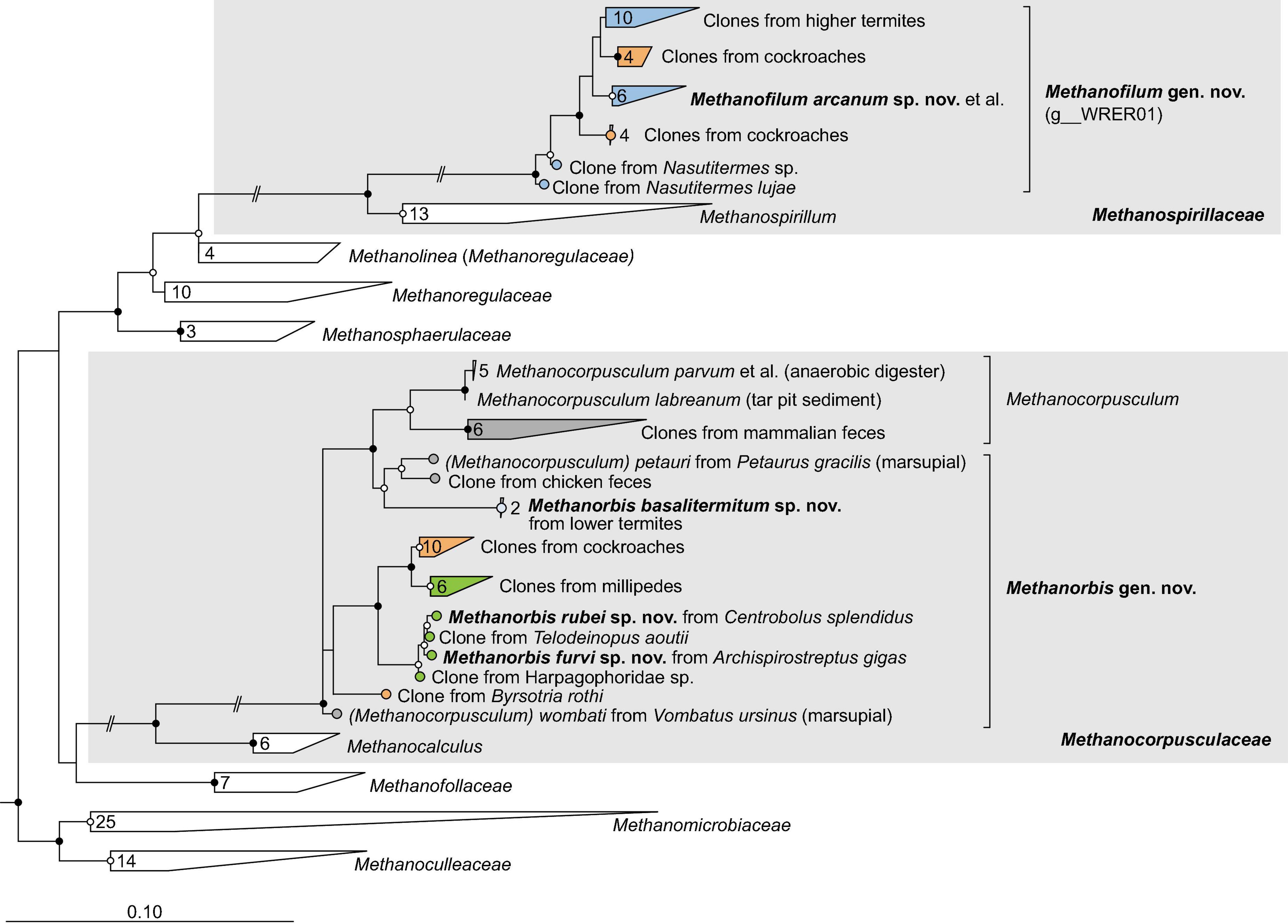
Figure 4. Phylogenetic tree of Methanomicrobiales, illustrating the position of the sequences from arthropods obtained in this and previous studies. Methanocellales were used as outgroup. Color coding and other details are the same as in Figure 1. A fully expanded version of the tree, including accession numbers, is included in Supplementary Figure 4.
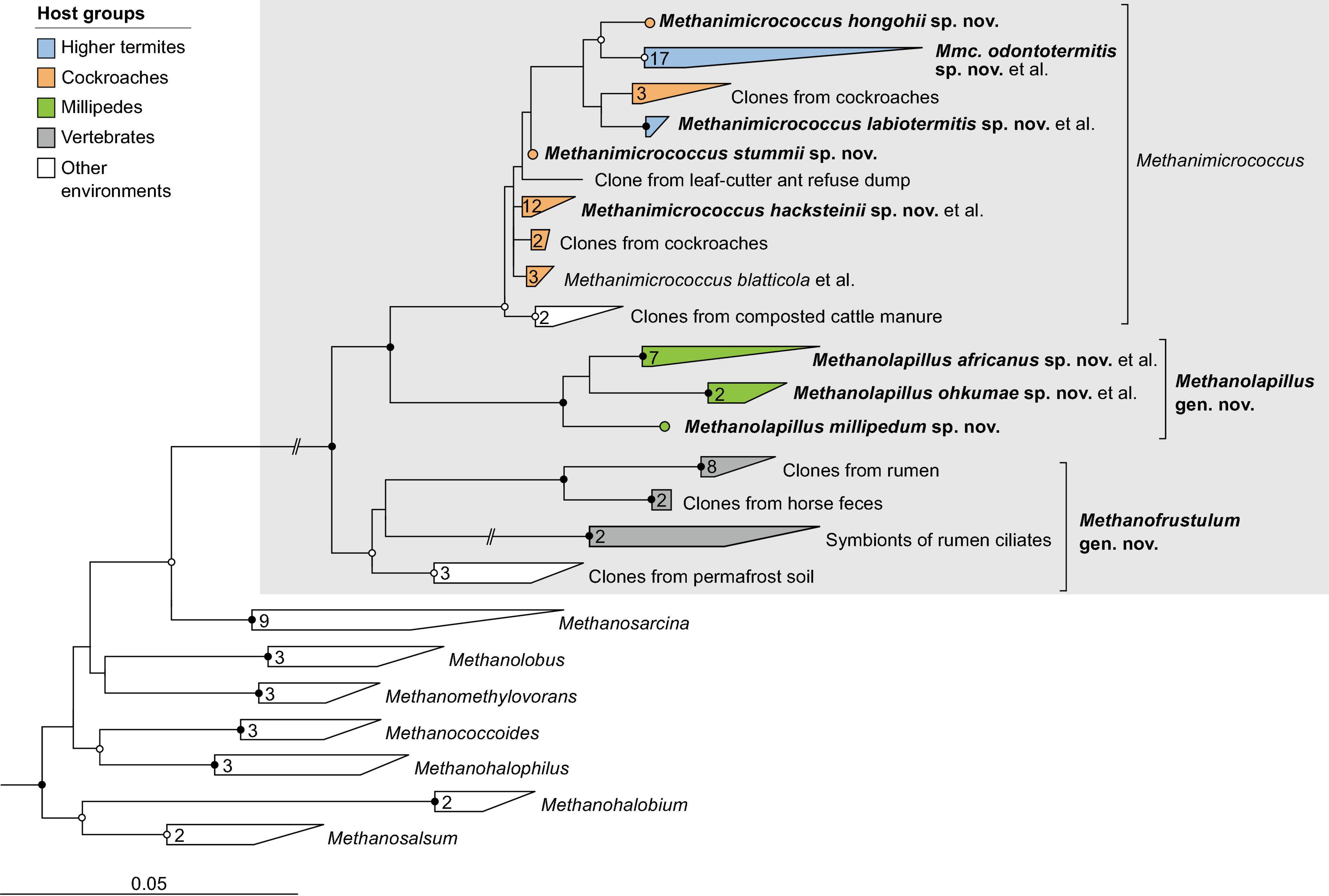
Figure 5. Phylogenetic tree of Methanosarcinaceae illustrating the position of the sequences from arthropods obtained in this and previous studies. Other Methanosarcinales were used as outgroup. Color coding and other details are the same as in Figure 1. A fully expanded version of the tree, including accession numbers, is included in Supplementary Figure 5.
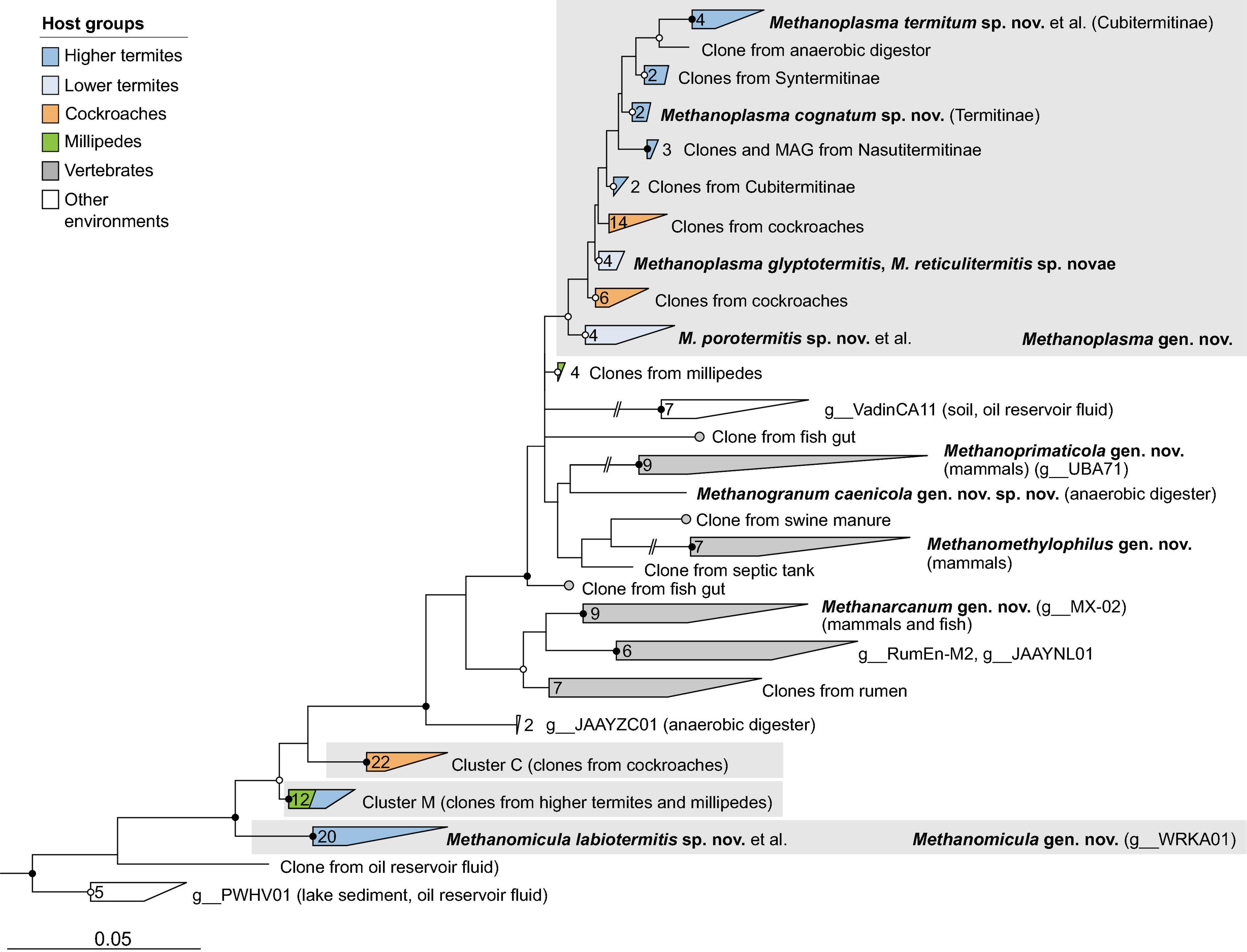
Figure 6. Phylogenetic tree of Methanomethylophilaceae, illustrating the position of the sequences from arthropods obtained in this and previous studies. Other Methanomassiliicoccales were used as outgroup. Color coding and other details are the same as in Figure 1. A fully expanded version of the tree, including accession numbers, is included in Supplementary Figure 6.
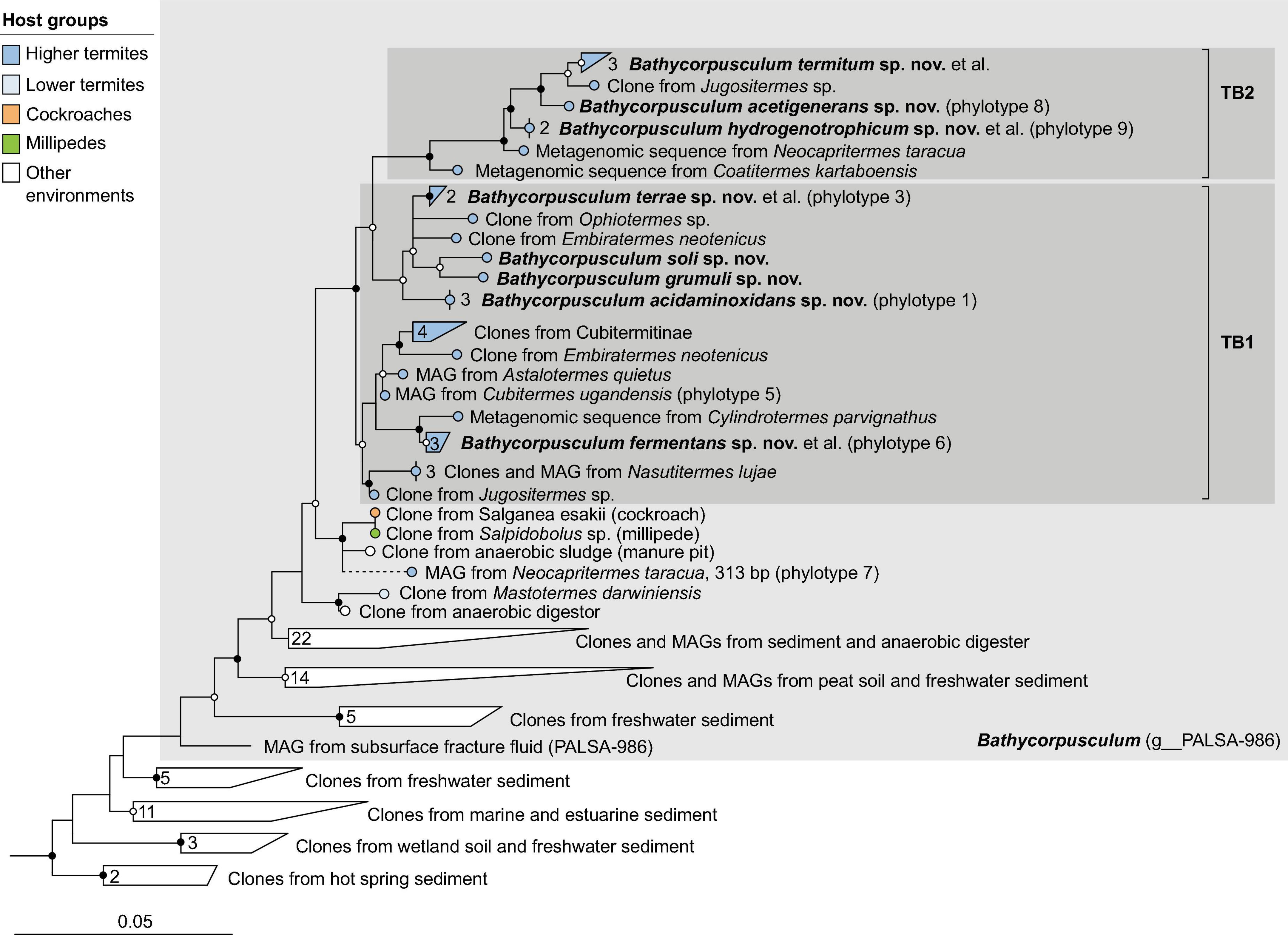
Figure 7. Phylogenetic tree of Bathycorpusculaceae, illustrating the position of the sequences from arthropods obtained in this and previous studies. Other Bathyarchaeales were used as an outgroup. Color coding and other details are the same as in Figure 1. A fully expanded version of the tree, including accession numbers, is included in Supplementary Figure 7.
Members of the order Methanobacteriales are the most common archaeal lineage in the intestinal tract of arthropods. All phylotypes fall within the radiation of the genus Methanobrevibacter sensu lato (Figure 3). Based on the RED values among members of this genus (Rinke et al., 2021), the current Genome Taxonomy Database (GTDB) distinguishes between Methanobrevibacter sensu stricto (which contains the type species, Methanobrevibacter ruminantium) and four additional genus-level lineages, Methanobrevibacter_A to Methanobrevibacter_D (hereafter Mbb_A–D). All sequences from arthropod guts fall into the radiation of Mbb_A, Mbb_C, and Mbb_D; the lineages Methanobrevibacter sensu stricto and Mbb_B are not represented in arthropods.
In the phylogenomic analysis, the new MAGs from termite guts further expanded the evolutionary divergence within the radiation of Mbb_C and Mbb_D, resulting in RED values for the internal nodes that require the introduction of additional genus-level taxa (Figure 2A). In accordance with the taxonomic ranks suggested by Rinke et al. (2021), we propose that the Methanobrevibacter species that do not fall within the radiation of Methanobrevibacter sensu stricto be placed in the new genera Methanocatella (Mbb_A), Methanarmilla (Mbb_B), Methanobaculum, Methanobinarius, and Methanorudis (Mbb_C), and Methanacia, Methanoflexus, and Methanovirga (Mbb_D), using the genomes of previously described species and uncultured archaea as nomenclatural type (see section “Taxonomy”).
The genera Methanocatella and Methanarmilla consist exclusively of isolates or uncultured archaea from the intestinal tract of mammals (Figure 3). Mbb_A comprises a large clade of 16S rRNA sequences from cockroaches and a few millipedes (Mbb_A2) and a smaller clade (Mbb_A1) of sequences from lower termites (Reticulitermes flavipes and Hodotermopsis sjoestedti), which are well separated from the genus Methanocatella but lack representatives with high- or medium-quality genomes. In the phylogenomic analysis, we identified a single low-quality genome (Hm464_bin.79) from the lower termite Hodotermes mossambicus that occupies a sister position to the genus Methanocatella, suggesting that the Mbb_A clade comprises additional genus-level taxa from arthropod guts (Figure 2B).
The remaining genera consist almost exclusively of representatives from the guts of termites, cockroaches, and millipedes (Figure 3). While the genera Methanacia, Methanobaculum, Methanobinarius, and Methanoflexus have cultured representatives, the genera Methanorudis and Methanovirga consist exclusively of uncultured archaea. Two clades in the radiation of Mbb_D that consist exclusively of clones from cockroaches (Mbb_D2) and higher termites (Mbb_D1) lack representatives with sequenced genomes. A few clones from the genus Methanobinarius were not obtained from arthropods guts but were recovered from a sapropelic ciliate or anaerobic bioreactors.
Representatives of the order Methanomicrobiales form several arthropod-specific clusters in the families Methanospirillaceae and Methanocorpusculaceae (Figure 4). The clones that fall into the radiation of Methanospirillaceae form a genus-level lineage that is sister to the genus Methanospirillum (Figure 4). The clade consists exclusively of uncultured methanogens from the intestinal tract of higher termites and several cockroaches. Since the MAGs from termite guts form a well-separated genus-level clade (WRER01 in GTDB) also in the phylogenomic tree (Figure 2B), we propose to classify them in the new genus Methanofilum (see section “Taxonomy”).
The clones that fall into the radiation of Methanocorpusculaceae form several lineages that occupy basal positions to the genus Methanocorpusculum. One lineage consists exclusively of sequences from millipedes, including three isolates from our laboratory (Protasov and Brune, unpublished results). It is loosely affiliated with additional lineages of uncultured representatives from cockroaches, millipedes, and termites. Members of the genus Methanocorpusculum form a well-supported cluster with a lineage of uncultured archaea from mammalian feces. In the phylogenomic analysis, however, only the genomes from mammalian feces fall into the genus Methanocorpusculum, whereas the MAGs from termites and the genomes of millipede isolates form a separate genus-level clade that also includes MAGs from wombat and chicken feces (Figure 2B). We propose to classify the members of this clade in the new genus Methanorbis (see section “Taxonomy”).
In the order Methanosarcinales, most sequences from arthropods guts fall into two genus-level clusters in the family Methanosarcinaceae (Figure 5). One of the clusters contains all representatives from termites and cockroaches, including Methanimicrococcus blatticola isolated from the cockroach Periplaneta americana (Sprenger et al., 2000; Figure 5). The cluster comprises the 16S rRNA genes of several MAGs from higher termites and three isolates from cockroaches that were obtained in our laboratory (Protasov and Brune, unpublished results); we propose to classify them as new species in the genus Methanimicrococcus (see section “Taxonomy”).
The second cluster consists exclusively of representatives from millipede guts, again including three isolates obtained in our laboratory (Protasov and Brune, unpublished results). Since the clade is well separated from the genus Methanimicrococcus also in the phylogenomic analysis (Figure 2C), we propose to classify the isolates as new species in the new genus Methanolapillus (see section “Taxonomy”).
The two clusters are sister to a clade of uncultured archaea from the rumen or feces of mammals, including endosymbionts of rumen ciliates. In the phylogenomic analysis (Figure 2C), the clade is represented by several genomes from ruminants and anaerobic digesters (Campanaro et al., 2020; Xie et al., 2021); we propose to classify these lineages in the new genus Methanofrustulum (see section “Taxonomy”).
With a few exceptions, the Methanomassiliicoccales from arthropod guts are representatives of the so-called intestinal clade, a family-level cluster that comprises several highly enriched cultures but no isolates. Phylogenetic analysis revealed the presence of several arthropod-specific lineages that are well separated from lineages found in the mammalian guts or anaerobic digesters (Figure 6). One of these lineages comprises numerous representatives from the guts of termites, cockroaches, and millipedes, including the previously characterized Candidatus Methanoplasma termitum (Lang et al., 2015). Based on several genomes from lower and higher termites that form a well-separated clade in the phylogenomic tree (Figure 2D), we propose to place the members of this lineage in the new genus Methanoplasma and the new family Methanomethylophilaceae (see section “Taxonomy”).
Three other genus-level clusters that split off at basal nodes in the Methanomethylophilaceae tree consist exclusively of uncultured methanogens from arthropods (Figure 6). One cluster consists exclusively of representatives from cockroaches (cluster C), and another is a mixed cluster comprising sequences from millipedes and higher termites (cluster M). The third cluster consists exclusively of representatives from higher termites, including the 16S rRNA genes of several MAGs. Members of this cluster form a genus-level clade also in the phylogenomic analysis (Figure 2D) and are assigned to the new genus Methanomicula (see section “Taxonomy”).
Members of the class Bathyarchaeia were represented exclusively in higher termites. In the 16S rRNA-based analysis, the clones fall within the radiation of two termite-specific clades previously described as Ca. Termiticorpusculum (TB1) and Ca. Termitimicrobium (TB2) in the recently described Bathyarchaeales (Loh et al., 2021; Khomyakova et al., 2023). Based on the 16S rRNA gene phylogeny, the phylotypes from termite guts represent a monophyletic group among various lineages of uncultured archaea from marine sediments, salt marshes, and anaerobic digesters (Figure 7). Phylogenomic analysis revealed that TB1 and TB2 are polyphyletic and separated by MAGs from hot spring sediments, anaerobic digesters, and permafrost soil (g__PALSA_986 in GTDB; Figure 2E). We propose to place members of the genus PALSA_986 in the new genus Bathycorpusculum, with Bathycorpusculum acidaminoxidans as type species (see section “Taxonomy”).
A small number of sequences from arthropod guts fall within the radiation of Nitrososphaerales, where they cluster with uncultured archaea in the genera Candidatus Nitrosocosmicus and g__UBA10452 (Nitrosophaeraceae) (Supplementary Figure 8). They were absent in most gut samples but were present in low abundance in several humivorous termites, millipedes, and the larva of the scarab beetle Pachnoda ephippiata (Figure 8 and Supplementary Tables 4, 5). In cases where individual compartments were sampled (soil-feeding termites of the genera Amitermes, Isognathotermes, Polyspathotermes, and Ophiotermes, and the humivorous larva of P. ephippiata), the same phylotypes dominated the clone libraries of food soil, nest material, and often also the anterior gut regions (Supplementary Table 3), suggesting that they are transient microbiota and originated from the environment.
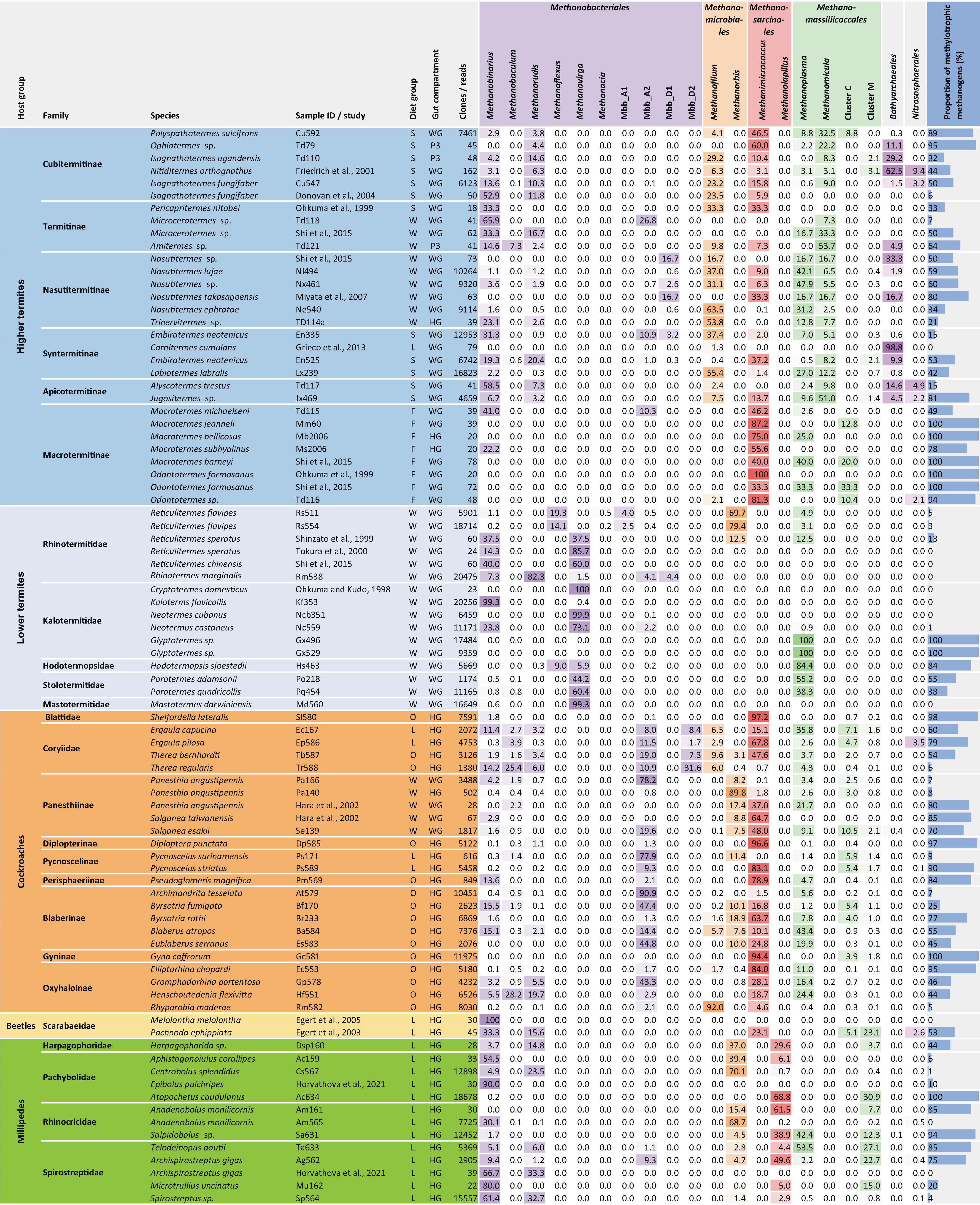
Figure 8. Relative abundance (%) of archaeal taxa in 16S rRNA gene libraries of various arthropods and the proportion of methylotrophs in the methanogenic community. The samples are sorted by host family and represent either whole guts (WG), entire hindguts (HG), or the largest of the proctodeal compartments (P3). Diet groups follow the classification of Arora et al. (2022). The number of clones/reads in the respective library is indicated. For more information and additional samples, see Supplementary Tables 3–5.
We assessed archaeal community structure in methane-emitting arthropods by classifying the 16S rRNA gene libraries obtained in this and previous studies using the phylogenetic framework of our curated reference database (Supplementary Tables 4, 5). A comparison of representative samples from all host groups revealed that the distribution of methanogenic taxa among arthropods is complex (Figure 8).
Members of Methanobacteriales are present in almost all arthropod species investigated but are unevenly distributed among host groups (Figure 8). While the genera Methanobaculum, Methanobinarius, and Methanorudis are present in all host groups, Methanovirga and Methanoflexus are present only in lower termites, and Methanacia only in Reticulitermes spp. The cockroach cluster (Mbb_A2), which is related to the genus Methanocatella, also contains representatives from several termites and millipedes. Although Methanobacteriales dominate the archaeal community in numerous representatives of each host group, they are frequently outnumbered by members of other orders even in closely related hosts. Most striking are the large differences in the occurrence of certain genera between independent samples of the same host species (e.g., Isognathotermes fungifaber, Embiratermes neotenicus, Panesthia angustipennis and Anadenobolus monilicornis), which corroborates that the specificity of both hosts and symbionts for their respective partners is not always strict. The complete absence of Methanobacteriales from the amplicon libraries of the cockroach Gyna caffrorum and two Glyptotermes species is noteworthy (Supplementary Table 4), whereas their absence from several clone libraries should be interpreted with caution because of insufficient sampling depth (Supplementary Table 5).
Representatives of Methanomicrobiales are common in higher termites and millipedes and of lower abundance in cockroaches (Figure 8). Members of the genus Methanorbis occur in millipedes and cockroaches, where they often dominate the archaeal community, but are absent in termites, with the notable exception of several Reticulitermes species. The genus Methanofilum, which occurs in all higher termites and, although in lower abundance, also in many cockroaches, is not encountered in lower termites and millipedes.
The order Methanosarcinales is represented in all host groups except lower termites (Figure 8). Members of the genus Methanimicrococcus are restricted to cockroaches and higher termites, where they often dominate the archaeal community, and the humivorous larva of the scarab beetle P. ephippiata. The genus Methanolapillus occurs exclusively in millipedes, where it frequently represents the predominant lineage of methanogens.
Members of the Methanomassiliicoccales occur in all host groups and can dominate the archaeal community in certain host species (Figure 8). Members of the genus Methanoplasma are found in termites, cockroaches, and millipedes, whereas the genus Methanomicula occurs exclusively in higher termites, typically in high relative abundance. Members of cluster C, which occur at low abundance in most cockroaches, are also found in some Macrotermitinae (a subfamily of fungus-cultivating higher termites), whereas members of cluster M occur in millipedes, cockroaches, and soil-feeding higher termites. Members of both clusters are present in the humivorous larva of the scarab beetle P. ephippiata.
Our comprehensive analysis of the archaeal diversity in the intestinal tract of terrestrial arthropods known to emit methane reveals distinct clades of methanogens from the orders Methanobacteriales, Methanomassiliicoccales, Methanomicrobiales, and Methanosarcinales. Almost all lineages exhibit a high specificity for a particular host group (i.e., termites, cockroaches, or millipedes) and occupy sister positions to lineages from vertebrates, indicating a common evolutionary origin of host-associated methanogens. Linking the 16S rRNA-based diversity data to a phylogenomic analysis of more than 80 archaeal MAGs from termite guts and the genomes of 9 isolates from cockroaches and millipedes allowed the description of novel genera for each order and a taxonomic revision of methanogens and other archaea in arthropod guts.
The first methanogens isolated from arthropod guts were members of the genus Methanobrevibacter (Leadbetter and Breznak, 1996; Leadbetter et al., 1998). Together with the 16S rRNA gene sequences of numerous uncultured representatives recovered from the intestinal tract of arthropods (reviewed in Brune, 2018), they represent lineages distinct from those that colonize the intestinal tract of vertebrates (Figure 3). Earlier studies using 16S rRNA and multi-locus gene sequence analyses had already suggested that the genus Methanobrevibacter is severely underclassified, comprising multiple genus-level clades that apparently coevolved with different host groups (i.e., ruminants, humans, and termites) (Dighe et al., 2004; Poehlein et al., 2018). This notion was then corroborated by a phylogenomic analysis that expanded the taxonomy of archaea to include genomes from uncultured lineages in the phylogenetic framework of GTDB and suggested that the high levels of RED within the genus Methanobrevibacter require the introduction of additional genus-level taxa (Rinke et al., 2021).
Our significantly expanded datasets of 16S rRNA gene sequences and MAGs from arthropod guts underscore the need for taxonomic revision. Based on the RED values of the internal nodes in the radiation of the genus Methanobrevibacter sensu lato, we propose to reclassify all species that do not fall into the M. ruminantium clade (comprising the type species of the genus Methanobrevibacter) into eight new genera: Methanocatella (Mbb_A), Methanarmilla (Mbb_B), Methanobaculum, Methanobinarius, and Methanorudis (Mbb_C), and Methanacia, Methanoflexus, and Methanovirga (Mbb_D) (Figure 2A). The presence of additional arthropod-specific clusters in the radiation of Mbb_A and Mbb_D (Figure 3), which lack representatives with sequenced genomes, suggests the presence of additional genus-level lineages that are candidates for future taxonomic revision. The same is true for the species Methanobrevibacter acididurans, which has no close relatives in public databases and whose genome remains to be sequenced.
Notably, each of the new genera is specific for a particular host group. While members of Methanobrevibacter sensu stricto, Methanocatella, and Methanarmilla are associated with the intestinal tract of ruminants and other vertebrates, all other genera are associated with arthropods and often include subclades restricted to either termites, cockroaches, or millipedes. The genera Methanacia, Methanoflexus, and Methanovirga currently consist exclusively of representatives from termites (Figure 3). The presence of a clade of unclassified phylotypes from higher termites (Mbb_D1) within the radiation of Mbb_D and another clade from cockroaches (Mbb_D2) in sister position to all clades from termites is in agreement with the evolutionary origin of termites among cockroaches (Inward et al., 2007) and a coevolutionary history of the members of the Mbb_D clade with their dictyopteran hosts.
The genera Methanobaculum, Methanobinarius, and Methanorudis also consist of lineages that are specific to particular arthropod host groups. In the genus Methanobinarius, the close relatedness among representatives from distantly related host lineages, i.e., termites (class Insecta) and millipedes (class Myriapoda), suggests an environmental transfer of methanogens between these soil-dwelling arthropods. This is underscored by the presence of Methanobinarius clones in anaerobic bioreactors (Carrillo-Reyes et al., 2014) and in the free-living, anaerobic ciliate Trimyema compressum (Shinzato et al., 2007).
Representatives of the genus Methanosphaera, which are typical for the intestinal tract of mammals (Hoedt et al., 2018; Thomas et al., 2022), were not detected in any arthropod host. Members of the genus Methanobacterium were only rarely encountered, indicating that the strains of Methanobacterium bryantii isolated from several higher termites (Deevong et al., 2004) are not common members of the archaeal microbiota of arthropods.
The order Methanomicrobiales comprises two so far unclassified arthropod-specific clades in the families Methanocorpusculaceae and Methanospirillaceae. In the phylogenomic analysis, the genomes from termites and millipedes are sister to the genus Methanocorpusculum (Figure 2B). A recent phylogenomic analysis revealed that the host-associated members of the genus Methanocorpusculum form two major clades (Volmer et al., 2023). Host clade 2, which is sister to the environmental clade comprising all described species of the genus Methanocorpusculum, consists entirely of uncultured archaea from mammalian feces. Host clade 1 consists of genomes from the feces of chickens and marsupials, including Ca. Methanocorpusculum faecipullorum and two isolates, Methanocorpusculum petauri and Methanocorpusculum wombati (Gilroy et al., 2021; Volmer et al., 2023). Since all members of host clade 1 fall within the radiation of genomes from millipedes and termites and are well separated from the remaining members of the genus Methanocorpusculum (Supplementary Figure 2), we propose to place them in the new genus Methanorbis (see section “Taxonomy”).
Amplicon sequencing of archaea in the intestinal tract of vertebrates has suggested that ancestral members of the genus Methanocorpusculum were present in the last common ancestor of ungulates (Thomas et al., 2022). Our results support a common ancestry of both arthropod-associated and vertebrate-associated Methanocorpusculaceae. Although the genera Methanorbis and Methanocorpusculum are not fully resolved in 16S rRNA-based analyses (Figure 4), phylogenomic analysis places all genomes from arthropods, including all MAGs from termite guts, into the genus Methanorbis (Supplementary Figure 2). The two sister clades of uncultured Methanocorpusculaceae from cockroaches and millipedes that lack representatives with sequenced genomes most likely belong also to the genus Methanorbis. The widespread distribution of Methanorbis in cockroaches and millipedes suggests that the representatives associated with chicken and marsupial feces were acquired by consuming arthropods. So far, it remains open whether the genus Methanocorpusculum, including the environmental clade, originated from a free-living or an arthropod-associated ancestor.
Members of the new genus Methanofilum occur exclusively in cockroaches and higher termites (Figures 4, 8). The absence of Methanospirillaceae in the intestinal tract of all other animals suggests that the genus arose within the order Blattodea, descending from free-living ancestors that occurred in their soil environment. This would mirror the situation with the genus Methanosphaera, which occurs exclusively in vertebrates and presumably evolved from an ancestral lineage of Methanobacteriaceae (Hoedt et al., 2018). The genus Methanomicrobium, which is common in the gut of ruminants (Janssen and Kirs, 2008; Henderson et al., 2015), is not represented in arthropods.
Arthropod-associated members of Methanosarcinales, which were first detected in archaeal clone libraries of higher termites (Ohkuma et al., 1999; Friedrich et al., 2001), cockroaches (Sprenger et al., 2000), and scarab beetle larvae (Egert et al., 2003), are abundant in all host groups except lower termites (Figure 8). The genus Methanimicrococcus comprises all representatives from cockroaches and higher termites, whereas the new genus Methanolapillus harbors those associated with millipedes (Figure 4), suggesting that the lineages in Insecta and Diplopoda evolved independently from each other.
Unlike the sister genus Methanosarcina, whose members have the widest substrate range among methanogens, all isolates and genomes in the host-associated genera Methanimicrococcus and Methanolapillus examined to date are obligately hydrogen-dependent methylotrophs. Genomic analysis of the uncultivated representatives of the mammal-associated genus Methanofrustulum is pending, but all arthropod-associated lineages have lost the methyl branch of the Wood–Ljungdahl pathway and use methanol and methylamines as substrates only in the presence of hydrogen (Sprenger et al., 2007; Thomas et al., 2021; Protasov and Brune, unpublished results).
The arthropod-specific genera occupy a sister position to the new genus Methanofrustulum, whose representatives were first detected in horses but are found also in ruminants and other ungulates (Lwin and Matsui, 2014; Huang et al., 2016), where they have been identified as endosymbionts of rumen ciliates (Regensbogenova et al., 2004). The wide distribution of Methanimicrococcus-related archaea (most likely representing Methanofrustulum) in short-read amplicon libraries of vertebrates (Thomas et al., 2022) suggests parallel evolution of gut-associated Methanosarcinaceae in vertebrates and arthropods.
The order Methanomassiliicoccales consists exclusively of obligately hydrogen-dependent methylotrophs (Zinke et al., 2021). It comprises two major clades that occur in contrasting environments. While members of the so-called environmental clade, represented by the family Methanomassiliicoccaceae, occur predominantly in anoxic soils, sediments, wetlands, and subsurface habitats, members of the so-called intestinal clade are found mostly in the guts of animals (Paul et al., 2012; Söllinger et al., 2016). The family name “Methanomethylophilaceae,” which was proposed based on a highly enriched culture from the human gut (Borrel et al., 2012; Gaci et al., 2014), has only candidate status so far because it contains no members with validly published names. Hence, we formally propose the new genus Methanomethylophilus as the type genus for the new family Methanomethylophilaceae, with the new species Methanomethylophilus alvi as the type species (see section “Taxonomy”). We also propose to include the new genera Methanarcanum, Methanogranum, Methanoplasma, and Methanoprimaticola in this family to accommodate other highly enriched cultures with sequenced genomes that so far have only Candidatus status (Iino et al., 2013; Lang et al., 2015; Weil et al., 2021; Chibani et al., 2022).
The genus Methanoplasma, which occurs exclusively in arthropod guts, belongs to an apical clade of the family Methanomethylophilaceae, with the genera Methanogranum, Methanomethylophilus, and Methanoprimaticola as its closest relatives (Figure 2). The same clade is also represented in the 16S rRNA-based analysis, although the branching order of its members is not fully resolved (Figure 6). Within the genus Methanoplasma, representatives from cockroaches and the phylogenetically older termite families branch more deeply than those from the phylogenetically younger higher termites, suggesting co-evolution between Methanoplasma and its blattodean hosts. An exception is the presence of an apical lineage in millipedes, which was most likely acquired by an environmental transfer from soil-feeding Cubitermitinae.
By contrast, the genus Methanomicula, which occurs exclusively in higher termites, occupies a basal position in the phylogeny of Methanomethylophilaceae (Figure 2). It contains no genomes from cockroaches and millipedes, but both host groups are represented in the 16S-based analyses (Figure 6). As in the case of the genus Methanimicrococcus, Methanomicula is also consistently absent in lower termites (Figure 8). While some older clone libraries described in the literature have been undersampled and suffer from primer bias against Methanomassiliicoccales (see discussion in Paul et al., 2012), the high abundance of Methanoplasma in amplicon libraries of lower termites suggests that the absence of Methanomicula is not an artifact.
Members of the “Bathyarchaeota,” a name coined by Meng et al. (2014) for the Miscellaneous Crenarchaeotal Group (MCG), are presently considered a class-level lineage in the phylum Thermoproteota. The class Bathyarchaeia was formally described only recently, based on its first cultured representative, Bathyarchaeum tardum isolated from the anaerobic sediment of a coastal lake (Khomyakova et al., 2023). The type genus, Bathyarchaeum, and its family, Bathyarchaeceae (formerly BA1), belong to the order Bathyarchaeales (formerly B26-1).
Members of the order Bathyarchaeales, at the time referred to as the “freshwater cluster” of the “Crenarchaeota,” were first detected in arthropod guts in archaeal clone libraries of soil-feeding termites (Friedrich et al., 2001). A genome-centric analysis of these termite-specific lineages identified them as members of the family UBA233 (also referred to as subgroup MCG-6 or Bathy-6) and tentatively assigned them to the candidate taxa “Termiticorpusculum” (TB1) and “Termitimicrobium” (TB2) (Loh et al., 2021). In the taxonomic framework of GTDB, they belong to a single genus-level lineage, described here as the new genus Bathycorpusculum (formerly PALSA-986), which also includes MAGs from peat soils, sediments, and anaerobic digesters, in the new family Bathycorpusculaceae (Figure 2E and Supplementary Figure 7).
Comparative genome analysis of Bathycorpusculum MAGs revealed a purely fermentative metabolism based on amino acids with the potential for reductive acetogenesis from H2 and CO2 (TB1) or possibly methylated compounds (TB2) (Loh et al., 2021) but ruled out the capacity for methanogenesis or alkane oxidation, which had been reported for other lineages of Bathyarchaeia (Evans et al., 2019). Until recently, host-associated Bathyarchaeia had been detected only in higher termites (Friedrich et al., 2001; Grieco et al., 2013; Shi et al., 2015). However, short-read amplicon libraries of archaea in the intestinal tract of animals documented the presence of Bathyarchaeia also in various species of vertebrates (Youngblut et al., 2021; Thomas et al., 2022). Thomas et al. (2022) demonstrated that the short reads from diverse vertebrates cluster in a sister position to representatives of the genus Bathycorpusculum. In contrast to the situation in higher termites, where members of the genus Bathycorpusculum appear to be part of the autochthonous microbiota (Loh et al., 2021), the vertebrate hosts are only distantly related, which supports the hypothesis that they were acquired independently from either host environment or diet (Youngblut et al., 2021; Thomas et al., 2022). Remarkably, amplicon libraries of cockroaches and millipedes also contain rare phylotypes that fall outside the radiation of TB1 and TB2 from termites in a lineage that also includes a clone from a manure pit (Figure 7 and Supplementary Figure 7).
Members of the order Nitrososphaerales were detected in archaeal clone libraries of soil-feeding termites more than 20 years ago (Friedrich et al., 2001). At the time referred to as the “Terrestrial Cluster” of “Crenarchaeota,” they were subsequently placed in the candidate phylum “Thaumarchaeota” (Brochier-Armanet et al., 2008), which was recently described as Nitrososphaerota (Oren and Garrity, 2021). The GTDB taxonomy classifies the class Nitrososphaeria in the phylum Thermoproteota.
Members of the order Nitrosophaerales are aerobic, ammonia-oxidizing archaea and occur in a wide range of marine and terrestrial ecosystems (Pester et al., 2011). In arthropod guts, they were detected in only a few soil or litter-feeding species of all host groups (Figure 8). The phylotypes do not form arthropod-specific lineages but fall into the radiation of uncultured representatives from soils and sediments (Supplementary Figure 8), suggesting that they are part of a transient microbiota taken up from the environment. This is supported by their prevalence in the food soil and midgut of the humivorous larva of P. ephippiata or in the food soil and/or nest material of soil-feeding termites (e.g., Isognathotermes spp., Ophiotermes sp., and Nitiditermes orthognatus), where they are abundant also in the anterior gut compartments but not in the hindgut (P1–P4; Supplementary Table 3). This is consistent with the consumption of nest material by soil-feeding termites (Nalepa et al., 2001).
Using the amoA gene as a functional marker, ammonia-oxidizing archaea have been detected particularly in the guts of soil-feeding termites and humivorous scarab beetle larvae (Majeed et al., 2014; Miambi et al., 2022). Considering the high ammonia concentrations in the guts of these host groups (Ji and Brune, 2006; Ngugi et al., 2011) and the considerable influx of oxygen into the peripheral regions of all gut compartments (Brune et al., 1995), Nitrososphaerales may be metabolically active during gut passage and contribute to the emissions of the greenhouse gas N2O by termites and scarab beetle larvae (Ngugi and Brune, 2012; Brauman et al., 2015).
Methanogens of the genera Methanocella and Methanosarcina were not detected in gut samples of arthropods but in the food soil of Cubitermes fungifaber and P. ephippiata. The latter were also absent in most gut samples of millipedes but highly abundant in their excreta (Šustr et al., 2014). A representative of the genus Methanothrix was detected in the gut of a single sample of I. fungifaber (Cu547). Members of these genera are typical for soil and sediment habitats but do not belong to the autochthonous archaeal microbiota in arthropod guts.
Methanogens colonizing the gut of arthropods reduce either CO2 or methyl groups to methane using hydrogen as electron donor. The former are hydrogenotrophic methanogens from the orders Methanobacteriales and Methanomicrobiales; the latter are obligately hydrogen-dependent methyl reducers from the orders Methanosarcinales and Methanomassiliicoccales. Obligately methyl-reducing Methanosarcinales, represented exclusively by the genera Methanimicrococcus and Methanolapillus (Thomas et al., 2021; Protasov and Brune, unpublished results), and all members of the order Methanomassiliicoccales lack the methyl branch of the Wood–Ljungdahl pathway and are therefore restricted to methylated substrates such as methanol or methylamines (e.g., Borrel et al., 2014; Lang et al., 2015; Söllinger et al., 2016). Members of the genus Methanosphaera, which are obligately methyl-reducing methanogens in the intestinal tract of mammals (Miller and Wolin, 1985; Fricke et al., 2006), are absent in arthropods.
The proportion of methylotrophic lineages in the methanogenic communities of arthropod guts differs substantially among host species (Figure 8). While each host family (and subfamily of higher termites) comprises species that harbor only hydrogenotrophic methanogens, all major host groups comprise representatives with a high abundance of methylotrophs. Even among lower termites, which were previously thought to harbor mainly hydrogenotrophic Methanobacteriales (reviewed by Brune, 2019), several species are heavily colonized by methylotrophs. Since methyl-reducing methanogens will always outcompete CO2-reducing hydrogenotrophs for H2, the abundance of methylotrophs in intestinal tracts is most likely regulated by the availability of methyl groups (Feldewert et al., 2020).
Methyl-disproportioning and aceticlastic methanogens are absent in the intestinal tract of arthropods. Although many members of the order Methanosarcinales can dismutate methyl groups to methane and CO2, their independence from external hydrogen is of little advantage in the intestinal tract of animals, where they are outcompeted by methyl-reducing methanogens owing to their low affinity for methanol and other methylated compounds (Sprenger et al., 2007; Feldewert et al., 2020). The absence of aceticlastic methanogens, however, is more puzzling, since acetate concentrations are much higher in intestinal tracts than in sediments. Although it is generally assumed that the rather slow-growing members of this guild cannot cope with the short residence times of the intestinal contents, it remains enigmatic why they do not avoid washout by attaching to intestinal surfaces or protists (see Brune, 2019).
The intestinal tracts of arthropods are characterized by steep radial gradients of oxygen and hydrogen between gut wall and lumen and strong axial dynamics of these and other physicochemical parameters (Brune, 2014, 2019). Hence, it is not surprising that their archaeal communities are diverse and differ between gut compartments (Friedrich et al., 2001). Hydrogenotrophic Methanobacteriaceae of the genera Methanacia, Methanobaculum, and Methanoflexus have been localized on the chitinous lining of the hindgut wall of termites (Leadbetter and Breznak, 1996; Leadbetter et al., 1998). The methyl-reducing M. blatticola colonizes the same microhabitat in the hindgut of cockroaches (Sprenger et al., 2000). Although attachment to the hindgut wall is thought to prevent washout, it comes at a cost. Hydrogenotrophs attached to the gut wall are not only severely hydrogen-limited but also exposed to the constant influx of oxygen into this microhabitat (Tholen and Brune, 2000). Both Methanobaculum cuticularis and M. blatticola actively remove oxygen from their environment (Leadbetter and Breznak, 1996; Sprenger et al., 2007). While Methanobinarius arboriphilus and M. cuticularis reduce oxygen using H2 via a F420-dependent oxidase (Seedorf et al., 2004; Tholen et al., 2007), the mechanism employed by M. blatticola is unclear.
Association with protists prevents washout and allows methanogens to position themselves in the anoxic lumen of the hindgut, where hydrogen supply is also better than at the gut wall (Brune, 2019). Moreover, most anaerobic protists in the hindgut of arthropods possess hydrogenosomes and provide a stable substrate source for their hydrogenotrophic symbionts. Associations between methanogens and protists are also common in sediments and are regarded as mutualistic because of the cross-feeding of H2 (Fenchel and Finlay, 2018; Treitli et al., 2023).
Ciliates of the genus Nyctotherus, which are found in the gut of cockroaches and millipedes, are commonly colonized by methanogens of the genera Methanobaculum and Methanobinarius, including Methanobinarius endosymbioticus from Nyctotherus ovalis (Gijzen et al., 1991; van Hoek et al., 2000; Lind et al., 2018; Supplementary Figure 3). Free-living relatives of these ciliates in sediments, however, are associated with hydrogenotrophic methanogens of the genera Methanoregula, Methanocorpusculum, Methanoplanus, or Methanobacterium (Fenchel and Finlay, 2018), suggesting that the endosymbionts are not host-specific but were acquired independently from the pool of methanogens present in the respective environment. This is also the case for rumen ciliates, which are associated with a close relative of M. ruminantium (Tokura et al., 1999).
Parabasalid flagellates of lower termites are also frequently associated with methanogens (Odelson and Breznak, 1985; Lee et al., 1987). They were identified as members of the genera Methanovirga and Methanoflexus (Ohkuma et al., 1995; Tokura et al., 2000; Hara et al., 2004; Inoue et al., 2008; Supplementary Figure 3). Both genera occur exclusively in the gut of lower termites, corroborating that flagellate-associated methanogens have also evolved multiple times from free-living lineages in their respective environment.
Although methanogens from arthropods typically form clusters specific for a particular host group, evidence of a co-cladogenesis is not always conclusive. In addition, there are numerous examples of host switching within a given clade (Figure 8). This suggests that the archaeal microbiota in arthropods exhibits a mixed mode of transmission, including both vertical transfer from parents to offspring and environmental exchange (e.g., through predation or co-habitation), as suggested already for the bacterial microbiota of termites (Bourguignon et al., 2018). Environmental transfer would also explain why millipede-associated archaea frequently cluster with those of termites, as millipedes are frequently found in termite nests (Mwabvu, 2005; Choosai et al., 2009).
Many lineages of methanogens present in millipedes, cockroaches, and higher termites are also found in the larva of P. ephippiata (Egert et al., 2003). With the exception of the genus Methanobinarius, all lineages found in the humivorous larva of P. ephippiata are absent in the root-feeding larva of the closely related Melolontha melolontha (Egert et al., 2005). An exchange of methanogens between arthropods that occur in the same habitat (e.g., via the feces) seems likely, especially given the close relatedness of the respective phylotypes (Supplementary Figures 3–7).
Arthropods harbor unique lineages of methanogens from several orders. They comprise both CO2-reducing and methyl-reducing hydrogenotrophs. Some lineages (Methanimicrococcus, Methanolapillus, Methanorbis, Methanomicula, and Methanoplasma) are sister groups of lineages from the intestinal tract of vertebrates, indicating a common evolutionary origin from non-intestinal ancestors, whereas other lineages must have arisen only in arthropods (Methanofilum). The deep-branching phylogenies of each host-associated clade (at least at the genus level) indicate that they have coevolved with their intestinal niches over a long period of time since acquisition from the environment. The occurrence of the same lineages in unrelated host groups suggests the presence of similar ecological niches in the gut of methane-emitting arthropods. However, the reason for the absence of methanogens in all other arthropod groups remains unclear.
Most archaea from the arthropod guts belong to genus-level lineages that are either unclassified or require reclassification. The presence of both high-quality genomes and 16S rRNA gene sequences for most lineages allow the proposal of new taxa under the Code of Nomenclature of Prokaryotes Described from Sequence Data (SeqCode) (Hedlund et al., 2022; Whitman et al., 2022). The new names and new combinations are listed in Table 1, along with the designated nomenclatural type. The authors of previously proposed Candidatus names are assigned as descriptors for the corresponding new taxa. The protologues including etymologies and the full descriptions are given in Supplementary Data File 1). The new isolates will be described also under ICNP in separate publications once genome analysis and phenotypic characterization are completed.
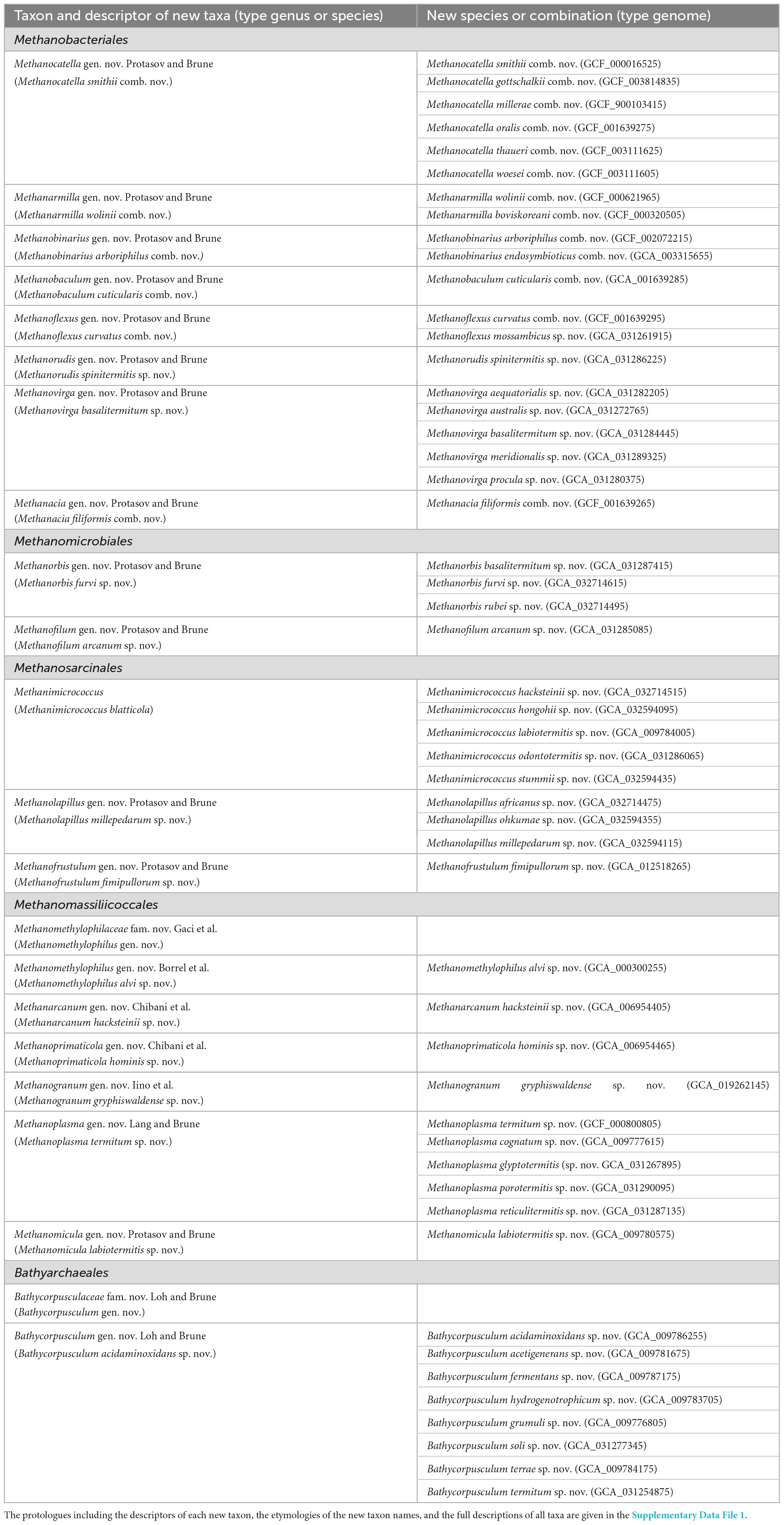
Table 1. New taxa and new combinations of archaea proposed under SeqCode and the designated nomenclatural type.
Termite colonies that were collected in the field were sampled within a week of collection. Samples from termite colonies maintained in other laboratories were processed within a few days after arrival. Species identity was confirmed by comparing their mitochondrial cytochrome oxidase II (COII) gene sequences (Pester and Brune, 2006) with those in public databases. COII gene sequences that were not represented in public databases have been submitted to NCBI GenBank. Cockroaches, beetles, and millipedes were obtained from commercial breeders and dissected within a few days after arrival. Detailed information on all samples is given in Supplementary Table 1.
Specimens were immobilized on ice, decapitated, and dissected with sterile forceps. Whole guts or individual gut sections were pooled and homogenized in phosphate buffer (Köhler et al., 2012; Schauer et al., 2012). The number of specimens included in each sample was adjusted to account for size differences between species (one gut or gut section for millipedes, scarab beetles and cockroaches, 3–10 for termites). DNA for clone libraries was extracted using a bead-beating protocol with subsequent phenol–chloroform purification (Paul et al., 2012). DNA for amplicon libraries was purified using the DNeasy soil kit (Qiagen), which also includes mechanical disruption with zirconium beads, according to the manufacturer’s instructions.
Archaeal 16S rRNA genes were amplified as previously described (Paul et al., 2012), using the archaea-specific forward primer Ar109f (5′-AMDGCTCAGTAACACGT-3′) with either the archaea-specific reverse primer Ar912r (5′-CTCCCCCGCCAATTCCTTTA-3′) or the prokaryote-specific reverse primer 1490R (5′-GGHTACCTTGTTACGACTT-3′). The PCR products were purified using the MinElute PCR Purification Kit (Qiagen) and cloned using the pGEM-T vector kit (Promega). For each library, between 20 and 50 clones with correctly sized inserts were bidirectionally sequenced with M13 primers on a capillary sequencer at GATC-Biotech (Konstanz, Germany).
Barcoded 16S rRNA amplicons were generated in two rounds of PCR. In the first round, 16S rRNA genes were amplified using primers Ar109F and 1490R (see above) tagged with M13 sequences at the 5′ end (M13F 5′-TGTAAAACGACGGCCAGT-3′; M13R 5′-GGAAACAGCTATGACCATG-3′). A 5′ block (5′-NH4-C6) was added to each primer to ensure that no untagged amplicons were carried over into the second PCR. In the second round, samples were multiplexed by attaching unique barcodes (16-mers) to each end of the amplicons using bar-coded M13 forward and reverse primers (Pacific Biosciences).
In both rounds, the PCR conditions followed standard PacBio amplicon generation protocols, except that the HiFi Hot Start DNA Polymerase (Roche Life Science) was replaced with Herculase (Agilent). Round 1: initial denaturation step (92°C for 2 min), followed by 35 cycles of denaturation (94°C for 20 s), annealing (52°C for 30 s), and extension (68°C for 45 s), and a final extension step (68°C for 7 min). Round 2: initial denaturation step (95°C for 3 min), followed by 12 cycles of denaturation (95°C for 30 s), annealing (57°C for 30 s), and extension (72°C for 1 min), and a final extension step (72°C for 7 min).
The barcoded amplicons were purified using AMPure PB beads (Beckman Coulter) following the manufacturer’s protocol, pooled at equimolar concentrations, and ligated with SMRTbell adapters following standard PacBio library preparation protocols. The library was sequenced on a Pacific Biosciences Sequel II platform at the Dresden Genome Center (DGC), Dresden, Germany, using one SMRT 8 M cell with the Sequel II Binding Kit 2.1 containing the Sequel Polymerase 2.0 and with a movie length of 600 min. Circular consensus (CCS) reads were generated using the CCS v. 6.4.0 Bioconda package (pbbioconda, Pacific Biosciences) (Grüning et al., 2018).
Read curation followed the pipeline of Martijn et al. (2017) with adaptations for single 16S rRNA gene sequencing. Briefly, the base-calling confidence of the raw CCS reads was assessed, and sequences with associated read quality scores below 0.99 were removed from the dataset. Sequences were curated using mothur software (Schloss, 2020), first by demultiplexing barcoded amplicons with the trim.seqs command and sorting the samples by host species, followed by removal of primer sequences. Chimerae were removed using the UCHIME package (Edgar et al., 2011) integrated in mothur with our curated reference database (see below). Quality-trimmed reads were clustered into operational taxonomic units (OTUs) at 97% sequence identity level using the VSEARCH tool (Rognes et al., 2016), again using our reference database as a template.
Sequences were imported into the ARB-SILVA database (v. 1381) using the ARB software package (v. 7.02), aligned with the SINA Aligner (v1.2.12) (Ludwig et al., 2004; Pruesse et al., 2012; Yilmaz et al., 2014), and placed into the phylogenetic framework of the guide tree using the ARB parsimony tool. The overall alignment was manually improved using the alignment editor integrated in ARB, taking into account secondary structures. Multiple sequence alignments comprising representative sequences of different archaeal orders or classes were exported with appropriate outgroup sequences. Maximum-likelihood trees were inferred using IQ-TREE 2 with the GTR + I + G4 substitution model suggested by the ModelFinder tool (Kalyaanamoorthy et al., 2017; Minh et al., 2020). Node support was assessed using the Shimodaira–Hasegawa approximate-likelihood ratio test (SH-aLRT) and ultrafast bootstrap analysis (Guindon et al., 2010; Hoang et al., 2018).
In this study, our in-house 16S rRNA reference database was expanded to include archaeal sequences from both host-associated and environmental samples. The current iteration of the Dictyopteran Gut Microbiota Reference Database (DictDb v. 5.1 Archaea) was built upon the framework of the latest release (v. 138.1) of the Silva 16S rRNA database (Quast et al., 2013) and includes only archaea. An extension of the previously published DictDb v. 3.0 (Mikaelyan et al., 2015), which covers only bacterial sequences, will be introduced in an upcoming publication. The curated database was further enriched with near-full-length 16S rRNA sequences from studies targeting archaeal diversity in arthropod guts, both from our research group and from the literature. These include the 16S rRNA gene sequences of our MAGs and metagenomes from termite guts (Hervé et al., 2020), and representative sequences obtained in the present study. We also included the curated 16S rRNA sequences provided by SBDI Sativa (Lundin and Andersson, 2021) to establish robust links to the GTDB taxonomy and the genome-based phylogenies. The curated taxon-specific trees of DictDb were used as sources for the reference alignment and taxonomy files for the analysis of next generation sequencing data with mothur (see above). The database was further enriched by adding sequences that are not included in the reference trees (mostly shorter sequences from this and previous studies) using the parsimony tool implemented in Arb.
High-molecular-weight DNA of pure cultures was isolated with the DNAEasy Blood & Tissue Kit (Qiagen) following the manufacturer’s protocol. The quality of isolated DNA was first checked by agarose gel electrophoresis and validated using an Agilent Bioanalyzer 2100 and the Agilent DNA 12000 kit as recommended by the manufacturer (Agilent Technologies, Waldbronn, Germany). The concentration and purity of the isolated DNA was first estimated with a Nanodrop ND-1000 instrument (PeqLab Erlangen, Germany), and the exact concentration was determined using the Qubit® dsDNA HS Assay kit as recommended by the manufacturer (Life Technologies GmbH, Darmstadt, Germany). Illumina sequencing libraries were prepared using the Nextera XT DNA Sample Preparation kit. To assess the quality and size of the libraries, samples were run on an Agilent Bioanalyzer 2100 using the Agilent High Sensitivity DNA kit according to the manufacturer’s instructions. DNA concentration of the libraries was determined using the Qubit® dsDNA HS Assay kit (Life Technologies GmbH). The libraries were sequenced using a MiSeq system and the reagent kit v3 with 600 cycles as recommended by the manufacturer (Illumina, San Diego, CA, USA). Quality control and quality-filtering of the generated Illumina reads were performed with FastQC v0.11.5 (Andrews, 2010) and Trimmomatic v0.39 (Bolger et al., 2014) using default parameters, respectively. Genomes were assembled with the SPAdes genome assembler software v3.15.2 with default parameters (Bankevich et al., 2012). The quality of the de novo assembly was validated using Qualimap v2.2.1 (García-Alcalde et al., 2012).
The genomes of strains Hf6, Ac7, Am2, and Es2 were additionally sequenced using Nanopore technology. Libraries were prepared with 1.5 μg high-molecular-weight DNA using the Ligation Sequencing lit 1D (SQK-LSK109) and the Native Barcode Expansion kit (EXP-NBD104 and EXP-NBD114) as recommended by the manufacturer (Oxford Nanopore Technologies). Libraries were sequenced for 72 h using a MinION device Mk1B and a SpotON Flow Cell R9.4.1 (Oxford Nanopore Technologies). Basecalling and demultiplexing was done with the MinKNOW software and Guppy in high accuracy mode. The generated reads were quality filtered using fastp v0.23.2 (Chen et al., 2018), and the remaining adapters were removed using porechop v0.2.4.3 Hybrids were assembled using Unicycler v0.5.0 with default settings.
Genes were predicted and the assembled genomes were annotated using Prokka v1.14.5 (Seemann, 2014) with default settings.
Genomes were classified using the GTDB toolkit (GTDB-Tk v2.3.0) with GTDB release 214 as reference (Chaumeil et al., 2022). The alignment of 53 archaeal marker genes generated by the GTDB toolkit was used to infer a phylogenomic tree with IQ-TREE 2 under the LG + F + I + G4 model suggested by modelfinder. Branch support was assessed by ultrafast bootstrap approximation (1,000 replicates). For rank normalization, RED values were calculated from the annotated tree according to Parks et al. (2018) using PhyloRank (v. 1.124). The archaeal phylogenomics tree (Figure 1) was rendered using iTOL v. 6.8 and edited in Inkscape v. 1.0.1 (Letunic and Bork, 2021).
Newly obtained representative OTU sequences were submitted to NCBI GenBank under the accession numbers OP851801–OP852117; OQ724653–OQ724818; OR354372–OR354382, and OR451225–OR451228. Clone library sequences from this study were submitted under the accession numbers OP713915–OP714075 and OR449907–OR449908. Binned small subunit (SSU) sequences extracted from MAGs were submitted under the accession numbers OQ730111–OQ730154; OR140526–OR140534, and OR359878–OR359882. The accession numbers of the new isolates and MAGs are listed in Supplementary Table 2. The Dictyopteran gut reference database (DictDb v. 5.1 Archaea) as Arb file and the accompanying mothur reference files are available on GitHub: https://github.com/brunelab/databases/.
The manuscript presents research on animals that do not require ethical approval for their study.
EP: Conceptualization, Resources, Investigation, Data curation, Formal analysis, Validation, Visualization, Writing – original draft, Writing – review and editing. JON: Conceptualization, Resources, Investigation, Data curation, Formal analysis, Validation, Visualization, Writing – original draft. JMKS: Conceptualization, Methodology, Investigation, Data curation, Formal analysis, Validation, Visualization, Writing – review and editing. USM: Data curation, Formal analysis, Visualization, Writing – review and editing. VH: Data curation, Formal analysis, Visualization, Writing – review and editing. CD: Data curation, Formal analysis, Visualization, Writing – review and editing. KL: Investigation, Formal analysis, Writing – review and editing. LM: Investigation, Formal analysis, Writing – review and editing. KP: Methodology, Investigation, Formal analysis, Writing – review and editing. AP: Methodology, Data curation, Writing – review and editing. TK-R: Data curation, Formal analysis, Writing – review and editing. EM: Resources, Investigation, Formal analysis, Writing – review and editing. HIB: Resources, Investigation, Formal analysis, Writing – review and editing. CF: Resources, Writing – review and editing. DKN: Resources, Writing – review and editing. RP: Resources, Writing – review and editing. DS-D: Resources, Writing – review and editing. JŠ: Resources, Writing – review and editing. RD: Resources, Methodology, Writing – review and editing. AB: Conceptualization, Funding acquisition, Project administration, Supervision, Resources, Data curation, Formal analysis, Validation, Visualization, Writing – original draft, Writing – review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Max Planck Society and a grant of the Deutsche Forschungsgemeinschaft (DFG) in the Collaborative Research Center SFB 987. EP, JMKS, and USM received scholarships from the International Max Planck Research School Principles of Microbial Life: From molecules to cells, from cells to interactions (IMPRS-μLife). JON received a scholarship from the Deutscher Akademischer Austauschdienst (DAAD). CD received a scholarship from the International Max Planck Research School for Environmental, Cellular and Molecular Microbiology (IMPRS-Mic). None of the funding bodies was involved in the design of the study, the collection, analysis, or interpretation of data, or in writing the manuscript.
We thank Kiyoto Maekawa (University of Toyama), Christine Nalepa (North Carolina State University), Rudolf H. Scheffrahn (University of Florida), and Gaku Tokuda (University of the Ryukyus) for insect samples, and Karen Brune for correcting the manuscript. We are grateful to the Kenya Wildlife Service (KWS) for the permission to collect termites from Kenya and to Rudolf H. Scheffrahn for identifying Alyscotermes trestus. We also thank Mechthild Bömeke and Melanie Heinemann for technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1281628/full#supplementary-material
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Cambridge: Babraham Bioinformatics.
Arora, J., Kinjo, Y., Šobotník, J., Buèek, A., Clitheroe, C., Stiblik, P., et al. (2022). The functional evolution of termite gut microbiota. Microbiome 10:78. doi: 10.1186/s40168-022-01258-3
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Borrel, G., Harris, H. M. B., Tottey, W., Mihajlovski, A., Parisot, N., Peyretaillade, E., et al. (2012). Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J. Bacteriol. 194, 6940–6941. doi: 10.1128/JB.01867-12
Borrel, G., Parisot, N., Harris, H. M. B., Peyretaillade, E., Gaci, N., Tottey, W., et al. (2014). Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genomics 15:679. doi: 10.1186/1471-2164-15-679
Bourguignon, T., Lo, N., Dietrich, C., Šobotník, J., Sidek, S., Roisin, Y., et al. (2018). Rampant host switching shaped the termite gut microbiome. Curr. Biol. 28, 649–654. doi: 10.1016/j.cub.2018.01.035
Brauman, A., Majeed, M. Z., Buatois, B., Robert, A., Pablo, A. L., and Miambi, E. (2015). Nitrous oxide (N2O) emissions by termites: Does the feeding guild matter? PLoS One 10:0144340. doi: 10.1371/journal.pone.0144340
Brochier-Armanet, C., Boussau, B., Gribaldo, S., and Forterre, P. (2008). Mesophilic Crenarchaeota: Proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 6, 245–252.
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 12, 168–180. doi: 10.1038/nrmicro3182
Brune, A. (2018). “Methanogens in the digestive tract of termites,” in (Endo)symbiotic Methanogenic Archaea, ed. J. Hackstein (Berlin: Springer), doi: 10.1007/978-3-642-13615-3_6
Brune, A. (2019). “Methanogenesis in the digestive tracts of insects and other arthropods,” in Handbook of Hydrocarbon and Lipid Microbiology, eds A. Stams and D. Sousa (Cham: Springer), doi: 10.1007/978-3-540-77587-4_56
Brune, A., Emerson, D., and Breznak, J. A. (1995). The termite gut microflora as an oxygen sink: Microelectrode determination of oxygen and pH gradients in guts of lower and higher termites. Appl. Environ. Microbiol. 61, 2681–2687.
Campanaro, S., Treu, L., Rodriguez-R, L. M., Kovalovszki, A., Ziels, R. M., Maus, I., et al. (2020). New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol. Biofuels 13, 1–18. doi: 10.1186/s13068-020-01679-y
Carrillo-Reyes, J., Celis, L. B., Alatriste-Mondragón, F., Montoya, L., and Razo-Flores, E. (2014). Strategies to cope with methanogens in hydrogen producing UASB reactors: Community dynamics. Int. J. Hydrogen Energy 39, 11423–11432. doi: 10.1016/j.ijhydene.2014.05.099
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2022). GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316. doi: 10.1093/bioinformatics/btac672
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chibani, C. M., Mahnert, A., Borrel, G., Almeida, A., Werner, A., Brugère, J.-F., et al. (2022). A catalogue of 1,167 genomes from the human gut archaeome. Nat. Microbiol. 7, 48–61. doi: 10.1038/s41564-021-01020-9
Choosai, C., Mathieu, J., Hanboonsong, Y., and Jouquet, P. (2009). Termite mounds and dykes are biodiversity refuges in paddy fields in north-eastern Thailand. Environ. Conserv. 36, 71–79. doi: 10.1017/S0376892909005475
Deevong, P., Hattori, S., Yamada, A., Trakulnaleamsai, S., Ohkuma, M., Noparatnaraporn, N., et al. (2004). Isolation and detection of methanogens from the gut of higher termites. Microbes Environ. 19, 221–226. doi: 10.1264/jsme2.19.221
Dighe, A. S., Jangid, K., González, J. M., Pidiyar, V. J., Patole, M. S., Ranade, D. R., et al. (2004). Comparison of 16S rRNA gene sequences of genus Methanobrevibacter. BMC Microbiol. 4:20. doi: 10.1186/1471-2180-4-20
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Egert, M., Stingl, U., Bruun, L. D., Pommerenke, B., Brune, A., and Friedrich, M. W. (2005). Structure and topology of microbial communities in the major gut compartments of Melolontha melolontha larvae (Coleoptera: Scarabaeidae). Appl. Environ. Microbiol. 71, 4556–4566. doi: 10.1128/AEM.71.8.4556-4566.2005
Egert, M., Wagner, B., Lemke, T., Brune, A., and Friedrich, M. W. (2003). Microbial community structure in midgut and hindgut of the humus-feeding larva of Pachnoda ephippiata (Coleoptera: Scarabaeidae). Appl. Environ. Microbiol. 69, 6659–6668. doi: 10.1128/AEM.69.11.6659-6668.2003
Evans, P. N., Boyd, J. A., Leu, A. O., Woodcroft, B. J., Parks, D. H., Hugenholtz, P., et al. (2019). An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 17, 219–232. doi: 10.1038/s41579-018-0136-7
Feldewert, C., Lang, K., and Brune, A. (2020). The hydrogen threshold of obligately methyl-reducing methanogens. FEMS Microbiol. Lett. 367:fnaa137. doi: 10.1093/femsle/fnaa137
Fenchel, T., and Finlay, B. J. (2018). “Free-living protozoa with endosymbiotic methanogens,” in (Endo) symbiotic methanogenic archaea, ed. J. Hackstein (Berlin: Springer), 1–11.
Fricke, W. F., Seedorf, H., Henne, A., Krüer, M., Liesegang, H., Hedderich, R., et al. (2006). The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J. Bacteriol. 188, 642–658. doi: 10.1128/JB.188.2.642-658.2006
Friedrich, M. W., Schmitt-Wagner, D., Lueders, T., and Brune, A. (2001). Axial differences in community structure of Crenarchaeota and Euryarchaeota in the highly compartmentalized gut of the soil-feeding termite Cubitermes orthognathus. Appl. Environ. Microbiol. 67, 4880–4890. doi: 10.1128/AEM.67.10.4880-4890.2001
Gaci, N., Borrel, G., Tottey, W., O’Toole, P. W., and Brugére, J. F. (2014). Archaea and the human gut: New beginning of an old story. World J. Gastroenterol. 20, 16062–16078. doi: 10.3748/wjg.v20.i43.16062
García-Alcalde, F., Okonechnikov, K., Carbonell, J., Cruz, L. M., Götz, S., Tarazona, S., et al. (2012). Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679. doi: 10.1093/bioinformatics/bts503
Gijzen, H. J., Broers, C. A. M., Barughare, M., and Stumm, C. K. (1991). Methanogenic bacteria as endosymbionts of the ciliate Nyctotherus ovalis in the cockroach hindgut. Appl. Environ. Microbiol. 57, 1630–1634. doi: 10.1128/aem.57.6.1630-1634.1991
Gilroy, R., Ravi, A., Getino, M., Pursley, I., Horton, D. L., Alikhan, N. F., et al. (2021). Extensive microbial diversity within the chicken gut microbiome revealed by metagenomics and culture. PeerJ 9, 1–142. doi: 10.7717/peerj.10941
Grieco, M. A. B., Cavalcante, J. J., Cardoso, A. M., Vieira, R. P., Machado, E. A., Clementino, M. M., et al. (2013). Microbial community diversity in the gut of the South American termite Cornitermes cumulans (Isoptera: Termitidae). Microb. Ecol. 65, 197–204.
Grüning, B., Dale, R., Sjödin, A., Rowe, J., Chapman, B. A., Tomkins-Tinch, C. H., et al. (2018). Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475–476. doi: 10.1038/s41592-018-0046-7
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321.
Hackstein, J. H. P., and Stumm, C. K. (1994). Methane production in terrestrial arthropods. Proc. Natl. Acad. Sci. U. S. A. 91, 5441–5445. doi: 10.1073/pnas.91.12.5441
Hackstein, J. H. P., and van Alen, T. A. (2018). “Methanogens in the gastro-intestinal tract of animals,” in (Endo)symbiotic Methanogenic Archaea, ed. J. H. P. Hackstein (Berlin: Springer), doi: 10.1007/978-3-642-13615-3_8
Hara, K., Shinzato, N., Oshima, T., and Yamagishi, A. (2004). Endosymbiotic Methanobrevibacter species living in symbiotic protists of the termite Reticulitermes speratus detected by fluorescent in situ hybridization. Microbes Environ. 19, 120–127. doi: 10.1264/jsme2.19.120
Hedlund, B. P., Chuvochina, M., Hugenholtz, P., Konstantinidis, K. T., Murray, A. E., Palmer, M., et al. (2022). SeqCode: a nomenclatural code for prokaryotes described from sequence data. Nat. Microbiol. 7, 1702–1708. doi: 10.1038/s41564-022-01214-9
Henderson, G., Cox, F., Ganesh, S., Jonker, A., Young, W., Janssen, P. H., et al. (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 5:14567. doi: 10.1038/srep14567
Hervé, V., Liu, P., Dietrich, C., Sillam-Dussès, D., Stiblik, P., Šobotník, J., et al. (2020). Phylogenomic analysis of 589 metagenome-assembled genomes encompassing all major prokaryotic lineages from the gut of higher termites. PeerJ 8:e8614.
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hoedt, E. C., Parks, D. H., Volmer, J. G., Rosewarne, C. P., Denman, S. E., McSweeney, C. S., et al. (2018). Culture- and metagenomics-enabled analyses of the Methanosphaera genus reveals their monophyletic origin and differentiation according to genome size. ISME J. 12, 2942–2953. doi: 10.1038/s41396-018-0225-7
Huang, X. D., Martinez-Fernandez, G., Padmanabha, J., Long, R., Denman, S. E., and McSweeney, C. S. (2016). Methanogen diversity in indigenous and introduced ruminant species on the Tibetan plateau. Archaea 2016:5916067. doi: 10.1155/2016/5916067
Iino, T., Tamaki, H., Tamazawa, S., Ueno, Y., Ohkuma, M., Suzuki, K. I., et al. (2013). Candidatus Methanogranum caenicola: A novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microbes Environ. 28, 244–250. doi: 10.1264/jsme2.ME12189
Inoue, J. I., Noda, S., Hongoh, Y., Ui, S., and Ohkuma, M. (2008). Identification of endosymbiotic methanogen and ectosymbiotic spirochetes of gut protists of the termite Coptotermes formosanus. Microbes Environ. 23, 94–97. doi: 10.1264/jsme2.23.94
Inward, D., Beccaloni, G., and Eggleton, P. (2007). Death of an order: A comprehensive molecular phylogenetic study confirms that termites are eusocial cockroaches. Biol. Lett. 3, 331–335. doi: 10.1098/rsbl.2007.0102
Janssen, P. H., and Kirs, M. (2008). Structure of the archaeal community of the rumen. Appl. Environ. Microbiol. 74, 3619–3625. doi: 10.1128/AEM.02812-07
Ji, R., and Brune, A. (2006). Nitrogen mineralization, ammonia accumulation, and emission of gaseous NH3 by soil-feeding termites. Biogeochemistry 78, 267–283. doi: 10.1007/s10533-005-4279-z
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Khomyakova, M. A., Merkel, A. Y., Mamiy, D. D., Klyukina, A. A., and Slobodkin, A. I. (2023). Phenotypic and genomic characterization of Bathyarchaeum tardum gen. nov., sp. nov., a cultivated representative of the archaeal class Bathyarchaeia. Front. Microbiol. 14:1214631. doi: 10.3389/fmicb.2023.1214631
Köhler, T., Dietrich, C., Scheffrahn, R. H., and Brune, A. (2012). High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Appl. Environ. Microbiol. 78, 4691–4701. doi: 10.1128/aem.00683-12
Lang, K., Schuldes, J., Klingl, A., Poehlein, A., Daniel, R., and Brune, A. (2015). New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of “Candidatus Methanoplasma termitum.” Appl. Environ. Microbiol. 81, 1338–1352. doi: 10.1128/AEM.03389-14
Leadbetter, J. R., and Breznak, J. A. (1996). Physiological ecology of Methanobrevibacter cuticularis sp. nov. and Methanobrevibacter curvatus sp. nov., isolated from the hindgut of the termite Reticulitermes flavipes. Appl. Environ. Microbiol. 62, 3620–3631. doi: 10.1128/aem.62.10.3620-3631.1996
Leadbetter, J. R., Crosby, L. D., and Breznak, J. A. (1998). Methanobrevibacter filiformis sp. nov., a filamentous methanogen from termite hindguts. Arch. Microbiol. 169, 287–292.
Lee, M. J., Schreurs, P. J., Messer, A. C., and Zinder, S. H. (1987). Association of methanogenic bacteria with flagellated protozoa from a termite hindgut. Curr. Microbiol. 15, 337–341. doi: 10.1007/BF01577591
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lind, A. E., Lewis, W. H., Spang, A., Guy, L., Embley, T. M., and Ettema, T. J. G. (2018). Genomes of two archaeal endosymbionts show convergent adaptations to an intracellular lifestyle. ISME J. 12, 2655–2667. doi: 10.1038/s41396-018-0207-9
Loh, H. Q., Hervé, V., and Brune, A. (2021). Metabolic potential for reductive acetogenesis and a novel energy-converting [NiFe] hydrogenase in Bathyarchaeia from termite guts – a genome-centric analysis. Front. Microbiol. 11:635786. doi: 10.3389/fmicb.2020.635786
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, A., et al. (2004). ARB: A software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Lundin, D., and Andersson, A. (2021). SBDI Sativa curated 16S GTDB database. SciLifeLab. Dataset. doi: 10.17044/scilifelab.14869077.v3
Lwin, K. O., and Matsui, H. (2014). Comparative analysis of the methanogen diversity in horse and pony by using mcrA gene and archaeal 16S rRNA gene clone libraries. Archaea 2014, 1–11. doi: 10.1155/2014/483574
Majeed, M. Z., Miambi, E., Barois, I., Randriamanantsoa, R., Blanchart, E., and Brauman, A. (2014). Contribution of white grubs (Scarabaeidae: Coleoptera) to N2O emissions from tropical soils. Soil Biol. Biochem. 75, 37–44. doi: 10.1016/j.soilbio.2014.03.025
Martijn, J., Lind, A. E., Spiers, I., Juzokaite, L., Bunikis, I., Vinnere Pettersson, O., et al. (2017). Amplicon sequencing of the 16S-ITS-23S rRNA operon with long-read technology for improved phylogenetic classification of uncultured prokaryotes. BioRxiv [Preprint]. doi: 10.1101/234690
Meng, J., Xu, J., Qin, D., He, Y., Xiao, X., and Wang, F. (2014). Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses. ISME J. 8, 650–659. doi: 10.1038/ismej.2013.174
Miambi, E., Jusselme, T. M. D., Châtelliers, C. C., des, Robert, A., Delort, A., et al. (2022). Potential gross and net N2O production by the gut of different termite species are related to the abundance of nitrifier and denitrifier groups. Front. Microbiomes 1:1017006. doi: 10.3389/frmbi.2022.1017006
Mikaelyan, A., Köhler, T., Lampert, N., Rohland, J., Boga, H., Meuser, K., et al. (2015). Classifying the bacterial gut microbiota of termites and cockroaches: A curated phylogenetic reference database (DictDb). Syst. Appl. Microbiol. 38, 472–482. doi: 10.1016/j.syapm.2015.07.004
Miller, T. L., and Wolin, M. J. (1985). Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch. Microbiol. 141, 116–122. doi: 10.1007/BF00423270
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Müller, N., Timmers, P., Plugge, C. M., Stams, A. J., and Schink, B. (2018). “Syntrophy in methanogenic degradation,” in (Endo)symbiotic methanogenic archaea, ed. J. H. Hackstein (Heidelberg: Springer), doi: 10.1007/978-3-642-13615-3
Mwabvu, T. (2005). The density and distribution of millipedes on termite mounds in miombo woodland, Zimbabwe. Afr. J. Ecol. 6, 101–103. doi: 10.1016/s1468-1641(10)60322-2
Nalepa, C. A., Bignell, D. E., and Bandi, C. (2001). Detritivory, coprophagy, and the evolution of digestive mutualisms in Dictyoptera. Insectes Soc. 48, 194–201. doi: 10.1007/PL00001767
Ngugi, D. K., and Brune, A. (2012). Nitrate reduction, nitrous oxide formation, and anaerobic ammonia oxidation to nitrite in the gut of soil-feeding termites (Cubitermes and Ophiotermes spp.). Environ. Microbiol. 14, 860–871. doi: 10.1111/j.1462-2920.2011.02648.x
Ngugi, D. K., Ji, R., and Brune, A. (2011). Nitrogen mineralization, denitrification, and nitrate ammonification by soil-feeding termites: A15N-based approach. Biogeochemistry 103, 355–369. doi: 10.1007/s10533-010-9478-6
Odelson, D. A., and Breznak, J. A. (1985). Nutrition and growth characteristics of Trichomitopsis termopsidis, a cellulolytic protozoan from termites. Appl. Environ. Microbiol. 49, 614–621. doi: 10.1128/aem.49.3.614-621.1985
Ohkuma, M., Noda, S., Horikoshi, K., and Kudo, T. (1995). Phylogeny of symbiotic methanogens in the gut of the termite Reticulitermes speratus. FEMS Microbiol. Lett. 134, 45–50. doi: 10.1016/0378-1097(95)00379-J
Ohkuma, M., Noda, S., and Kudo, T. (1999). Phylogenetic relationships of symbiotic methanogens in diverse termites. FEMS Microbiol. Lett. 171, 147–153. doi: 10.1016/S0378-1097(98)00593-X
Oren, A., and Garrity, G. M. (2021). Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 71:005056.
Parks, D. H., Chuvochina, M., Rinke, C., Mussig, A. J., Chaumeil, P., and Hugenholtz, P. (2021). GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 202, 1–10. doi: 10.1093/nar/gkab776
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36:996. doi: 10.1038/nbt.4229
Paul, K., Nonoh, J. O., Mikulski, L., and Brune, A. (2012). Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Appl. Environ. Microbiol. 78, 8245–8253. doi: 10.1128/aem.02193-12
Pester, M., and Brune, A. (2006). Expression profiles of fhs (FTHFS) genes support the hypothesis that spirochaetes dominate reductive acetogenesis in the hindgut of lower termites. Environ. Microbiol. 8, 1261–1270. doi: 10.1111/j.1462-2920.2006.01020.x
Pester, M., Schleper, C., and Wagner, M. (2011). The Thaumarchaeota: An emerging view of their phylogeny and ecophysiology. Curr. Opin. Microbiol. 14, 300–306. doi: 10.1016/j.mib.2011.04.007
Poehlein, A., Schneider, D., Soh, M., Daniel, R., and Seedorf, H. (2018). Comparative genomic analysis of members of the genera Methanosphaera and Methanobrevibacter reveals distinct clades with specific potential metabolic functions. Archaea 2018:7609847. doi: 10.1155/2018/7609847
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Regensbogenova, M., McEwan, N. R., Javorsky, P., Kisidayova, S., Michalowski, T., Newbold, C. J., et al. (2004). A re-appraisal of the diversity of the methanogens associated with the rumen ciliates. FEMS Microbiol. Lett. 238, 307–313. doi: 10.1016/j.femsle.2004.07.049
Rinke, C., Chuvochina, M., Mussig, A. J., Chaumeil, P.-A., Davín, A. A., Waite, D. W., et al. (2021). A standardized archaeal taxonomy for the Genome Taxonomy Database. Nat. Microbiol. 6, 946–959. doi: 10.1038/s41564-021-00918-8
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 1–22. doi: 10.7717/peerj.2584
Schauer, C., Thompson, C. L., and Brune, A. (2012). The bacterial community in the gut of the cockroach Shelfordella lateralis reflects the close evolutionary relatedness of cockroaches and termites. Appl. Environ. Microbiol. 78, 2758–2767. doi: 10.1128/aem.07788-11
Seedorf, H., Dreisbach, A., Hedderich, R., Shima, S., and Thauer, R. K. (2004). F420H2 oxidase (FprA) from Methanobrevibacter arboriphilus, a coenzyme F420-dependent enzyme involved in O2 detoxification. Arch. Microbiol. 182, 126–137. doi: 10.1007/s00203-004-0675-3
Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shi, Y., Huang, Z., Han, S., Fan, S., and Yang, H. (2015). Phylogenetic diversity of Archaea in the intestinal tract of termites from different lineages. J. Basic Microbiol. 55, 1021–1028.
Shinzato, N., Watanabe, I., Meng, X. Y., Sekiguchi, Y., Tamaki, H., Matsui, T., et al. (2007). Phylogenetic analysis and fluorescence in situ hybridization detection of archaeal and bacterial endosymbionts in the anaerobic ciliate Trimyema compressum. Microb. Ecol. 54, 627–636. doi: 10.1007/s00248-007-9218-1
Söllinger, A., Schwab, C., Weinmaier, T., Loy, A., Tveit, A. T., Schleper, C., et al. (2016). Phylogenetic and genomic analysis of Methanomassiliicoccales in wetlands and animal intestinal tracts reveals clade-specific habitat. FEMS Microbiol. Ecol. 92, 1–12. doi: 10.1093/femsec/fiv149
Sprenger, W. W., Hackstein, J. H. P., and Keltjens, J. T. (2007). The competitive success of Methanomicrococcus blatticola, a dominant methylotrophic methanogen in the cockroach hindgut, is supported by high substrate affinities and favorable thermodynamics. FEMS Microbiol. Ecol. 60, 266–275. doi: 10.1111/j.1574-6941.2007.00287.x
Sprenger, W. W., Van Belzen, M. C., Rosenberg, J., Hackstein, J. H. P., and Keltjens, J. T. (2000). Methanomicrococcus blatticola gen. nov., sp. nov., a methanol- and methylamine-reducing methanogen from the hindgut of the cockroach Periplaneta americana. Int. J. Syst. Evol. Microbiol. 50, 1989–1999. doi: 10.1099/00207713-50-6-1989
Šustr, V., Chroňáková, A., Semanová, S., Tajovský, K., and Šimek, M. (2014). Methane production and methanogenic archaea in the digestive tracts of millipedes (Diplopoda). PLoS One 9:e102659. doi: 10.1371/journal.pone.0102659
Tholen, A., and Brune, A. (2000). Impact of oxygen on metabolic fluxes and in situ rates of reductive acetogenesis in the hindgut of the wood-feeding termite Reticulitermes flavipes. Environ. Microbiol. 2, 436–449. doi: 10.1046/j.1462-2920.2000.00127.x
Tholen, A., Pester, M., and Brune, A. (2007). Simultaneous methanogenesis and oxygen reduction by Methanobrevibacter cuticularis at low oxygen fluxes. FEMS Microbiol. Ecol. 62, 303–312. doi: 10.1111/j.1574-6941.2007.00390.x
Thomas, C. M., Quéméner, E. D., Gribaldo, S., and Borrel, G. (2022). Factors shaping the abundance and diversity of the gut archaeome across the animal kingdom. Nat. Commun. 13:3358. doi: 10.1038/s41467-022-31038-4
Thomas, C. M., Taib, N., Gribaldo, S., and Borrel, G. (2021). Comparative genomic analysis of Methanimicrococcus blatticola provides insights into host-adaptation in archaea and the evolution of methanogenesis. ISME Commun. 1:47. doi: 10.1038/s43705-021-00050-y
Tokura, M., Ohkuma, M., and Kudo, T. (2000). Molecular phylogeny of methanogens associated with flagellated protists in the gut and with the gut epithelium of termites. FEMS Microbiol. Ecol. 33, 233–240. doi: 10.1016/S0168-6496(00)00065-9
Tokura, M., Tajima, K., and Ushida, K. (1999). Isolation of Methanobrevibacter sp. as a ciliate-associated ruminal methanogen. J. Gen. Appl. Microbiol. 45, 43–47. doi: 10.2323/jgam.45.43
Treitli, S., Hanousková, P., Benes, V., Brune, A., Čepička, I., and Hampl, V. (2023). Hydrogenotrophic methanogenesis is the key process in the obligately syntrophic consortium of the anaerobic amoeba Pelomyxa schiedti. ISME J. 17, 1884–1894. doi: 10.1038/s41396-023-01499-6
van Hoek, A. H. A. M., van Alen, T. A., Sprakel, V. S. I., Leunissen, J. A. M., Brigge, T., Vogels, G. D., et al. (2000). Multiple acquisition of methanogenic archaeal symbionts by anaerobic ciliates. Mol. Biol. Evol. 17, 251–258. doi: 10.1093/oxfordjournals.molbev.a026304
Volmer, J. G., Soo, R. M., Evans, P. N., Hoedt, E. C., Alsina, A. L. A., Woodcroft, B. J., et al. (2023). Isolation and characterisation of novel Methanocorpusculum species indicates the genus is ancestrally host - associated. BMC Biol. 21:59. doi: 10.1186/s12915-023-01524-2
Weil, M., Hoff, K. J., Meißner, W., Schäfer, F., Söllinger, A., Wang, H., et al. (2021). Full genome sequence of a Methanomassiliicoccales representative enriched from peat soil. Microbiol. Resour. Announc. 10, e443–e421.
Whitman, W. B., Chuvochina, M., Hedlund, B. P., Hugenholtz, P., Konstantinidis, K. T., Murray, A. E., et al. (2022). Development of the SeqCode: A proposed nomenclatural code for uncultivated prokaryotes with DNA sequences as type. Syst. Appl. Microbiol. 45:126305. doi: 10.1016/j.syapm.2022.126305
Xie, F., Jin, W., Si, H., Yuan, Y., Tao, Y., Liu, J., et al. (2021). An integrated gene catalog and over from the gastrointestinal microbiome of ruminants. Microbiome 9, 1–20.
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “all-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, 643–648. doi: 10.1093/nar/gkt1209
Youngblut, N. D., Reischer, G. H., Dauser, S., Maisch, S., Walzer, C., Stalder, G., et al. (2021). Vertebrate host phylogeny influences gut archaeal diversity. Nature microbiology 6, 1443–1454.
Keywords: archaea, methanogens, gut microbiota, termites, cockroaches, millipedes, Bathyarchaeia, Nitrososphaerales
Citation: Protasov E, Nonoh JO, Kästle Silva JM, Mies US, Hervé V, Dietrich C, Lang K, Mikulski L, Platt K, Poehlein A, Köhler-Ramm T, Miambi E, Boga HI, Feldewert C, Ngugi DK, Plarre R, Sillam-Dussès D, Šobotník J, Daniel R and Brune A (2023) Diversity and taxonomic revision of methanogens and other archaea in the intestinal tract of terrestrial arthropods. Front. Microbiol. 14:1281628. doi: 10.3389/fmicb.2023.1281628
Received: 22 August 2023; Accepted: 13 October 2023;
Published: 15 November 2023.
Edited by:
Michel Geovanni Santiago-Martínez, University of Connecticut, United StatesReviewed by:
Nahui Olin Medina-Chavez, University of Minnesota Twin Cities, United StatesCopyright © 2023 Protasov, Nonoh, Kästle Silva, Mies, Hervé, Dietrich, Lang, Mikulski, Platt, Poehlein, Köhler-Ramm, Miambi, Boga, Feldewert, Ngugi, Plarre, Sillam-Dussès, Šobotník, Daniel and Brune. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Brune, YnJ1bmVAbXBpLW1hcmJ1cmcubXBnLmRl
†Present address: James O. Nonoh, Department of Biomedical Sciences and Technology, School of Public Health, Maseno University, Kisumu, Kenya
‡These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.