 Juan Zhao
Juan Zhao Yanli Cheng1,3
Yanli Cheng1,3 Wentao Qin
Wentao Qin- 1Institute of Plant Protection, Beijing Academy of Agriculture and Forestry Sciences, Beijing, China
- 2Beijing Key Laboratory of Environment Friendly Management on Fruit Diseases and Pests in North China, Beijing Academy of Agriculture and Forestry Sciences, Beijing, China
- 3College of Life Sciences, Yangtze University, Jingzhou, Hubei, China
- 4College of Horticulture and Plant Protection, Inner Mongolia Agricultural University, Hohhot, Inner Mongolia, China
Introduction: Verticillium wilt is the most devastating soil-borne disease affecting Cotinus coggygria in the progress of urban landscape construction in China.
Methods: To assess the variability of the rhizosphere-associated soil microbiome in response to Verticillium wilt occurrence, we investigated the microbial diversity, taxonomic composition, biomarker species, and co-occurrence network of the rhizosphere-associated soil in Verticillium wilt-affected C. coggygria using Illumina sequencing.
Results: The alpha diversity indices of the rhizosphere bacteria in Verticillium wilt-affected plants showed no significant variability compared with those in healthy plants, except for a moderate increase in the Shannon and Invsimpson indices, while the fungal alpha diversity indices were significantly decreased. The abundance of certain dominant or crucial microbial taxa, such as Arthrobacter, Bacillus, Streptomyces, and Trichoderma, displayed significant variations among different soil samples. The bacterial and fungal community structures exhibited distinct variability, as evidenced by the Bray–Curtis dissimilarity matrices. Co-occurrence networks unveiled intricate interactions within the microbial community of Verticillium wilt-affected C. coggygria, with greater edge numbers and higher network density. The phenomenon was more evident in the fungal community, showing increased positive interaction, which may be associated with the aggravation of Verticillium wilt with the aid of Fusarium. The proportions of bacteria involved in membrane transport and second metabolite biosynthesis functions were significantly enriched in the diseased rhizosphere soil samples.
Discussion: These findings suggested that healthy C. coggygria harbored an obviously higher abundance of beneficial microbial consortia, such as Bacillus, while Verticillium wilt-affected plants may recruit antagonistic members such as Streptomyces in response to Verticillium dahliae infection. This study provides a theoretical basis for understanding the soil micro-ecological mechanism of Verticillium wilt occurrence, which may be helpful in the prevention and control of the disease in C. coggygria from the microbiome perspective.
Introduction
Cotinus coggygria Scop., also known as the smoke tree, is one of the most important landscape trees worldwide with ornamental, medicinal, and economic values (Alexandra-Gabriela et al., 2023). It is widely planted as a well-known red leaf plant in China, especially in Fragrant Hill and other famous scenic spots in the Beijing municipality. Verticillium wilt caused by Verticillium dahliae Kleb. is considered the most devastating disease in many regions where C. coggygria is cultivated (Lei, 1993). The main manifestations of Verticillium wilt in C. coggygria is vascular bundle blockage, early deciduous growth, and stunted growth, which not only destroy the red leaf landscape but also cause massive tree mortality (Zhou et al., 2019). As a typical soil-borne pathogen, V. dahliae is characterized by strong temperature adaptation and a wide host range, and can harm more than 200 species of plants (Tian et al., 2016). Owing to V. dahliae’s long latency and it being prone to develop drug resistance, conventional prevention and control measures were inefficient or difficult to implement on Verticillium wilt (Xiong et al., 2014). With the continuous expansion of forest areas and the reduction of tree species, the Verticillium wilt disease is becoming more serious, which negatively influences the sustainable cultivation of C. coggygria (Tian et al., 2016).
Plants select and shape the rhizosphere-associated soil microbiome, stimulating or repressing certain members of the indigenous microbial communities, which usually act as the first defense line against soil-borne pathogens through a range of mechanisms, including direct pathogen inhibition, space and nutrition competition, and antibiotic production (Berg et al., 2017; Park et al., 2023). Numerous previous studies have demonstrated the variability of plant rhizosphere-associated soil microbiome, such as olives with Verticillium wilt occurrence (Fernández-González et al., 2020), ficus trees affected by brown root rot (Liu et al., 2022), and poplars under different genotypes and soil conditions (Karliński et al., 2020). Rhizosphere-associated microbiota, which closely interact with plant roots, are important for plant health maintenance and disease resistance induction (Wei et al., 2019). It becomes a central issue for present research in both forestry and ecology fields to unravel how soil microbial communities change in response to pathogen infection (Liao et al., 2022).
Understanding the association between disease occurrence and rhizosphere-associated microbiome may provide a basis for manipulating soil microbiome to promote plant health (Wei et al., 2021). Zhou et al. (2019) have reported the dynamic changes in bacterial diversity and community structure of C. coggygria plants from different years, geographic areas, and landscape configurations. However, studies were rarely investigated to understand the nature of the rhizosphere-associated soil microbiome in C. coggygria in response to Verticillium wilt occurrence. Therefore, we aimed to characterize the microbial diversity, taxonomic composition, biomarker species, and co-occurrence network of the rhizosphere and non-rhizosphere soil microbiome of C. coggygria under Verticillium wilt-affected and healthy conditions in the hope of providing a theoretical foundation for the scientific management of Verticillium wilt.
Materials and methods
Site description and sample collection
The soil samples were collected from the northern slope of Fragrant Hill (39°99′61′′N and 116°19′50′′E) in the Haidian district of Beijing, China, in August 2021. Fragrant Hills is the best-known Cotinus coggygria planting region in China, which is also a typical region of Verticillium wilt occurrence. This region has a sub-humid, warm temperate continental monsoon climate with an annual precipitation of approximately 600 mm. The typical symptoms of Verticillium wilt were foliar chlorosis and necrosis, stunting, vascular discoloration, die-back of individual branches, and death of the entire tree (Wang et al., 2013). For each plant, the Verticillium wilt disease rating was assessed on a scale of 0 to 4, based on the proportion of yellowing or wilting leaves on the entire plant (Wang et al., 2013). In this study, the C. coggygria plants that showed distinct Verticillium wilt symptoms on more than two-thirds of the stems based on disease rating and were positive for pathogen isolation were classified as diseased plants, while the plants with no wilting symptoms and were negative for pathogen isolation were considered healthy plants.
Typical diseased and healthy plants of C. coggygria with similar ages and sizes in the same C. coggygria forest district were selected to obtain their rhizosphere-associated soil samples (Edwards et al., 2015; Zhou et al., 2021), with three replications each group and five plants each replication. After carefully digging out the fibrous root systems of plants, the loose soil particles and large clods removed from the roots were collected as non-rhizosphere soil. The soils tightly adhered to the surface of the roots were washed with phosphate-buffered saline, and then the rhizosphere soil was reserved after the centrifugation of the soil slurry (Zhou et al., 2021). In addition, the belowground soil in the C. coggygria forest district, approximately 80–100 cm horizontally away from plant roots, was sampled as bulk soil. The soil samples from five plants in each replication were combined as a single composite sample. Therefore, five soil sample groups, including the rhizosphere soil from healthy and Verticillium wilt-affected plants (RHS and RPS), the non-rhizosphere soil from healthy and affected plants (NRHS and NRPS), as well as the bulk soil in the C. coggygria forest (LS), were obtained with three replications. All samples were placed in aseptic bags, transported back to the lab, and treated using a 2 mm sieve. Each sample was divided into two parts: one part was air-dried to measure the physicochemical properties, and the other part was stored at −80°C for DNA extraction.
Measurement of soil physicochemical properties
The physicochemical properties of the C. coggygria soil samples, such as soil organic carbon, total nitrogen, total phosphorus, total potassium, ammonium nitrogen, nitrate nitrogen, available phosphorus, available potassium, and pH, were tested following the methods of Bao (2000). Total organic carbon (TOC) was measured by the K2Cr2O7-H2SO4 oxidation method in an automated TOC analyzer (TOC-VCPH; Shimadzu, Shimane-ken, Japan) following the manufacturer’s instructions. Total nitrogen (TN) was determined by the Kjeldahl procedure with a carbon–hydrogen–nitrogen (CHN) elemental analyzer (Perkin Elmer; Boston, MA, USA). Total phosphorus (TP) was measured by the molybdenum antimony colorimetric method with an ultraviolet–visible spectrophotometer (UV-2550; Shimadzu, Kyoto, Japan). Total potassium (TK) was measured by the NaOH fusion flame emission technique with a flame photometer (FP6400; Drawell, Shanghai, China). Soil-available phosphorus (AVP) was extracted with 0.5 mol·L−1 of NaHCO3 and determined using the molybdenum blue method. Soil-available potassium (AVK) was extracted with 1 mol·L−1 of NH4Ac and determined using the flame emission technique. The soil ammonium nitrogen (NH4+-N) and nitrate nitrogen (NO3−-N) concentrations were determined by an automated flow injection analyzer (FS3100; Xylem, Washington, United States) after the extraction of the soil samples with 2 mol·L−1 of KCl. Soil pH was determined with a pH meter (ST2100; OHRUS, Jiangsu, China) with a soil-to-water ratio of 1:2.5 (Marcos et al., 2019).
DNA extraction, library preparation, and sequencing
Total genomic DNA was extracted using the Power Soil DNA Isolation Kit (Mo Bio Laboratories, Inc., USA) according to the standard protocols. The concentration and purity of the extracted DNA were determined using a NanoDrop 2000 UV–Vis spectrophotometer (Thermo Fisher Scientific, Wilmington, United States). The DNA quality was checked by 1% agarose gel electrophoresis. The V3–V4 variable region of the bacterial 16S rRNA gene was amplified using the primers 338F and 806R (Zhou et al., 2017). The fungal rDNA-ITS sequence was amplified using the primers ITS1F and ITS2R (Adams et al., 2013; Mcguire et al., 2013). High-throughput sequencing was performed using the Illumina MiSeq sequencing platform at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China).
Data processing and sequence analysis
Raw sequence files were analyzed and quality-filtered with the Quantitative Insights Into Microbial Ecology (QIIME 1.9.1 software, http://qiime.org/install/index.html). Sequences were screened at 97% similarity using UPRASE (version 7.0.1090) with the help of an agglomerative clustering algorithm (Edgar, 2013). The taxonomic identities of the bacteria and fungi were determined using the SILVA database1 and the Unite database,2 respectively. All sequences were deposited in the NCBI Sequence Read Archive with the submission accession number SAMN35564492-SAMN35564506 under BioProject ID PRJNA 979017 for the bacterial community and SAMN35564971-SAMN35564985 under BioProject ID PRJNA 979102 for the fungal community. Functional prediction of the microbial community was performed using PICRUSt2 based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Douglas et al., 2019), Bugbase, and FUNGuild (Nguyen et al., 2016) on the cloud platform of Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China.3
Statistical analyses
All the data were displayed as the mean ± standard deviation of three replications. The significance of the differences (p < 0.05) was evaluated by the least significant difference (LSD) test after a one-way analysis of variance (ANOVA) using the SPSS 17.0 software. Alpha diversity analysis of the microbial community was performed by Mothur software version 1.30.2 (Schloss et al., 2009). The beta diversity was calculated based on Bray–Curtis distance metrices and visualized by principal coordinate analysis (PCoA) in QIIME (Caporaso et al., 2010). Linear discriminant analysis was conducted by LEfSe software4 to identify the microbial biomarkers at different taxonomic levels (Segata et al., 2011). The strong (ρ > 0.8 or < −0.8) and significant (adjusted p-value < 0.01) correlations were retained for use in co-occurrence network construction at the genus level. Network visualization and topological analysis were performed using Gephi v0.9.2.
Results
Sequencing data analyses
Through high-throughput sequencing analyses of 16S rRNA and the ITS region, a total of 6,133 operational taxonomic units (OTUs) were obtained for the bacteria, which were further assigned into 39 phyla, 127 classes, 300 orders, 479 families, and 876 genera, including some unclassified groups, whereas 2,579 OTUs attributed to 14 phyla, 51 classes, 125 orders, 282 families, and 604 genera were obtained for the fungi. According to the Venn diagrams, a total of 2,212 bacterial OTUs were shared across all the soil samples, while 113~246 unique bacterial OTUs were exclusively detected in each sample. In addition, a total of 396 fungal OTUs were shared, and 72~286 fungal OTUs were uniquely detected (Supplementary Figure S1).
Alpha diversity variation in different soil samples
Alpha diversity of the bacterial OTUs indicated that significant differences in richness indices were detected between the NRHS and NRPS soil samples; the Sobs, Chao, and Ace indices decreased significantly in the non-rhizosphere soil of Verticillium wilt-affected C. coggygria. For evenness and diversity estimates, we found a clear separation between the RHS and RPS soil samples (p < 0.05), with higher values observed for the RPS sample (Figure 1A). In comparison to the alpha diversity indices of the fungal community, the Sobs, Chao, and Ace indices were significantly decreased in Verticillium wilt-affected C. coggygria soil samples (p < 0.05). The Shannoneven and Shannon indices of the RPS sample, as well as the Invsimpson index of the NRPS sample, were also obviously reduced (Figure 1B).
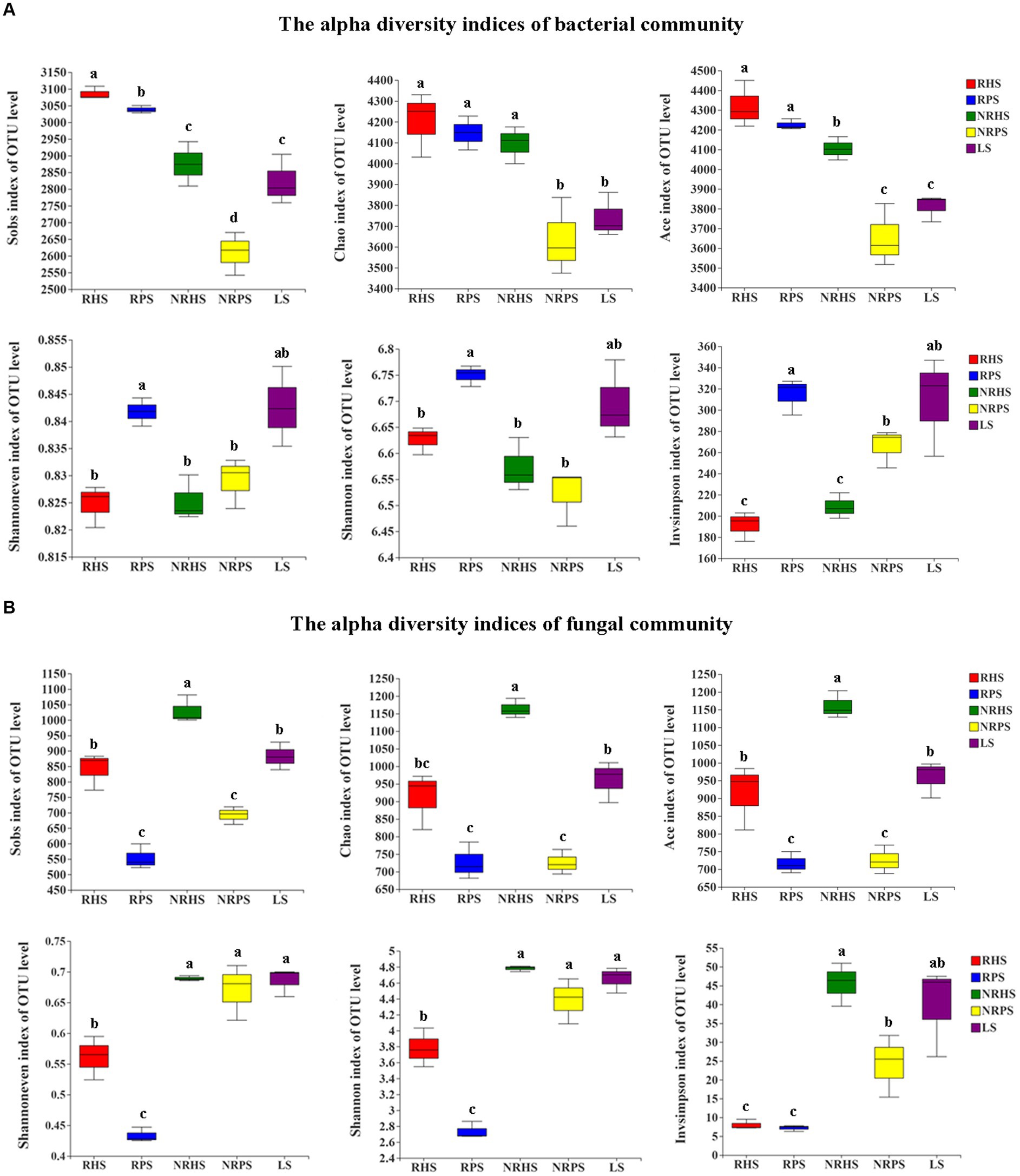
Figure 1. Boxplots of the alpha diversity indices of bacterial (A) and fungal (B) OTUs in the Cotinus coggygria soil microbiome under different conditions. Different lowercase letters denoted significant differences at p-values < 0.05 levels among different soil samples. RHS, rhizosphere soil from healthy plants; RPS, rhizosphere soil from Verticillium wilt-affected plants; NRHS, non-rhizosphere soil from healthy plants; NRPS, non-rhizosphere soil from Verticillium wilt-affected plants; LS, bulk soil in the C. coggygria forest.
Community composition variation in different soil samples
Considering the relative abundances of the bacterial taxa, Actinobacteria (30.5%–46.5%) was the major phylum, followed by Proteobacteria (16.9%–25.3%), Acidobacteria (7.1%–21.6%), Chloroflexi (6.2%–12.5%), Firmicutes (1.1%–8.9%), and Gemmatimonadota (2.2%–3.9%), accounting for more than 87% of the total bacterial abundance (Supplementary Table S1). The primary bacteria at the class level consisted of Actinobacteria, Alphaproteobacteria, Thermoleophilia, Vicinamibacteria, and Gammaproteobacteria (Supplementary Figure S2). The predominant fungal phyla were Ascomycota (47.6%–82.6%), followed by Basidiomycota (9.9%–50.5%) and Mortierellomycota (0.5%–10.2%), accounting for more than 92% of the total abundance (Supplementary Table S1). It was clear that the abundance difference in some fungal taxa explained the obvious variation in the community composition of Verticillium wilt-affected soil samples (Supplementary Figure S2).
Heatmap analysis of the top 30 bacterial taxa at the genus level (Figure 2A) showed that the bacterial genera mainly consisted of Arthrobacter, Bacillus, Streptomyces, and Rubrobacter, whose abundance significantly differed between rhizosphere soil samples of Verticillium wilt-affected and healthy C. coggygria, despite some similar soil physicochemical characteristics such as total nitrogen, total phosphorus, total potassium, available phosphorus, nitrate nitrogen, and pH (Supplementary Figure S3). As shown in Figure 2B, the genus Bacillus was significantly enriched in the RHS soil sample, while Streptomyces was obviously enriched in the RPS and NRPS samples.
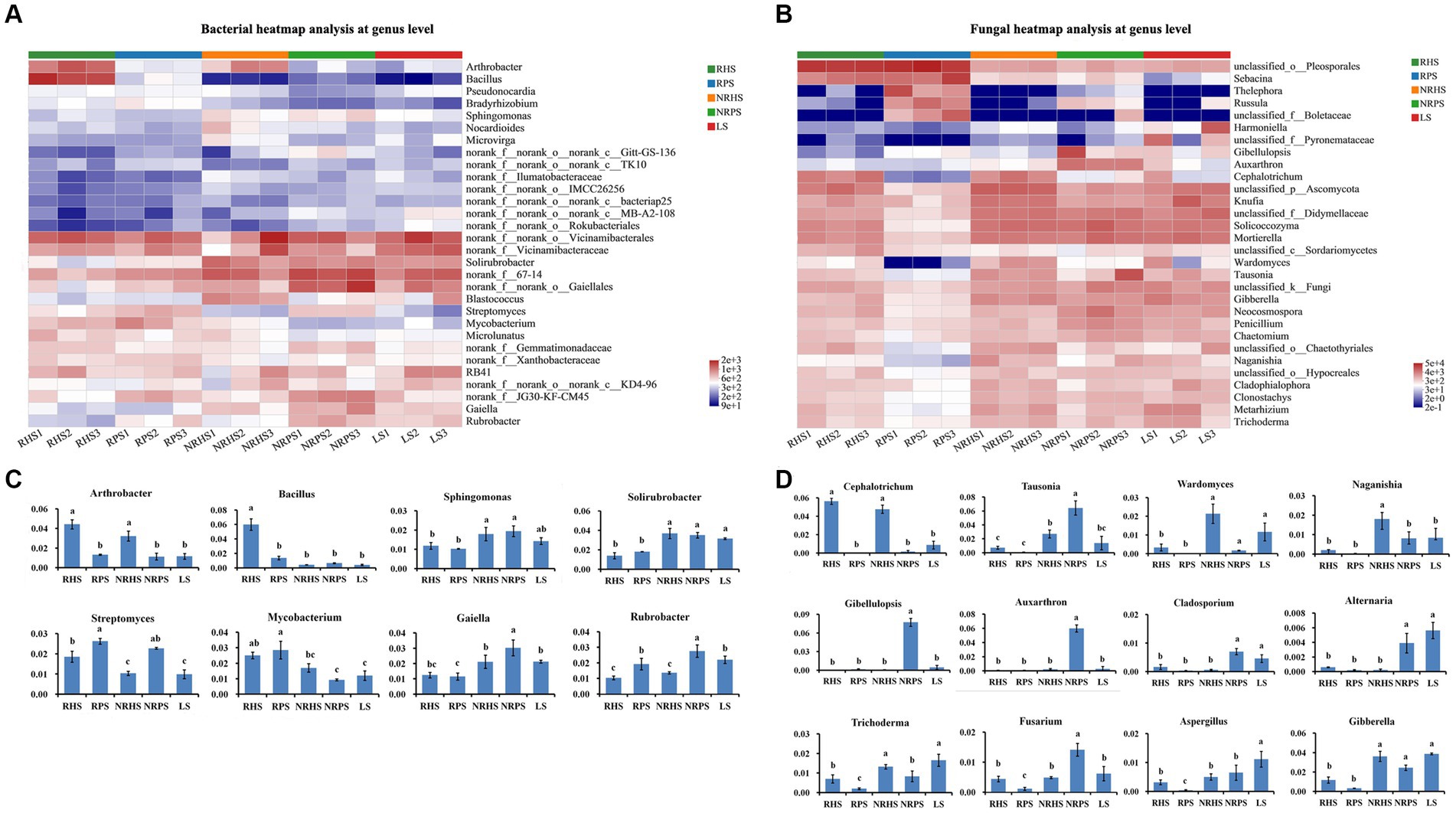
Figure 2. Relative abundances of the dominant bacterial and fungal taxa in the Cotinus coggygria soil microbiome under different conditions. (A) Heatmap representing the relative abundance of the top 30 bacterial taxa at the genus level, (B) average relative abundance of bacterial genera with significant differences among different soil samples, (C) heatmap representing the relative abundance of the top 30 fungal taxa at the genus level, and (D) average relative abundance of fungal genera with crucial features or significant differences among the soil samples. The x-axis represented samples, and the y-axis represented relative abundance. The error bar represented the standard deviation of mean values. Different lowercase letters showed significant differences in abundance among different soil samples based on the LSD test (p < 0.05). RHS, rhizosphere soil from healthy plants; RPS, rhizosphere soil from Verticillium wilt-affected plants; NRHS, non-rhizosphere soil from healthy plants; NRPS, non-rhizosphere soil from Verticillium wilt-affected plants; LS, bulk soil in the C. coggygria forest.
The relative abundances of genera Cephalotrichum and Wardomyces were lower in the soil samples of Verticillium wilt-affected C. coggygria, while the genus Russula obviously increased in abundance (Figure 2C). We also compared some well-known fungal genera and found that Trichoderma was significantly increased in both rhizosphere and non-rhizosphere soils of the healthy plants (Figure 2D). The variations in some crucial microbial species were further compared among soil samples based on a 95% confidence interval and Welch’s t-test (Supplementary Figures S4, S5). Principal coordinate analysis (PCoA) indicated that the bacterial community structure was distinctly separated based on soil compartments and plant health status, with the PC1 axis showing 32.81% variation and the PC2 axis explaining 17.91% (Figure 3A). Soil compartments and plant health status also explained 39.62% and 19.05% of the variability in fungal community composition, respectively (Figure 3B).
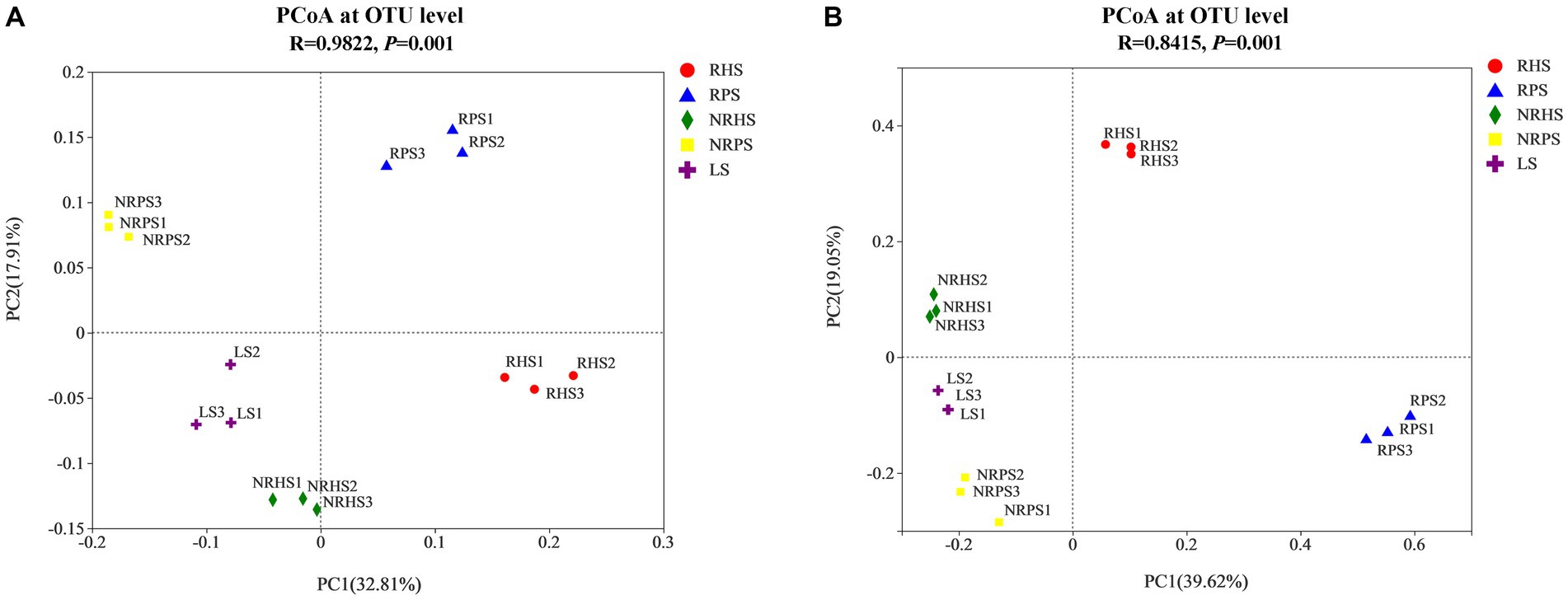
Figure 3. Principal coordinate analysis (PCoA) of bacterial (A) and fungal (B) communities of the Cotinus coggygria soil microbiome under different conditions. The PCoA plots were based on the Bray–Curtis dissimilarity of bacterial and fungal communities at the OTU levels according to the ANOSIM test. RHS, rhizosphere soil from healthy plants; RPS, rhizosphere soil from Verticillium wilt-affected plants; NRHS, non-rhizosphere soil from healthy plants; NRPS, non-rhizosphere soil from Verticillium wilt-affected plants; LS, bulk soil in the C. coggygria forest.
Microbial biomarkers variation in different soil samples
Linear discriminant analysis effect size (LEfSe) was used to scan the biomarkers in the soil microbiome of C. coggygria from different soil compartments and plant health status. At the phylum level, Gemmatimonadota was significantly enriched in the RHS sample, while Cyanobacteria and Myxococcota were enriched in the RPS sample. The phylum Actinobacteriota, Chloroflexia, and Methylomirabilota represented the biomarkers of the NRHS, NRPS, and LS samples, respectively. At the genus level, Bacillus, Arthrobacter, and Microlunatus were significantly enriched in the RHS sample, while Mycobacterium, Streptomyces, and Bradyrhizobium were more abundant in the RPS sample (Figure 4). LEfSe analysis based on the fungal genera also displayed some unique biomarkers in different soil samples (Figure 5).
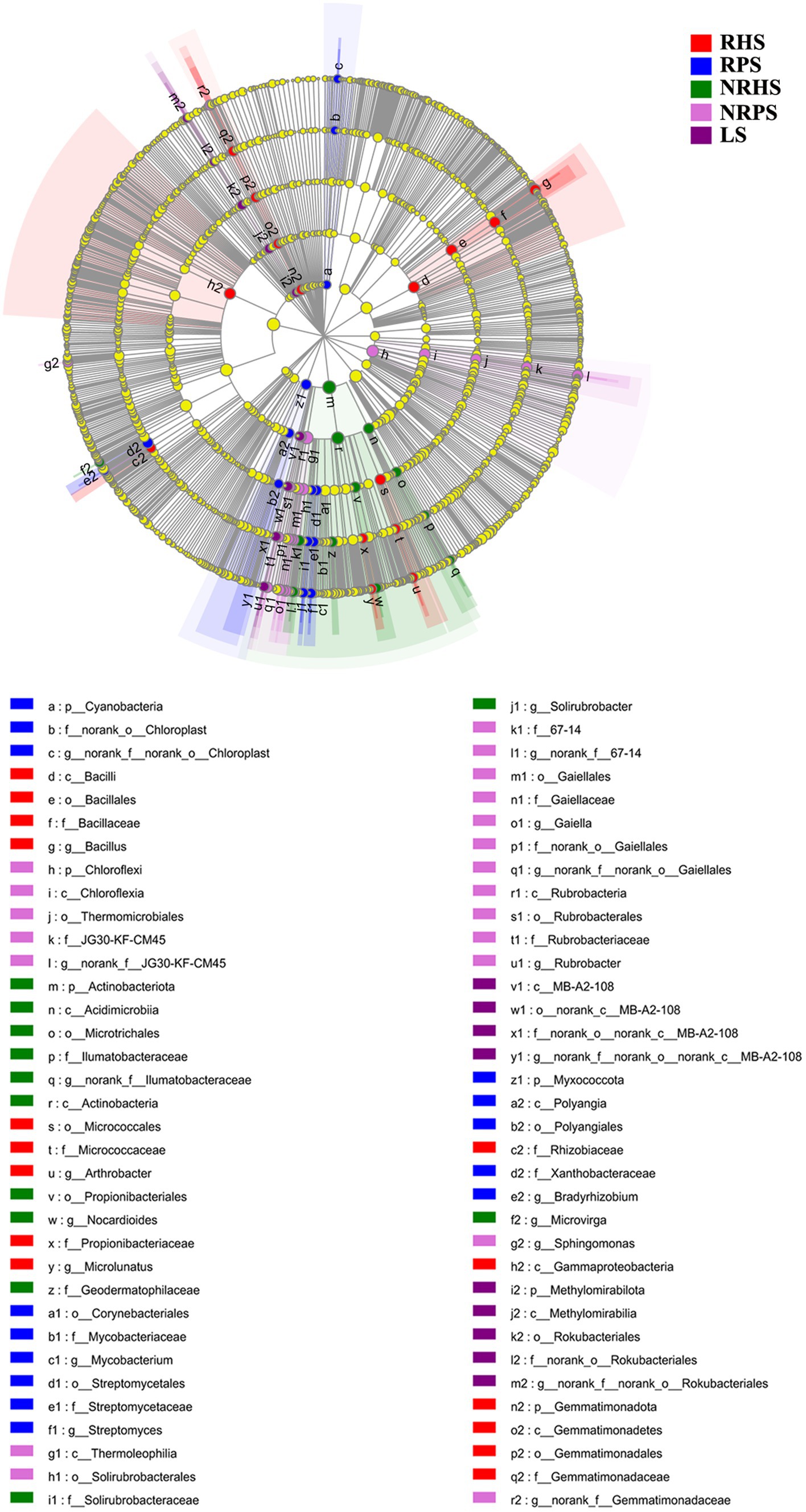
Figure 4. Linear discriminant analysis effect size (LEfSe) of bacterial taxa in the Cotinus coggygria soil microbiome under different conditions. The non-parametric factorial Kruskal–Wallis test with p < 0.05 and logarithmic LDA score > 3.5 was used to identify the significantly enriched bacterial taxa (using the all-against-all comparisons parameter). Different colors depicted different soil samples, while circles from inside to outside showed phylogenetic levels from phylum to genera. The sizes of the circles were proportional to the mean relative abundance of each taxon. The yellow circles represented the absence of significantly different taxa. Genera with a relative abundance of less than 0.1% were not included.
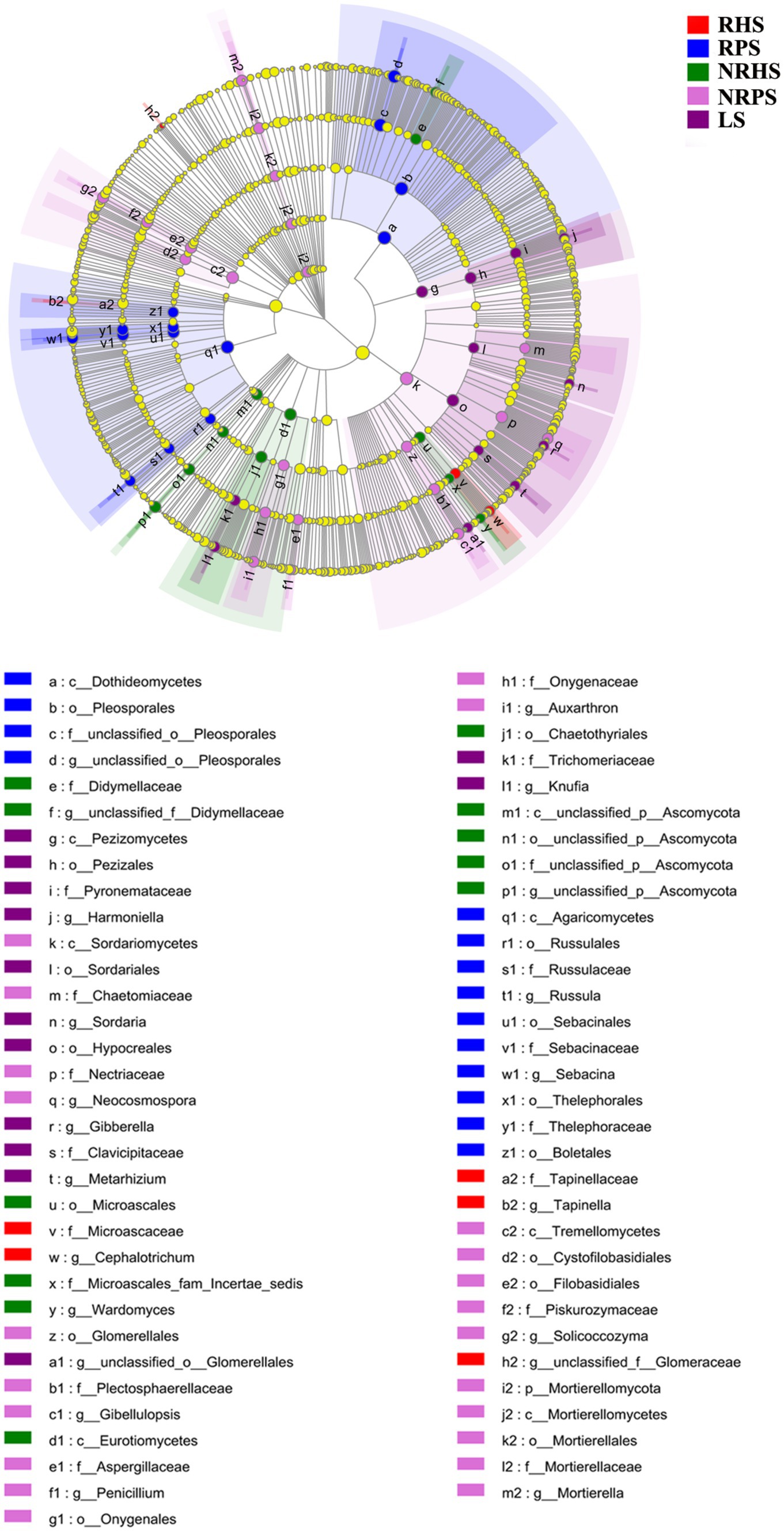
Figure 5. Linear discriminant analysis effect size (LEfSe) of fungal taxa in the Cotinus coggygria soil microbiome under different conditions. The non-parametric factorial Kruskal–Wallis test with p < 0.05 and logarithmic LDA score > 3.5 was used to identify the significantly enriched fungal taxa (using the all-against-all comparisons parameter). The notes were the same as Figure 4.
The histogram of the LDA effect value indicated that Bacillus (from class to genus), Micrococcaceae (from order to family), the phylum Firmicutes, and the genus Arthrobacter were the most decisive bacterial members in the RHS soil sample, while Mycobacterium (from family to genus) and Streptomyces (from order to genus) were obviously the biomarkers in the RPS sample. In addition, the relative abundance of Actinobacteria (from phylum to class) and Thermomicrobiales (from class to order) were the most apparent taxa in NRHS and NRPS soil samples, respectively (Supplementary Figure S6). On the other hand, the genera Cephalotrichum and Tapinella were the most influential members, leading to a distinct fungal community in RHS, while the genera Sebacina and Russula were the biomarkers in RPS (Supplementary Figure S7).
Interaction network variation in different soil samples
To gain a deeper insight into the interaction variability of the soil microbiome in Verticillium wilt-affected Cotinus coggygria, the microbial communities in the rhizosphere and non-rhizosphere soil samples were merged. Results showed that the microbial networks of Verticillium wilt-affected plants differed profoundly from the healthy networks, with higher average degrees, degree centrality, closeness centrality, and network density (Supplementary Table S2). More positive interactions occurred in the healthy bacterial network (BHS, 68.6%, 450 edges) than in the diseased soil samples (BPS, 52.9%, 546 edges). In the fungal network, the positive interactions of the diseased network (FPS, 84.6%, 981 edges) were significantly increased compared to the healthy samples (FHS, 62.5%, 450 edges). The ratio of positive to negative interactions (P/N ratio) was decreased in the bacterial network of Verticillium wilt-affected plants, while those of the fungal network were obviously increased from 1.7 to 5.5 (Supplementary Figure S8). All these suggested that the disease status strengthened the microbial nodes and their associations in the microbial community, especially the fungal co-occurrence network.
We also found that the nodes representing genera Blastococcus, Solirubrobacter, Marmoricola, Nocardioides, and Microbacterium belonging to Actinobacteria and genus Lysobacter from Proteobacteria were closely related to other bacterial genera with higher degrees in the healthy network (Supplementary Table S3; Figure 6A). The genera Streptomyces, Solirubrobacter, and Actinomadura belonging to Actinobacteriota, as well as Vicinamibacter (Acidobacteriota), Sphingomonas (Proteobacteria), and Bacillus (Firmicutes), occupied critical positions in the diseased network and had important impacts on other bacterial groups (Supplementary Table S4; Figure 6B). In the soil fungal co-occurrence network of healthy plants (Figure 6C), the genera Phialocephala, Fusicolla, Mycoarthris, Clonostachys, and Cladophialophora belonging to Ascomycota, as well as the genus Mortierella belonging to Basidiomycota, displayed the highest degree, degree centrality, or betweenness centrality (Supplementary Table S5). The nodes representing the genera Fusarium, Neocosmospora, Mortierella, Trichoderma, Aspergillus, and Scytalidium were closely related to other genera, and their degrees of connection were the highest in the diseased network (Supplementary Table S6). Among which the genus Fusarium had 29 positively related genera, such as Metarhizium, Penicillium, Wardomyces, Mortierella, and Cephalotrichum (Figure 6D).
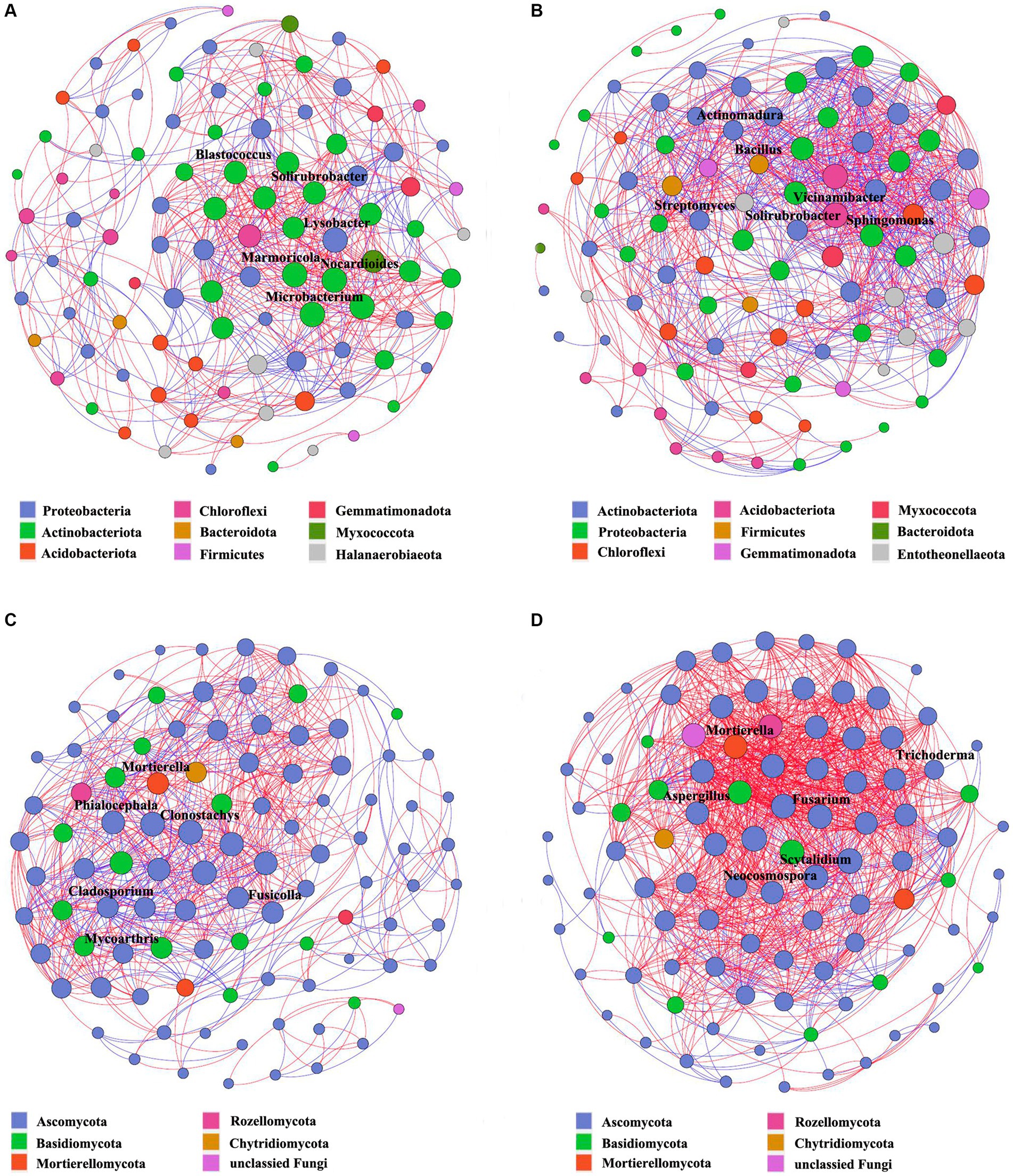
Figure 6. Co-occurrence networks constructed based on bacterial and fungal communities in the Cotinus coggygria soil microbiome under different conditions. (A) Bacterial networks of soil samples from healthy plants, BHS, (B) bacterial networks of soil samples from Verticillium wilt-affected plants, BPS, (C) fungal networks of soil samples from healthy plants, FHS, and (D) fungal networks of soil samples from Verticillium wilt-affected plants, FPS. The nodes were colored based on phyla, and each node size was proportional to the degree of centrality. A connection denotes a strong (Spearman’s ρ > 0.8 or < −0.8, red or blue edges) and significant (p < 0.05) correlation. Potential keystone taxa were shown with bold genera names.
Microbial function variation in different soil samples
The distribution of bacterial function was significantly different in the soil microbiome of Verticillium wilt-affected Cotinus coggygria. The functions connected with transport and catabolism, folding, sorting, and degradation, as well as energy metabolism, were significantly higher in the RHS soil sample (t-test, corrected p < 0.01), while bacteria from the diseased soil samples had more functions related to membrane transport and second metabolite biosynthesis. At level 3, peroxisome, protein processing in the endoplasmic reticulum, and photosynthesis were significantly higher in RHS (p < 0.01), while the phosphotransferase system, tropane, piperidine, and pyridine alkaloid biosynthesis were in higher proportions in RPS (Figure 7A). The pathways related to the phosphotransferase system were significantly enhanced in the NRPS sample, while pantothenate and CoA biosynthesis, peptidoglycan biosynthesis, and fatty acid biosynthesis functions were obviously increased in the NRHS (Figure 7B). Functions related to antioxidant capacity were higher in the soil samples of Verticillium wilt-affected plants without a significant difference. The biofilm-forming function was significantly increased in bacteria from the rhizosphere soil compartments (Figure 7C). In addition, the trophic modes of the C. coggygria soil microbiome were dominated by saprotrophs, followed by pathotrophs and symbiotrophs (Supplementary Figure S9).
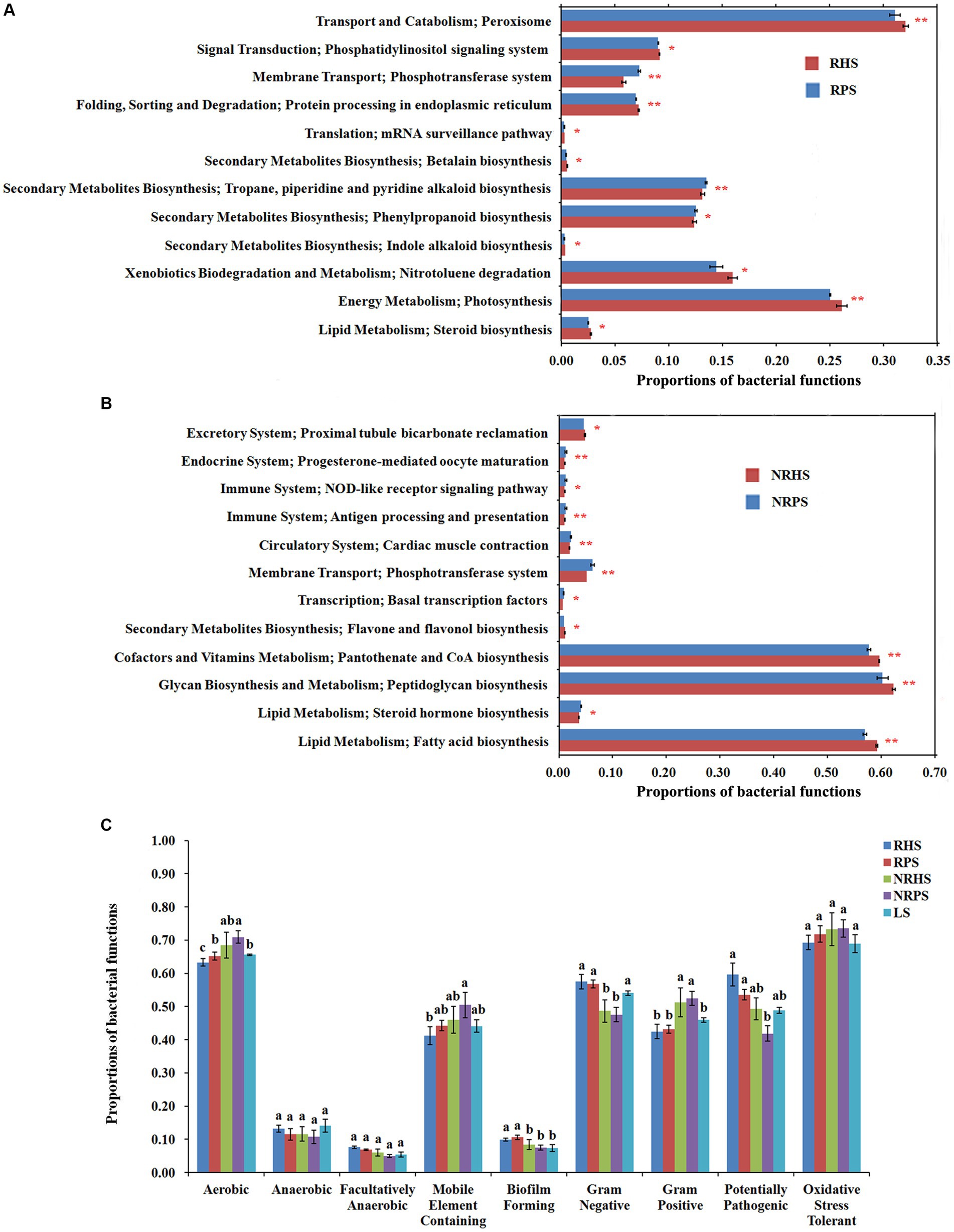
Figure 7. Proportions of bacterial functions in the Cotinus coggygria soil microbiome under different conditions based on KEGG and Bugbase databases. (A,B) KEGG ortholog function for bacteria in the rhizosphere (A) and non-rhizosphere (B) soil samples, (C) bacteria function profiles in different soil samples based on Bugbase prediction. RHS, rhizosphere soil from healthy plants; RPS, rhizosphere soil from Verticillium wilt-affected plants; NRHS, non-rhizosphere soil from healthy plants; NRPS, non-rhizosphere soil from Verticillium wilt-affected plants; LS, bulk soil in C. coggygria forest.
Discussion
The occurrence of soil-borne diseases is closely related to the imbalance of the soil microecosystem; therefore, understanding the variability of the rhizosphere-associated soil microbiome is of great importance in plant health maintenance (Zheng et al., 2022). Rhizosphere-associated soil microbiome has been previously observed on a series of crops and plants (Dudenhöffer et al., 2016; Zhou et al., 2021). In order to determine whether the soil microbiome functioned to sustain Cotinus coggygria health, we investigated the microbial diversity, community composition, biomarker species, and interaction network of the rhizosphere and non-rhizosphere soil samples of Verticillium wilt-affected and healthy C. coggygria in an urban area of Beijing, China. Results indicated that the bacterial Shannoneven, Shannon, and InvSimpson indices in the rhizosphere soil of Verticillium wilt-affected C. coggygria exhibited a significant increase in comparison to those observed in healthy plants. These findings were consistent with the previous study, as the alpha diversity indices, including Chao1 and Shannon index, were significantly higher in root-rotted samples of areca nut palm (Li et al., 2021). However, the Shannon and Simpson indices of soil samples obtained from healthy cotton plants exhibited higher values compared to those derived from diseased plants (Wei et al., 2021). The fungal diversity indices, including Sobs, Chao, Ace, Shannoneven, and Shannon indices, exhibited a distinct reduction in the rhizosphere soil of diseased samples, which may be related to a decrease in species richness and an increase in abundance of some specific fungi (Li et al., 2023).
Plants select and shape the rhizosphere-associated soil microbiome, stimulating or repressing certain members of the indigenous microbial communities, which can act as the first defense line against soil-borne pathogens through diverse mechanisms (Berg et al., 2017; Fernández-González et al., 2020). In a comparison of the taxon abundance of soil microbiome across different soil samples, a significant reduction in specific bacteria such as Acidobacteria and Gemmatimonadetes was observed in the rhizosphere of Verticillium wilt-affected plants. This finding aligns with the recent research that the decline of Acidobacteria is associated with wilt development in olive (Fernández-González et al., 2020) and cotton (Wei et al., 2021), also caused by V. dahliae. The relative abundance of Gemmatimonadetes in the rhizosphere exhibited a negative correlation with V. dahliae infection, as documented by Fernández-González et al. (2020).
In this study, the genera Arthrobacter, Bacillus, and Trichoderma exhibited evident abundance in healthy soil samples; the phenomenon may involve the enrichment of beneficial microbes (Andric et al., 2020). Trichoderma was previously reported to promote plant growth or elicit disease resistance through the production of bioactive substances, regulation of related defensive pathways, or competition for space and nutrients with pathogens (Wei et al., 2019; Zhou et al., 2021). However, the relative abundance of Streptomyces was found to be increased in the Verticillium wilt-affected C. coggygria, which may be related to the recruitment of antagonistic microbes driven by diseased host plants (Yin et al., 2021), since Streptomyces usually produce secondary metabolites with biological activities and have been isolated from soil to inhibit phytopathogen growth (Barka et al., 2016; Takahashi and Nakashima, 2018). These were in accordance with the results of rhizosphere soil studies in rusty root-affected Ginseng (Wei et al., 2020) and Fusarium wilt-infected watermelon (Meng et al., 2019). The increased abundance of Fusarium in diseased plants may be associated with the occurrence and aggravation of Verticillium wilt in C. coggygria, agreeing with previous research that Fusarium contributed to the exacerbation of grapevine trunk disease (Li et al., 2023) and Zanthoxylum bungeanum root rot (Liao et al., 2022).
In-depth analyses of co-occurrence networks within microbial communities may facilitate the elucidation of the functional roles of microbiota and ascertain the stability of the ecosystem (Barberán et al., 2011; Fernández-González et al., 2020). In this study, certain genera, such as Bacillus, Streptomyces, Solirubrobacter, and Sphingomonas, exhibited higher clustering coefficients and occupied pivotal positions within the bacterial ecology. These keystone taxa frequently co-occur with other microbes and potentially play primary roles in microbial ecosystems (Agler et al., 2016; Banerjee et al., 2016, 2018). The enhanced tolerance to pathogens may be related to the intricate interaction network in soil ecology, implying that pathogen suppression is driven by a microbial consortium rather than individual microorganisms (Gómez et al., 2017; Zhou et al., 2021). The bacterial network illustrated higher network density and more edge numbers in diseased soil samples, while those were particularly pronounced in the fungal communities. Certain pathogenic or harmful fungi may interact with each other to exacerbate the disease severity of Verticillium wilt. For example, the genus Fusarium acted as the keystone taxon in the diseased network, exhibiting the highest clustering coefficient values and displaying positive correlations with 29 genera. The microbial community may respond to the perturbation and increase resistance against pathogen infection through more efficient information dissemination (Banerjee et al., 2018).
The rhizosphere-associated soil microbiome primarily referred to the microorganisms residing in soil particles that were closely related to the C. coggygria roots. Soil ecological niche-mediated microbiome variability has been demonstrated in different plant species. Similar to previous investigations conducted on other plants such as potato (Shi et al., 2019), rice (Edwards et al., 2015), and ficus trees (Liu et al., 2022), the rhizosphere soil was more likely to be influenced by root exudates, exhibiting distinct niche variability, which showed significant enrichment of Bacillus and Mycobacterium. Together, our findings demonstrated that C. coggygria possessed the ability to modulate the rhizosphere-associated soil microbiome under different plant health statuses. The healthy plants selectively recruited a consortium of beneficial microbes such as Arthrobacter, Bacillus, and Trichoderma; these microbial species may exert their effects on the establishment of a favorable rhizosphere micro-ecological environment through positive interaction (Berendsen et al., 2018). Understanding the correlation between plant health and soil microbiome holds the potential for manipulating soil microbiome to enhance plant health and manage plant disease (Kwak et al., 2018; Pascale et al., 2020). Further studies will focus on the isolation and screening of the enriched beneficial species or keystone taxa, and on the evaluation of their consistency in plant growth promotion and disease suppression abilities.
Conclusion
The composition and structure patterns of the rhizosphere-associated soil microbiome in C. coggygria were not well understood in the forest ecosystem, nor were those from different plant health status or soil compartments. Our study provides a comprehensive study of the rhizosphere-associated soil microbiome variability of Verticillium wilt-affected C. coggygria in Beijing, China. The microbial diversity, community composition, and abundance of certain microbial taxa varied in accordance with the plants’ healthy status and soil compartments. The disease status of Verticillium wilt strengthened the microbial nodes and their interactions in the co-occurrence network, especially the fungal community. The results improved our understanding of the association between the rhizosphere-associated soil microbiome and Verticillium wilt occurrence in the C. coggygria plant, which has important implications for soil-borne disease management in forest tree species in future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
JZ: Data curation, Investigation, Project administration, Writing – original draft, Writing – review & editing. YC: Investigation, Methodology, Validation, Writing – review & editing. NJ: Investigation, Methodology, Validation, Writing – review & editing. GQ: Investigation, Methodology, Validation, Writing – review & editing. WQ: Project administration, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Youth Research Fund of the Beijing Academy of Agriculture and Forestry Sciences, China (QNJJ202313), the Innovation Capacity Foundation of the Beijing Academy of Agriculture and Forestry Sciences, China (KJCX201910), and the Beijing Natural Science Foundation Project, China (6232009).
Acknowledgments
The authors thank Bin Zhang (College of Life and Environmental Sciences, Minzu University of China, Beijing) for her assistance in article review and statistical analysis guidance. The authors also thank those personnel who offer help in our samples collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1279096/full#supplementary-material
Footnotes
References
Adams, R., Miletto, M., Taylor, J., and Thomas, D. B. (2013). Dispersal in microbes: fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 7, 1262–1273. doi: 10.1038/ismej.2013.28
Agler, M. T., Ruhe, J., Kroll, S., Morhenn, C., Kim, S. T., Weigel, D., et al. (2016). Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 14:e1002352. doi: 10.1371/journal.pbio.1002352
Alexandra-Gabriela, C., Victorita, T., Cristian, E. P., Elena, M. M., Florenta, E. H., Tatiana, V. D., et al. (2023). Phenological and environmental factors' impact on secondary metabolites in medicinal plant Cotinus coggygria Scop. Plan. Theory 12:1762. doi: 10.3390/plants12091762
Andric, S., Meyer, T., and Ongena, M. (2020). Bacillus responses to plant-associated fungal and bacterial communities. Front. Microbiol. 11:1350. doi: 10.3389/fmicb.2020.01350
Banerjee, S., Kirkby, C. A., Schmutter, D., Bissett, A., Kirkegaard, J. A., and Richardson, A. E. (2016). Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 97, 188–198. doi: 10.1016/j.soilbio.2016.03.017
Banerjee, S., Schlaeppi, K., Marcel, G. A., and Heijden, V. D. (2018). Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576. doi: 10.1038/s41579-018-0024-1
Barberán, A., Bater, S. T., Casamayor, E. O., and Fierer, N. (2011). Using network analysis to explore co-occurrence patterns in soil microbial community. ISME J. 6, 343–351. doi: 10.1038/ismej.2011.119
Barka, E. A., Vatsa, P., Sanchez, L., Gaveau-Vaillant, N., Jacquard, C., Klenk, H. P., et al. (2016). Taxonomy, physiology, and natural products of Actinobacteria. Microbiol. Mol. Biol. Rev. 80, 1–43. doi: 10.1128/MMBR.00019-15
Berendsen, R. L., Vismans, G., Yu, K., Song, Y., de Jonge, R., Burgman, W. P., et al. (2018). Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 12, 1496–1507. doi: 10.1038/s41396-018-0093-1
Berg, G., Köberl, M., Rybakova, D., Müller, H., Grosch, R., and Smalla, K. (2017). Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol. Ecol. 93:5. doi: 10.1093/femsec/fix050
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Frederic, D. B., and Elizabeth, K. C. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Douglas, G. M., Maffei, V. J., Zaneveld, J., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2019). PICRUSt 2: an improved and extensible approach for metagenome inference. BioRxiv [Preprint]. BioRxiv: 672295
Dudenhöffer, J. H., Scheu, S., Jousset, A., and Cahill, J. (2016). Systemic enrichment of antifungal traits in the rhizosphere microbiome after pathogen attack. J. Ecol. 104, 1566–1575. doi: 10.1111/1365-2745.12626
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edwards, J., Johnson, C., and Santos-Medellin, C. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U. S. A. 112, 911–920. doi: 10.1073/pnas.1414592112
Fernández-González, A. J., Cardoni, M., Gómez-Lama, C. C., Valverde-Corredor, A., Villadas, P. J., Fernández-López, M., et al. (2020). Linking belowground microbial network changes to different tolerance level towards verticillium wilt of olive. Microbiome. 8:11. doi: 10.1186/s40168-020-0787-2
Gómez, E. R., Bruijnde, I., Postma, J., and Raaijmakers, J. M. (2017). Current insights into the role of rhizosphere bacteria in disease suppressive soils. Front. Microbiol. 8:2529. doi: 10.3389/fmicb.2017.02529
Karliński, L., Ravnskov, S., and Rudawska, M. (2020). Soil microbial biomass and community composition relates to poplar genotypes and environmental conditions. Forests 11:262. doi: 10.3390/f11030262
Kwak, M. J., Kong, H. G., Choi, K., Kwon, S. K., Song, J. Y., Lee, J., et al. (2018). Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 36, 1100–1109. doi: 10.1038/nbt.4232
Lei, Z. P. (1993). Study of Verticillium causing Cotinus coggygria wilt in the Beijing area. J. Beijing Forestry Univ. 15, 88–93.
Li, Y. H., Li, X. H., Zhang, W., Zhang, J., Wang, H., Peng, J. B., et al. (2023). Belowground microbiota analysis indicates that fusarium spp. exacerbate grapevine trunk disease. Environ. Microbiome. 18:29. doi: 10.1186/s40793-023-00490-0
Li, H., Ma, X., Tang, Y. Q., Yan, C. L., Hu, X. W., Huang, X., et al. (2021). Integrated analysis reveals an association between the rhizosphere microbiome and root rot of areca nut palm. Pedosphere 31, 725–735. doi: 10.1016/S1002-0160(21)60022-X
Liao, L. B., Chen, X. X., Xiang, J., Zhang, N. N., Wang, E. T., and Shi, F. S. (2022). Zanthoxylum bungeanum root-rot associated shifts in microbiomes of root endosphere, rhizosphere, and soil. PeerJ. 10:e13808. doi: 10.7717/peerj.13808
Liu, T. Y., Chen, C. H., Yang, Y. L., Tsai, I. J., Ho, Y. N., and Chung, C. L. (2022). The brown root rot fungus phellinus noxius affects microbial communities in different root-associated niches of Ficus trees. Environ. Microbiol. 24, 276–297. doi: 10.1111/1462-2920.15862
Marcos, M. S., Bertiller, M. B., and Olivera, N. L. (2019). Microbial community composition and network analyses in arid soils of the Patagonian Monte under grazing disturbance reveal an important response of the community to soil particle size. Appl. Soil Ecol. 138, 223–232. doi: 10.1016/j.apsoil.2019.03.001
Mcguire, K. L., Payne, S. G., Palmer, M. I., Gillikin, C. M., Keefe, D., Kim, S. J., et al. (2013). Digging the New York city skyline: soil fungal communities in green roofs and city parks. PloS One 8:e58020. doi: 10.1371/journal.pone.0058020
Meng, T. Z., Wang, Q. J., Abbasi, P., and Ma, Y. (2019). Deciphering differences in the chemical and microbial characteristics of healthy and fusarium wilt-infected watermelon rhizosphere soils. Appl. Microbiol. Biotechnol. 103, 1497–1509. doi: 10.1007/s00253-018-9564-6
Nguyen, N. H., Song, Z. W., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Park, I., Seo, Y. S., and Mannaa, M. (2023). Recruitment of the rhizo-microbiome army: assembly determinants and engineering of the rhizosphere microbiome as a key to unlocking plant potential. Front. Microbiol. 14:1163832. doi: 10.3389/fmicb.2023.1163832
Pascale, A., Proietti, S., Pantelides, I. S., and Stringlis, I. A. (2020). Modulation of the root microbiome by plant molecules: the basis for targeted disease suppression and plant growth promotion. Front. Plant Sci. 10:1741. doi: 10.3389/fpls.2019.01741
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Shi, W. C., Li, M. C., Wei, G. S., Tian, R. M., Li, C. P., Wang, B., et al. (2019). The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome. Microbiome. 7:14. doi: 10.1186/s40168-019-0629-2
Takahashi, Y., and Nakashima, T. (2018). Actinomycetes, an inexhaustible source of naturally occurring antibiotics. Antibiotics. 7:45. doi: 10.3390/antibiotics7020045
Tian, L. Y., Wang, Y. L., Yu, J., Xiong, D., Zhao, H., and Tian, C. (2016). The mitogen-activated protein kinase VdPbs2 of Verticillium dahliae regulates microsclerotia formation, stress response, and plant infection. Front. Microbiol. 7:1532. doi: 10.3389/fmicb.2016.01532
Wang, Y. L., Wang, Y., and Tian, C. M. (2013). Quantitative detection of pathogen DNA of Verticillium wilt on smoke tree Cotinus coggygria. Plant Dis. 97, 1645–1651. doi: 10.1094/PDIS-04-13-0406-RE
Wei, F., Feng, H. J., Zhang, D. Z., Feng, Z. L., Zhao, L. H., Zhang, Y. L., et al. (2021). Composition of rhizosphere microbial communities associated with healthy and Verticillium wilt diseased cotton plants. Front. Microbiol. 12:618169. doi: 10.3389/fmicb.2021.618169
Wei, X. M., Wang, X. Y., Cao, P., Gao, Z. T., Chen, A. J., and Han, J. P. (2020). Microbial community changes in the rhizosphere soil of healthy and rusty Panax ginseng and discovery of pivotal fungal genera associated with rusty roots. Biomed. Res. Int. 2020, 1–13. doi: 10.1155/2020/8018525
Wei, F., Zhao, L. H., Xu, X. M., Feng, H. J., Shi, Y. Q., Deakin, G., et al. (2019). Cultivar dependent variation of the cotton rhizosphere and endosphere microbiome under field conditions. Front. Plant Sci. 10:1659. doi: 10.3389/fpls.2019.01659
Xiong, D. G., Wang, Y. L., Ma, J., Klosterman, S. J., Xiao, S. X., and Tian, C. M. (2014). Deep mRNA sequencing reveals stage-specific transcriptome alterations during microsclerotia development in the smoke tree vascular wilt pathogen, Verticillium dahliae. BMC Genomics 15:324. doi: 10.1186/1471-2164-15-324
Yin, C. T., Vargas, C. J. M., Schlatter, D. C., Hagerty, C. H., Hulbert, S. H., and Paulitz, T. C. (2021). Rhizosphere community selection reveals bacteria associated with reduced root disease. Microbiome. 9:86. doi: 10.1186/s40168-020-00997-5
Zheng, Y. X., Wang, J. M., Zhao, W. L., Cai, X. J., Xu, Y. L., Chen, X. L., et al. (2022). Effect of bacterial wilt on fungal community composition in rhizosphere soil of tobaccos in tropical Yunnan. Plant Pathol J. 38, 203–211. doi: 10.5423/PPJ.OA.03.2022.0035
Zhou, Y. J., Li, J. H., Friedman, R. C., and Wang, H. F. (2017). Variation of soil bacterial communities in a chronosequence of rubber tree (Hevea brasiliensis) plantations. Front. Plant Sci. 8:849. doi: 10.3389/fpls.2017.00849
Zhou, X., Wang, J. T., Wang, W. H., Tsui, C. K. M., and Cai, L. (2021). Changes in bacterial and fungal microbiomes associated with tomatoes of healthy and infected by fusarium oxysporum f. sp. lycopersici. Microb. Ecol. 81, 1004–1017. doi: 10.1007/s00248-020-01535-4
Keywords: Cotinus coggygria, Verticillium wilt, soil microbiome, rhizosphere, plant health, Illumina sequencing
Citation: Zhao J, Cheng Y, Jiang N, Qiao G and Qin W (2023) Rhizosphere-associated soil microbiome variability in Verticillium wilt-affected Cotinus coggygria. Front. Microbiol. 14:1279096. doi: 10.3389/fmicb.2023.1279096
Edited by:
Entao Wang, National Polytechnic Institute (IPN), MexicoReviewed by:
Martha Helena Ramírez-Bahena, University of Salamanca, SpainJinshui Yang, China Agricultural University, China
Copyright © 2024 Zhao, Cheng, Jiang, Qiao and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wentao Qin, cWlud2VudGFvQGlwcGJhYWZzLmNu