 Zhongxing Xu
Zhongxing Xu Xuan Wang3,4
Xuan Wang3,4 Jia Meng
Jia Meng Lin Zhang
Lin Zhang Bowen Song
Bowen Song- 1Department of Public Health, School of Medicine and Holistic Integrative Medicine, Nanjing University of Chinese Medicine, Nanjing, China
- 2School of AI and Advanced Computing, Xi'an Jiaotong-Liverpool University, Suzhou, China
- 3Department of Biological Sciences, Xi'an Jiaotong-Liverpool University, Suzhou, China
- 4Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, United Kingdom
- 5AI University Research Centre, Xi'an Jiaotong-Liverpool University, Suzhou, China
- 6School of Information and Control Engineering, China University of Mining and Technology, Xuzhou, China
5-Methyluridine (m5U) is one of the most common post-transcriptional RNA modifications, which is involved in a variety of important biological processes and disease development. The precise identification of the m5U sites allows for a better understanding of the biological processes of RNA and contributes to the discovery of new RNA functional and therapeutic targets. Here, we present m5U-GEPred, a prediction framework, to combine sequence characteristics and graph embedding-based information for m5U identification. The graph embedding approach was introduced to extract the global information of training data that complemented the local information represented by conventional sequence features, thereby enhancing the prediction performance of m5U identification. m5U-GEPred outperformed the state-of-the-art m5U predictors built on two independent species, with an average AUROC of 0.984 and 0.985 tested on human and yeast transcriptomes, respectively. To further validate the performance of our newly proposed framework, the experimentally validated m5U sites identified from Oxford Nanopore Technology (ONT) were collected as independent testing data, and in this project, m5U-GEPred achieved reasonable prediction performance with ACC of 91.84%. We hope that m5U-GEPred should make a useful computational alternative for m5U identification.
Introduction
To date, over 170 types of RNA modifications have been identified, occurring on various RNA molecules and influencing nearly every stage of RNA's lifecycle. Scientific research has revealed that these chemical modifications play pivotal roles in numerous critical biological processes (Ontiveros et al., 2019), such as embryonic development (Zhong et al., 2008), cancer development (Zhang et al., 2016a,b), gene-expression regulation (Carlile et al., 2014), and stress response (Wang et al., 2017). Studies have consistently highlighted the significant role of RNA modification in the field of microbiology, encompassing a wide range of aspects, such as the host's m6A-marked transcriptome response to the presence of microbiota in mice (Wang et al., 2019), the maintenance of homeostasis between hosts and microbes through modification statuses (Zhuo et al., 2022), and the modulation of host–cell interactions driven by RNA modification (Kostyusheva et al., 2021).
Among over 170 types of chemical markers, RNA 5-methyluridine (m5U) is one of the most prevalent and plays a significant role in RNA stability, transcription, and translation. For instance, m5U contributes positively to the stability of RNA structures, enhancing their function by modifying base stacking and shaping secondary structures (Agris et al., 2007). Moreover, research studies have demonstrated that m5U modification may be associated with virus replication, antiviral immunity, and the development of certain diseases (Väre et al., 2017). Therefore, accurate identification of m5U holds profound implications for comprehending fundamental biological processes and functions across different species.
Wet-lab experimental approaches combined with high-throughput sequencing techniques have offered experimentally validated m5U sites in multiple species (Xuan et al., 2018; Carter et al., 2019). However, wet-lab approaches can be a costly and time-consuming process; thus, an increasing number of computational efforts have been made, targeting different aspects of biological problems, including phosphorylation prediction (Zhang G. et al., 2023), protein structure prediction (Jumper et al., 2021), drug discovery (Chen et al., 2023), and microbiome studies (Goodswen et al., 2021; Jiang et al., 2022; Yuan et al., 2023). For epitranscriptomic field, a number of bioinformatics databases (Boccaletto et al., 2018; Luo et al., 2021; Song et al., 2021, 2023; Bao et al., 2023; Liang et al., 2023) and in silico prediction frameworks (Qiu et al., 2017; Zhai et al., 2018; Chen et al., 2019; Körtel et al., 2021; Xiong et al., 2021; Liang et al., 2022; Song et al., 2022; Yao et al., 2023) have been widely applied. For example, SRAMP was the first sequence-based framework for m6A prediction (Zhou et al., 2016), which was also capable of predicting the binding sites of YTHDF1 and YTHDF2. In addition to SRAMP, m6A-Reader (Zhen et al., 2020) was developed specifically to unveil the target specificity and regulatory function of six m6A reader proteins (YTHDF1-3, YTHDC1-2, and EIF3A), from which users can identify the putative m6A sites involving specific m6A enzymes. In terms of m5U RNA modification, Jiang et al. (2020) proposed the first sequence-based human m5U prediction framework m5UPred, followed by iRNA-m5U targeting yeast transcriptome (Feng and Chen, 2022). The prediction performance of human m5U has been further improved by m5U-SVM (Ao et al., 2023) and m5U-autoBio (Yu et al., 2023). In addition, RNADSN was developed by learning the common features between tRNA m5U and mRNA m5U (Li et al., 2022). These studies together have greatly facilitated the in silico identification of m5U modification. However, the predictive performance of most computational models is limited by methods that rely on primary sequence-based feature encoding, which does not account for nucleotide frequencies in the training dataset (Hebsgaard et al., 1996), so it is difficult to obtain more complete information from the entire dataset.
To complement sequence-derived features with a more comprehensive understanding of sample information, here, we present m5U-GEPred, the first m5U prediction framework that combines sequence-derived features and graph embeddings to identify putative m5U modification site. Specifically, m5U-GEPred applies a feature extraction strategy of graph embedding techniques for m5U identification, which uses neighborhood-based node embedding technology to obtain feature representations containing information related to other samples through unsupervised learning. With more refined feature extraction, m5U-GEPred outperformed the state-of-the-art m5U predictors built on human and yeast transcriptome, with an average AUROC of 0.984 and 0.985, respectively. In addition, we further collected the human m5U modification sites deriving from Oxford Nanopore Technology (ONT) as independent testing datasets, and the proposed m5U-GEPred achieved a reasonable prediction performance with ACC of 91.84%. The overall framework of m5U-GEPred is presented in Figure 1.
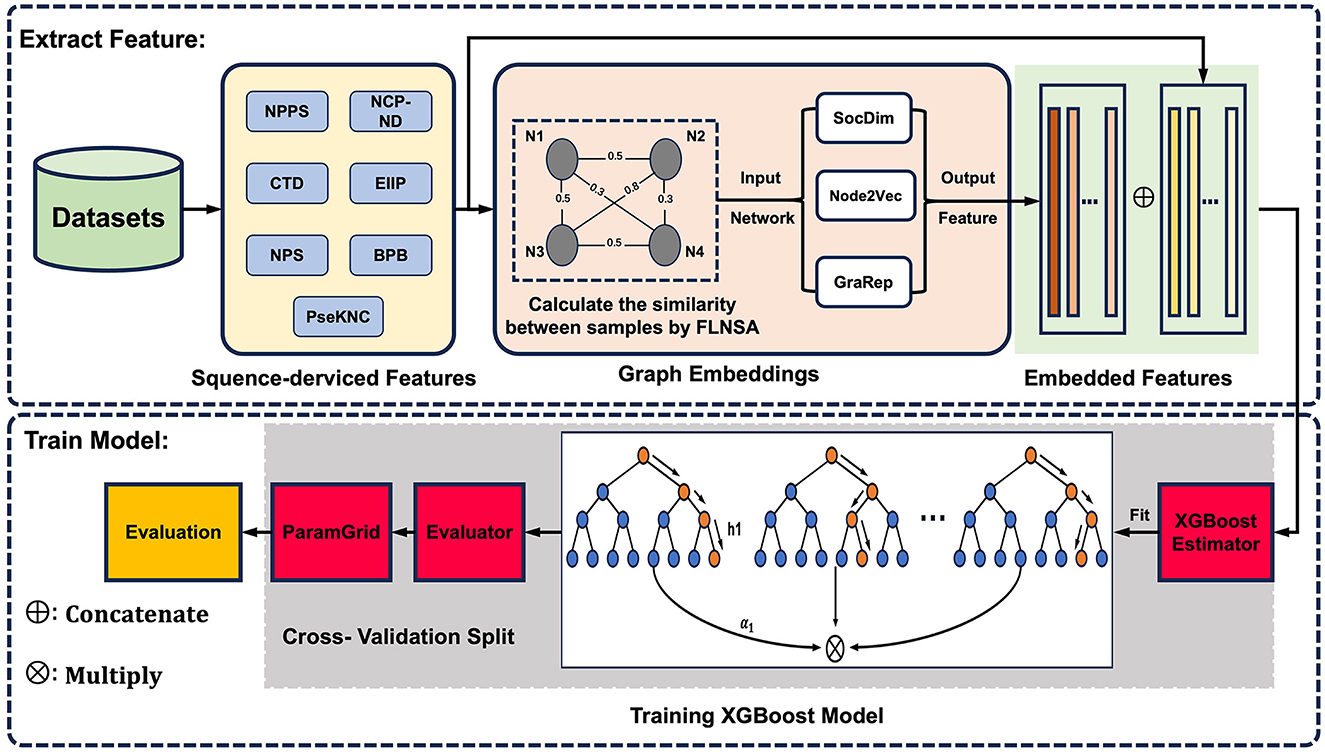
Figure 1. Overall framework of m5U-GEPred. m5U-GEPred was developed by merging sequence-derived information and graph embeddings for accurate m5U identification, which used neighborhood-based node embedding through unsupervised learning to extract feature representation containing information from other samples.
Materials and methods
Benchmark datasets
To build the prediction framework, we obtained the human and yeast m5U modification sites from previously published m5UPred (Jiang et al., 2020) and iRNA-m5U (Feng and Chen, 2022), respectively. The experimentally validated human m5U sites separated by techniques (miCLIP-seq/FICC-seq) and cell lines (HEK293/HAP1) were extracted for cross-techniques and cross-cell-type validations. Specifically, we used m5U sites identified in miCLIP-seq for model development and tested on FICC-seq and vice versa. A total of 3,696 m5U sites were obtained from m5UPred to extract the global information of m5U data under full transcript mode. In addition to human datasets, the training and testing yeast m5U sites were derived from iRNA-m5U, from which 744 positive/negative sites were collected. The positive and negative data were all 41 nt sequences with m5Us or unmodified Us in the center.
To further test the performance of our newly proposed framework, the experimentally validated m5U sites identified from Oxford Nanopore Technology (ONT) were collected from DirectRMDB (Zhang Y. et al., 2023) and used as independent testing data. Detailed m5U datasets used in this study are presented in Supplementary material.
Model architecture
Inspired by previous studies targeting sequence extraction and graph embedding learning (Zheng et al., 2018; Wang et al., 2021b; Hu et al., 2023), the newly proposed m5U-GEPred can be divided into two main phases (see Figure 1). In phase one, feature extraction involved extracting the sequence-derived information and learning graph embeddings. Seven sequence-based encoding methods were used to convert RNA sequences into numerical vectors. Next, by combining the entire dataset and sequence-derived features, a fast linear neighborhood similarity method constructs a global information network, where samples represent network nodes and edges signify the similarity relationships between the samples. Three unsupervised neighborhood-based node embedding methods, namely, SocDim, Node2Vec, and GraRep, are utilized to learn the characteristics of each node within the global information network, ensuring that graph embedding features of RNA sequences contain relevant information from other samples. Finally, these two types of features are integrated through a feature fusion strategy.
Phase two focused on model building. The data were divided into training and testing datasets, maintaining an 8:2 ratio. The training set was used to train the XGBoost model, while the test set was employed to assess the performance of the predictor. In addition, the cross-technique and cross-cell-type validation were further employed for performance evaluation, where the predictor was trained by m5U sites obtained from one technique/cell type and tested on another one. In this project, all the scripts used to build m5U-GEPred are freely accessible, as shown in Supplementary material.
Sequence-based information
Sequence composition and frequency
The nucleotide pair spectrum (NPS) encoding method captures the RNA-seq environment at a specific position by counting the frequency of occurrence of all k-spacer nucleotide pairs in the RNA-seq (Zhou et al., 2016). The k-spaced nucleotide pair is L1{N}L2. Taking sequence “AXXTXG” as an example, “AT” represents a nucleotide pair with a 2 spacing, and “TG” represents a nucleotide pair with a 1 spacing. The window W is the distance from L2 to L1. There are N arbitrary nucleotide pairs between L1 and L2, so the frequency can be calculated as follows:
where C(L1{N}L2) is the count of L1{N}L2 inside a flanking window, and d is the space between two nucleotides ranging from 0 to dmax. The encoding method converts a gene sequence into a vector DNPS with a dimension of 4 × 4 × (dmax+1) = 48. The optimized dmax was 2 for prediction modes.
The composition, transition, and distribution (CTD) method (Tong and Liu, 2019) is employed to represent global transcribed sequence descriptors. CTD features encompass nucleotide composition, nucleotide transition, and nucleotide distribution, with the latter two serving as RNA secondary structure features essential for classifying coding RNAs. Nucleotide composition (first index C) refers to the percentage composition of each nucleotide present in a transcribed RNA sequence. Nucleotide transitions (second index T) denote the percentage frequency of four nucleotide transitions occurring between adjacent positions. Finally, nucleotide distribution (third index D) illustrates the five relative positions of each nucleotide along the transcribed RNA sequence, specifically at 0% (first), 25%, 50%, 75%, and 100% (last). Both nucleotide transition and nucleotide distribution characteristics play crucial roles in the classification of coding RNAs.
The Bi-profile Bayes feature extraction method describes a Bayesian decision function (Shao et al., 2009). Given an RNA-seq sample S = {s1, s2, s3, ⋯ , sn}, S can be divided into two categories, namely, f+ and f−, where f+ and f− represent the nucleotide sequence data of known modified sites (positive dataset) and unmodified sites (negative dataset). For each position in the positive and negative datasets, the probability of occurrence of each base (A, C, G, and U) is calculated. According to Bayes' rule, the posterior probability of S for these two categories can be given as follows:
Assuming that the prior distribution of category is uniform, namely, P(f+) = P(f−), the formula is as follows:
where is weigh vector, and is the posterior probability vector. With respect to training sample, S, f(S) = 1 corresponds to class f+ and f(S) = −1 corresponds to class f−. In this study, represents the posterior probability of each nucleotide at each position in f+ (positive feature space) and represents the posterior probability of each nucleotide at each position in f− (negative feature space), which we call Bi-profile.
Physical and chemical properties
Electron–ion interaction pseudo-potential (EIIP) encoding method (Nair and Sreenadhan, 2006) converts the nucleotides A, G, C, and U in the RNA sequence into their corresponding electron–ion interaction pseudo-potentials by using a simple “EIIP indicator sequence” potential value. Specifically, the EIIP values of nucleotides are as follows: A (adenosine) 0.1260, C (cytosine) 0.1340, G (guanine) 0.0806, and U (uracil) 0.1335. In this method, each nucleotide is assigned a real number associated with its corresponding EIIP value.
Local structure information
Pseudo-k-component nucleotide assemblies (PseKNC) are inspired by the PseAAC approach in computational proteomics to represent RNA sequence samples by incorporating global or long-range sequence order effects (Guo et al., 2014).
Converting a gene sequence into vector
where
In the above equation, is the frequency of k-tuple nucleotide composition (i.e., the combination of k consecutive nucleotides). w is the weight factor. λ is the number of RNA sequence-associated cascades. θj and Θ(RiRi+1,Ri+jRi+j+1) are given as follows:
where μ is the number of selected local RNA structural features. For a given dinucleotide RiRi+1 at position i, we assign a numerical value Pv(RiRi+1) for the v-th local RNA structural property [where (v = 1, 2, …, μ)]. Pν(Ri+jRi+j+1) represents the corresponding value for the dinucleotide Ri+jRi+j+1 at position i + j. We consider six local RNA structural properties. The detailed values used for the six physical structural properties were extracted from a previous study (Guo et al., 2014) and are presented in Supplementary Table S1.
Periodicity features
The nucleotide chemical properties and nucleotide distribution (NCP-ND) feature coding approach combines the chemical properties of nucleotides and their distribution (Bari et al., 2013). Nucleotide distribution is used to measure the density dj of a specific nucleotide Hj at position j and can be derived as follows:
where
n is an RNA sequence of length, j = 1, 2, 3, …n . DNCP−ND is an n×4 dimensional vector.
In the nucleotide chemical property coding scheme, each nucleotide in the RNA sequence exhibits a different function according to its unique chemical structure, thus defining the three coordinate values of the coding scheme.
Nucleotide pair features in sequence
NPPS is a feature representation algorithm based on the position specificity of multi-interval nucleotide pairs (Xing et al., 2017). The frequencies of occurrences of different nucleic acid types are stored at different positions of negative datasets in arrays , :
and are calculated similarly in the positive dataset. Suppose the k-th nucleotide is “U” and the (k+ξ)-th nucleotide is “G”, can be calculated using the conditional probability formula and the frequency matrix as follows:
The dimension of the vector DNPPSis C2,as pk=-.
Graph embedding
To obtain the graph embedding features for each RNA sequence, we build a network encompassing the entire dataset. Within this network, each RNA sequence is considered a node, and the connections between RNA sequences are represented by edges, which typically connect two similar sample nodes. The fast linear neighbor similarity approach (FLNSA) is an efficient method for extracting “sample–sample” similarity (Zhang et al., 2018).
The sequence-derived features, which we extracted above, are converted into n-dimensional feature vectors x1, x2, ⋯ , xm, and each row represents a sample vector and converts the vector into an m*n matrix:
C represents an indicator matrix, where C(i, j) = 1, if xj is a neighbor of xi, and C(i,j) = 0 otherwise, with C(i,i) = 0. The set of neighbors for xi, denoted as N(xi), is determined based on the Euclidean distance between xi and other data points.
Generally, a portion of xi's neighbors is selected based on distance, and the ratio of neighbor points to all data points is referred to as the neighborhood ratio, denoted as K. The Frobenius norm is represented by ∥·∥F. The column vector e, with all elements equal to 1, is denoted as (1, 1, …, 1)T, while ⊙ signifies the Hadamard product. The tradeoff parameter, μ, is set to 3. W is an m × m weight matrix, where the i-th row of W indicates the reconstruction contributions of other data points to the data pointxi.
Wij can be re-calculated as follows:
Let λ=μe.
Finally, an undirected and unweighted graph is constructed with w as the adjacency matrix.
SocDim
When multiple relationships are associated with the same network, the SocDim method (Tang and Liu, 2009) can extract the social dimensions of different affiliations of participants hidden in the network and convert them into features for discriminative learning. The method measures the effective amount of community structure in a complex network by measuring the degree of offset (modularity) between interactive platforms and platforms in the network, which involves community detection, a fundamental task in social network analysis.
The modular is defined as follows:
Modularity can be reformulated as follows:
When Q > 0, it means that soft clustering captures a certain degree of community structure. The modularity matrix is defined as follows:
where A is the interaction matrix, m is the number of nodes, and d is the column vector of nodes. SocDim can extract dimensions (B) on the top of the module matrix of the network.
Node2Vec
Node2Vec is a graph embedding algorithm designed to learn continuous feature representations of nodes within networks (Grover and Leskovec, 2016). This algorithm aims to learn the mapping of nodes to a low-dimensional feature space, maximizing the preservation of neighborhood information within the network. It employs a biased random walk procedure for efficient exploration of diverse communities, enabling the acquisition of richer representations. Node2Vec formulates the learning of feature vectors in the network as a maximum likelihood optimization problem, which is addressed through the Skip-gram architecture. The objective function is the logarithmic probability of the network neighborhood Ns(u) by maximizing the observed node u as follows:
Node2Vec employs two sampling strategies (Figure 2): breadth-first sampling (BFS) and depth-first sampling (DFS), which are based on the network community (nodes directly adjacent to the starting node) and the structural role of the node (the distance from the source node gradually increasing nodes) principle. For example, in a neighborhood of size 3, BFS will sample three nodes N1, N2, and N3, while DFS will sample three nodes N4, N5, and N6.
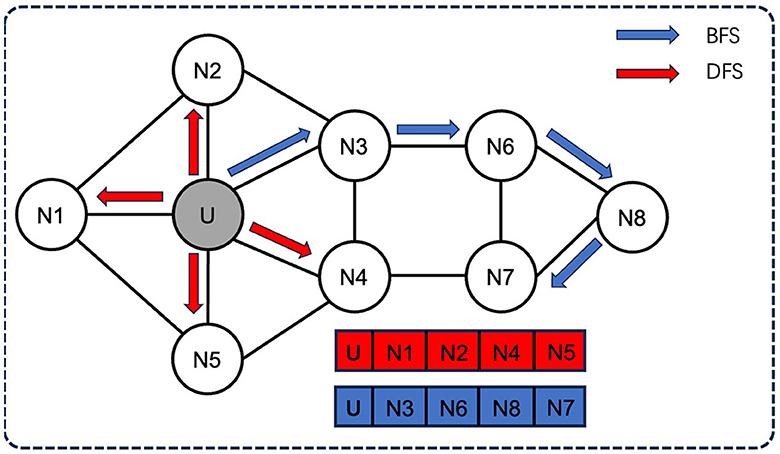
Figure 2. Two sampling strategies of Node2Vec: BFS and DFS. Node2Vec employs two sampling strategies: breadth-first sampling (BFS) and depth-first sampling (DFS), both of which aim to capture different aspects of the network structure. For example, in a neighborhood of size 3, BFS will sample three nodes N1, N2, and N3, while DFS will sample three nodes N4, N5, and N6.
GraRep
GraRep is an algorithm that captures relevant global structural information of a graph by learning the latent representation of vertices on a weighted graph (Cao et al., 2015). The algorithm manipulates different global transformation matrices and extracts various k-step relationship information between vertices with different k values directly from the graph. First, the k-step probability matrix Ak is calculated using the inverse matrix of the degree matrix D and the adjacency matrix S. Then, the k-step logarithmic probability matrix Xkis calculated and adjusted appropriately, and the positive-logarithmic probability matrix Xk is factorized by SVD to construct the representation vector Wk rows, thereby obtaining the k-step representation of each vertex. Finally, all k-step representations are concatenated into a global representation.
Furthermore, GraRep designs an accurate on-graph loss function by incorporating non-linear combinations of different local relational information and extending it to support weighted graphs.
Model construction and performance evaluation
XGBoost is an advanced gradient-boosting algorithm that has consistently displayed outstanding performance (Chen and Guestrin, 2016). Compared with other state-of-the-art gradient boosting techniques, such as CatBoost (Dorogush et al., 2018; Prokhorenkova et al., 2018) and LightGBM (Ke et al., 2017), XGBoost offers the following advantages: (1) It employs a regularized learning framework that prevents overfitting and enhances model generalization; (2) XGBoost utilizes an efficient and parallelized tree construction algorithm to accelerate the training process; (3) it supports the handling of sparse data and missing values, making it suitable for various real-world applications; (4) XGBoost has an extensive range of hyperparameters for tuning, allowing for flexibility and customization to fit specific tasks and datasets. By leveraging these advantages, XGBoost has consistently proven to be a powerful and versatile tool for a wide array of machine learning problems.
For performance evaluation, we applied the following evaluation metrics. In general, the receiver operating characteristic (ROC) curve (sensitivity against 1-specificity) and the area under the ROC curve (AUROC) were used as the primary performance evaluation metrics. In addition, we also calculated sensitivity (Sn), specificity (Sp), Matthews correlation coefficient (MCC), and overall accuracy (ACC) as additional indicators for evaluating the reliability of the model. A five-fold cross-validation was applied on training datasets, while the testing datasets were used for independent testing. Only the m5U sites that were not included as part of the training data were selected for independent testing purposes.
Among them, TP represents true positive, while TN represents true negative; FP stands for the number of false positive, and FN stands for the number of false negative.
Results
Performance evaluation in the human transcriptome
The prediction performance of m5U-GEPred in human transcriptome was first evaluated by independent testing datasets and compared with previously published models. For a fair comparison, m5UPred and m5U-autoBio were selected as baseline models based on the same datasets employed. As shown in Table 1, the proposed m5U-GEPred achieved reasonable improvements in prediction performances.
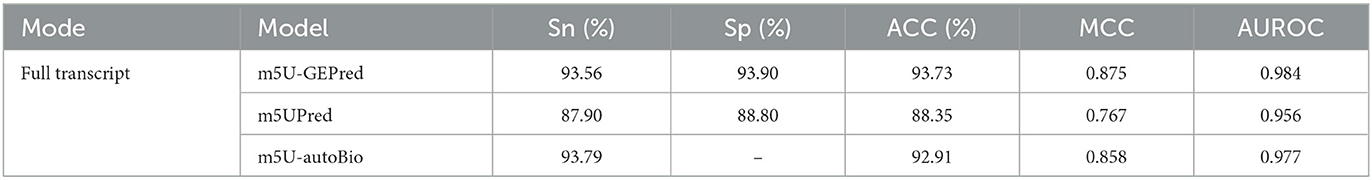
Table 1. Prediction performance using independent testing dataset.
Performance evaluation by cross-technique and cross-cell-type validation
Following m5UPred, we, then, divided the experimentally validated m5U sites according to their profiling techniques (miCLIP and FICC-seq) and cell lines (HEK293 and HAP1). For five-fold cross-validation (see Table 2), our newly proposed m5U-GEPred achieved an average AUROC of 0.964 and 0.968 under cross-technique and cross-cell-type validation, respectively, marking reasonable improvements in accuracy compared with the baseline model m5UPred (0.956 and 0.955). In terms of independent testing, m5U-GEPred (0.952 and 0.967) outperformed m5UPred (0.882 and 0.899) with increasing improvements. We also compared the performance of m5U-GEPred with the recently published model m5U-autoBio. When tested on an independent dataset, the performance of m5U-GEPred also outperformed m5U-autoBio (0.883 and 0.921), suggesting the reliability of our newly proposed approach.
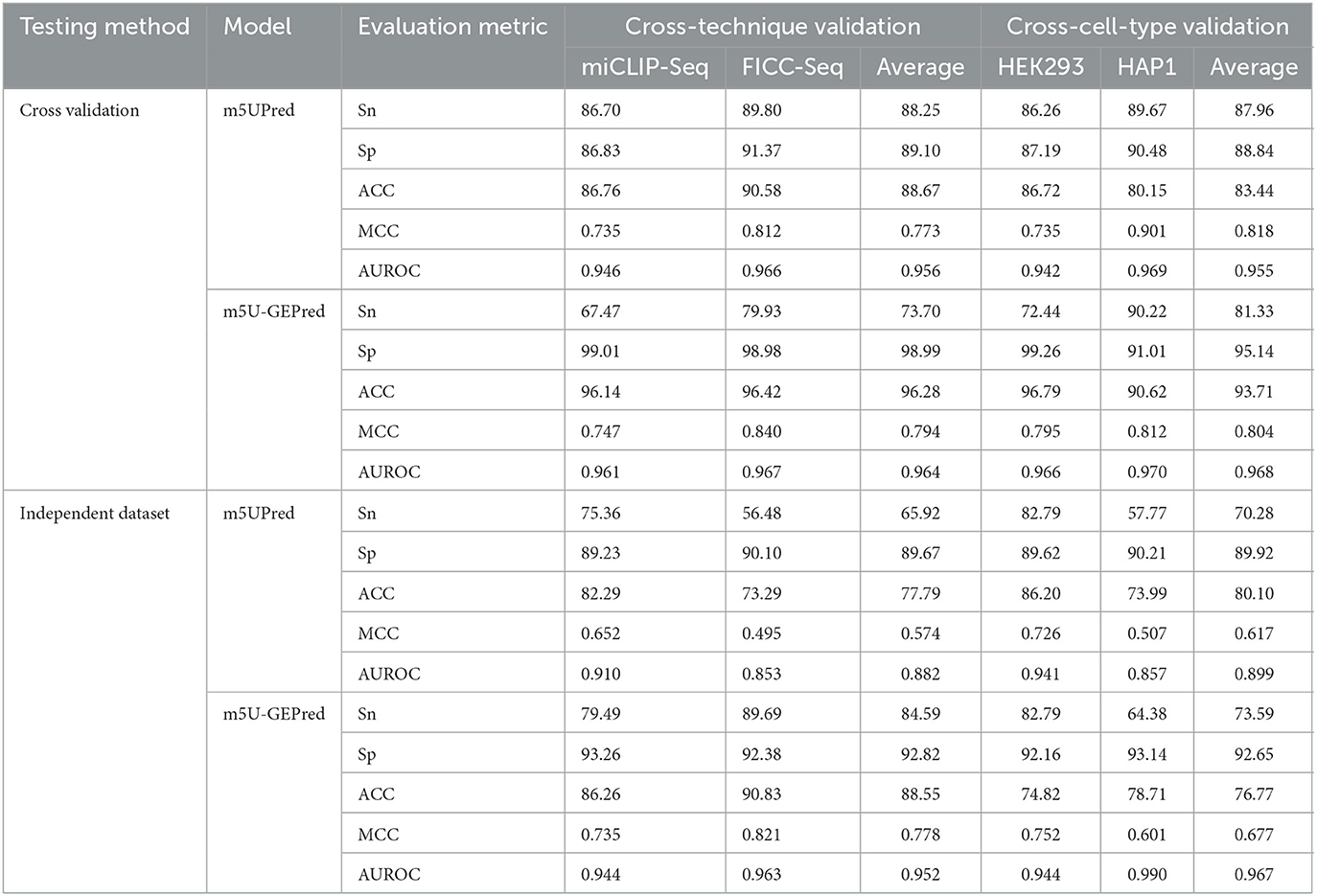
Table 2. Cross-technique and cross-cell-type validation on full transcript mode.
Independent testing by m5U sites generated from nanopore direct RNA sequencing
To further test the performance of our newly proposed framework, the m5U sites identified by Oxford Nanopore Technology (ONT) were collected as independent testing data. A total of 98 ONT-derived m5U sites were extracted from DirectRMDB, and m5U-GEPred successfully identified 90 of them with an ACC of 91.84%.
Performance evaluation of m5U-GEPred in yeast transcriptome
In addition to human datasets, the proposed framework was also evaluated by yeast datasets. As shown in Table 3, the performance of m5U-GEPred systemically outperformed iRNA-m5U, which was, to the best of our knowledge, the only available m5U predictor in yeast transcriptome. For a fair comparison, the training and testing datasets used to build yeast predictor were exactly the same as iRNA-m5U.
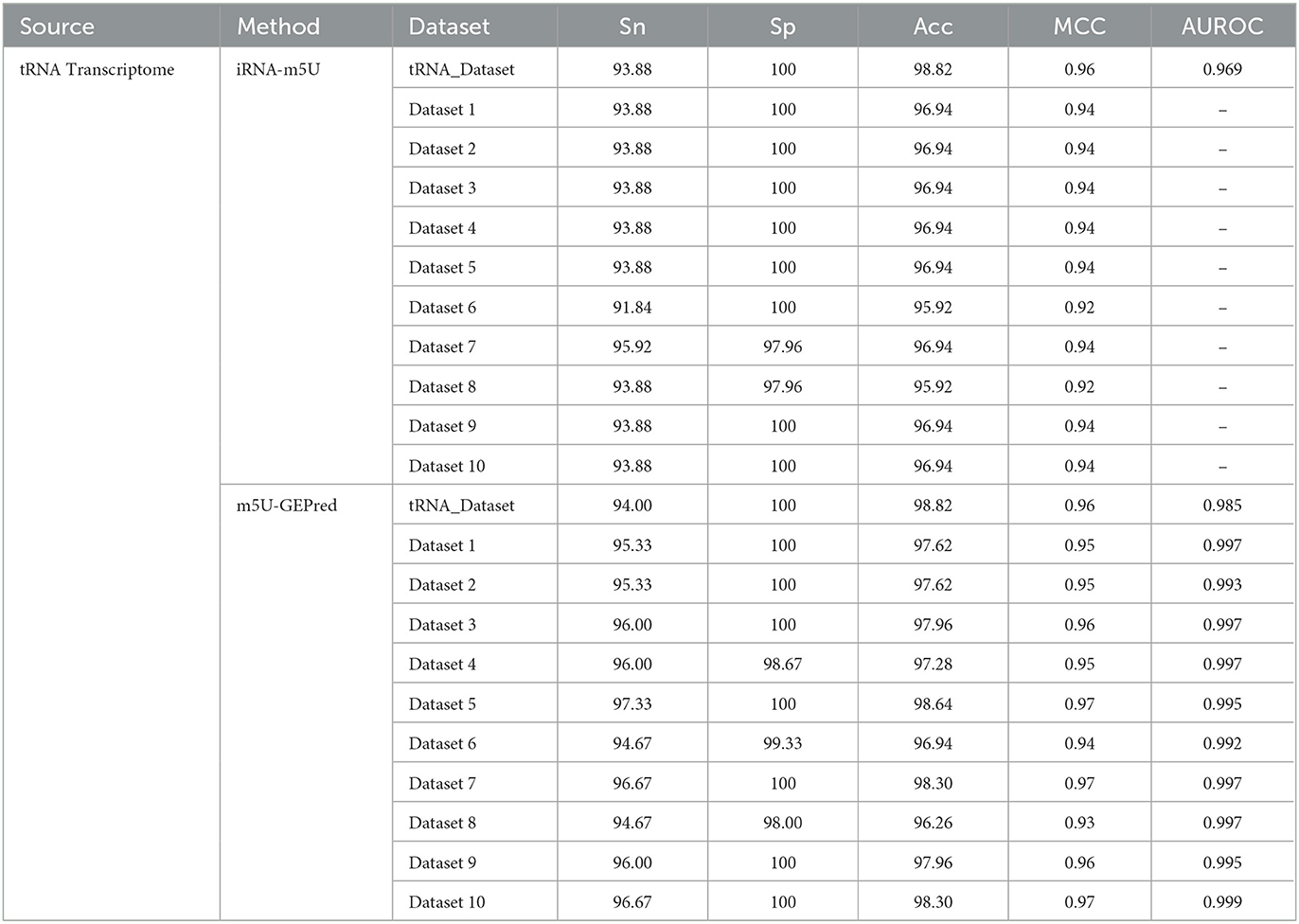
Table 3. Prediction performance of yeast m5U dataset.
In addition, we conducted cross-species validation using human and yeast m5U datasets. As shown in Supplementary Table S2, the results indicated that m5U modification may exhibit distinct patterns in yeast and human transcriptomes, respectively, suggesting the need to develop species-specific models for m5U identification. These findings are consistent with a previous study iRNA-m5U (Feng and Chen, 2022), which only correctly identified 22.45% of human m5U sites using yeast training datasets.
Functional characterization of the predicted m5U modification sites using m5U-GEPred
To try to further interpret the prediction results related to biological aspects, we performed a transcriptome-wide prediction of putative m5U sites using the newly proposed model. Specifically, we randomly selected 10,000 Us from various types of RNAs of human transcripts and predicted their m5U probabilities. Using 0.5 as a cutoff, 224 putative m5U modification sites were identified. First, we tried to interpret the prediction results by plotting the overall distribution of the putative m5U modification sites using MetaTX (Wang et al., 2021a). The results suggested that putative m5U sites were enriched in the 5′UTR (Supplementary Figure S1A). We further performed the gene ontology enrichment analysis of their hosting genes, and as shown in Supplementary Figure S1B, the top 10 biological processes enriched with the predicted m5U sites. It may be worth noting that the reason for selecting 0.5 as a general cutoff threshold is that machine learning classifiers usually obtain the lowest empirical rate at a value of 0.5. We further examined the predicted m5U sites using different cutoff thresholds (Supplementary Table S3). Additionally, the above results were observed by screening a small portion of the transcriptome (10,000 sites), and these results (~2% of positive results) only suggest a high m5U probability at the sequence level (learned from the sequences of positive samples), which should be combined with customized cutoff thresholds (Supplementary Table S3) and wet-lab approaches for final determination. In conclusion, a computational model combined with functional analysis can be a valuable alternative for target identification and result interpretation.
Conclusion
The accurate identification of 5-methyluridine (m5U) modification sites within RNAs holds profound biological significance. In this study, a novel computational approach m5U-GEPred was proposed for m5U identification across two independent species. m5U-GEPred combined sequence characteristics and graph embedding-based information, extracting the global information of training data that complemented the local information represented by conventional sequence features. In addition, it may be worth noting that the m5U sites detected by experimental approaches may directly relate to reads mapped to expressed genes. This limited the detecting power of modified residues located on low expressed genes under specific conditions. The proposed framework m5U-GEPred accepts sequence information solely as input, which could serve as a useful alternative for m5U identification. In addition, the results showed that our newly proposed framework achieved reasonable improvements in prediction performance, compared with the state-of-the-art models developed in human and yeast transcriptome, respectively.
Nevertheless, the proposed m5U-GEPred was developed by combining sequence-based features and graph embedding information, which achieved enhanced prediction performance. The enhanced results suggest that the experimentally identified m5U modification sites may have a strong sequence pattern, but the reverse may not necessarily be true (the sequence may be just one of the key features for determining m5U). Consequently, machine learning models provide suggestion for the potentially modified residues based on their learned features, which would significantly narrow down the range of target interests (but still a wider range than final experimental identification) for further wet-lab experiments. Consequently, the m5U prediction framework and its applications can be further expanded by incorporating the latest sequencing data and binding regions of m5U-related enzymes to enable accurate m5U identification under different biological contexts, such as target-specific m5U prediction.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
ZX: Methodology, Writing–original draft. XW: Software, Writing–review and editing. JM: Conceptualization, Writing–review and editing. LZ: Conceptualization, Writing–review and editing. BS: Conceptualization, Data curation, Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Supercomputing Platform of Xi'an Jiaotong-Liverpool University, the National Natural Science Foundation of China (31671373 and 61971422), the Scientific Research Foundation of Nanjing University of Chinese Medicine (Grant No. 013038030001), and the XJTLU Key Program Special Fund (KSF-E-51 and KSF-P-02).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1277099/full#supplementary-material
References
Agris, P. F., Vendeix, F. A., and Graham, W. D. (2007). tRNA's wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 366, 1–13. doi: 10.1016/j.jmb.2006.11.046
Ao, C., Ye, X., Sakurai, T., Zou, Q., and Yu, L. (2023). m5U-SVM: identification of RNA 5-methyluridine modification sites based on multi-view features of physicochemical features and distributed representation. BMC Biol. 21, 93. doi: 10.1186/s12915-023-01596-0
Bao, X., Zhang, Y., Li, H., Teng, Y., Ma, L., Chen, Z., et al. (2023). RM2Target: a comprehensive database for targets of writers, erasers and readers of RNA modifications. Nucleic Acids Res. 51(D1), D269–D279. doi: 10.1093/nar/gkac945
Bari, A. G., Reaz, M. R., Choi, H. J., and Jeong, B. S. (2013). “DNA encoding for splice site prediction in large DNA sequence,” in Database Systems for Advanced Applications: 18th International Conference, DASFAA 2013, International Workshops: BDMA, SNSM, SeCoP, Wuhan, China, April 22-25, 2013. Proceedings 18 (Cham: Springer Berlin Heidelberg), 46–58.
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Bagiński, B., Wirecki, T. K., et al. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307. doi: 10.1093/nar/gkx1030
Cao, S., Lu, W., and Xu, Q. (2015). “GraRep: learning graph representations with global structural information,” in Proceedings of the 24th ACM International on Conference on Information and Knowledge Management (New York, NY: ACM), 891–900. doi: 10.1145/2806416.2806512
Carlile, T. M., Rojas-Duran, M. F., Zinshteyn, B., Shin, H., Bartoli, K. M., Gilbert, W. V., et al. (2014). Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146. doi: 10.1038/nature13802
Carter, J. M., Emmett, W., Mozos, I. R., Kotter, A., Helm, M., Ule, J., et al. (2019). FICC-Seq: a method for enzyme-specified profiling of methyl-5-uridine in cellular RNA. Nucleic Acids Res. 47, e113–e113. doi: 10.1093/nar/gkz658
Chen, T., and Guestrin, C. (2016). “XGBoost: a scalable tree boosting system,” in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (New York, NY: ACM), 785–794. doi: 10.1145/2939672.2939785
Chen, W., Liu, X., Zhang, S., and Chen, S. (2023). Artificial intelligence for drug discovery: resources, methods, and applications. Mol. Ther.-Nucleic Acids 31, 691–702. doi: 10.1016/j.omtn.2023.02.019
Chen, W., Song, X., Lv, H., and Lin, H. (2019). Irna-m2g: identifying n2-methylguanosine sites based on sequence-derived information. Mol. Ther.-Nucleic Acids 18, 253–258. doi: 10.1016/j.omtn.2019.08.023
Dorogush, A. V., Ershov, V., and Gulin, A. (2018). CatBoost: gradient boosting with categorical features support. arXiv. [preprint]. doi: 10.48550/arXiv.1810.11363
Feng, P., and Chen, W. (2022). iRNA-m5U: a sequence based predictor for identifying 5-methyluridine modification sites in saccharomyces cerevisiae. Methods 203, 28–31. doi: 10.1016/j.ymeth.2021.04.013
Goodswen, S. J., Barratt, J. L., Kennedy, P. J., Kaufer, A., Calarco, L., Ellis, J. T., et al. (2021). Machine learning and applications in microbiology. FEMS Microbiol. Rev. 45, fuab015. doi: 10.1093/femsre/fuab015
Grover, A., and Leskovec, J. (2016). “node2vec: scalable feature learning for networks,” in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (New York, NY: ACM), 855–864. doi: 10.1145/2939672.2939754
Guo, S. H., Deng, E. Z., Xu, L. Q., Ding, H., Lin, H., Chen, W., et al. (2014). iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 30, 1522–1529. doi: 10.1093/bioinformatics/btu083
Hebsgaard, S. M., Korning, P. G., Tolstrup, N., Engelbrecht, J., Rouzé, P., Brunak, S., et al. (1996). Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 24, 3439–3452. doi: 10.1093/nar/24.17.3439
Hu, K., Zhu, X., Liu, H., Qu, Y., Wang, F. L., Hao, T., et al. (2023). Convolutional neural network-based entity-specific common feature aggregation for knowledge graph embedding learning. IEEE Trans. Consum. Electron. doi: 10.1109/TCE.2023.3302297
Jiang, J., Song, B., Tang, Y., Chen, K., Wei, Z., Meng, J., et al. (2020). m5UPred: a web server for the prediction of RNA 5-methyluridine sites from sequences. Mol. Ther.-Nucleic Acids 22, 742–747. doi: 10.1016/j.omtn.2020.09.031
Jiang, Y., Luo, J., Huang, D., Liu, Y., and Li, D. D. (2022). Machine learning advances in microbiology: a review of methods and applications. Front. Microbiol. 13, 925454. doi: 10.3389/fmicb.2022.925454
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Ke, G., Meng, Q., Finley, T., Wang, T., Chen, W., Ma, W., et al. (2017). Lightgbm: a highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 30.
Körtel, N., Rücklé, C., Zhou, Y., Busch, A., Hoch-Kraft, P., Sutandy, F. R., et al. (2021). Deep and accurate detection of m6A RNA modifications using miCLIP2 and m6Aboost machine learning. Nucleic Acids Res. 49, e92–e92. doi: 10.1093/nar/gkab485
Kostyusheva, A., Brezgin, S., Glebe, D., Kostyushev, D., and Chulanov, V. (2021). Host-cell interactions in HBV infection and pathogenesis: the emerging role of m6A modification. Emerg. Microbes Infect. 10, 2264–2275. doi: 10.1080/22221751.2021.2006580
Li, Z., Mao, J., Huang, D., Song, B., and Meng, J. (2022). RNADSN: transfer-learning 5-methyluridine (m5U) modification on mRNAs from common features of tRNA. Int. J. Mol. Sci. 23, 13493. doi: 10.3390/ijms232113493
Liang, Z., Ye, H., Ma, J., Wei, Z., Wang, Y., Zhang, Y., et al. (2023). m6A-Atlas v2.0: updated resources for unraveling theN6-methyladenosine (m6A) epitranscriptome amongmultiple species. Nucleic Acids Res. gkad691. doi: 10.1093/nar/gkad691
Liang, Z., Zhang, L., Chen, H., Huang, D., and Song, B. (2022). m6A-Maize: weakly supervised prediction of m6A-carrying transcripts and m6A-affecting mutations in maize (Zea mays). Methods 203, 226–232. doi: 10.1016/j.ymeth.2021.11.010
Luo, X., Li, H., Liang, J., Zhao, Q., Xie, Y., Ren, J., et al. (2021). RMVar: an updated database of functional variants involved in RNA modifications. Nucleic Acids Res. 49, D1405–D1412. doi: 10.1093/nar/gkaa811
Nair, A. S., and Sreenadhan, S. P. (2006). A coding measure scheme employing electron-ion interaction pseudopotential (EIIP). Bioinformation 1, 197.
Ontiveros, R. J., Stoute, J., and Liu, K. F. (2019). The chemical diversity of RNA modifications. Biochem. J. 476, 1227–1245. doi: 10.1042/BCJ20180445
Prokhorenkova, L., Gusev, G., Vorobev, A., Dorogush, A. V., and Gulin, A. (2018). CatBoost: unbiased boosting with categorical features. Adv. Neural Inf. Process. Syst. 31.
Qiu, W. R., Jiang, S. Y., Xu, Z. C., Xiao, X., and Chou, K. C. (2017). iRNAm5C-PseDNC: identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget 8, 41178. doi: 10.18632/oncotarget.17104
Shao, J., Xu, D., Tsai, S. N., Wang, Y., and Ngai, S. M. (2009). Computational identification of protein methylation sites through bi-profile Bayes feature extraction. PLoS ONE 4, e4920. doi: 10.1371/journal.pone.0004920
Song, B., Chen, K., Tang, Y., Wei, Z., Su, J., De Magalhães, J. P., et al. (2021). ConsRM: collection and large-scale prediction of the evolutionarily conserved RNA methylation sites, with implications for the functional epitranscriptome. Brief. Bioinform. 22, bbab088. doi: 10.1093/bib/bbab088
Song, B., Huang, D., Zhang, Y., Wei, Z., Su, J., de Magalhães, J. P., et al. (2022). m6A-TSHub: unveiling the context-specific m6A methylation and m6A-affecting mutations in 23 human tissues. Genomics Proteomics Bioinformatics. doi: 10.1016/j.gpb.2022.09.001
Song, B., Wang, X., Liang, Z., Ma, J., Huang, D., Wang, Y., et al. (2023). RMDisease V2.0 an updated database of genetic variants that affect RNA modifications with disease and trait implication. Nucleic Acids Res. 51(D1), D1388–D1396. doi: 10.1093/nar/gkac750
Tang, L., and Liu, H. (2009). “Relational learning via latent social dimensions,” in Proceedings of the 15th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (New York, NY: ACM), 817–826. doi: 10.1145/1557019.1557109
Tong, X., and Liu, S. (2019). CPPred: coding potential prediction based on the global description of RNA sequence. Nucleic Acids Res. 47, e43–e43. doi: 10.1093/nar/gkz087
Väre, V. Y., Eruysal, E. R., Narendran, A., Sarachan, K. L., and Agris, P. F. (2017). Chemical and conformational diversity of modified nucleosides affects tRNA structure and function. Biomolecules 7, 29. doi: 10.3390/biom7010029
Wang, X., Li, Y., Chen, W., Shi, H., Eren, A. M., Morozov, A., et al. (2019). Transcriptome-wide reprogramming of N6-methyladenosine modification by the mouse microbiome. Cell Res. 29, 167–170. doi: 10.1038/s41422-018-0127-2
Wang, Y., Chen, K., Wei, Z., Coenen, F., Su, J., Meng, J., et al. (2021a). MetaTX: deciphering the distribution of mRNA-related features in the presence of isoform ambiguity, with applications in epitranscriptome analysis. Bioinformatics 37, 1285–1291. doi: 10.1093/bioinformatics/btaa938
Wang, Y., Guo, R., Huang, L., Yang, S., Hu, X., He, K., et al. (2021b). m6AGE: a predictor for n6-methyladenosine sites identification utilizing sequence characteristics and graph embedding-based geometrical information. Front. Genet. 12, 670852. doi: 10.3389/fgene.2021.670852
Wang, Y., Pang, C., Li, X., Hu, Z., Lv, Z., Zheng, B., et al. (2017). Identification of tRNA nucleoside modification genes critical for stress response and development in rice and Arabidopsis. BMC Plant Biol. 17, 1–15. doi: 10.1186/s12870-017-1206-0
Xing, P., Su, R., Guo, F., and Wei, L. (2017). Identifying N6-methyladenosine sites using multi-interval nucleotide pair position specificity and support vector machine. Sci. Rep. 7, 46757. doi: 10.1038/srep46757
Xiong, Y., He, X., Zhao, D., Tian, T., Hong, L., Jiang, T., et al. (2021). Modeling multi-species RNA modification through multi-task curriculum learning. Nucleic Acids Res. 49, 3719–3734. doi: 10.1093/nar/gkab124
Xuan, J. J., Sun, W. J., Lin, P. H., Zhou, K. R., Liu, S., Zheng, L. L., et al. (2018). RMBase v2. 0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 46(D1), D327–D334. doi: 10.1093/nar/gkx934
Yao, J., Hao, C., Chen, K., Meng, J., and Song, B. (2023). “Pseudouridine identification and functional annotation with PIANO,” in Computational Epigenomics and Epitranscriptomics (New York, NY: Springer US), 153–162. doi: 10.1007/978-1-0716-2962-8_11
Yu, L., Zhang, Y., Xue, L., Liu, F., Jing, R., Luo, J., et al. (2023). Evaluation and development of deep neural networks for RNA 5-methyluridine classifications using autoBioSeqpy. Front. Microbiol. 14, 1175925. doi: 10.3389/fmicb.2023.1175925
Yuan, H., Wang, Z., Wang, Z., Zhang, F., Guan, D., Zhao, R., et al. (2023). Trends in forensic microbiology: from classical methods to deep learning. Front. Microbiol. 14, 1163741. doi: 10.3389/fmicb.2023.1163741
Zhai, J., Song, J., Cheng, Q., Tang, Y., and Ma, C. (2018). PEA: an integrated R toolkit for plant epitranscriptome analysis. Bioinformatics 34, 3747–3749. doi: 10.1093/bioinformatics/bty421
Zhang, C., Samanta, D., Lu, H., Bullen, J. W., Zhang, H., Chen, I., et al. (2016a). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Nat. Acad. Sci. 113, E2047–E2056. doi: 10.1073/pnas.1602883113
Zhang, C., Zhi, W. I., Lu, H., Samanta, D., Chen, I., Gabrielson, E., et al. (2016b). Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217-and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget 7, 64527. doi: 10.18632/oncotarget.11743
Zhang, G., Tang, Q., Feng, P., and Chen, W. (2023). IPs-GRUAtt: an attention-based bidirectional gated recurrent unit network for predicting phosphorylation sites of SARS-CoV-2 infection. Mol. Ther. Nucleic Acids 32, 28–35. doi: 10.1016/j.omtn.2023.02.027
Zhang, W., Tang, G., Wang, S., Chen, Y., Zhou, S., Li, X., et al. (2018). “Sequence-derived linear neighborhood propagation method for predicting lncRNA-miRNA interactions,” in 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) (Madrid: IEEE), 50–55. doi: 10.1109/BIBM.2018.8621184
Zhang, Y., Jiang, J., Ma, J., Wei, Z., Wang, Y., Song, B., et al. (2023). DirectRMDB: a database of post-transcriptional RNA modifications unveiled from direct RNA sequencing technology. Nucleic Acids Res. 51, D106–D116. doi: 10.1093/nar/gkac1061
Zhen, D., Wu, Y., Zhang, Y., Chen, K., Song, B., Xu, H., et al. (2020). m6A reader: epitranscriptome target prediction and functional characterization of N 6-methyladenosine (m6A) readers. Front. Cell Dev. Biol. 8, 741. doi: 10.3389/fcell.2020.00741
Zheng, Y., Nie, P., Peng, D., He, Z., Liu, M., Xie, Y., et al. (2018). m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. 46(D1), D139–D145. doi: 10.1093/nar/gkx895
Zhong, S., Li, H., Bodi, Z., Button, J., Vespa, L., Herzog, M., et al. (2008). MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20, 1278–1288. doi: 10.1105/tpc.108.058883
Zhou, Y., Zeng, P., Li, Y. H., Zhang, Z., and Cui, Q. (2016). SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 44, e91. doi: 10.1093/nar/gkw104
Keywords: RNA modification, 5-methyluridine, graph embedding, multi-species, sequence feature
Citation: Xu Z, Wang X, Meng J, Zhang L and Song B (2023) m5U-GEPred: prediction of RNA 5-methyluridine sites based on sequence-derived and graph embedding features. Front. Microbiol. 14:1277099. doi: 10.3389/fmicb.2023.1277099
Received: 14 August 2023; Accepted: 02 October 2023;
Published: 23 October 2023.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Assaf Katz, University of Chile, ChilePengmian Feng, North China University of Science and Technology, China
Copyright © 2023 Xu, Wang, Meng, Zhang and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bowen Song, Ym93ZW4uc29uZyYjeDAwMDQwO25qdWNtLmVkdS5jbg==