 Amy Pham1,2
Amy Pham1,2 James G. Volmer3
James G. Volmer3 Daniel C. Chambers2,4Daniel J. Smith4,5David W. Reid5,6,7Lucy Burr8,9
Daniel C. Chambers2,4Daniel J. Smith4,5David W. Reid5,6,7Lucy Burr8,9 Timothy J. Wells1,6*
Timothy J. Wells1,6*- 1Frazer Institute, The University of Queensland, Brisbane, QLD, Australia
- 2Queensland Lung Transplant Service, The Prince Charles Hospital, Brisbane, QLD, Australia
- 3Centre for Microbiome Research, Queensland University of Technology, Brisbane, QLD, Australia
- 4Faculty of Medicine, The University of Queensland, Brisbane, QLD, Australia
- 5The Adult Cystic Fibrosis Centre and Department of Thoracic Medicine, The Prince Charles Hospital, Brisbane, QLD, Australia
- 6Australian Infectious Diseases Research Centre, The University of Queensland, Brisbane, QLD, Australia
- 7QIMR Berghofer Medical Research Institute, Brisbane, QLD, Australia
- 8Department of Respiratory Medicine, Mater Health, South Brisbane, QLD, Australia
- 9Mater Research, The University of Queensland, Brisbane, QLD, Australia
Introduction: The Burkholderia cepacia complex (BCC) encompasses a group of at least 22 genetically distinct gram-negatives bacterial species ubiquitous in nature. Recognised as a group of genetically and phenotypically flexible species, the BCC inhabits diverse ecological niches causing both plant and human diseases. Comparative genomic analysis provides an in depth understanding into the population biology, phylogenetic relationship, and genomic architecture of species.
Methods: Here, we genomically characterise Burkholderia anthina isolated from patients with chronic lung infections, an understudied pathogen within the Burkholderia cepacia complex.
Results: We demonstrate that B. anthina is polyphyletic and constitutes two distinct evolutionary lineages. Core- and pan-genome analyses demonstrated substantial metabolic diversity, with B. anthina Clade I enriched in genes associated with microbial metabolism in diverse environments, including degradation of aromatic compounds and metabolism of xenobiotics, while B. anthina Clade II demonstrated an enhanced capability for siderophore biosynthesis.
Discussion: Based on our phylogenetic and comparative genomic analyses, we suggest stratifying B. anthina to recognise a distinct species harbouring increased potential for iron metabolism via siderophore synthesis, for which we propose the name Burkholderia anthinoferum (sp. nov.).
Introduction
The Burkholderia cepacia complex (BCC) represents a group of metabolically versatile and highly adaptable bacteria found inhabiting diverse ecological niches. In the early 1980’s, the BCC emerged as an opportunistic pathogen in patients with cystic fibrosis (CF), gaining notoriety due to its intrinsic resistance to antibiotics and propensity to spread rapidly within the community causing devasting outbreaks (Speert et al., 2002; Vandamme et al., 2003). Treatment and management of Burkholderia infections still represents a long-standing challenge for clinicians, with infection considered a contraindication for lung transplantation due to the significant post-operative mortality associated with the bacteria (Aris et al., 2001; Hauser et al., 2011; Ramos et al., 2016). Infections by the BCC pose a serious therapeutic challenge, and hence, understanding their genomic architecture and metabolic potential is critical to understand their pathogenesis. Encompassing a group of at least 22 genetically distinct species, the BCC features some of the largest and most complex bacterial genomes described to date, characterised by a multireplicon structure ranging from 6 to 9 Mb. Harbouring bacteriophages, several genomic islands, and an extensive repository of insertion sequence elements, the large genome size of the species underpins their genetic and metabolic versatility (Baldwin et al., 2004; Holden et al., 2009).
The taxonomy of Burkholderia has undergone extensive reclassification over the last few decades and continues to expand as new lineages and species are proposed (Vandamme et al., 1997; Coenye et al., 2001a,b; Vandamme et al., 2002; Mullins and Mahenthiralingam, 2021). Historically, the BCC was thought to constitute a single species known as Pseudomonas cepacia, however in 1992 was renamed to recognise several distinct genomovars. Following the implementation of recA sequence analysis, taxonomic distinction of species within the BCC expanded with new species being rapidly described. The genetic diversity within species was also recognised, with the taxonomy of B. cenocepacia extended to include four distinct subgroups within the bacterial population known as lineage III-A, III-B, III-C and III-D (Mahenthiralingam et al., 2000; Vandamme et al., 2003). Likewise, B. cepacia was also divided into separate sub lineages following the identification of types AD, AW, and K, with group K now consisting of two validly named species, B. contaminans and B. lata (Vermis et al., 2002; Vanlaere et al., 2009; Depoorter et al., 2020). Although the recA based identification scheme has been widely implemented in epidemiological investigations its utility to resolve phylogenetic relationships among other closely related species is limited (LiPuma et al., 1999). The advent of sequencing technologies has provided a better means to genomically characterise species and evaluate phylogenetic relationships. Indeed, recent comparative genomic analyses revealed novel genomic taxa within the BCC, with the proposal of new species B. paludism, B. reimsis, and B. orbicola (Ong et al., 2016; Esmaeel et al., 2018; Wallner et al., 2019; Morales-Ruiz et al., 2022).
B. anthina represents an understudied pathogen within the BCC. From the 2,425 sequenced BCC genomes deposited in the National Center for Biotechnology Information (NCBI) database to date, only 31 (1.27%) correspond to B. anthina strains, with most isolates originating from an environmental source. In the current study, we examined phylogenetic relationships among Burkholderia species within the BCC and conducted comparative genomic analyses of B. anthina. Whole genome sequencing of Burkholderia isolates collected from patients with CF in Queensland, Australia identified a further five strains of B. anthina. Genomic characterisation of B. anthina genomes identified two evolutionarily distinct clades with considerable genomic divergence. Based on phylogenetic evidence, whole genome average nucleotide identity, and comparative genome analyses, we propose the division of B. anthina into two separate species: B. anthina and B. anthinoferrum (sp. nov.).
Methods
Isolation of Burkholderia spp. from sputum and culture conditions
Cases of Burkholderia infections detected in patients with CF attending The Prince Charles Hospital and the Mater Hospital, Queensland, Australia, between 2017 and 2022 were included in the study. Clinical specimens were collected from the Prince Charles Hospital as per the Research Collaboration Agreement HREC/17/QPCH/277, and the Mater Hospital as per HREC/14/QPAH/275. Bacterial isolation was performed from expectorated sputum samples. Briefly, a direct inoculum from sputum was plated out onto Burkholderia cepacia selective agar (BCSA) and incubated at 37°C under aerobic conditions for 48 h. The medium, supplemented with crystal violet, vancomycin and gentamicin, inhibits growth of gram-positive bacteria and gram-negative bacilli Pseudomonas, for increased selectivity of Burkholderia. Bacteria were grown under aerobic conditions in lysogeny broth (LB) (1% (w/v) tryptone, 1% (w/v) yeast extract, 0.5% (w/v) sodium chloride) at 37°C, with shaking at 220 rpm overnight.
Whole genome sequencing and assembly of Burkholderia spp. isolate genomes
For genomic DNA extraction, bacterial cultures were grown in 5 mL of LB, followed by overnight incubation at 37°C with shaking at 220 rpm. Genomic DNA was extracted using a QIAamp DNA Mini Kit (Qiagen) as per the manufacturer’s instructions. DNA concentration and purity were assessed using a Nanodrop 1,000 spectrophotometer. Whole genome sequencing (WGS) was performed by MicrobesNG1 using the Illumina sequencing platform with 2 × 250 bp pair-end reads. Genome assembly was conducted by MicrobesNG using their standard analysis pipeline. Briefly, Kraken was used to identify the closet available reference genome (Wood and Salzberg, 2014). The reads were mapped to the reference genome using BWA mem to assess the quality of the data (Li and Durbin, 2009). De novo assembly of the reads was conducted using SPAdes (Bankevich et al., 2012). Genome annotation was completed using Prokka (Seemann, 2014). Reference genomes were downloaded from the NCBI database2 (Supplementary Table 1).
Quality assessment, phylogeny, and average nucleotide analysis of Burkholderia spp. isolate genomes
Genome quality assessment and assembly metrics were assessed using CheckM (v1.1.2) (Parks et al., 2015). Genomes with an estimated completeness of ≥90% and contamination of ≤5% were determined high-quality and included for comparative analysis. Multi-locus sequence typing (MLST) was determined by querying genomes against the Burkholderia cepacia complex typing database3 available on PubMLST (Jolley et al., 2018). Under the MLST scheme seven independent loci comprising of genes atpD, gltB, gyrB, recA, lepA, phaC, and trpB are assessed to identify differences in the allelic profile to define the sequence type (ST). Taxonomic classification was assigned using the Genome Taxonomy Database Toolkit (GTDB-Tk; v2.2.4) with reference database r207. A concatenated set of 975 core genes were produced using BPGA and the resultant output used to predict phylogeny (Parks et al., 2018; Supplementary Data 1). Phylogenetic analysis was conducted using RAxML (v8.2.13) (Stamatakis, 2014) and visualised in iToL (v6.8.1).4 Average nucleotide identity (ANI) was determined using FastANI (v1.1) and visualised using the ‘pheatmap’ (v1.0.12) and ‘ggplot’ (v3.1.3) packages in Rstudio (v4.1.3) and R (v4.2.3). Genomes were annotated through EnrichM (v0.4.155) using the ‘annotate’ function, with the input parameter ‘--orthologs’ to annotate orthologous genes.
Comparative genomic analysis of Burkholderia anthina genomes
A total number of 34 B. anthina genomes were included in the comparative genomic analyses. Statistical analyses for comparing distribution of genome size, number of CDSs, and coding density were determined using an unpaired t-test. The Bacterial Pan Genome Analysis (BPGA) pipeline using the USEARCH tool was used to cluster protein sequences into gene families (Chaudhari et al., 2016). A sequence identity cut-off value of 80% was used for orthologous clustering. The pan-genome profile analysis tool was used to determine the pan and core genome size, calculated with 100 iterations. Data are calculated as a function of genomes introduced into the analysis. The pan-genome functional analysis tool was used to assign KEGG Ortholog pathways to protein sequences, and execute comparative functional analysis for core, accessory, and unique genes. Principle component analysis (PCA) of KEGG orthologs was performed using the ‘factoextra’ (v1.0.7), ‘ggplot2’ (v3.4.4), ‘purrr’ (v1.0.2), ‘scatterplot3d’ (v0.3-44), ‘ade4’ (v1.7-22), and ‘gdata’ (v3.0.0) packages in Rstudio (v4.1.3) and R (v4.2.3). Comparative genomics based on KEGG Orthology was completed using EnrichM (v0.4.15; see footnote 5). Genomes were annotated using the ‘annotate’ function, with the input parameter ‘--ko’ used to annotate KEGG Orthology. Statistical analyses between clades were performed in EnrichM by Fisher’s exact test. Corrected p-values of <0.05 were considered significant. Secondary metabolite gene clusters were predicted using anti-SMASH (v7.0.06) using the parameter ‘relaxed’ to identify hits (Blin et al., 2023). In silico characterisation of the ethylenediaminesuccinic acid hydroxyarginine (EDHA) biosynthetic gene cluster was conducted through BLASTn searches. Streptomyces sp. MA5143a was used as the reference sequence and encodes for an EDHA operon containing four genes denoted AesA, AesB, AesC, and AesD. The genetic organisation of the EDHA gene cluster was visualised using the ‘gggenes’ (v0.5.1) and ‘ggplot2’ (v3.4.4) packages in Rstudio (v4.1.3) and R (v4.2.3). Identification of iron genes and iron gene operons was predicted using the bioinformatic tool FeGenie (Garber et al., 2020). The RAST server (v2.0) was used to annotate genomes and identify genes associated with iron metabolism, specifically siderophore biosynthesis (Aziz et al., 2008; Overbeek et al., 2014; Brettin et al., 2015).
Results
Characterisation of Burkholderia isolated from patients with CF in South-East Queensland identifies clonal strains shared among patients.
Taxonomic analysis of Burkholderia previously relied on recA gene sequence analysis, with the molecular approach proving to be a valuable tool in the early distinction between species. Although widely implemented and used in epidemiological investigations, its utility to resolve in depth phylogenetic relationships among closely related species within the BCC is limited. To further understand the evolutionary relationships of Burkholderia, we performed genomic analyses among and within species of the BCC. Here, we genome sequenced 18 clinical isolates cultured from patients with CF in Queensland, Australia. The quality of each genome assembly was assessed using CheckM, with genomes more than 95% complete, and < 5% contaminated retained for analysis (Table 1). A total of 177 Burkholderia genomes were included as part of this study, including 159 publicly available genomes, with B. gladioli selected as the outgroup species. The genome size of Burkholderia strains isolated in this study ranged from 6.3 to 8.3 Mbp, with a G + C content of 67%, and predicted number of coding genes ranging from 5,600–7,600 genes (Table 1).
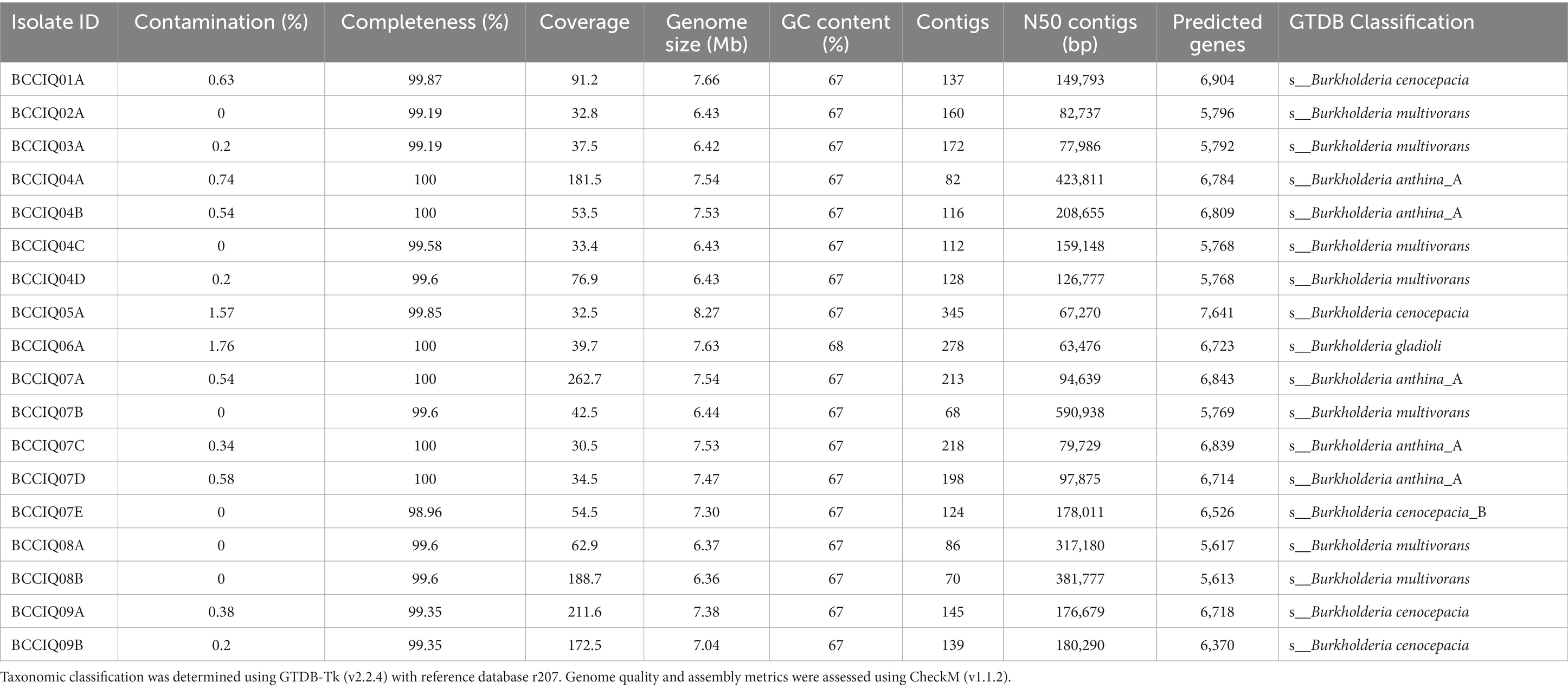
Table 1. Genome statistics of Burkholderia isolates included in this study.
Phylogenetic analysis was performed using a set of 975 core genes produced using BPGA and inferred using RAxML. MLST analysis was performed to inform strain relatedness among isolates. As part of this study, we obtained four isolates belonging to B. cenocepacia III-A, and one isolate belonging to B. cenocepacia III-B. All B. cenocepacia isolates cultured in this study clustered separate from each other and displayed a unique ST (Figure 1). While five of the seven cultured B. multivorans isolates (BCCIQ02A, BCCIQ03A, BCCIQ04C/D and BCCIQ07B) displayed the same ST-622 and clustered tightly together. Phylogenetic placement of Burkholderia genomes identified several distinct phylogenetic clades within B. anthina. Notably, all five B. anthina isolates clustered within Clade II, displayed the same ST-2133, and represent the first clinical isolates reported within the group. Based on a well-supported phylogeny, it is evident that B. anthina Clade II represents a truly distinct group to B. anthina Clade I. As shown in Figure 1, B. anthina is polyphyletic with Clade I, as well as B. vietnamiensis, and B. ambifaria, diverging earlier than B. anthina Clade II. Phylogenetic placement of B. anthina Clade II suggest that this lineage shares a more recent common ancestor with B. cenocepacia, B. cepacia, B. stabilis, and B. pyrrocinia.
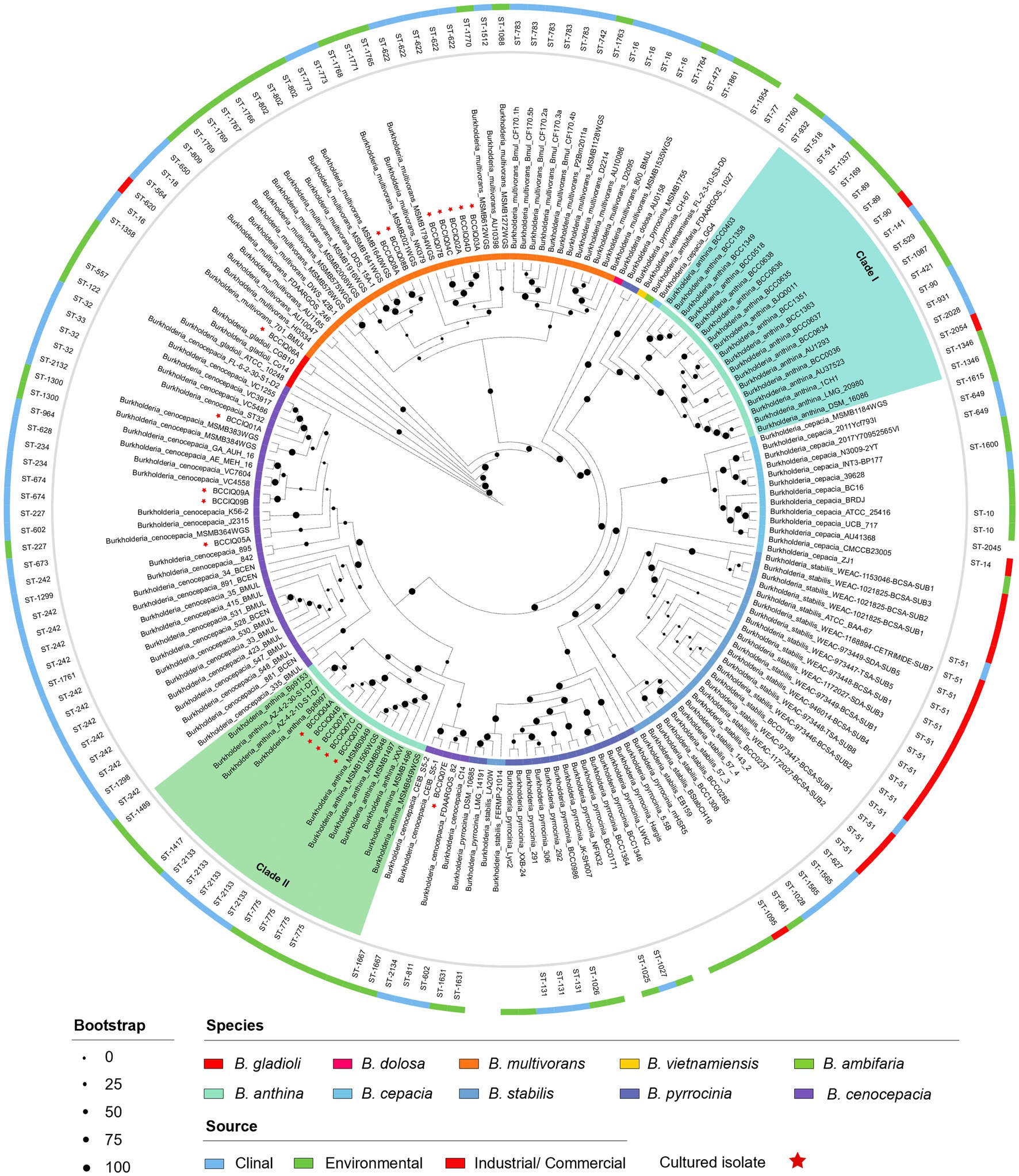
Figure 1. Phylogenetic distribution of Burkholderia spp. genomes. Concatenated core gene files were produced using BPGA, with B. gladioli used as the outgroup. Phylogeny was inferred using RAxML (v8.2.13) and visualised by iToL (v6.8.1). Cultured isolates recovered from this study are denoted BCCIQ, boldfaced in red, and identified by a red star. Phylogenetic placement of B. anthina (shaded green) indicates two distinct evolutionary clades within the species group.
Comparative genomic analyses reveal hidden genetic diversity of B. anthina
The phylogenetic separation in B. anthina is further reflected in the genomic architecture between the two clades. B. anthina Clade I genomes have a significantly larger genome than B. anthina Clade II (p < 0.0001), with a mean size of 7.7 Mbp as compared to 7.4 Mbp (Figure 2A). Accordingly, B. anthina Clade I have a significantly greater number of predicted coding sequences than B. anthina Clade II (p = 0.0029), with an average of 472 more coding sequences (Figure 2B). The coding density of both clades was comparable with an average of 87% (Figure 2C). The pan-genome of B. anthina demonstrated characteristics of an “open” pan genome, composed of a core genome of 3,371 orthologous genes (12.2%) shared by all strains, and a pan genome of 24,274 genes (87.8%) (Figure 2D). The gene accumulation rarefaction curve increased with the addition of each new genome, reflecting an increase in the number of gene families, and expanding the gene pool diversity. When analysed within their respective clades, Clade I (Figure 2E) and Clade II (Figure 2F) genomes also revealed an “open” profile. At an analysis of 16 genomes, Clade I demonstrated the greatest diversity among strains with only 4,237 core genes (19.4%) and 17,624 (80.6%) pan genes. This contrasts with Clade II which had 5,464 core genes (38%) and 9,020 (62%) pan genes. The pan and core genome profile of B. anthina Clade I highlight the genetic variability within the group and reflects the differences among genomes.
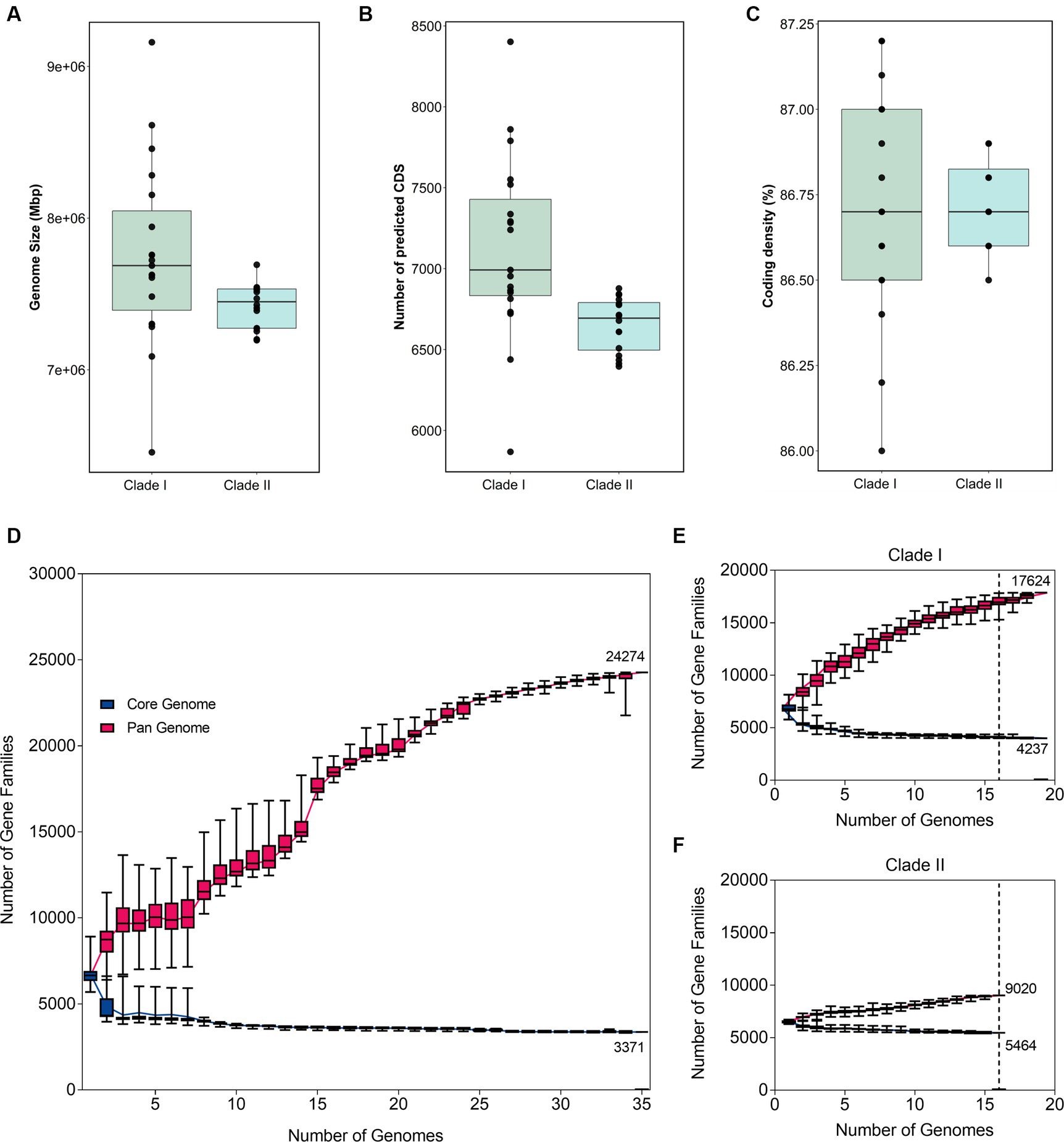
Figure 2. Variations in genomic architecture between B. anthina genomes. Comparison of (A) genome size, (B) coding density, and (C) predicted coding sequences in B. anthina Clade I (19 genomes) compared with B. anthina Clade II (16 genomes). Genomic statistics were assessed using CheckM (v1.1.2) and the BPGA analysis tool. Comparative analysis of the pan and core genome of (D) all B. anthina genomes, (E) B. anthina Clade I, and (F) B. anthina Clade II. The pan-genome profile was determined using the BPGA software, calculated with 100 iterations. The number of gene families is graphed as a function of the number of genomes sequentially added. The dotted line denotes the reference point at which the pan-core genome profile can be accurately compared across the clades. At 16 genomes, the pan-core plot of Clade I showed 17,624 genes in the pan-genome, compared to Clade II which contained 9,020 genes.
Phylogenomic analysis indicates two evolutionary distinct clades within B. anthina
Whole genome average nucleotide identity (ANI) was calculated to determine strain relatedness and assess the species boundaries of B. anthina and closely related species as indicated by phylogenetic placement. A threshold of 98% ANI is considered the cut-off for strain classification within species, whereas a threshold of 95% ANI is considered the cut-off for species delineation (Goris et al., 2007; Jain et al., 2018). Consistent with the relationships inferred by phylogenetic analyses, ANI evaluation confirmed the identification of two distinct lineages within B. anthina (Figure 3). Pairwise ANI of B. anthina Clade I and Clade II demonstrated 90% pairwise identity, indicating that the two groups represent distinct species according to the operational 95% ANI threshold commonly used for species designation and appropriate for Burkholderia. Genomes clustering with B. anthina Clade I appeared to be genetically variable with further clusters designated I-A, I-B, and I-C identified within the group. Among these clusters, genomes displayed a pairwise ANI between 94.7–95.8%, bordering the threshold for species demarcation. In comparison B. anthina Clade II harboured highly similar strains with a pairwise ANI ≥ 98%. Assessment of species boundaries between B. anthina Clade II and B. cenocepacia confirmed that these groups are distinct to each other, with less than 95% pairwise identity (Supplementary Table 2). Taken together, these analyses corroborate our findings that there are two distinct lineages within B. anthina.
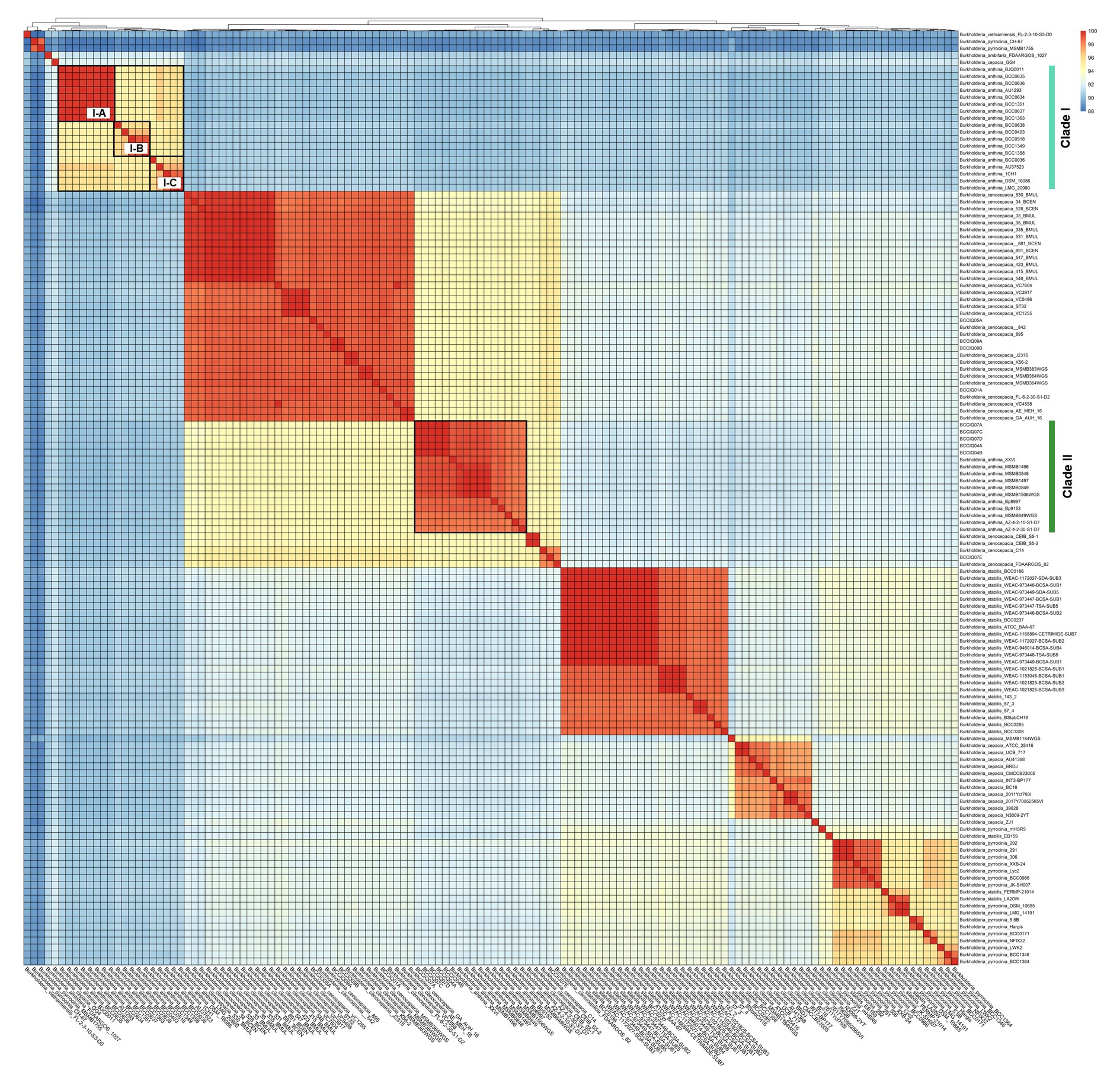
Figure 3. Pairwise average nucleotide identity comparison between B. anthina genomes and closely related Burkholderia species. Pairwise ANI was calculated using FastANI (v1.1) and visualised using the ‘pheatmap’ (v1.0.12) and ‘ggplot’ (v3.1.3) packages in Rstudio (v4.1.3) and R (v4.2.3). The heatmap depicts phylogenetic relationships, with ANI values clustered according to their distance patterns. ANI score is calculated from BLAST hits between orthologous genes of the core genome.
Analysis of secondary metabolites reveals diverse metabolic potential
To explore the functional differences of the different B. anthina clades, the pangenome was analysed according to their KEGG ortholog pathways. Both groups demonstrated a similar KEGG ortholog distribution profile regarding the relative proportion of core, accessory, and unique genes assigned to each KEGG pathway. However, B. anthina Clade II, displayed a higher relative proportion of unique genes associated with cellular processes and motility. KEGG categories associated with metabolism, including amino acid and carbohydrate metabolism were the most abundant categories across both B. anthina clades for core, accessory, and unique genes (Figure 4A). Interestingly, B. anthina genomes displayed substantial variance in KEGG orthologs between and within the identified clade groups (Figure 4B). To highlight clade specific adaptations and identify distinct biological interactions, we performed a KEGG ortholog enrichment analysis using a Fisher’s exact test. An examination of enriched KEGG orthologs revealed 722 genes differentially enriched among the two groups, with 451 (62.5%) genes enriched in Clade I and 271 (37.5%) genes enriched in Clade II (Figure 4B; Supplementary Table 3). KEGG orthologs associated with metabolism represented the most prominent KEGG category enriched in both groups. KEGG categories associated with genetic information processing, environmental information processing, and cellular processes were also enriched, albeit at a lower proportion. Genes associated with global overview and map, carbohydrate metabolism, amino acid metabolism, and xenobiotics biodegradation and metabolism showed frequent enrichment in both groups. In terms of genes encoding functions for metabolism, Clade I showed greater functional enrichments linked to metabolic pathways, microbial metabolism in diverse environments, and degradation of aromatic compounds. Of ecological importance, xenobiotics biodegradation and metabolism were largely enriched in Clade I, with gene functions specific to benzoate degradation, xylene degradation, chlorocyclohexane and chlorobenzene degradation, dioxin degradation, toluene degradation, steroid degradation, and styrene degradation.
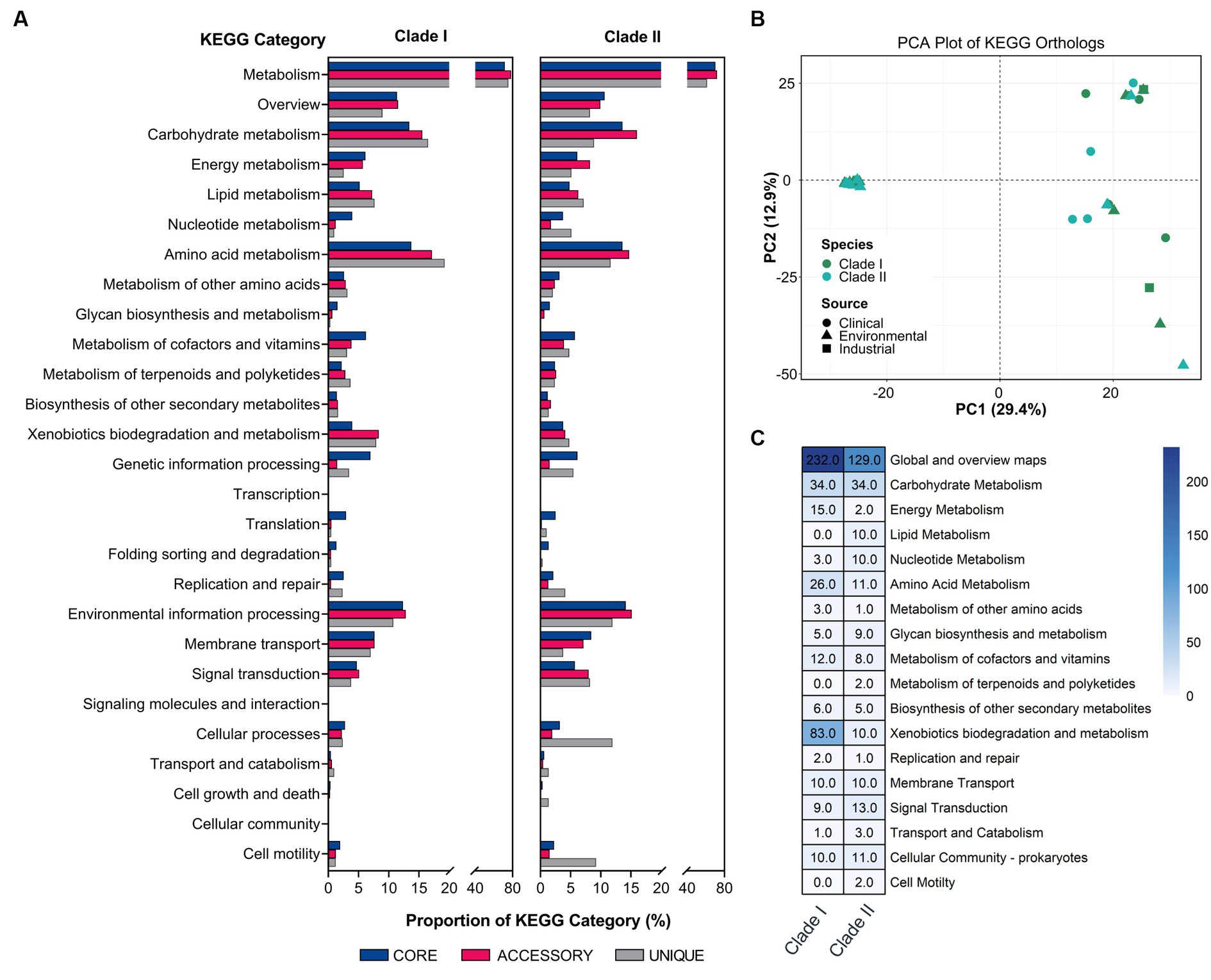
Figure 4. Functional analysis of major KEGG categories in B. anthina. (A) Comparison of KEGG distribution profile of core, accessory, and unique gene sets in Clade I and Clade II represented as a percentage. (B) Principal component analysis of KEGG orthologs between clades. Gene annotations were generated using EnrichM (v0.4.15; https://github.com/geronimp/enrichM) using the ‘—KEGG’ analyses. Genomes are coloured according to species classification, with the shape of the symbol referring to the source of the isolate, as indicated by the legend. (C) Differential enrichment of KEGG pathways in Burkholderia anthina displayed. KO annotation and statistical analyses were performed using the ‘annotate’ and ‘enrichment’ functions of EnrichM (v0.4.15; https://github.com/geronimp/enrichM). B. anthina genomes were grouped by clades and compared by Fisher’s exact test, where KOs with corrected p values of <0.05 were retained and considered significant.
Analysis of secondary metabolites identified 18 different biosynthetic gene clusters (BGC) in B. anthina with clear differences between Clades I and II (Figure 5). Genes clusters inferred to encode the biosynthetic pathway for terpene production (186), non-ribosomal peptide synthases (NRPS) (72), type I polyketide synthase (T1PKs) (65), and NRP-metallophone (56) were most abundant. BGC counts associated with terpene production, NRPS, and T1PKs were higher in Clade II than that of Clade I. While Clade I demonstrated a broader distribution of specialised metabolite cluster types, with ranthipeptide, betalactone, and butyrolactone exclusively present in Clade I, albeit not detected across all genomes. Aminopolycarboxylic-acid and other were exclusively present in all genomes within Clade II. The aminopolycarboxylic-acid gene cluster is predicted to encode the biosynthesis of ethylenediaminesuccinic acid hydroxyarginine (EDHA), an iron responsive regulated zwitterionic siderophore. To validate the presence of the predicted EDHA gene cluster we bioinformatically identified the operon within the genome, using Streptomyces sp. MA5143a as the reference sequence. As predicted, all Clade II B. anthina genomes harboured homologues for all four genes, allowing for the biosynthesis of EDHA a newly described siderophore (Supplementary Figure 1). To gain insight into the overall iron metabolism potential of B. anthina, the bioinformatic tool FeGenie was used identify genes related to iron acquisition, storage, and redox cycling. Genes associated with iron gene regulation and iron storage were comparable across the two clades, and gene families related with heme oxygenase, iron reduction, or magnetosome formation were absent across all genomes (Figure 6A). Generally, a greater number of gene families associated with iron transport were identified in Clade I (Figure 6B), while gene families associated with siderophore synthesis were higher in B. anthina Clade II (Figure 6C). Genome annotations by RAST predicted on average 31 more genes associated with siderophore synthesis in Clade II compared to Clade I, highlighting the extended siderophore assembly capabilities of the group (Supplementary Table 4). All B. anthina genomes encoded genes required for ornibactin synthesis, while only Clade II genomes encoded genes for pyochelin synthesis, and at least 20 genes putatively involved in siderophore biosynthesis. Interestingly, four phylogenetically closely related strains (B. anthina 1CH1, B. anthina AU37523, B. anthina DSM 16086, and B. anthina LMG 20980) identified in Clade I harboured the genes FhuB, FhuC, and FhuD required for uptake of ferric aerobactin. The synthesis and transport of the aerobactin siderophore is frequently produced by several Enterobacteriaceae, including Escherichia, Vibrio, Salmonella, and Shigella (Waters and Crosa, 1988; Sheldon et al., 2016). Given that aerobactin biosynthesis is not an established feature of Burkholderia it is likely these genes were horizontally acquired. Overall, these results highlight clear genomic and metabolic differences between the two clades identified in B. anthina and provides strong evidence that they should be considered two different species.
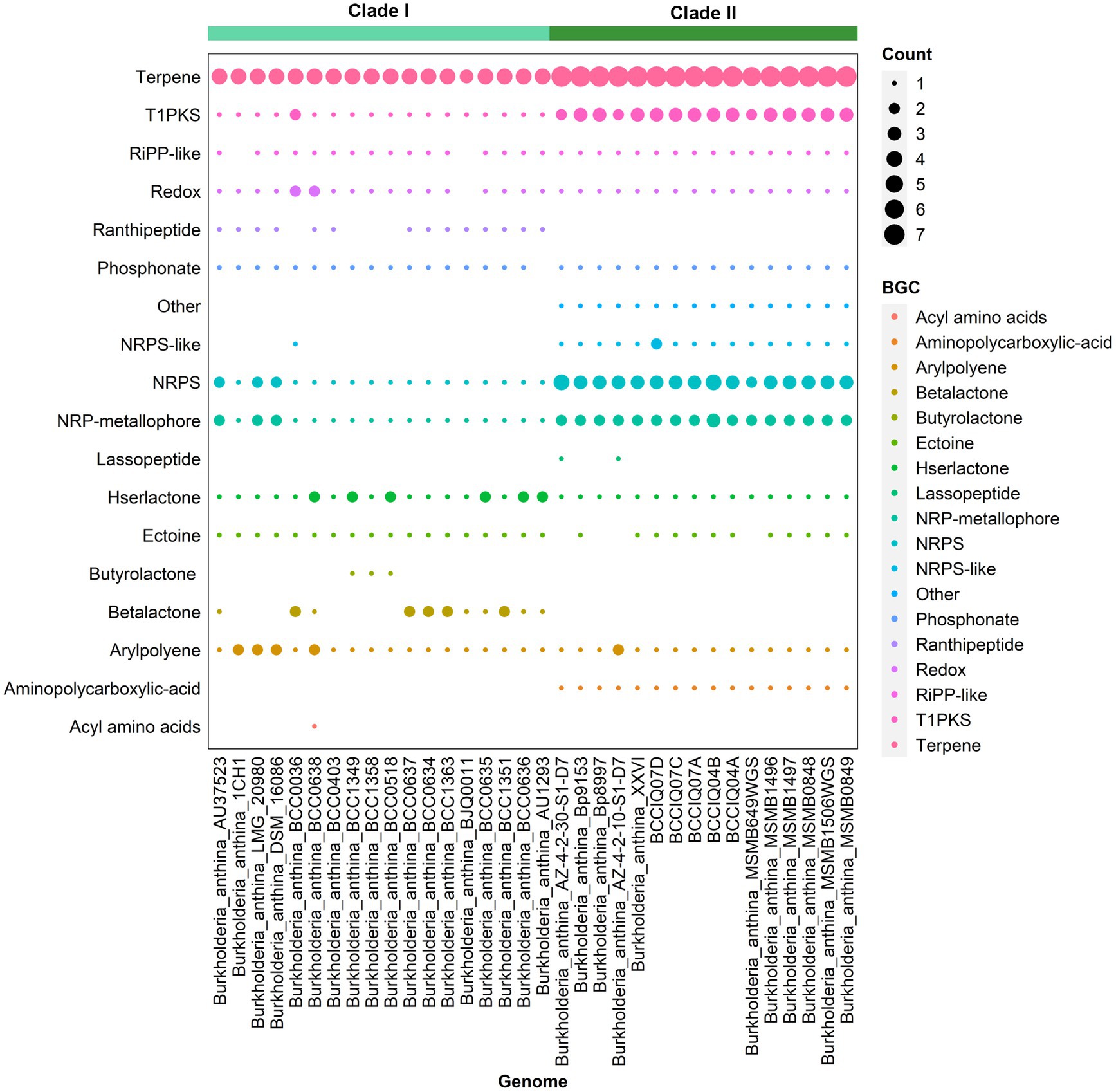
Figure 5. Presence of biosynthetic gene clusters in B. anthina genomes. BGCs were detected using Anti-SMASH (v7.0.0). The size of the dot represents BGC count. Genomes are clustered according to clade designation.
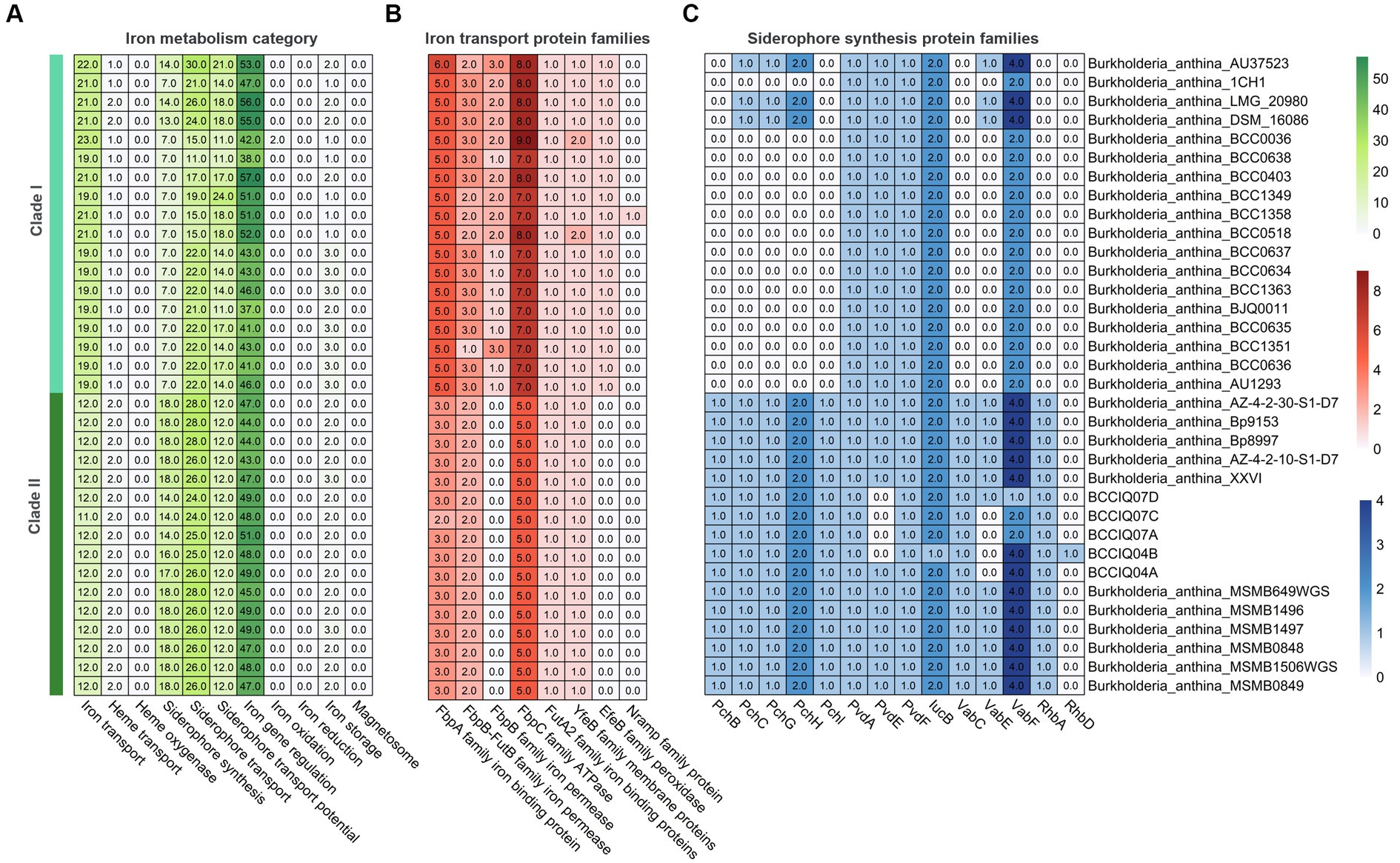
Figure 6. Heatmap showing gene count of different iron metabolism categories in B. anthina genomes. (A) Gene count involved in iron transport, iron oxidation and reduction, iron storage, and iron regulation were predicted using FeGenie. Protein families associated with (B) iron transport and (C) siderophore synthesis.
Discussion
The Burkholderia cepacia complex represents a significant group of organisms heterogeneously distributed in the natural environment, and contains species of ecological, pathogenic, and biotechnological interest (Coenye and Vandamme, 2003; de Los Santos-Villalobos et al., 2018). Harbouring species that are metabolically versatile and highly adaptable, the BCC can inhabit diverse ecological niches, and tolerate environmental conditions subject to nutrient limitation, toxic compounds, antibiotics, and antimicrobial peptides. The genomic and phenotypic flexibility of these microbes is in part explained by the large coding capacity of these genomes. The advent of next-generation sequencing technologies has facilitated the advancement of comparative genomics to elucidate the genomic architecture, population biology, and phylogenetic relationships among species (De Smet et al., 2015). Through genomic analysis of the BCC, we have identified a distinct clade within B. anthina which ancestrally diverged from its sister clade. Comparative analysis uncovered remarkable genetic and metabolic diversity between the two clades. Based on our phylogenetic analyses, ANI, and comparative genomic studies, we suggest amending B. anthina to recognise a new species. We propose that the name B. anthina is retained to represent all strains clustering with the type strain B. anthina LMG 20980, identified in this study as Clade I. While strains forming Clade II be recognised as a new species, for which we propose the name Burkholderia anthinoferrum sp. nov. (an.thin.no.ferrum: anthino, meaning ‘from the garden’ or ‘from flowers’, referring to the origin of the majority of the isolates as previously proposed, ferrum; iron, referring to the species iron acquisition potential for siderophore synthesis). Herein, Clade I will be referred to as B. anthina, and Clade II will be referred to as B. anthinoferrum.
Since its first description in 1997, B. anthina has been isolated from several sources including the rhizosphere soil of gardens and house plants, commercial solutions, and from the sputum of patients with CF (Vandamme et al., 2002; Coenye et al., 2003). Despite the rapid expansion of Burkholderia genome sequences in various national databases, there are currently a paucity of genomes corresponding to B. anthina, with the few that do primarily originating from an environmental source. The difficulties associated with resolving the taxonomy of Burkholderia is particularly evident when considering the taxonomic complexity that exists with the genus. In part, this is complicated by the high level of homologous recombination between species which further blurs taxonomic boundaries (Zhou et al., 2020). In the study by Wallner et al. (2019), genomic analyses of B. cenocepacia revealed two distinct clades within the species that harboured differential host-specific genetic adaptations to humans and plants. Despite being closely related (>95% ANI) according to the threshold for species delimitation by ANI, it was proposed that B. cenocepacia be amended to recognise a new species B. oribcola sp. nov. From our analyses, B. anthina and B. anthinoferrum were found to be polyphyletic, having ancestrally diverged from each other, which is further reflected by differences in their genetic and functional profiles. Pan-genomic analysis demonstrated substantial genetic variance within the population. The low number of shared genes observed in the population is consistent with the pan-genome of several other Burkholderia species, which are considered to be highly heterogenous (Seo et al., 2015; Bochkareva et al., 2018; Zhou et al., 2020; Bach et al., 2022). Demonstrating a high evolutionary rate, it could be suggested that the necessity of constant genomic adaptation is driven by selective pressures to adapt to the environment, which corroborates the diverse environments of isolates observed in B. anthina (Zhou et al., 2020). In contrast, lower intra-species diversity was apparent in B. anthinoferrum which supports a more recent evolution in addition to phylogenetic reconstruction.
In terms of biological capabilities, B. anthina were significantly enriched in KEGG orthologs associated with global overview and maps (metabolic pathways, microbial metabolism in diverse environments and degradation of aromatic compounds), and xenobiotic degradation and metabolism (benzoate degradation, xylene degradation, chlorocyclohexane and chlorobenzene degradation, dioxin degradation, toluene degradation, steroid degradation, and styrene degradation). The ability to metabolise and utilise xenobiotic contaminants as a carbon or nitrogen source serves as a competitive advantage for microorganisms, particularly in nutrient-harsh environments (Miglani et al., 2022). Several bacteria including Burkholderia, Pseudomonas, Achromobacter, Bacillus, Enterobacter and Alcaligenes have remarkable bioremediation potential to degrade xenobiotic compounds from contaminated environments (Tang et al., 2016; Morya et al., 2020; Mishra et al., 2021; Miglani et al., 2022; Tirkey et al., 2022). Burkholderia xenovorans LB400 has been extensively studied for its ability to degrade xenobiotic compounds, including halogenated aromatics such as polychlorinated biphenyls; a highly persistent and carcinogenic pollutant resistant to environmental degradation (Erickson and Mondello, 1992; Gibson et al., 1993; Hofer et al., 1993; Furukawa, 2000; Denef et al., 2004). Similarly, Burkholderia vietnamiensis G4 is well studied for its potential in the degradation of trichloroethylene, a common organic ground water contaminant (Nelson et al., 1986; O'Sullivan and Mahenthiralingam, 2005).
The low bioavailability of iron and other essential metal ions in the environment is a significant nutrient limiting factor, and as such microorganisms must be equipped with robust iron acquisition mechanisms. Siderophores are small chelating compounds secreted under iron limiting conditions to sequester metal ions from the environment to increase bioavailability (Hider and Kong, 2010; Kramer et al., 2020). Siderophore-mediated iron uptake by Burkholderia generally consists of four types and includes ornibactin, pyochelin, cepabactin, and cepaciachelin (Thomas, 2007; Butt and Thomas, 2017). B. anthina is known to encode the biosynthesis of two of these iron chelating compounds, ornibactin and pyochelin, with the former siderophore displaying a peptide structure similar to that of pyoverdine produced by fluorescent pseudomonads, including Pseudomonas aeruginosa (Sokol et al., 2000; Agnoli et al., 2006; Butt and Thomas, 2017). In line with the literature, B. anthina genomes encoded the genes required for ornibactin synthesis, while B. anthinoferrum encoded the genes for ornibactin and pyochelin synthesis. Analysis of secondary metabolite potential revealed that B. anthinoferrum possessed an even wider capacity for siderophore biosynthesis than B. anthina, with the former encoding the biosynthetic gene clusters required for siderophore synthesis using the classical NRP pathway and the NRP-independent siderophore pathway (Challis, 2005). Specifically, an aminopolycarboxylic acid siderophore gene cluster (NRP-independent pathway) encoding the biosynthesis of EDHA was identified and exclusively present in B. anthinoferrum genomes. This novel iron-responsive siderophore has recently been reported in the genomes of Streptomyces scabies, S. avermitilis, Corynebacterium pseudotuberculosis, C. ulcerans, and Nocardia brasiliensis (Spohn et al., 2018). The identification of an additional siderophore system and several putative genes for siderophore assemble reveals an extended metabolic repertoire of B. anthinoferrum, which may confer an advantage in environments with insufficient iron bioavailability, such as the CF lung (Drevinek et al., 2008). Indeed, the production of both ornibactin and pyochelin siderophores has previously been associated with increased virulence of Burkholderia strains in respiratory infections and correlated with morbidity in patients with CF (Sokol, 1986; Sokol and Woods, 1988; Agnoli et al., 2006). From an agricultural-management perspective, siderophores also provide a microbial-based alternative for effective control against plant pathogens. Previous investigations demonstrated that B. anthinoferrum XXVI produced a hydroxamate siderophore with biocontrol potential against the causal agent of anthracnose, Colletotrichum lindemuthianumn (de Los Santos-Villalobos et al., 2012). Recent characterisation of B. anthinoferrum XXVI genome also identified several putative genes for siderophore synthesis, suggesting increased potential for iron acquisition via the production of chelating compounds (de Los Santos-Villalobos et al., 2018). Although several novel siderophore assembly genes were genomically identified in the genomes of B. anthinoferrum, it is unknown whether these siderophore systems are functional and involved in iron acquisition. Future functional studies are required to confirm that EDHA is synthesised and understand its role in scavenging iron. Taken together, our analyses show distinct differences in the genetic architecture and metabolic capabilities between B. anthina and B. anthinoferrum, and along with their polyphyletic origin, provide further evidence that these groups represent distinct species.
Description of Burkholderia anthinoferrum sp. nov.
Burkholderia anthinoferrum sp. nov. (an.thin.no.ferrum: anthino, meaning ‘from the garden’ or ‘from flowers’, ferrum; iron). Cells are gram negative, non-motile, non-sporulating, coccobacilli bacteria. Growth is observed in LB, MacConkey and BCSA agar at 30, 37, and 42°C. Colonies in LB and BCSA are circular, convex, and cream coloured, while colonies in MacConkey are light pink coloured. Growth in BCSA produces an alkaline medium due the fermentation of lactose and sucrose and is strain dependent. The G + C content for the members of the species is 67.0%. Strains have been isolated predominately from environmental samples, but recently from clinical samples as described in this study. We propose that Burkholderia anthinoferrum sp. nov. XXVI be used as the type strain, a rhizosphere bacterium isolated from a mango orchid in Mexico. The strain holds a central position in the phylogeny of its clade and has been characterised in the literature (de Los Santos-Villalobos et al., 2012, 2018).
Data availability statement
The datasets generated during this study are available on the NCBI database under the BioProject PRJNA1033429. Whole-genome sequences for Burkholderia isolates are under the Biosample accession SAMN38033600-SAMN38033617.
Ethics statement
The studies involving humans were approved by The Prince Charles Hospital and QLD Princess Alexandra Hospital Research Ethics Committees. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from primarily isolated as part of your previous study for which ethical approval was obtained. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
AP: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. JV: Writing – review & editing, Investigation. DC: Resources, Writing – review & editing. DS: Resources, Writing – review & editing. DR: Resources, Writing – review & editing. LB: Resources, Writing – review & editing. TW: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
Samples were obtained from the David Serisier Research Biobank at Mater Misericordiae Ltd. The Translational Research Institute is supported by grants from the Australian and Queensland Governments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1274280/full#supplementary-material
Footnotes
References
Agnoli, K., Lowe, C. A., Farmer, K. L., Husnain, S. I., and Thomas, M. S. (2006). The ornibactin biosynthesis and transport genes of Burkholderia cenocepacia are regulated by an extracytoplasmic function sigma factor which is a part of the Fur regulon. J. Bacteriol. 188, 3631–3644. doi: 10.1128/JB.188.10.3631-3644.2006
Aris, R. M., Routh, J. C., LiPuma, J. J., and Heath, D. G., and Gilligan, P. H. (2001). Lung transplantation for cystic fibrosis patients with Burkholderia cepacia complex. Survival linked to genomovar type. Am J Respir Crit Care Med, 164, 2102–2106. doi: 10.1164/ajrccm.164.11.2107022
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bach, E., Sant'Anna, F. H., Seger, G., and Passaglia, L. M. P. (2022). Pangenome inventory of Burkholderia sensu lato, Burkholderia sensu stricto, and the Burkholderia cepacia complex reveals the uniqueness of Burkholderia catarinensis. Genomics 114, 398–408. doi: 10.1016/j.ygeno.2021.11.011
Baldwin, A., Sokol, P. A., Parkhill, J., and Mahenthiralingam, E. (2004). The Burkholderia cepacia epidemic strain marker is part of a novel genomic island encoding both virulence and metabolism-associated genes in Burkholderia cenocepacia. Infect. Immun. 72, 1537–1547. doi: 10.1128/IAI.72.3.1537-1547.2004
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol, 19, 455–477. doi: 10.1089/cmb.2012.0021
Blin, K., Shaw, S., Augustijn, H. E., Reitz, Z. L., Biermann, F., Alanjary, M., et al. (2023). antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51, W46–W50. doi: 10.1093/nar/gkad344
Bochkareva, O. O., Moroz, E. V., Davydov, I. I., and Gelfand, M. S. (2018). Genome rearrangements and selection in multi-chromosome bacteria Burkholderia spp. BMC Genomics 19:965. doi: 10.1186/s12864-018-5245-1
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Butt, A. T., and Thomas, M. S. (2017). Iron acquisition mechanisms and their role in the virulence of Burkholderia species. Front. Cell. Infect. Microbiol. 7:460. doi: 10.3389/fcimb.2017.00460
Challis, G. L. (2005). A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. Chembiochem 6, 601–611. doi: 10.1002/cbic.200400283
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Coenye, T., LiPuma, J. J., Henry, D., Hoste, B., Vandemeulebroecke, K., Gillis, M., et al. (2001a). Burkholderia cepacia genomovar VI, a new member of the Burkholderia cepacia complex isolated from cystic fibrosis patients. Int. J. Syst. Evol. Microbiol. 51, 271–279. doi: 10.1099/00207713-51-2-271
Coenye, T., Mahenthiralingam, E., Henry, D., LiPuma, J. J., Laevens, S., Gillis, M., et al. (2001b). Burkholderia ambifaria sp. nov., a novel member of the Burkholderia cepacia complex including biocontrol and cystic fibrosis-related isolates. Int. J. Syst. Evol. Microbiol. 51, 1481–1490. doi: 10.1099/00207713-51-4-1481
Coenye, T., and Vandamme, P. (2003). Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ. Microbiol. 5, 719–729. doi: 10.1046/j.1462-2920.2003.00471.x
Coenye, T., Vandamme, P., LiPuma, J. J., Govan, J. R., and Mahenthiralingam, E. (2003). Updated version of the Burkholderia cepacia complex experimental strain panel. J. Clin. Microbiol. 41, 2797–2798. doi: 10.1128/JCM.41.6.2797-2798.2003
de Los Santos-Villalobos, S., Barrera-Galicia, G. C., Miranda-Salcedo, M. A., and Pena-Cabriales, J. J. (2012). Burkholderia cepacia XXVI siderophore with biocontrol capacity against Colletotrichum gloeosporioides. World J. Microbiol. Biotechnol. 28, 2615–2623. doi: 10.1007/s11274-012-1071-9
de Los Santos-Villalobos, S., Kremer, J. M., Parra-Cota, F. I., Hayano-Kanashiro, A. C., Garcia-Ortega, L. F., Gunturu, S. K., et al. (2018). Draft genome of the fungicidal biological control agent Burkholderia anthina strain XXVI. Arch. Microbiol. 200, 803–810. doi: 10.1007/s00203-018-1490-6
De Smet, B., Mayo, M., Peeters, C., Zlosnik, J. E. A., Spilker, T., Hird, T. J., et al. (2015). Burkholderia stagnalis sp. nov. and Burkholderia territorii sp. nov., two novel Burkholderia cepacia complex species from environmental and human sources. Int. J. Syst. Evol. Microbiol. 65, 2265–2271. doi: 10.1099/ijs.0.000251
Denef, V. J., Park, J., Tsoi, T. V., Rouillard, J. M., Zhang, H., Wibbenmeyer, J. A., et al. (2004). Biphenyl and benzoate metabolism in a genomic context: outlining genome-wide metabolic networks in Burkholderia xenovorans LB400. Appl. Environ. Microbiol. 70, 4961–4970. doi: 10.1128/AEM.70.8.4961-4970.2004
Depoorter, E., De Canck, E., Peeters, C., Wieme, A. D., Cnockaert, M., Zlosnik, J. E. A., et al. (2020). Burkholderia cepacia complex taxon K: where to Split? Front. Microbiol. 11:1594. doi: 10.3389/fmicb.2020.01594
Drevinek, P., Holden, M. T., Ge, Z., Jones, A. M., Ketchell, I., Gill, R. T., et al. (2008). Gene expression changes linked to antimicrobial resistance, oxidative stress, iron depletion and retained motility are observed when Burkholderia cenocepacia grows in cystic fibrosis sputum. BMC Infect. Dis. 8:121. doi: 10.1186/1471-2334-8-121
Erickson, B. D., and Mondello, F. J. (1992). Nucleotide sequencing and transcriptional mapping of the genes encoding biphenyl dioxygenase, a multicomponent polychlorinated-biphenyl-degrading enzyme in Pseudomonas strain LB400. J. Bacteriol. 174, 2903–2912. doi: 10.1128/jb.174.9.2903-2912.1992
Esmaeel, Q., Issa, A., Sanchez, L., Clement, C., Jacquard, C., and Barka, E. A. (2018). Draft genome sequence of Burkholderia reimsis BE51, a plant-associated bacterium isolated from agricultural rhizosphere. Microbiol. Resour. Announc. 7, e00978–00918. doi: 10.1128/MRA.00978-18
Furukawa, K. (2000). Biochemical and genetic bases of microbial degradation of polychlorinated biphenyls (PCBs). J. Gen. Appl. Microbiol. 46, 283–296. doi: 10.2323/jgam.46.283
Garber, A. I., Nealson, K. H., Okamoto, A., McAllister, S. M., Chan, C. S., Barco, R. A., et al. (2020). FeGenie: a comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front. Microbiol. 11:37. doi: 10.3389/fmicb.2020.00037
Gibson, D. T., Cruden, D. L., Haddock, J. D., Zylstra, G. J., and Brand, J. M. (1993). Oxidation of polychlorinated biphenyls by Pseudomonas sp. strain LB400 and Pseudomonas pseudoalcaligenes KF707. J. Bacteriol. 175, 4561–4564. doi: 10.1128/jb.175.14.4561-4564.1993
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Hauser, A. R., Jain, M., Bar-Meir, M., and McColley, S. A. (2011). Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin Microbiol Rev. 24, 29–70. doi: 10.1128/CMR.00036-10
Hider, R. C., and Kong, X. (2010). Chemistry and biology of siderophores. Nat. Prod. Rep. 27, 637–657. doi: 10.1039/b906679a
Hofer, B., Eltis, L. D., Dowling, D. N., and Timmis, K. N. (1993). Genetic analysis of a Pseudomonas locus encoding a pathway for biphenyl/polychlorinated biphenyl degradation. Gene 130, 47–55. doi: 10.1016/0378-1119(93)90345-4
Holden, M. T., Seth-Smith, H. M., Crossman, L. C., Sebaihia, M., Bentley, S. D., Cerdeno-Tarraga, A. M., et al. (2009). The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 191, 261–277. doi: 10.1128/JB.01230-08
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3:124. doi: 10.12688/wellcomeopenres.14826.1
Kramer, J., Ozkaya, O., and Kummerli, R. (2020). Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 18, 152–163. doi: 10.1038/s41579-019-0284-4
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
LiPuma, J. J., Dulaney, B. J., McMenamin, J. D., Whitby, P. W., Stull, T. L., Coenye, T., et al. (1999). Development of rRNA-based PCR assays for identification of Burkholderia cepacia complex isolates recovered from cystic fibrosis patients. J. Clin. Microbiol. 37, 3167–3170. doi: 10.1128/JCM.37.10.3167-3170.1999
Mahenthiralingam, E., Bischof, J., Byrne, S. K., Radomski, C., Davies, J. E., Av-Gay, Y., et al. (2000). DNA-based diagnostic approaches for identification of Burkholderia cepacia complex, Burkholderia vietnamiensis, Burkholderia multivorans, Burkholderia stabilis, and Burkholderia cepacia genomovars I and III. J. Clin. Microbiol. 38, 3165–3173. doi: 10.1128/JCM.38.9.3165-3173.2000
Miglani, R., Parveen, N., Kumar, A., Ansari, M. A., Khanna, S., Rawat, G., et al. (2022). Degradation of xenobiotic pollutants. An environmentally sustainable approach. Metabolites 12:818. doi: 10.3390/metabo12090818
Mishra, S., Lin, Z., Pang, S., Zhang, W., Bhatt, P., and Chen, S. (2021). Recent advanced technologies for the characterization of xenobiotic-degrading microorganisms and microbial communities. Front. Bioeng. Biotechnol. 9:632059. doi: 10.3389/fbioe.2021.632059
Morales-Ruiz, L. M., Rodriguez-Cisneros, M., Kerber-Diaz, J. C., Rojas-Rojas, F. U., Ibarra, J. A., and Estrada-de Los Santos, P. (2022). Burkholderia orbicola sp. nov., a novel species within the Burkholderia cepacia complex. Arch. Microbiol. 204:178. doi: 10.1007/s00203-022-02778-0
Morya, R., Salvachua, D., and Thakur, I. S. (2020). Burkholderia: an untapped but promising bacterial genus for the conversion of aromatic compounds. Trends Biotechnol. 38, 963–975. doi: 10.1016/j.tibtech.2020.02.008
Mullins, A. J., and Mahenthiralingam, E. (2021). The hidden genomic diversity, specialized metabolite capacity, and revised taxonomy of Burkholderia Sensu Lato. Front. Microbiol. 12:726847. doi: 10.3389/fmicb.2021.726847
Nelson, M. J., Montgomery, S. O., O'Neill, E. J., and Pritchard, P. H. (1986). Aerobic metabolism of trichloroethylene by a bacterial isolate. Appl. Environ. Microbiol. 52, 383–384. doi: 10.1128/aem.52.2.383-384.1986
Ong, K. S., Aw, Y. K., Lee, L. H., Yule, C. M., Cheow, Y. L., and Lee, S. M. (2016). Burkholderia paludis sp. nov., an antibiotic-siderophore producing novel Burkholderia cepacia complex species, isolated from Malaysian tropical peat swamp soil. Front. Microbiol. 7:2046. doi: 10.3389/fmicb.2016.02046
O'Sullivan, L. A., and Mahenthiralingam, E. (2005). Biotechnological potential within the genus Burkholderia. Lett. Appl. Microbiol. 41, 8–11. doi: 10.1111/j.1472-765X.2005.01758.x
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Ramos, K. J., Quon, B. S., Psoter, K. J., Lease, E. D., Mayer-Hamblett, N., Aitken, M. L., and Goss, C. H. (2016). Predictors of non-referral of patients with cystic fibrosis for lung transplant evaluation in the United States. J Cyst Fibros. 15, 196–203. doi: 10.1016/j.jcf.2015.11.005
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics, 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seo, Y. S., Lim, J. Y., Park, J., Kim, S., Lee, H. H., Cheong, H., et al. (2015). Comparative genome analysis of rice-pathogenic Burkholderia provides insight into capacity to adapt to different environments and hosts. BMC Genomics 16:349. doi: 10.1186/s12864-015-1558-5
Sheldon, J. R., Laakso, H. A., and Heinrichs, D. E. (2016). Iron acquisition strategies of bacterial pathogens. Microbiol. Spectr. 4, 43–85. doi: 10.1128/9781555819286.ch3
Sokol, P. A. (1986). Production and utilization of pyochelin by clinical isolates of Pseudomonas cepacia. J. Clin. Microbiol. 23, 560–562. doi: 10.1128/jcm.23.3.560-562.1986
Sokol, P. A., Darling, P., Lewenza, S., Corbett, C. R., and Kooi, C. D. (2000). Identification of a siderophore receptor required for ferric ornibactin uptake in Burkholderia cepacia. Infect. Immun. 68, 6554–6560. doi: 10.1128/IAI.68.12.6554-6560.2000
Sokol, P. A., and Woods, D. E. (1988). Effect of pyochelin on Pseudomonas cepacia respiratory infections. Microb. Pathog. 5, 197–205. doi: 10.1016/0882-4010(88)90022-8
Speert, D. P., Henry, D., Vandamme, P., Corey, M., and Mahenthiralingam, E. (2002). Epidemiology of Burkholderia cepacia complex in patients with cystic fibrosis, Canada. Emerg. Infect. Dis. 8, 181–187. doi: 10.3201/eid0802.010163
Spohn, M., Edenhart, S., Alanjary, M., Ziemert, N., Wibberg, D., Kalinowski, J., et al. (2018). Identification of a novel aminopolycarboxylic acid siderophore gene cluster encoding the biosynthesis of ethylenediaminesuccinic acid hydroxyarginine (EDHA). Metallomics 10, 722–734. doi: 10.1039/c8mt00009c
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tang, S., Yin, H., Zhou, S., Chen, S., Peng, H., Liu, Z., et al. (2016). Simultaneous Cr(VI) removal and 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47) biodegradation by Pseudomonas aeruginosa in liquid medium. Chemosphere 150, 24–32. doi: 10.1016/j.chemosphere.2016.02.010
Thomas, M. S. (2007). Iron acquisition mechanisms of the Burkholderia cepacia complex. Biometals 20, 431–452. doi: 10.1007/s10534-006-9065-4
Tirkey, S. R., Ram, S., Mitra, M., and Mishra, S. (2022). Performance analysis of Pseudomonas sp. strain SA3 in naphthalene degradation using phytotoxicity and microcosm studies. Biodegradation 33, 169–180. doi: 10.1007/s10532-022-09972-3
Vandamme, P., Henry, D., Coenye, T., Nzula, S., Vancanneyt, M., LiPuma, J. J., et al. (2002). Burkholderia anthina sp. nov. and Burkholderia pyrrocinia, two additional Burkholderia cepacia complex bacteria, may confound results of new molecular diagnostic tools. FEMS Immunol. Med. Microbiol. 33, 143–149. doi: 10.1111/j.1574-695X.2002.tb00584.x
Vandamme, P., Holmes, B., Coenye, T., Goris, J., Mahenthiralingam, E., LiPuma, J. J., et al. (2003). Burkholderia cenocepacia sp. nov.--a new twist to an old story. Res. Microbiol. 154, 91–96. doi: 10.1016/S0923-2508(03)00026-3
Vandamme, P., Holmes, B., Vancanneyt, M., Coenye, T., Hoste, B., Coopman, R., et al. (1997). Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int. J. Syst. Bacteriol. 47, 1188–1200. doi: 10.1099/00207713-47-4-1188
Vanlaere, E., Baldwin, A., Gevers, D., Henry, D., De Brandt, E., LiPuma, J. J., et al. (2009). Taxon K, a complex within the Burkholderia cepacia complex, comprises at least two novel species, Burkholderia contaminans sp. nov. and Burkholderia lata sp. nov. Int. J. Syst. Evol. Microbiol. 59, 102–111. doi: 10.1099/ijs.0.001123-0
Vermis, K., Coenye, T., Mahenthiralingam, E., Nelis, H. J., and Vandamme, P. (2002). Evaluation of species-specific recA-based PCR tests for genomovar level identification within the Burkholderia cepacia complex. J. Med. Microbiol. 51, 937–940. doi: 10.1099/0022-1317-51-11-937
Wallner, A., King, E., Ngonkeu, E. L. M., Moulin, L., and Bena, G. (2019). Genomic analyses of Burkholderia cenocepacia reveal multiple species with differential host-adaptation to plants and humans. BMC Genomics 20:803. doi: 10.1186/s12864-019-6186-z
Waters, V. L., and Crosa, J. H. (1988). Divergence of the aerobactin iron uptake systems encoded by plasmids pColV-K30 in Escherichia coli K-12 and pSMN1 in Aerobacter aerogenes 62-1. J. Bacteriol. 170, 5153–5160. doi: 10.1128/jb.170.11.5153-5160.1988
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Keywords: Burkholderia cepacia complex, comparative genomics, cystic fibrosis, Burkholderia anthina , siderophore
Citation: Pham A, Volmer JG, Chambers DC, Smith DJ, Reid DW, Burr L and Wells TJ (2023) Genomic analyses of Burkholderia respiratory isolates indicates two evolutionarily distinct B. anthina clades. Front. Microbiol. 14:1274280. doi: 10.3389/fmicb.2023.1274280
Edited by:
Apichai Tuanyok, University of Florida, United StatesReviewed by:
Dinesh Sriramulu, Independent Researcher, Chennai, IndiaLeonardo Gabriel Panunzi, CEA Saclay, France
Copyright © 2023 Pham, Volmer, Chambers, Smith, Reid, Burr and Wells. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy J. Wells, dGltb3RoeS53ZWxsc0B1cS5lZHUuYXU=