 Katherine C. Crosby1†
Katherine C. Crosby1† Mariah Rojas1†Parul Sharma1,2
Mariah Rojas1†Parul Sharma1,2 Marcela A. Johnson1,2Reza Mazloom3
Marcela A. Johnson1,2Reza Mazloom3 Brian H. Kvitko4
Brian H. Kvitko4 Theo H. M. Smits5
Theo H. M. Smits5 Stephanus N. Venter6
Stephanus N. Venter6 Teresa A. Coutinho6,7
Teresa A. Coutinho6,7 Lenwood S. Heath3
Lenwood S. Heath3 Marike Palmer8,9*‡
Marike Palmer8,9*‡ Boris A. Vinatzer1*‡
Boris A. Vinatzer1*‡- 1School of Plant and Environmental Sciences, Virginia Tech, Blacksburg, VA, United States
- 2Graduate Program in Genetics, Bioinformatics, and Computational Biology, Virginia Tech, Blacksburg, VA, United States
- 3Department of Computer Science, Virginia Tech, Blacksburg, VA, United States
- 4Department of Plant Pathology, University of Georgia, Athens, GA, United States
- 5Environmental Genomics and System Biology Research Group, Institute of Natural Resource Sciences, Zurich University of Applied Sciences, Wädenswil, Switzerland
- 6Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute, University of Pretoria, Pretoria, South Africa
- 7Centre for Microbial Ecology and Genomics, University of Pretoria, Pretoria, South Africa
- 8School of Life Sciences, University of Nevada, Las Vegas, Las Vegas, NV, United States
- 9Department of Microbiology, University of Manitoba, Winnipeg, MB, Canada
As the name of the genus Pantoea (“of all sorts and sources”) suggests, this genus includes bacteria with a wide range of provenances, including plants, animals, soils, components of the water cycle, and humans. Some members of the genus are pathogenic to plants, and some are suspected to be opportunistic human pathogens; while others are used as microbial pesticides or show promise in biotechnological applications. During its taxonomic history, the genus and its species have seen many revisions. However, evolutionary and comparative genomics studies have started to provide a solid foundation for a more stable taxonomy. To move further toward this goal, we have built a 2,509-gene core genome tree of 437 public genome sequences representing the currently known diversity of the genus Pantoea. Clades were evaluated for being evolutionarily and ecologically significant by determining bootstrap support, gene content differences, and recent recombination events. These results were then integrated with genome metadata, published literature, descriptions of named species with standing in nomenclature, and circumscriptions of yet-unnamed species clusters, 15 of which we assigned names under the nascent SeqCode. Finally, genome-based circumscriptions and descriptions of each species and each significant genetic lineage within species were uploaded to the LINbase Web server so that newly sequenced genomes of isolates belonging to any of these groups could be precisely and accurately identified.
Introduction
The goal of taxonomy is to develop an organized system of named groups (taxa) of described organisms that is conducive to meaningful identification of those organisms and permits clear communication about them (Baron, 1996). From medical, veterinary, and plant pathology perspectives, it is also important that the identification of an unknown as a member of a taxon enables inference of the pathogenic potential of the organism. In fact, identification plays a critical role in the choice of medical treatment and determination of contagion risk in the case of disease diagnostics, decision-making on disease control and management in the case of disease outbreaks, or in decision-making on biosafety levels in microbiology research (Bayot and King, 2022). It also plays a key role in environmental applications when assessing risks and deciding on regulatory actions concerning the issuance of a field permit or approval for the commercialization of a microbial product (OECD, 2003; Chandler et al., 2011). Furthermore, the name of any taxon to which an organism belongs should not raise any doubts about which group the name refers to.
The current evolutionary, rank-based, hierarchical taxonomy of prokaryotes from the phylum level to the species/subspecies level and the established rules of nomenclature provide a framework that generally allows us to reach the described goal. In particular, species names are associated with type strains (Parker et al., 2019), or more recently, type genomes under the SeqCode (Hedlund et al., 2022; Whitman et al., 2022), to provide anchoring and stability of names. However, the evolutionary relationships among prokaryotes are impossible to establish based on phenotypes. Furthermore, sequencing one or a small number of marker genes, such as the 16S rRNA gene, is not always sufficient to correctly determine how bacteria are related to each other (Hauben et al., 1997). This is one of the reasons why several named species that were validly published in the past were later revised and assigned to different genera or families, including species that are, at present, assigned to the genus Pantoea (Gavini et al., 1989; Palmer et al., 2018a), the focus of this article. With the advent of cost-effective next-generation sequencing, whole-genome sequencing and constructing accurate core genome phylogenies (where all genes present only once in each analyzed genome are used) have become possible. Moreover, genome sequence comparisons currently allow us to easily assign bacteria to species based on average nucleotide identity (ANI), whereby 95% ANI is the generally agreed-upon guideline for the delineation of bacteria belonging to the same species (Konstantinidis and Tiedje, 2005; Jain et al., 2018). In recent years, the Genome Taxonomy Database (GTDB) has leveraged core genome phylogenies and ANI to establish exclusively genome-based taxa from phylum to species rank (Parks et al., 2020) that reflect evolutionary relationships and have standardized breadths of diversity (Parks et al., 2018).
However, there is another problem inherent to prokaryotic taxonomy that cannot be solved when using the species and subspecies as the smallest units: Important phenotypes, for example, the host range of plant pathogens (Cai et al., 2011), can vary within species, are sometimes linked to the accessory genome, and, therefore, do not always align with phylogeny (Venter et al., 2022). In these cases, identification of an unknown at the species or subspecies level is not sufficient to lead to meaningful phenotype prediction.
Typically, organisms with a recent common ancestor are more likely to share phenotypes with each other than with more distant relatives because they have vertically inherited a higher number of shared genes and alleles. Moreover, frequent recombination within populations of closely related organisms that occupy the same ecological niche further increases the number of shared genes and alleles among the members of these populations (Arevalo et al., 2019; Palmer et al., 2019). Therefore, identification of an unknown as a member of a group that has a very recent common ancestor can be expected to provide a more accurate prediction of the phenotype of the unknown than identification at the species level. The Life Identification Number (LIN) approach takes advantage of this fact by continuing hierarchical rank-based taxonomy from the species rank toward the strain level using a strictly ANI-based approach (Marakeby et al., 2014; Vinatzer et al., 2017). LINs consist of a series of positions, each representing a different ANI threshold, with ANI thresholds increasing from the left to the right. Any group of related genomes can be easily named by the LIN prefix they share (Vinatzer et al., 2017). The more similar the genomes of the groups' members are and the further their LIN prefix extends to the right, the more phenotypes they can be expected to share. Therefore, as long as the reciprocal ANI of the members of a group can be accurately measured because it is not too low (~70%) and not too high (~99.95%; depending on genome quality), the group can be clearly circumscribed. We call such groups “LINgroups”.
The genus Pantoea is a taxon that has proven to be particularly difficult to classify based on phenotypes (Tambong, 2019) as it shares many characteristics with other genera such as Erwinia, Enterobacter, and Pectobacterium (Brady et al., 2007). With the advent of DNA analysis tools, some species in the genera Erwinia, Enterobacter, and Pectobacterium were transferred to the genus Pantoea (Gavini et al., 1989; Mergaert et al., 1993; Brady et al., 2008, 2010). By the end of 2022, 19 names of species in the genus Pantoea were validly published as summarized on the LPSN website (Parte et al., 2020) and described in the Bergey's Manual chapter for Pantoea (Palmer and Coutinho, 2022), two named species were effectively published but the names could not be validly published under the International Code of Nomenclature of Prokaryotes; and an additional 29 species clusters were found to qualify as species in the GTDB (release 207) based on core genome phylogeny and ANI but have not been named (Parks et al., 2018, 2020, 2022).
The name of the genus Pantoea (“of all sorts and sources”) reflects that this genus includes bacteria that have been found in a wide range of environments, including plants, animals, soils, components of the water cycle, humans, and the built environment (Palmer and Coutinho, 2022). Mechan Llontop et al. (2021) found that bacteria in the genus are among the most common and efficient rain-borne colonizers of tomato leaves, suggesting that members of the genus frequently migrate between compartments of the water cycle and plants, as well as possibly other environments, without having adapted to a particular environment. However, some Pantoea species or subspecies have adapted and live in more restricted environments, for example, the insect-transmitted plant pathogen P. stewartii subsp. stewartii (Mergaert et al., 1993). For other species in the genus, it is not clear if all lineages within the same species have a similar pathogenic potential or if some lineages are pathogenic and others are not. Most notably, within P. agglomerans, Rezzonico et al. (2009) found isolates from humans, plants, and the environment. Some P. agglomerans strains appear to be opportunistic human pathogens, while others have been found to have biocontrol activity against plant pathogens (Rezzonico et al., 2009; Lorenzi et al., 2022). To date, it has not been possible to distinguish between strains with different phenotypes based on their genomes (Sulja et al., 2022). This is a challenge with practical implications since determining if a potentially beneficial P. agglomerans strain could at the same time present a risk to crops and even agricultural workers is a key question for which regulators need an answer when deciding on field permits and commercialization (Bonaterra et al., 2014). Another problem is that gene sequences and genome sequences sometimes have the wrong taxonomic assignment in NCBI, in particular at the species rank, at which up to 10% of genomes of Pantoea strains were found to be misidentified or mislabeled (Tambong, 2019), thus confusing NCBI users about the identity of their query sequences.
To contribute to the advancement of the taxonomy of the genus Pantoea and to facilitate the correct and precise identification of newly sequenced genomes, we compared 559 genome sequences of the genus. We computed pairwise ANI values between all genomes and built a highly resolved core genome tree of 437 representative genomes. We also determined which clades correspond to population clusters based on recent horizontal gene transfer events and analyzed differences in gene content between some of the genetic lineages that belong to the same species to help us determine if these lineages may occupy different ecological niches. We also compiled ecological and phenotypic information from the literature and from genome metadata, and we determined which clades in the core genome tree correspond to named species or yet-unnamed species clusters circumscribed in GTDB, 15 of which we named here under the SeqCode (SeqCode Registry List: seqco.de/r:xfaladud). We finally circumscribed all these genetic lineages as LINgroups in LINbase (Tian et al., 2020) and added the corresponding descriptions so that any newly sequenced Pantoea genome can be identified as a member of any of these groups.
Methods
Genomes classified by GTDB release 207 (Parks and Hugenholtz, 2022) as Pantoea and with over 90% completeness were downloaded from the NCBI Assembly database on 4 October 2022, while taxonomy data and genome quality data were downloaded directly from GTDB. Associated metadata about sources of isolation were downloaded from NCBI's BioSample and BioProject databases on 5 October 2022. Additional genomes of strains isolated from precipitation that had been identified as Pantoea based on 16S rRNA sequencing (Failor et al., 2017) were also analyzed and submitted to NCBI under Project PRJNA445714. Genomes and metadata were then uploaded to LINbase, and ANI-based LINs were assigned as previously described (Tian et al., 2020).
When multiple genomes were over 99.95% identical to each other based on LINflow (Tian et al., 2021) analysis, only one genome representative for such a group was used for further analysis. These genomes were annotated using Bakta version 1.4.0 (Schwengers et al., 2021), and pangenome analysis was performed using the software PIRATE (Bayliss et al., 2019). Core genes were identified based on their presence in 95% of all genomes. Two core genome trees were constructed using (i) maximum likelihood (ML) with ultrafast bootstrapping in IQ-TREE (version 2.2.0.3) (Minh et al., 2020) and (ii) approximate ML with the generalized time-reversible model and SH-like branch support in FastTree version 2.1.11 (Price et al., 2010). Differences in gene content between clades were determined by creating a presence–absence matrix for all genomes for all gene families from the PIRATE output file, -PIRATE.gene_families.ordered.tsv, whereby presence was marked as 1 and absence as 0. This file was then queried in R (version 4.2.1) to determine the differences in gene content between clades within various Pantoea species.
The same genomes used for the pangenome and core genome analyses were also used as input for PopCOGenT (Arevalo et al., 2019) to determine population clusters using a reverse ecology approach. PopCOGenT detects recent gene flow events between genomes by searching for regions without SNPs. Two or more genomes that have more frequent and longer SNP-free regions are considered to belong to the same ecologically and genetically distinct population.
Finally, all named species found in the List of Prokaryotic Names with Standing in Nomenclature (Parte et al., 2020), unnamed species clusters in GTDB release 207 (Parks and Hugenholtz, 2022), and other clades identified in the core genome tree were circumscribed in LINbase, and descriptions from the Results section of this study were added to these circumscriptions.
Results
ANI clustering, phylogenomics, and population genomics lead to largely congruent results
A total of 550 genomes with over 90% completeness were found to be classified in GTDB release 207 (Parks and Hugenholtz, 2022) as members of the genus Pantoea. Nine genomes of strains isolated from precipitation and identified as members of the genus Pantoea (Failor et al., 2017) were also included. Supplementary Table 1 lists these 559 genomes together with their taxonomic lineages based on NCBI and GTDB, with their ANI-based LINs expressing their precise genomic similarity to each other, with metadata NCBI BioSample data, NCBI BioProject data, and research articles, and a variety of additional metadata from NCBI and GTDB. Genomes of Pantoea species that were later reclassified as members of the genera Mixta and Tatumella or that were found by GTDB to belong to other genera were not included in our analysis.
Based on their assigned LINs (see Supplementary Table 1), 559 putative Pantoea genomes were found to have at least 80% pairwise ANI values and to belong to 437 groups with pairwise ANI values of over 99.95%. Only one genome for each of these 437 groups was retained for further analysis to provide a more balanced representation of the genus Pantoea than the original 559 genomes.
At the 95% ANI species threshold (LIN position F), 49 LINgroups were identified. These species-level LINgroups comprised between 1 and 147 genomes each. Eighteen of the LINgroups corresponded to validly published species under the genus Pantoea, four to effectively published named Pantoea species, two to effectively published species outside of the genus, and another twenty-five were provisionally named in GTDB as species clusters and do not correspond to either effectively or validly published species (Table 1; Supplementary Table 3). Three GTDB species clusters were not considered here since they had representative genomes that were <90% complete and had been excluded from our analysis (Table 1). Within some of the species-level LINgroups, LINgroups at higher ANI levels stood out. Such groups are discussed further in the next section, and their corresponding LINs are reported in Supplementary Table 3.
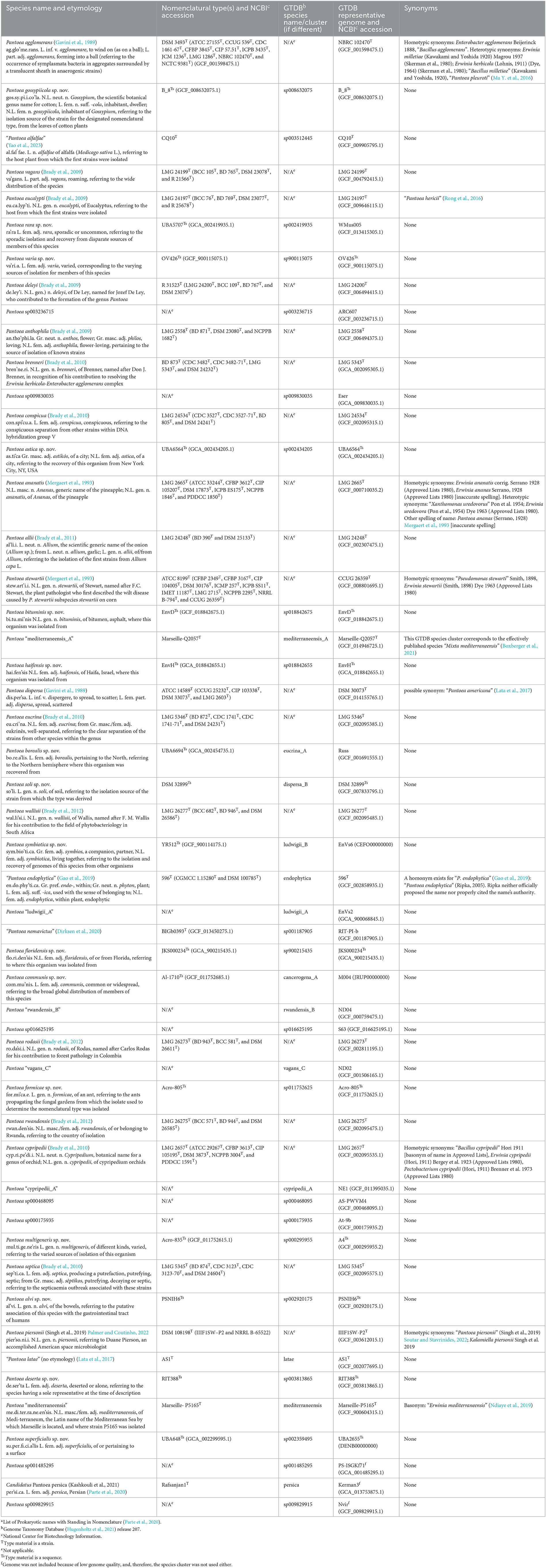
Table 1. Pantoea species names and etymology from the LPSNa or the new species names proposed here, the nomenclatural type from the LPSNa or proposed here, the name/number of the corresponding GTDBb species/cluster, the GTDBb representative and NCBIc accession number, and synonyms.
The core genome and pangenome were then determined. The pangenome was found to be composed of 36,144 orthologous groups, and the core genome was found to consist of 2,509 orthologous groups (based on their presence in 95% of the analyzed genomes) with a total length of 2,737,734 nucleotides. From this data, two robust core genome trees were constructed using ML approaches, i.e., a ML tree constructed with IQ-TREE (Supplementary Figure 1) and an approximate ML tree constructed with FastTree (Supplementary Figure 2). A pangenome tree using all 36,144 orthologous genes was also constructed (Supplementary Figure 3). The ML core genome tree in which the major clades are highlighted is shown in Figure 1. There was overall high congruence among all three trees, and individual phylogenetic lineages are described below.
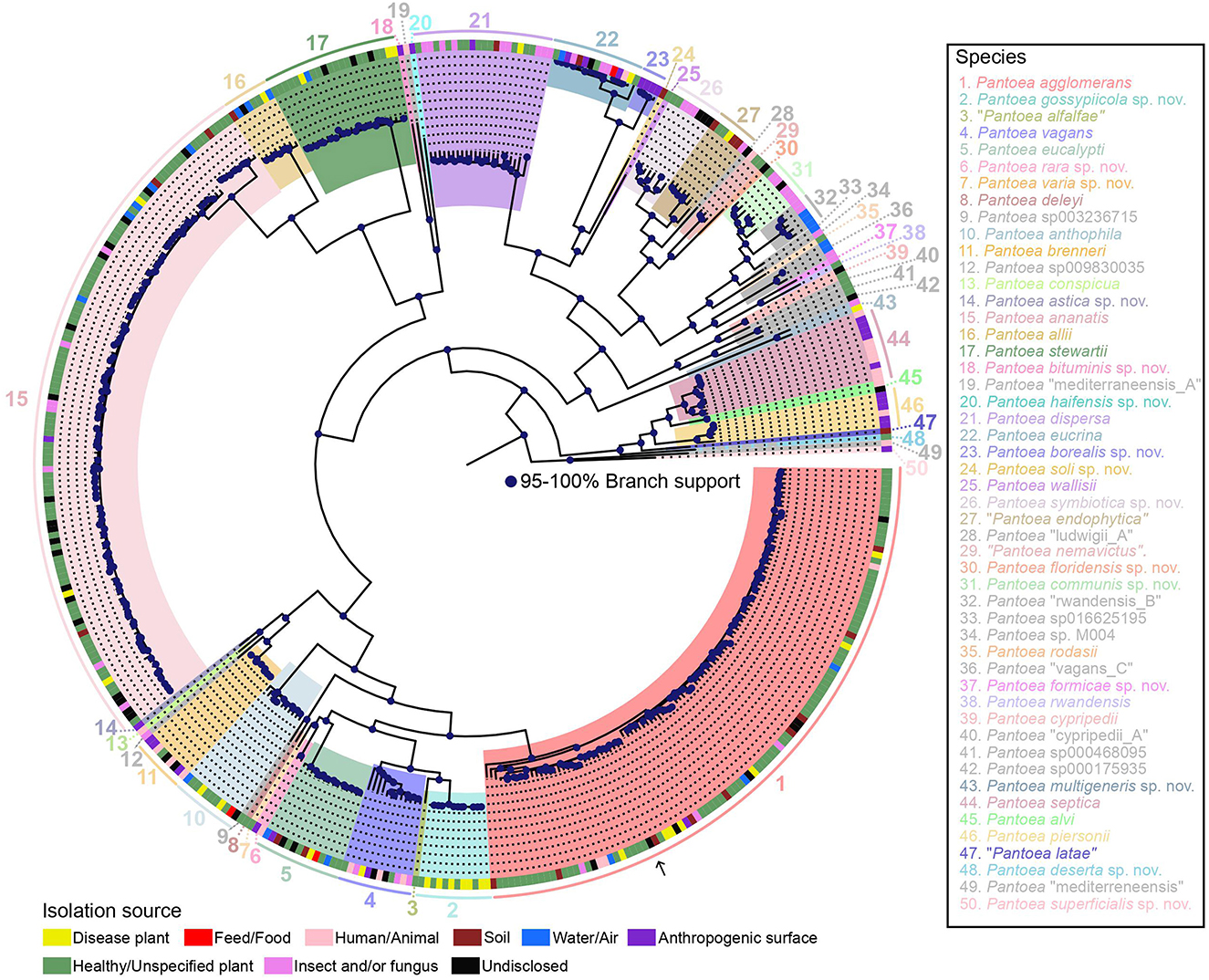
Figure 1. ML phylogeny of 437 representative genomes of the genus Pantoea based on the alignment of 2,509 core genes and rooted with Mixta calida DSM 22759T (outgroup not shown). Species names (or GTDB species cluster identifiers for yet-unnamed species) as well as isolation sources are provided in the legend. Species newly named under the SeqCode are indicated with “sp. nov.” following the species epithet. The nomenclatural type for the genus, P. agglomerans DSM 3493T, is indicated with an arrow. Strain names were suppressed but are included in the same tree shown in Supplementary Figure 1, which also highlights type strains or type sequences, respectively, and includes within-species lineage information. Strain isolation sources from the NCBI BioSample database and other strain metadata are provided in Supplementary Table 1, which also includes the 122 genomes omitted from the tree because of high similarity to the genomes that were included.
To identify population clusters in the genus Pantoea, the reverse ecology tool PopCOGent (Arevalo et al., 2019) was used to identify populations based on recent horizontal gene transfer (HGT) events. Genomes inferred to belong to the same populations were denoted with the same population identifiers (Supplementary Figure 1). In most cases, species contained two or more population clusters. Only a small number of species, mostly species for which only a few genomes have been sequenced, belonged to the same cluster (see details below).
Finally, strain source information available in NCBI's BioSample and BioProject databases and in the published literature were correlated with genome similarity, phylogeny, and ecology. Sources were classified into the following categories: healthy or unspecified plants, diseased plants, humans/animals, insects, soil, fungi, water/air, anthropogenic surfaces, feed/food, and undisclosed. Categories are listed for all individual genomes in Figure 1, Supplementary Figure 1, and Supplementary Table 1 and are summarized by species in Supplementary Table 2.
A detailed description of 49 Pantoea species follows in the order in which they are located in the tree in Figure 1 (clockwise, starting with P. agglomerans). For each species, taxonomic details (species name, etymology, type strain or genome, and/or representative genome in GTDB, and synonyms) are provided in Table 1, and LINs are provided in Supplementary Table 3. Yet-unnamed species clusters were named under the SeqCode (Hedlund et al., 2022; Whitman et al., 2022) when raw reads for at least one genome sequence, chosen as the type, were available in the NCBI SRA database, as required by the SeqCode. New species names are labeled with “sp. nov.” following species epithets.
Pantoea agglomerans (76A6B2C0D0E3F)
Phylogeny, genomics, and reverse ecology
Out of 122 genomes identified as P. agglomerans (Gavini et al., 1989) in the GTDB Release 207, 104 representative genomes were used in our analysis (the other 17 genomes were omitted to limit redundancy since they had >99.95% ANI in the genomes that were included). The chosen genomes cluster with the genome of the P. agglomerans-type strain DSM 3493T in a single clade with 100% bootstrap support. Genomes in this clade have a minimum pairwise ANI of 97%, indicating their conspecificity. All genomes in this clade are thus members of P. agglomerans, even though some are misidentified or mislabeled in NCBI as P. stewartii, P. vagans, or the non-validly published heterotypic synonyms “P. pleuroti” and “Curtobacterium plantarum” (Ma Y. et al., 2016) (see Supplementary Table 1 for NCBI organism names, GTDB names, and species names assigned based on our analysis). The ANI between the genome of DSM 3493T and the most similar genome outside of the clade, that of strain DE0421 (P. eucalypti), is approximately 91%, confirming this taxon's genomic exclusivity from genomes in other clades. Genomes misidentified as P. agglomerans in NCBI are located in numerous other clades (see Supplementary Table 1).
The P. agglomerans clade is divided into three subclades (Figure 1). Clades 1 and 2 are fully supported in the ML analysis (Supplementary Figure 1). Clade 3 consists of a single genome, P5. The same subclades are present in the approximate ML core genome tree (Supplementary Figure 2) and in the pangenome tree (Supplementary Figure 3). Genomes in subclade 1 have a minimum pairwise ANI of 98.5%, and genomes in subclade 2 have a minimum pairwise ANI of 98.0%. The approximate ANI between the subclades is 98%. The subclades also represent distinct population clusters based on HGT, with subclade 2 including two genomes comprising the sole members of distinct and separate populations. When comparing the gene content of the subclades, we found 13 genes (encoding electron transport chain-related proteins, transporters, a carrier protein, and pilus/fimbriae-associated proteins) to be present in over 95% of subclade 2 genomes and absent from subclade 1 genome. Genome P5 contains 75 genes (encoding CRISPR-associated proteins, transporters, enzymes, and hypothetical proteins) not present in any of the genomes of subclades 1 and 2. However, no genes were present in over 95% of subclade 1 genomes and absent from subclade 2 genomes (Supplementary Table 4). Therefore, gene content differences do not strongly support distinct ecological differentiation of the identified subclades, which is also in agreement with the similar isolation sources reported for strains in the two clades.
The genome of strain P. agglomerans pv. gypsophilae 824-1 is located in P. agglomerans subclade 1, and the genome of P. agglomerans pv. betae 4188 is located in P. agglomerans subclade 2. These strains are representatives of a pathogen of baby's breath (Gypsophila paniculata) and of beet (Beta vulgaris), respectively. Since only one genome is available for each of the two pathogens, it is not possible to determine if the baby's breath pathogen is specific to subclade 1 and the beet pathogen is specific to subclade 2.
P. agglomerans genomes share a most recent common ancestor (MRCA) with genomes in a clade that includes P. vagans, P. eucalypti, and a number of genomes of other species listed below. P. deleyi splits off at a node immediately basal to this clade. Based on previous phylogenomic analysis (Palmer and Coutinho, 2022), the effectively published species “Curtobacterium plantarum” and “P. pleuroti” cluster with the P. agglomerans-type strains and are heterotypic synonyms of P. agglomerans.
Ecology, lifestyle, and biotechnological uses
Most genome-sequenced strains in the P. agglomerans species were isolated as epiphytes or endophytes from a broad range of plants (92/120; see Supplementary Table 1 for details). Out of these 92 strains, eight are listed in the NCBI BioSample database as having been isolated from plants with disease symptoms. However, it is not clear whether all these strains caused the observed symptoms. P. agglomerans is thus more likely to be a common plant-associated bacterium instead of a plant pathogen. The exceptions are bacteria in the pathovars gypsophilae and betae mentioned above, since the genomes of these bacteria contain plasmids encoding hrp type III secretion systems (T3SS) and effectors that were shown to be necessary for the formation of galls on their host plants (Nissan et al., 2019). Some strains of P agglomerans have also been shown to cause disease on onion leaves and bulbs (Edens et al., 2006; Moloto et al., 2020). One isolate of P. agglomerans from a corn leaf (named Erwinia herbicola in the original publication, a heterotypic synonym of the species) was one of the very first bacteria found to have ice nucleation activity (INA) (Lindow et al., 1978). INA bacteria are hypothesized to travel through the water cycle and possibly contribute to precipitation (Failor et al., 2017). In fact, two of the genome-sequenced P. agglomerans strains were isolated from precipitation. Another eight strains were isolated from the soil. This is consistent with early reports on the presence of P. agglomerans in water (Gavini et al., 1989) and soil (Kageyama et al., 1992). On a regular basis, P. agglomerans has also been reported as a clinical isolate (Milanowski et al., 1995; Bujdáková et al., 2001; Koester et al., 2020; Lorenzi et al., 2022). However, in most cases, isolates were not genome sequenced, and their identity as P. agglomerans could not be confirmed. While the genome-sequenced P. agglomerans-type strain ICMP 12534T was isolated from a human knee laceration and other genome-sequenced strains were isolated from the sputum of a cystic fibrosis (CF) patient and from a cervix (based on NCBI BioSample data; Supplementary Table 1), in none of the cases of human infection could the observed symptoms be unequivocally attributed to P. agglomerans; see also (Rezzonico et al., 2010, 2012a,b).
Biotechnologically relevant properties include the production of antibiotics (Smith et al., 2013; Smits et al., 2019; Robinson et al., 2020), biocontrol of plant diseases (Vanneste et al., 2002; Kim et al., 2012; Smits et al., 2015; Lorenzi et al., 2022), and many others. Another potential biotechnological application could be the use of P. agglomerans with INA in the production of frozen foods and in the control of insects by making larvae more susceptible to frost damage (Coutinho and Venter, 2009). In conclusion, the entire P. agglomerans species appears to have been adapted to life in association with plants, and its members survive well in various environments. Therefore, bacteria in this species may frequently migrate between plants, soil, and aquatic environments and occasionally colonize animals and humans. Without strong evidence for or against human pathogenicity of the species or within-species lineages and the absence of genomic signatures to predict pathogenicity (Lorenzi et al., 2022), implementation in biotechnological applications, in particular as a biocontrol organism, remains negatively affected.
Pantoea gossypiicola sp. nov. (76A6B2C0D0E2F)
Based on the BioSample metadata available for genomes in this species (34 in total, 12 of them included in our analysis), all strains (including the GTDB representative and nomenclatural type B_8Ts) were recovered as epiphytes from diseased cotton leaves (Gossypium hirsutum). To determine if P. gossypiicola sp. nov. is a pathogen of cotton, Koch's postulates would need to be completed. The minimum pairwise ANI of genomes in this species is 99.25%. The high intraclade ANI shows that the diversity within the species cluster has been poorly sampled. Although three clonal lineages can be distinguished based on phylogeny (each with 100% branch support) and ANI, all strains of the species belong to the same population, suggesting that the clonal lineages have not adapted to separate ecological niches. P. gossypiicola sp. nov. shares an MRCA with “Pantoea alfalfae”.
“Pantoea alfalfae” (76A6B2C0D0E0F)
This species had two known representatives in GTDB at the time genome sequences were downloaded for analysis: SRS89 (the only genome included in the tree), isolated from Stevia rebaudiana seeds, and a metagenome-assembled genome (MAG) of medium quality (80.3% completeness), UBA8265, recovered from a laboratory enrichment culture for Salmonella. SRS89 is misidentified in the NCBI as P. vagans. Recently, the species was effectively published as “P. alfalfae” with strain CQ10, isolated from alfalfa (Medicago sativa) seeds, as a proposed type strain (Yao et al., 2023). The conspecificity of CQ10 and SRS89 is supported by phylogenomics in the recent release of GTDB. As stated above, “Pantoea alfalfae” shares an MRCA with P. gossypiicola sp. nov.
Pantoea vagans (76A6B2C0D0E1F)
Phylogeny, genomics, and reverse ecology
P. vagans (Brady et al., 2009) includes the genomes of 13 strains, including the type strain LMG 24199T. One additional genome of the species was omitted from the phylogenetic analysis. In NCBI, these strains are either identified as P. vagans or only at the genus level. Strains misidentified in the NCBI as P. vagans are located in the phylogenetic tree as members of other species, for example, P. agglomerans and P. eucalypti. P. vagans genomes have a minimal pairwise ANI of 96%, revealing a genomically diverse group of genome-sequenced strains. This diversity is also reflected in the reverse ecology analysis, which identified six population clusters. One cluster (cluster 24, corresponding to subclade 1 in Supplementary Figure 1) has eight members, and the other four clusters only have one member each. Gene content comparison revealed that the genomes in population cluster 24 share 11 genes (encoding a type II toxin–antitoxin system CcdA family antitoxin and a hemolysin activator protein) that are absent from the other four genomes (Supplementary Table 4). Furthermore, the clade corresponding to population cluster 24 is also conserved in the other trees, further suggesting that population cluster 24 represents an ecologically distinct population (Supplementary Figures 2–4). The strains in the P. vagans clade share an MRCA with P. gossypiicola sp. nov. and “P. alfalfae” described above.
Ecology, lifestyle, and biotechnological uses
The P. vagans-type strain and other non–genome-sequenced strains of the species were isolated from eucalyptus presenting bacterial blight and dieback symptoms and from maize with brown stalk rot (Brady et al., 2009), but there is no evidence of being the causal agent of the observed disease symptoms. P. vagans has also been isolated from other plants, insects, and occasionally from humans in a medical context. However, as for the plant isolates, there is no indication that P. vagans caused these infections.
One example of the beneficial and biotechnological properties of P. vagans is the biocontrol organism P. vagans C9-1 (Ishimaru et al., 1988; Smits et al., 2011), which produces three antibiotics: the peptide-derived antibiotic Pantocin A (Smits et al., 2019), the antibiotic herbicolin I (Kamber et al., 2012a), and a third antibiotic, recently identified as PNP-3 (Williams and Stavrinides, 2020), which is an antibiotic not produced by any other strain of P. vagans currently sequenced (Sulja et al., 2022).
Pantoea eucalypti (76A6B2C0D0E6F)
Phylogeny, genomics, and reverse ecology
The 15 genomes of P. eucalypti (14 of which were used in the phylogenetic analysis, including the genome of the type strain LMG 24197T) have a minimal pairwise ANI of over 99% and belong to the same population cluster, revealing that the P. eucalypti strains sequenced to date are members of a single clonal lineage. Some of these members are mislabeled in NCBI as they were misidentified as other Pantoea species, but their very high ANI to the P. eucalypti-type strain genome and their phylogenetic position in the same clade clearly identify them as P. eucalypti. The P. eucalypti clade shares an MRCA with P. rara sp. nov. and P. varia sp. nov. Recently, it was reported (Palmer and Coutinho, 2022) that the “Pantoea hericii”-type strain represents a heterotypic synonym of P. eucalypti.
Ecology, lifestyle, and biotechnological uses
The P. eucalypti-type strain was isolated from a eucalyptus plant with symptoms of blight and dieback in Uruguay (Brady et al., 2009). P. eucalypti was also isolated from other plant species, but no information is available about whether these isolates caused symptoms in any of these plants, and a T3SS gene cluster was not detected in any of the eight P. eucalypti genomes previously analyzed by Moretti et al. (2021). Other genome-sequenced P. eucalypti strains were isolated from precipitation, soil, and commercial poultry feed. Beneficial and biotechnological properties of P. eucalypti were described for P. eucalypti FBS 135, which promotes Masson pine (Pinus massoniana) seedling growth and increases seedling survival rates (Song et al., 2020).
Pantoea rara sp. nov. (76A6B2C0D0E5F)
This species consists of the genomes of strain WMus005 isolated from the oral cavity of a wild house mouse (Mus musculus) and the metagenome-assembled genome (MAG) UBA5707Ts, chosen as the nomenclatural type, recovered from a metagenomic sample taken from the New York City subway system. The pairwise ANI of the two genomes is over 99%, and they thus clearly belong to the same species. P. rara sp. nov. shares an MRCA with P. varia sp. nov.
Pantoea varia sp. nov. (76A6B2C0D0E4F)
This species consists of the genomes of two strains, of which OV426Ts was chosen as the nomenclatural type, isolated during the characterization of Populus root and rhizosphere microbial communities and from an indoor dust sample (the genome of this strain, AF015A5, is not included in the GTDB and not in our analysis but is deposited in the RefSeq database with accession number GCF_004798195.1 and misidentified as P. allii). The two genomes have a pairwise ANI of approximately 97%. As stated above, P. varia sp. nov. shares an MRCA with P. rara sp. nov.
Pantoea deleyi (76A6B2C0D1E0F)
The genome of the P. deleyi-type strain, LMG 24200T, the only genome of this species included in our analysis, shares an MRCA with P. sp003236715. The P. deleyi-type strain was isolated from Eucalyptus (Brady et al., 2009). The symptoms of infection on leaves and shoots include bacterial blight and dieback. The genome of P. deleyi contains two gene clusters encoding T3SS: an uncharacterized Hrp-2b type and the SPI-1 type, highly similar to the SPI T3SSs of the plant pathogenic strains P. stewartii subsp. stewartii DC283 or E. amylovora CFBP 1430 (Smits et al., 2010; Correa et al., 2012; Kamber et al., 2012b; Moretti et al., 2021), suggesting that the symptoms observed on the host plant may have been caused by P. deleyi.
Pantoea sp003236715 (76A6B2C0D1E1F)
In GTDB release 207, this species cluster encompasses a single genome-sequenced strain, ARC607, isolated from rice (Oryza sativa) seeds. As mentioned above, this species shares an MRCA with P. deleyi.
Pantoea anthophila (76A6B2C0D3E0F)
P. anthophila consists of the genome of the type strain LMG 2558T and 13 additional genomes (three of which were omitted from the phylogenetic analysis). The P. anthophila clade splits at a node that is basal to all clades described to date. It includes strains that are misidentified or mislabeled as P. agglomerans and P. vagans in the NCBI. However, the minimal pairwise ANI of strains within the clade is 98%, clearly showing that all members belong to P. anthophila. There are seven separate population clusters in this species. The four genomes that belong to the same population share seven genes (including, among others, genes coding for bacteriocin and hypothetical proteins) that are absent from the seven genomes in the other six clusters (Supplementary Table 4), supporting the idea that this population may occupy a separate ecological niche. However, this is not evident based on isolation source information since the majority of strains in the species, including the type strain, were isolated from plants. There is currently no indication of pathogenicity, and only three out of seven P. anthophila genomes analyzed by Moretti et al. (2021) had a gene cluster coding for a T3SS. Furthermore, one P. antophila strain was isolated from hypersaline lake water and one from commercial poultry feed, suggesting that, similar to P. agglomerans and P. vagans, this species may not only be adapted to life in association with plants but also survive well as a free-living organism in the environment.
Pantoea brenneri (76A6B2C0D2E2F)
Phylogeny, genomics, and reverse ecology
Nine genomes of P. brenneri were used in the phylogenetic analysis, including the genome of the type strain LMG 5343T, and another 15 genomes were not included. The minimal pairwise ANI in the P. brenneri clade is 99%, suggesting that this species has limited diversity or that sampling has been limited. This is supported by reverse ecology analysis, which assigned all genomes in the clade to the same population cluster. The genome most closely related to P. brenneri is that of strain Eser (P. sp009830035), with which the P. brenneri clade shares an MRCA.
Ecology, lifestyle, and biotechnological uses
The type strain, P. brenneri, was isolated from a human urethra. Another two strains were isolated from human sputum and an unidentified source in Canada (Brady et al., 2010). Sixteen (two of them included in the phylogenetic analysis) almost identical genome-sequenced P. brenneri strains were isolated from environmental surfaces on the International Space Station. Other genome-sequenced strains were isolated from human samples and anthropogenic environments, including a sample of brown algae (Ectocarpus subulatus) and an endophyte sample from gray willow (Salix atrocinerea). Overall, P. brenneri appears to have a much closer association with humans and is more commonly found in anthropogenic environments than P. agglomerans, P. vagans, and P. anthophila (among others). Whether it is a human commensal or an opportunistic human pathogen cannot be concluded at this point.
Pantoea sp009830035 (76A6B2C0D2E3F)
This species cluster consists of the genome of a single strain, Eser, isolated from a brown stink bug (Euschistus servus). It shares an MRCA with the P. brenneri clade (see above).
Pantoea conspicua (76A6B2C0D2E1F)
The only genome in the P. conspicua clade is of the species-type strain LMG 24534T. It shares an MRCA with Pantoea astica sp. nov. The type strain was isolated from human blood (Brady et al., 2010).
Pantoea astica sp. nov. (76A6B2C0D2E0F)
This species consists of a single MAG, UBA6564Ts in GTDB release 207, which was chosen as a nomenclatural type. It was assembled from a DNA sample of a metal surface in New York City. P. conspicua is the most closely related species (see above).
Pantoea ananatis (76A6B13C0D1E0F)
Phylogeny, genomics, and reverse ecology
The 114 genomes in the P. ananatis species that were used in the phylogenetic analysis, including the genome of the type strain LMG 2665T, have a minimal pairwise ANI of 96%. Another 35 genomes were omitted because they were highly similar to the 114 genomes that were included. The P. ananatis clade is divided into two main subclades, each fully supported in core genome phylogenies (labeled 1 and 2 in Supplementary Figure 1) and also maintained in the pangenome tree (Supplementary Figure 3). The minimal pairwise ANI is 98.5% for genomes within subclade 1 and 99% for genomes within subclade 2. The ANI between the two subclades is ~97%. The two subclades represent two separate population clusters, with subclade 1 strain FDAARGOS680 being the only member of a third population. This suggests that strains in the two subclades may occupy separate ecological niches. This is also supported by gene content differences. Two genes (one related to ion transport and a putative beta-carotene dioxygenase) are present in over 95% of subclade 1 genomes, but neither in a single genome of subclade 2 nor FDAARGOS680. Additionally, nine genes (associated with cytochrome C, membrane proteins, and hypothetical proteins) are present in over 95% of subclade 2 genomes but completely absent from subclade 1 genomes and from FDAARGOS680. The genome of strain FDAARGOS680 contains 160 genes not present in any of the genomes of subclades 1 and 2 (Supplementary Table 4). P. ananatis shares a MRCA with P. allii. We note that some genomes in the P. ananatis clade are misidentified or mislabeled in NCBI as P. vagans, while other genomes located in the phylogenetic tree in clades corresponding to other species are misidentified or mislabeled as P. ananatis in NCBI.
Ecology, lifestyle, and biotechnological uses
P. ananatis LMG 2665T was isolated from the diseased Ananas comosus (pineapple). Bacteria in the P. ananatis species have been isolated over the years as pathogens from a growing number of crops and forest plants (Coutinho and Venter, 2009), for example, as a causative agent of the bacterial leaf blight of rice (Yu et al., 2021). P. ananatis has received extensive attention as a pathogen of onions, where its capacity to cause disease is dependent on the HiVir biosynthetic genes for the phosphonate phytotoxin Pantaphos. P. ananatis onion virulence is also supported by alt (allicin tolerance) genes conferring increased resistance to Allium thiosulfinate defensive chemistry (Asselin et al., 2018; Stice et al., 2020; Agarwal et al., 2021a; Polidore et al., 2021; Shin et al., 2023). P. ananatis has also been isolated as an endophyte and an epiphyte from many healthy plants, a symptomatic button mushroom, insects, soil, precipitation, and an air sample. It was also reported to cause bacteremia in humans, but identification was only based on partial 16S rRNA sequencing (De Baere et al., 2004); thus, the taxonomic affiliation of these strains remains uncertain. Not a single genome-sequenced isolate originates from human tissues or organs. Therefore, isolation sources and pathogenicity assays of P. ananatis clearly indicate pathogenic potential in plants, but pathogenicity in humans and animals remains uncertain.
There are several examples of biotechnological applications of P. ananatis. For example, a genetically engineered P. ananatis strain was used to synthesize the amino acids cysteine (Takumi et al., 2017) and L-glutamic acid (Hara et al., 2012) and isoprenoids (Nitta et al., 2021). Ma J. et al. (2016) demonstrated that another P. ananatis strain metabolizes rice straw by degrading lignocellulose, and Schulz et al. (2017) showed that P. ananatis, in the presence of the yeast Papiliotrema baii, detoxifies hydroxylated benzoxaxolinones. Deivanai et al. (2014) instead described that rice seedlings have higher fitness in the presence of P. ananatis 13 SE 1, and Mechan Llontop et al. (2020) found a strain isolated from rain to have biocontrol activity against the fire blight pathogen E. amylovora. Furthermore, the crtI carotene desaturase gene of P. ananatis (referred to in the publication under its heterotypic synonym Erwinia uredovora) was incorporated into the vitamin A biosynthetic pathway of Golden Rice (Paine et al., 2005).
Pantoea allii (76A6B13C0D0E0F)
Phylogeny, genomics, and reverse ecology
The eight genomes in the P. allii clade, including that of the type strain LMG 24248T, have a minimal pairwise ANI of at least 98.5%, revealing genomic homogeneity significantly above the 95% ANI species threshold. However, the clade has a substructure well-supported by the ML phylogeny, and genomes were assigned to three distinct population clusters consisting of four genomes, three genomes, and one genome, respectively, possibly suggesting the occupation of separate ecological niches. However, this was not supported by the other trees (Supplementary Figures 2, 3), which did not maintain the same clade substructure, and was only partially supported by differences in gene content: no genes were found to be uniquely present in the largest population, 14 genes were found to be uniquely present in the second population, and 153 genes were found to be present only in the genome of strain BAV3077 (Supplementary Table 4). P. allii shares an MRCA with P. ananatis and P. stewartii.
Ecology, lifestyle, and biotechnological uses
The Pantoea allii-type strain was isolated from onion seed (Allium cepa) (Brady et al., 2011). The capacity of the type strain to cause necrosis on onion leaves was shown to be dependent on the halophos phosphonate biosynthetic gene cluster (Zhao et al., 2023). Other P. allii strains were isolated from onions with center rot (Brady et al., 2011). Genome-sequenced P. allii strains were also isolated from the seed and rhizosphere soils of other plants and from rainfall. While P. allii is a confirmed onion pathogen, it is not yet clear if it causes disease on any of the other plants from which it was isolated.
Pantoea stewartii (76A6B13C1D0E0F)
Phylogeny, comparative genomics, and reverse ecology
The P. stewartii clade consists of 22 genomes, including the genome of the type strain CCUG 26359T, while another 11 genomes were omitted from the phylogenetic analysis. The P. stewartii clade splits off at a node immediately basal to P. allii and P. ananatis. The maximal pairwise ANI among genomes within the clade is 98.5%, revealing limited genomic diversity. The clade has an extensive substructure with a dozen small clades that are highly supported by all phylogenies. However, these clades do not correlate with the six population clusters identified by reverse ecology analysis. In particular, population cluster 4 includes the genomes of many separate clades, which could be explained by frequent recombination events.
The assignment of genomes to subspecies stewartii, the pathogen that causes Stewart's Wilt of corn (Zea mays), and subspecies indologenes, the pathogen described as the causative agent of leaf spot on millet (Mergaert et al., 1993), is incomplete in NCBI. Furthermore, based on isolation sources reported in NCBI's BioSample database, neither the genomes of strains recovered from corn nor the genomes of strains recovered from millet can be assigned to separate clades. Moreover, these genomes are interspersed with the genomes of strains recovered from other plants and the environment. Therefore, based on isolation sources alone, neither subspecies appears to be monophyletic. Further studies will be necessary to clarify how subspecies are related to each other, including the use of discriminatory primers (Shin et al., 2022).
Ecology, lifestyle, and biotechnological uses
Stewart's Wilt disease was first described by Stewart (1897), and the causative agent was later described as P. stewartii subsp. stewartii (Mergaert et al., 1993), which is transmitted by the corn flea beetle (Chaetocnema pulicaria) (Rand and Cash, 1937). P. stewartii subsp. indologenes was originally described as a pathogen of leaf spot on pearl millet (Pennisetum glaucum) and foxtail millet (Setaria italica) (Mergaert et al., 1993) but, since then, it has been found to cause disease on several additional plant species, including onion (Allium cepa) (Agarwal et al., 2021b; Koirala et al., 2021). P. stewartii subsp. stewartii is a T3SS-dependent pathogen of corn. After identifying subspecies indologenes as pathogens of center rot of onion (Allium cepa) and other Allium species (Stumpf et al., 2018), two pathovars were described, cepacicola and setariae, based on their capacity to cause disease on both millet and Allium species or only on millet, respectively (Koirala et al., 2021). While P. stewartii subsp. indologenes pv. cepacicola pathogenicity on millet is T3SS-dependent, pathogenicity on onion is dependent on either the HiVir or halophos phosphonate biosynthetic gene clusters (Zhao et al., 2023). Based on NCBI BioSample information, P. stewartii was also isolated from several additional plant species, a waterfall, and an unspecified environmental source. In most cases, the NCBI BioSample data do not indicate if the plants were diseased, but several publications confirm that P. stewartii is in fact a pathogen of these plants (Azizi et al., 2019; Abidin et al., 2021). We conclude that P. stewartii as a species is potentially a pathogen of a wide range of plants but that more host range testing, similar to the study published by Koirala et al. (2021), needs to be done to understand the degree to which different lineages within the species adapt to different plant species.
Pantoea bituminis sp. nov. (76A6B2C3D0E0F)
In GTDB release 207, this species consists of the genome of a single strain, EnvDTs, the nomenclatural type, which was isolated from asphalt. P. “mediterraneensis_A” is the most closely related species cluster (see below).
Pantoea “mediterraneensis_A” (76A6B2C2D0E0F)
This species cluster was effectively published as “Mixta mediterraneensis” (Boxberger et al., 2021). However, both our phylogenetic analysis and GTDB place this taxon in the genus Pantoea. The species cluster currently consists of the genome of a single strain, Marseille-Q2057, which was isolated from the skin swabs of a healthy human (Ndiaye et al., 2019; Boxberger et al., 2021). As stated above, P. “mediterraneensis_A” shares an MRCA with P. bituminis sp. nov.
Pantoea haifensis sp. nov. (76A6B2C4D0E0F)
This species currently consists of the genome of a single strain, EnvHTs, the nomenclatural type, which was isolated from asphalt in Haifa, Israel. P. haifensis sp. nov., P. “mediterraneensis_A”, and P. bituminis sp. nov. share an MRCA that splits off at a node basal to all clades described to date.
Pantoea dispersa (76A6B5C3D0E0F)
Phylogeny, genomics, and reverse ecology
The clade corresponding to P. dispersa consists of 23 genomes, including the genome of the type strain DSM 30073T (with an additional 12 genomes that were not included in the phylogenetic analysis). The clade shares an MRCA with a clade corresponding to P. eucrina, P. borealis sp. nov., P. wallisii, and P. soli sp. nov. The minimal pairwise ANI among P. dispersa genomes is 96%. Within P. dispersa, many subclades can be seen; however, many of them are unsupported and inconsistent across trees. Only one subclade, consisting of two genomes, is clearly distinct from the other subclades based on ANI since these two genomes have an ANI below 97% for all other P. dispersa genomes.
Ecology, lifestyle, and biotechnological uses
Genome-sequenced P. dispersa strains have been isolated from sources as diverse as soil, plants, insects (including fungal gardens of fungus-farming ants), a fermentation starter, and a nursing call button in a hospital intensive care unit. There are also reports of P. dispersa as a potential pathogen of humans and plants (Yang et al., 2023) but, because identification was limited to partial 16S rRNA sequencing and/or Koch's postulates were not completed, any conclusions about the pathogenicity of P. dispersa to plants, animals, and humans would be premature. Biotechnological properties of P. dispersa, again identified based on partial 16S rRNA sequencing, include the growth promotion of sugarcane (Saccharum) (Singh et al., 2020).
Pantoea eucrina (76A6B5C5D0E1F)
Phylogeny, genomics, and reverse ecology
The P. eucrina clade consists of 15 genomes, including the genome of the type strain LMG 5346T (plus one genome that was omitted). The clade shares an MRCA with P. borealis sp. nov. The maximal pairwise ANI among P. eucrina genomes is ~96%, showing that the diversity within this species has been sampled more broadly than many other Pantoea species. Within the species, it is possible to distinguish four clades, each fully supported in the core genome phylogenies (Supplementary Figures 1, 2) and also present in the pangenome tree (Supplementary Figure 3). Three of the clades have multiple members with pairwise ANI within each clade above 98%. Clade 4 is presented by a single strain, M_9. Based on reverse ecology analysis, the genomes of the first three clades belong to the same population cluster, and only the single genome of clade 4 represents a separate population. No genes were found to distinguish the clusters. This suggests that, although based on ANI and phylogeny P. eucrina appears diverse, its analyzed members share a similar ecological niche.
Ecology, lifestyle, and biotechnological uses
The P. eucrina-type strain was isolated from a human trachea. Brady et al. (2010) also reported isolations from a human cyst, human urine, and human spinal fluid. Genome-sequenced strains were isolated from a wide variety of sources, including plants, a fungal garden of a fungus-farming ant, a mosquito, precipitation, and even a ready-to-eat vegetable soup in a nursing home. Therefore, P. eucrina strains appear to survive well in many environments without having adapted to any specific environment. While the species description includes several human-associated sources, only one of the genome-sequenced strains is human-associated. Two plant sources are listed as symptomatic, but P. eucrina is not described as the causative agent. Therefore, it is premature to draw any conclusions about the risk that bacteria in this species are opportunistic human or plant pathogens.
Biotechnological properties involve a strain identified as P. eucrina based on 16S rRNA sequencing that was isolated from a marine sponge (Chondrosia reniformis), which was found to produce N-lipoamino acids, some with potential antimicrobial activity (Vitale et al., 2020); however, more robust identification of the strain is required to confirm its taxonomic affiliation.
Pantoea borealis sp. nov. (76A6B5C5D0E0F)
Four genomes of this species were included in our analysis. The genome of the MAG UBA6694Ts, the nomenclatural type, and three other MAGs were sourced from environmental samples of metallic or plastic surfaces of the New York City subway. The fourth genome is of the strain Russ, isolated from an indoor trash can. Pairwise ANI among the four genomes is over 99.25%, revealing that they belong to a single clonal lineage. The P. borealis clade shares an MRCA with the P. eucrina clade. One publication about the strain Russ discusses its potential for being an opportunistic human pathogen (Moghadam et al., 2016). Other genomes have not been described in the literature. Therefore, at this point, the species appears prevalent in anthropogenic environments, but there is no evidence for it being a pathogen.
Pantoea soli sp. nov. (76A6B5C4D0E0F)
The genome of a single strain, DSM 32899Ts, which is the nomenclatural type, belongs to the species P. soli. The type strain was isolated from the soil. It splits off at a node that is basal to the P. eucrina/P. borealis clade.
Pantoea wallisii (76A6B5C6D0E0F)
The genome of the only sequenced strain of P. wallisii, type strain LMG 26277T, is located on a branch that splits off basally to form P. soli sp. nov. The P. wallisii-type strain was isolated from diseased Eucalyptus seedlings, but there was no evidence that it caused the observed disease symptoms (Brady et al., 2012).
Pantoea symbiotica sp. nov. (76A6B5C1D0E2F)
This species includes nine genomes, one of which is the nomenclatural type YR512Ts. Another two genomes were not included in the phylogenetic analysis. The clade corresponding to this species is a sister clade of “P. endophytica”. Pairwise ANI between the eight genomes is over 97%. The strains were isolated from tree roots (Brown et al., 2012), fungi (Wong et al., 2020), and endophytes of grapevine. Based on these isolation sources, this species appears to be adapted to the rhizosphere.
“Pantoea endophytica” (76A6B5C1D0E1F)
This effectively published species, “P. endophytica” (Gao et al., 2019), consists of six genomes, including that of the type strain 596T. The genomes have a reciprocal ANI of over 98%, and one more divergent genome, B9002, is basal to the other genomes and has an ANI of ~96% to the former. The same subclade structure is found in all phylogenies (Supplementary Figures 2, 3). The six highly similar genomes belong to three distinct population clusters, and the more divergent genome is the only member of a fourth population. Comparing gene content revealed that these population clusters contain 4, 9, 255, and 545 unique genes, respectively, further suggesting that they may have adapted to different ecological niches (Supplementary Table 4). “P. endophytica” shares an MRCA with P. symbiotica sp. nov. “P. endophytica” strains have been isolated from plant roots and soil. Based on the small sample of genome-sequenced strains, this species may thus be mainly plant root- and soil-associated.
Pantoea “ludwigii_A” (76A6B5C1D0E0F)
This species cluster consists of the genome of a single strain, EnVs2, which was isolated from a grapevine (Vitis vinifera) stem (Lòpez-Fernàndez et al., 2016). In the core genome tree, the genome of EnVs2 is located on a branch that shares an MRCA with the MRCA of the clade that includes “P. endophytica” and P. symbiotica sp. nov.
“Pantoea nemavictus” (76A6B5C1D2E0F)
This effectively published species (Dirksen et al., 2020) currently consists of the genomes of three strains, including the genome of the type strain BIGb0393T, with a pairwise ANI of over 98%. The corresponding clade splits off at a node that is basal to the P. symbiotica sp. nov. and “P. endophytica” clades. Isolation sources are a roundworm (Caenorhabditis elegans) from a rotting plant stem, the internal stem tissue of poison ivy (Toxicodendron radicans), and poplar (Populus deltoides) roots. This species may thus be a plant endophyte.
Pantoea floridensis sp. nov. (76A6B5C1D1E0F)
The clade corresponding to this species consists of the genome of a single strain, JKS000234Ts, chosen as the nomenclatural type. It was isolated from a fungus-farming ant garden in Florida, USA. One additional, very similar genome from the same source was omitted from the tree. This species shares an MRCA with “P. nemavictus”.
Pantoea communis sp. nov. (76A6B5C0D1E0F)
This species consists of seven genome-sequenced strains, including the nomenclatural type Al-1710Ts. The minimal pairwise ANI among the seven genomes is 97%. The species shares an MRCA with the genome of P. “rwandensis_B”. Isolation sources include insects, an ant fungus garden, plants, and other environmental sources. Therefore, this species shares plants and insects as isolation sources with many other Pantoea species, and no adaptation to any specific environment is evident.
Note: There is an eighth genome in the same LINgroup as P. communis sp. nov., M004, which is the GTDB representative of the corresponding species cluster “Pantoea cancerogena_A” and is identified as Enterobacter cancerogenus in NCBI, isolated from a waterfall. This genome is located on a branch that splits off at a basal node to the P. “rwandensis_B” clade in the core genome tree (in both the IQ-Tree and FastTree), although it has an ANI of over 95% with the seven P. communis sp. nov. genomes and clusters with the other seven P. communis sp. nov. genomes in the same clade with 100% branch support in the pangenome tree (Supplementary Figure 3). This is a rare case where ANI and core genome phylogeny do not correlate with each other. Therefore, although this genome shares a high ANI with members of P. communis sp. nov., the assignment of this genome to any species remains uncertain as phylogeny and genome metrics are not in agreement.
Pantoea “rwandensis_B” (76A6B5C0D1E2F)
Five genome-sequenced strains belong to this species cluster, including the GTDB representative ND04. Pairwise ANI among the five genomes is 98% or higher. As described above, the clade corresponding to this species shares an MRCA with seven of the Pantoea communis sp. nov. genomes. Isolation sources of P. “rwandensis_B” include the coffee berry borer (Hypothenemus hampei), a river, a natural lake, and a waterfall. Therefore, this species may be more adapted to aquatic environments compared to other Pantoea species.
Pantoea sp016625195 (76A6B5C0D1E1F)
Only the genome of a single strain, GTDB representative S63, belongs to this species cluster. Strain S63 was isolated from the coffee borer beetle (Hypothenemus hampei). The genome is located on a branch that splits off at a node that is basal to the P. “rwandensis_B”/P. communis sp. nov. clade.
Pantoea rodasii (76A6B5C0D3E0F)
The P. rodasii clade is represented by the single genome sequence of the type strain LMG 26273T and is located on the branch splitting off at the most basal node of the P. “rwandensis_B”/P. comunis sp. nov./P. sp016625195 clade. The type strain was isolated from Eucalyptus leaves with bacterial blight and dieback symptoms. However, based on the data provided in the species description (Brady et al., 2012), it is not clear if it was the causative agent of the observed symptoms.
Pantoea “vagans_C” (76A6B5C0D0E0F)
This species cluster consists of three genome-sequenced strains: ND02 (the GTDB representative) and ND03, both isolated from a waterfall located in a tropical rainforest, and Ps-ISGKm56, obtained from the midgut of a stink bug. These genomes share an ANI of over 97%. Note that genome ND03 is designated in NCBI as P. rodasii. In the core genome tree, the clade corresponding to this group is a sister to the P. rwandensis/P. formicae sp. nov. clade (see below).
Pantoea formicae sp. nov. (76A6B5C0D2E0F)
This species is represented by the genome of a single strain, Acro-805Ts, the nomenclatural type, isolated from the fungal garden of an ant. It shares an MRCA with the type strain of P. rwandensis (see below).
Pantoea rwandensis (76A6B5C0D2E1F)
The genome of the type strain, LMG 26275T, is the only available genome sequence for this species. It shares an MRCA with the P. formicae strain Acro-805. Bacteria in the P. rwandensis species, including the type strain, were isolated from Eucalyptus seedlings with bacterial blight or dieback symptoms (Brady et al., 2012), but based on the species description, it is not clear if they represent the causative agent.
Pantoea cypripedii (76A6B5C7D0E1F)
The P. cypripedii clade consists of the type strain LMG 2657T and one other strain of unknown origin, which has an ANI of over 99.9% compared to the type strain. The two genomes, which are both in the tree, also belong to the same population cluster. P. cypripedii shares an MRCA with the species cluster P. “cypripedii_A”. The P. cypripedii-type strain was isolated from a Lady Slipper Orchid (Cypripedium) (Brady et al., 2010), but there is scarce knowledge about the ecology and life cycle of this species.
Pantoea “cypripedii_A” (76A6B5C7D0E0F)
This species consists of a single genome-sequenced strain, NE1 (the GTDB representative), isolated as an endophyte from an Egyptian Riverhemp (Sesbania spp.) root nodule and found to be plant growth promoting based on the provided metadata.
Pantoea sp000468095 (76A6B5C7D1E1F)
This species cluster consists of the genomes of two strains, AS-PWVM4 (the GTDB representative) and MSR2. ANI between the two genomes is above 98.5%. The corresponding clade shares an MRCA with the GTDB species cluster P. sp000175935. Strain AS-PWVM4 was isolated from a plant rhizosphere sample and found to have biofertilizer and biocontrol activities (Khatri et al., 2013), while MSR2 was obtained from the roots of a legume plant. This species cluster may thus be a root endophyte based on the two available genomes.
Pantoea sp000175935 (76A6B5C7D1E0F)
This species consists of the genome of a single strain, At-9b, the GTDB representative. Based on the genome's BioSample data, it was isolated from a corn (Zea mays) plant with Stewart's Wilt symptoms.
Pantoea multigeneris sp. nov. (76A6B34C0D0E0F)
This species consists of the genomes of two strains, A4 and the nomenclatural type Acro-835Ts, which have a pairwise ANI of ~96%. Strain A4 was isolated from a decaying flower bud, while strain Acro-835 was isolated from an ant fungus garden. This species is located on the branch splitting off at the most basal node of the large clade that includes the species from P. dispersa to P. sp000175935 described above.
Pantoea septica (76A6B2C1D1E1F)
Phylogeny, genomics, and reverse ecology
The 13 genomes (six of them MAGs) in this species (12 of them were included in the phylogenetic analysis) have pairwise genome similarity as low as 95% ANI, revealing a genomically diverse species. The corresponding clade consists of four subclades, each consisting of two to five genomes. Each clade is supported in all trees and corresponds to a separate population cluster, suggesting that strains of P. septica may possibly occupy at least four different ecological niches. Gene content analysis found 15, 50, 35, and 47 genes, respectively, to be unique to each subclade, further supporting that these are ecologically relevant populations. Genes unique to the four clades include genes coding for transporters, regulators, various enzymes, type VI secretion systems, flagella, chemotaxis, and hypothetical proteins (Supplementary Table 4).
Ecology, lifestyle, and biotechnological uses
The type strain of P. septica, LMG 5345T, was isolated from a human stool sample. Other strains were isolated from the human blood, skin, and gut, including one isolated from a preterm neonatal blood sepsis patient and one isolated from a human host sourced by an intensive care unit. The six MAGs were all assembled from metagenomes derived from metal and plastic surfaces in New York City. Therefore, P. septica clearly appears to be a human-associated species, but insufficient evidence exists to support that it is a pathogen.
Pantoea alvi sp. nov. (76A6B2C1D1E0F)
This species comprises two genomes, the nomenclatural type PSNIH6Ts and UMGS54, with a pairwise ANI of over 99.75%. The corresponding clade is a sister clade of P. septica. Strain PSNIH6 was isolated from hospital plumbing, while MAG UMGS54 was assembled from the metagenome of a human gut sample. Therefore, P. alvi sp. nov. may occupy a similar ecological niche to its sister species, P. septica.
Pantoea piersonii (76A6B2C1D0E0F)
Phylogeny, genomics, and reverse ecology
The nine genomes, including that of the type strain DSM 108198T, in this species (including three genomes not used in the phylogenetic analysis), have over 96% ANI to each other. The corresponding branch splits off at a node that is basal to the MRCA of P. septica and P. alvi sp. nov. The species contains two population clusters corresponding to separate clades that are supported in all trees (Supplementary Figures 1–3), one with five members and one with one member. The five genomes of the first cluster share 172 genes that are absent in the genome of the other cluster, while this latter genome has 116 genes absent from the other five genomes, adding further evidence that they represent two distinct populations. Genes unique to either of the two clades include genes encoding phage components, the toxin–antitoxin systems, transporters, multidrug resistance, various enzymes, regulators, antibiotic biosynthesis, and hypothetical proteins (Supplementary Table 4).
Ecology, lifestyle, and biotechnological uses
The type strain and some other strains of the species were isolated from environmental samples collected inside the International Space Station, while one strain was isolated from human urine, and three MAGs were sourced from surfaces in New York City. Therefore, bacteria in this species may have an ecological niche associated with humans and may be commonly found in anthropogenic environments without causing disease, similar to bacteria in the related P. septica and P. alvi species.
“Pantoea latae” (76A6B2C1D3E0F)
This effectively published species (Lata et al., 2017) only has one member, the genome of the type strain AS1T, which is located on the branch splitting off at a basal node for P. piersonii. Strain AS1T was isolated from the rhizosphere of a Florida Coontie (Zamia floridana) plant in Florida, USA (Lata et al., 2017).
Pantoea deserta sp. nov. (76A6B2C1D2E0F)
This species includes the genome of a single strain, the nomenclatural type RIT388Ts, isolated from an Ayan tree (Distemonanthus benthamianus), known for its antifungal and antibacterial properties (Soutar and Stavrinides, 2019; Gan et al., 2020). The genome is located on the branch splitting off at the most basal node of the clade, which includes P. septica, P. alvi, P. piersonii, and P. latae.
Pantoea “mediterraneensis” (76A6B24C0D0E0F)
This species consists of the genome of a single strain, the type strain Marseille-P5165T, isolated from human skin swabs (Ndiaye et al., 2019; Boxberger et al., 2021). Based on GTDB, previous phylogenomic analyses of the Erwiniaceae family (Soutar and Stavrinides, 2019, 2022), and our core genome tree, this species, whose basonym is “Erwinia mediterraneensis”, belongs to the genus Pantoea. In fact, the genome of Marseille-P5165 shares an MRCA with MAG UBA648TS of P. superficialis sp. nov on a branch splitting off at a basal node to all other Pantoea genomes but does not cluster with the outgroup Mixta calida. We note that, because of their position in the tree, we cannot exclude that P. “mediterraneensis” and P. superficialis sp. nov. are members of a related genus instead. Additional phylogenetic reconstruction with the genomes of additional genera will need to be performed to confirm their genus affiliation.
Pantoea superficialis sp. nov. (76A6B24C1D0E0F)
This species consists of two MAGs, UBA2655 and the nomenclatural type UBA648Ts, both assembled from a metagenome sourced from a metallic or plastic surface in New York City. Only the MAG UBA648Ts was used in the phylogenetic analysis. It shares an MRCA with P. “mediterraneensis”.
Conclusion
By combining phylogenetic core genome reconstruction, genome similarity analysis, a reverse ecology approach, selected gene content comparisons based on a pangenome analysis, and a review of the provenances of samples and the available literature, we have here compiled the description of 49 Pantoea species and 25 within-species lineages. After this thorough analysis of 559 genome-sequenced strains, Pantoea remains true to its name by being “of all sorts and sources”, with most species and within-species lineages showing little to no obvious specialization to a particular lifestyle or environment.
Only four species include bona-fide plant pathogens based on available data at this point (P. ananatis, P. stewartii, P. allii, and P. agglomerans). Since three of these species (P. ananatis, P. stewartii, and P. allii) belong to the same larger clade (LINgroup 76A6B13C), the MRCA of this clade may have made the switch to a pathogenic lifestyle. Therefore, any strain that belongs to this clade, even if not a member of the three currently described species, may be more likely to be a plant pathogen than other members of the genus. However, different virulence mechanisms and sporadic plant pathogenicity among some of these species would support caution. Genes specific to this clade were previously identified in a comparative genomics study (Palmer et al., 2018b), where pathogenicity-associated genes included an additional copy of the rfb locus for the synthesis of lipopolysaccharides of the O-antigen and a locus encoding aerobactin that may aid in survival in hosts during iron-limiting conditions, among numerous other poorly characterized genes. Although some strains of P. agglomerans have been validated as hrp1 T3SS-dependent gall-forming plant pathogens or pathogens of onion, plant pathogenicity appears to be largely sporadic within the group. Furthermore, other Pantoea species include members that were isolated from symptomatic plants; however, Koch's postulates have not been completed, and the sporadic nature of their isolation makes it likely that they live in association with diseased plants but are not the causative agents, such as P. eucalypti, P. vagans, P. eucrina, or P. gossypiicola.
Three Pantoea species, P. septica, P. piersonii, and P. alvi, appear instead to be mostly human-associated. These three species are also members of a single clade (corresponding to LINgroups 76A6B2C1D0E and 76A6B2C1D1E). Therefore, other strains that belong to this clade may be more likely to have a human/animal-associated lifestyle. There is currently no indication that they are opportunistic human pathogens. Another clade with above-average human-associated isolation sources is the P. brenneri, P. conspicua, P. astica sp. nov., and P. sp009830035 clade (76A6B2C0D1/2/3E), whose MRCA may thus also have adapted to a more human/animal-associated niche. However, for this clade, there is no indication of a pathogenic lifestyle. With future increased genomic representation of these groups, further comparative genomics analyses may shed light on the adaptations associated with shifts in lifestyles.
All other species with more than one or two members appear to be associated with multiple sources, mainly plants, soil, insects, fungi, air, and water. Furthermore, the distinct within-species lineages, whose circumscription is supported by phylogenomics, recent HGT events, and differences in gene content, still seem to be generalists. Therefore, there appears to be little adaptation and specialization to different ecological niches in most species of the genus, even when a within-species lineage becomes evolutionarily isolated. Bacteria may instead be frequently migrating between environments, for example, from plants to the water cycle and/or soil and back to plants, as suggested by the finding that rain-borne Pantoea bacteria are efficient colonizers of plant leaves (Mechan Llontop et al., 2021).
P. agglomerans deserves particular attention since it is considered to be a biosecurity risk based on reports indicating a potential for causing human infections (Bonaterra et al., 2014). However, only three out of over 120 genome-sequenced members of P. agglomerans were isolated from human tissues or wounds, and it is unclear if they were causative agents of infection. Therefore, based on the data analyzed here, we are not able to further support or reject an elevated biosecurity risk for P. agglomerans overall or for any of its three within-species lineages.
In general, a lack in the clear correlation of isolation sources with taxonomic assignment and a lack of evidence for pathogenicity based on currently available data of genome-sequenced isolates indicate lower pathogenic potential for several species in the genus compared to what one would expect based on literature. For example, several recent reviews report the pathogenicity of some Pantoea species to plants and/or humans (Bartlett et al., 2022; Yang et al., 2023). However, when one looks at the evidence, in several cases, either Koch's postulates were not completed and/or bacteria were only identified based on partial 16S rRNA sequencing, which is insufficient for the identification at the species rank, particularly in the Enterobacteriaceae.
Another problem in assessing the biosecurity risk of Pantoea species is the misidentification or mislabeling of genes and genomes in NCBI databases. GTDB (Parks et al., 2022) has significantly reduced this confusion by assigning genomes to a solid genome-based taxonomy. However, GTDB does not provide descriptions of species, and many species clusters only have provisional names. Furthermore, being a taxonomy database, phylogenetic information between the genus rank and species rank and within species is not provided. By reconstructing phylogenetic relationships from the rank of genus to the level of genetic lineages within species, naming new species using the SeqCode (Hedlund et al., 2022), and delineating and describing these lineages, we have further improved the taxonomy of the genus Pantoea. Note that naming under SeqCode was not possible for some species clusters because raw sequencing data were not available for any of the member genomes, and, therefore, none of the genomes could be used as a nomenclatural type for those species.
Importantly, any newly sequenced genomes in the Pantoea genus that will be added to LINbase in future will be automatically classified based on ANI, including the genomes that have been added to GTDB as part of release 214 after the analysis for this study was completed. Even if these genomes do not belong to any of the species and lineages delineated here or any newly named species, it would still be possible to identify them since the LINs assigned to these new genomes will precisely express their similarity to all previously sequenced Pantoea genomes. The caveat is that, since we did not observe the specialization of lineages to specific lifestyles, most importantly, pathogenicity to plants or animals, such precise identification, may still not allow the prediction of any relevant phenotypes.
In conclusion, our analysis establishes a solid genome-based framework for the precise identification of Pantoea isolates. As a next step, already available isolates will need to be thoroughly phenotyped with regard to their pathogenicity to plants and animals to determine the biosecurity risk they present; genome comparisons need to be extended to the analysis of confirmed or putative virulence genes; and sampling needs to continue to further our knowledge of the phenotypic and genotypic diversity that exists within the genus (note that since the writing of this study and completion of genome analyses, new genomes, and species clusters have already been added to GTDB Release 214, and we will update LINbase with these genomes and species clusters). Making progress in regard to all three of these aspects will lead us toward a more meaningful classification and identification of the genus, which will allow for better protection of human, animal, and plant health from pathogenic members, while expanding the deployment of beneficial members in biotechnology and biocontrol.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA445714.
Author contributions
KC: Data curation, Investigation, Writing—original draft, Writing—review & editing. MR: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing—review & editing. PS: Investigation, Methodology, Supervision, Writing—review & editing. MJ: Investigation, Methodology, Supervision, Writing—review & editing. RM: Data curation, Investigation, Software, Writing—review & editing. BK: Validation, Writing—original draft, Writing—review & editing. TS: Validation, Writing—original draft, Writing—review & editing. SV: Writing—original draft, Writing—review & editing. TC: Writing—original draft, Writing—review & editing. LSH: Conceptualization, Writing—review & editing. MP: Data curation, Validation, Visualization, Writing—original draft, Writing—review & editing. BAV: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project Writing—review & editing.
Funding
This research was funded by the National Science Foundation (DBI-2018522 and IOS-1754721) and the United States Department of Agriculture—National Institute of Food and Agriculture (2021-67021-34343). Funding for BAV was also provided in part by the Virginia Agricultural Experiment Station and the Hatch Program of USDA NIFA. MR was partly funded by the National Institute of Health (GM072767-14).
Conflict of interest
Life Identification Number and LIN are registered trademarks of this Genomic Life Inc. LSH and BAV report in accordance with Virginia Tech policies and procedures and their ethical obligation as researchers, that they have a financial interest in this Genomic Life Inc. that may be affected by the publication of this manuscript. They have disclosed those interests fully to Virginia Tech, and they have in place an approved plan for managing any potential conflicts arising from this relationship.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1254999/full#supplementary-material
Supplementary Figure 1. Core genome phylogenetic tree prepared with IQ-TREE. Phylogeny shown in Figure 1 with strain names included together with population cluster assignment (in brackets next to strain names). In addition to clades corresponding to species, subclades within species are also annotated. Branch support is inferred with ultrafast bootstrapping from 1,000 replicates. The same color scheme to Figure 1 is used to denote species. Type strains and type sequences are followed by a “T” or “Ts”, respectively.
Supplementary Figure 2. Core genome phylogenetic tree prepared with FastTree. Approximate ML phylogenetic tree of the genus Pantoea of the same genomes used in Supplementary Figure 1. Shimodaira-Hasegawa-like branch support values from 1,000 replicates are indicated as colored dots at nodes.
Supplementary Figure 3. Pangenome phylogenetic tree made with IQ-Tree. ML phylogenetic pangenome tree of Pantoea constructed with IQ-TREE. Branch support was inferred with ultrafast bootstrapping from 1,000 replicates and are indicated as colored dots at the nodes.
Supplementary Table 1. Metadata file. Information on the strains and genomes within the genus Pantoea, including phylogenetic tree assignment, Life Identification Number (LIN) assignment, species names, National Center for Biotechnology Information (NCBI) metadata, including taxonomic lineage information, Genome Taxonomy Database (GTDB) metadata, including taxonomic lineage information, CheckM results, and genomic information (Parks et al., 2015; Hugenholtz et al., 2021).
Supplementary Table 2. Distribution of isolation sources for each Pantoea species. Information on the species within the genus Pantoea, including genomic information from the National Center of Biotechnology (NCBI) and general information from the List of Prokaryotic Standing in Nomenclature (LPSN) and Genome Taxonomy Database (GTDB) (Parte et al., 2020; Hugenholtz et al., 2021).
Supplementary Table 3. Species and lineages circumscribed as LINgroups in LINbase. This table includes LINs of Pantoea species, lineages above species rank, and within-species lineages.
Supplementary Table 4. Gene differences between selected within-species subclades.
References
Abidin, N., Ismail, S. I., Vadamalai, G., Yusof, M. T., Hakiman, M., Karam, D. S., et al. (2021). Pathogenic variability of the jackfruit-bronzing bacterium Pantoea stewartii subspecies stewartii infection to jackfruit varieties and its pivotal plant hosts in Malaysia. Agronomy 11, 2113. doi: 10.3390/agronomy11112113
Agarwal, G., Choudhary, D., Stice, S. P., Myers, B. K., Gitaitis, R. D., Venter, S. N., et al. (2021a). Pan-genome-wide analysis of Pantoea ananatis identified genes linked to pathogenicity in onion. Front. Microbiol. 12, 684756. doi: 10.3389/fmicb.2021.684756
Agarwal, G., Gitaitis, R. D., and Dutta, B. (2021b). Pan-genome of novel Pantoea stewartii subsp. indologenes reveals genes involved in onion pathogenicity and evidence of lateral gene transfer. Microorganisms 9, 1761. doi: 10.20944/preprints202107.0400.v1
Arevalo, P., VanInsberghe, D., Elsherbini, J., Gore, J., and Polz, M. F. (2019). A reverse ecology approach based on a biological definition of microbial populations. Cell 178, 820–834.e14. doi: 10.1016/j.cell.2019.06.033
Asselin, J. A. E., Bonasera, J. M., and Beer, S. V. (2018). Center rot of onion (Allium cepa) caused by Pantoea ananatis Requires pepM, a predicted phosphonate-related gene. Mol. Plant Microbe Interact. 31, 1291–1300. doi: 10.1094/MPMI-04-18-0077-R
Azizi, M. M. F., Ismail, S. I., Hata, E. M., Zulperi, D., Ina-Salwany, M. Y., and Abdullah, M. A. F. (2019). First report of Pantoea stewartii subsp. indologenes causing leaf blight on rice in Malaysia. Plant Dis. 103, 1407–1407. doi: 10.1094/PDIS-08-18-1403-PDN
Baron, E. J. (1996). Classification. University of Texas Medical Branch at Galveston. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK8406/ (accessed February 25, 2023).
Bartlett, A., Padfield, D., Lear, L., Bendall, R., and Vos, M. (2022). A comprehensive list of bacterial pathogens infecting humans. Microbiology 168, 001269. doi: 10.1099/mic.0.001269
Bayliss, S. C., Thorpe, H. A., Coyle, N. M., Sheppard, S. K., and Feil, E. J. (2019). PIRATE: a fast and scalable pangenomics toolbox for clustering diverged orthologues in bacteria. Gigascience 8, giz119. doi: 10.1093/gigascience/giz119
Bayot, M. L., and King, K. C. (2022). Biohazard Levels. StatPearls Publishing. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK535351/ (accessed April 19, 2023).
Bonaterra, A., Badosa, E., Rezzonico, F., Duffy, B., and Montesinos, E. (2014). Phenotypic comparison of clinical and plant-beneficial strains of Pantoea agglomerans. Int. Microbiol. 17, 81–90. doi: 10.2436/20.1501.01.210
Boxberger, M., Antezack, A., Magnien, S., Cassir, N., and La Scola, B. (2021). Draft genome and description of Mixta mediterraneensis strain Marseille-Q2057T sp.nov., a new bacterium isolated from human healthy skin. New Microbes New Infect. 40, 100840. doi: 10.1016/j.nmni.2021.100840
Brady, C., Cleenwerck, I., Venter, S., Vancanneyt, M., Swings, J., and Coutinho, T. (2008). Phylogeny and identification of Pantoea species associated with plants, humans and the natural environment based on multilocus sequence analysis (MLSA). Syst. Appl. Microbiol. 31, 447–460. doi: 10.1016/j.syapm.2008.09.004
Brady, C., Venter, S., Cleenwerck, I., Vancanneyt, M., Swings, J., and Coutinho, T. (2007). A FAFLP system for the improved identification of plant-pathogenic and plant-associated species of the genus Pantoea. Syst. Appl. Microbiol. 30, 413–417. doi: 10.1016/j.syapm.2007.01.005
Brady, C. L., Cleenwerck, I., van der Westhuizen, L., Venter, S. N., Coutinho, T. A., and De Vos, P. (2012). Pantoea rodasii sp. nov., Pantoea rwandensis sp. nov. and Pantoea wallisii sp. nov., isolated from eucalyptus. Int. J. Syst. Evol. Microbiol. 62, 1457–1464. doi: 10.1099/ijs.0.032615-0
Brady, C. L., Cleenwerck, I., Venter, S. N., Engelbeen, K., De Vos, P., and Coutinho, T. A. (2010). Emended description of the genus Pantoea, description of four species from human clinical samples, Pantoea septica sp. nov., Pantoea eucrina sp. nov., Pantoea brenneri sp. nov. and Pantoea conspicua sp. nov., and transfer of Pectobacterium cypripedii (Hori 1911) Brenner et al. 1973 emend. Hauben et al. 1998 to the genus as Pantoea cypripedii comb. nov. Int. J. Syst. Evol. Microbiol. 60, 2430–2440. doi: 10.1099/ijs.0.017301-0
Brady, C. L., Goszczynska, T., Venter, S. N., Cleenwerck, I., De Vos, P., Gitaitis, R. D., et al. (2011). Pantoea allii sp. nov., isolated from onion plants and seed. Int. J. Syst. Evol. Microbiol. 61, 932–937. doi: 10.1099/ijs.0.022921-0
Brady, C. L., Venter, S. N., Cleenwerck, I., Engelbeen, K., Vancanneyt, M., Swings, J., et al. (2009). Pantoea vagans sp. nov., Pantoea eucalypti sp. nov., Pantoea deleyi sp. nov. and Pantoea anthophila sp. nov. Int. J. Syst. Evol. Microbiol. 59, 2339–2345. doi: 10.1099/ijs.0.009241-0
Brown, S. D., Utturkar, S. M., Klingeman, D. M., Johnson, C. M., Martin, S. L., Land, M. L., et al. (2012). Twenty-one genome sequences from Pseudomonas species and 19 genome sequences from diverse bacteria isolated from the rhizosphere and endosphere of Populus deltoides. J. Bacteriol. 194, 5991–5993. doi: 10.1128/JB.01243-12
Bujdáková, H., Hanzen, J., Jankovicová, S., Klimácková, J., Moravcíková, M., Milosovic, P., et al. (2001). Occurrence and transferability of beta-lactam resistance in Enterobacteriaceae isolated in Children's University Hospital in Bratislava. Folia Microbiol. 46, 339–344. doi: 10.1007/BF02815624
Cai, R., Yan, S., Liu, H., Leman, S., and Vinatzer, B. A. (2011). Reconstructing host range evolution of bacterial plant pathogens using Pseudomonas syringae pv. tomato and its close relatives as a model. Infect. Genet. Evol. 11, 1738–1751. doi: 10.1016/j.meegid.2011.07.012
Chandler, D., Bailey, A. S., Tatchell, G. M., Davidson, G., Greaves, J., and Grant, W. P. (2011). The development, regulation and use of biopesticides for integrated pest management. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 366, 1987–1998. doi: 10.1098/rstb.2010.0390
Correa, V. R., Majerczak, D. R., Ammar, E.-D., Merighi, M., Pratt, R. C., Hogenhout, S. A., et al. (2012). The bacterium Pantoea stewartii uses two different type III secretion systems to colonize its plant host and insect vector. Appl. Environ. Microbiol. 78, 6327–6336. doi: 10.1128/AEM.00892-12
Coutinho, T. A., and Venter, S. N. (2009). Pantoea ananatis: an unconventional plant pathogen. Mol. Plant Pathol. 10, 325–335. doi: 10.1111/j.1364-3703.2009.00542.x
De Baere, T., Verhelst, R., Labit, C., Verschraegen, G., Wauters, G., Claeys, G., et al. (2004). Bacteremic infection with Pantoea ananatis. J. Clin. Microbiol. 42, 4393–4395. doi: 10.1128/JCM.42.9.4393-4395.2004
Deivanai, S., Bindusara, A. S., Prabhakaran, G., and Bhore, S. J. (2014). Culturable bacterial endophytes isolated from mangrove tree (Rhizophora apiculata Blume) enhance seedling growth in Rice. J. Nat. Sci. Biol. Med. 5, 437–444. doi: 10.4103/0976-9668.136233
Dirksen, P., Assié, A., Zimmermann, J., Zhang, F., Tietje, A.-M., Marsh, S. A., et al. (2020). CeMbio - the Caenorhabditis elegans microbiome resource. G3 10, 3025–3039. doi: 10.1534/g3.120.401309
Edens, D. G., Gitaitis, R. D., Sanders, F. H., and Nischwitz, C. (2006). First report of Pantoea agglomerans causing a leaf blight and bulb rot of onions in Georgia. Plant Dis. 90, 1551. doi: 10.1094/PD-90-1551A
Failor, K. C., Schmale, D. G. III., Vinatzer, B. A., and Monteil, C. L. (2017). Ice nucleation active bacteria in precipitation are genetically diverse and nucleate ice by employing different mechanisms. ISME J. 11, 2740–2753. doi: 10.1038/ismej.2017.124
Gan, H. M., Parthasarathy, A., Henry, K. R., Savka, M. A., Thomas, B. N., and Hudson, A. O. (2020). Whole-genome sequencing of Pantoea sp. strain RIT388, a potential oral opportunistic pathogen isolated from a chewing stick (Distemonanthus benthamianus). Microbiol. Resour. Announc. 9, e01468-19. doi: 10.1128/MRA.01468-19
Gao, J. L., Xue, J., Yan, H., Tong, S., Sayyar Khan, M., Wang, L. W., et al. (2019). Pantoea endophytica sp. nov., novel endophytic bacteria isolated from maize planting in different geographic regions of northern China. Syst. Appl. Microbiol. 42, 488–494. doi: 10.1016/j.syapm.2019.06.001
Gavini, F., Mergaert, J., Beji, A., Mielcarek, C., Izard, D., Kersters, K., et al. (1989). Transfer of Enterobacter agglomerans (Beijerinck 1888) Ewing and Fife 1972 to Pantoea gen. nov. as Pantoea agglomerans comb. nov. and description of Pantoea dispersa sp. nov. Int. J. Syst. Evol. Microbiol. 39, 337–345. doi: 10.1099/00207713-39-3-337
Hara, Y., Kadotani, N., Izui, H., Katashkina, J. I., Kuvaeva, T. M., Andreeva, I. G., et al. (2012). The complete genome sequence of Pantoea ananatis AJ13355, an organism with great biotechnological potential. Appl. Microbiol. Biotechnol. 93,331–341. doi: 10.1007/s00253-011-3713-5
Hauben, L., Vauterin, L., Swings, J., and Moore, E. R. (1997). Comparison of 16S ribosomal DNA sequences of all Xanthomonas species. Int. J. Syst. Bacteriol. 47, 328–335. doi: 10.1099/00207713-47-2-328
Hedlund, B. P., Chuvochina, M., Hugenholtz, P., Konstantinidis, K. T., Murray, A. E., Palmer, M., et al. (2022). SeqCode: a nomenclatural code for prokaryotes described from sequence data. Nat. Microbiol. 7, 1702–1708. doi: 10.1038/s41564-022-01214-9
Hugenholtz, P., Chuvochina, M., Oren, A., Parks, D. H., and Soo, R. M. (2021). Prokaryotic taxonomy and nomenclature in the age of big sequence data. ISME J. 15, 1879–1892. doi: 10.1038/s41396-021-00941-x
Ishimaru, C. A., Klos, E. J., and Brubaker, R. R. (1988). Multiple antibiotic production by Erwinia herbicola. Phytopathology 78, 746. doi: 10.1094/Phyto-78-746
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5514. doi: 10.1038/s41467-018-07641-9
Kageyama, B., Nakae, M., Yagi, S., and Sonoyama, T. (1992). Pantoea punctata sp. nov., Pantoea citrea sp. nov., and Pantoea terrea sp. nov. isolated from fruit and soil samples. Int. J. Syst. Bacteriol. 42, 203–210. doi: 10.1099/00207713-42-2-203
Kamber, T., Lansdell, T. A., Stockwell, V. O., Ishimaru, C. A., Smits, T. H. M., and Duffy, B. (2012a). Characterization of the biosynthetic operon for the antibacterial peptide herbicolin in Pantoea vagans biocontrol strain C9-1 and incidence in Pantoea species. Appl. Environ. Microbiol. 78, 4412–4419. doi: 10.1128/AEM.07351-11
Kamber, T., Smits, T. H. M., Rezzonico, F., and Duffy, B. (2012b). Genomics and current genetic understanding of Erwinia amylovora and the fire blight antagonist Pantoea vagans. Trees 26, 227–238. doi: 10.1007/s00468-011-0619-x
Khatri, I., Kaur, S., Devi, U., Kumar, N., Sharma, D., Subramanian, S., et al. (2013). Draft genome sequence of plant growth-promoting rhizobacterium Pantoea sp. Strain AS-PWVM4. Genome Announc. 1, e00947-13. doi: 10.1128/genomeA.00947-13
Kim, I.-Y., Pusey, P. L., Zhao, Y., Korban, S. S., Choi, H., and Kim, K. K. (2012). Controlled release of Pantoea agglomerans E325 for biocontrol of fire blight disease of apple. J. Control. Release 161, 109–115. doi: 10.1016/j.jconrel.2012.03.028
Koester, T., Kusano, T., Eijer, H., Escher, R., and Waldegg, G. (2020). Septic arthritis of the knee due to Pantoea agglomerans: look for the thorn. J. Bone Jt. Infect. 6, 51–55. doi: 10.5194/jbji-6-51-2020
Koirala, S., Zhao, M., Agarwal, G., Gitaitis, R., Stice, S., Kvitko, B., et al. (2021). Identification of two Novel Pathovars of Pantoea stewartii subsp. indologenes affecting Allium sp. and Millets. Phytopathology 111, 1509–1519. doi: 10.1094/PHYTO-11-20-0508-R
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Lata, P., Govindarajan, S. S., Qi, F., Li, J.-L., Maurya, S. K., and Sahoo, M. K. (2017). De novo whole-genome sequence of Pantoea latae strain AS1, isolated from Zamia floridana rhizosphere in central Florida, USA. Genome Announc. 5, e00640-17. doi: 10.1128/genomeA.00640-17
Lindow, S. E., Arny, D. C., and Upper, C. D. (1978). Erwinia herbicola: a bacterial ice nucleus active in increasing frost injury to corn. Phytopathology 68, 523. doi: 10.1094/Phyto-68-523
Lòpez-Fernàndez, S., Compant, S., Vrhovsek, U., Bianchedi, P. L., Sessitsch, A., Pertot, I., et al. (2016). Grapevine colonization by endophytic bacteria shifts secondary metabolism and suggests activation of defense pathways. Plant Soil 405, 155–175. doi: 10.1007/s11104-015-2631-1
Lorenzi, A. S., Bonatelli, M. L., Chia, M. A., Peressim, L., and Quecine, M. C. (2022). Opposite sides of Pantoea agglomerans and its associated commercial outlook. Microorganisms 10, 2072. doi: 10.3390/microorganisms10102072
Ma, J., Zhang, K., Huang, M., Hector, S. B., Liu, B., Tong, C., et al. (2016). Involvement of Fenton chemistry in rice straw degradation by the lignocellulolytic bacterium Pantoea ananatis Sd-1. Biotechnol. Biofuels 9, 211. doi: 10.1186/s13068-016-0623-x
Ma, Y., Yin, Y., Rong, C., Chen, S., Liu, Y., Wang, S., et al. (2016). Pantoea pleuroti sp. nov., isolated from the fruiting bodies of Pleurotus eryngii. Curr. Microbiol. 72, 207–212. doi: 10.1007/s00284-015-0940-5
Marakeby, H., Badr, E., Torkey, H., Song, Y., Leman, S., Monteil, C. L., et al. (2014). A system to automatically classify and name any individual genome-sequenced organism independently of current biological classification and nomenclature. PLoS ONE 9, e89142. doi: 10.1371/journal.pone.0089142
Mechan Llontop, M. E., Hurley, K., Tian, L., Bernal Galeano, V. A., Wildschutte, H. K., Marine, S. C., et al. (2020). Exploring rain as source of biological control agents for fire blight on apple. Front. Microbiol. 11, 199. doi: 10.3389/fmicb.2020.00199
Mechan Llontop, M. E., Tian, L., Sharma, P., Heflin, L., Bernal-Galeano, V., Haak, D. C., et al. (2021). Experimental evidence pointing to rain as a reservoir of tomato phyllosphere microbiota. Phytobiomes J. 5, 382–399. doi: 10.1094/PBIOMES-04-21-0025-R
Mergaert, J., Verdonck, L., and Kersters, K. (1993). Transfer of Erwinia ananas (synonym, Erwinia uredovora) and Erwinia stewartii to the Genus Pantoea emend. as Pantoea ananas (Serrano 1928) comb. nov. and Pantoea stewartii (Smith 1898) comb. nov., respectively, and description of Pantoea stewartii subsp. indologenes subsp. nov. Int. J. Syst. Bacteriol. 43, 162–173. doi: 10.1099/00207713-43-1-162
Milanowski, J., Sorenson, W. G., Lewis, D. M., and Dutkiewicz, J. (1995). Chemotaxis of alveolar macrophages and neutrophils in response to microbial products derived from organic dust. J. Investig. Allergol. Clin. Immunol. 5, 221–227.
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Moghadam, F., Couger, M. B., Russ, B., Ramsey, R., Hanafy, R. A., Budd, C., et al. (2016). Draft genome sequence and detailed analysis of Pantoea eucrina strain Russ and implication for opportunistic pathogenesis. Genom Data 10, 63–68. doi: 10.1016/j.gdata.2016.09.006
Moloto, V. M., Goszczynska, T., Hassen, A. I., Pierneef, R., and Coutinho, T. (2020). Draft genome sequences of Pantoea agglomerans strains BD1274 and BD1212, isolated from onion seeds, reveal major differences in pathogenicity and functional genes. Microbiol. Resour. Announc. 9, e01507-19. doi: 10.1128/MRA.01507-19
Moretti, C., Rezzonico, F., Orfei, B., Cortese, C., Moreno-Pérez, A., van den Burg, H. A., et al. (2021). Synergistic interaction between the type III secretion system of the endophytic bacterium Pantoea agglomerans DAPP-PG 734 and the virulence of the causal agent of olive knot Pseudomonas savastanoi pv. savastanoi DAPP-PG 722. Mol. Plant Pathol. 22, 1209–1225. doi: 10.1111/mpp.13105
Ndiaye, C., Lo, C. I., Bassene, H., Raoult, D., Lagier, J.-C., and Sokhna, C. (2019). Lysinibacillus timonensis sp. nov., Microbacterium timonense sp. nov., and Erwinia mediterraneensis sp. nov., three new species isolated from the human skin. New Microbes New Infect. 31, 100579. doi: 10.1016/j.nmni.2019.100579
Nissan, G., Chalupowicz, L., Sessa, G., Manulis-Sasson, S., and Barash, I. (2019). Two Pantoea agglomerans type III effectors can transform nonpathogenic and phytopathogenic bacteria into host-specific gall-forming pathogens. Mol. Plant Pathol. 20, 1582–1587. doi: 10.1111/mpp.12860
Nitta, N., Tajima, Y., Yamamoto, Y., Moriya, M., Matsudaira, A., Hoshino, Y., et al. (2021). Fermentative production of enantiopure (S)-linalool using a metabolically engineered Pantoea ananatis. Microb. Cell Fact. 20, 54. doi: 10.1186/s12934-021-01543-0
OECD (2003). Guidance Document on the Use of Taxonomy in Risk Assessment of Micro-Organisms: Bacteria. Paris: Organisation for Economic Co-operation and Development.
Paine, J. A., Shipton, C. A., Chaggar, S., Howells, R. M., Kennedy, M. J., Vernon, G., et al. (2005). Improving the nutritional value of Golden Rice through increased pro-vitamin A content. Nat. Biotechnol. 23, 482–487. doi: 10.1038/nbt1082
Palmer, M., and Coutinho, T. A. (2022). Pantoea. Bergey's Manual of Systematics of Archaea and Bacteria. New York, NY: John Wiley & Sons, 1–25.
Palmer, M., Steenkamp, E. T., Coetzee, M. P. A., Avontuur, J. R., Chan, W.-Y., van Zyl, E., et al. (2018a). Mixta gen. nov., a new genus in the Erwiniaceae. Int. J. Syst. Evol. Microbiol. 68, 1396–1407. doi: 10.1099/ijsem.0.002540
Palmer, M., Steenkamp, E. T., Coetzee, M. P. A., Blom, J., and Venter, S. N. (2018b). Genome-based characterization of biological processes that differentiate closely related bacteria. Front. Microbiol. 9, 113. doi: 10.3389/fmicb.2018.00113
Palmer, M., Venter, S. N., Coetzee, M. P. A., and Steenkamp, E. T. (2019). Prokaryotic species are sui generis evolutionary units. Syst. Appl. Microbiol. 42, 145–158. doi: 10.1016/j.syapm.2018.10.002
Parker, C. T., Tindall, B. J., and Garrity, G. M. (eds)., (2019). International code of nomenclature of prokaryotes. Int. J. Syst. Evol. Microbiol. 69, S1–S111. doi: 10.1099/ijsem.0.000778
Parks, D., and Hugenholtz, P. (2022). Genome Taxonomy Database r207. Available online at: https://gtdb.ecogenomic.org/
Parks, D. H., Chuvochina, M., Chaumeil, P.-A., Rinke, C., Mussig, A. J., and Hugenholtz, P. (2020). A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086. doi: 10.1038/s41587-020-0501-8
Parks, D. H., Chuvochina, M., Rinke, C., Mussig, A. J., Chaumeil, P.-A., and Hugenholtz, P. (2022). GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794. doi: 10.1093/nar/gkab776
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., and Chaumeil, P.-A. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parte, A. C., Sardà Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C., and Göker, M. (2020). List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Polidore, A. L. A., Furiassi, L., Hergenrother, P. J., and Metcalf, W. W. (2021). A phosphonate natural product made by Pantoea ananatis is necessary and sufficient for the hallmark lesions of onion Center Rot. MBio 12, e03402-20. doi: 10.1128/mBio.03402-20
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. doi: 10.1371/journal.pone.0009490
Rand, F. V., and Cash, L. C. (1937). Bacterial Wilt of Corn. Washington, DC: U.S. Department of Agriculture.
Rezzonico, F., Smits, T. H. M., and Duffy, B. (2012a). Misidentification slanders Pantoea agglomerans as a serial killer. J. Hosp. Infect. 81, 137–139. doi: 10.1016/j.jhin.2012.02.013
Rezzonico, F., Smits, T. H. M., Montesinos, E., Frey, J. E., and Duffy, B. (2009). Genotypic comparison of Pantoea agglomerans plant and clinical strains. BMC Microbiol. 9, 204. doi: 10.1186/1471-2180-9-204
Rezzonico, F., Stockwell, V. O., Tonolla, M., Duffy, B., and Smits, T. H. M. (2012b). Pantoea clinical isolates cannot be accurately assigned to species based on metabolic profiling. Transpl. Infect. Dis. 14, 220–221. doi: 10.1111/j.1399-3062.2011.00684.x
Rezzonico, F., Vogel, G., Duffy, B., and Tonolla, M. (2010). Application of whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry for rapid identification and clustering analysis of Pantoea species. Appl. Environ. Microbiol. 76, 4497–4509. doi: 10.1128/AEM.03112-09
Robinson, L. J., Verrett, J. N., Sorout, N., and Stavrinides, J. (2020). A broad-spectrum antibacterial natural product from the cystic fibrosis isolate, Pantoea agglomerans Tx10. Microbiol. Res. 237, 126479. doi: 10.1016/j.micres.2020.126479
Rong, C., Ma, Y., Wang, S., Liu, Y., Chen, S., Huang, B., et al. (2016). Pantoea hericii sp. nov., isolated from the fruiting bodies of Hericium erinaceus. Curr. Microbiol. 72, 738–743. doi: 10.1007/s00284-016-1011-2
Schulz, M., Sicker, D., Schackow, O., Hennig, L., Yurkov, A., Siebers, M., et al. (2017). Interspecies-cooperations of Abutilon theophrasti with root colonizing microorganisms disarm BOA-OH allelochemicals. Plant Signal. Behav. 12, e1358843. doi: 10.1080/15592324.2017.1358843
Schwengers, O., Jelonek, L., Dieckmann, M. A., Beyvers, S., Blom, J., and Goesmann, A. (2021). Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 7, 685. doi: 10.1099/mgen.0.000685
Shin, G. Y., Dutta, B., and Kvitko, B. H. (2023). The genetic requirements for HiVir-mediated onion necrosis by Pantoea ananatis, a necrotrophic plant pathogen. Mol. Plant Microbe Interact. 36, 381–391. doi: 10.1094/MPMI-11-22-0246-R
Shin, G. Y., Smith, A., Coutinho, T. A., Dutta, B., and Kvitko, B. H. (2022). Validation of species-specific PCR assays for the detection of Pantoea ananatis, P. agglomerans, P. allii, and P. stewartii. Plant Dis. 106, 2563–2570. doi: 10.1094/PDIS-08-21-1810-SC
Singh, P., Singh, R. K., Li, H.-B., Guo, D.-J., Sharma, A., Lakshmanan, P., et al. (2020). Diazotrophic bacteria Pantoea dispersa and Enterobacter asburiae promote sugarcane growth by inducing nitrogen uptake and defense-related gene expression. Front. Microbiol. 11, 600417. doi: 10.3389/fmicb.2020.600417
Smith, D. D. N., Kirzinger, M. W. B., and Stavrinides, J. (2013). Draft genome sequence of the antibiotic-producing cystic fibrosis isolate Pantoea agglomerans Tx10. Genome Announc. 1, e00904-13. doi: 10.1128/genomeA.00904-13
Smits, T. H. M., Duffy, B., Blom, J., Ishimaru, C. A., and Stockwell, V. O. (2019). Pantocin A, a peptide-derived antibiotic involved in biological control by plant-associated Pantoea species. Arch. Microbiol. 201, 713–722. doi: 10.1007/s00203-019-01647-7
Smits, T. H. M., Rezzonico, F., Blom, J., Goesmann, A., Abelli, A., Morelli, R. K., et al. (2015). Draft genome sequence of the commercial biocontrol strain Pantoea agglomerans P10c, e01448-15. Genome Announc. 3. doi: 10.1128/genomeA.01448-15
Smits, T. H. M., Rezzonico, F., Kamber, T., Blom, J., Goesmann, A., Ishimaru, C. A., et al. (2011). Metabolic versatility and antibacterial metabolite biosynthesis are distinguishing genomic features of the fire blight antagonist Pantoea vagans C9-1. PLoS ONE 6, e22247. doi: 10.1371/journal.pone.0022247
Smits, T. H. M., Rezzonico, F., Kamber, T., Goesmann, A., Ishimaru, C. A., Stockwell, V. O., et al. (2010). Genome sequence of the biocontrol agent Pantoea vagans strain C9-1. J. Bacteriol. 192, 6486–6487. doi: 10.1128/JB.01122-10
Song, Z., Lu, Y., Liu, X., Wei, C., Oladipo, A., and Fan, B. (2020). Evaluation of Pantoea eucalypti FBS135 for pine (Pinus massoniana) growth promotion and its genome analysis. J. Appl. Microbiol. 129, 958–970. doi: 10.1111/jam.14673
Soutar, C. D., and Stavrinides, J. (2019). Molecular validation of clinical Pantoea isolates identified by MALDI-TOF. PLoS ONE 14, e0224731. doi: 10.1371/journal.pone.0224731
Soutar, C. D., and Stavrinides, J. (2022). Phylogenomic analysis of the Erwiniaceae supports reclassification of Kalamiella piersonii to Pantoea piersonii comb. nov. and Erwinia gerundensis to the new genus Duffyella gen. nov. as Duffyella gerundensis comb. nov. Mol. Genet. Genom. 297, 213–225. doi: 10.1007/s00438-021-01829-3
Stewart, F. C. (1897). A bacterial disease of sweet corn. New York Agr. Exp. Stat. Bull. 130, 422–439.
Stice, S. P., Thao, K. K., Khang, C. H., Baltrus, D. A., Dutta, B., and Kvitko, B. H. (2020). Thiosulfinate tolerance is a virulence strategy of an atypical bacterial pathogen of onion. Curr. Biol. 30, 3130–3140.e6. doi: 10.1016/j.cub.2020.05.092
Stumpf, S., Kvitko, B., Gitaitis, R., and Dutta, B. (2018). Isolation and characterization of novel Pantoea stewartii subsp. indologenes strains exhibiting center rot in onion. Plant Dis. 102, 727–733. doi: 10.1094/PDIS-08-17-1321-RE
Sulja, A., Pothier, J. F., Blom, J., Moretti, C., Buonaurio, R., Rezzonico, F., et al. (2022). Comparative genomics to examine the endophytic potential of Pantoea agglomerans DAPP-PG 734. BMC Genom. 23, 742. doi: 10.1186/s12864-022-08966-y
Takumi, K., Ziyatdinov, M. K., Samsonov, V., and Nonaka, G. (2017). Fermentative production of cysteine by Pantoea ananatis. Appl. Environ. Microbiol. 83, e02502-16. doi: 10.1128/AEM.02502-16
Tambong, J. T. (2019). Taxogenomics and systematics of the genus Pantoea. Front. Microbiol. 10, 2463. doi: 10.3389/fmicb.2019.02463
Tian, L., Huang, C., Mazloom, R., Heath, L. S., and Vinatzer, B. A. (2020). LINbase: a web server for genome-based identification of prokaryotes as members of crowdsourced taxa. Nucleic Acids Res. 48, W529–W537. doi: 10.1093/nar/gkaa190
Tian, L., Mazloom, R., Heath, L. S., and Vinatzer, B. A. (2021). LINflow: a computational pipeline that combines an alignment-free with an alignment-based method to accelerate generation of similarity matrices for prokaryotic genomes. PeerJ 9, e10906. doi: 10.7717/peerj.10906
Vanneste, J. L., Cornish, D. A., Yu, J., and Voyle, M. D. (2002). P10c: a new biological control agent for control of fire blight which can be sprayed or distributed using honey bees. Acta Hortic. 590, 231–235. doi: 10.17660/ActaHortic.2002.590.33
Venter, S. N., Palmer, M., and Steenkamp, E. T. (2022). Relevance of prokaryotic subspecies in the age of genomics. New Microbes New Infect. 48, 101024. doi: 10.1016/j.nmni.2022.101024
Vinatzer, B. A., Weisberg, A. J., Monteil, C. L., Elmarakeby, H. A., Sheppard, S. K., and Heath, L. S. (2017). A proposal for a genome similarity-based taxonomy for plant-pathogenic bacteria that is sufficiently precise to reflect phylogeny, host range, and outbreak affiliation applied to Pseudomonas syringae sensu lato as a proof of concept. Phytopathology 107, 18–28. doi: 10.1094/PHYTO-07-16-0252-R
Vitale, G. A., Sciarretta, M., Cassiano, C., Buonocore, C., Festa, C., Mazzella, V., et al. (2020). Molecular network and culture media variation reveal a complex metabolic profile in Pantoea cf. eucrina D2 associated with an acidified marine sponge. Int. J. Mol. Sci. 21, 6307. doi: 10.3390/ijms21176307
Whitman, W. B., Chuvochina, M., Hedlund, B. P., Hugenholtz, P., Konstantinidis, K. T., Murray, A. E., et al. (2022). Development of the SeqCode: a proposed nomenclatural code for uncultivated prokaryotes with DNA sequences as type. Syst. Appl. Microbiol. 45, 126305. doi: 10.1016/j.syapm.2022.126305
Williams, A. N., and Stavrinides, J. (2020). Pantoea natural product 3 is encoded by an eight-gene biosynthetic gene cluster and exhibits antimicrobial activity against multi-drug resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Microbiol. Res. 234, 126412. doi: 10.1016/j.micres.2020.126412
Wong, N. H., Rosato, A. J., Rose, Y. M., Penix, T. S., Fung, J. B., Vanitski, A. L., et al. (2020). Isolation and whole-genome sequencing of 12 mushroom-associated bacterial strains: An inquiry-based laboratory exercise in a genomics course at the Rochester Institute of Technology. Microbiol. Resour. Announc. 9, e01457-19. doi: 10.1128/MRA.01457-19
Yang, W.-T., Yi, Y.-J., and Xia, B. (2023). Unveiling the duality of Pantoea dispersa: a mini review. Sci. Total Environ. 873, 162320. doi: 10.1016/j.scitotenv.2023.162320
Yao, B., Huang, R., Zhang, Z., and Shi, S. (2023). Diverse virulence attributes of Pantoea alfalfae sp. nov. CQ10 responsible for bacterial leaf blight in alfalfa revealed by genomic analysis. Int. J. Mol. Sci. 24, 8138. doi: 10.3390/ijms24098138
Yu, L., Yang, C., Ji, Z., Zeng, Y., Liang, Y., and Hou, Y. (2021). First report of new bacterial leaf blight of rice caused by Pantoea ananatis in Southeast China. Plant Dis. 406, 310. doi: 10.1094/PDIS-05-21-0988-PDN
Keywords: Pantoea, taxonomy, genomics, reverse ecology, core genome, pangenome analysis
Citation: Crosby KC, Rojas M, Sharma P, Johnson MA, Mazloom R, Kvitko BH, Smits THM, Venter SN, Coutinho TA, Heath LS, Palmer M and Vinatzer BA (2023) Genomic delineation and description of species and within-species lineages in the genus Pantoea. Front. Microbiol. 14:1254999. doi: 10.3389/fmicb.2023.1254999
Received: 08 July 2023; Accepted: 10 October 2023;
Published: 09 November 2023.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Carrie Brady, University of the West of England, United KingdomMargarita Gomila, University of the Balearic Islands, Spain
Copyright © 2023 Crosby, Rojas, Sharma, Johnson, Mazloom, Kvitko, Smits, Venter, Coutinho, Heath, Palmer and Vinatzer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marike Palmer, bWFyaWtlLnBhbG1lckBnbWFpbC5jb20=; Boris A. Vinatzer, dmluYXR6ZXJAdnQuZWR1
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work