 Katie Vigil
Katie Vigil Tiong Gim Aw
Tiong Gim Aw
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 22 September 2023
Sec. Virology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1248323
This article is part of the Research TopicAdvanced Bioinformatic Approaches in Veterinary VirologyView all 5 articles
Introduction: Viral diseases of marine mammals are difficult to study, and this has led to a limited knowledge on emerging known and unknown viruses which are ongoing threats to animal health. Viruses are the leading cause of infectious disease-induced mass mortality events among marine mammals.
Methods: In this study, we performed viral metagenomics in stool and serum samples from California sea lions (Zalophus californianus) and bottlenose dolphins (Tursiops truncates) using long-read nanopore sequencing. Two widely used long-read de novo assemblers, Canu and Metaflye, were evaluated to assemble viral metagenomic sequencing reads from marine mammals.
Results: Both Metaflye and Canu assembled similar viral contigs of vertebrates, such as Parvoviridae, and Poxviridae. Metaflye assembled viral contigs that aligned with one viral family that was not reproduced by Canu, while Canu assembled viral contigs that aligned with seven viral families that was not reproduced by Metaflye. Only Canu assembled viral contigs from dolphin and sea lion fecal samples that matched both protein and nucleotide RefSeq viral databases using BLASTx and BLASTn for Anelloviridae, Parvoviridae and Circoviridae families. Viral contigs assembled with Canu aligned with torque teno viruses and anelloviruses from vertebrate hosts. Viruses associated with invertebrate hosts including densoviruses, Ambidensovirus, and various Circoviridae isolates were also aligned. Some of the invertebrate and vertebrate viruses reported here are known to potentially cause mortality events and/or disease in different seals, sea stars, fish, and bivalve species.
Discussion: Canu performed better by producing the most viral contigs as compared to Metaflye with assemblies aligning to both protein and nucleotide databases. This study suggests that marine mammals can be used as important sentinels to surveil marine viruses that can potentially cause diseases in vertebrate and invertebrate hosts.
Worldwide, viruses are reported to cause 72% of the infectious disease-induced mass mortality events (ID MME) in marine mammals from 1955 to 2018, specifically morbilliviruses and Influenza A viruses (Sanderson and Alexander, 2020). The U.S. National Oceanic and Atmospheric Administration (NOAA) reports that around 49% of marine mammal unusual mortality events (UME) from 1991 to 2021 are classified as undetermined (Onens et al., 2023). Marine mammals infected with viruses may be more susceptible to other oceanic algal toxins and harmful bacteria such as Vibrio and Klebsiella exacerbated by climate change (Bogomolni et al., 2016; Siebert et al., 2017; Sanderson and Alexander, 2020). For example, increased water temperatures enhance the survival of Vibrio parahaemolyticus in marine environments, which was documented to cause die offs of northern sea otters in Alaska due to septicemia and enteritis (Burek et al., 2008). Furthermore, Klebsiella pneumoniae was introduced into the New Zealand sea lion population in 1998 and is known to cause endemic pup mortality (Castinel et al., 2007; Roe et al., 2015). Harmful algal blooms that produce brevotoxins (Flewelling et al., 2005), domoic acid (Lefebvre et al., 1999; Gulland, 2000; Lefebvre et al., 2016), and saxitoxin (Lefebvre et al., 2016; Fire et al., 2020) are documented to cause mortality in marine mammals and are becoming more prevalent globally due to climate change (Hendrix et al., 2021).
The monitoring of viruses in ocean ecosystems can increase the likelihood of detecting emerging infectious diseases. Calicivirus is a prime example of a zoonotic virus, with ocean origin, that spilled over from sea lion to swine, and is known to cause vesicular disease in marine mammals (Neill et al., 1995; Smith et al., 1998). Furthermore, UME first responders were documented to contract sealpox from marine mammals infected with Parapoxvirus, with symptoms presented as contagious pustular dermatitis or lesion (Clark et al., 2005; Roess et al., 2011). In 1988, a phocine distemper Morbillivirus caused the biggest ID MME, with over 18,000 harbor seals (Phoca vitulina) stranded in Europe (Dietz et al., 1989). Morbilliviruses have caused more than half of the ID MME’s in marine mammals since 1955 (Sanderson and Alexander, 2020) with symptoms of skin lesions, pneumonia, brain infections and pup abortions (Pomeroy et al., 2005; Duignan et al., 2014). Although distemper viruses are not readily transmissible to humans, it was documented that canine distemper virus can adapt to use human cell receptors suggesting potential zoonotic spillover (Bieringer et al., 2013; Sakai et al., 2013). Influenza A virus (IAV) is the second leading cause of viral ID MME’s in marine mammals after being reported in harbor seal die offs since 1979, causing acute hemorrhagic pneumonia (Webster et al., 1981b; Sanderson and Alexander, 2020). Several cases of conjunctivitis caused by an IAV virus spillover event from seals to humans were documented in 1981 (Webster et al., 1981a). Therefore, there is a critical need to develop more comprehensive, rapid, and affordable detection methods for zoonotic viruses in marine environments to prevent the spread of emerging and endemic infectious diseases.
Viral diversity in marine environments is immense, and most viruses cannot be identified using traditional culturing techniques (Noble and Fuhrman, 1997; Munang'andu et al., 2017; Arya, 2020). Early metagenomics studies revealed that most of the viral diversity remains uncharacterized (Breitbart et al., 2002). Next generation sequencing (NGS) technologies combined with reliable bioinformatic pipelines are critical for the detection and characterization of novel and existing viral pathogens in marine mammals that could cross over to other vertebrate populations. Although the second generation short-read (200–400 bp) technologies, such as Illumina or Ion Torrent have high read throughput, the shorter DNA fragments can be difficult to assemble and annotate (Pop and Salzberg, 2008; Hu et al., 2021). The third generation nanopore sequencing technology is providing a new opportunity to develop more rapid, portable, and cost-effective genomic sequencing assays for viruses. For example, the nanopore-based sequencer MinION can produce read lengths of over 10 kB, which can overcome annotation of genomic repeat regions and structural variations that are difficult to assemble (Tørresen et al., 2019; Hu et al., 2021). Nanopore sequencing has been used to detect various viruses from diverse clinical samples (Quick et al., 2017; Hayashida et al., 2019; Cohen et al., 2020; Brown et al., 2021). This technology has been shown to sequence the full genome of four variants of herpes simplex viruses in single read, with read lengths ranging from 100 kb to 2.3 Mb (Saranathan et al., 2022). To our knowledge, the nanopore long-read sequencing technology has not been evaluated for non-targeted metagenomic sequencing of viruses in marine mammals.
De novo assemblers are programs that assemble shorter nucleotide sequences into longer fragments called contigs without a reference database. Many de novo assemblers that exist for long-read sequencing technologies have been evaluated only using bacteria (Chin et al., 2013; Zimin et al., 2013; Kamath et al., 2017; Koren et al., 2017; Kolmogorov et al., 2020), plants (Chin et al., 2016; Ruan and Li, 2020), and fungi (Chin et al., 2016) samples. Choosing the right de novo assembler is important for constructing error-free and artifact-less genome assemblies. De novo assembly algorithms include overlap-layout-consensus (OLC), de-Bruijn-graph (DBG), string-graph (SG) and hybrid approaches (Dida and Yi, 2021). Briefly, the OLC algorithm finds overlaps between reads, creates a read layout, then a consensus sequence is produced (Idury and Waterman, 1995; Li et al., 2012; Liao et al., 2019). DBG is an algorithm that chops reads up into short k-mers (substrings of length k), where overlapping edges (k−1) are found, resulting in an Eulerian (edges) or Hamiltonian (nodes) path to create a graph (Pevzner et al., 2001; Li et al., 2012) where contigs are constructed (Compeau et al., 2011). SG is a simplified OLC where sequence reads (nodes) and non-transitive edges produce suffix to prefix overlaps (Liao et al., 2019). Canu (Koren et al., 2017) is an upgraded long-read OLC assembler algorithm that integrates newer computational procedures to overcome noisy overlapping reads and decreases assembly time compared to the now unsupported Celera Assembler (Myers et al., 2000; Miller et al., 2008). Metaflye is a long-read DBG assembler algorithm that constructs repeat graphs from arbitrary paths called disjoints that are stringed together to construct contigs (Kolmogorov et al., 2019, 2020). Both Metaflye and Canu have been evaluated to construct genomes of plant (Arabidopsis thaliana), bacteria (Escherichia coli, Bacillus cereus, and Staphylococcus aureus), human, and yeast (Saccharomyces cerevisiae) (Dida and Yi, 2021). Metaflye outperformed Canu by generating larger contigs and higher N50 values, but is prone to more mis-assemblies and mismatches (Dida and Yi, 2021).
The objectives of this study were to (i) use metagenomics to characterize viruses in stool and serum samples from bottlenose dolphins (Tursiops truncates) and California sea lions (Zalophus californiansus) with a long-read nanopore sequencing approach; (ii) compare two de novo assemblers, Canu v2.2 and Metaflye v2.9.1, in generating viral contigs for annotation. The improved knowledge on marine viruses and successful protocol development will lead to a translational science that informs protection of animals and public health under a One Health framework.
Five sea lion fecal, four sea lion serum, four dolphin fecal, and four dolphin serum samples were collected from the U.S. Navy’s dolphin and sea lion clinic facility in Point Loma, CA (Lat 32.746021, Long −117.237030) in 2018 and 2019, respectively. Samples were collected in 10 mL conical tubes and frozen at –80°C until analysis.
Samples from Navy animals were collected during their routine care and under the authority codified in U.S. Code, Title 10, Section 7524. Secretary of Navy Instruction 3900.41H directs that Navy marine mammals be provided the highest quality of care. The U.S. Navy Marine Mammal Program (MMP), Naval Information Warfare Center (NIWC) Pacific, houses, and cares for a population of bottlenose dolphins and California sea lions in San Diego Bay (CA, United States). NIWC Pacific is accredited by The Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International and adheres to the national standards of the U.S. Public Health Service policy on the Humane Care and Use of Laboratory Animals and the Animal Welfare Act.
Animal fecal samples were weighed in 50 mL sterile conical tubes. Sterile phosphate-buffered saline (PBS) was added at 1 mL/g and vortexed for 5 min. Sterile PBS was also used as a negative control. The samples were centrifuged at 15,000g for 10 min, and the supernatant was filtered through 0.45 μm and 0.22 μm filters. Serum samples were filtered directly through 0.45 μm and 0.22 μm filters. In order to remove any free DNA, 1 mL aliquot of each sample was incubated with Ambion™ DNase I (RNase Free) (Thermo Fisher Scientific, United States) at 37°C for 1 h in a water bath at a final concentration of 0.1 U (unit) per μL. The DNase was deactivated by treating with 50 mM EDTA at 75°C in a heat block for 10 min.
Viral nucleic acids were extracted using the Invitrogen™ PureLink™ Viral RNA/DNA Extraction kit according to manufacturer’s instructions (Thermo Fisher Scientific, United States). In this study, 200 μL of pre-treated samples were extracted and eluted into 50 μL sterile RNase-free water. Nucleic acid samples were stored in −80°C until further processing.
To generate sufficient material for sequencing, a random reverse transcription/amplification protocol was used to amplify both viral DNA and RNA. Briefly, 5 μL of the nucleic acid samples were mixed with 1uL of primer-A (5’-GTTTCCCAGTCACGATCNNNNNNNNN) (40 μM) (Wang et al., 2002, 2003), 4 μL nuclease-free water, and was heated to 65°C for 5 min and 22°C for 5 min. The 10 μL reaction was then added to 4 μL 5X First Strand Buffer [250 mM Tris–HCl (pH 8.3), 375 mM KCl, 15 mM MgCl2], 0.4 μL deoxynucleotide Solution Mix (dNTP) (10 mM) (New England Biolabs, United States), 0.6 μL nuclease-free water, 2 μL DTT (100 mM), 2uL SuperScript™ III Reverse Transcriptase (200 U/uL) (Thermo Fisher Scientific, United States), 1uL RNaseOUT™ (40 U/uL) (Thermo Fisher Scientific, United States) and was heated to 42°C for 60 min, 94°C for 2 min, and 10°C for 5 min. The 20 μL reaction was added to 2 μL 5X Sequenase™ Reaction Buffer, 7.7 μL nuclease-free water, 0.3 μL Sequenase™ Version 2.2 DNA Polymerase (13 U/μL) (Thermo Fisher Scientific, United States), and was heated to 10°C for 5 min, 37°C for 8 min with 1°C/s ramping, 94°C for 2 min, 10°C for 5 min (1.2 μL of 2.5 μL Sequenase™ Version 2.2 DNA Polymerase (13 U/uL) with 7.5 μL Sequenase™ Enzyme Dilution Buffer was added to each tube during this step), 37°C for 8 min with 1°C/s ramping, and 94°C for 8 min. 6 μL of the cDNA template was added to 8 μL MgCl2, 10 μL Buffer II (100 mM Tris–HCl, pH 8.3, 500 mM KCl), 1 μL dNTP (10 mM), 1 μL Primer-B (5’-GTTTCCCAGTCACGATC) (100 μM) (Wang et al., 2002, 2003), 1 μL AmpliTaq Gold™ DNA Polymerase with Gold Buffer and MgCL2 (Thermo Fisher Scientific, United States), and 73 μL nuclease-free water. The cDNA was amplified under the following conditions, 94°C for 15 min, 40 cycles of 94°C for 30 s, 40°C for 30 s, 50°C for 30 s, and 72°C for 1 min. The resulting PCR product was run on a 1% agarose gel and a smear between 500 bp to 1 kb was considered positive for sequencing. Positive samples were purified using the Wizard SV Gel and PCR Clean-Up System according to manufacturer’s instructions (Promega, Madison, WI, United States), where 100 μL cDNA was purified and eluted into 50 μL nuclease-free water. Samples were not normalized to a specific concentration prior to barcoding for sequencing.
Amplified samples were checked for DNA concentration using Qubit® 4 dsDNA HS Assay kit according to manufacturer’s instructions (Thermo Fisher Scientific, United States), and 1 μL samples were tested for concentration (ng/μL). 50 μL of purified rPCR samples were barcoded using the Oxford Nanopore PCR Barcoding Kit (SQK-PBK004) Version: RPB_9059_v1_revN_14Aug2019 according to the manufacturer’s instructions (Oxford Nanopore, United Kingdom). All samples were sequenced on the flow cell MinION Mk1b with R9.4.1 flow cell chemistry. Due to the frequent software updates, different version of MinKNOW and Guppy basecaller were used (MinKNOW v20.10.3 and v22.05.5 with Guppy basecaller v4.4.1 and v6.1.7 for fecal samples; MinKNOW v21.02.1 with Guppy basecaller v4.3.4 for all serum samples). The libraries were analyzed in the MinKNOW software under the following parameters: PCR Barcoding kit (SQK-PBK004), Native Barcoding Expansion 1–12 (EXP-PBC001), fast basecalling, trim barcodes, mid-read barcode filtering and barcode trimming. Fastq files were concatenated after sequencing for data analysis and bioinformatics.
The in silico pipeline analysis for nanopore sequencing data is shown in (Figure 1). Fastq files were uploaded to the Oxford Nanopore Technology (ONT) EPI2ME Software and analyzed using the “Whats in my pot?” WIMP (Humane + Viral) workflow to obtain quality control statistics on reads analyzed, total yield (Mb), average quality score, and average sequence length. Fastq sequencing files were analyzed in the Ubuntu 20.04 LTS 64-bit on Tulane Universities’ Cypress high performance computing (HPC) 124-node cluster with dual 10-core 2.8 GHz Intel Xeon E5-2680 v2 CPUs, 64 GB or RAM, and dual Xeon Phi 7120P coprocessor system. Fastq files were de novo assembled with Canu v2.2 and Metaflye v2.9.1 (Koren et al., 2017; Kolmogorov et al., 2020) pipeline and polished with Medaka v1.6.01 (Lee et al., 2021). Canu 2.2 program parameters for low coverage reads were genomeSize = 2 m maxinputCoverage = 10,000 corOutCoverage = 10,000 corMhapSensitivity = high corMinCoverage = 0 redMemory = 32 oeaMemory = 32 batMemory = 64 minInputCoverage = 0 stopOnLowCoverage = 0, and genomeSize = 2 m maxInputCoverage = 100 –nanopore fecal samplesDNA.fastq for high coverage data, respectively. Metaflye v2.9.1 program parameters were –meta –threads 20 –nano-raw. Medaka v1.6.0 program parameters were medaka_consensus -i ${BASECALLS} -d ${DRAFT} -o ${OUTDIR} -t ${NPROC} -m r941_min_high_g330. All contigs were annotated using Basic Local Alignment Search Tool (BLAST) against the National Center for Biotechnology Information (NCBI) GenBank protein database (viral RefSeq). BLASTx hits with e-values ≤10−4 were used for analysis. BLASTx output files were analyze to viral families using MEGAN-LR (long-read) Community Edition 6.24.1 with default parameters, min score 50.0, max expected 0.0001, min percent identity 10.0, top percent 10.0, min support percent 0.01, min support 1, min read length 0, LCA algorithm longReads, percent to cover 80.0, and read assignment mode readCount. Viral family contigs that were positive for viral families that infect invertebrate and vertebrate hosts were subject to manual BLASTn analysis for confirmation. Version 5 BLAST+ 2.10.0 was used for both BLASTx and BLASTn database queries. The Quality Assessment Tool for Genome Assemblies (QUAST) v5.2 MetaQUAST was used to analyze contig output files. To determine if Canu v2.22 assembles more viral family reads compared to MetaFlye v2.91,3 a one-tailed Wilcoxon rank-sum test (also known as the Mann–Whitney U test) for non-parametric data was used. Statistical analysis was performed using R version 4.2.3 (R Core Team, 2013).
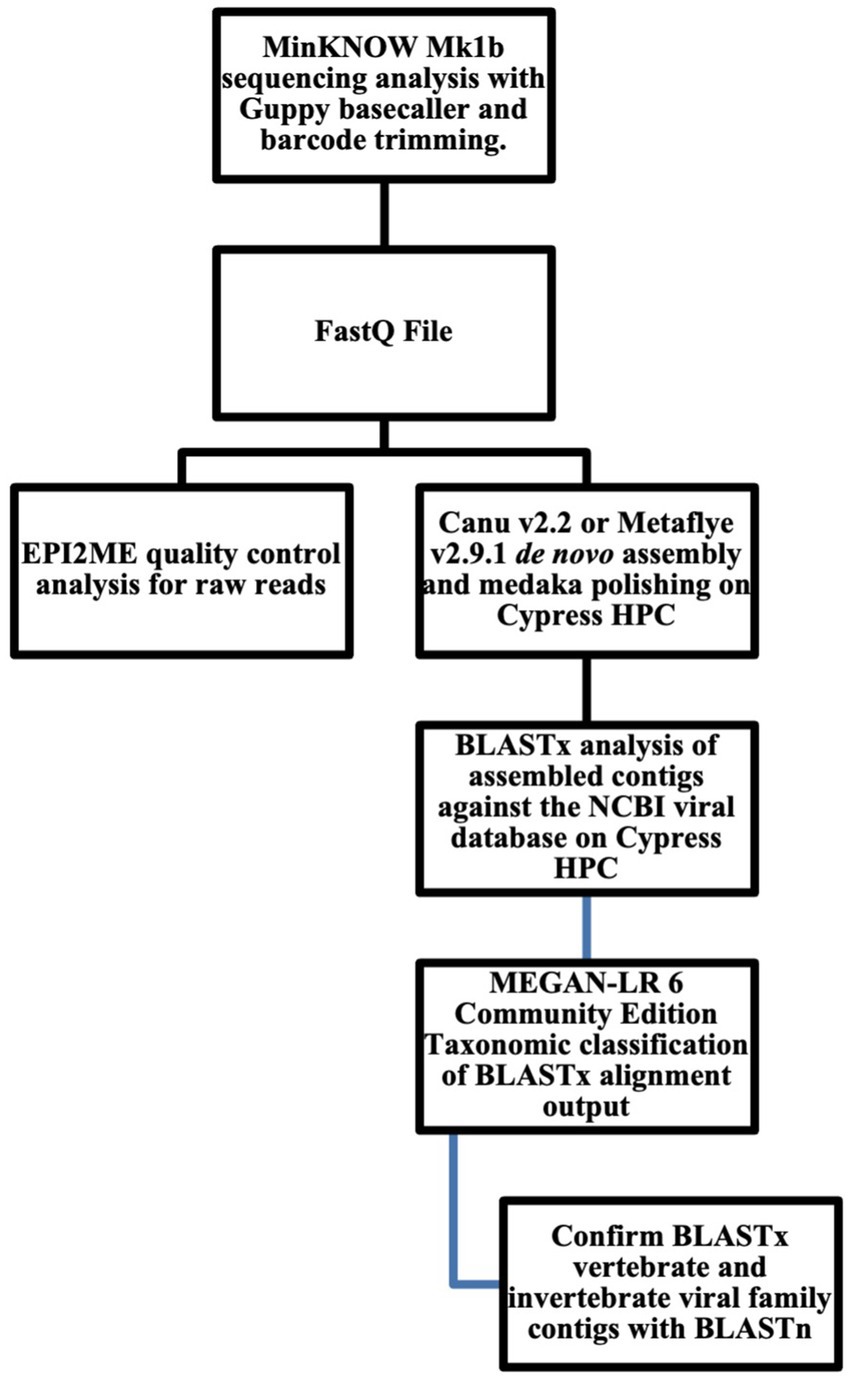
Figure 1. Bioinformatics workflow for long-read viral metagenomics.
The original nucleotide sequences described in this study have been deposited in the GenBank database under the Bioproject accession numbers PRJNA998092.
A long-read sequencing of randomly primed amplicons using the nanopore MinION generated a total of 1,698,981 sequencing reads, yielding 1,557 Mb after quality filtering of “passed” reads. The highest number of sequencing reads at 1,120,197 reads after basecalling, were produced from sea lion fecal samples, followed by dolphin fecal samples with 456,126 sequence reads after basecalling (Table 1). The average quality score of all the sequences ranged from 9 to 15, with the average sequence length (bases) ranging from 627 to 769 bases (Table 1).
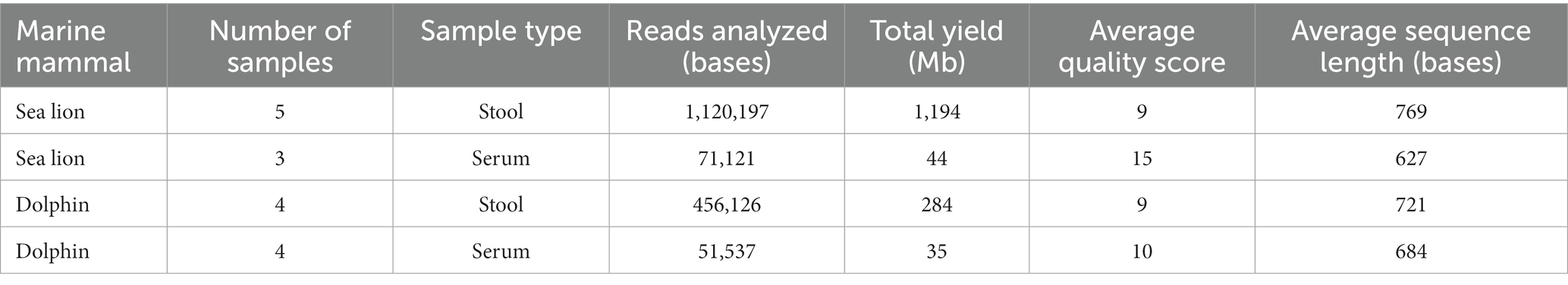
Table 1. EPI2ME quality control analysis of passed raw read fastq files generated from the MinKNOW software.
Canu v2.2 assembled a total of 333 contigs, with 118 viral contigs ranging from 1,029 to 13,513 nucleotides (nt) long for dolphin samples and 593 total contigs, with 224 viral contigs ranging from 1,026 to 8,114 nt for sea lion samples. In this study, viral contigs are the assembled sequences that aligned with the reference sequences from the NCBI Viral RefSeq database using BLASTx. Metaflye v2.9.1 assembled a total of 177 contigs, with 76 viral contigs ranging from 262 to 3,631 nt for dolphin samples and a total of 130 contigs, with 46 viral contigs ranging from 1,128 to 4,740 for sea lion samples (Table 2). Canu v2.2 produced the longest contig at 13,513 nt from dolphin fecal samples. Metaflye v2.9.1 produced higher N50 values compared to Canu v2.2 for both dolphin and sea lion fecal and serum samples. Canu v2.2 produced more contigs that could be annotated as viruses using the NCBI database compared to Metaflye v2.9.1. The mean viral contig size was higher for dolphin fecal samples, but lower in sea lion fecal samples with Canu v2.2 compared to Metaflye v2.9.1. The mean viral contig size was lower for dolphin serum samples, but higher for sea lion serum samples with Canu v2.2 compared to Metaflye v2.9.1. Metaflye v2.9.1 did not produce viral contigs for sea lion serum samples. Canu v2.2 produced lower percent viral contigs for dolphin fecal samples, but higher percent viral contigs for sea lion fecal samples, while Metaflye v2.9.1 produced higher percent viral contigs for dolphin serum. Overall, Canu v2.2 used lower central processing units (CPUs) compared to Metaflye v2.9.1 but took a longer time to assemble contigs compared to Metaflye v2.9.1 (Table 2).
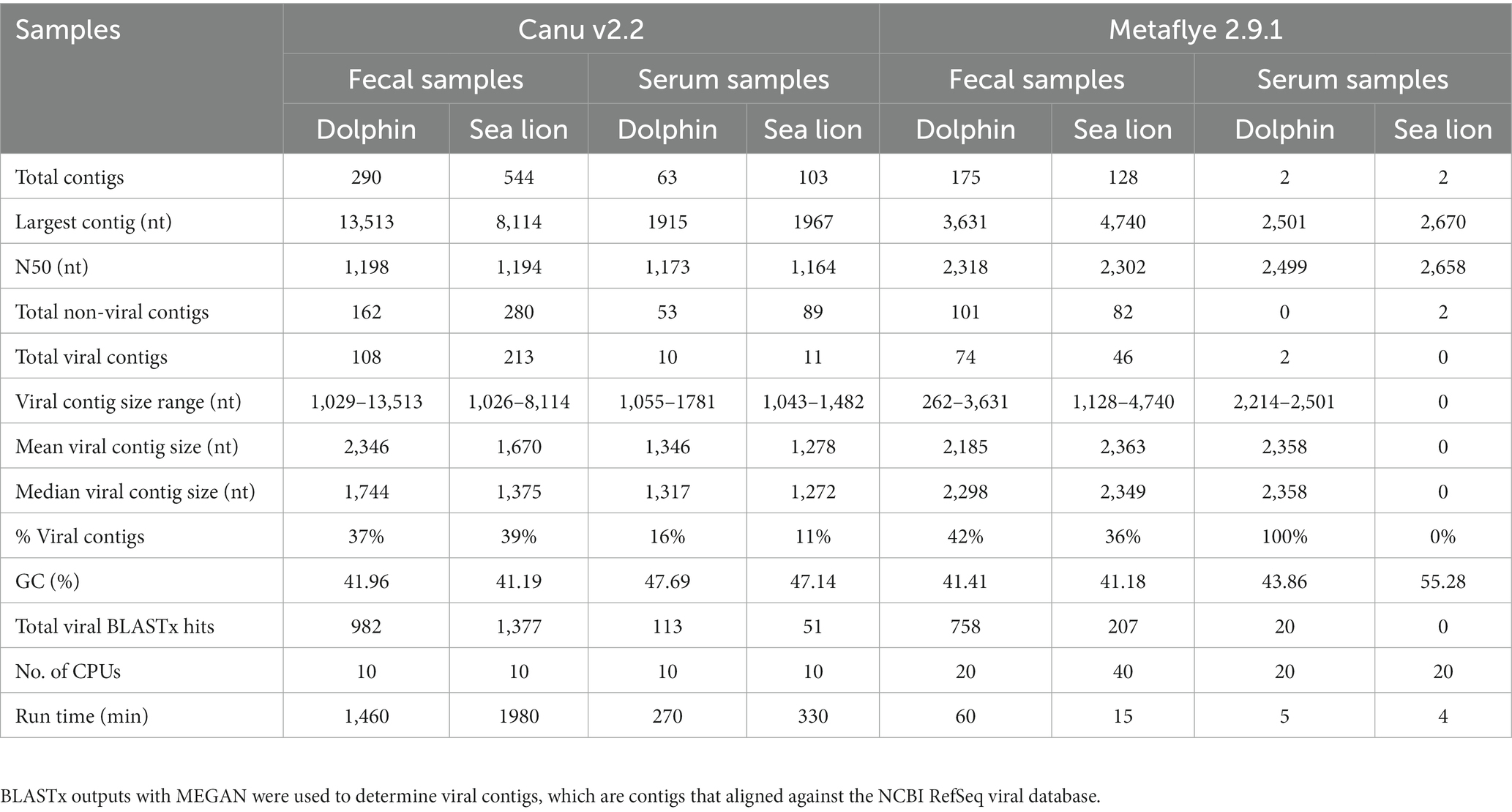
Table 2. A comparison of the performance and annotation outputs for de novo assemblers Canu v2.2 and Metaflye v2.9.1.
Using different de novo assemblers, contigs from diverse viral host distributions were observed. For dolphin fecal samples, both Metaflye v2.9.1 and Canu v2.2 assembled viral contigs that aligned with bacteriophages, invertebrate, and vertebrate viruses, but only Canu v2.2 produced contigs that aligned with algal viruses (Figure 2). Overall, Canu v2.2 produced viral contigs from more diverse hosts in fecal samples compared to Metaflye v2.9.1 (Figure 2). Vertebrate hosts were dominant in dolphin serum using Canu v2.2, while invertebrate hosts were dominant in dolphin serum using Metaflye v2.9.1. Contigs aligning with viruses from amoeba hosts were only detected in sea lion fecal and serum samples using Canu v2.2. Contigs aligning to plant viral hosts were only detected in sea lion fecal samples with Canu v2.2. Distribution of virus types between samples and de novo assemblers were similar. All serum samples had contigs that aligned 100% with dsDNA type viruses. Both dolphin and sea lion fecal samples had contigs that aligned with dsDNA, RNA and ssDNA virus types using either Canu v2.2 or Metaflye v2.9.1. Contigs that aligned with RNA viruses had slightly higher distribution in sea lion fecal samples compared to dolphin fecal samples, while ssDNA types had similar distribution (Figure 3).
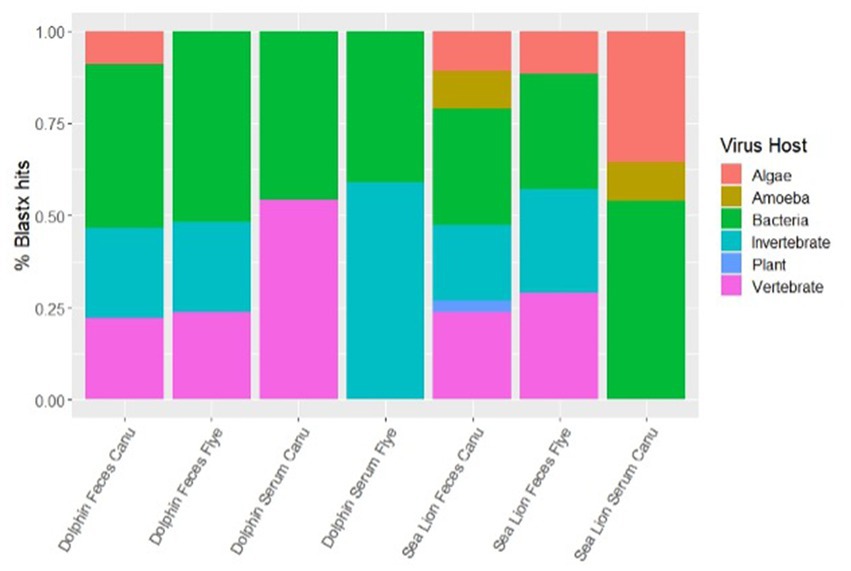
Figure 2. Percent distribution of contigs by types of viral host in dolphin and sea lion fecal and serum samples. Contigs were assembled using Canu v2.2 and Metaflye v2.9.1 and annotated using BLASTx program against the NCBI Virus database.
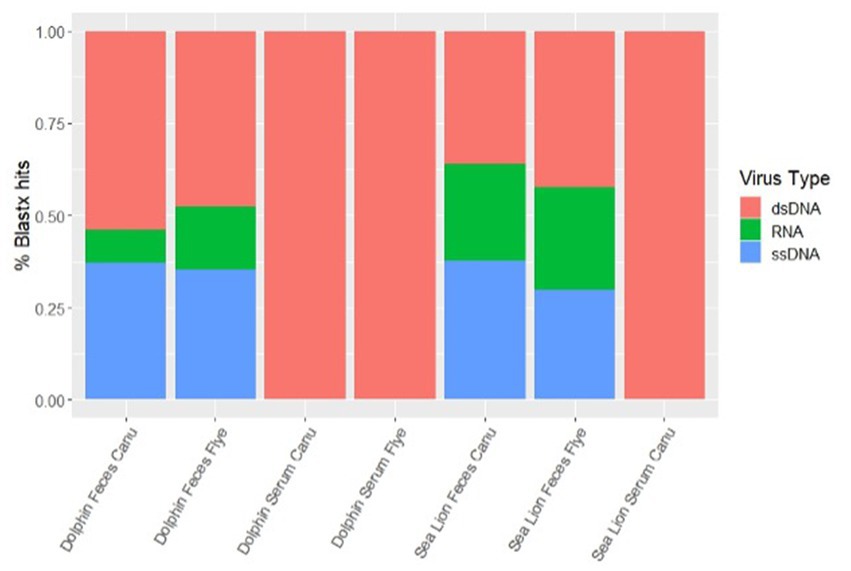
Figure 3. Percent distribution of contigs by types of viral genome in dolphin and sea lion fecal and serum samples. Contigs were assembled using Canu v2.2 and Metaflye v2.9.1 and annotated using BLASTx program against the NCBI Virus database.
Canu v2.2 assembled significantly higher numbers of viral contigs compared to Metaflye v2.9.1 for dolphin fecal samples, sea lion fecal samples and sea lion serum samples (p-value = <0.05) (Figure 4). Assembled viral contigs aligned with several vertebrate viral families such as Anelloviridae, Parvoviridae, Poxviridae, Smacoviridae using the protein BLASTx program (Figure 4). Invertebrate viruses detected in fecal samples of dolphin and sea lion aligned to the families of Baculoviridae, Circoviridae, Iridoviridae, and Parvoviridae. Baculoviridae viral reads only aligned with Canu v2.2 assembled viral contigs from dolphin fecal samples. Circoviridae, Parvoviridae, Riboviria (realm) viral reads were higher in sea lion fecal samples with Canu v2.2.
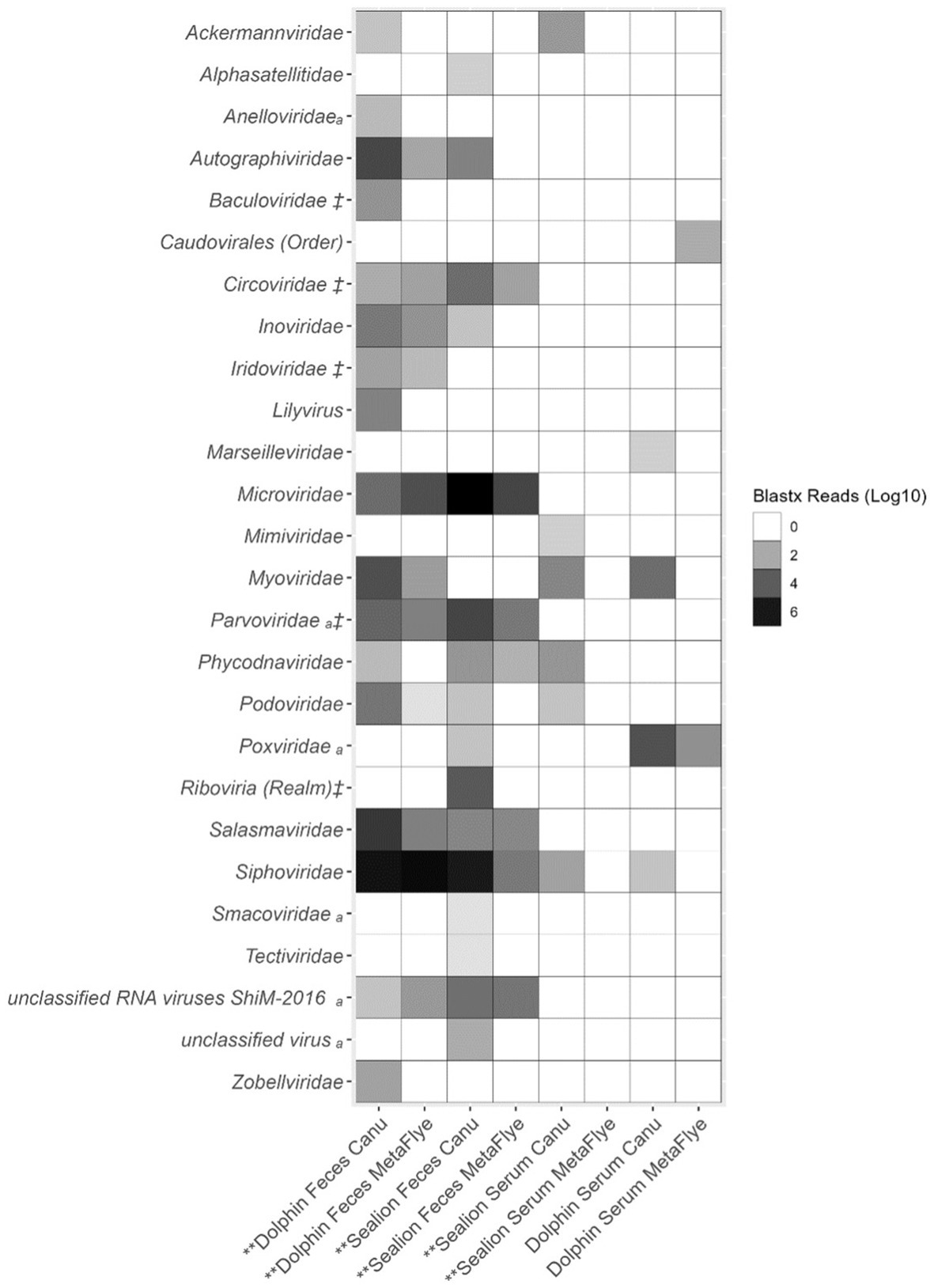
Figure 4. Total viral family reads from BLASTx (e-value = ≤10−4) output files of assembled contigs using Canu v2.2 and Metaflye v2.9.1 against the NCBI Virus Refseq database from dolphin and sea lion fecal samples and serum. avertebrate viral hosts and ‡ invertebrate viral hosts. Asterisks (**) represent p-value = <0.05 for the one-tailed Wilcoxon Rank Sum test for non-parametric log10 data specifying that the alternative hypothesis is that Canu v2.2 assembled viral reads were significantly greater than MetaFlye v2.9.1.
Bacteriophage families include Autographiviridae, Inoviridae, Myoviridae, Podoviridae, Salasmaviridae, Siphoviridae, and Zobellviridae were also detected in dolphin fecal samples using Canu v2.2 (Figure 4). In sea lion fecal samples, bacteriophage families included Microviridae and Tectiviridae were detected.
Although Canu v2.2 and Metaflye v2.9.1 assembled viral contigs from similar viral families, some discrepancies were found. Canu v2.2 produced viral contigs of seven viral families that were absent from the Metaflye v2.9.1 output (Figure 5). Overall, most of the viruses that were detected with both Canu v2.2 and Metaflye v2.9.1 were linear, non-enveloped dsDNA viruses (Supplementary Data_Sheet_1_virus_genome excel file).
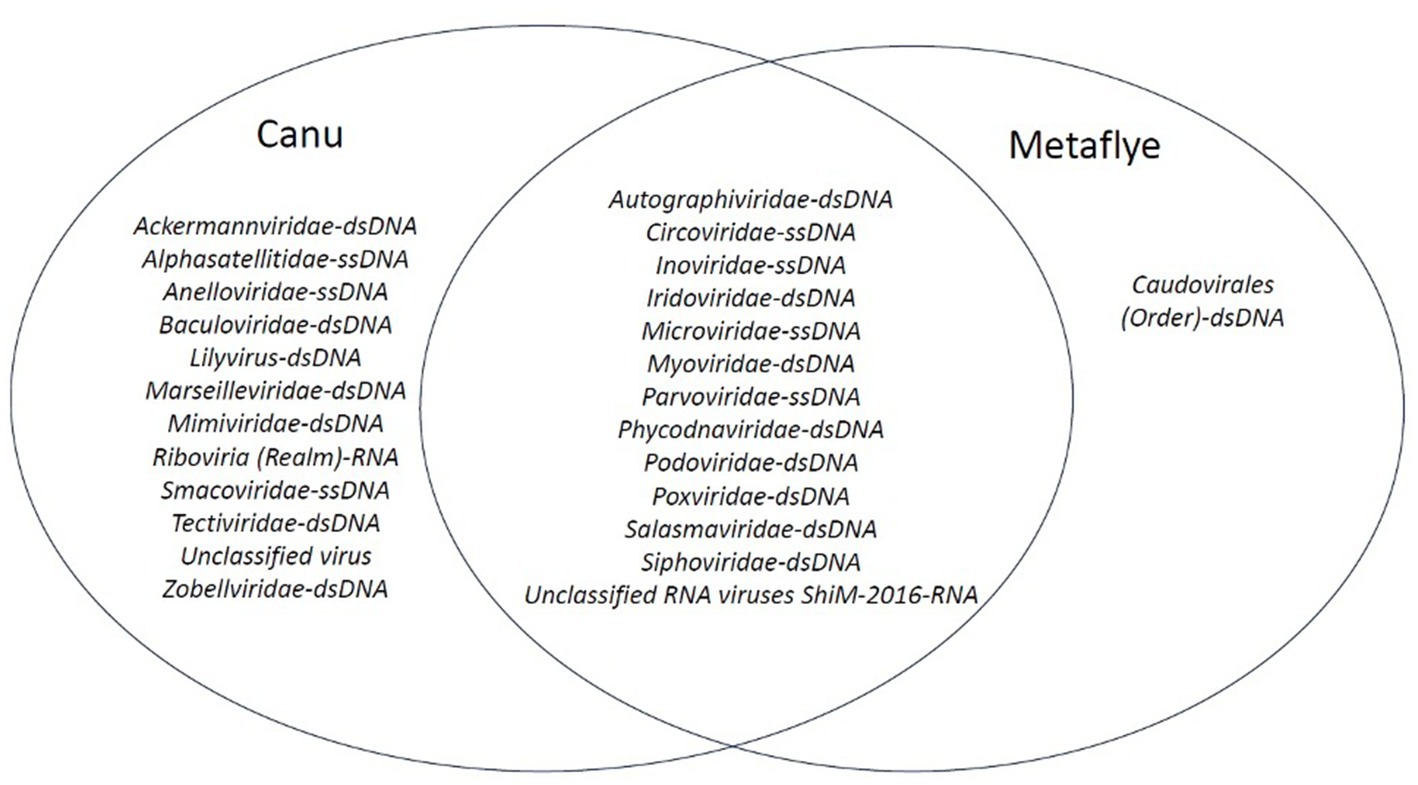
Figure 5. Venn diagram of viral families that aligned with contigs assembled by Canu v2.2 and Metaflye v2.9.1 from BLASTx (e-value = ≤10−4) output files against the NCBI Virus Refseq database.
Viral contigs that were aligned using the protein (amino acid, aa) BLASTx program were further confirmed using the nucleotide BLASTn program. Canu v2.2 generated contigs of vertebrate and invertebrate viruses that were positive for both protein and nucleotide NCBI RefSeq viral databases, while Metaflye v2.9.1 only generated viral contigs that had positive viral families for protein, not nucleotide. No viral contigs from vertebrate and invertebrate hosts were identified in serum samples using both assemblers with both BLASTx and BLASTn searches. Dolphin fecal samples contained viral contigs that were found to be associated with seals, sea stars, and oysters, as confirmed by both protein and nucleotide databases. These viruses included torque teno midi virus (TTMDV), seal annellovirus, and sea star densoviruses (DNV) (Table 3). Sea lion fecal samples contained viral contigs that were also detected in oysters, fish, sea stars, crayfish, and clams. These viruses included sea star DNV, Cherax quadricarinatus DNV, Circoviridae species., and Ambidensovirus (AmDNV) (Table 4).
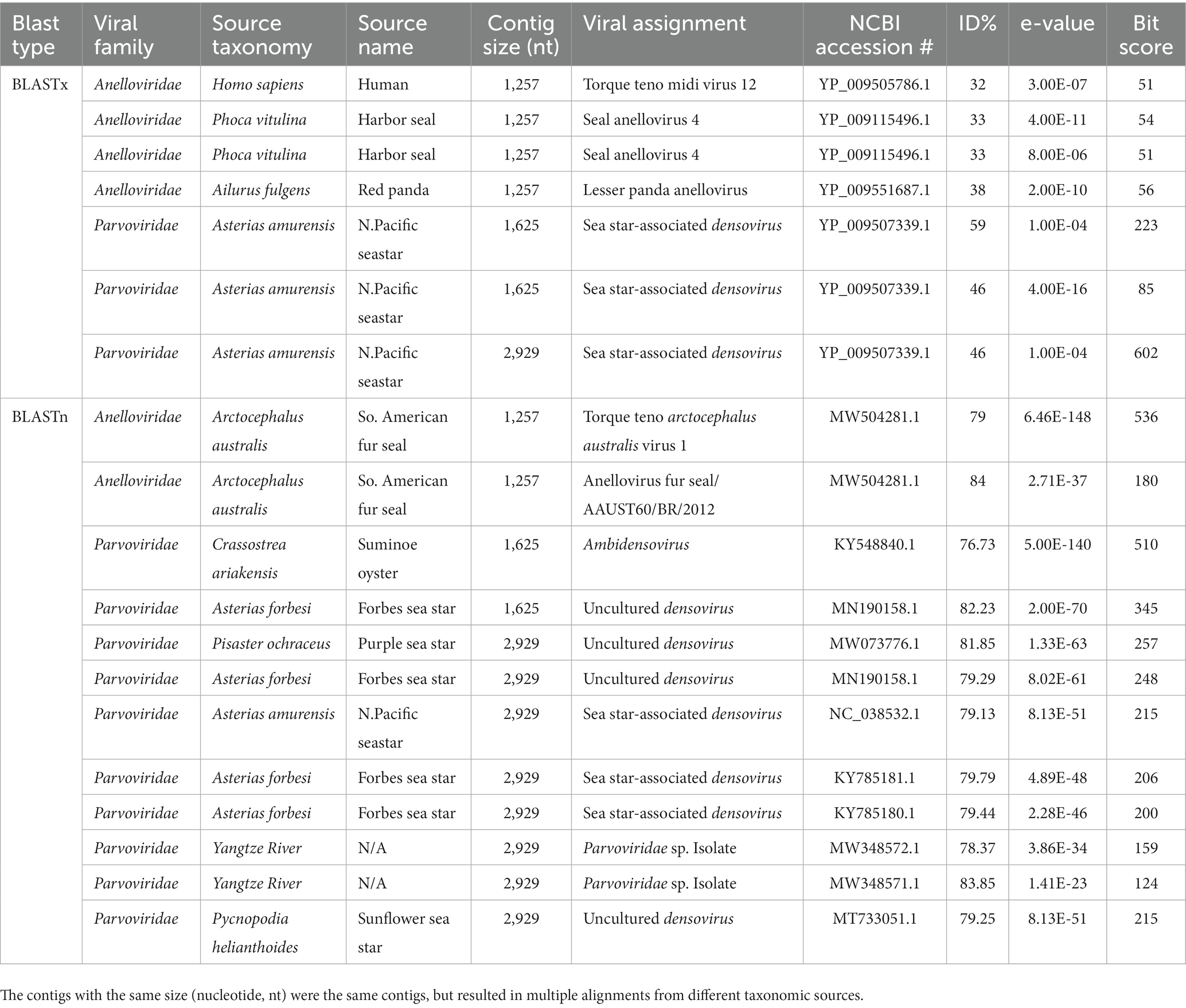
Table 3. Canu v2.2 assembled vertebrate and invertebrate viral contigs from pooled dolphin fecal samples (n = 4) that shared the same viral families between BLASTx and BLASTn.
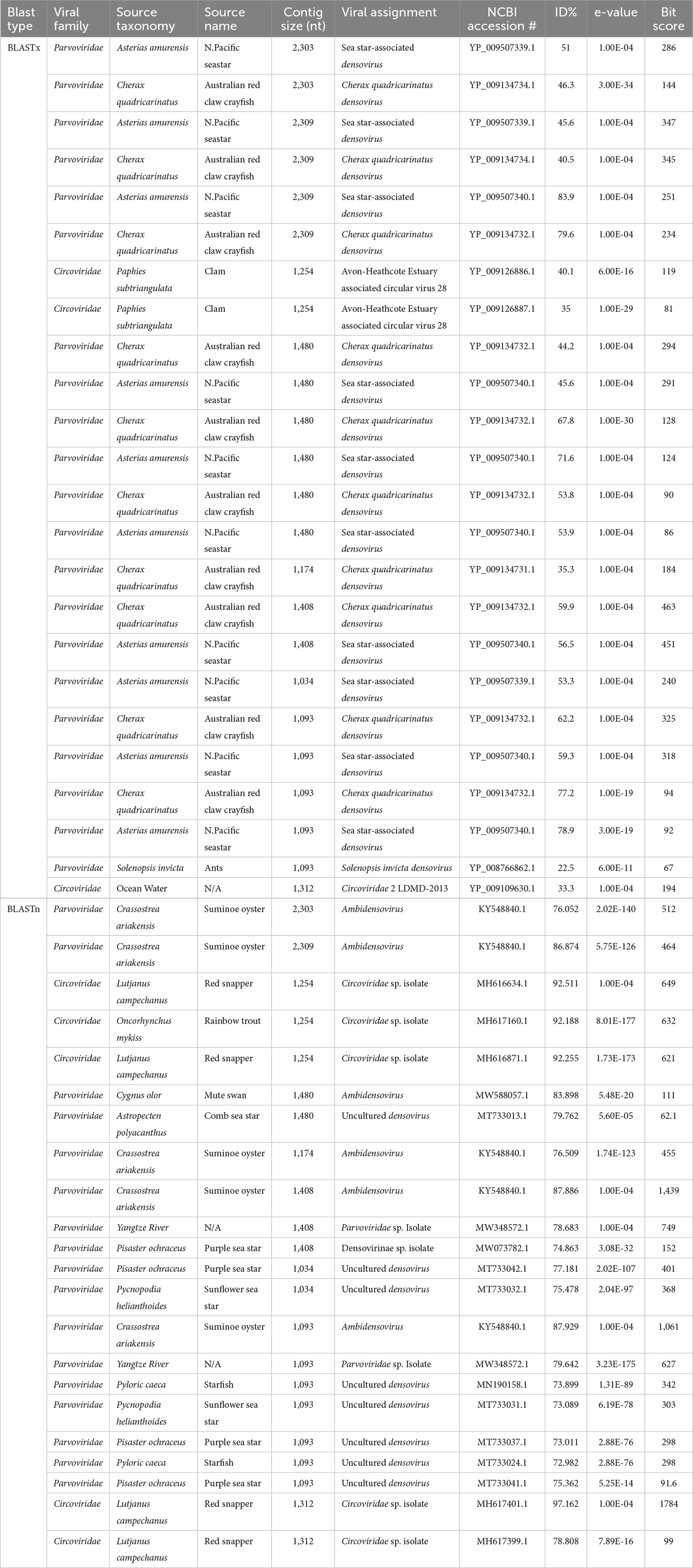
Table 4. Canu v2.2 assembled vertebrate and invertebrate viral contigs from pooled sea lion fecal samples (n = 5) that shared the same viral families between BLASTx and BLASTn.
Assembling reliable metagenomic sequencing data is critical to characterizing viral diversity in marine environments. De novo assembly is important because it allows researchers to construct genomes without the need for a reference genome or when reference genomes are not available. De novo assembly can also discover novel genes and genetic variants of viruses (Allen et al., 2011; Chiu, 2013; Carbo et al., 2020). Several nanopore-based sequencing bioinformatic tools have been developed to handle the long-read sequencing reads, but many lack performance testing on unknown viral metagenomic sequencing dataset (Li, 2016; Kamath et al., 2017; Koren et al., 2017; Wang et al., 2018; Kolmogorov et al., 2020; Ruan and Li, 2020). This work aimed at filling this gap by using viral metagenomics data generated from marine mammal fecal and serum specimens with the nanopore sequencing platform.
Long-read sequencing technologies, such as the MinION nanopore sequencer, have several advantages over short-read sequencing technologies, e.g., it is portable, it does not require large imaging equipment to detect DNA nucleotides, lower cost, it can be powered through a Universal Serial Bus (USB) port, and it can be used in the field (Kono and Arakawa, 2019). In addition, nanopore sequencing can sequence longer stretches of DNA (>500 bp) (Adewale, 2020), can pick up long repetitive sequences (Kono and Arakawa, 2019), does not require fragmentation, and can directly sequence RNA molecules (Garalde et al., 2018). Although short-read sequencing can produce more reads with shorter lengths (<300 bp) (Hu et al., 2021), long-read sequencing is capable of sequencing full viral genomes, thus making assembly less error prone (Kono and Arakawa, 2019). Our results showed variation in results between the two de novo assemblers, Canu and Metaflye. Although Metaflye was a faster assembler that generated higher N50, it produced less viral alignments against the NCBI RefSeq viral protein database using BLASTx as compared to Canu. A study comparing Canu and Metaflye using sequences of bacteria, mammal, plant and fungi revealed that Metaflye outcompeted Canu with larger N50 values, but high error rates when assembling full genomes against reference sequences (Dida and Yi, 2021). Other studies show Canu and Metaflye performing similarly (Jung et al., 2020; Wang et al., 2021). Recent genome assembly pipelines created for viruses are using Canu for preprocessing of reads before reference alignment (Roach et al., 2022; Yu et al., 2023), suggesting that this assembler could produce quality assemblies for metagenomics analysis. De novo algorithms that generate large contigs and high N50s are considered good quality for genome assembly, but sometimes the results are inaccurate and produce more mismatches (Dida and Yi, 2021), which could be why Metaflye produced mismatches between viral contigs aligned against protein and nucleotide sequence databases for invertebrate and vertebrate viral families.
Canu was the only assembler that produced contigs that matched the same viral families between the protein and nucleotide databases from marine mammal fecal samples. This suggests that Canu could assemble contigs that are more accurate for viral annotation from environmental samples as compared to Metaflye. Canu is an OLC algorithm based de novo assembler, and is documented to have less error rates compared to DBG assemblers for long-reads (Kono and Arakawa, 2019). Vertebrate and invertebrate viruses in fecal samples of Parvoviridae, Anelloviridae, and Circoviridae families were confirmed with Canu assembler for both protein and nucleotide BLAST searches. In this study, there was a discrepancy between protein BLASTx and nucleotide BLASTn results for serum samples. For example, all the invertebrate and vertebrate viral contigs in serum samples identified using BLASTx against the protein database aligned with bacteriophages when using BLASTn against the nucleotide database. This could be that the lowest amount of sequence reads were generated in serum samples and both Canu and Metaflye are more accurate with higher sequencing read counts. De novo assembly could be less accurate with low sequencing depth due to insufficient genome coverage and limited redundancy for genome regions. De novo assemblers rely on read overlapping for OLC (Koren et al., 2017) or DBG (Kolmogorov et al., 2020) graph construction for contig or scaffold assembly. A low sequencing depth may result in reduced overlapping information making it harder for the algorithms to accurately detect sequencing overlaps. In addition, low read count may produce shorter contigs resulting in a fragmented assembly that may not accurately represent true structure of the sequenced genome regions. As seen in this study, the shortest mean nucleotide contig lengths were observed in serum samples and this could cause discrepancy in results between the two BLAST search programs. A future study could include a mock viral community to establish a baseline for sequencing reads prior to de novo assembly to better understand the relationship between sequencing depth and de novo assembly for viral metagenomics.
In this study, several viral contigs from dolphin and sea lion fecal samples aligned with viruses that were also isolated from human, mammal, seals, sea stars, bivalves, fish, birds and crayfish. These viruses include annelloviruses (AV), torque teno viruses (TTV), circoviruses and densoviruses. Multiple taxonomic sources aligned with the same viral contigs, which indicates that some viruses may infect multiple hosts (Tables 3 4). These results suggest that marine mammals can be used as important sentinel species (Bossart, 2006, 2011) to monitor marine environments for viruses that may spillover to different organisms.
First discovered in a serum samples from Hepatitis B (HBV) and Hepatitis C (HCV) patients (Khudair et al., 2019), TTV are single-stranded circular DNA (ssDNA) viruses, with a ~ 3.8 kb genome size and are currently classified into the Anelloviridae family under the genus Alphatorquevirus (Lolomadze and Rebrikov, 2020). TTV are known to be a diverse viral group with over 20 genotypes and 40% viral genome heterogeneity with frequent recombination in the N22 region of the open reading frame (ORF) 1 gene (Manni et al., 2002; Hino and Miyata, 2007; Hsiao et al., 2021). The TTV-like mini virus (TTMV) was added to the TTV group in 2000 (Takahashi et al., 2000) and has a genome size of ~2.9 kb (Hino and Miyata, 2007). Torque teno midi virus (TTMDV) is generally considered a non-pathogenic virus, and is commonly found in the virome of human blood (Cebriá-Mendoza et al., 2021). TTV have also been proposed as a fecal viral indicator for monitoring water quality (Griffin et al., 2008; Hamza et al., 2011; Haramoto et al., 2018; Tavakoli Nick et al., 2019). Viral contigs detected in dolphin fecal samples aligned with TTV’s from human (32% aa identity), South American fur seal (Arctocephalus australis) (79%–84% nt identities), and harbor seal (Phoca vitulina) (33% aa identity) sources. The South American fur seal torque teno arctecephalus australis virus 1 that aligned at 79% nt identity with the dolphin viral contig was detected in 2018 from seals found dead on the Rio Grande do Sul State shore in Brazil (Canova et al., 2021). The TTV that aligned with human could be indicative of a multi-host virus and possible sources from human wastewater or stormwater runoff. TTV are known to be highly resistant to wastewater treatment processes and are frequently detected in wastewater influent samples (Carducci et al., 2006; Haramoto et al., 2008; Plummer et al., 2014; Tavakoli Nick et al., 2019).
The Circoviridae family are circular ssDNA viruses with genome sizes of 1.7–2.1 kb and are composed of two genera Circovirus and Cyclovirus (Breitbart et al., 2017). Viral contigs from sea lion fecal samples aligned with viruses from the Circoviridae family of clams (Paphies subtriangulata) (35%–40.1% aa identity), red snappers (Lutjanus campechanus) (79%–97% nt identity) and rainbow trout (Oncorhynchus mykiss) (92% nt identity) sources. The Avon-Heathcote Estuary associated circular virus 28 from clams that aligned with a sea lion viral contig is from a group of single-stranded DNA (ssDNA) viruses encoding a replication-associated protein (Rep) (CRESS) viruses (Dayaram et al., 2015). Circoviridae species have unknown implications of disease but can cause infections in fish. In fish, circoviruses have been associated with skin and fin infections, called cauliflower disease (Doszpoly et al., 2014).
The Parvoviridae family are non-enveloped ssDNA viruses with linear genomes of 4–6 kb, and are split up into two subfamilies Parvovirinae and Densovirinae (Cotmore et al., 2019). The subfamily, Densovirinae (commonly referred to as densoviruses) infect insects and invertebrates, notably decapod crustaceans (shrimp and crayfish) (Jackson et al., 2020) Viral contigs from dolphin and sea lion fecal samples aligned with sea star associated densovirus (SSaDV). SSaDV is associated with sea star wasting disease (SSWD) that caused mass mortality events in >20 species of asteroids since 2013 from Alaska to Southern California (Hewson et al., 2014, 2018). SSaDV from dolphin fecal samples aligned with SSaDV from the North Pacific sea star (46%–59% aa identity; 79% nt identitiy). Viral contigs from sea lion fecal samples aligned with diverse densoviruses from comb sea star (Astropcten polyacanthus) (80% nt identity), purple sea star (Piesaster ochraceus) (77% nt identity), sunflower sea star (75% nt identity), and starfish (Pyloric caeca) (74% nt identity) sources. Sea stars are considered keystone species that play a role in regulating other species, such as bivalves, snails and other invertebrates in marine ecosystems (Paine, 1966; Collinge et al., 2008; Menge and Sanford, 2013). Viral contigs from sea lion fecal samples aligned with Cherax quadricarinatus densovirus (CqDV) from the Australian red claw crayfish (Cherax quadricarinatus) (35.3%–79.6% aa identity). CqDV has been linked to mortalities in red claw crayfish, which is a significant threat to the aquaculture industry (Saoud et al., 2013; Bochow et al., 2015). In addition, crayfish has an important ecological role in maintaining water quality (Chen et al., 2022), economic importance by providing food and jobs (McClain and Romaire, 2004), cultural importance with festivals (Gutierrez, 1998), and scientific importance for studying neurobiology, behavior and genetics (Huber et al., 2011; Jiang et al., 2014; Bacqué-Cazenave et al., 2017; D’Agnese et al., 2020). Viral contigs from dolphin fecal samples aligned with AmDNV from the Suminoe oyster (Crassostrea ariakensis) (76.7% nt identity). AmDNV was first isolated in 2017 from the Suminoe oysters (Crassostrea ariakensis) and shared similar amino acid identities with SSaDV (76–89% nt identity). But AmDNV has not been documented to cause mortality in oysters, instead most likely originated from the surrounding ocean ecosystem (Kang et al., 2017). Viral contigs from sea lion fecal samples aligned with AmDNV from Suminoe oysters (76%–88% nt identity) from Wuxi City, Jiangsu province, China (Kang et al., 2017). Another viral contig from sea lion fecal samples aligned with a AmDNV from a mute swan (Cygnus olor) (84% nt identity) and is not associated with disease in this bird species and more research is needed to understand the potential impact on bird health.
The use of filtration and DNase treatment could potentially reduce viral concentrations and diversity due to losses during these sample processing steps. DNase is an enzyme that breaks down extracellular “naked” or “free” DNA that are present in fecal and serum samples. In theory, intact viral particles should be resistant to DNase treatment through their protective protein coat such as a capsid and/or envelope, but some DNA viruses may be sensitive to DNase treatments. Studies have shown decreasing viral particles between samples with and without DNase treatment (Bettarel et al., 2000; Briese et al., 2015). Future studies could incorporate the use of biotinylated oligonucleotide probes (targeted viral enrichment) to capture viruses from complex matrices after sequencing library preparation to avoid upstream enzymatic treatments (Briese et al., 2015; Martínez-Puchol et al., 2020, 2022; Bonny et al., 2021). Although several studies use filtration (Allen et al., 2011; Fontenele et al., 2019; Garcia-Heredia et al., 2021; Patterson et al., 2021; Crum et al., 2023; LaRocca et al., 2023) to reduce background host and bacterial nucleic acids, it too can cause losses of viruses in filtration.
Although the third generation nanopore sequencing produces longer sequencing reads, it can be prone to higher error rates and low-quality scores, compared to short-read sequencing. The use of polishing after de novo assembly for error correction of substitutions, insertions and deletions has proved to increase assembly accuracy (Huang et al., 2021; Lee et al., 2021; Liu et al., 2022). Medaka polisher has been shown to decrease error rates after assemblies with MetaFlye or Canu (Goldsmith et al., 2020, 2021; Brancaccio et al., 2021; Wick et al., 2021). Future studies could include different combinations or rounds of polishing with Medaka or a combination of multiple types of polishers to enhance genome assembly accuracy. Furthermore, deeper long-read nanopore sequencing flow-cells such as the ONT PromethION (Yahara et al., 2021) with the updated Dorado4 (Pugh, 2023) base-caller and flow-cell chemistries that can reach quality scores of ~20 could be used in future studies to increase sequencing reads. In this study, different versions of the MinKNOW and Guppy basecaller software were used, due to frequent updates of the software packages by ONT. The different software updates would not affect the results of this study, because the same sequence files were used to compare Canu and MetaFlye assemblies. However, this may produce varying basecalling and/or read depth results when sequencing of viruses in environmental samples.
In this study, long-read metagenomics sequencing using a portable nanopore MinION sequencer allowed the detection of viruses in fecal and serum samples of marine mammals that are known to cause diseases in vertebrate and invertebrate hosts. This suggests that marine mammals could be used as sentinel species of ocean and human health by monitoring for emerging pathogens under a One Health framework. This sequencing method coupled with the sequence assembler, Canu identified Parvoviridae, Annelloviridae and Circoviridae that were confirmed with the NCBI viral protein (BLASTx) and nucleotide (BLASTn) Refseq databases. The data analysis approach presented here will be useful for virus surveillance using a long-read metagenomics sequencing.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI—PRJNA998092.
Samples from Navy animals were collected during their routine care and under the authority codified in U.S. Code, Title 10, Section 7524. Secretary of Navy Instruction 3900.41H directs that Navy marine mammals be provided the highest quality of care. The U.S. Navy Marine Mammal Program (MMP), Naval Information Warfare Center (NIWC) Pacific, houses, and cares for a population of bottlenose dolphins and California sea lions in San Diego Bay (CA, United States). NIWC Pacific is accredited by The Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International and adheres to the national standards of the U.S. Public Health Service policy on the Humane Care and Use of Laboratory Animals and the Animal Welfare Act. The study was conducted in accordance with the local legislation and institutional requirements.
KV: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, visualization. TA: conceptualization, resources, writing—review and editing, supervision, project administration, funding acquisition. All authors contributed to the article and approved the submitted version.
This research was funded by the Office of Naval Research (ONR), U.S. Department of the Navy under grant no. N00014-20-1-2117.
This research was supported in part using high performance computing (HPC) resources and services provided by Information Technology at Tulane University, New Orleans, LA. We would like to thank Carl Baribault for his bioinformatics expertise throughout this project and troubleshooting different in silico pipelines.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1248323/full#supplementary-material
1. ^ https://github.com/nanoporetech/medaka
2. ^ https://github.com/marbl/canu
Adewale, B. A. (2020). Will long-read sequencing technologies replace short-read sequencing technologies in the next 10 years? Afr. J. Lab. Med. 9, 1–5. doi: 10.4102/ajlm.v9i1.1340
Allen, L. Z., Ishoey, T., Novotny, M. A., McLean, J. S., Lasken, R. S., and Williamson, S. J. (2011). Single virus genomics: a new tool for virus discovery. PLoS One 6:e17722. doi: 10.1371/journal.pone.0017722
Arya, P., Metagenomics based approach to reveal the secrets of unculturable microbial diversity from aquatic environment, in S. MandalDe and P. Bhatt, eds., Recent advancements in microbial diversity. (2020), Amsterdam: Elsevier. p. 537–559.
Bacqué-Cazenave, J., Cattaert, D., Delbecque, J. P., and Fossat, P. (2017). Social harassment induces anxiety-like behaviour in crayfish. Sci. Rep. 7:39935. doi: 10.1038/srep39935
Bettarel, Y., Sime-Ngando, T., Amblard, C., and Laveran, H. (2000). A comparison of methods for counting viruses in aquatic systems. Appl. Environ. Microbiol. 66, 2283–2289. doi: 10.1128/AEM.66.6.2283-2289.2000
Bieringer, M., Han, J. W., Kendl, S., Khosravi, M., Plattet, P., and Schneider-Schaulies, J. (2013). Experimental adaptation of wild-type canine distemper virus (CDV) to the human entry receptor CD150. PLoS One 8:e57488. doi: 10.1371/journal.pone.0057488
Bochow, S., Condon, K., Elliman, J., and Owens, L. (2015). First complete genome of an Ambidensovirus; Cherax quadricarinatus densovirus, from freshwater crayfish Cherax quadricarinatus. Mar. Genomics 24, 305–312. doi: 10.1016/j.margen.2015.07.009
Bogomolni, A. L., Bass, A. L., Fire, S., Jasperse, L., Levin, M., Nielsen, O., et al. (2016). Saxitoxin increases phocine distemper virus replication upon in-vitro infection in harbor seal immune cells. Harmful Algae 51, 89–96. doi: 10.1016/j.hal.2015.10.013
Bonny, P., Schaeffer, J., Besnard, A., Desdouits, M., Ngang, J. J. E., and le Guyader, F. S. (2021). Human and animal RNA virus diversity detected by metagenomics in Cameroonian clams. Front. Microbiol. 12:770385. doi: 10.3389/fmicb.2021.770385
Bossart, G. D. (2006). Marine mammals as sentinel species for oceans and human health. Oceanography 19, 134–137. doi: 10.5670/oceanog.2006.77
Bossart, G. D. (2011). Marine mammals as sentinel species for oceans and human health. Vet. Pathol. 48, 676–690. doi: 10.1177/0300985810388525
Brancaccio, R. N., Robitaille, A., Dutta, S., Rollison, D. E., Tommasino, M., and Gheit, T. (2021). MinION nanopore sequencing and assembly of a complete human papillomavirus genome. J. Virol. Methods 294:114180. doi: 10.1016/j.jviromet.2021.114180
Breitbart, M., Delwart, E., Rosario, K., Segalés, J., and Varsani, A., ICTV Report Consortium (2017). ICTV virus taxonomy profile: circoviridae. J. Gen. Virol. 98, 1997–1998. doi: 10.1099/jgv.0.000871
Breitbart, M., Salamon, P., Andresen, B., Mahaffy, J. M., Segall, A. M., Mead, D., et al. (2002). Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. 99, 14250–14255. doi: 10.1073/pnas.202488399
Briese, T., Kapoor, A., Mishra, N., Jain, K., Kumar, A., Jabado, O. J., et al. (2015). Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. MBio 6, e01491–e01415. doi: 10.1128/mBio.01491-15
Brown, E., Freimanis, G., Shaw, A. E., Horton, D. L., Gubbins, S., and King, D. (2021). Characterising foot-and-mouth disease virus in clinical samples using nanopore sequencing. Front. Vet. Sci. 8:656256. doi: 10.3389/fvets.2021.656256
Burek, K. A., Gulland, F. M., and O'Hara, T. M. (2008). Effects of climate change on Arctic marine mammal health. Ecol. Appl. 18, S126–S134. doi: 10.1890/06-0553.1
Canova, R., Budaszewski, R. F., Weber, M. N., da Silva, M. S., Puhl, D. E., Battisti, L. O., et al. (2021). Spleen and lung virome analysis of South American fur seals (Arctocephalus australis) collected on the southern Brazilian coast. Infect. Genet. Evol. 92:104862. doi: 10.1016/j.meegid.2021.104862
Carbo, E. C., Sidorov, I. A., Zevenhoven-Dobbe, J. C., Snijder, E. J., Claas, E. C., Laros, J. F. J., et al. (2020). Coronavirus discovery by metagenomic sequencing: a tool for pandemic preparedness. J. Clin. Virol. 131:104594. doi: 10.1016/j.jcv.2020.104594
Carducci, A., Verani, M., Battistini, R., Pizzi, F., Rovini, E., Andreoli, E., et al. (2006). Epidemiological surveillance of human enteric viruses by monitoring of different environmental matrices. Water Sci. Technol. 54, 239–244. doi: 10.2166/wst.2006.475
Castinel, A., Duignan, P. J., Pomroy, W. E., López-Villalobos, N., Gibbs, N. J., Chilvers, B. L., et al. (2007). Neonatal mortality in New Zealand sea lions (Phocarctos hookeri) at Sandy Bay, Enderby Island, Auckland islands from 1998 to 2005. J. Wildl. Dis. 43, 461–474. doi: 10.7589/0090-3558-43.3.461
Cebriá-Mendoza, M., Bracho, M. A., Arbona, C., Larrea, L., Díaz, W., Sanjuán, R., et al. (2021). Exploring the diversity of the human blood virome. Viruses 13:2322. doi: 10.3390/v13112322
Chen, L., Xu, J., Wan, W., Xu, Z., Hu, R., Zhang, Y., et al. (2022). The microbiome structure of a rice-crayfish integrated breeding model and its association with crayfish growth and water quality. Microbiol. Spect. 10, e02204–e02221. doi: 10.1128/spectrum.02204-21
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Chin, C.-S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
Chiu, C. Y. (2013). Viral pathogen discovery. Curr. Opin. Microbiol. 16, 468–478. doi: 10.1016/j.mib.2013.05.001
Clark, C., McIntyre, P. G., Evans, A., McInnes, C. J., and Lewis-Jones, S. (2005). Human sealpox resulting from a seal bite: confirmation that sealpox virus is zoonotic. Br. J. Dermatol. 152, 791–793. doi: 10.1111/j.1365-2133.2005.06451.x
Cohen, J. M., Sauer, E. L., Santiago, O., Spencer, S., and Rohr, J. R. (2020). Divergent impacts of warming weather on wildlife disease risk across climates. Science 370:eabb1702. doi: 10.1126/science.abb1702
Collinge, S. K., Ray, C., and Cully, J. (2008). Effects of disease on keystone species, dominant species, and their communities. eds. R. Ostfeld, F. Keesing, and V. Eviner. Infect. Dis. Ecol., 129–144. doi: 10.1515/9781400837885.129
Compeau, P. E., Pevzner, P. A., and Tesler, G. (2011). How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 29, 987–991. doi: 10.1038/nbt.2023
Cotmore, S. F., Agbandje-McKenna, M., Canuti, M., Chiorini, J. A., Eis-Hubinger, A. M., Hughes, J., et al. (2019). ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 100, 367–368. doi: 10.1099/jgv.0.001212
Crum, E., Merchant, Z., Ene, A., Miller-Ensminger, T., Johnson, G., Wolfe, A. J., et al. (2023). Coliphages of the human urinary microbiota. PLoS One 18:e0283930. doi: 10.1371/journal.pone.0283930
D’Agnese, E., Lambourn, D., Rice, J., Duffield, D., Huggins, J., Spraker, T., et al. (2020). Reemergence of Guadalupe fur seals in the U.S. Pacific northwest: the epidemiology of stranding events during 2005–2016. Mar. Mamm. Sci. 36, 828–845. doi: 10.1111/mms.12678
Dayaram, A., Goldstien, S., Argüello-Astorga, G. R., Zawar-Reza, P., Gomez, C., Harding, J. S., et al. (2015). Diverse small circular DNA viruses circulating amongst estuarine molluscs. Infect. Genet. Evol. 31, 284–295. doi: 10.1016/j.meegid.2015.02.010
Dida, F., and Yi, G. (2021). Empirical evaluation of methods for de novo genome assembly. PeerJ Comput. Sci. 7:e636. doi: 10.7717/peerj-cs.636
Dietz, R., Heide-Jørgensen, M. P., and Härkönen, T. (1989). Mass deaths of harbor seals (Phoca vitulina) in Europe. Ambio (Sweden) 18, 258–264.
Doszpoly, A., Tarján, Z. L., Glávits, R., Müller, T., and Benkő, M. (2014). Full genome sequence of a novel circo-like virus detected in an adult European eel Anguilla anguilla showing signs of cauliflower disease. Dis. Aquat. Org. 109, 107–115. doi: 10.3354/dao02730
Duignan, P. J., van Bressem, M. F., Baker, J., Barbieri, M., Colegrove, K., de Guise, S., et al. (2014). Phocine distemper virus: current knowledge and future directions. Viruses 6, 5093–5134. doi: 10.3390/v6125093
Fire, S. E., Browning, J. A., Durden, W. N., and Stolen, M. K. (2020). Comparison of during-bloom and inter-bloom brevetoxin and saxitoxin concentrations in Indian river lagoon bottlenose dolphins, 2002–2011. Aquat. Toxicol. 218:105371. doi: 10.1016/j.aquatox.2019.105371
Flewelling, L. J., Naar, J. P., Abbott, J. P., Baden, D. G., Barros, N. B., Bossart, G. D., et al. (2005). Red tides and marine mammal mortalities. Nature 435, 755–756. doi: 10.1038/nature435755a
Fontenele, R. S., Lacorte, C., Lamas, N. S., Schmidlin, K., Varsani, A., and Ribeiro, S. G. (2019). Single stranded DNA viruses associated with capybara faeces sampled in Brazil. Viruses 11:710. doi: 10.3390/v11080710
Garalde, D. R., Snell, E. A., Jachimowicz, D., Sipos, B., Lloyd, J. H., Bruce, M., et al. (2018). Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206. doi: 10.1038/nmeth.4577
Garcia-Heredia, I., Bhattacharjee, A. S., Fornas, O., Gomez, M. L., Martínez, J. M., and Martinez-Garcia, M. (2021). Benchmarking of single-virus genomics: a new tool for uncovering the virosphere. Environ. Microbiol. 23, 1584–1593. doi: 10.1111/1462-2920.15375
Goldsmith, C., Cohen, D., Duboi, A., Martinez, M.-G., Petitjean, K., Corlu, A., et al. (2020). Long read sequencing and de novo assembly of hepatitis B virus identifies 5mCpG in CpG islands. bioRxiv. doi: 10.1101/2020.05.29.122259
Goldsmith, C., Cohen, D., Dubois, A., Martinez, M. G., Petitjean, K., Corlu, A., et al. (2021). Cas9-targeted nanopore sequencing reveals epigenetic heterogeneity after de novo assembly of native full-length hepatitis B virus genomes. Microbial. Genomics 7:000507. doi: 10.1099/mgen.0.000507
Griffin, J. S., Plummer, J. D., and Long, S. C. (2008). Torque Teno virus: an improved indicator for viral pathogens in drinking waters. Virol. J. 5:112. doi: 10.1186/1743-422X-5-112
Gulland, F. M., (2000) Domoic acid toxicity in California Sea lions (Zalophus Californianus) stranded along the Central California coast, may-October 1998: Report to the National Marine Fisheries Service Working Group on unusual marine mammal mortality events. Vol. 1. US Department of Commerce, National Oceanic and Atmospheric Administration
Gutierrez, C. P. (1998). “Cajuns and crawfish” in The taste of American place: a reader on regional and ethnic foods. eds. B. G. Shortridge and J. R. Shortridge (Lanham, MD: Rowman & Littlefield), 139–144.
Hamza, I. A., Jurzik, L., Überla, K., and Wilhelm, M. (2011). Evaluation of pepper mild mottle virus, human picobirnavirus and torque Teno virus as indicators of fecal contamination in river water. Water Res. 45, 1358–1368. doi: 10.1016/j.watres.2010.10.021
Haramoto, E., Katayama, H., and Ohgaki, S. (2008). Quantification and genotyping of torque Teno virus at a wastewater treatment plant in Japan. Appl. Environ. Microbiol. 74, 7434–7436. doi: 10.1128/AEM.01605-08
Haramoto, E., Kitajima, M., Hata, A., Torrey, J. R., Masago, Y., Sano, D., et al. (2018). A review on recent progress in the detection methods and prevalence of human enteric viruses in water. Water Res. 135, 168–186. doi: 10.1016/j.watres.2018.02.004
Hayashida, K., Orba, Y., Sequeira, P. C., Sugimoto, C., Hall, W. W., Eshita, Y., et al. (2019). Field diagnosis and genotyping of chikungunya virus using a dried reverse transcription loop-mediated isothermal amplification (LAMP) assay and MinION sequencing. PLoS Negl. Trop. Dis. 13:e0007480. doi: 10.1371/journal.pntd.0007480
Hendrix, A. M., Lefebvre, K. A., Quakenbush, L., Bryan, A., Stimmelmayr, R., Sheffield, G., et al. (2021). Ice seals as sentinels for algal toxin presence in the Pacific Arctic and subarctic marine ecosystems. Mar. Mamm. Sci. 37, 1292–1308. doi: 10.1111/mms.12822
Hewson, I., Bistolas, K. S. I., Quijano Cardé, E. M., Button, J. B., Foster, P. J., Flanzenbaum, J. M., et al. (2018). Investigating the complex association between viral ecology, environment, and Northeast Pacific Sea star wasting. Front. Mar. Sci. 5:77. doi: 10.3389/fmars.2018.00077
Hewson, I., Button, J. B., Gudenkauf, B. M., Miner, B., Newton, A. L., Gaydos, J. K., et al. (2014). Densovirus associated with sea-star wasting disease and mass mortality. Proc. Natl. Acad. Sci. 111, 17278–17283. doi: 10.1073/pnas.1416625111
Hino, S., and Miyata, H. (2007). Torque teno virus (TTV): current status. Rev. Med. Virol. 17, 45–57. doi: 10.1002/rmv.524
Hsiao, K.-L., Wang, L. Y., Cheng, J. C., Cheng, Y. J., Lin, C. L., and Liu, H. F. (2021). Detection and genetic characterization of the novel torque Teno virus group 6 in Taiwanese general population. R. Soc. Open Sci. 8:210938. doi: 10.1098/rsos.210938
Hu, T., Chitnis, N., Monos, D., and Dinh, A. (2021). Next-generation sequencing technologies: an overview. Hum. Immunol. 82, 801–811. doi: 10.1016/j.humimm.2021.02.012
Huang, Y.-T., Liu, P.-Y., and Shih, P.-W. (2021). Homopolish: a method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 22, 1–17. doi: 10.1186/s13059-021-02282-6
Huber, R., Panksepp, J. B., Nathaniel, T., Alcaro, A., and Panksepp, J. (2011). Drug-sensitive reward in crayfish: an invertebrate model system for the study of SEEKING, reward, addiction, and withdrawal. Neurosci. Biobehav. Rev. 35, 1847–1853. doi: 10.1016/j.neubiorev.2010.12.008
Idury, R. M., and Waterman, M. S. (1995). A new algorithm for DNA sequence assembly. J. Comput. Biol. 2, 291–306. doi: 10.1089/cmb.1995.2.291
Jackson, E. W., Wilhelm, R. C., Johnson, M. R., Lutz, H. L., Danforth, I., Gaydos, J. K., et al. (2020). Diversity of sea star-associated densoviruses and transcribed endogenous viral elements of densovirus origin. J. Virol. 95, e01594–e01520. doi: 10.1128/JVI.01594-20
Jiang, H., Xing, Z., Lu, W., Qian, Z., Yu, H., and Li, J. (2014). Transcriptome analysis of red swamp crawfish Procambarus clarkii reveals genes involved in gonadal development. PLoS One 9:e105122. doi: 10.1371/journal.pone.0105122
Jung, H., Jeon, M. S., Hodgett, M., Waterhouse, P., and Eyun, S. I. (2020). Comparative evaluation of genome assemblers from long-read sequencing for plants and crops. J. Agric. Food Chem. 68, 7670–7677. doi: 10.1021/acs.jafc.0c01647
Kamath, G. M., Shomorony, I., Xia, F., Courtade, T. A., and Tse, D. N. (2017). HINGE: long-read assembly achieves optimal repeat resolution. Genome Res. 27, 747–756. doi: 10.1101/gr.216465.116
Kang, Y.-J., Huang, W., Zhao, A. L., Lai, D. D., Shao, L., Shen, Y. Q., et al. (2017). Densoviruses in oyster Crassostrea ariakensis. Arch. Virol. 162, 2153–2157. doi: 10.1007/s00705-017-3343-z
Khudair, E. A., Al-Shuwaikh, A. M., and Farhan, N. M. (2019). Detection of TTV antigen in patients with hepatitis HBV and HCV. Iraqi J. Med. Sci. 17, 43–49. doi: 10.22578/IJMS.17.1.7
Kolmogorov, M., Bickhart, D. M., Behsaz, B., Gurevich, A., Rayko, M., Shin, S. B., et al. (2020). metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110. doi: 10.1038/s41592-020-00971-x
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kono, N., and Arakawa, K. (2019). Nanopore sequencing: review of potential applications in functional genomics. Develop. Growth Differ. 61, 316–326. doi: 10.1111/dgd.12608
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
LaRocca, C. J., Jacobsen, K. L., Inoko, K., Zakharkin, S. O., Yamamoto, M., and Davydova, J. (2023). Viral shedding in mice following intravenous adenovirus injection: impact on biosafety classification. Viruses 15:1495. doi: 10.3390/v15071495
Lee, J. Y., Kong, M., Oh, J., Lim, J. S., Chung, S. H., Kim, J. M., et al. (2021). Comparative evaluation of nanopore polishing tools for microbial genome assembly and polishing strategies for downstream analysis. Sci. Rep. 11:20740. doi: 10.1038/s41598-021-00178-w
Lefebvre, K. A., Powell, C. L., Busman, M., Doucette, G. J., Moeller, P. D. R., Silver, J. B., et al. (1999). Detection of domoic acid in northern anchovies and California sea lions associated with an unusual mortality event. Nat. Toxins 7, 85–92. doi: 10.1002/(SICI)1522-7189(199905/06)7:3<85::AID-NT39>3.0.CO;2-Q
Lefebvre, K. A., Quakenbush, L., Frame, E., Huntington, K. B., Sheffield, G., Stimmelmayr, R., et al. (2016). Prevalence of algal toxins in Alaskan marine mammals foraging in a changing arctic and subarctic environment. Harmful Algae 55, 13–24. doi: 10.1016/j.hal.2016.01.007
Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. doi: 10.1093/bioinformatics/btw152
Li, Z., Chen, Y., Mu, D., Yuan, J., Shi, Y., Zhang, H., et al. (2012). Comparison of the two major classes of assembly algorithms: overlap–layout–consensus and de-Bruijn-graph. Brief. Funct. Genomics 11, 25–37. doi: 10.1093/bfgp/elr035
Liao, X., Li, M., Zou, Y., Wu, F. X., Yi-Pan, and Wang, J. (2019). Current challenges and solutions of de novo assembly. Quant. Biol. 7, 90–109. doi: 10.1007/s40484-019-0166-9
Liu, L., Yang, Y., Deng, Y., and Zhang, T. (2022). Nanopore long-read-only metagenomics enables complete and high-quality genome reconstruction from mock and complex metagenomes. Microbiome 10:209. doi: 10.1186/s40168-022-01415-8
Lolomadze, E. A., and Rebrikov, D. V. (2020). Constant companion: clinical and developmental aspects of torque Teno virus infections. Arch. Virol. 165, 2749–2757. doi: 10.1007/s00705-020-04841-x
Manni, F., Rotola, A., Caselli, E., Bertorelle, G., and di Luca, D. (2002). Detecting recombination in TT virus: a phylogenetic approach. J. Mol. Evol. 55, 563–572. doi: 10.1007/s00239-002-2352-y
Martínez-Puchol, S., Cardona, L., Drago, M., Gazo, M., and Bofill-Mas, S. (2022). Viral metagenomics reveals persistent as well as dietary acquired viruses in Antarctic fur seals. Sci. Rep. 12:18207. doi: 10.1038/s41598-022-23114-y
Martínez-Puchol, S., Rusiñol, M., Fernández-Cassi, X., Timoneda, N., Itarte, M., Andrés, C., et al. (2020). Characterisation of the sewage virome: comparison of NGS tools and occurrence of significant pathogens. Sci. Total Environ. 713:136604. doi: 10.1016/j.scitotenv.2020.136604
McClain, W. R., and Romaire, R. P. (2004). Crawfish culture: a Louisiana aquaculture success story. World Aquac. 35, 31–35.
Menge, B. A., and Sanford, E. (2013). “Ecological role of sea stars from populations” in Starfish: Biology and ecology of the Asteroidea. ed. J. M. Lawrence (Asteroidea: JHU Press), 67.
Miller, J. R., Delcher, A. L., Koren, S., Venter, E., Walenz, B. P., Brownley, A., et al. (2008). Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24, 2818–2824. doi: 10.1093/bioinformatics/btn548
Munang'andu, H., Mugimba, K. K., Byarugaba, D. K., Mutoloki, S., and Evensen, Ø. (2017). Current advances on virus discovery and diagnostic role of viral metagenomics in aquatic organisms. Front. Microbiol. 8:406. doi: 10.3389/fmicb.2017.00406
Myers, E. W., Sutton, G. G., Delcher, A. L., Dew, I. M., Fasulo, D. P., Flanigan, M. J., et al. (2000). A whole-genome assembly of drosophila. Science 287, 2196–2204. doi: 10.1126/science.287.5461.2196
Neill, J. D., Meyer, R. F., and Seal, B. S. (1995). Genetic relatedness of the caliciviruses: San Miguel sea lion and vesicular exanthema of swine viruses constitute a single genotype within the Caliciviridae. J. Virol. 69, 4484–4488. doi: 10.1128/jvi.69.7.4484-4488.1995
Noble, R. T., and Fuhrman, J. (1997). Virus decay and its causes in coastal waters. Appl. Environ. Microbiol. 63, 77–83. doi: 10.1128/aem.63.1.77-83.1997
Onens, P., Wilkin, S., Fauquier, D., Spradlin, T., Manley, S., Greig, D., et al. (2023). 2019 Report of marine mammal Strandings in the United States: National Overview.
Paine, R. T. (1966). Food web complexity and species diversity. Am. Nat. 100, 65–75. doi: 10.1086/282400
Patterson, Q. M., Kraberger, S., Martin, D. P., Shero, M. R., Beltran, R. S., Kirkham, A. L., et al. (2021). Circoviruses and cycloviruses identified in Weddell seal fecal samples from McMurdo Sound, Antarctica. Infect. Genet. Evol. 95:105070. doi: 10.1016/j.meegid.2021.105070
Pevzner, P. A., Tang, H., and Waterman, M. S. (2001). An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. 98, 9748–9753. doi: 10.1073/pnas.171285098
Plummer, J. D., Long, S. C., Liu, Z., and Charest, A. A. (2014). Torque Teno virus occurrence and relationship to bacterial and viral indicators in feces, wastewaters, and waters in the United States. Environ. Eng. Sci. 31, 671–680. doi: 10.1089/ees.2014.0091
Pomeroy, P., Hammond, J. A., Hall, A. J., Lonergan, M., Duck, C. D., Smith, V. J., et al. (2005). Morbillivirus neutralising antibodies in Scottish grey seals Halichoerus grypus: assessing the effects of the 1988 and 2002 PDV epizootics. Mar. Ecol. Prog. Ser. 287, 241–250. doi: 10.3354/meps287241
Pop, M., and Salzberg, S. L. (2008). Bioinformatics challenges of new sequencing technology. Trends Genet. 24, 142–149. doi: 10.1016/j.tig.2007.12.006
Pugh, J. (2023). The current state of nanopore sequencing. Methods Mol. Biol. 2632, 3–14. doi: 10.1007/978-1-0716-2996-3_1
Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith, A. D., Gangavarapu, K., et al. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276. doi: 10.1038/nprot.2017.066
Roach, M. J., Beecroft, S. J., Mihindukulasuriya, K. A., Wang, L., Paredes, A., Henry-Cocks, K., et al. (2022). Hecatomb: an end-to-end research platform for viral metagenomics. bio Rxiv :2022.05.15.492003. doi: 10.1101/2022.05.15.492003
Roe, W. D., Rogers, L., Pinpimai, K., Dittmer, K., Marshall, J., and Chilvers, B. L. (2015). Septicaemia and meningitis caused by infection of New Zealand sea lion pups with a hypermucoviscous strain of Klebsiella pneumoniae. Vet. Microbiol. 176, 301–308. doi: 10.1016/j.vetmic.2015.01.019
Roess, A. A., Levine, R. S., Barth, L., Monroe, B. P., Carroll, D. S., Damon, I. K., et al. (2011). Sealpox virus in marine mammal rehabilitation facilities, North America, 2007-2009. Emerg. Infect. Dis. 17, 2203–2208. doi: 10.3201/eid1712.101945
Ruan, J., and Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158. doi: 10.1038/s41592-019-0669-3
Sakai, K., Yoshikawa, T., Seki, F., Fukushi, S., Tahara, M., Nagata, N., et al. (2013). Canine distemper virus associated with a lethal outbreak in monkeys can readily adapt to use human receptors. J. Virol. 87, 7170–7175. doi: 10.1128/JVI.03479-12
Sanderson, C. E., and Alexander, K. A. (2020). Unchartered waters: climate change likely to intensify infectious disease outbreaks causing mass mortality events in marine mammals. Glob. Chang. Biol. 26, 4284–4301. doi: 10.1111/gcb.15163
Saoud, I. P., Ghanawi, J., Thompson, K. R., and Webster, C. D. (2013). A review of the culture and diseases of redclaw crayfish Cherax quadricarinatus (von martens 1868). J. World Aquacult. Soc. 44, 1–29. doi: 10.1111/jwas.12011
Saranathan, R., Asare, E., Leung, L., de Oliveira, A. P., Kaugars, K. E., Mulholland, C. V., et al. (2022). Capturing structural variants of herpes simplex virus genome in full length by Oxford nanopore sequencing. Microbiol. Spectr. 10:e0228522. doi: 10.1128/spectrum.02285-22
Siebert, U., Rademaker, M., Ulrich, S. A., Wohlsein, P., Ronnenberg, K., and Prenger-Berninghoff, E. (2017). Bacterial microbiota in harbor seals (Phoca vitulina) from the North Sea of Schelswig-Holstein, Germany, around the time of morbillivirus and influenza epidemics. J. Wildl. Dis. 53, 201–214. doi: 10.7589/2015-11-320
Smith, A. W., Skilling, D. E., Cherry, N., Mead, J. H., and Matson, D. O. (1998). Calicivirus emergence from ocean reservoirs: zoonotic and interspecies movements. Emerg. Infect. Dis. 4, 13–20. doi: 10.3201/eid0401.980103
Takahashi, K., Iwasa, Y., Hijikata, M., and Mishiro, S. (2000). Identification of a new human DNA virus (TTV-like mini virus, TLMV) intermediately related to TT virus and chicken anemia virus. Arch. Virol. 145, 979–993. doi: 10.1007/s007050050689
Tavakoli Nick, S., Mohebbi, S. R., Hosseini, S. M., Mirjalali, H., and Alebouyeh, M. (2019). Occurrence and molecular characterization of torque Teno virus (TTV) in a wastewater treatment plant in Tehran. J. Water Health 17, 971–977. doi: 10.2166/wh.2019.137
Tørresen, O. K., Star, B., Mier, P., Andrade-Navarro, M. A., Bateman, A., Jarnot, P., et al. (2019). Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 47, 10994–11006. doi: 10.1093/nar/gkz841
Wang, J., Chen, K., Ren, Q., Zhang, Y., Liu, J., Wang, G., et al. (2021). Systematic comparison of the performances of de novo genome assemblers for oxford nanopore technology reads from piroplasm. Front. Cell. Infect. Microbiol. 11:696669. doi: 10.3389/fcimb.2021.696669
Wang, D., Coscoy, L., Zylberberg, M., Avila, P. C., Boushey, H. A., Ganem, D., et al. (2002). Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. U. S. A. 99, 15687–15692. doi: 10.1073/pnas.242579699
Wang, H., Marcišauskas, S., Sánchez, B. J., Domenzain, I., Hermansson, D., Agren, R., et al. (2018). RAVEN 2.0: a versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 14:e1006541. doi: 10.1371/journal.pcbi.1006541
Wang, D., Urisman, A., Liu, Y. T., Springer, M., Ksiazek, T. G., Erdman, D. D., et al. (2003). Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 1:e2:E2. doi: 10.1371/journal.pbio.0000002
Webster, R. G., Geraci, J., Petursson, G., and Skirnisson, K. (1981a). Conjunctivitis in human beings caused by influenza a virus of seals. N. Engl. J. Med. 304:911. doi: 10.1056/NEJM198104093041515
Webster, R. G., Hinshaw, V. S., Bean, W. J., van Wyke, K. L., Geraci, J. R., St. Aubin, D. J., et al. (1981b). Characterization of an influenza a virus from seals. Virology 113, 712–724. doi: 10.1016/0042-6822(81)90200-2
Wick, R. R., Judd, L. M., Cerdeira, L. T., Hawkey, J., Méric, G., Vezina, B., et al. (2021). Trycycler: consensus long-read assemblies for bacterial genomes. Genome Biol. 22, 1–17. doi: 10.1186/s13059-021-02483-z
Yahara, K., Suzuki, M., Hirabayashi, A., Suda, W., Hattori, M., Suzuki, Y., et al. (2021). Long-read metagenomics using PromethION uncovers oral bacteriophages and their interaction with host bacteria. Nat. Commun. 12:27. doi: 10.1038/s41467-020-20199-9
Yu, R., Cai, D., and Sun, Y. (2023). AccuVIR: an ACCUrate VIRal genome assembly tool for third-generation sequencing data. Bioinformatics 39:btac827. doi: 10.1093/bioinformatics/btac827
Keywords: nanopore sequencing, metagenomics, viruses, marine mammals, bioinformatics
Citation: Vigil K and Aw TG (2023) Comparison of de novo assembly using long-read shotgun metagenomic sequencing of viruses in fecal and serum samples from marine mammals. Front. Microbiol. 14:1248323. doi: 10.3389/fmicb.2023.1248323
Edited by:
Sara Louise Cosby, Queen's University Belfast, United KingdomReviewed by:
Massimiliano Orsini, Experimental Zooprophylactic Institute of the Venezie (IZSVe), ItalyCopyright © 2023 Vigil and Aw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiong Gim Aw, dGF3QHR1bGFuZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.