
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 31 August 2023
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1236208
 Edgar I. Campos-Madueno1,2
Edgar I. Campos-Madueno1,2 Claudia Aldeia1Vincent Perreten3Parham Sendi1Aline I. Moser1
Claudia Aldeia1Vincent Perreten3Parham Sendi1Aline I. Moser1 Andrea Endimiani1*
Andrea Endimiani1*We implemented culture- and shotgun metagenomic sequencing (SMS)-based methods to assess the gut colonization with extended-spectrum cephalosporin-resistant Enterobacterales (ESC-R-Ent) in 42 volunteers. Both methods were performed using native and pre-enriched (broth supplemented with cefuroxime) stools. Native culture screening on CHROMID® ESBL plates resulted in 17 positive samples, whereas the pre-enriched culture (gold-standard) identified 23 carriers. Overall, 26 ESC-R-Ent strains (24 Escherichia coli) were identified: 25 CTX-M and 3 DHA-1 producers (2 co-producing CTX-Ms). Using the SMS on native stool (“native SMS”) with thresholds ≥60% for both identity and coverage, only 7 of the 23 pre-enriched culture-positive samples resulted positive for blaCTX-M/blaDHA genes (native SMS reads mapping to blaCTX-M/blaDHAs identified in gold-standard: sensitivity, 59.0%; specificity 100%). Moreover, an average of 31.5 and 24.6 antimicrobial resistance genes (ARGs) were detected in the 23 pre-enriched culture-positive and the 19 negative samples, respectively. When the pre-enriched SMS was implemented, more blaCTX-M/blaDHA genes were detected than in the native assay, including in stools that were pre-enriched culture-negative (pre-enriched SMS reads mapping to blaCTX-M/blaDHAs identified in gold-standard: sensitivity, 78.3%; specificity 75.0%). In addition, the pre-enriched SMS identified on average 38.6 ARGs/sample, whereas for the corresponding native SMS it was 29.4 ARGs/sample. Notably, stools resulting false-negative by using the native SMS had lower concentrations of ESC-R-Ent (average: ~105 vs. ~107 CFU/g) and E. coli classified reads (average: 193,959 vs. 1.45 million) than those of native SMS positive samples. Finally, the detection of blaCTX-M/blaDHA genes was compared with two well-established bioinformatic tools. In conclusion, only the pre-enriched SMS assured detection of most carriers of ESC-R-Ent. However, its performance was not comparable to the pre-enriched culture-based approach.
The prevalence of people colonized at intestinal level with extended-spectrum cephalosporin-resistant Enterobacterales (ESC-R-Ent) has reached concerning levels (Bezabih et al., 2021). For instance, 21.1% of patients and 17.6% of healthy individuals worldwide carry extended-spectrum β-lactamase (ESBL)-producing Escherichia coli in their intestinal tract. Moreover, during the period 2001–2020, colonization rates increased 3-fold (from 7 to 25.7%) and 10-fold (from 2.6 to 26.4%) in health-care and community settings, respectively (Bezabih et al., 2022).
It is well-known that hospitalized individuals who carry at gut level ESC-R-Ent may contribute to the spread of these pathogens and, more importantly, can develop difficult-to-treat infections with high morbidity and mortality rates (Schwaber and Carmeli, 2007; Ling et al., 2021; Campos-Madueno et al., 2023). Conversely, data regarding the ESC-R-Ent intestinal colonization in healthy people is limited to prevalence surveys, mostly focusing on travelers returning from endemic regions (Bernasconi et al., 2016; Pires et al., 2016; Voor In 't Holt et al., 2020; Moser et al., 2021a,b). These individuals are also at risk of developing infections (e.g., urinary tract infections) and/or transmitting their ESC-R-Ent to people who are in close contact with them (e.g., in the household) (Valverde et al., 2008; Banerjee et al., 2013; Soraas et al., 2013; Tham et al., 2013; Arcilla et al., 2017). In this overall scenario, rapid and accurate recognition of ESC-R-Ent intestinal carriers represents an important public-health task. In fact, since current strategies to decolonize ESC-R-Ent carriers are not effective (Campos-Madueno et al., 2023), prompt implementation of countermeasures to prevent transmission of ESC-R-Ent are essential in both community and hospitals (e.g., infection prevention and control, IPC) (Tacconelli et al., 2014; Tschudin-Sutter et al., 2017).
For patients referring to the hospital setting, rapid and valid multiplex molecular screening assays for ESC-R-Ent may be implemented [e.g., those PCR-based, (Campos-Madueno et al., 2023)]. However, in most clinical microbiology laboratories, routine screening of the intestinal tract is still performed using culture-based methods; this is particularly true for individuals in the community (e.g., returning travelers) (Campos-Madueno et al., 2023). In these culture-based approaches, native stool or perianal/perirectal swab samples are directly plated on selective chromogenic media (Blane et al., 2016; Girlich et al., 2019). A broth pre-enrichment step (with or without antibiotics) may also be added before plating to increase the detection rate of ESC-R-Ent (Blane et al., 2016; Jazmati et al., 2016, 2017; Rondinaud et al., 2020; Gallah et al., 2023). Though, this approach is seldom implemented due to its increased hands-on time and turnaround time (TAT).
Recently, shotgun metagenomic sequencing (SMS) has been proposed as an alternative to culture-based methods with promising diagnostic capability (Ko et al., 2022; Purushothaman et al., 2022). However, most studies focusing on the detection of antimicrobial resistance genes (ARGs) have been conducted in the hospital setting (e.g., samples from patients with comorbidities) (Gigliucci et al., 2018; Mu et al., 2019; Leggett et al., 2020; Afridi et al., 2021; Yee et al., 2021). In contrast, few studies have directly analyzed the presence of ARGs in fecal samples of healthy people in the community by SMS (Bengtsson-Palme et al., 2015; Guernier-Cambert et al., 2021). In an Australian study, Guernier-Cambert et al. used the SMS to screen for ARGs in healthy women (n = 7) and infants (n = 2), identifying 64 unique ARGs shared across all samples. However, no culture-based methods were used to confirm the resistance phenotypes of the detected ARGs (Guernier-Cambert et al., 2021). In another analysis, Bengtsson-Palme et al. studied the resistome of 35 Swedish travelers by both culture and SMS methods finding an increased abundance of ARGs after travel. Nevertheless, ESBL encoding genes (i.e., blaCTX-Ms) were not detected by SMS, despite the fact that stool samples were culture-positive for CTX-M-producing E. coli strains (Bengtsson-Palme et al., 2015). In the UK, Dallman et al. used SMS to investigate the prevalence of ARGs and gut microbiota patterns in travelers (n = 50) at different time points (pre-travel, post-travel and follow-up). However, culture-based methods were not used to confirm the findings of the SMS analysis (Dallman et al., 2023).
In the present study, we simultaneously used culture-, PCR-, and SMS-based methods to assess the intestinal ESC-R-Ent colonization status of healthy individuals. In particular, we evaluated the ability of SMS as a potential tool to detect ESBL and plasmid-mediated AmpC (pAmpC) bla genes directly from the stool samples. Results were contextualized considering several variables such as pre-enrichment of the stool sample, gut microbiota pattern, concentrations of the colonizing ESC-R-Ent (if any), and detection threshold cutoffs.
Forty-two stool samples from volunteers were collected throughout 2021 and included in the present study. This analysis is part of an ongoing study on intestinal colonization of Swiss expats.1 Individuals living abroad for ≥3 months and who were ≥ 18 years old met the study inclusion criteria. At enrollment, volunteers received instructions, consent forms, shipping material and an epidemiological questionnaire. Volunteers were invited to ship their samples to the Institute for Infectious Diseases (IFIK; Bern, Switzerland) via ordinary post in collection vessels and transport containers within maximum 7 days from the return to Switzerland (e.g., to spend holidays). The study was approved by the ethical committee of the Canton Bern (ID: 2020-01683).
At arrival to the IFIK, stool samples were screened for ESC-R-Ent by both direct and pre-enriched culture-based methods (for simplicity referred hereafter as “native culture” and “pre-enriched culture,” respectively). First, a stool aliquot (~50–100 μg) was resuspended in 1 mL saline solution (sodium chloride, 0.85%), then an undiluted 100 μL aliquot was directly plated onto a CHROMID® ESBL (bioMérieux) agar plate and incubated overnight at 36 ± 1°C. This approach has an estimated limit of detection (LOD) for ESC-R-Ent of 1–2 101 colony forming units (CFU)/100 mg of stool (Naas et al., 2011).
In parallel to the native culture, a second stool aliquot (~50–100 μg) was pre-enriched for 6 h in 10 mL Luria-Bertani (LB) broth supplemented with cefuroxime (30 μg disk, BD BBL™ Sensi-Disc™) as previously done (Bernasconi et al., 2016; Budel et al., 2019; Moser et al., 2021a,b). The pre-enrichment culture was then diluted to 0.5 McFarland, plated (100 μL) onto a CHROMID® ESBL plate and incubated overnight at 36 ± 1°C. CHROMID® ESBL selective plates are referred henceforward as “ESBL-plates.”
ID for growing colonies (at least 1 on the ESBL-plates) was achieved using the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS; Bruker). ASTs were performed on broth microdilution with the Sensititre GNX2F panel (Thermo Fisher Scientific). Results were interpreted according to the 2022 EUCAST breakpoints (v12.0).2 Ent confirmed to be ESC-R (i.e., MICs >1 μg/mL for ceftriaxone, ceftazidime, cefotaxime, or cefepime) underwent whole-genome sequencing.
ESC-R-Ent carriage was determined directly from the native stool. CFU/g counts were performed in 10−1 serial dilutions in 1 mL saline sterile solution and plated (100 μL) onto ESBL-plates followed by overnight incubation at 36 ± 1°C. An average of 3 independent measurements was used for the final CFU/g calculation per stool sample.
Genomic DNA (gDNA) for WGS was extracted from the isolated ESC-R-Ent strains with the Invitrogen™ PureLink™ Microbiome DNA purification kit (ThermoFisher Scientific) as previously described (Campos-Madueno et al., 2021). Quality and quantity of the gDNA were assessed by Nanodrop™ and Qubit™ 3 (ThermoFisher Scientific). WGS was conducted using Illumina libraries prepared with the NEBNext® Ultra™ II DNA library prep kit on the NovaSeq 6000 platform (2 × 150 bp paired-end output) by Eurofins Genomics (Ebersberg, Germany). Briefly, raw reads were quality checked and trimmed with FastQC v0.11.93 and Trimmomatic v0.364 with default parameters, respectively. Draft assemblies were generated with Unicycler v0.4.85 using the trimmed Illumina paired-end reads as input.
gDNA isolation from native stool was performed as described in the section above, but following the protocol for fecal samples from the Invitrogen™ PureLink™ Microbiome DNA purification kit using ~200 mg of starting material. For the gDNA isolation from the pre-enriched stool, an 800 μL aliquot of the cefuroxime pre-enriched LB broth was used as starting material following the above protocol, but with an initial spin down step (10 min, 14,000 × g) to pellet the sample. Quality and quantity of stool and enriched gDNA isolations were assessed as described above.
SMS libraries for gDNA obtained from native and pre-enriched stool samples (referred hereafter as “native SMS” and “pre-enriched SMS,” respectively) were prepared with the NEBNext® Ultra™ II FS DNA Library Prep Kit and sequenced on a NovaSeq 6000 platform (2 × 150-bp paired-end output, minimum depth of raw reads, 10 million, M) by Eurofins Genomics. Downstream analyses were conducted as suggested by the general guidelines described by Quince et al. (2017). The subsequent SMS raw reads were quality checked with FastQC v0.11.9. Optical duplicates were removed with BBMap v38.186 using clumpify.sh with the parameters “dedupe optical dist = 12,000” followed by read trimming with Trimmomatic v0.36 set to minimum length of 60-bp. The trimmed reads were decontaminated from human reads with Kneaddata v0.10.07 using both GRCh37 (hg37dec) and GRCh38 (GRCh38_noalt_decoy_as) human genome assemblies with decoy sequences. Downstream SMS analyses were conducted using only decontaminated reads.
To simulate the minimum sequence depth required to detect blaCTX-M or blaDHA genes (see below), the decontaminated reads of each stool (i.e., the paired FASTQ files) were sub-sampled without replacement at 10, 20, 40, 60, and 80% using the 100, 150, 200, 250, and 300 seeds with Seqtk v1.3-r1068, respectively. Furthermore, the same dataset (i.e., subsampled reads) was used to generate the rarefaction curves targeting all ARGs.
A positive SMS result was defined when reads corresponding to blaCTX-M or blaDHA genes were mapped at ≥60% coverage and ≥ 60% identity, whereas a negative SMS result corresponded to less than <60% for both (see section below).
The draft assemblies generated from the WGS of culture strains (ESC-R-Ent) were screened for ARGs and multi-locus sequence type (MLST) using the ResFinder v4.1 and MLST v2.0 databases from the Center for Genomic Epidemiology (CGE)9 using default parameters. Additionally, species identification was confirmed with the TYGS server.10
SMS decontaminated reads were used to screen for ARGs using the command line version of ResFinder set to paired-end reads (coverage and identity thresholds set to 60%), which uses KMA v1.3.22 (CGE) to map reads to the ResFinder database by k-mer alignment (KMA) (Clausen et al., 2018). The ResFinder KMA pipeline was chosen to analyze the metagenomic reads because of its speed (i.e., in contrast to assembly-based methods) and good performance, making it an ideal candidate for fast screening tests (i.e., lower TATs) (Clausen et al., 2018). Perfect matches corresponded to ARG hits with 100% coverage and identity, whereas partial matches (indicated as “-like”) corresponded to ARG hits thresholds ≥60% but <100% for both coverage and identity.
Read counts, mapping depth and coverage statistics were calculated automatically by the output of the KMA software. Bar plots and heatmap visualizations were generated with R v4.1.2 (2021-11-01)11 using “ggplot2” v3.3.6 and “pheatmap” v1.0.12 packages.
A secondary screen to support the detection of blaCTX-M and blaDHA genes directly from reads using the ResFinder pipeline was performed using the Comprehensive Antibiotic Resistance Database (CARD) Resistance Gene Identifier (RGI)12 command line version to screen metagenomic short reads (bwt) with KMA as the default read aligner (RGI v5.2.0; CARD database v3.2.7).
Decontaminated reads from all native and pre-enriched SMS samples were classified at the species level using exact k-mer matches with Kraken2 v2.1.1 (NCBI taxonomic database build on: 03/28/2022)13 with default parameters. The Kraken2 classified reads were further processed with Bracken v2.6.1 (Bracken-build: 150-bp read length and 35 k-mer length)14 to estimate relative abundance at the species level (Lu et al., 2022). Individual Bracken output files were merged with the “combine_bracken_outputs.py” python script from the Bracken software. The merged Bracken data was processed with the “phyloseq” v1.38.0 R package to determine and visualize the top 10 bacterial species relative abundance in all SMS samples. The Bracken classified reads corresponding to E. coli, Klebsiella pneumoniae, and Citrobacter freundii were extracted with “phyloseq.”
Screening of blaCTX-M and blaDHA genes was also carried out by standard PCR (FastStart™ Taq-DNA-Polymerase; Roche) using gDNA isolated from native and pre-enriched stool samples (referred hereafter as “native PCR” and “pre-enriched PCR,” respectively). In particular, stools that carried culture strains possessing blaCTX-M and/or blaDHA were screened (30 cycles) with previously reported primers (Perez-Perez and Hanson, 2002; Pitout et al., 2004). According to the intensity of the bands in the agarose gels, positive PCRs were visually defined as strong (+++), intermediate (++), and weak (+).
Sensitivity and specificity for the detection of blaCTX-M and/or blaDHA genes with native and pre-enriched SMS (positive SMS) were calculated using as gold-standard the results of the pre-enriched culture (i.e., positive or negative) together with its corresponding strain WGS characterization (i.e., presence of blaCTX-M and/or blaDHA genes). A false-positive result in the SMS analyses was defined as the detection of blaCTX-M and/or blaDHA genes that were not detected with the gold-standard.
The draft WGS assemblies for the detected strains (n = 26) are available in the National Center for Biotechnology Information (NCBI) under BioProject accession PRJNA931234 (BioSamples: SAMN33052292-SAMN33052317; Genomes: JAQQZM000000000-JAQQYN000000000). The human-decontaminated SMS reads for native and pre-enriched experiments (n = 42 and n = 21, respectively) are available in the NCBI Sequence Read Archive under BioProject accession PRJNA931814 (experiment run accessions: SRR23345838- SRR23345884, SRR24519239-SRR24519254).
A total of 42 healthy volunteers (21 males and 21 females with an average age of 52.3 and 46.0 years, respectively) were included in the study. The stool samples were sent to the IFIK by the participants within an average of 2 days upon their return to Switzerland. Volunteers reported living for an average duration of 3.3 years in a foreign country located in Africa, Asia, or Europe (n = 11, n = 17, and n = 14, respectively). Although this is not an epidemiological study aiming to establish prevalence and risk factors for ESC-R-Ent gut colonization, a summary of the volunteers’ demographic and clinical characteristics is depicted in Supplementary Table S1.
As shown in Table 1, screening for ESC-R-Ent by native culture resulted in 17 (40.5%) positive samples, whereas 23 (54.8%) positives were identified using the pre-enriched culture. Overall, ESC-R-Ent showed an average concentration of 3.13 × 107 (range: 1.78 × 102–3.97 × 108) CFU/g per stool sample. A total of 26 ESC-R-Ent strains (24 E. coli, one C. freundii and one K. pneumomiae) were isolated. All 26 ESC-R-Ent were fully susceptible to carbapenems, while 12 (46.2%), 7 (26.9%), and 18 (69.2%) were resistant to ciprofloxacin, gentamicin and trimethoprim-sulfamethoxazole, respectively (Supplementary Table S2).
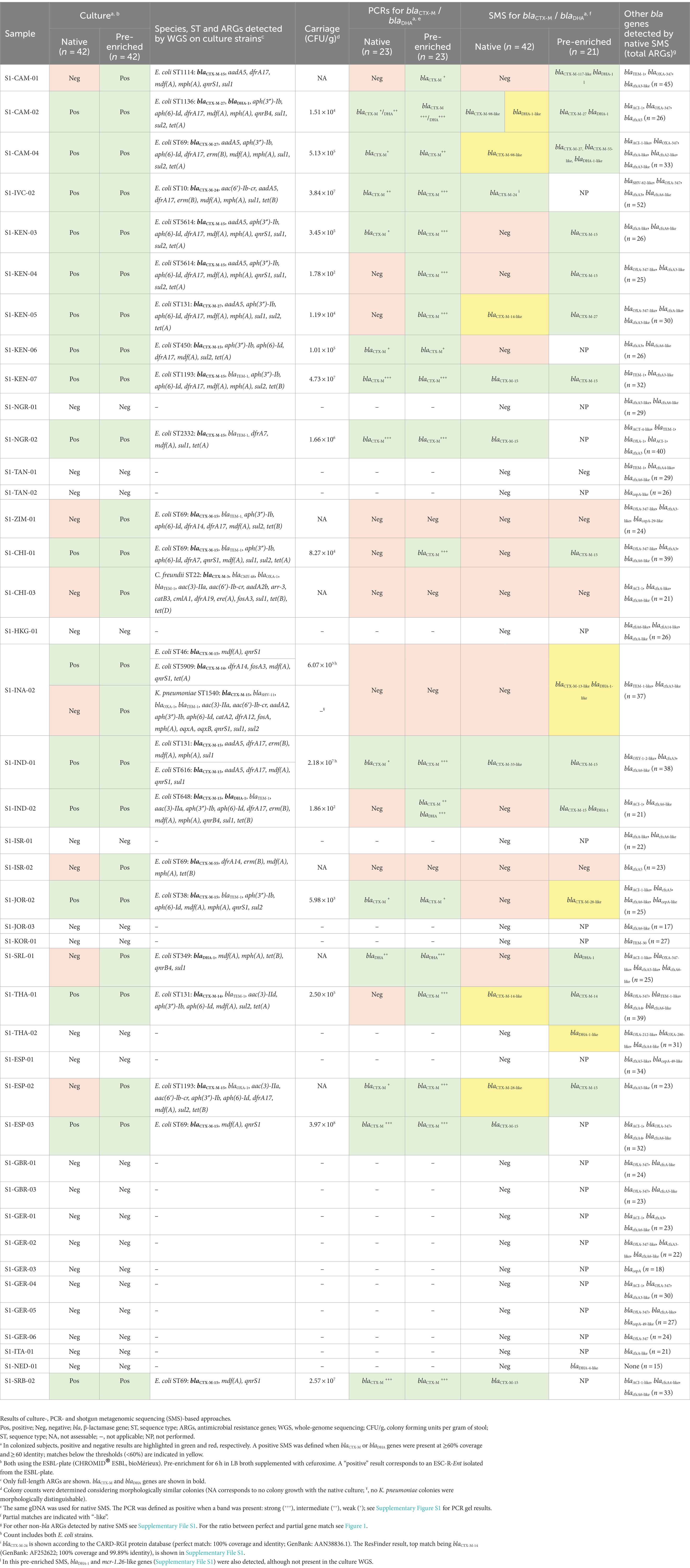
Table 1. Screening of stool sample for the 42 healthy Swiss people living abroad.
WGS on culture strains revealed that 6 out of 24 ESC-R E. coli strains were of ST69, but further pandemic clones were also detected (e.g., ST131, n = 3). In particular, 23 blaCTX-Ms- and 3 blaDHA-1-positives (2 co-harboring blaCTX-Ms) were found. Moreover, 2 volunteers carried CTX-M-producing C. freundii or K. pneumoniae strains (Table 1).
For the 23 pre-enriched culture-positive samples, native PCRs detected only 13 (56.5%) positives for blaCTX-Ms and/or blaDHA (one co-harboring blaCTX-M) (Table 1, highlighted in green). The band intensities of the PCR products for 12 blaCTX-M-positives were strong/intermediate (n = 5) and weak (n = 7), while intermediate bands were present for 2 blaDHA-positives (Supplementary Figure S1).
The 23 pre-enriched culture-positive samples were also tested by pre-enriched PCRs. As a result, 6 out of 10 samples that resulted negative for blaCTX-M/blaDHA genes by using the native PCR were actually positive using the pre-enriched approach (Table 1). As shown in Supplementary Figure S1, the band intensities for the PCR products were mostly strong/intermediate (5 out of 6).
The native SMS for the 42 stool samples resulted in an average of 13.8 M reads [phred quality score (Q) range for forward and reverse reads: Q26-Q37 (~99% of reads ≥Q30)] that were used for downstream analyses (Supplementary Table S3; Supplementary Table S4). On average, each of the 23 pre-enriched culture-positive samples contained 31.5 ARGs, whereas those of the 19 that were pre-enriched culture-negative possessed 24.6 ARGs (Figure 1; Supplementary Figure S2; Supplementary File S1).
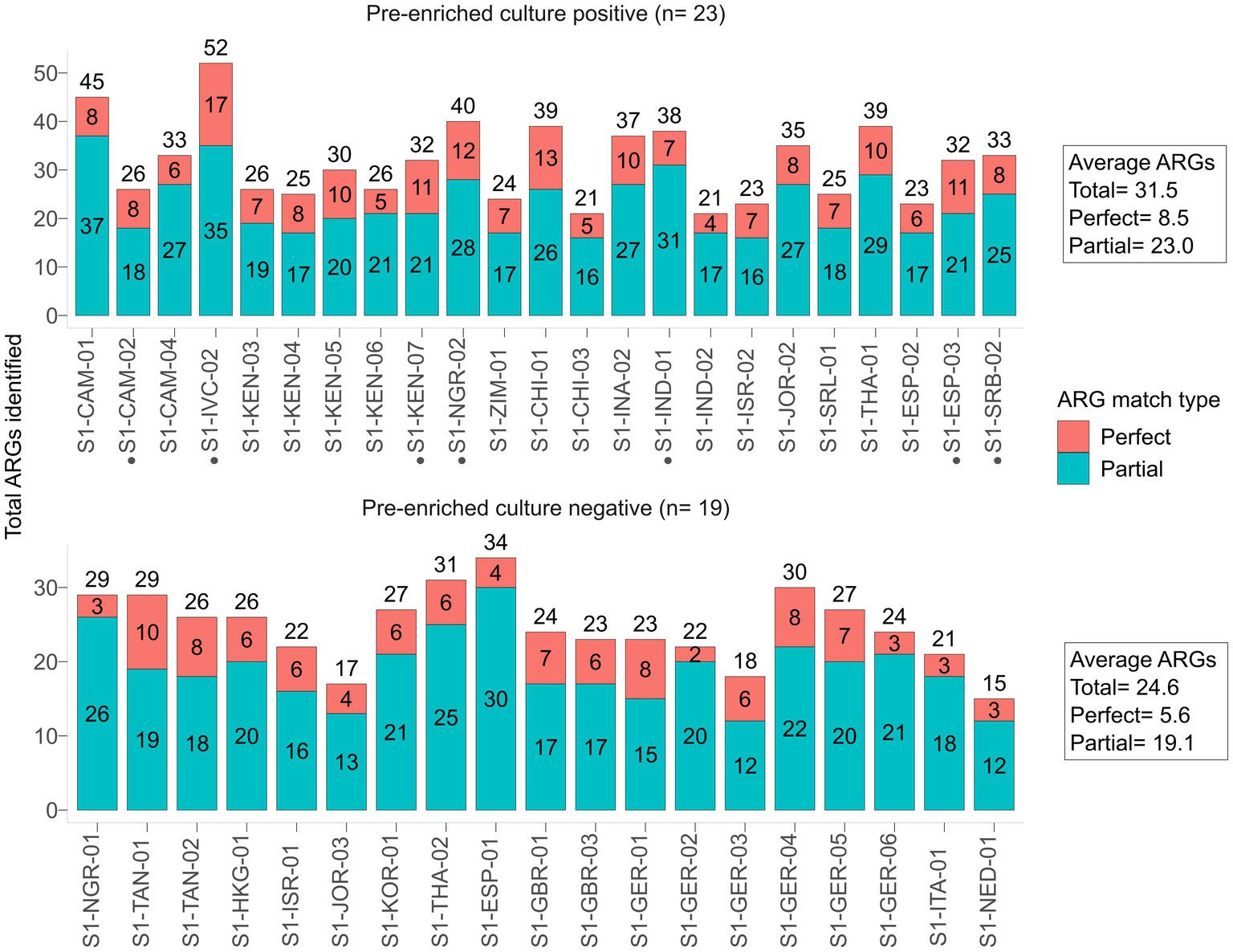
Figure 1. Antibiotic resistance genes (ARGs) identified by native SMS, plotted by pre-enriched culture screening result. The total number of ARGs counts and the sample are shown on the y-axis and x-axis, respectively. ARG counts are separated by match type according to the legend and shown in the bars (total count at top); a perfect match corresponds to 100% coverage and 100% nucleotide identity, while a partial match corresponds to ≥60% coverage and ≥ 60% nucleotide identity. See Supplementary File S1 for the list of all identified ARGs. Samples with a black circle (•) are those in which blaCTX-M genes were identified by SMS at ≥60% coverage and identity threshold (n = 7).
Only 7 samples that were pre-enriched culture blaCTX-M-positives were also detected by the native SMS (Table 1, highlighted in green; Figure 2A). In particular, 5 of these 7 SMS positive samples showed perfect matches (SI-IVC-02, S1-KEN-07, S1-NGR-02, S1-ESP-03, and S1-SRB-02 with blaCTX-M-15/24 genes). Two other samples with partial blaCTX-M matches were S1-CAM-02 (blaCTX-M-98-like) and S1-IND-01 (blaCTX-M-33-like) (Table 1; Supplementary File S1). Overall, sensitivity and specificity for the native SMS were 59.0 and 100%, respectively.
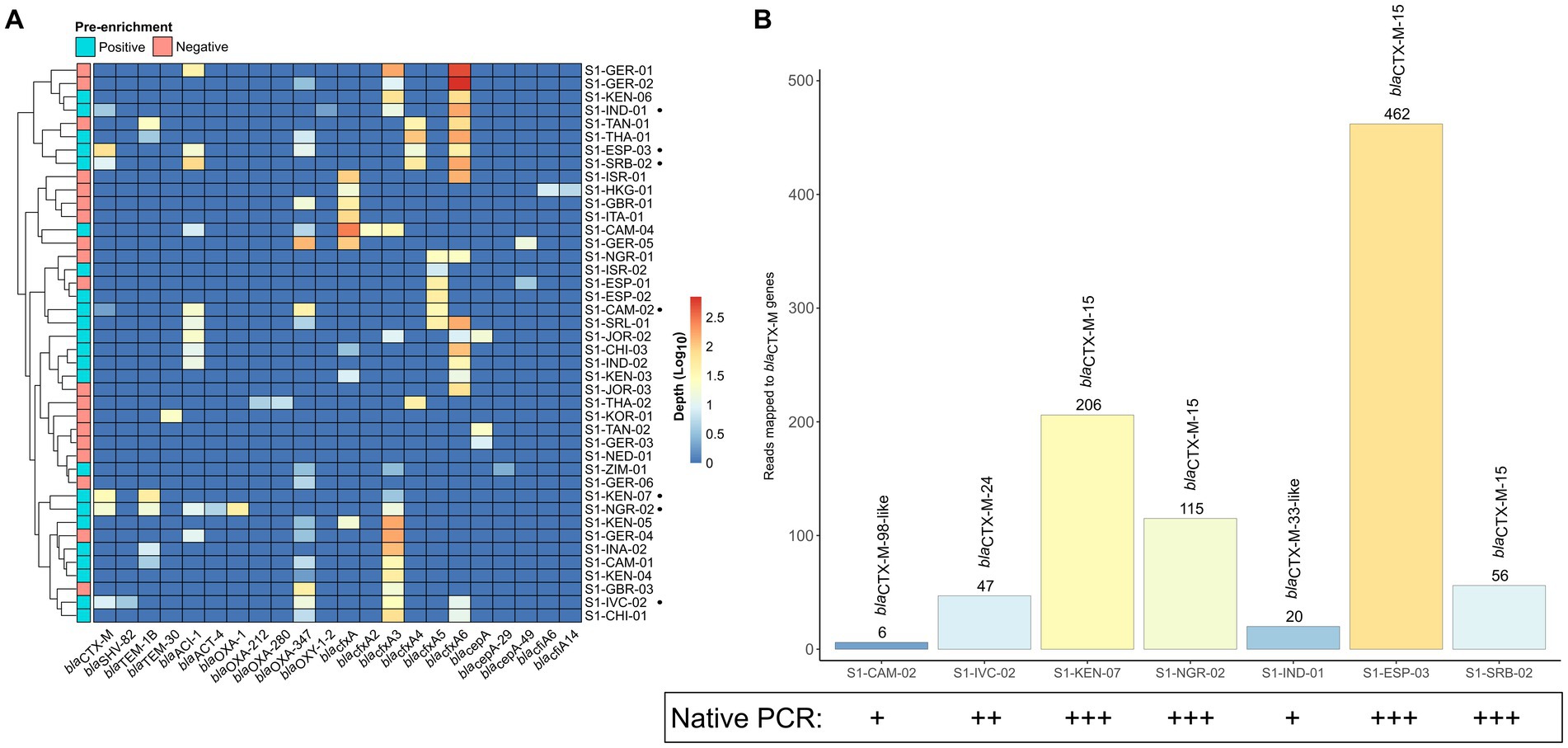
Figure 2. β-lactamase (bla) coding genes identified by native shotgun metagenomic sequencing (SMS). (A) Cluster matrix showing the mapping depth of the bla genes identified per sample. Pre-enrichment culture screening results are noted on the legend. Sample names and bla genes are shown on the y- and x-axis, respectively. Sample names marked by a black circle (•) correspond to blaCTX-M-positive samples by native SMS (≥60% identity and coverage threshold; n = 7). (B) Total number of mapped reads to the blaCTX-M genes identified by native SMS. The total number of mapped reads to a blaCTX-M gene per sample is shown above the bar and are colored by mapping depth (See Supplementary File S1 for depth, nucleotide identify and coverage). Corresponding native PCR results (intensity of bands) is also shown.
Notably, at below the defined threshold (≤60% identity and coverage), 5 native SMS negative samples turned out to be positive for blaCTX-Ms or blaDHAs, but with 24–46% identity and coverage (Table 1, highlighted in yellow).
Finally, to confirm our results obtained using the ResFinder platform, a supplementary screening with the CARD database (RGI using protein homology models) also detected the same blaCTX-M and/or blaDHA genes with good agreement, except for two samples (i.e., S1-IND-01 and S1-SRB-02) where the blaCTX-Ms were not identified (Supplementary File S1).
Using the same gDNA, PCR yielded blaCTX-M amplicons with band intensities proportional to the presence (read mapping) of blaCTX-Ms in native SMS (Figure 2B).
In the 5 out of the 7 native SMS blaCTX-M-positive samples, strong/intermediate bands corresponded to perfect blaCTX-M-15/−24 matches in the SMS analysis. In these 5 samples, the number of SMS mapped reads for blaCTX-Ms were at least 47 and the ESC-R-Ent concentrations were 1.66 × 106 CFU/g to 3.97 × 108 CFU/g. In contrast, weak native PCR bands were observed in the 2 native SMS blaCTX-M-positive samples with partial blaCTX-M coverage. Moreover, the number of SMS mapped reads for these 2 samples were ≤ 20 and the ESC-R-Ent stool concentration were 1.51 × 104 CFU/g and 2.18 × 107 CFU/g, respectively (Figure 2B; Table 1).
In the 5 native SMS samples that were negative, but contained partial blaCTX-M or blaDHA matches, native PCRs were intermediate/weak positive for 3 and negative for 2 of them. The ESC-R-Ent stool concentration of these 2 samples were 1.19 × 104 CFU/g and 5.13 × 105 CFU/g, respectively (Table 1).
Overall, in the native SMS, blaCTX-Ms were detected at an average minimum depth of 976,719 reads in 3 samples (S1-KEN-07, S1-NGR-02 and S1-ESP-03) corresponding to strong PCR bands, whereas in the other 4 native SMS samples, the average minimum depth was ~5.5 M reads corresponding to intermediate/weak bands (Supplementary Figure S1; Supplementary File S1).
In the rarefaction plot, we show that the total number of ARGs per native SMS sample reached saturation at approximately ≥80–100% of the total library size (Supplementary Figure S4A; Supplementary File S1). The minimum sequencing depth (% of total library size) required to detect the blaCTX-M genes was ≤20% for the 5 samples with strong/intermediate native PCR bands, whereas the 2 samples with weak bands needed ≥40% (Supplementary Figure S4B).
To investigate whether the sensitivity of SMS to detect blaCTX-M/DHA genes could be improved using enriched stool gDNA, we conducted SMS on 21 of the 42 samples. For the 21 samples, the pre-enriched SMS resulted in an average of ~13.1 M high quality reads (~99% of reads ≥Q30) (Supplementary Table S3; Supplementary Table S4; Supplementary Figure S3). On average, this approach identified 38.6 ARGs per sample, whereas the corresponding native SMS samples had an average of 29.4 ARGs (Figure 3A; Supplementary File S1). Notably, 18 of the 21 samples were pre-enriched culture-positive for blaCTX-M and/or blaDHA-possessing strains but, as mentioned above, only part of them (i.e., 3/18) were correctly identified as positive with the native SMS analysis (Table 1).
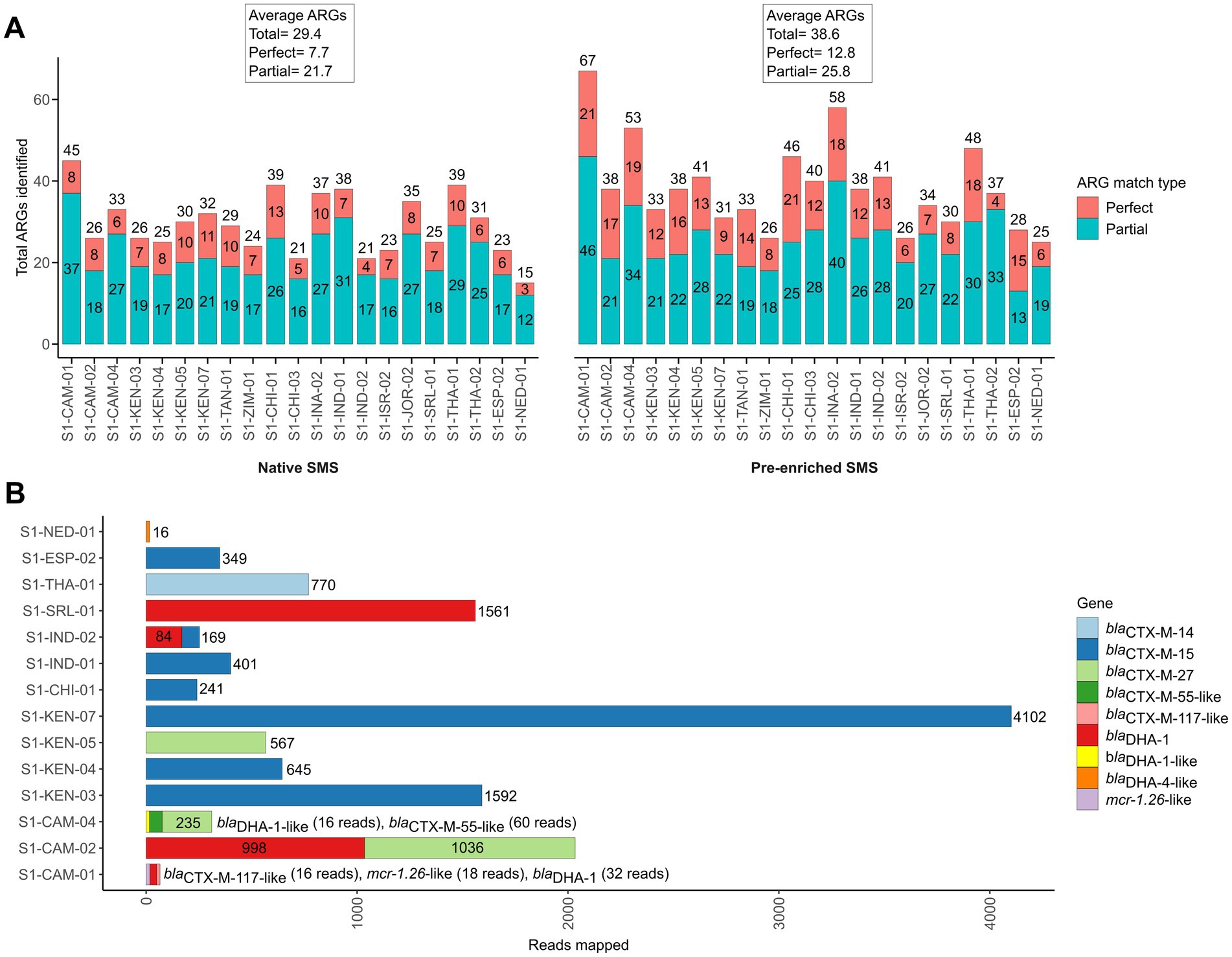
Figure 3. Antibiotic resistance genes (ARGs) identified by pre-enriched shotgun metagenomic sequencing (SMS) in 21 representative samples. (A) Comparison of ARGs identified by native and pre-enriched SMS. The total number of ARGs counts and the sample are shown on the y-axis and x-axis for both native and pre-enriched SMS, respectively. ARG counts are separated by match type according to the legend and shown in the bars (total count at top); a perfect match corresponds to 100% coverage and 100% nucleotide identity, while a partial match corresponds to ≥60% coverage and ≥ 60% nucleotide identity. See Supplementary File S1 for the list of all identified ARGs. (B) The blaCTX-M-, blaDHA-1- and mcr-1-like genes identified by using pre-enriched SMS. The total number of mapped reads per sample is shown in the bars and colored by gene (for a complete list of identified ARGs and coverage, see Supplementary File S1). Note, blaDHA-4-like was identified in S1-NED-01 (64.04 and 64.21% identity and coverage, respectively), while below threshold (≤60% identity and coverage) reads mapping to blaCTX-M-13-like (2 reads; 34.47% identity/coverage) and blaDHA-1-like (2 reads; 20.26% identity/coverage), blaCTX-M-28-like (2 reads; 34.47% identity/coverage), and blaDHA-1-like (9 reads; 38.42% identity/coverage) were detected in S1-INA-02, SI-JOR-02, and S1-THA-02, respectively (data not shown).
With the pre-enriched SMS approach, 13 out of the 18 samples turned out to be positive for blaCTX-M/blaDHA genes (Table 1, highlighted in green). Interestingly, 1 out of the remaining 3 samples that were pre-enriched culture-negative (S1-NED-01) resulted false-positive for a blaDHA-4-like gene. Overall, sensitivity and specificity for the pre-enriched SMS were 78.3 and 75.0%, respectively.
Of note, two blaDHA genes were not identified by culture in S1-CAM-01 and S1-CAM-04; the first also contained an mcr-1 (mcr-1.26-like) gene at 86.6% identity and coverage, respectively (Table 1; Figure 3B). Furthermore, two samples that resulted negative by pre-enriched SMS (S1-INA-02 and S1-JOR-02) were actually harboring blaCTX-M/blaDHA genes, but with only 20.3–34.4% identity and coverage (Table 1, highlighted in yellow; Figure 3).
Overall, blaCTX-M and/or blaDHA genes were detected at an average minimum depth of 1.2 M reads in the pre-enriched SMS. In one sample (S1-CAM-01) corresponding to weak PCR bands, blaCTX-M and blaDHA genes were detected at an average minimum depth of 2.21 M and 4.42 M reads, respectively (Supplementary Figure S1; Supplementary File S1).
Similarly to the native SMS, the rarefaction plot shows that the total number of ARGs per pre-enriched SMS sample reached saturation at approximately ≥80–100% of the total library size (Supplementary Figure S4A). For the 13 positive samples, the minimum sequencing depth (% of total library size) required to detect blaCTX-Ms was ≤20%, whereas that to detect blaDHA genes ranged between 10 and 80% (Supplementary Figure S4B; Supplementary File S1).
In the decontaminated reads from both native and pre-enriched SMS, the top 10 bacterial species included E. coli (Figure 4A).
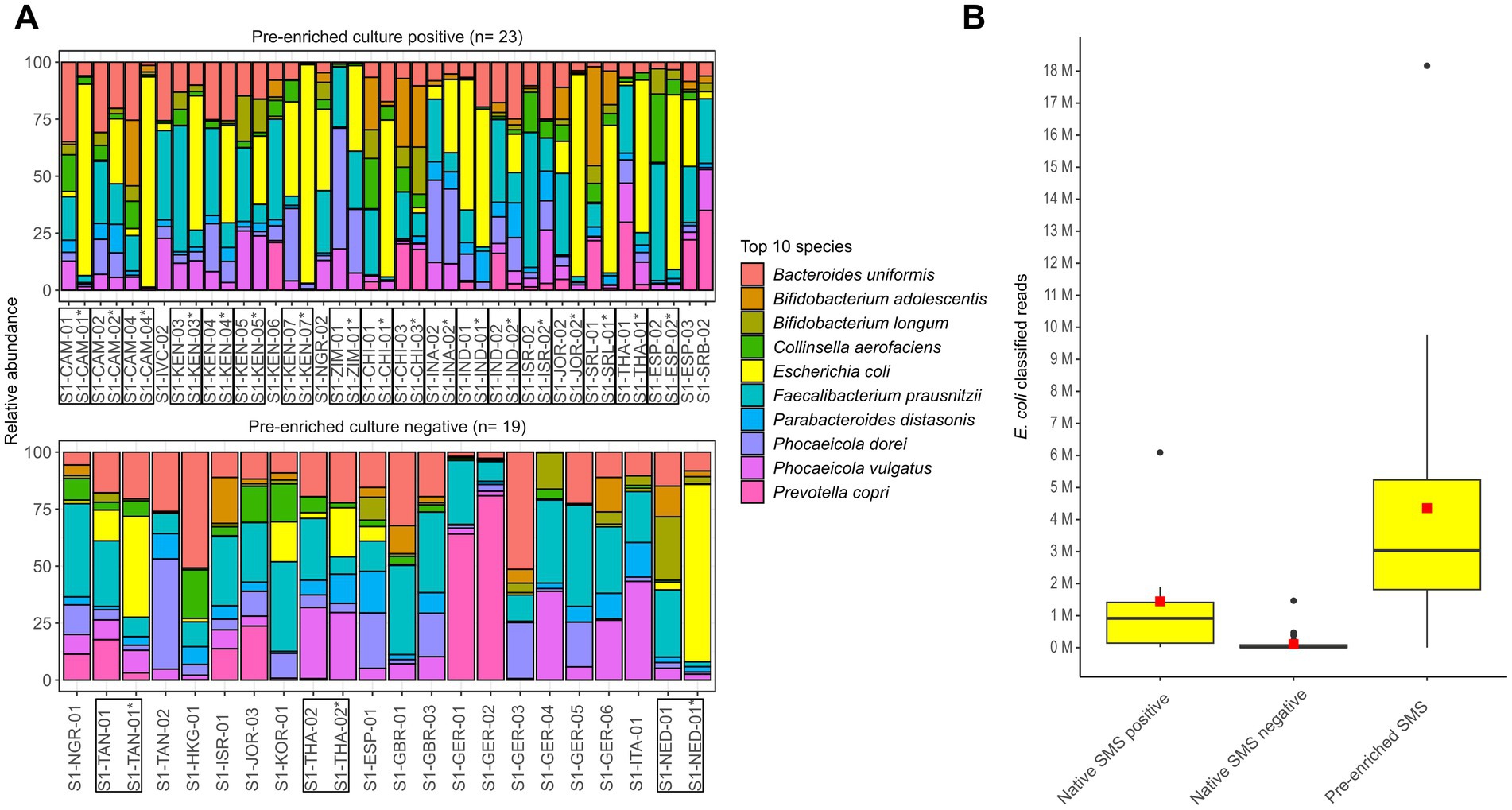
Figure 4. Microbiome characterization at the species level by shotgun metagenomic sequencing (SMS). (A) Native and pre-enriched SMS relative abundance for the top 10 bacterial species in the 23 and 19 pre-enrichment culture positive and negative stools, respectively. Results of the pre-enriched SMS are marked with an asterisk (*); both pre-enriched and matching native SMS samples are marked with a box. (B) Distribution of E. coli classified reads in the 42 native (positive: n = 7; negative: n = 35) and 21 pre-enriched SMS (positive: n = 14; negative: n = 7). The red square and black line in the box plot correspond to the average and median number of classified E. coli reads, respectively: 1,449,006.7 and 916,144 (native SMS positive), 113,465.7 and 20,449 (native SMS negative), 4,358,135.9 and 3,030,602 (pre-enriched SMS). See Supplementary File S1 for raw data.
In native SMS samples that where blaCTX-M-positive (n = 7), the number of E. coli classified reads was on average of ~1.45 M, while in the 35 samples resulting native SMS negatives it was 113,466. In 10 of the 35 native SMS negatives that were both native and pre-enriched culture-positives, an average of 193,959 classified E. coli reads were detected. Furthermore, in the 21 pre-enriched SMS samples, the number of E. coli classified reads was on average of ~4.36 M (Figure 4B; Supplementary File S1).
The SMS-based approach has been suggested as a tool for the non-targeted identification of genes from clinical samples (Chiu and Miller, 2019). However, data regarding the implementation of this technique on complex samples like the stool of people living in the community is scarce.
In this work, we evaluated the potential of SMS as a screening tool for intestinal colonization with ESC-R-Ent in healthy individuals and compared it with the standard culture-based methods. Specifically, we focused on the detection of blaCTX-Ms and blaDHAs and explored potential factors that may influence their detection with SMS. Other common acquired ESBL and AmpC bla genes in Enterobacterales (e.g., blaTEM, blaSHV and blaCMY) were not considered due to their absence or rarity in the genome of our 26 isolated ESC-R-Ent (Bush and Bradford, 2020).
Identification of intestinal carriers of ESBL or AmpC producers is usually achieved by native culture screened on selective chromogenic media. However, considering that the concentration of some ESC-R-Ent in the gut may be lower than the technical LOD (~101 CFU/100 mg) and the commensal bacteria (e.g., Bacteroides spp.), the native culture approach may not provide sufficient resolution (e.g., resulting in visible bacterial growth in culture plates) to detect these pathogens, unless a pre-enrichment step is performed (Naas et al., 2011; Girlich et al., 2014; Rondinaud et al., 2020; Sadek et al., 2020; Gallah et al., 2023).
Our analysis confirmed that even a short selective pre-enrichment (6 h) increases the capacity to identify the ESC-R-Ent colonized individuals of about 15% when compared to the direct screening alone (Table 1). For this reason, the pre-enriched culture approach was used as gold-standard to evaluate the performance of the SMS method in detecting blaCTX-M and/or blaDHA genes.
Recently, SMS has been used to identify bacterial species and ARGs such as Clostridioides difficile, KPC-producing K. pneumoniae, Shiga toxin- and CTX-M-producing E. coli in stool samples from hospitalized patients (Andersen et al., 2016; Schneeberger et al., 2016; Ward et al., 2016; Zhou et al., 2016; Grall et al., 2017; Gigliucci et al., 2018; Mu et al., 2019; Leggett et al., 2020; Yee et al., 2021). In contrast, only Guernier-Cambert et al. (2021), Bengtsson-Palme et al. (2015), and Dallman et al. (2023) analyzed the stool of healthy people (n = 9, n = 35, and n = 50, respectively) by using SMS.
As we have done in the present work, most of the herein cited studies have used the Illumina short-read native SMS, which is a well-established method. Nevertheless, despite the ability of short-read SMS to detect genetic targets, the method has a long TAT (e.g., up to 40 h sequencing run). This time lag is inappropriate for implementing IPC measures in clinical practice (Liu et al., 2021). Cost is also an issue, as most clinical laboratories do not have the infrastructure, equipment, or funding to implement short-read SMS. In addition, there are no standardized bioinformatics pipelines for pre-processing, analyzing and interpreting SMS data, making it difficult to implement in the clinical laboratory (Chiu and Miller, 2019). Therefore, because differences in results may be due only to differences in the technical approaches, comparing the performance (i.e., sensitivity) between SMS studies may not be appropriate.
In our study, comparison of the native SMS and the pre-enriched culture approaches showed that the native SMS has a very low sensitivity (59.0%) for the detection of blaCTX-M/DHA genes in stool (Table 1, Figure 2). Other bla genes, such as the blacfxA-type from Bacteroides spp. were identified (Eitel et al., 2013). The presence of such bla genes is common due to their high abundance in the gut flora (Gatica et al., 2019). In fact, 3 species belonging to the Bacteroidaceae family were among the top 10 species in all native SMS samples tested in the present study (Figure 4A). Overall, considering the amount of ARGs detected (Figure 1; Supplementary File S1), SMS can be used as a non-targeted method to describe the overall resistome (Bengtsson-Palme et al., 2015). Nevertheless, in view of the arguments outlined above, gDNA from less represented species (e.g., the ESBL producers) might not be abundant enough to be detected by native SMS.
Despite the suggested low sensitivity of native SMS, it is important to note that several factors such as the sequencing depth could influence the detection of important ARGs (Santiago-Rodriguez et al., 2020). However, although various sequencing depths have been proposed (e.g., 5–50 M, including 2.5 billion ultradeep sequencing), it is not yet clear what is the optimal value required to detect targeted ARGs in stool (Hillmann et al., 2018; Zaheer et al., 2018; Liu et al., 2022).
In this study, native SMS resulted in variable sequencing depths, as well as the identification of blaCTX-M genes which was possible at an average minimum depth between 996,719 to 5.5 M reads (Supplementary Figure S2; Supplementary Table S3; Supplementary File S1). Furthermore, blaCTX-M genes were identified earlier at lower sequencing depths than other ARGs, confirming a high concentration of bacteria carrying such bla genes (Table 1; Supplementary Figure S4). Nevertheless, such differences were expected and not found to be a major factor affecting SMS sensitivity in this study. In fact, the average of reads for the native SMS corresponding to the pre-enriched culture-positive and culture-negative samples were in the same range (~13–15 M). This observation leads to the hypothesis that other factors such as the bacterial load (CFU/g) in native stool might play a more important role in the detection of ARGs, as their presence (e.g., copy number) might be reflected by the ESC-R-Ent carrying them.
E. coli is estimated to be present in a wide range (3 × 102 to 109 CFU/g) in healthy individuals (Wotzka et al., 2018). Intestinal carriage of 3.6 × 107 CFU/g of ESC-R E. coli (average) and up to 1 × 108 CFU/g (e.g., CTX-M-15-producing E. coli), for example, have been reported in travelers to Vietnam and in a university hospital in Morocco, respectively (Girlich et al., 2014; Nakayama et al., 2020). Moreover, in the study by Grall et al., the blaCTX-M genes detected by native SMS were from stool samples of adults under imipenem treatment with a median carriage load of log 9.4 CFU/g (equivalent to 2.51 × 109 CFU/g) (Grall et al., 2017).
In line with the above surveys, the ESC-R-Ent stool concentration in the native SMS positive and negative samples in our study varied between 104 to 108 and 102 to 105 CFU/g, respectively (Table 1). Consistently, the number of E. coli classified reads was also variable between native SMS positive and negative samples (on average 1.45 M vs. 113,466, respectively) (Figure 4B; Supplementary File S1).
The proportion of the target bacterial pathogens to other species in the stool needs to be large enough to be captured during the gDNA isolation and following SMS processing (Thomas et al., 2012). We found that 5 positive native SMS samples mapped in full (100% coverage, range, 47–462 reads) to blaCTX-M genes (Table 1; Figure 2B). Importantly, a simple native PCR result was indicative of a following positive native SMS result: strong bands corresponded to complete coverage of the blaCTX-M genes (Supplementary Figure S1). Moreover, in most samples, the ESC-R-Ent concentration in stool was proportional to the intensity of the PCR results and to the number of mapped reads (Table 1; Figure 2B). Therefore, considering that the abundance of ESC-R-Ent in gDNA is variable, the predictability of a successful native SMS screening to identify important ARGs (e.g., blaCTX-Ms) may be evaluated by PCR-based tests commonly used in the diagnostic laboratory (Endimiani et al., 2020).
Another important factor to consider is the sensitivity of the parameters used (e.g., coverage cutoffs) for the in silico screening as they might influence the results (e.g., low abundance reads may be discarded during metagenome assembly) (Thomas et al., 2012).
We detected blaCTX-M at 100% (n = 5) and at 77–90% (n = 2) coverage (Figure 2B; Supplementary File S1). On this note, at lower cutoffs (≤60%), 4 samples that were native SMS negative actually revealed reads mapping to blaCTX-Ms. Therefore, because of the low abundance of important ARGs, it is essential to consider that stricter cutoffs might lead to false-negative results. In this case, using the ResFinder default parameters (i.e., ≥90% identity and ≥ 60% coverage) we would have missed 2 out of 7 blaCTX-M-positives (S1-CAM-02 and S1-IND-01) in the native SMS (Table 1; Supplementary Table S1).
Remarkably, the use of different screening tools such as CARD-RGI, which uses a protein homolog-based database as opposed to the nucleotide database used by ResFinder, may affect the detection sensitivity for the blaCTX-M/DHA genes. In fact, we found that CARD-RGI failed to detect 2 blaCTX-M-positives (S1-IND-01 and S1-SRB-02; Supplementary File S1) at ≥60% coverage as our ResFinder pipeline.
An alternative and cheap strategy to increase the sensitivity of SMS is to consider SMS from pre-enriched stool samples. Few studies have explored this approach with the aim of enhancing the detection of specific pathogens and ARGs present at low concentrations in stool. For example, in the study by Raymond et al., the enriched stool (7 days in both aerobic and anaerobic broths with and without cefoxitin) of 18 participants treated with cefprozil for 78 days were analyzed by SMS. They found that the long stool enrichments were necessary for the detection of less abundant bacteria and an increase in overall ARGs compared to native SMS (Raymond et al., 2019). In another recent study, a shorter stool enrichment (6 h) in combination with 3 antibiotics (vancomycin, metronidazole, and cefpodoxime) was necessary to detect carbapenemase-producing Ent (blaNDM-1- and blaKPC-2-positive) in spiked stools from 3 inpatients under ceftazidime exposure and one healthy volunteer (Peto et al., 2019).
Results of these exploratory studies clearly indicated that the choice of the antibiotic(s) used for the enrichment select the group of pathogens and ARGs that the SMS can detect during the analysis (i.e., narrowing down the number of hypothetical perceivable targets present in the stool). Therefore, a preliminary strategic decision based on what to look for in the sample of interest should be done prior to SMS analysis.
In our case, a pre-enrichment with cefuroxime for 6 h was specifically implemented to target the ESC-R-Ent. As a result, this approach was sufficient to increase the sensitivity of SMS of about 20%. In particular, the pre-enriched SMS led to the identification of blaCTX-Ms in many samples that were not identified by native SMS (an increase of about 50–55%). An additional side benefit of the pre-enriched SMS approach was the overall lower requirement of sequencing depth (~1.2 M reads for both blaCTX-M and blaDHA genes) to detect blaCTX-Ms/DHAs vs. 996,719 to 5.5 M reads in the native SMS, potentially speeding up sequencing times (Koh et al., 2023). Both blaCTX-M and blaDHA genes were also detected earlier at lower sequencing depths than other ARGs, supporting the observation of a high concentration of bacteria carrying such bla genes (Table 1; Supplementary Figure S4).
Furthermore, pre-enrichment also resulted in the identification of other important ARGs not observed in the genome of isolated ESC-R-Ent such as the blaDHA genes (Table 1; Figure 3). In this regard, it can be speculated that such blaDHA genes (especially those with thresholds ≤60%) were mostly carried by natural hosts (e.g., Morganella morganii) that overgrowth under the selective pressure of cefuroxime affecting the final specificity of the pre-enriched SMS analysis. This highlights the importance of the concurrent use of culture-based methods as a confirmatory test, as findings detected by SMS may not be clinically relevant (i.e., false-positives).
We finally emphasize that the stool pre-enrichment could be beneficial to improve the detection of clinically important ARGs with other DNA-based methods as well. For instance, we previously demonstrated that testing pre-enriched stools (LB broth supplemented with colistin) increased the detection rate of mcr-1 compared to native stools by real-time PCR (Dona et al., 2017).
Our results indicate that the native SMS analysis is unable to reveal the presence of the clinically important blaCTX-M/blaDHA ARGs in the stool of colonized individuals who have a low concentration of ESC-R-Ent (e.g., in healthy people in the community). With the pre-enriched SMS, a short selective incubation time and the use of detection cutoffs lower than those set by default in available analytical platforms may significantly increase the sensitivity of SMS. However, the native culture method still offers the same results, whereas the pre-enriched culture-based approach is performing significantly better in detecting ESC-R-Ent producing CTX-Ms and/or DHAs β-lactamases.
Although pre-enriched SMS is a promising alternative to non-targeted native SMS, further studies should investigate the effect of deep sequencing (beyond >10 M reads) with and without non-targeted pre-enriched SMS (i.e., no addition of antibiotic) and native SMS alone, since performance [i.e., sensitivity to detect specific ARGs (blaCTX-M or blaDHA) or all present ARGs] may simply increase due to the availability of more reads or vice versa, as may have been the case in this study. Furthermore, the pre-analytical effects (e.g., stool collection methods, gDNA isolation kit) should be considered as they may impact the sensitivity of SMS analyses (Thomas et al., 2012; Quince et al., 2017; Guan et al., 2021).
We also emphasize that, at the moment, the routine implementation of the Illumina short-read SMS platform presents logistical challenges, high costs, and long TATs. In this context, the Nanopore long-read sequencing may represent a cheaper, faster (<6 h sequencing run) and easy to implement alternative to Illumina. Moreover, the Nanopore metagenomic output could further link the identified ARG with the corresponding bacterial species and mobile genetic element (e.g., plasmid) (Mu et al., 2019; Leggett et al., 2020). However, as opposed to Illumina technology, Nanopore requires high DNA inputs for library preparations (e.g., up to 400 ng depending on the kit implemented), which might affect the overall detection sensitivity of important ARGs (Petersen et al., 2019). Therefore, the potential of Nanopore SMS stool analysis should be evaluated in the near future with large and comparative studies, as we have done for the Illumina SMS in the present work.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA931234; https://www.ncbi.nlm.nih.gov/, PRJNA931814.
The studies involving humans were approved by Ethical Committee of the Canton Bern (ID: 2020-01683). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
AE: conception and design. EC-M, CA, AM, PS, and AE: acquisition of data. EC-M and AE: drafting the work. All authors: analysis and interpretation of data, critical revision for important intellectual content, final approval of the version to be published, agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
This work was supported by the Swiss National Science Foundation (SNF) grant No. 192514 (to AE). EC-M is a PhD student (2021–2024) supported by SNF.
We thank Pablo Porragas Paseiro for his technical help.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1236208/full#supplementary-material
1. ^https://data.snf.ch/grants/grant/192514
2. ^https://www.eucast.org/clinical_breakpoints
3. ^https://github.com/s-andrews/FastQC
4. ^https://github.com/usadellab/Trimmomatic
5. ^https://github.com/rrwick/Unicycler
6. ^https://sourceforge.net/projects/bbmap/
7. ^https://github.com/biobakery/kneaddata
8. ^https://github.com/lh3/seqtk
9. ^https://www.genomicepidemiology.org/
11. ^https://www.r-project.org/
12. ^https://github.com/arpcard/rgi
Afridi, O. K., Ali, J., and Chang, J. H. (2021). Fecal microbiome and resistome profiling of healthy and diseased Pakistani individuals using next-generation sequencing. Microorganisms 9:616. doi: 10.3390/microorganisms9030616
Andersen, H., Connolly, N., Bangar, H., Staat, M., Mortensen, J., Deburger, B., et al. (2016). Use of shotgun metagenome sequencing to detect fecal colonization with multidrug-resistant bacteria in children. J. Clin. Microbiol. 54, 1804–1813. doi: 10.1128/JCM.02638-15
Arcilla, M. S., Van Hattem, J. M., Haverkate, M. R., Bootsma, M. C. J., Van Genderen, P. J. J., Goorhuis, A., et al. (2017). Import and spread of extended-spectrum β-lactamase-producing Enterobacteriaceae by international travellers (COMBAT study): a prospective, multicentre cohort study. Lancet Infect. Dis. 17, 78–85. doi: 10.1016/S1473-3099(16)30319-X
Banerjee, R., Strahilevitz, J., Johnson, J. R., Nagwekar, P. P., Schora, D. M., Shevrin, I., et al. (2013). Predictors and molecular epidemiology of community-onset extended-spectrum β-lactamase-producing Escherichia coli infection in a midwestern community. Infect. Control Hosp. Epidemiol. 34, 947–953. doi: 10.1086/671725
Bengtsson-Palme, J., Angelin, M., Huss, M., Kjellqvist, S., Kristiansson, E., Palmgren, H., et al. (2015). The human gut microbiome as a transporter of antibiotic resistance genes between continents. Antimicrob. Agents Chemother. 59, 6551–6560. doi: 10.1128/AAC.00933-15
Bernasconi, O. J., Kuenzli, E., Pires, J., Tinguely, R., Carattoli, A., Hatz, C., et al. (2016). Travelers can import colistin-resistant Enterobacteriaceae, including those possessing the plasmid-mediated mcr-1 gene. Antimicrob. Agents Chemother. 60, 5080–5084. doi: 10.1128/AAC.00731-16
Bezabih, Y. M., Bezabih, A., Dion, M., Batard, E., Teka, S., Obole, A., et al. (2022). Comparison of the global prevalence and trend of human intestinal carriage of ESBL-producing Escherichia coli between healthcare and community settings: a systematic review and meta-analysis. JAC Antimicrob Resist. 4:dlac048. doi: 10.1093/jacamr/dlac048
Bezabih, Y. M., Sabiiti, W., Alamneh, E., Bezabih, A., Peterson, G. M., Bezabhe, W. M., et al. (2021). The global prevalence and trend of human intestinal carriage of ESBL-producing Escherichia coli in the community. J. Antimicrob. Chemother. 76, 22–29. doi: 10.1093/jac/dkaa399
Blane, B., Brodrick, H. J., Gouliouris, T., Ambridge, K. E., Kidney, A. D., Ludden, C. M., et al. (2016). Comparison of 2 chromogenic media for the detection of extended-spectrum β-lactamase producing Enterobacteriaceae stool carriage in nursing home residents. Diagn. Microbiol. Infect. Dis. 84, 181–183. doi: 10.1016/j.diagmicrobio.2015.11.008
Budel, T., Kuenzli, E., Clement, M., Bernasconi, O. J., Fehr, J., Mohammed, A. H., et al. (2019). Polyclonal gut colonization with extended-spectrum cephalosporin- and/or colistin-resistant Enterobacteriaceae: a normal status for hotel employees on the island of Zanzibar, Tanzania. J. Antimicrob. Chemother. 74, 2880–2890. doi: 10.1093/jac/dkz296
Bush, K., and Bradford, P. A. (2020). Epidemiology of β-lactamase-producing pathogens. Clin. Microbiol. Rev. 33. doi: 10.1128/CMR.00047-19
Campos-Madueno, E. I., Moradi, M., Eddoubaji, Y., Shahi, F., Moradi, S., Bernasconi, O. J., et al. (2023). Intestinal colonization with multidrug-resistant Enterobacterales: screening, epidemiology, clinical impact, and strategies to decolonize carriers. Eur. J. Clin. Microbiol. Infect. Dis. 42, 229–254. doi: 10.1007/s10096-023-04548-2
Campos-Madueno, E. I., Moser, A. I., Risch, M., Bodmer, T., and Endimiani, A. (2021). Exploring the global spread of Klebsiella grimontii isolates possessing blaVIM-1 and mcr-9. Antimicrob. Agents Chemother. 65:e0072421. doi: 10.1128/AAC.00724-21
Chiu, C. Y., and Miller, S. A. (2019). Clinical metagenomics. Nat. Rev. Genet. 20, 341–355. doi: 10.1038/s41576-019-0113-7
Clausen, P., Aarestrup, F. M., and Lund, O. (2018). Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19:307. doi: 10.1186/s12859-018-2336-6
Dallman, T. J., Neuert, S., Fernandez Turienzo, C., Berin, M., Richardson, E., Fuentes-Utrilla, P., et al. (2023). Prevalence and persistence of antibiotic resistance determinants in the gut of travelers returning to the United Kingdom is associated with colonization by pathogenic Escherichia coli. Microbiol. Spectr. e0518522. doi: 10.1128/spectrum.05185-22
Dona, V., Bernasconi, O. J., Kasraian, S., Tinguely, R., and Endimiani, A. (2017). A SYBR(R) green-based real-time PCR method for improved detection of mcr-1-mediated colistin resistance in human stool samples. J. Glob. Antimicrob Resist. 9, 57–60. doi: 10.1016/j.jgar.2017.01.007
Eitel, Z., Soki, J., Urban, E., and Nagy, E., ESCMID Study Group on Anaerobic Infection (2013). The prevalence of antibiotic resistance genes in Bacteroides fragilis group strains isolated in different European countries. Anaerobe 21, 43–49. doi: 10.1016/j.anaerobe.2013.03.001
Endimiani, A., Ramette, A., Rhoads, D. D., and Jacobs, M. R. (2020). The evolving role of the clinical microbiology laboratory in identifying resistance in gram-negative bacteria: an update. Infect. Dis. Clin. N. Am. 34, 659–676. doi: 10.1016/j.idc.2020.08.001
Gallah, S., Scherer, M., Collin, T., Gomart, C., Veziris, N., Benzerara, Y., et al. (2023). Five-hour detection of intestinal colonization with extended-spectrum-β-lactamase-producing Enterobacteriaceae using the beta-lacta phenotypic test: the BLESSED study. Microbiol. Spectr. 11:e0295922. doi: 10.1128/spectrum.02959-22
Gatica, J., Jurkevitch, E., and Cytryn, E. (2019). Comparative metagenomics and network analyses provide novel insights into the scope and distribution of β-lactamase homologs in the environment. Front. Microbiol. 10:146. doi: 10.3389/fmicb.2019.00146
Gigliucci, F., Von Meijenfeldt, F. A. B., Knijn, A., Michelacci, V., Scavia, G., Minelli, F., et al. (2018). Metagenomic characterization of the human intestinal microbiota in fecal samples from STEC-infected patients. Front. Cell. Infect. Microbiol. 8:25. doi: 10.3389/fcimb.2018.00025
Girlich, D., Bouihat, N., Poirel, L., Benouda, A., and Nordmann, P. (2014). High rate of faecal carriage of extended-spectrum β-lactamase and OXA-48 carbapenemase-producing Enterobacteriaceae at a university hospital in Morocco. Clin. Microbiol. Infect. 20, 350–354. doi: 10.1111/1469-0691.12325
Girlich, D., Grosperrin, V., Naas, T., and Dortet, L. (2019). CHROMagar ESBL/mSuperCARBA bi-plate medium for detection of ESBL- and carbapenemase-producing Enterobacteriaceae from spiked stools. Diagn. Microbiol. Infect. Dis. 95, 107–112. doi: 10.1016/j.diagmicrobio.2019.05.002
Grall, N., Lazarevic, V., Gaia, N., Couffignal, C., Laouenan, C., Ilic-Habensus, E., et al. (2017). Unexpected persistence of extended-spectrum β-lactamase-producing Enterobacteriaceae in the faecal microbiota of hospitalised patients treated with imipenem. Int. J. Antimicrob. Agents 50, 81–87. doi: 10.1016/j.ijantimicag.2017.02.018
Guan, H., Pu, Y., Liu, C., Lou, T., Tan, S., Kong, M., et al. (2021). Comparison of fecal collection methods on variation in gut metagenomics and untargeted. Metabolomics 6:e0063621. doi: 10.1128/mSphere.00636-21
Guernier-Cambert, V., Chamings, A., Collier, F., and Alexandersen, S. (2021). Diverse bacterial resistance genes detected in Fecal samples from clinically healthy women and infants in Australia-a descriptive pilot study. Front. Microbiol. 12:596984. doi: 10.3389/fmicb.2021.596984
Hillmann, B., Al-Ghalith, G. A., Shields-Cutler, R. R., Zhu, Q., Gohl, D. M., Beckman, K. B., et al. (2018). Evaluating the information content of shallow shotgun metagenomics 3:e00069-18. doi: 10.1128/mSystems.00069-18,
Jazmati, N., Hein, R., and Hamprecht, A. (2016). Use of an enrichment broth improves detection of extended-spectrum-β-lactamase-producing Enterobacteriaceae in clinical stool samples. J. Clin. Microbiol. 54, 467–470. doi: 10.1128/JCM.02926-15
Jazmati, N., Jazmati, T., and Hamprecht, A. (2017). Importance of pre-enrichment for detection of third-generation cephalosporin-resistant Enterobacteriaceae (3GCREB) from rectal swabs. Eur. J. Clin. Microbiol. Infect. Dis. 36, 1847–1851. doi: 10.1007/s10096-017-3000-1
Ko, K. K. K., Chng, K. R., and Nagarajan, N. (2022). Metagenomics-enabled microbial surveillance. Nat. Microbiol. 7, 486–496. doi: 10.1038/s41564-022-01089-w
Koh, W. L. C., Poh, S. E., Lee, C. K., Chan, T. H. M., Yan, G., Kong, K. W., et al. (2023). Towards a rapid-turnaround low-depth unbiased metagenomics sequencing workflow on the Illumina platforms. Bioengineering 10:10. doi: 10.3390/bioengineering10050520
Leggett, R. M., Alcon-Giner, C., Heavens, D., Caim, S., Brook, T. C., Kujawska, M., et al. (2020). Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat. Microbiol. 5, 430–442. doi: 10.1038/s41564-019-0626-z
Ling, W., Furuya-Kanamori, L., Ezure, Y., Harris, P. N. A., and Paterson, D. L. (2021). Adverse clinical outcomes associated with infections by Enterobacterales producing ESBL (ESBL-E): a systematic review and meta-analysis. JAC Antimicrob Resist. 3:dlab068. doi: 10.1093/jacamr/dlab068
Liu, J., Wang, X., Xie, H., Zhong, Q., and Xia, Y. (2022). Analysis and evaluation of different sequencing depths from 5 to 20 million reads in shotgun metagenomic sequencing, with optimal minimum depth being recommended. Genome 65, 491–504. doi: 10.1139/gen-2021-0120
Liu, D., Zhou, H., Xu, T., Yang, Q., Mo, X., Shi, D., et al. (2021). Multicenter assessment of shotgun metagenomics for pathogen detection. EBioMedicine 74:103649. doi: 10.1016/j.ebiom.2021.103649
Lu, J., Rincon, N., Wood, D. E., Breitwieser, F. P., Pockrandt, C., Langmead, B., et al. (2022). Metagenome analysis using the kraken software suite. Nat. Protoc. 17, 2815–2839. doi: 10.1038/s41596-022-00738-y
Moser, A. I., Kuenzli, E., Budel, T., Campos-Madueno, E. I., Bernasconi, O. J., Decrom-Beer, S., et al. (2021a). Travellers returning from the island of Zanzibar colonized with MDR Escherichia coli strains: assessing the impact of local people and other sources. J. Antimicrob. Chemother. 76, 330–337. doi: 10.1093/jac/dkaa457
Moser, A. I., Kuenzli, E., Campos-Madueno, E. I., Budel, T., Rattanavong, S., Vongsouvath, M., et al. (2021b). Antimicrobial-resistant Escherichia coli strains and their plasmids in people, poultry, and chicken meat in Laos. Front. Microbiol. 12:708182. doi: 10.3389/fmicb.2021.708182
Mu, A., Kwong, J. C., Isles, N. S., Goncalves Da Silva, A., Schultz, M. B., Ballard, S. A., et al. (2019). Reconstruction of the genomes of drug-resistant pathogens for outbreak investigation through metagenomic sequencing. mSphere 4:4. doi: 10.1128/mSphere.00529-18
Naas, T., Ergani, A., Carrer, A., and Nordmann, P. (2011). Real-time PCR for detection of NDM-1 carbapenemase genes from spiked stool samples. Antimicrob. Agents Chemother. 55, 4038–4043. doi: 10.1128/AAC.01734-10
Nakayama, T., Kumeda, Y., Kawahara, R., and Yamamoto, Y. (2020). Quantification and long-term carriage study of human extended-spectrum/AmpC β-lactamase-producing Escherichia coli after international travel to Vietnam. J. Glob. Antimicrob. Resist. 21, 229–234. doi: 10.1016/j.jgar.2019.11.001
Perez-Perez, F. J., and Hanson, N. D. (2002). Detection of plasmid-mediated AmpC β-lactamase genes in clinical isolates by using multiplex PCR. J. Clin. Microbiol. 40, 2153–2162. doi: 10.1128/JCM.40.6.2153-2162.2002
Petersen, L. M., Martin, I. W., Moschetti, W. E., Kershaw, C. M., and Tsongalis, G. J. (2019). Third-generation sequencing in the clinical laboratory: exploring the advantages and challenges of nanopore sequencing. J. Clin. Microbiol. 58:e01315-19. doi: 10.1128/JCM.01315-19
Peto, L., Fawcett, N. J., Crook, D. W., Peto, T. E. A., Llewelyn, M. J., and Walker, A. S. (2019). Selective culture enrichment and sequencing of feces to enhance detection of antimicrobial resistance genes in third-generation cephalosporin resistant Enterobacteriaceae. PLoS One 14:e0222831. doi: 10.1371/journal.pone.0222831
Pires, J., Kuenzli, E., Kasraian, S., Tinguely, R., Furrer, H., Hilty, M., et al. (2016). Polyclonal intestinal colonization with extended-spectrum cephalosporin-resistant Enterobacteriaceae upon traveling to India. Front. Microbiol. 7:1069. doi: 10.3389/fmicb.2016.01069
Pitout, J. D., Hossain, A., and Hanson, N. D. (2004). Phenotypic and molecular detection of CTX-M-β-lactamases produced by Escherichia coli and Klebsiella spp. J. Clin. Microbiol. 42, 5715–5721. doi: 10.1128/JCM.42.12.5715-5721.2004
Purushothaman, S., Meola, M., and Egli, A. (2022). Combination of whole genome sequencing and metagenomics for microbiological diagnostics. Int. J. Mol. Sci. 23. doi: 10.3390/ijms23179834
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J., and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844. doi: 10.1038/nbt.3935
Raymond, F., Boissinot, M., Ouameur, A. A., Deraspe, M., Plante, P. L., Kpanou, S. R., et al. (2019). Culture-enriched human gut microbiomes reveal core and accessory resistance genes. Microbiome 7:56. doi: 10.1186/s40168-019-0669-7
Rondinaud, E., Ruppe, E., Matheron, S., Lucet, J. C., and Armand-Lefevre, L., VOYAG-R study group (2020). Screening methods for intestinal carriage of multidrug-resistant Enterobacterales: interest of enrichment broth. Diagn. Microbiol. Infect. Dis. 97:115079. doi: 10.1016/j.diagmicrobio.2020.115079
Sadek, M., Poirel, L., and Nordmann, P. (2020). Optimal detection of extended-spectrum β-lactamase producers, carbapenemase producers, polymyxin-resistant Enterobacterales, and vancomycin-resistant enterococci from stools. Diagn. Microbiol. Infect. Dis. 96:114919. doi: 10.1016/j.diagmicrobio.2019.114919
Santiago-Rodriguez, T. M., Garoutte, A., Adams, E., Nasser, W., Ross, M. C., La Reau, A., et al. (2020). Metagenomic information recovery from human stool samples is influenced by sequencing depth and profiling method. Genes 11:11. doi: 10.3390/genes11111380
Schneeberger, P. H. H., Becker, S. L., Pothier, J. F., Duffy, B., N'goran, E. K., Beuret, C., et al. (2016). Metagenomic diagnostics for the simultaneous detection of multiple pathogens in human stool specimens from cote d'Ivoire: a proof-of-concept study. Infect. Genet. Evol. 40, 389–397. doi: 10.1016/j.meegid.2015.08.044
Schwaber, M. J., and Carmeli, Y. (2007). Mortality and delay in effective therapy associated with extended-spectrum β-lactamase production in Enterobacteriaceae bacteraemia: a systematic review and meta-analysis. J. Antimicrob. Chemother. 60, 913–920. doi: 10.1093/jac/dkm318
Soraas, A., Sundsfjord, A., Sandven, I., Brunborg, C., and Jenum, P. A. (2013). Risk factors for community-acquired urinary tract infections caused by ESBL-producing enterobacteriaceae--a case-control study in a low prevalence country. PLoS One 8:e69581. doi: 10.1371/journal.pone.0069581
Tacconelli, E., Cataldo, M. A., Dancer, S. J., De Angelis, G., Falcone, M., Frank, U., et al. (2014). ESCMID guidelines for the management of the infection control measures to reduce transmission of multidrug-resistant gram-negative bacteria in hospitalized patients. Clin. Microbiol. Infect. 20, 1–55. doi: 10.1111/1469-0691.12427
Tham, J., Odenholt, I., Walder, M., Andersson, L., and Melander, E. (2013). Risk factors for infections with extended-spectrum β-lactamase-producing Escherichia coli in a county of southern Sweden. Infect. Drug Resist. 6, 93–97. doi: 10.2147/IDR.S46290
Thomas, T., Gilbert, J., and Meyer, F. (2012). Metagenomics - a guide from sampling to data analysis. Microb. Inform. Exp. 2:3. doi: 10.1186/2042-5783-2-3
Tschudin-Sutter, S., Lucet, J. C., Mutters, N. T., Tacconelli, E., Zahar, J. R., and Harbarth, S. (2017). Contact precautions for preventing nosocomial transmission of extended-spectrum β-lactamase-producing Escherichia coli: a point/counterpoint review. Clin. Infect. Dis. 65, 342–347. doi: 10.1093/cid/cix258
Valverde, A., Grill, F., Coque, T. M., Pintado, V., Baquero, F., Canton, R., et al. (2008). High rate of intestinal colonization with extended-spectrum-β-lactamase-producing organisms in household contacts of infected community patients. J. Clin. Microbiol. 46, 2796–2799. doi: 10.1128/JCM.01008-08
Voor In 't Holt, A. F., Mourik, K., Beishuizen, B., Van Der Schoor, A. S., Verbon, A., Vos, M. C., et al. (2020). Acquisition of multidrug-resistant Enterobacterales during international travel: a systematic review of clinical and microbiological characteristics and meta-analyses of risk factors. Antimicrob. Resist. Infect. Control 9:71. doi: 10.1186/s13756-020-00733-6
Ward, D. V., Scholz, M., Zolfo, M., Taft, D. H., Schibler, K. R., Tett, A., et al. (2016). Metagenomic sequencing with strain-level resolution implicates uropathogenic E. coli in necrotizing enterocolitis and mortality in preterm infants. Cell Rep. 14, 2912–2924. doi: 10.1016/j.celrep.2016.03.015
Wotzka, S. Y., Kreuzer, M., Maier, L., Zund, M., Schlumberger, M., Nguyen, B., et al. (2018). Microbiota stability in healthy individuals after single-dose lactulose challenge-a randomized controlled study. PLoS One 13:e0206214. doi: 10.1371/journal.pone.0206214
Yee, R., Breitwieser, F. P., Hao, S., Opene, B. N. A., Workman, R. E., Tamma, P. D., et al. (2021). Metagenomic next-generation sequencing of rectal swabs for the surveillance of antimicrobial-resistant organisms on the Illumina Miseq and Oxford MinION platforms. Eur. J. Clin. Microbiol. Infect. Dis. 40, 95–102. doi: 10.1007/s10096-020-03996-4
Zaheer, R., Noyes, N., Ortega Polo, R., Cook, S. R., Marinier, E., Van Domselaar, G., et al. (2018). Impact of sequencing depth on the characterization of the microbiome and resistome. Sci. Rep. 8:5890. doi: 10.1038/s41598-018-24280-8
Keywords: ESBL, AmpC, stool, metagenomics, Illumina, enrichment
Citation: Campos-Madueno EI, Aldeia C, Perreten V, Sendi P, Moser AI and Endimiani A (2023) Detection of blaCTX-M and blaDHA genes in stool samples of healthy people: comparison of culture- and shotgun metagenomic-based approaches. Front. Microbiol. 14:1236208. doi: 10.3389/fmicb.2023.1236208
Edited by:
Andreas Erich Zautner, University Hospital Magdeburg, GermanyReviewed by:
Anna Gorska, University of Verona, ItalyCopyright © 2023 Campos-Madueno, Aldeia, Perreten, Sendi, Moser and Endimiani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Endimiani, YW5kcmVhLmVuZGltaWFuaUB1bmliZS5jaA==; YWVuZGltaWFuaUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.