 Ping Zhu
Ping Zhu Tejas Somvanshi
Tejas Somvanshi Jichen Bao
Jichen Bao Silvan Scheller
Silvan Scheller
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 12 December 2023
Sec. Biology of Archaea
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1235616
This article is part of the Research TopicThe Methane Moment – Cross-Boundary Significance of MethanogensView all 20 articles
Methanogenic archaea play an important role in the global carbon cycle and may serve as host organisms for the biotechnological production of fuels and chemicals from CO2 and other one-carbon substrates. Methanosarcina acetivorans is extensively studied as a model methanogen due to its large genome, versatile substrate range, and available genetic tools. Genome editing in M. acetivorans via CRISPR/Cas9 has also been demonstrated. Here, we describe a user-friendly CRISPR/Cas12a toolbox that recognizes T-rich (5′-TTTV) PAM sequences. The toolbox can manage deletions of 3,500 bp (i.e., knocking out the entire frhADGB operon) and heterologous gene insertions with positive rates of over 80%. Cas12a-mediated multiplex genome editing was used to edit two separate sites on the chromosome in one round of editing. Double deletions of 100 bp were achieved, with 8/8 of transformants being edited correctly. Simultaneous deletion of 100 bp at one site and replacement of 100 bp with the 2,400 bp uidA expression cassette at a separate site yielded 5/6 correctly edited transformants. Our CRISPR/Cas12a toolbox enables reliable genome editing, and it can be used in parallel with the previously reported Cas9-based system for the genetic engineering of the Methanosarcina species.
Methanogenic archaea are significant global contributors to the production of the potent greenhouse gas methane, which is estimated to be about 1 Gt/year (Thauer et al., 2010). At the same time, these microorganisms are increasingly utilized for the biotechnological reduction of atmospheric CO2, and therefore may help mitigate global warming. Methanosarcina species are among the best studied, as many details about their genomic information (Deppenmeier et al., 2002; Galagan et al., 2002; Maeder et al., 2006) and versatile methanogenesis (Costa and Leigh, 2014; Kurth et al., 2020) have been earlier revealed. Metabolic engineering of Methanosarcina facilitates many diverse biotechnological applications. For instance, the engineered expression of an esterase in Methanosarcina acetivorans allowed cells to utilize methyl esters for growth and methane production (Lessner et al., 2010). Additionally, isoprene production was demonstrated by engineering the expression of an isoprene synthase gene in Methanosarcina (Aldridge et al., 2021).
Genome manipulations in M. acetivorans are typically more challenging than those in well-established host microorganisms (e.g., Escherichia coli) because of the multiple copies of its genome (Hildenbrand et al., 2011). If not all genome copies are uniformly edited, heterozygous mutants that still retain a wild-type chromosome may persist. For metabolic engineering projects requiring several rounds of editing, marker-free edits are necessary. In Methanosarcina, the puromycin acetyltransferase (pac) gene for puromycin resistance and the isoleucyl-tRNA synthetase (ileS) gene for pseudomonic acid resistance are the two selective markers currently used (Metcalf et al., 1997; Boccazzi et al., 2000). The hypoxanthine phosphoribosyltransferase (hpt) gene has been widely utilized as a counterselectable marker in Methanosarcina Δhpt hosts (Guss et al., 2008). Traditional gene editing, which involves introducing linear DNA fragments followed by homologous recombination, generally shows lower transformation and editing efficiencies than CRISPR (clustered regularly interspaced short palindromic repeats)-based methods (Ronda et al., 2016). The ΦC31 integrase-mediated site-specific recombination system, which has a fixed target site on the genome (Guss et al., 2008), is efficient for introducing a singular piece of DNA, but lacks the ability to edit multiple sites or perform repeated edits. As an alternative, CRISPR system-mediated genome editing tools have been developed for methanogens. The CRISPR/Cas9 system was initially generated for M. acetivorans (Nayak and Metcalf, 2017), followed by the construction of the CRISPRi-dCas9 tool for gene regulation (Dhamad and Lessner, 2020) in the same methanogen.
Cas12a (also known as Cpf1) is a class 2 type V endonuclease (Paul and Montoya, 2020) that recognizes a thymine (T)-rich (5′-TTTV, V = A, G, and C) protospacer adjacent motif (PAM) (Safari et al., 2019), as opposed to the guanine (G)-rich (NGG-3′) PAM of Cas9. Consequently, Cas12a increases the number of potential targeting sites along the genome, especially for T-rich hosts. The double-stranded breaks (DSB) caused by Cas12a will generate sticky ends, which are helpful for DNA repair and genome stability during genetic manipulation (Vanegas et al., 2019). Since the gRNA (guide RNA, also called spacer) does not rely on tracrRNA (trans-activating CRISPR RNA) for maturation (Nakade et al., 2017), edited constructions become simplified, particularly when multiplex genome editing is employed (Bandyopadhyay et al., 2020). More recently, a Cas12a-based genome editing system has been successfully implemented for genetic engineering in Methanococcus maripaludis (Bao et al., 2022).
To increase the versatility of CRISPR editing in M. acetivorans, we constructed a system that expresses the Cas12a endonuclease from Lachnospiraceae bacterium (LbCas12a). To evaluate the performance of this editing toolbox, single gRNA-mediated gene knockout and heterologous gene integration approaches were used. As a tool to expedite metabolic engineering, Cas12a-mediated multiplex genome editing was to establish gene edits at two independent sites along the genome with only one round of editing.
To generate a Cas12a-gRNA expression system that is cloneable in most E. coli strains, the E. coli/Methanosarcina shuttle vector pM000 (Supplementary Figure S1B) was constructed from the pWM321 plasmid (Metcalf et al., 1997) by replacing the origin of replication (ori) from plasmid R6K with the ori from plasmid ColE1. To establish markerless editability, the hpt gene that confers sensitivity to the purine analog 8-aza-2,6-diaminopurine (8ADP) for counter selection (Ehlers et al., 2011) was inserted downstream of the pac gene, yielding plasmid pM001 (Supplementary Figure S1C). The LbCas12a gene was amplified from pY016 (Zetsche et al., 2015), fused with the tetracycline-regulated promoter PmcrB (tetO1), and inserted into the multiple cloning site (MCS) of pM001 to yield the plasmid pMCp4. To obtain the final Cas12a-gRNA expression system, the gRNA cassette is introduced into pMCp4 (Figure 1A). It is worth mentioning that tetracycline was not required for the expression of Cas12a, as in the previous studies with Cas9, where no significant difference in the genome editing efficiency was observed in M. acetivorans (Nayak and Metcalf, 2017; Dhamad and Lessner, 2020).
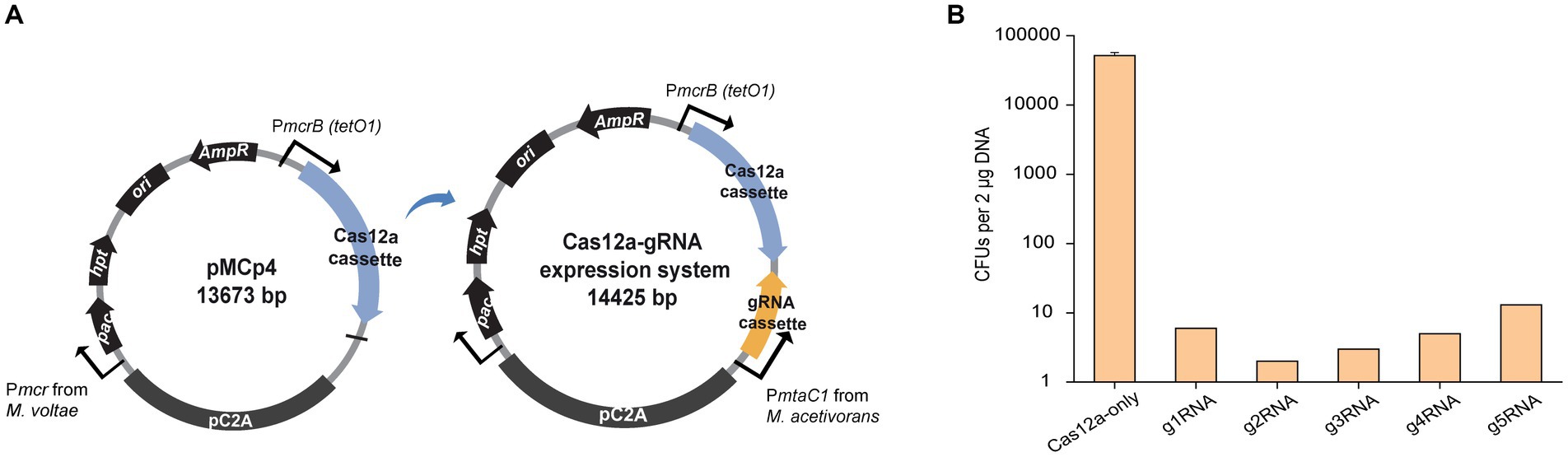
Figure 1. Construction of the Cas12a-gRNA expression system for M. acetivorans. (A) Schematic diagram of pMCp4 and Cas12a-gRNA expression system. The plasmid pMCp4 expresses the Cas12a protein in M. acetivorans. The Cas12a-gRNA expression system expresses the Cas12a-gRNA complex in M. acetivorans. Cas12a and gRNA cassettes are equipped with tetracycline-regulated promoter PmcrB (tetO1) and the promoter PmtaC1 from M. acetivorans, respectively. (B) Targeting efficiency of the Cas12a-gRNA expression system. Cas12a-only, plasmid pMCp4. g1RNA, g2RNA, g3RNA, g4RNA, and g5RNA, five gRNAs designed for targeting various locations along the ssuC gene. Error bar represents the standard deviation of triplicate measurements. Standard deviations were not determined for gRNA-expressing transformation data, as all cells were plated out to analyze the lethal efficiency of the Cas12a-gRNA complex.
To verify whether the Cas12a protein is not cytotoxic, the shuttle vector pM001 and the Cas12a-expressing plasmid pMCp4 were each transformed into M. acetivorans, and then the transformation efficiencies were compared. When 2 μg of plasmid DNA was used for the transformations, pM001 yielded 40,000 ± 20,000 CFUs of PurR transformants and pMCp4 yielded 34,000 ± 19,000 CFUs of PurR transformants (p = 0.75 in two-tailed t-test, Supplementary Figure S2), which indicates the expression of Cas12a protein is non-toxic to M. acetivorans cells.
To evaluate the targeting efficiency of the Cas12a-gRNA expression system, five different gRNAs were used to target various locations along the non-essential ssuC gene (which encodes the permease subunit of the sulfonate ABC transporter) for introducing DSBs on the genome. All five gRNAs resulted in less than 15 CFUs (per 2 μg DNA) in the transformation, thus demonstrating the high gene-targeting efficiency of the constructed system (Figure 1B).
To investigate the efficiency of gene deletion, the homologous repair (HR) arms for gene editing were inserted into the Cas12a-gRNA expression system to generate the Cas12a-gRNA gene editing system (Figure 2A). The g1RNA (5’-TTGCGATTCCCTCAGCCATG CCC-3′) was used to target the ssuC locus. For gRNA cassette construction, PCR amplification was used to synthesize the gRNA and direct repeat (DR) sequences that contain promoter (PmtaC1) and terminator (TmtaB1) regions (Supplementary Figure S3A). Deletion lengths varied from 100 bp to 2000 bp (Figure 2B) and a 1,000 bp length of flanking HR sequence was chosen as optimal based on previous work (Nayak and Metcalf, 2017).
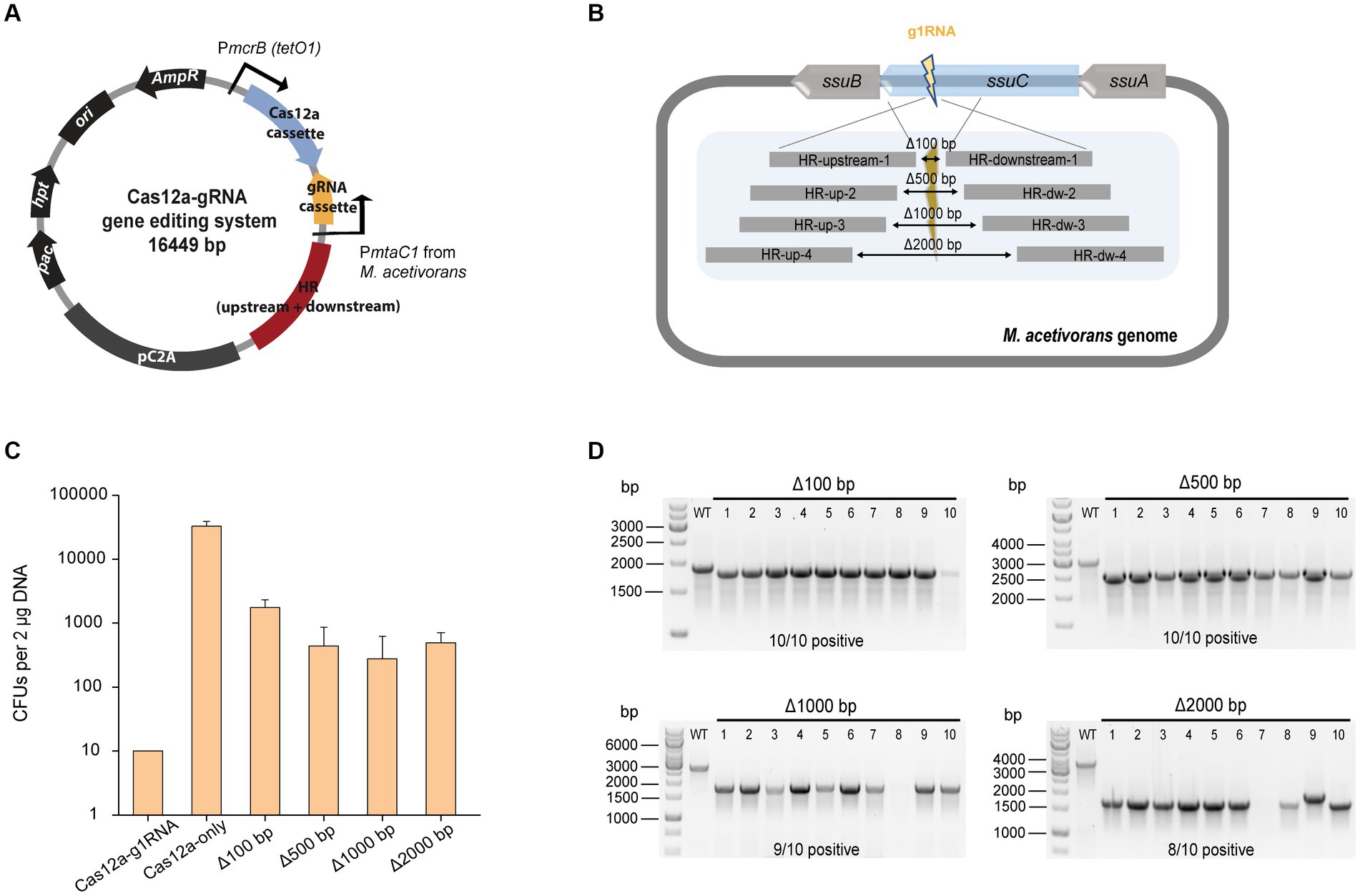
Figure 2. CRISPR/Cas12a-mediated gene knockout. (A) Schematic diagram of Cas12a-gRNA gene editing system. Cas12a and gRNA cassettes are equipped with promoters PmcrB (tetO1) and PmtaC1, respectively. Gene editing was achieved by the introduced upstream and downstream homologous repair (HR) arms. (B) Scheme for generating gene deletions in ssuC. g1RNA was designed to target ssuC to form a double-stranded break (DSB). Various sizes of gene knockouts were generated by introducing a 1,000-bp length of flanking HR sequence near the leakage site. Δ100 bp, Δ500 bp, Δ1000 bp, and Δ2000 bp, plasmids generating 100 bp, 500 bp, 1,000 bp, and 2000 bp of gene knockouts while repairing the DSB. (C) Transformation efficiency of deletion-generating plasmids. Cas12a-g1RNA, plasmid expressing Cas12a-g1RNA complex that targets ssuC to produce the DSB on the genome. Cas12a-only, plasmid pMCp4 expresses Cas12a. Error bars represent the standard deviation of triplicate measurements. Standard deviations were not determined for Cas12a-g1RNA transformation data, as all cells were plated out to analyze the lethal efficiency of the Cas12a-g1RNA complex. (D) Editing efficiency of deletion-generating plasmids. Ten PurR transformants were randomly selected for colony PCR. WT, wild type M. acetivorans strain. Thermo Scientific™ GeneRuler 1 kb DNA ladder was used for sizing DNA fragments.
When the plasmid pMCp2-g1RNA (Table 1) was transformed into M. acetivorans, the Cas12a-g1RNA complex was expressed and only 10 PurR transformants were observed (Figure 2C). Transformation of plasmid pMCp4 (Cas12a-only) yielded 33,000 ± 6,000 CFUs of PurR transformants (per 2 μg DNA). Among the various lengths of deletions tested, the 100 bp-deletion from the genome gave the highest repairing efficiency (1,800 ± 600 CFUs). This higher transformation efficiency obtained when only a small fragment of the chromosome is deleted also matches a previous Cas9-mediated gene knockout study, which concluded shorter deletions (i.e., Δ100 bp and Δ500 bp) are more stable and reliable (Nayak and Metcalf, 2017). For the plasmids that led to larger fragment deletions (i.e., Δ1000 bp and Δ2000 bp), similar transformant yields were obtained. To verify the knockout (or editing) efficiency, 10 transformants were randomly selected in each trial and subjected to colony PCR (Figure 2D). The primers involved are presented in Supplementary Figure S4A and Supplementary Table S1. Higher positive rates were observed with the shorter deletions, in which 100% of the 100 bp-deleted transformants were positive. On the other hand, 80% of positive clones were observed in the 2000 bp-deletion experiments. Three isolates from each transformation set were randomly selected for Sanger sequencing and all were edited correctly (Supplementary Figure S4B). The Sanger sequencing results were provided in Supplementary Table S2.
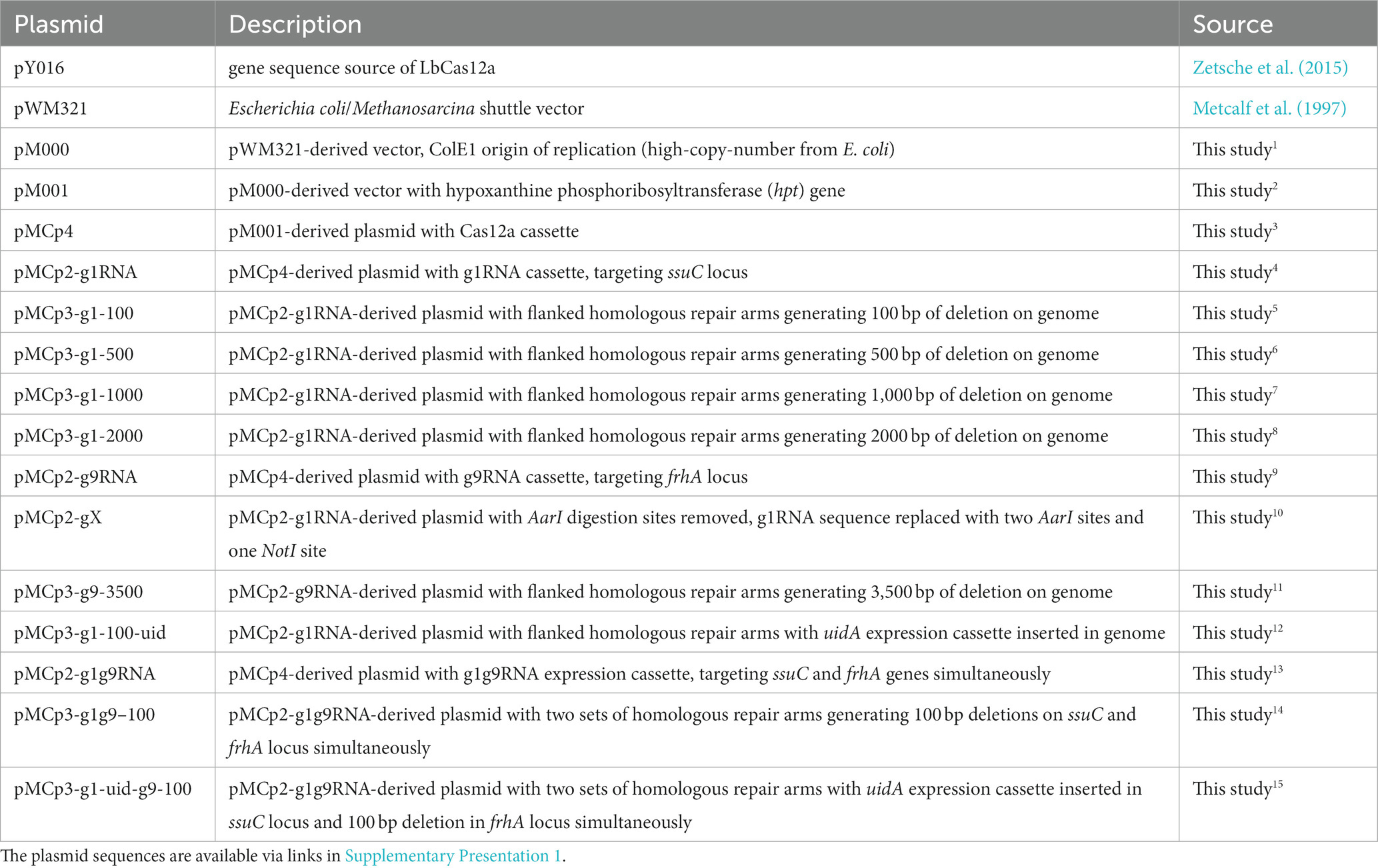
Table 1. Plasmids used in this study.
In order to assess the gene insertion performance of our Cas12a-gRNA gene editing system, a heterologous gene cassette for the expression of β-glucuronidase (uidA) gene was placed within the HR arms, given its previous successful use in Methanosarcina (Guss et al., 2008). For uidA cassette construction, the uidA gene was PCR amplified from E. coli BL21 genomic DNA and fused with the mcr promoter (Pmcr) and terminator (Tmcr) from M. barkeri (Supplementary Figure S5A). The insertion efficiency was obtained by comparing the number of PurR transformants from the editing plasmid pMCp3-g1-100-uid (8,800 ± 2,400 CFUs) and plasmid pMCp4 (117,000 ± 12,000 CFUs) (Figure 3A). The positive rate for 20 transformants was determined by colony PCR using the primers veri2/veri8 (Figure 3B). To ensure the genome copies within one transformant were all edited (homozygous chromosome), a second set of primers (veri5/veri12) was used to detect the presence of possible mixed transformants (Supplementary Figure S5B). 100% of the transformants were identified with uidA cassette and no heterozygous genotypes were detected. Three isolates were randomly selected for Sanger sequencing to detect the uidA gene cassette and all the edits were positive (Supplementary Figure S5C and Supplementary Table S2), demonstrating the high performance for Cas12a-mediated gene insertion.
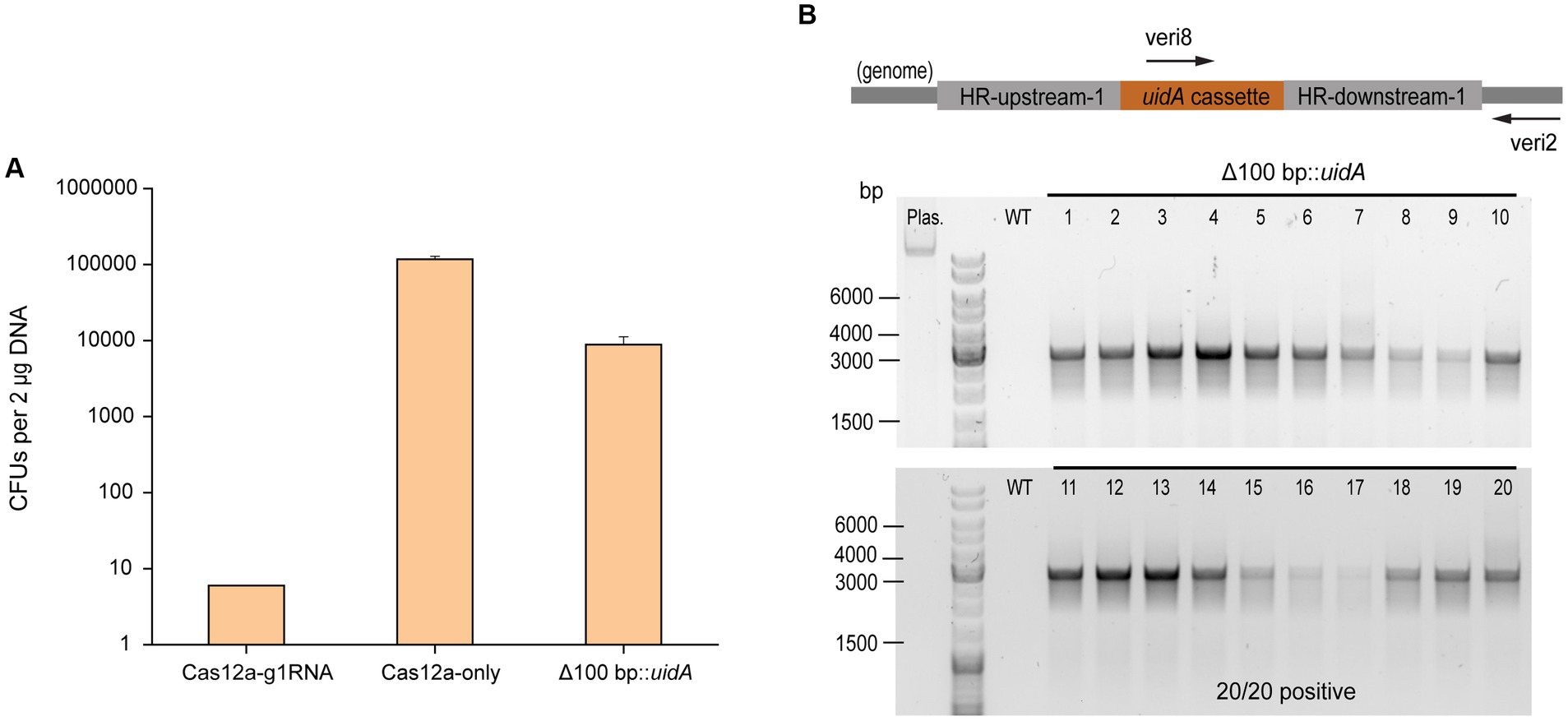
Figure 3. CRISPR/Cas12a-mediated gene insertion. (A) Transformation efficiency of Cas12a-mediated gene insertion. Cas12a-g1RNA, plasmid pMCp2-g1RNA expressing Cas12a-g1RNA complex that targets ssuC to produce the DSB on the genome. Cas12a-only, plasmid pMCp4 expresses Cas12a. Δ100 bp::uidA, plasmid pMCp3-g1-100-uid replaced 100 bp with the uidA expressing cassette in genome. Error bars represent the standard deviation of triplicate measurements. Standard deviations were not determined for Cas12a-g1RNA transformation data, as all cells were plated out to analyze the lethal efficiency of the Cas12a-g1RNA complex. (B) Editing efficiency of gene insertion-generating plasmid. Upper panel, scheme for the engineered genome containing uidA cassette and the detecting primers used in colony PCR. veri8 and veri2, forward and reverse primer target uidA and genome. HR-upstream-1 and HR-downstream-1, the flanking HR sequence identical to the ones used in Figure 2B. Bottom panel, 20 PurR transformants were randomly selected for colony PCR to verify the existance of the uidA gene. Plas. and WT are plasmid pMCp3-g1-100-uid and wild type M. acetivorans genome, respectively, and served as negative controls. Thermo Scientific™ GeneRuler DNA Ladder Mix was used for sizing DNA fragments.
First, plasmid pMCp2-g1g9RNA was constructed to simultaneously target the ssuC and frhA (encoding the coenzyme F420 hydrogenase alpha subunit) genes (Figure 4A), where the gRNA cassette contains two tandem gRNA sequences (g1RNA and g9RNA) equipped with promoter PmtaC1 and terminator TmtaB1 (Supplementary Figure S6). Next, to assess the effect of double-site deletions, two sets of HR sequences that generate 100-bp deletions in each of the genes (see above) were designed and assembled into pMCp2-g1g9RNA, producing the plasmid pMCp3-g1g9–100. Among the obtained PurR transformants (107 ± 23 CFUs per 2 μg DNA), eight were randomly selected for colony PCR. Primers veri1/veri2 and veri11/Cp54 were used to target the genome regions flanked the ssuC-and frhA-breakages, separately (Figure 4B). All eight transformants were positive and the desired gene deletions were detected in both ssuC and frhA regions. Three isolates were randomly picked for Sanger sequencing and all clones were correctly edited (Supplementary Figure S7A).
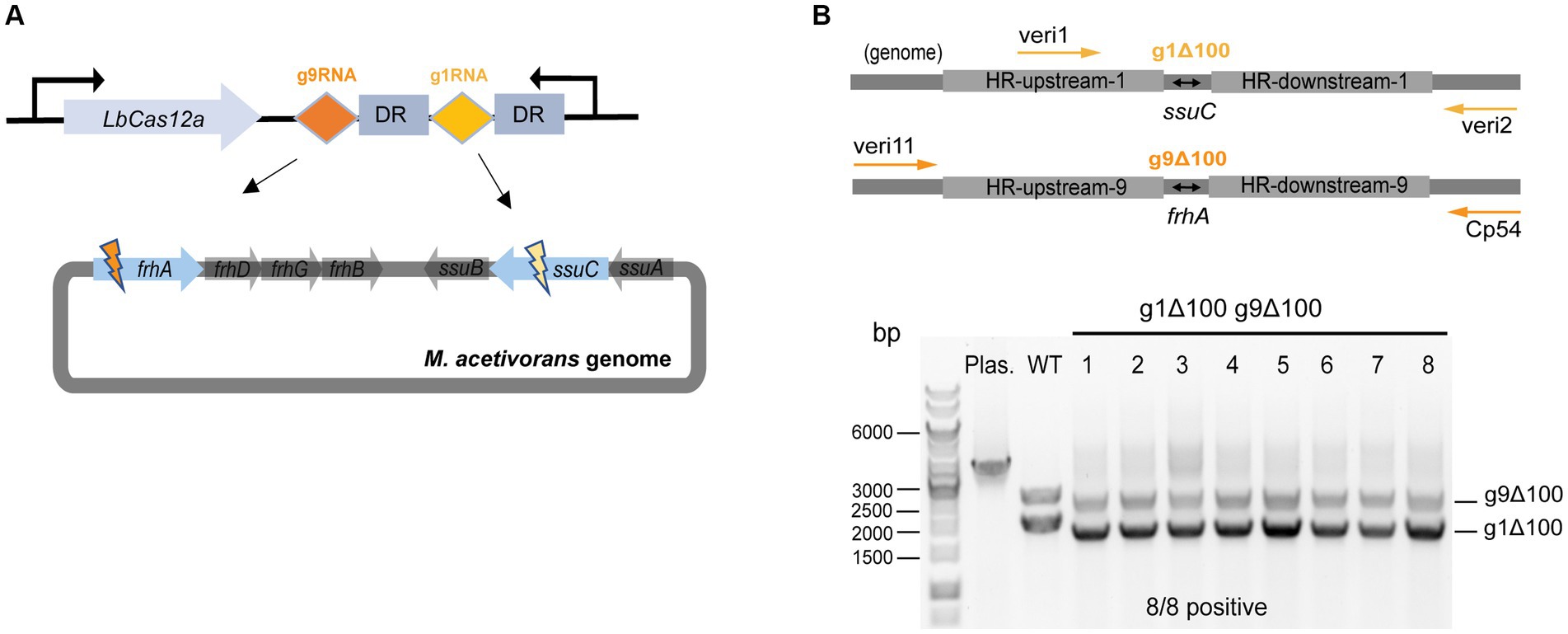
Figure 4. CRISPR/Cas12a-mediated multiplex genome editing. (A) The CRISPR array designed for targeting two sites on the genome. g1RNA and g9RNA target ssuC and frhA, generating two DSBs in the genome simultaneously. DR, direct repeat sequences in the gRNA cassette. (B) Editing efficiency of the multiplex gene knockout. Upper panel, scheme for the engineered genome with 100 bp-deletions in both ssuC and frhA sites and the primers used in colony PCR. veri1 and veri2, forward and reverse primer target HR sequence and the downstream of ssuC. veri11 and Cp54, forward and reverse primer target the upstream and the downstream of frhA. Bottom panel, positive rate of multiplex gene knockout. Eight transformants were randomly selected for colony PCR to verify the gene knockout efficiency. Plas. and WT are plasmid pMCp3-g1g9–100 and wild type M. acetivorans strain, respectively, and served as the negative controls. g1Δ100, gene deletion in ssuC. g9Δ100, gene deletion in frhA. Thermo Scientific™ GeneRuler 1 kb DNA ladder was used for sizing DNA fragments.
To evaluate the versatility of our CRISPR/Cas12a toolbox for multiplex editing, simultaneous gene deletion and insertion at two independent sites were attempted with the plasmid pMCp3-g1-uid-g9-100. This construct facilitates the insertion of the uidA cassette into the ssuC locus as well as makes a 100-bp deletion in the frhA gene in one round of transformation (Supplementary Figure S7B). Out of the 280 CFUs of PurR transformants, six were randomly selected for colony PCR verification using primers veri2/veri8 and veri9/veri13 (Supplementary Figure S7C). 5/6 were edited correctly in both sites (i.e., contain the uidA cassette within the ssuC gene and exhibit a 100 bp-deletion within the frhA gene), which indicates the potential of using CRISPR/Cas12a system in multi-tasks genome editing in metabolic engineering applications.
To demonstrate iterative editing with the CRISPR/Cas12a system, the plasmid curing efficiency was calculated for the positively engineered M73-uidA isolate. For plasmid removal, the isolate was cultivated in HS-methanol medium without puromycin for five serial transfers (i.e., 1% inoculum of cells in early stationary phase was utilized in each transfer). Subsequently, cells were streaked out on HS solid medium containing 20 μg/mL 8ADP for counterselection. Seven 8ADPR transformants were randomly selected to detect the curing efficiency and the consistency of gene editing. As a result, 7/7 of the isolates exhibited outright plasmid removal, while 5/7 had correctly inserted uidA cassettes (Supplementary Figure S8). To explore a faster procedure, plasmids removal efficiency after only one round of transfer (same conditions as above) was evaluated. Same plasmid curing efficiency was obtained with the randomly selected 8ADPR transformants, demonstrating the reliable application of the CRISPR/Cas12a system for performing multiple rounds of gene editing in M. acetivorans.
We developed a CRISPR/Cas12a toolbox to facilitate the genetic engineering of M. acetivorans. No significant effects on cell growth were observed with LbCas12a protein expression in M. acetivorans, allowing CRISPR/Cas12a to be a useful tool in this methanogen. Achieving a higher targeting efficiency usually requires the simultaneous screening of multiple gRNA sequences, which often involves using laborious and multi-step methods to construct several plasmids. As a simplification, we constructed AarI-free plasmid pMCp2-gX (Supplementary Figure S9), thereby allowing for quick preparation of multiple Cas12a-gRNA-expressing plasmids through the Gibson assembly method.
Many factors can affect the editing performance of CRISPR/Cas systems, such as the genome regions to be edited, gRNA targeting efficiency and precision, and HR sequence length. For the present study, a 23-nt gRNA was empirically chosen, as the longer sequence length displayed improved targeting specificity compared to a shorter 20-nt gRNA (Bin Moon et al., 2018; Gao et al., 2018). Conversely, shorter gRNA sequence lengths in the Cas9-mediated system proved to be more effective (Fu et al., 2014). It is worth noting that transformation efficiencies varied when using polyethylene glycol (PEG) 4,000-mediated transformation, although there was no significant effect on the positive rates.
Long fragment deletions are reported to be more efficient with the CRISPR/Cas12a system than the Cas9-based one (Wang et al., 2022), which is consistent with our experiments where the Cas12a-mediated long fragment deletion of 2000 bp resulted in a higher number of transformants than obtained with CRISPR/Cas9 editing (Nayak and Metcalf, 2017). To establish the possible length limits of gene deletion with our CRISPR/Cas12a system, the plasmid pMCp3-g9-3500 was constructed for removing the entire frhADGB operon, resulting in a 3,500 bp gene knockout from the genome. A higher number of PurR transformants (2,800 ± 1,400 CFUs) was obtained in comparison to the 2000 bp-deletion trial. Colony PCR and Sanger sequencing proved that all the transformants selected were edited correctly and no heterozygous genotypes were observed (Supplementary Figure S10), which suggests that our CRISPR/Cas12a editing tool is efficient for making long fragment deletions.
To assess the accuracy and effectiveness of our toolbox, five randomly selected engineered strains as well as a parent strain were sent for whole genome sequencing (WGS). All genomes contained the desired mutations. It must be mentioned, however, that intrachromosomal translocations occurred in 3/5 of the tested strains (Supplementary Table S3), while no structural variants were detected in the parent strain. These intrachromosomal translocations have not been observed in previous mutational techniques used in M. acetivorans. Intrachromosomal translocations arise due to the mistakes made by the repair mechanism of the cells (Brunet and Jasin, 2018). As non-homologous end joining (NHEJ) is absent in M. acetivorans, the possible reasons for genomic aberrations are either alternative NHEJ as known as microhomology-mediated end joining (MMEJ), or homologous recombination pathways (Agarwal et al., 2006; Nayak and Metcalf, 2017). The possible presence of MMEJ in Methanosarcina has been hypothesized given the presence of PARP-like enzyme (Raychaudhuri et al., 2000). Additionally, M. acetivorans genome contains annotated flap endonuclease (FEN) which is also involved in MMEJ pathway. Detailed understanding of MMEJ in archaea would require additional studies (Marshall and Santangelo, 2020), but MMEJ being one of the leading causes of translocation has been shown in mouse embryonic stem cells (Brunet and Jasin, 2018). The tendency of all CRISPR systems (Cas9, Cas12a, and Cas12f) to induce intrachromosomal translocations was also revealed in the editing of eukaryotic cells (Xin et al., 2022). Therefore, CRISPR tools-triggered genomic structural variations should be considered when using them for genome editing in M. acetivorans and as such we would recommend users for WGS after using the toolbox. Genome translocations have been important factors for the evolution and adaption of Methanosarcinales, where many organisms were reported with rich transposable elements and transposases (Gehlert et al., 2023). Genome annotations of M. acetivorans have been shown to carry more than 70 transposases (Galagan et al., 2002), indicating its potentially active transposon activity. If not the DNA repair mechanisms, another possible cause for intra-chromosomal translocations could be the transposases. By coupling it with WGS, this toolbox provides an additional tool in the repertoire for editing M. acetivorans.
High-salt (HS) medium containing 125 mM methanol was used for cultivating M. acetivorans in single-cell form at 37°C (Sowers et al., 1993). M. acetivorans WWM73 was used as the host strain in this study and relevant derivatives are listed in Table 2. HS solid medium containing 1.4% agar (Bacto™ Dehydrated, Fisher Scientific) and 2 μg/mL puromycin (InvivoGen, Inc.) was used for screening M. acetivorans transformants. HS solid medium containing 1.4% agar and 20 μg/mL 8ADP (Sigma-Aldrich) was used for screening M. acetivorans transformants in the plasmid curing experiment. E. coli DH10B (Fisher Scientific) and E. coli XL10-Gold (Agilent Technologies) were used as the cloning hosts for constructing CRISPR plasmids. E. coli transformants were selected using lysogeny broth (LB) solid medium supplemented with 100 μg/mL ampicillin.

Table 2. Strains used in this study.
All plasmids used in this study are listed in Table 1. All primers are listed in Supplementary Table S1. PrimeSTAR® Max DNA Polymerase (Takara Bio) was used for amplifying gene fragments by PCR. SapphireAmp Fast PCR Master Mix (Takara Bio) was used for verifying E. coli and M. acetivorans transformants by colony PCR. HiFi DNA Assembly Master Mix (New England BioLabs) was used for Gibson assembly and T4 DNA Ligase (Promega) was used for ligation. crRNA sequences for multiplex genome targeting were synthesized and combined using the Gibson assembly method. Donor DNA with upstream and downstream of HR arms was inserted immediately downstream of the gRNA cassette using either Gibson assembly or ligation to construct the CRISPR/Cas12a genome editing system, known as plasmid pMCp3-X in Table 1.
Polyethylene glycol (PEG) 4,000-mediated transformation of M. acetivorans was performed with 2 μg DNA transformed according to the method described previously (Oelgeschläger and Rother, 2009). M. acetivorans transformants were grown on plates of HS solid medium with required antibiotics and incubated at 37°C for 10–15 days in an anaerobic jar with a controlled headspace of N2/CO2/1% H2S (75/20/5). Chemically competent E. coli cells were used for the transformation of assembled and ligated plasmids.
The genomic DNA was extracted with Wizard® Genomic DNA Purification Kit (Promega). The Microbial WGS was performed using Illumina NovaSeq 6,000 by Novogene (UK) Company. Samples were sequenced in a paired-end 150 bp sequencing strategy and aligned with the M. acetivorans (C2A) reference genome (NC_003552.1) through BWA (Li and Durbin, 2009) software (parameters: mem-t 4-k 32-M). Structural variants (large deletions, insertions and translocations) were predicted using BreakDancer (Chen et al., 2009). Relevant data has been uploaded in National Center for Biotechnology Information (NCBI) with accession no. PRJNA1026875.
The original contributions presented in the study are included in the article/Supplementary materials, further inquiries can be directed to the corresponding authors.
PZ, JB, and SS conceived the study. PZ and JB performed the experiments. TS and PZ analyzed the whole genome sequencing results. PZ wrote the manuscript. JB and SS supervised the experiments and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by Novo Nordisk Foundation (grant NNF19OC0054329 to SS; grant NNF20OC0065032 to JB) and the Academy of Finland (grant 326020 to SS).
We thank the Novo Nordisk Foundation (grant NNF19OC0054329 to SS; grant NNF20OC0065032 to JB) and the Academy of Finland (grant 326020 to SS) for funding this research. We thank the Aalto Science Institute (AScI) project student Shreyash Borkar for helping with the plasmid transformation. We thank Prof. Michael Rother and Dr. Christian Schöne for testing the editing efficiency of our CRISPR/Cas12a system in their lab. We thank Dr. Ingemar von Ossowski for his helpful comments and suggestions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1235616/full#supplementary-material
Agarwal, S., Tafel, A. A., and Kanaar, R. (2006). DNA double-strand break repair and chromosome translocations. DNA Repair 5, 1075–1081. doi: 10.1016/j.dnarep.2006.05.029
Aldridge, J., Carr, S., Weber, K. A., and Buan, N. R. (2021). Anaerobic production of isoprene by engineered Methanosarcina species Archaea. Appl. Environ. Microbiol. 87, e02417–e02420. doi: 10.1128/AEM.02417-20
Bandyopadhyay, A., Kancharla, N., Javalkote, V. S., Dasgupta, S., and Brutnell, T. P. (2020). CRISPR-Cas12a (Cpf1): a versatile tool in the plant genome editing tool box for agricultural advancement. Front. Plant Sci. 11:584151. doi: 10.3389/fpls.2020.584151
Bao, J., de Dios Mateos, E., and Scheller, S. (2022). Efficient CRISPR/Cas12a-based genome-editing toolbox for metabolic engineering in Methanococcus maripaludis. ACS Synth. Biol. 11, 2496–2503. doi: 10.1021/acssynbio.2c00137
Bin Moon, S., Lee, J. M., Kang, J. G., Lee, N.-E., Ha, D.-I., Kim, D. Y., et al. (2018). Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3′-overhang. Nat. Commun. 9:3651. doi: 10.1038/s41467-018-06129-w
Boccazzi, P., Zhang, K., and Metcalf, W. W. (2000). Generation of dominant selectable markers for resistance to Pseudomonic acid by cloning and mutagenesis of the ileS gene from the archaeon Methanosarcina barkeri Fusaro. J. Bacteriol. 182, 2611–26118. doi: 10.1128/JB.182.9.2611-2618.2000
Brunet, E., and Jasin, M. (2018). Induction of chromosomal translocations with CRISPR-Cas9 and other nucleases: understanding the repair mechanisms that give rise to translocations. Adv. Exp. Med. Biol. 1044, 15–25. doi: 10.1007/978-981-13-0593-1_2
Chen, K., Wallis, J. W., McLellan, M. D., Larson, D. E., Kalicki, J. M., Pohl, C. S., et al. (2009). BreakDancer: an algorithm for high resolution mapping of genomic structural variation. Nat. Methods 6, 677–681. doi: 10.1038/nmeth.1363
Costa, K. C., and Leigh, J. A. (2014). Metabolic versatility in methanogens. Curr. Opin. Biotechnol. 29, 70–75. doi: 10.1016/j.copbio.2014.02.012
Deppenmeier, U., Johann, A., Hartsch, T., Merkl, R., Schmitz, R. A., Martinez-Arias, R., et al. (2002). The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J. Mol. Microbiol. Biotechnol. 4, 453–461.
Dhamad, A. E., and Lessner, D. J. (2020). A CRISPRi-dCas9 system for Archaea and its use to examine gene function during nitrogen fixation by Methanosarcina acetivorans. Appl. Environ. Microbiol. 86:e01402-20. doi: 10.1128/AEM.01402-20
Ehlers, C., Jäger, D., and Schmitz, R. A. (2011). Establishing a markerless genetic exchange system for Methanosarcina mazei strain Gö1 for constructing chromosomal mutants of small RNA genes. Archaea 2011, 1–7. doi: 10.1155/2011/439608
Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M., and Joung, J. K. (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 32, 279–284. doi: 10.1038/nbt.2808
Galagan, J. E., Nusbaum, C., Roy, A., Endrizzi, M. G., Macdonald, P., FitzHugh, W., et al. (2002). The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 12, 532–542. doi: 10.1101/gr.223902
Gao, Z., Herrera-Carrillo, E., and Berkhout, B. (2018). Improvement of the CRISPR-Cpf1 system with ribozyme-processed crRNA. RNA Biol. 15, 1458–1467. doi: 10.1080/15476286.2018.1551703
Gehlert, F. O., Nickel, L., Vakirlis, N., Hammerschmidt, K., Vargas Gebauer, H. I., Kießling, C., et al. (2023). Active in vivo translocation of the Methanosarcina mazei Gö1 Casposon. Nucleic Acids Res. 51, 6927–6943. doi: 10.1093/nar/gkad474
Guss, A. M., Rother, M., Zhang, J. K., Kulkkarni, G., and Metcalf, W. W. (2008). New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2, 193–203. doi: 10.1155/2008/534081
Hildenbrand, C., Stock, T., Lange, C., Rother, M., and Soppa, J. (2011). Genome copy numbers and gene conversion in methanogenic archaea. J. Bacteriol. 193, 734–743. doi: 10.1128/JB.01016-10
Kurth, J. M., Op den Camp, H. J. M., and Welte, C. U. (2020). Several ways one goal—methanogenesis from unconventional substrates. Appl. Microbiol. Biotechnol. 104, 6839–6854. doi: 10.1007/s00253-020-10724-7
Lessner, D. J., Lhu, L., Wahal, C. S., and Ferry, J. G. (2010). An engineered methanogenic pathway derived from the domains Bacteria and Archaea. mBio 1, e00243–e00210. doi: 10.1128/mBio.00243-10
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Maeder, D. L., Anderson, I., Brettin, T. S., Bruce, D. C., Gilna, P., Han, C. S., et al. (2006). The Methanosarcina barkeri genome: comparative analysis with Methanosarcina acetivorans and Methanosarcina mazei reveals extensive rearrangement within methanosarcinal genomes. J. Bacteriol. 188, 7922–7931. doi: 10.1128/JB.00810-06
Marshall, C. J., and Santangelo, T. J. (2020). Archaeal DNA repair mechanisms. Biomolecules 10:1472. doi: 10.3390/biom10111472
Metcalf, W. W., Zhang, J. K., Apolinario, E., Sowers, K. R., and Wolfe, R. S. (1997). A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc. Natl. Acad. Sci. 94, 2626–2631. doi: 10.1073/pnas.94.6.2626
Nakade, S., Yamamoto, T., and Sakuma, T. (2017). Cas9, Cpf1 and C2c1/2/3—What’s next? Bioengineered 8, 265–273. doi: 10.1080/21655979.2017.1282018
Nayak, D. D., and Metcalf, W. W. (2017). Cas9-mediated genome editing in the methanogenic archaeon Methanosarcina acetivorans. Proc. Natl. Acad. Sci. U. S. A. 114, 2976–2981. doi: 10.1073/pnas.1618596114
Oelgeschläger, E., and Rother, M. (2009). In vivo role of three fused corrinoid/methyl transfer proteins in Methanosarcina acetivorans. Mol. Microbiol. 72, 1260–1272. doi: 10.1111/j.1365-2958.2009.06723.x
Paul, B., and Montoya, G. (2020). CRISPR-Cas12a: functional overview and applications. Biom. J. 43, 8–17. doi: 10.1016/j.bj.2019.10.005
Raychaudhuri, S., Karmakar, P., and Thakur, A. R. (2000). γ-Ray-induced DNA damage and repair in Methanosarcina barkeri. Anaerobe 6, 325–331. doi: 10.1006/anae.2000.0359
Ronda, C., Pedersen, L. E., Sommer, M. O. A., and Nielsen, A. T. (2016). CRMAGE: CRISPR optimized MAGE recombineering. Sci. Rep. 6:19452. doi: 10.1038/srep19452
Safari, F., Zare, K., Negahdaripour, M., Barekati-Mowahed, M., and Ghasemi, Y. (2019). CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell Biosci. 9, 36–21. doi: 10.1186/s13578-019-0298-7
Sowers, K. R., Boone, J. E., and Gunsalus, R. P. (1993). Disaggregation of Methanosarcina spp. and growth as single cells at elevated Osmolarity. Appl. Environ. Microbiol. 59, 3832–3839. doi: 10.1128/aem.59.11.3832-3839.1993
Thauer, R. K., Kaster, A.-K., Goenrich, M., Schick, M., Hiromoto, T., and Shima, S. (2010). Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu. Rev. Biochem. 79, 507–536. doi: 10.1146/annurev.biochem.030508.152103
Vanegas, K. G., Jarczynska, Z. D., Strucko, T., and Mortensen, U. H. (2019). Cpf1 enables fast and efficient genome editing in aspergilli. Fungal Biol. Biotechnol. 6:6. doi: 10.1186/s40694-019-0069-6
Wang, Z., Wang, Y., Wang, Y., Chen, W., and Ji, Q. (2022). CRISPR/Cpf1-mediated multiplex and large-fragment gene editing in Staphylococcus aureus. ACS Synth. Biol. 11, 3049–3057. doi: 10.1021/acssynbio.2c00248
Xin, C., Yin, J., Yuan, S., Ou, L., Liu, M., Zhang, W., et al. (2022). Comprehensive assessment of miniature CRISPR-Cas12f nucleases for gene disruption. Nat. Commun. 13:5623. doi: 10.1038/s41467-022-33346-1
Keywords: CRISPR/Cas12a, genome editing, Methanosarcina acetivorans, methanogens, synthetic biology
Citation: Zhu P, Somvanshi T, Bao J and Scheller S (2023) CRISPR/Cas12a toolbox for genome editing in Methanosarcina acetivorans. Front. Microbiol. 14:1235616. doi: 10.3389/fmicb.2023.1235616
Edited by:
Zhe Lyu, North Carolina State University, United StatesReviewed by:
Dipti D. Nayak, University of California, Berkeley, United StatesCopyright © 2023 Zhu, Somvanshi, Bao and Scheller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvan Scheller, c2lsdmFuLnNjaGVsbGVyQGFhbHRvLmZp; Jichen Bao, amljaGVuLmJhb0BhYWx0by5maQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.