
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 22 August 2023
Sec. Biology of Archaea
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1214631
 Maria A. Khomyakova1*
Maria A. Khomyakova1* Alexander Y. Merkel1
Alexander Y. Merkel1 Dana D. Mamiy2
Dana D. Mamiy2 Alexandra A. Klyukina1
Alexandra A. Klyukina1 Alexander I. Slobodkin1
Alexander I. Slobodkin1Bathyarchaeia are widespread in various anoxic ecosystems and are considered one of the most abundant microbial groups on the earth. There are only a few reports of laboratory cultivation of Bathyarchaeia, and none of the representatives of this class has been isolated in pure culture. Here, we report a sustainable cultivation of the Bathyarchaeia archaeon (strain M17CTs) enriched from anaerobic sediment of a coastal lake. The cells of strain M17CTs were small non-motile cocci, 0.4–0.7 μm in diameter. The cytoplasmic membrane was surrounded by an S-layer and covered with an outermost electron-dense sheath. Strain M17CTs is strictly anaerobic mesophile. It grows at 10–45°C (optimum 37°C), at pH 6.0–10.0 (optimum 8.0), and at NaCl concentrations of 0–60 g l−1 (optimum 20 g l−1). Growth occurred in the presence of methoxylated aromatic compounds (3,4-dimethoxybenzoate and vanillate) together with complex proteinaceous substrates. Dimethyl sulfoxide and nitrate stimulated growth. The phylogenomic analysis placed strain M17CTs to BIN-L-1 genus-level lineage from the BA1 family-level lineage and B26-1 order-level lineage (former subgroup-8) within the class Bathyarchaeia. The complete genome of strain M17CTs had a size of 2.15 Mb with a DNA G + C content of 38.1%. Based on phylogenomic position and phenotypic and genomic properties, we propose to assign strain M17CTs to a new species of a novel genus Bathyarchaeum tardum gen. nov., sp. nov. within the class Bathyarchaeia. This is the first sustainably cultivated representative of Bathyarchaeia. We propose under SeqCode the complete genome sequence of strain M17CTs (CP122380) as a nomenclatural type of Bathyarchaeum tardum, which should be considered as a type for the genus Bathyarchaeum, which is proposed as a type for the family Bathyarchaeaceae, order Bathyarchaeales, and of the class Bathyarchaeia.
Bathyarchaeia is a cosmopolitan group of Archaea inhabiting diverse environments (Xiang et al., 2017; Zhou et al., 2018). Most often Bathyarchaeia are found in freshwater and marine sediments, where they could constituent a large portion of microbial communities. They have also been detected in soils, subsurface petroleum reservoirs, animal-associated habitats, and extreme environments such as terrestrial hot springs and deep-sea hydrothermal vents (Kubo et al., 2012; Fillol et al., 2016; Zhou et al., 2019). In many sediments, Bathyarchaeia are numerically dominant among Archaea, and it was argued that they are the most abundant Archaea on the planet (~2.0–3.9 × 1028 cells) (Kubo et al., 2012; Lloyd et al., 2013; He et al., 2016). The wide distribution and high abundance suggest that Bathyarchaeia may play an important role in the global biogeochemical cycles (Harris et al., 2018; Zhou et al., 2018). Bathyarchaeia are capable of degrading diverse organic compounds such as detrital proteins, plant-derived carbohydrates, lignin, and aromatic compounds (Meng et al., 2014; Lazar et al., 2016; Yu et al., 2018; Zhou et al., 2018; Yin et al., 2022). Moreover, members of this archaeal group have genomic markers for acetogenesis and methane metabolism, which are the most fundamental and ancient microbial biochemical energy-conservation processes (Evans et al., 2015; He et al., 2016; Loh et al., 2021). Several studies found genomic evidence that Bathyarchaeia could be involved in the nitrogen and sulfur cycles (Evans et al., 2015; Lazar et al., 2016; Zhang et al., 2016; Harris et al., 2018; Zhou et al., 2018). It was noticed that the distribution of different groups of Bathyarchaeia is related to geochemical parameters such as concentrations of total organic carbon, ammonium, and nitric oxide as well as to salinity and pH (Pan et al., 2019; Zou et al., 2020).
Current genome-based taxonomy considers Bathyarchaeia as a class, consisting of seven orders, within the phylum Thermoproteota (GTDB, Rinke et al., 2021). More recently, re-classification of Bathyarchaeia into eight order-level units was proposed (Hou et al., 2023). However, for a long time, Bathyarchaeia were classified based on 16S rRNA gene phylogeny into 25 subgroups, which demonstrated versatile physiological capabilities and possessed distinct ecological niches (Kubo et al., 2012; Fillol et al., 2016; Zhou et al., 2018). No pure cultures of Bathyarchaeia have been obtained so far, and the conclusions on their physiology and possible ecological roles are based on metagenomic data. Attempts to cultivate Bathyarchaeia are described only in a few studies. The enrichment cultures from freshwater estuarine sediment with a variety of carbon substrates were set up, and Bathyarchaeia affiliated with subgroup-4 and subgroup-8 were subcultured in these enrichments up to six transfers (Gagen et al., 2013). In another study, a 10-time increase in the gene copy numbers of Bathyarchaeia was demonstrated in incubations with lignin compared to the original sample. It was concluded that Bathyarchaeia of subgroup-8 were capable of assimilating CO2 autotrophically while utilizing lignin as an energy source (Yu et al., 2018). The combination of cell extraction, size fractionation, and roll-bottle technique resulted in the enrichment of Bathyarchaeia and two bacterial species on lignin (Hu et al., 2021). Stimulation of Bathyarchaeia in the initial enrichments by syringaldehyde, 4-hydroxybenzaldehyde, and vanillin under anaerobic conditions has been recently shown (Lin et al., 2022).
In this study, we obtained a highly purified culture of the representative of Bathyarchaeia (strain M17CTs). The culture has high cell density and is amendable to regular subsequent transfers. Based on the results of cultivation experiments and metagenomic analysis, we determined the main cellular, physiological, and genomic characteristics of strain M17CTs. This is the first study describing the cellular morphology and cell wall structure of the defined representative of Bathyarchaeia; also for the first time, a circular genome of Bathyarchaeia becomes available. To sustain the stability of nomenclature currently used for Bathyarchaeia, we formally describe strain M17CTs as Bathyarchaeum tardum gen. nov., sp. nov. under the SeqCode.
Enrichment culture was obtained from a mixed sample of sediment and water collected in May 2017 from Lake Golubitskoe, located 150 m off the coast of the Azov Sea, Taman Peninsula, Russia. Temperature and pH values at the sampling site (45°19′47.7“N 37°15′48.7”E) were 20°C and 8.0–8.5, respectively, and NaCl concentration was 11 g l−1. Samples were taken anaerobically in tightly stoppered bottles and transported to the laboratory. An enrichment culture was initiated by inoculation of 10% (w/v) of the sample into anaerobically prepared (gas phase - N2, 100%), sterile (135°C, 1h), liquid medium of the following composition (per liter distilled water): KH2PO4, NH4Cl, KCl, MgCl2·6H2O (0.33 g each); CaCl2·6H2O, 0.033 g; NaCl, 20.00 g; NaHCO3, 2.00 g; 1 ml trace element solution (Slobodkin et al., 2012), 1 ml vitamin solution (Wolin et al., 1963), and resazurin solution (0.001g l−1). NaHCO3, vitamins, and Na2S·9H20 (0.5 g l−1) were added after boiling and cooling the medium. The pH of the autoclaved medium was 8.0–8.5 at 20°C. 3,4-dimethoxybenzoic acid (DMB, 10 mM) was added as the substrate from anaerobically prepared sterile stock solution before inoculation. Ampicillin (1 g l−1) was used for the suppression of bacterial growth. The incubation temperature was 30°C.
Growth of the enrichment cultures was monitored by direct cell counting using a phase-contrast microscope (Olympus CX-43) followed by high-throughput sequencing of 16S rRNA gene amplicons for the prospective samples. Transmission electron microscopy was performed to determine cell morphology using JEM-100 electron microscope (JEOL, Japan).
All the cultivation experiments were performed in triplicate using Hungate tubes. Temperature (from 10 to 60°C) and pH growth ranges were determined using the same medium as mentioned before, with 10 mM DMB as a substrate and additives necessary for growth, inter alia yeast extract (0.1 g l−1), antibiotic mixture of streptomycin and lincomycin (0.05 g l−1 each), and sterilized (121°C, 30 min) sample of the natural sediment 0.2% (v/v). pH of the medium was adjusted to different values in the range of 5.0–10.0 with sterile anaerobic solutions of HCl (2 M) or NaOH (5%, w/v). To determine the NaCl requirement, varying amounts of NaCl (0–10.0%, w/v) were added directly into the Hungate tubes from pre-sterilized anaerobic stock solutions. Microaerobic growth was tested using the medium lacking the reducing agent, under 2% O2 (in N2) in the gas phase.
Electron donors and acceptors were added from sterile anaerobic stock solutions before inoculation. All the organic substrates (peptides, carbohydrates, organic acids, and methoxylated aromatic compounds) were filter-sterilized using 0.2 μm pore size syringe filters (Millipore). Growth with molecular hydrogen (10% in the gas phase) and carbon monoxide as the electron donor was tested in the presence of carbon dioxide as an electron acceptor. Molecular hydrogen, dimethyl sulfide, and methane formation were monitored by gas chromatography on a HayeSep N 80/100 mesh column at 40°C with argon as the carrier gas at 20 ml min−1 flow rate. Determination of nitrate and nitrite concentrations was held on a Stayer ion chromatograph (Aquilon) equipped with an IonPac AS22 column (Dionex, USA) and conductivity detector. Concentrations of DMB and 3,4-dihydroxybenzoate (DHB) were determined by ultra-performance liquid chromatography (UPLC)–UV analysis. The measurements were carried out on the equipment of the Shared-Access Equipment Center “Industrial Biotechnology” of the Federal Research Center “Fundamentals of Biotechnology” Russian Academy of Sciences. 0.1 ml of the sample was diluted with 0.9 ml of deionized water and used for UPLC analysis. UPLC-MS analysis was performed on Azura UVD 2.1S single wavelength UV/VIS detector (Knauer, Germany) equipped with A4061XB 10 μl analytical flow cell (Knauer, Germany) coupled to Elute UPLC (Bruker Daltonik, Germany) on Triart PFP 3 μm 2.1 × 150 mm 120 Å reverse phase column (YMC, Japan) with following conditions: gradient elution at 0.25 ml min−1 from 5% to 95% B in 10 min (A: 0.1% formic acid in water, B: 0.1% formic acid in acetonitrile), column at 30°C, 5 μl injection volume, and detection at 254 nm at 1 Hz rate. Data processing was performed in Compass DataAnalysis 5.1 (Bruker Daltonik, Germany).
DNA from mud samples and enrichment cultures was isolated using FastDNA Spin Kit for Soil according to the manufacturer's protocol (MP Biomedicals, Santa Ana, California, USA). The V4 region of the 16S rRNA amplicon libraries preparation, sequencing, and analysis was performed as previously described (Khomyakova et al., 2022). A shotgun metagenome library preparation and sequencing were done in BioSpark Ltd., Moscow, Russia, using KAPA HyperPlus Library Preparation Kit (KAPA Biosystems, UK), according to the manufacturer's protocol and NovaSeq 6000 system (Illumina, San Diego, CA, USA) with the reagent kit, which can read 100 nucleotides from each end. Raw reads were processed with Cutadapt (Martin, 2011) and Trimmomatic (Bolger et al., 2014) for adapter removal and quality filtering. Reads were processed in MetaWRAP (Uritskiy et al., 2018) using MetaSPAdes (Nurk et al., 2017) and MEGAHIT (Li et al., 2015) for assembly, MaxBin 2 (Wu et al., 2016), MetaBAT 2 (Kang et al., 2019), and CONCOCT (Alneberg et al., 2014) for binning and Salmon (Patro et al., 2017) for coverage calculation. Bin completeness and contamination were evaluated using CheckM (Parks et al., 2015). Taxonomies were assigned to each bin using GTDB-Tk (Parks et al., 2018). In the case when a more accurate phylogenetic analysis was needed, de novo phylogenetic trees were built using the list of 122 archaeal marker genes which were taken from GTDB (Parks et al., 2018). The trees were built using the IQ-TREE 2 program (Minh et al., 2020) with fast model selection via ModelFinder (Kalyaanamoorthy et al., 2017) and ultrafast bootstrap approximation (Minh et al., 2013) as well as approximate likelihood-ratio test for branches (Anisimova and Gascuel, 2006). All the sequencing data are deposited in NCBI BioProject PRJNA864620. Gene search and annotation were performed by BLAST (Altschul et al., 1990) and KEGG (Kanehisa et al., 2023) services. The MEROPS database (Rawlings et al., 2018) was used to identify putative proteases/peptidases in MAG M17C. The sequences identified as peptidases were collected and used as input for SignalP 6.0 (Teufel et al., 2022), to identify export signals. HydDB (Søndergaard et al., 2016) was used to classify the catalytic subunits of the hydrogenases.
For the quantification of Bathyarchaeia, a primer system for qPCR was designed using Primer-BLAST (Ye et al., 2012). The sequences of representatives of Bathyarchaeia that were used as targets for primers designed were obtained from our results of 16S rRNA gene-based profiling and metagenome sequencing. Sequences of the resulting primers were Bath2022F 5′- GGTAGGGGTGAAATCCTATAATCCCG−3′ and Bath2022R 5′- CCTCACCGTCGRGCGCGTTCTAG−3′. Product size−78 bp. According to TestPrime 1.0 (Klindworth et al., 2013), this primer pair is highly specific to a group of sequences from Bathyarchaeia class. The specificity of the primer system was also verified by sequencing the PCR product. For the calibration curve, linearized plasmid pAL2-T (Evrogen, Russia) with cloned M17C-01 16S rRNA gene fragment was used. qPCR analyses were carried out on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) and qPCRmix-HS SYBR (Evrogen, Russia). The amplification program consisted of the following steps: primary denaturation at 95°C for 5 min, followed by 30 cycles of denaturation at 95°C for 15 s, annealing at 64°C for 15 s, and elongation at 72°C for 12 s. The concentration of the primers was 0.25 μM each. Standards, samples, and negative controls were run in triplicate.
Cells growing in the logarithmic phase were harvested by centrifugation and resuspended in 0.5 ml of phosphate-buffered saline (PBS) containing, in grams per liter, NaCl, 8.0; KCl, 0.2; Na2HPO4, 1.44; and NaH2PO4, 0.2 (pH 7.0). The cell suspension was mixed with 1.5 ml of 4% (w/v) freshly prepared paraformaldehyde solution (Sigma, Deisenhofen, Germany) and fixed for 1 h at room temperature. The cells were then collected by centrifugation (6,600 g for 1 min) and washed twice with PBS to ensure the removal of paraformaldehyde. The resulting pellet was resuspended in 0.5 ml of 50% ethanol–PBS (v/v). The Bathyarchaeia-specific oligonucleotide probe MCG493 (Kubo et al., 2012; 5′- CTTGCCCTCTCCTTATTCC-3′) was purchased from Syntol (Moscow, Russia). Hybridization was done on gelatin-coated (0.1%, w/v) and dried Teflon-laminated slides. The fixed cell sample was applied to these wells, hybridized to the fluorescent probe, and stained with the universal DNA stain 4',6-diamidino-2-phenylindole (DAPI, 1 mM) as described earlier (Dedysh et al., 2001). The specimens were examined with a ZeissAxioplan2 microscope (Zeiss, Jena, Germany) equipped with the Zeiss Filters No20 and 02 for Cy3-labeled probe and DAPI staining, respectively.
Enrichment cultures from anaerobic sediment of the lake Golubitskoe were set up by adding three different methoxylated aromatic compounds–2-methoxybenzoate, 2-methoxyphenol, and DMB. To suppress the growth of bacteria, ampicillin was added to the cultivation medium. Microbial growth was monitored by light-microscopic examinations followed by high-throughput sequencing of 16S rRNA gene amplicons as well as by parallel qPCR detection of Bathyarchaeia with specific primers. After 2 months of incubation, only the enrichment with DMB showed visible optical turbidity and growth of cells, mostly small non-motile cocci. The relative abundance of Bathyarchaeia in this enrichment was 43%. Attempts to obtain separate colonies were unsuccessful either with 1% Gelrite gellan gum or with 1.5% agar as the solidifying agents. We tried to purify Bathyarchaeia by serial dilution-to-extinction technique on a liquid medium. Overall, ten consequent 10% (v/v) transfers were made since the initial enrichment without loss of Bathyarchaeia. A detailed diagram of the transfers is shown in Supplementary Figure 1. Since the third transfer, we consider the bathyarchaeal component present in the cultures as a single population and designate it as strain M17CTs. Finally, we obtain two highly purified cultures of strain M17CTs growing with DMB and casamino acids in the presence of antibiotics (mixture of lincomycin and streptomycin) and sterilized natural sediment (Supplementary material 1). The first culture contained only two archaeal species—strain M17CTs and Methanocalculus alkaliphilus (96% and 3% of relative abundance, respectively) (16S rRNA gene profiling data; BioSample SAMN34140869). This culture had an extremely slow growth rate (up to 6 months to reach the maximal cell density) and a low cell yield (1–2 x 106 cells ml−1). The second culture grew faster (within 2–3 months) but contained more archaeal species. In addition to strain M17CTs (97.87%) and Methanocalculus (0.16%), the species of Methanolobus (0.38%), uncultured Thermoplasmatota (0.88% in total), and Methanomicrobiaceae (0.71%) were detected (shotgun metagenomic data; BioSample SAMN33973097). The doubling time for this second culture was estimated to be 7.2 days (Figure 1). Variations in substrate combinations and concentrations could probably improve growth rate or cell yield. At least the rates of growth were strongly dependent on the age and viability of the inoculum and could also vary depending on the composition of the microbial community, which was obvious for our cultivation lines.
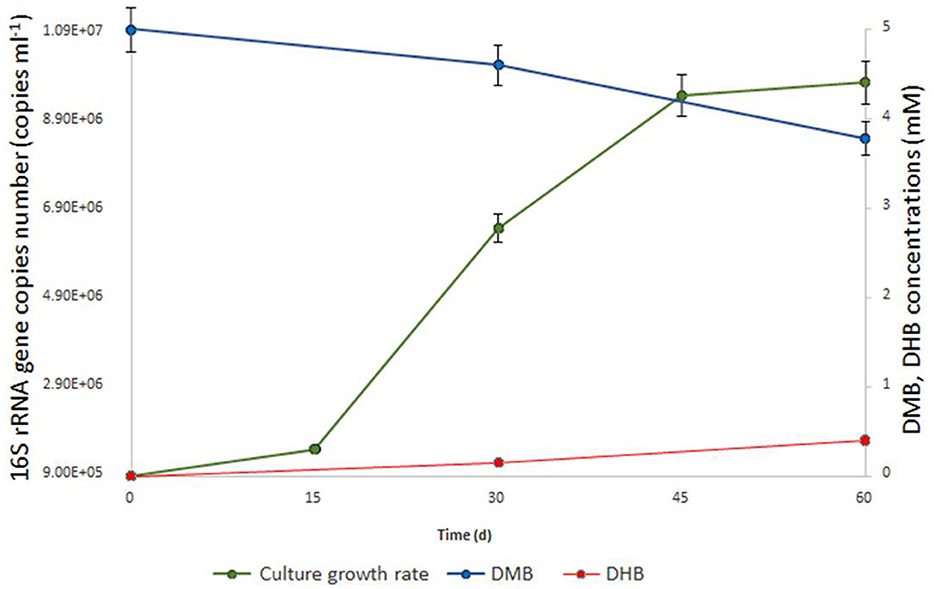
Figure 1. Growth dynamics of M17CTs cultivated with DMB (5mM). Culture was also supplemented with 0.1 g l−1 yeast extract, 0.20 ml 10 ml −1 (v/v) of sterilized sediment from a natural sample and antibiotics mixture of streptomycin and lincomycin (0.05 g l−1 each). 1.2 mM of DMB (blue line) was consumed, and 0.4 mM of DHB (red line) was produced (in addition to acetate and ethanol) during 2 months of cultivation. No DHB, acetate, or ethanol was detected in un-inoculated controls after 2 months of incubation. The doubling time was estimated to be 7.2 days.
FISH analysis employing Bathyarchaeia-specific oligonucleotide probe MCG493 yielded a strong hybridization signal and demonstrated that the culture was predominantly represented by Bathyarchaeia (Figure 2). Transmission electron microscopy revealed that the cells of strain M17CTs were regular-shaped cocci with a diameter of 0.4–0.7 μm (Figure 3A). Flagella, pili, or other appendages on the surface of the cell were not observed, and motility was not detected. Chains of 3–5 cells covered by a common layer were occasionally formed (Figure 3B). The ultrathin section showed that the cells had an electron-dense layer, superimposing the cytoplasmic membrane, probably the S-layer typical for many Archaea (Figure 3C). In addition, the cells were covered by the outermost electron-dense layer, especially well-distinguishable during binary fission (Figures 3D, E). This outermost layer is probably a protein or polysaccharide sheath covering several individual cells. The cells of the strain M17CTs had a well-defined cytoplasmic membrane and no visible organelle-like structures. The cells of M17CTs were morphologically different from their co-culture partner Methanocalculus alkaliphilus, 0.4–0.7 μm in diameter regular cocci with binary division vs. 0.8–1.2 μm in diameter irregular cocci with division by septa, correspondingly (Figure 3F) (Zhilina et al., 2013; Sorokin et al., 2015).
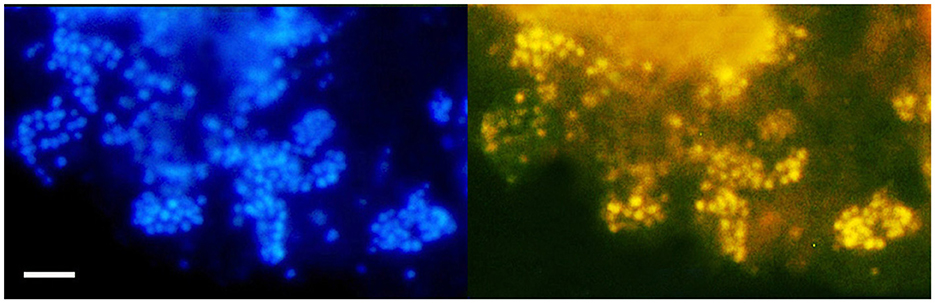
Figure 2. Fluorescence in situ hybridization (FISH) image of strain M17CTs cultivated with DMB. For FISH, M17CTs cells were hybridized with a Bathyarchaeia 16S rRNA targeted probe MCG493 (yellow fluorescence, right panel). After hybridization, the cells were counterstained with DAPI (blue fluorescence, left panel). Bar 1 μm.
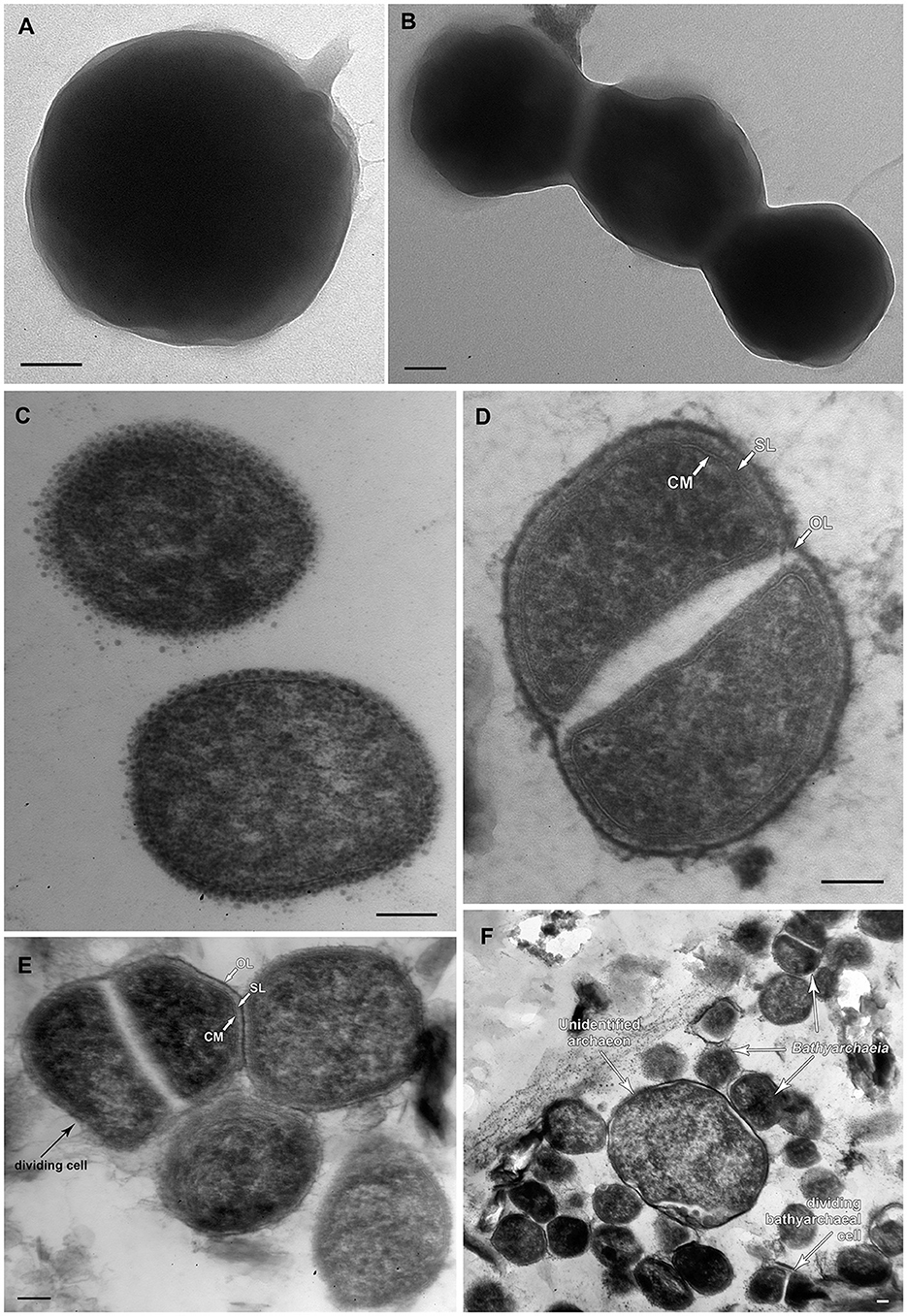
Figure 3. Morphological characteristics of strain M17CTs. Electron micrographs of negatively stained cells showing (A) single cell of coccoid morphology with small areas of mucus on the surface of the cell; (B) chain of cells without surface appendages covered by unidentified common layer. Ultrathin section of (C) typical coccoid cells of strain M17CTs of regular shape; (D, E) dividing cells, (F) multiple cells of strain M17CTs and the single cell of the unidentified archaeon present in culture. CM—cytoplasmic membrane; SL—S-layer; OL—outermost surface layer. Bar 0.1 μm.
Strain M17CTs was mesophilic and had an optimum growth temperature of 37°C and a temperature range for growth from 10°C to 45°C (Supplementary Figure 2A). The pH range for growth was 6.0–10.0, with an optimum at pH 8.0 (Supplementary Figure 2B). Growth of strain M17CTs was observed at NaCl concentrations of 0–6.0% (w/v) with an optimum at 2.0% NaCl (Supplementary Figure 2C).
Initial enrichments of strain M17CTs were performed in the medium containing DMB as a substrate, without the addition of other organic supplements. During the purification process, strain M17CTs gradually lost the ability to grow solely on DMB; however, the addition of 0.1 g l−1 yeast extract restored sustainable growth. Strain M17CTs was not subcultured if only yeast extract was added to the cultural medium.
To determine the compounds that can be used as growth substrates instead of DMB/yeast extract, we performed physiological experiments, in which the growth of strain M17CTs was monitored by real-time PCR along with high-throughput profiling of the 16S rRNA gene (Table 1).
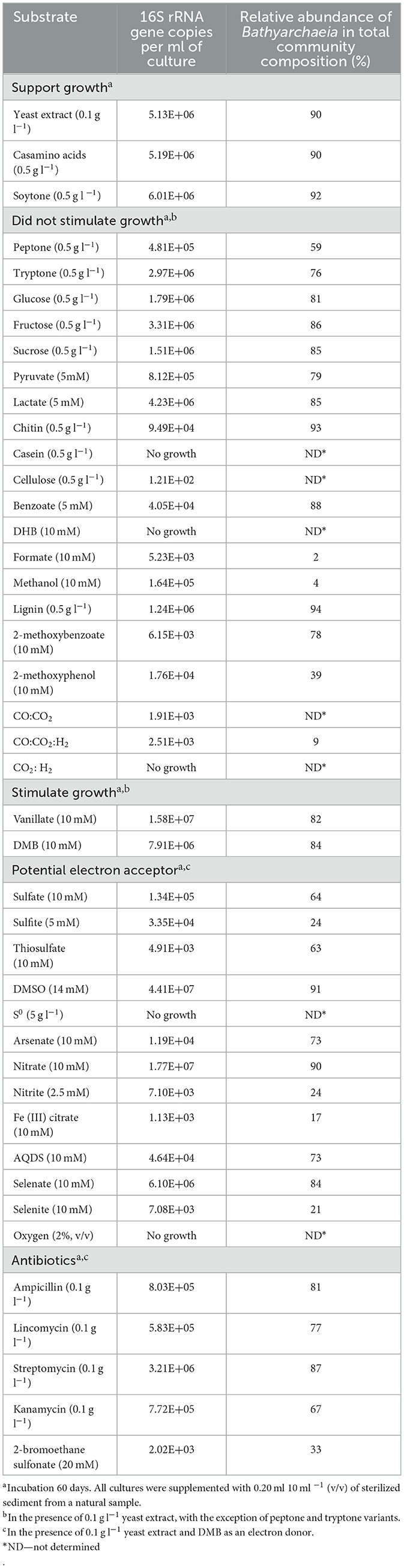
Table 1. Effect of different compounds on growth of strain M17CTs.
No growth stimulation was observed when the concentration of yeast extract was increased up to 0.5 g l−1 compared to 0.1 g l−1. Yeast extract could be replaced by casamino acids or soytone but not by tryptone or peptone. Glucose, fructose, sucrose, lactate, pyruvate, lignin, chitin, casein, cellulose, formate, methanol, benzoate, DHB, 2-methoxyphenol, 2-methoxybenzoate, molecular hydrogen, and carbon monoxide did not stimulate growth (Table 1). From tested compounds, only DMB and vanillate stimulated the growth of strain M17CTs. Products of growth on DMB and yeast extract were DHB, acetate, and ethanol. 1.2 mM of DMB was consumed, and 0.4 mM of DHB was produced during 2 months of cultivation. No DHB, acetate, or ethanol was detected in non-inoculated controls after 2 months of incubation. Small amounts of methane (<0.5 mM) were produced by methanogenic partners.
Among tested potential electron acceptors, dimethyl sulfoxide (DMSO) and nitrate stimulated the growth of M17CTs. Sulfate, thiosulfate, antraquinone-2,6-disulfonate (AQDS), selenate, selenite, arsenate, Fe (III)-citrate, elemental sulfur, sulfite, nitrite, or oxygen (2% (v/v) in the gas phase did not stimulate growth. Oxygen and elemental sulfur had a profound inhibitory effect (Table 1).
Of the antibiotics tested, strain M17CTs showed the highest resistance to streptomycin (0.1 g l−1) (Table 1).
According to our phylogenomic reconstruction based on 122 archaeal single copy conserved marker genes (Parks et al., 2018), M17CTs (M17C-01 MAG) is a part of BIN-L-1 genus-level lineage (Figure 4A) which belongs to the BA1 family-level lineage and B26-1 order-level lineage according to GTDB (Parks et al., 2018). The B26-1 group is formerly referred to as Bathy-8 (Yu et al., 2017). BIN-L-1 cluster contains several MAGs from enrichment cultures growing with lignin as well as MAG from sediments associated with petroleum seepage and freshwater sediments (Yu et al., 2018). These data indicate the possible specialization of representatives of this genus-level group to growth on natural methoxylated compounds. The phylogenetic position of M17CTs revealed by 16S rRNA gene-based phylogenetic reconstruction agrees well with the phylogenomic data (Figure 4B).
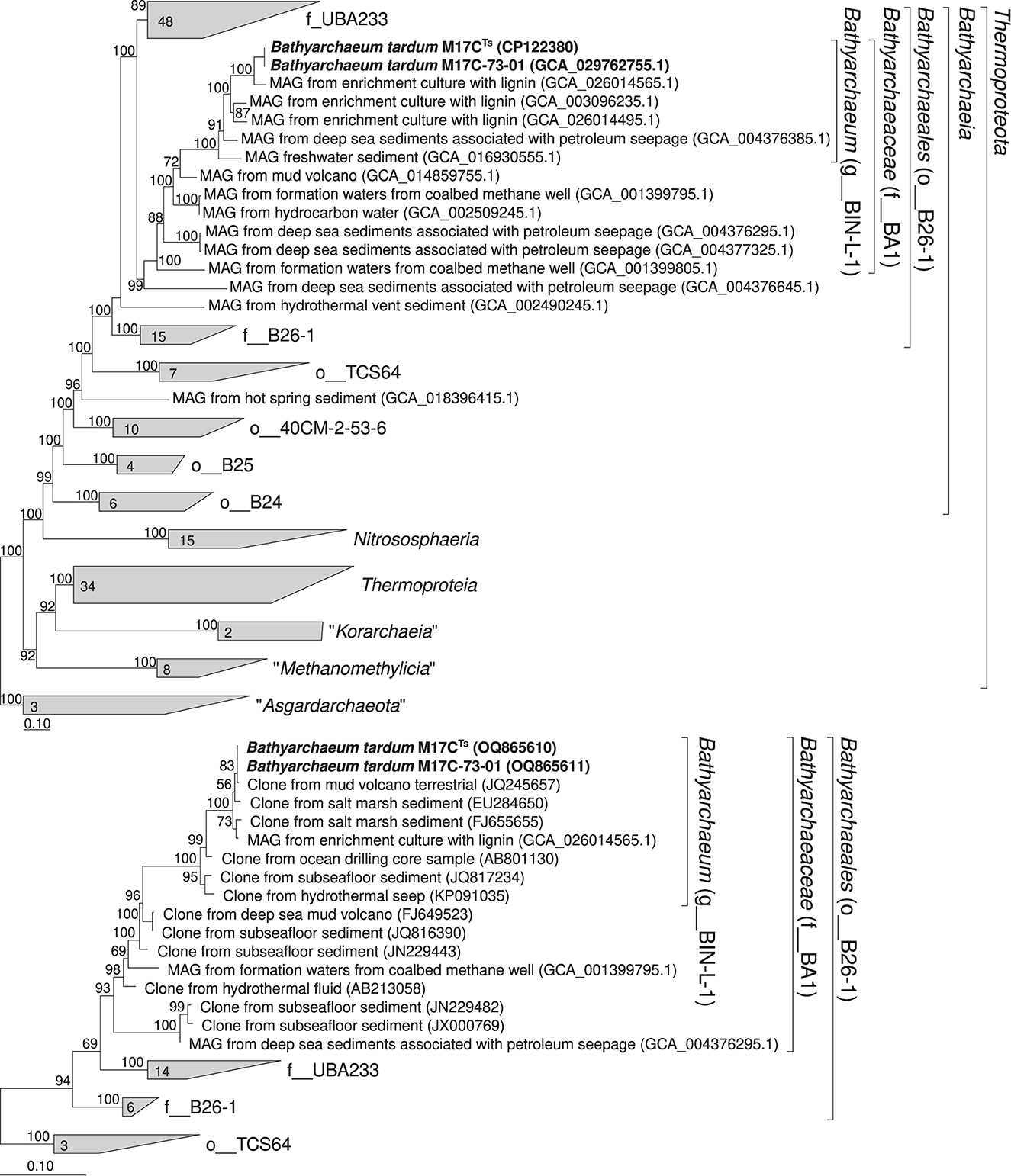
Figure 4. Phylogenomic placement of M17C-01Ts and M17C-73-01 MAGs based on (A) concatenated partial amino acid sequences of 122 archaeal single copy conserved marker genes with taxonomic designations according to the GTDB (Parks et al., 2018); (B) on 16S rRNA gene sequences. The “g_” stands for genus level, “f_”—for family level, and “o_”—order level. The trees were built using the IQ-TREE 2 program (Minh et al., 2020) with fast model selection via ModelFinder (Kalyaanamoorthy et al., 2017) and ultrafast bootstrap approximation (Minh et al., 2013) as well as approximate likelihood-ratio test for branches (Anisimova and Gascuel, 2006). Bootstrap consensus tree is shown with values placed at the nodes. Bar, 0.1 changes per position.
We analyzed the distribution of BIN-L-1 cluster members based on 16S rRNA gene sequence data available from public databases. For this purpose, we selected sequences in GenBank that have more than 94.5% similarity to the 16S rRNA gene sequences of MAGs of this group (Yarza et al., 2014). This selection consisted of 457 sequences. Most of the sites where these microorganisms were found belonged to the marine sediments often associated with the presence of terrigenous organic matter, e.g., terrigenous deposits, estuary (Li et al., 2012), and sunken woods (Fagervold et al., 2012). The second largest group of ecotopes where BIN-L-1-related sequences have been found are the sites associated with methane seepages, e.g., hydrate-bearing sediments (Dang et al., 2009), cold seep sediment (Li et al., 1999), and mud volcanoes (Cheng et al., 2012). Soils and freshwater sediments were less significant environments for BIN-L-1-related sequences.
For metagenomic analysis, two enrichment cultures from different cultivation stages, grown on DMB and antibiotics mixture with different proteinaceous additives, were taken (Supplementary Figure 1). The first metagenome (M17C) was analyzed after the third transfer on DMB and ampicillin, where the relative abundance of Bathyarchaeia group according to 16S rRNA gene-based profiling reached 74%, and the second metagenome (M17C-73) was taken from the eight transfer on DMB, antibiotics, and casamino acids (here Bathyarchaeia group built up 98%) since the richness by the group of interest as well as its growth rates was the highest of the achieved.
The main assembly quality indicators for M17C and M17C-73 metagenomes are shown in Supplementary Table 1. An overview of all MAGs is shown in Table 2. As a result of M17C metagenomic analysis, we were able to assemble the genome of the Bathyarchaeia (MAG M17C-01) to the level of a complete circular chromosome of the size of 2,152,572 bp (98.28% completeness and 0.934% contamination according to CheckM). The nomenclatural type is CP122380Ts according to SeqCode requirements (Hedlund et al., 2022; Whitman et al., 2022). Its relative abundance in the metagenome was 83.38%. In addition to the M17C-01 Bathyarchaeia, the M17C metagenome also contained uncultured representatives of Acholeplasmataceae (isolated later as Mariniplasma sp.) and Dethiobacteraceae families (4.51% and 3.03%, respectively) as well as representatives of the Methanothrix and Soehngenia genera (3.29% and 3.61%, respectively).
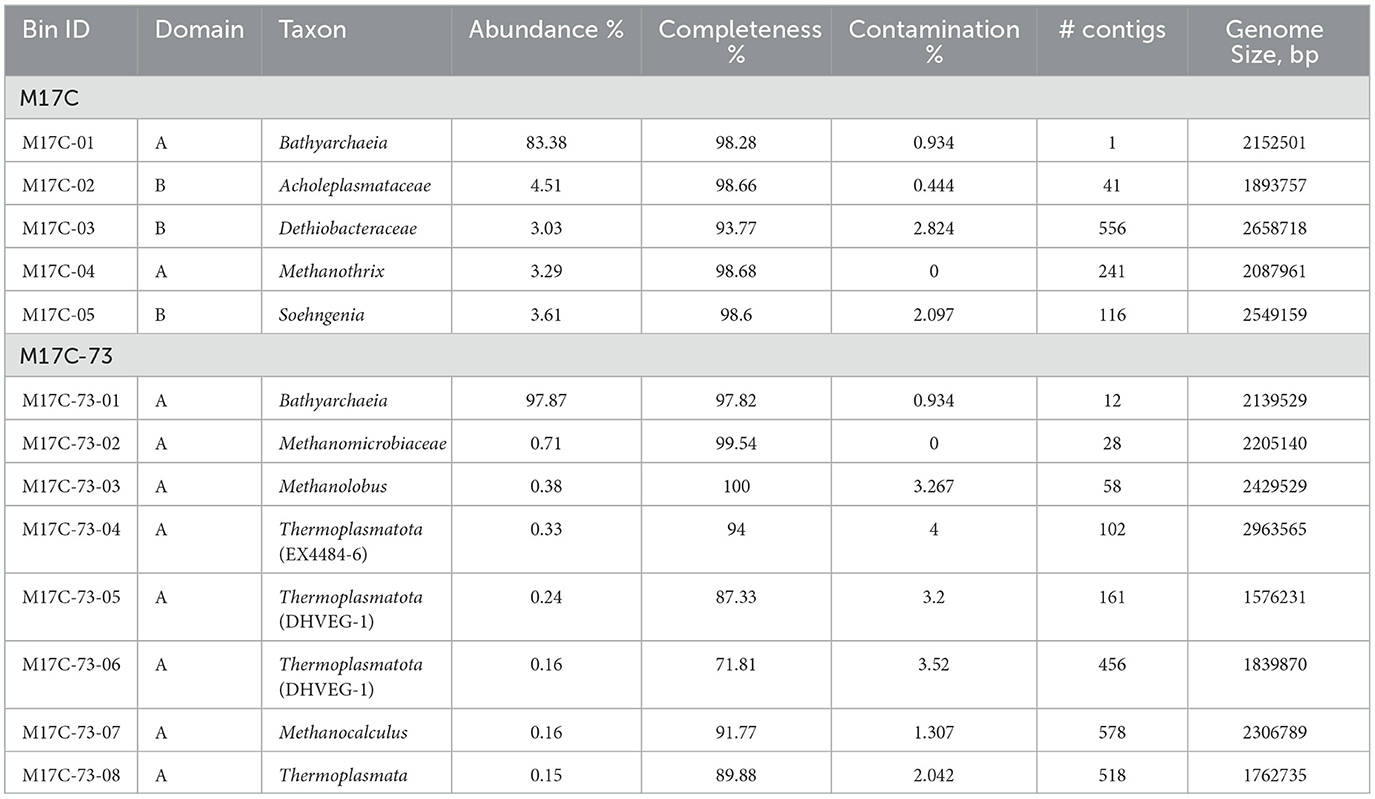
Table 2. Overview of all MAGs >50% complete and with <5% contamination.
The genome of the Bathyarchaeia from the M17C-73 metagenome (MAG M17C-73-01) was very close but not identical to M17C-01: ANI value is 99.31%; in addition, both MAGs contain identical 16S rRNA genes. Apparently, we see here a case of microdiversity in Bathyarchaeia population. The relative abundance of the M17C-73-01 MAG in the M17C-73 metagenome reached 97.87%. The rest of the community was represented only by archaea: by methanogens of the Methanolobus and Methanocalculus genera as well as of Methanomicrobiaceae family and by uncultivated representatives of the “Candidatus Thermoplasmatota” phylum including archaea of the DHVEG-1 and EX4484-6 phylogenetic clusters. They all represented only a minor (<1%) part of the community. M17C-01 genome was chosen as a type genome and further analyzed to explain the revealed physiological features of the M17CTs isolate.
Our physiological experiments demonstrate the ability of strain M17CTs to metabolize proteinaceous substrates. Growth on amino acids typically requires the gluconeogenic pathway for carbohydrate synthesis (Danson et al., 2007) and in line with that all genes for a reverse glycolytic pathway, including pyruvate:ferredoxin oxidoreductase and fructose 1,6-bisphosphatase, have been identified in the genome. However, glucose-6-phosphatase is absent, which makes it impossible to synthesize glucose from phosphorylated form. The genome of strain M17CTs contained genes for the degradation of detrital proteins (i.e., multiple membrane ABC transporters, peptidases, aminotransferases, and ferredoxin oxidoreductases) as well as genes encoding intracellular archaeal proteasome complex (WGM88999; WGM89238; WGM90643) and several presumably extracellular peptidases of M24 and M42 families (WGM88659 and WGM89452 correspondingly). All these proteins are intracellular which is consistent with the data on the deficiency of extracellular proteases in Bathyarchaeia (Berben et al., 2022). Apparently, utilization of protein compounds by strain M17CTs is limited to amino acids, which are small enough for immediate transmembrane uptake. Numerous solute-binding protein subunits of ABC-type transporters were encoded in the genome of strain M17CTs; their physiological substrates are difficult to predict based solely on the sequence information. The genome of strain M17CTs encoded several genes of aminotransferases, in particular, aromatic amino acid aminotransferase and histidinol-phosphate aminotransferase (WGM89691; WGM89080). Degradation of the amino acid backbone proceeds through donor:ferredoxin oxidoreductases, yielding reduced ferredoxin as the final product. In the genome of strain M17CTs, we identified genes of aldehyde:ferredoxin oxidoreductase, 2-oxoacid:ferredoxin oxidoreductase, and pyruvate:ferredoxin oxidoreductase (Supplementary Table 2). Pyruvate produced in aminotransferase reaction can be further oxidized by pyruvate:ferredoxin oxidoreductase (WGM89686-89) to acetyl-CoA, which can be oxidized with ATP production by acetyl-CoA synthetase (WGM89748). Acetate kinase and phosphate acetyltransferase responsible for substrate-level phosphorylation in fermenting and acetogenic bacteria are absent from the genome strain M17CTs. The pathways for the formation of ethanol, which was found along with acetate as a metabolic product of strain M17CTs, remain unclear. Genes of alcohol dehydrogenase were absent from the genome (Supplementary Table 2); however, genes of aldehyde:ferredoxin oxidoreductase playing an important role in alcohol-producing anaerobes (Nissen and Basen, 2019) were present (WGM89453). Acetogenesis may fulfill the role of the redox sink, recycling reduced ferredoxin produced by amino acid degradation.
Inspite of the presence of the genes for a complete Embden–Meyerhof–Parnas (glycolysis) pathway (Supplementary Table 2), in physiological experiments, strain M17CTs did not show the ability to metabolize carbohydrates. The reason for this could be the absence of sugar transporters.
The isolation process together with physiological experiments has demonstrated the ability of strain M17CTs to grow on methoxylated aromatic compounds such as DMB and vanillate. We conducted a manual BLASTP search for O-demethylase genes involved in methoxydotrophic growth and found a putative cluster of three neighboring genes (WGM89983-84; WGM89986), previously described in Bacteria and Archaea (Kurth et al., 2021; Welte et al., 2021). Activating enzyme (AE), required for the reduction of corrinoid protein after an inadvertent oxidation of the corrinoid cofactor, was not found in the genome. On the other hand, its presence is not a prerequisite for the O-demethylase reaction. For example, Moorella thermoacetica does not possess AE, and its O-demethylase is a three-component system; in this case, the reductive activation system is not required (Naidu and Ragsdale, 2001). As the methyltransferases and corrinoid protein are cytosolic, strain M17CTs needs transporters for the uptake of methoxylated aromatic compounds. Previous studies have characterized archaeal putative aromatic acid:H+ symporter belonging to the major facilitator superfamily from Archaeoglobus fulgidus grown on 2-methoxyphenol (Welte et al., 2021). We also found in the genome of strain M17CTs gene WGM90668, which demonstrates high amino acids similarity with one of MFS transporters from Archaeoglobus fulgidus, probably involved in the import of the methoxylated aromatic compounds and export of the hydroxylated derivatives. However, in strain M17CTs genome, this gene is located apart from a putative O-demethylase cluster.
Attempts to detect in the genome of strain M17CTs genes of benzoyl-CoA reductase (Bcr), a key enzyme of anaerobic benzoate catabolism, revealed the presence of a cluster of three adjacent genes, encoding BcrACD, while gene of BcrB, a part of substrate-activating module, was not detected (Supplementary Table 2). In our growth experiments, it was shown that benzoate and DHB inhibited the growth of strain M17CTs (Table 1). Genes of corresponding aromatic ligases and reductases were absent from the genome. Several putative transporters of aromatic compounds were found (WGM90668; WGM89538).
The methyl group from the methoxylated substrate most probably enters the Wood–Ljungdahl pathway (WLP), leading to acetogenesis, since a key enzyme of the methanogenic pathway (McrA) was not encoded in the genome. The strain M17CTs genome contained a complete set of WLP genes in the archaeal version, involving methanofuran and tetrahydromethanopterin (THMPT) as C1 methyl group carriers (Supplementary Table 2). The THMPT branch might function in either oxidation or reduction directions. In the oxidation direction, methylene is oxidized to CO2, producing ATP while reducing NAD+ and ferredoxin. Hox hydrogenase, found in the genome (WGM89841-43; WGM90709), can catalyze hydrogen production for cofactor regeneration and disposal of excess reductants, including those that would be produced by the THMPT branch working in the oxidation direction. In the reduction direction, the branch could serve for carbon fixation.
During physiological experiments, we found that the addition of DMSO and nitrate stimulated the growth of strain M17CTs. Dimethyl sulfide was formed as a product of DMSO reduction (up to 2.3 mM). The enzymes involved in DMSO and nitrate reduction in strain M17CTs remain unclear. Electron shuttling via a cysteine–cystine couple, characteristic for Thermococcus species, growing on peptides and DMSO (Choi et al., 2016), seems to be incomplete, and only WGM89435 gene encoding thioredoxin reductase was identified. Genes of DMSO reductase of acetogenic bacterium M. thermoacetica, composed of three subunits (Rosenbaum et al., 2022), were not found except the genes encoding putative subunit DmsB (WGM89217; WGM89773).
The consumption of nitrate was relatively low, and 0.5–0.7 mM of nitrate was consumed from the initial concentration of 10 mM. Nitrite and N2O were not formed. Genes coding for periplasmic nitrate reductase (NapAB) were not found in the genome of strain M17CTs. The genes of catalytic and quinol-oxidizing subunits (NarGI) of another dissimilatory nitrate reductase, a membrane-bound Nar-type nitrate reduction complex, were also absent from the genome. One gene (WGM89217) demonstrated 33% amino acid sequence identity (52.7% similarity) to the electron-transfer subunit NarH from archaeon Haloferax mediterranei. A gene homologous to assimilatory nitrate-reductase NasA was found (WGM89749), which demonstrated 22.5% amino acid sequence identity (51.7% similarity) to NasA from Haloferax medditerranii (Martínez-Espinosa et al., 2001). No other Nas subunits were identified. The genome of strain M17CTs harbors several other nitrogen-related genes encoding ammonium transporter (amt, WGM90598) and hydroxylamine reductase (hcp, WGM90665).
The genome of strain M17CTs had a gene cluster with high homology to NADH:quinone oxidoreductases (Nuo); only NuoJ-encoding gene was not found in the genome. On the other hand, several genes from this cluster resemble catalytic subunits of [NiFe] hydrogenases of subgroup-4. It was proposed recently that in Bathyarchaeia of subgroup 6, this cluster is not a respiratory complex but is instead a novel energy-converting Hfo hydrogenase (Loh et al., 2021). Both coordination sites of the [NiFe] cofactor (L1 and L2 motifs; Vignais and Billoud, 2007), which are no longer conserved in NuoD, are present in WGM88738 (annotated as NuoD in GenBank, Supplementary Figure 3). Genes encoding the NuoEFG subunits known to form the domain involved in NADH binding and oxidation seem to be present in the genome of strain M17CTs and are organized in one cluster, suggesting the ability of this complex to accept electrons from NADH (WGM89840-42; Sazanov, 2007). The genome of strain M17CTs also has genes, encoding six of the eight subunits of the archaeal V-type ATP synthase, and a primitive respiration can be possible as described for another archaeon (Sapra et al., 2003).
Since the discovery of Bathyarchaeia two decades ago, versatile metabolic capabilities such as acetogenesis, methanogenesis, and fermentation have been suggested for its members; however, none of these physiological functions have been directly confirmed due to the lack of laboratory cultures (Inagaki et al., 2003; Evans et al., 2015; Zhou et al., 2018). We have obtained the first highly purified culture with a relative abundance of Bathyarchaeia 98%. To date, morphological characteristics of Bathyarchaeia were not investigated. We have demonstrated that the cells of the bathyarchaeal component of the culture (strain M17CTs) are small non-motile cocci, and the chains of the cells are covered with an outer sheath of unknown composition. Combining physiological studies with genome analyses, we found that strain M17CTs is an anaerobic, slow-growing archaeon, capable of utilization of complex proteinaceous substrates and methoxylated aromatic compounds.
The ability of archaea to transform methoxylated aromatic compounds, which are important components of lignin, the second most abundant polymer on the earth, was unknown for a long time and was demonstrated only recently (Mayumi et al., 2016; Welte et al., 2021). The central carbon metabolism of strain M17CTs during methoxydotrophic growth most probably involves the oxidation of methyl groups of 3,4-dimethoxybenzoate or vanillate, reduction of CO2, and production of acetate. Utilization of DMB and vanillate as growth substrates is strongly supported by the presence of O-demethylase gene cluster in the genome. The confinement of the closest relatives of strain M17CTs to the sources rich in lignin, as well as to petroleum seepages, confirms the dependence of this group on aromatic compounds. Strain M17CTs does not use carbohydrates and is not capable of lithotrophic growth. Most probably, its ecological function in anaerobic sediments is the utilization of low concentrations of oligomers derived from plants and animals necromass.
According to metagenomic analyses, anaerobic respiration is not common in Bathyarchaeia and is only suggested for nitrogen compounds (Lazar et al., 2016; Harris et al., 2018; Deb and Das, 2022). In particular, genes encoding periplasmic nitrate reductase (narH) and nitrite reductase (nrfHA) were found in the genome of “Candidatus Bathyarchaeota” BE326-BA-RLH, suggesting the potential for dissimilatory nitrate reduction (Harris et al., 2018). We have observed ~2.5-time stimulation of growth of strain M17CTs by nitrate. However, genes encoding catalytic subunits of dissimilatory nitrate reductases were not found in the genome. The reason for growth stimulation by nitrate is not clear, and low amounts of nitrate were consumed which may indicate its assimilation or use as an electron acceptor in energy metabolism by yet unknown biochemical mechanisms.
Another potential electron acceptor that stimulates the growth of strain M17CTs is DMSO. This methylated sulfur compound plays a significant role in the biogeochemical cycle of dimethyl sulfide (Xiong et al., 2016). In M. thermoacetica, the reduction of DMSO may be associated with bacterial acetogenesis where DMSO can be used alongside CO2 as an electron acceptor (Rosenbaum et al., 2022). We did not find genetic determinants of this process (neither Dms-type nor Dor-type) in the genome of strain M17CTs. No obvious genomic determinants of terminal reductases and of electron transport chain components such as cytochrome oxidases, quinones, or c-type cytochromes were detected, so the ability of strain M17CTs to conserve energy by anaerobic respiration is not evident.
Strain M17CTs probably has mutualistic relationships with methanogenic component of the cultures. In nature, Bathyarchaeia are often co-occurred with Methanomicrobia, where they are localized in close association (Collins et al., 2005; Xiang et al., 2017). Multiple deficiencies in purine and pyrimidine biosynthesis as well as in the synthesis of cofactors, including CoF420 (taking part in WLP) and vitamins such as cobalamin which is necessary for O-demethylase reaction, were found in the genome of strain M17CTs (Supplementary Table 2). Our experiments show that the addition of an inhibitor of methanogenesis 2-bromoethane sulfonate suppressed the growth of strain M17CTs (Table 1). Thus, strain M17CTs could be obligatory dependent on a methanogenic partner, Methanocalculus alkaliphilus, in acquiring the growth factors necessary for metabolism. Meanwhile, Methanocalculus alkaliphilus receives molecular hydrogen produced by strain M17CTs as a growth substrate.
This study expands our knowledge of the diversity, metabolic functions, and possible ecological role of Bathyarchaeia. Based on phylogenetic position, phenotypic, physiological, and genomic properties, we propose to assign strain M17CTs to the novel taxa on the species, genus, family, order, and class levels. To preserve the currently widely used and stable name for this group of archaea, we also propose that the term Bathyarchaeia (Meng et al., 2014) be retained for all new taxa.
Bathyarchaeum tardum (tar'dum. L. neut. adj. tardum, slow, referring to its slow growth).
Cells are small, non-motile cocci, with a diameter of 0.4–0.7 μm. Occasionally forms chains of 3–5 cells covered by a common sheath. The cytoplasmic membrane is surrounded by an S-layer. Strictly anaerobic. Mesophilic. Grows at 10–45°C (optimum 37°C), at pH 6.0–10.0 (optimum 8.0), and at NaCl concentrations of 0–60 g l−1 (optimum 20 g l−1). Grows on 3,4-dimethoxybenzoic acid, vanillate in the presence of yeast extract, casamino acids, or soytone. Does not utilize glucose, fructose, sucrose, lactate, pyruvate, lignin, chitin, peptone, tryptone, casein, cellulose, formate, methanol, benzoate, 3,4-dihydroxybenzoate, 2-methoxyphenol, 2-methoxybenzoate, molecular hydrogen, and carbon monoxide. Dimethyl sulfoxide and nitrate stimulate growth, while oxygen, elemental sulfur, carbon monoxide, and molecular hydrogen inhibit growth. Does not reduce sulfate, thiosulfate, antraquinone-2,6-disulfonate, selenate, selenite, arsenate, Fe(III)-citrate, elemental sulfur, sulfite, nitrite, or oxygen. Growth depends on addition of sterile natural sediment. The complete genome of strain M17CTs, available under the GenBank assembly accession number (CP122380Ts), is the designated nomenclatural type for the species and was recovered from an enrichment culture, cultivated on 3,4-dimethoxybenzoic acid and established from the anaerobic sediment of a coastal lake at the Taman Peninsula, Russian Federation. The genome has the size of 2.15 Mb and a G + C content of 38.1%. Completeness is estimated by CheckM at 98.28% with 0.934% contamination. Bathyarchaeum tardum is also presented by high-quality MAG M17C-73-01 (GCA_029762755.1; Genome size-2139529bp, # of contigs–12, completeness–97.82%, contamination–0.934%). Both MAGs have identical full-length 16S rRNA genes, and ANI value between MAGs is 99.31%. With the other MAGs of the Bathyarchaeum genus, which were included in this genus based on our phylogenetic reconsideration (Figure 4), they have an ANI value no higher than 84.5%.
Ba.thy.ar.chae'um (Gr. masc.adj. bathys, deep as it locates deep phylogenetic branching within Archaea; N.L. neut. n. archaeum, ancient one, archaeon; from Gr. masc. adj. archaîos, ancient; N.L. neut. n. Bathyarchaeum, deeply branched archaeon).
High-quality MAGs of this genus have been assembled from enrichment cultures with 3,4-dimethoxybenzoic acid inoculated from anaerobic sediment of a coastal lake at the Taman Peninsula, from enrichment cultures with lignin inoculated from coastal sediments of northern East China Sea, from deep-sea sediments associated with petroleum seepage (Atlantic Ocean), and from sediment of high-sulfide freshwater Zodletone spring (Oklahoma, USA). Based on 16S rRNA gene sequence data, microorganisms of this genus were often found in the marine sediments associated with the presence of terrigenous organic matter, e.g., terrigenous deposits, estuary (Li et al., 2012), and sunken woods (Fagervold et al., 2012) and in ecotopes associated with methane seepages, e.g., hydrate-bearing sediments (Dang et al., 2009), cold seep sediment (Li et al., 1999), and mud volcanoes (Cheng et al., 2012). AAI values among genomes representing separate species within the genus range between 71.87% and 83.83%. Our phylogenomic reconstruction (Figure 4) supports delineation of the genus Bathyarchaeum. The Relative Evolutionary Divergence (RED) in phylogenomic reconstruction of GTDB 207 for the genus Bathyarchaeum (g__BIN-L-1) is 0.931.
The nomenclatural type of the genus is BathyarchaeumtardumTs.
Ba.thy.ar.chae.a.ce'ae (N.L.fem.n. Bathyarchaeum, type genus of the family; L. fem. pl. suff. -aceae, ending to denote a family; N.L. fem. pl. n. Bathyarchaeaceae, family of the genus Bathyarchaeum).
Our phylogenomic reconstruction (Figure 4) supports delineation of the family Bathyarchaeaceae. RED in phylogenomic reconstruction of GTDB 207 for the family Bathyarchaeaceae (f__BA1) is 0.691. AAI values range between 59.6 and 70.8% among members of different genera of this family. The nomenclatural type of the family is the genus Bathyarchaeum.
Ba.thy.ar.chae.a'les (N.L. fem. n. Bathyarchaeum, type genus of the order; N.L. fem. pl. suff. -ales, ending denoting an order; N.L. fem. pl. n. Bathyarchaeales, order of the genus Bathyarchaeum, type genus of the order).
Our phylogenomic reconstruction (Figure 4) supports delineation of the order Bathyarchaeales. RED in phylogenomic reconstruction of GTDB 207 for the order Bathyarchaeales (o__B26-1) is 0.514. The nomenclatural type of the order is the genus Bathyarchaeum.
Ba.thy.ar.chae'ia (N.L. fem. n. Bathyarchaeum, type genus of the class; N.L. neut. n. suff. -ia, ending to denote a class; N.L.neut. pl. n. Bathyarchaeia, class of the genus Bathyarchaeum).
GTDB 207 supports delineation of the class Bathyarchaeia (f__BA1) with RED being equal to 0.514. The nomenclatural type of the class is the genus Bathyarchaeum.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
MK, AM, DM, and AK: investigation. MK, AM, and AS: writing—original draft preparation. AS and MK: writing—reviewing and editing. MK and AM: visualization. AM: funding acquisition. All authors contributed to the article and approved the submitted version.
The study was supported by the Russian Science Foundation, project no. 22-24-00868.
The authors are grateful to Sorokin D. Y. for the sincere scientific interest shown in this study and for valuable advices regarding cultivation tricks. They also want to thank Dedysh S. N. and Kulichevskaya I. S. for help with FISH and Kevbrin V. V. for assistance with the chromatographic analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1214631/full#supplementary-material
Alneberg, J., Bjarnason, B. S., Bruijn, d. e., Schirmer, I., Quick, M., Ijaz, J. U.Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Altschul, S., Gish, W., Miller, W., Myers, E., and Lipman, D. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anisimova, M., and Gascuel, O. (2006). Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552. doi: 10.1080/10635150600755453
Berben, T., Forlano Bó, F., In 't Zandt, M. H., Yang, S., Liebner, S., and Welte, C. U. (2022). The Polar Fox Lagoon in Siberia harbours a community of Bathyarchaeota possessing the potential for peptide fermentation and acetogenesis. Anton Leeuw. Int. J. 115, 1229–1244. doi: 10.1007/s10482-022-01767-z
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cheng, T. W., Chang, Y. H., Tang, S. L., Tseng, C. H., Chiang, P. W., Chang, K. T., et al. (2012). Metabolic stratification driven by surface and subsurface interactions in a terrestrial mud volcano. ISME J. 6, 2280–2290. doi: 10.1038/ismej.2012.61
Choi, A. R., Kim, M. S., Kang, S. G., and Lee, H. S. (2016). Dimethyl sulfoxide reduction by a hyperhermophilic archaeon Thermococcus onnurineus NA1 via a cysteine-cystine redox shuttle. J. Microbiol. 54, 31–38. doi: 10.1007/s12275-016-5574-1
Collins, G., O'Connor, L., Mahony, T., Gieseke, A., de Beer, D., and O'Flaherty, V. (2005). Distribution, localization, and phylogeny of abundant populations of Crenarchaeota in anaerobic granular sludge. Appl. Environ. Microbiol. 71, 7523–7527. doi: 10.1128/AEM.71.11.7523-7527.2005
Dang, H., Luan, X., Zhao, J., and Li, J. (2009). Diverse and novel nifH and nifH-like gene sequences in the deep-sea methane seep sediments of the Okhotsk Sea. Appl. Environ. Microbiol. 75, 2238–2245. doi: 10.1128/AEM.02556-08
Danson, M. J., Lamble, H. J., and Hough, D. W. (2007). “Central metabolism,” in Archaea: Molecular and Cellular Biology, ed. R. Cavicchioli, (Washington DC: ASM Press), 260–287. doi: 10.1128./9781555815516.ch12
Deb, S., and Das, S. K. (2022). Phylogenomic analysis of metagenome-assembled genomes deciphered novel acetogenic nitrogen-fixing Bathyarchaeota from hot spring sediments. Microbiol. Spectr. 10, e0035222. doi: 10.1128/spectrum.00352-22
Dedysh, S. N., Derakshani, M., and Liesack, W. (2001). Detection and enumeration of methanotrophs in acidic Sphagnum peat by 16S rRNA fluorescence in situ hybridisation, including the use of newly developed oligonucleotide probes for Methylocella palustris. Appl. Environ. Microbiol. 67, 4850–4857. doi: 10.1128/AEM.67.10.4850-4857.2001
Evans, P. N., Parks, D. H., Chadwick, G. L., Robbins, S. J., Orphan, V. J., Golding, S. D., et al. (2015). Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science. 350, 434–438. doi: 10.1126/science.aac7745
Fagervold, S. K., Galand, P. E., Zbinden, M., Gaill, F., Lebaron, P., Palacios, C., et al. (2012). Sunken woods on the ocean floor provide diverse specialized habitats for microorganisms. FEMS Microbiol. Ecol. 82, 616–628. doi: 10.1111/j.1574-6941.2012.01432.x
Fillol, M., Auguet, J-. C., Casamayor, E. O., and Borrego, C. M. (2016). Insights in the ecology and evolutionary history of the Miscellaneous Crenarchaeotic Group lineage. ISME J. 10, 665–677. doi: 10.1038/ismej.2015.143
Gagen, E. J., Huber, H., Meador, T., Hinrichs, K. U., and Thomm, M. (2013). Novel cultivation-based approach to understanding the miscellaneous crenarchaeotic group (MCG) archaea from sedimentary ecosystems. Appl. Environ. Microbiol. 79, 6400–6406. doi: 10.1128/AEM.02153-13
Harris, R. L., Lau, M. C. Y., Cadar, A., Bartlett, D. H., Cason, E., van Heerden, E., et al. (2018). Draft genome sequence of “Candidatus Bathyarchaeota” archaeon BE326-BA-RLH, an uncultured denitrifier and putative anaerobic methanotroph from South Africa's deep continental biosphere. Microbiol. Resour. Announc. 7, e01295–18. doi: 10.1128/MRA.01295-18
He, Y., Li, M., Perumal, V., Feng, X., Fang, J., Xie, J., et al. (2016). Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat. Microbiol. 1, 16035. doi: 10.1038/nmicrobiol.2016.35
Hedlund, B. P., Chuvochina, M., Hugenholtz, P., Konstantinidis, K. T., Murray, A. E., Palmer, M., et al. (2022). SeqCode: a nomenclatural code for prokaryotes described from sequence data. Nat. Microbiol. 7, 1702–1708. doi: 10.1038/s41564-022-01214-9
Hou, J., Wang, Y., Zhu, P., Yang, N., Liang, L., Yu, T., et al. (2023). Taxonomic and carbon metabolic diversification of Bathyarchaeia during its coevolution history with early Earth surface environment. Sci. Adv. 9, eadf5069. doi: 10.1126./sciadv.adf5069
Hu, H., Natarajan, V. P., and Wang, F. (2021). Towards enriching and isolation of uncultivated archaea from marine sediments using a refined combination of conventional microbial cultivation methods. Mar. Life Sci. Technol. 3, 231–242. doi: 10.1007/s42995-021-00092-0
Inagaki, F., Suzuki, M., Takai, K., Oida, H., Sakamoto, T., Aoki, K., et al. (2003). Microbial communities associated with geological horizons in coastal subseafloor sediments from the sea of okhotsk. Appl. Environ. Microbiol. 69, 7224–7235. doi: 10.1128/AEM.69.12.7224-7235.2003
Kalyaanamoorthy, S., Minh, B. Q., Wong, T., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M., and Ishiguro-Watanabe, M. (2023). KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. doi: 10.1093/nar/gkac963
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. Peer J. 7, e7359. doi: 10.7717/peerj.7359
Khomyakova, M. A., Merkel, A. Y., Kopitsyn, D. S., and Slobodkin, A. I. (2022). Pelovirga terrestris gen. nov., sp. nov., anaerobic, alkaliphilic, fumarate-, arsenate-, Fe(III)- and sulfur-reducing bacterium isolated from a terrestrial mud volcano. Syst. Appl. Microbiol. 45, 126304. doi: 10.1016/j.syapm.2022.126304
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. doi: 10.1093/nar/gks808
Kubo, K., Lloyd, K. G., Biddle, J. F., Amann, R., Teske, A., Knittel, K., et al. (2012). Archaea of the Miscellaneous Crenarchaeotal Group are abundant, diverse and widespread in marine sediments. ISME J. 6, 1949–1965. doi: 10.1038/ismej.2012.37
Kurth, J. M., Nobu, M. K., Tamaki, H., Jonge, d. e., Berger, N., Jetten, S., et al. (2021). Methanogenic archaea use a bacteria-like methyltransferase system to demethoxylate aromatic compounds. ISME J. 15, 3549–3565. doi: 10.1038/s41396-021-01025-6
Lazar, C. S., Baker, B. J., Seitz, K., Hyde, A. S., Dick, G. J., Hinrichs, K. U., et al. (2016). Genomic evidence for distinct carbon substrate preferences and ecological niches of Bathyarchaeota in estuarine sediments. Environ. Microbiol. 18, 1200–1211. doi: 10.1111/1462-2920.13142
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinform. 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, L., Kato, C., and Horikoshi, K. (1999). Microbial diversity in sediments collected from the deepest cold-seep area, the Japan Trench. Mar. Biotechnol (NY). 1, 391–400. doi: 10.1007/pl00011793
Li, Q., Wang, F., Chen, Z., Yin, X., and Xiao, X. (2012). Stratified active archaeal communities in the sediments of Jiulong River estuary, China. Front. Microbiol. 3, 311. doi: 10.3389/fmicb.2012.00311
Lin, D. D., Liu, Y. F., Zhou, L., Yang, S. Z., Gu, J. D., Mu, B. Z., et al. (2022). Stimulation of Bathyarchaeota in enrichment cultures by syringaldehyde, 4-hydroxybenzaldehyde and vanillin under anaerobic conditions. Int. Biodeterior. Biodegradation 171, 105409. doi: 10.1016/j.ibiod.2022.105409
Lloyd, K. G., Schreiber, L., Petersen, D. G., Kjeldsen, K. U., Lever, M. A., Steen, A. D., et al. (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature. 496, 215–218. doi: 10.1038/nature12033
Loh, H. Q., Hervé, V., and Brune, A. (2021). Metabolic potential for reductive acetogenesis and a novel energy-converting [NiFe] hydrogenase in Bathyarchaeia from termite guts—A genome-centric analysis. Front. Microbiol. 11, 635786. doi: 10.3389/fmicb.2020.635786
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 17.1, 10–12. doi: 10.14806/ej.17.1.200
Martínez-Espinosa, R. M., Marhuenda-Egea, F. C., and Bonete, M. J. (2001). Assimilatory nitrate reductase from the haloarchaeon Haloferax mediterranei: purification and characterisation. FEMS Microbiol. Lett. 204, 381–385. doi: 10.1016/s0378-1097(01)00431-1
Mayumi, D., Mochimaru, H., Tamaki, H., Yamamoto, K., Yoshioka, H., Suzuki, Y., et al. (2016). Methane production from coal by a single methanogen. Science 354, 222–225. doi: 10.1126/science.aaf8821
Meng, J., Xu, J., Qin, D., He, Y., Xiao, X., Wang, F., et al. (2014). Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses. ISME J. 8, 650–659. doi: 10.1038/ismej.2013.174
Minh, B. Q., Nguyen, M. A., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. E30, 1188–1195. doi: 10.1093/molbev/mst024
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. E37, 1530–1534. doi: 10.1093/molbev/msaa015
Naidu, D., and Ragsdale, S. W. (2001). Characterization of a three-component vanillate O-demethylase from Moorella thermoacetica. J. Bacteriol. 183, 3276–3281. doi: 10.1128/JB.183.11.3276-3281.2001
Nissen, L. S., and Basen, M. (2019). The emerging role of aldehyde: ferredoxin oxidoreductases in microbially-catalyzed alcohol production. J. Biotechnol. 306, 105–117. doi: 10.1016/j.jbiotec.2019.09.005
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 5, 824–834. doi: 10.1101/gr.213959.116
Pan, J., Chen, Y., Wang, Y., Zhou, Z., and Li, M. (2019). Vertical distribution of bathyarchaeotal communities in mangrove wetlands suggests distinct niche preference of Bathyarchaeota subgroup 6. Microb Ecol. 77, 417–428. doi: 10.1007/s00248-018-1309-7
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of tran-script expression. Nat. methods 14, 417–419. doi: 10.1038/nmeth.4197
Rawlings, N. D., Barrett, A. J., Thomas, P. D., Huang, X., Bateman, A., Finn, R. D., et al. (2018). The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632. doi: 10.1093/nar/gkx1134
Rinke, C., Chuvochina, M., Mussig, A. J., Chaumeil, P. A., Davín, A. A., Waite, D. W., et al. (2021). A standardized archaeal taxonomy for the Genome Taxonomy Database. Nat. Microbiol. 6, 946–959. doi: 10.1038/s41564-021-00918-8
Rosenbaum, F. P., Poehlein, A., Daniel, R., and Müller, V. (2022). Energy-conserving dimethyl sulfoxide reduction in the acetogenic bacterium Moorella thermoacetica. Environ. Microbiol. 24, 2000–2012. doi: 10.1111/1462-2920.15971
Sapra, R., Bagramyan, K., and Adams, M. W. W. (2003). A simple energy-conserving system: Proton reduction coupled to proton translocation. Proc. Natl. Acad. Sci. USA. 100, 7545–7550. doi: 10.1073/pnas.1331436100
Sazanov, L. A. (2007). Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry 46, 2275–2288. doi: 10.1021/bi602508x
Slobodkin, A. I., Reysenbach, A-. L., Slobodkina, G. B., Baslerov, R. V., Kostrikina, N. A., Wagner, I. D., et al. (2012). Thermosulfurimonas dismutans gen. nov., sp. nov., an extremely thermophilic sulfur-disproportionating bacterium from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 62, 2565–2571. doi: 10.1099/ijs.0.034397-0
Søndergaard, D., Pedersen, C. N., and Greening, C. (2016). HydDB: A web tool for hydrogenase classification and analysis. Sci, Rep.6, 34212. doi: 10.1038/srep34212
Sorokin, D. Y., Abbas, B., Merkel, A. Y., Rijpstra, W. I. C., Damsté, J. S. S., Sukhacheva, M. V., et al. (2015). Methanosalsum natronophilum sp. nov., and Methanocalculus alkaliphilus sp. nov., haloalkaliphilic methanogens from hypersaline soda lakes. Int. J. Syst. Evol. Microbiol. 65, 3739–3745. doi: 10.1099/ijsem.0.000488
Teufel, F., Almagro Armenteros, J. J., Johansen, A. R. Gíslason, M. H., Pihl, S. I., Tsirigos, K. D., et al. (2022). SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025. doi: 10.1038/s41587-021-01156-3
Uritskiy, G. V., DiRuggiero, J., and Taylor, J. (2018). MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158. doi: 10.1186/s40168-018-0541-1
Vignais, P. M., and Billoud, B. (2007). Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272. doi: 10.1021/cr050196r
Welte, C. U., Graaf, d. e., Dalcin Martins, R., Jansen, P., Jetten, R. S. M. S. M., and Kurth, J. M. (2021). A novel methoxydotrophic metabolism discovered in the hyperthermophilic archaeon Archaeoglobus fulgidus. Environ. Microbiol. 23, 4017–4033. doi: 10.1111/1462-2920.15546
Whitman, W. B., Chuvochina, M., Hedlund, B. P., Hugenholtz, P., Konstantinidis, K. T., Murray, A., et al. (2022). Development of the SeqCode: a proposed nomenclatural code for uncultivated prokaryotes with DNA sequences as type. Syst. Appl. Microbiol. 126305. doi: 10.1016./j.syapm.2022.126305
Wolin, E. A., Wolin, M. J., and Wolfe, R. S. (1963). Formation of methane by bacterial extracts. J. Biol. Chem. 238, 2882–2886.
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Xiang, X., Wang, R., Wang, H., Gong, L., Man, B., Xu, Y., et al. (2017). Distribution of Bathyarchaeota communities across different terrestrial settings and their potential ecological functions. Sci. Rep. 7, 45028. doi: 10.1038/srep45028
Xiong, L., Jian, H., Zhang, Y., and Xiao, X. (2016). The two sets of DMSO respiratory systems of Shewanella piezotolerans WP3 are involved in deep sea environmental adaptation. Front. Microbiol. 7, 1418. doi: 10.3389/fmicb.2016.01418
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K. H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 9, 635–645. doi: 10.1038/nrmicro3330
Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S., Madden, T. L., et al. (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 13, 134. doi: 10.1186/1471-2105-13-134
Yin, X., Zhou, G., Cai, M., Zhu, Q. Z., Richter-Heitmann, T., Aromokeye, D. A., et al. (2022). Catabolic protein degradation in marine sediments confined to distinct archaea. ISME J. 16, 1617–1626. doi: 10.1038/s41396-022-01210-1
Yu, T., Liang, Q., Niu, M., and Wang, F. (2017). High occurrence of Bathyarchaeota (MCG) in the deep-sea sediments of South China Sea quantified using newly designed PCR primers. Environ. Microbiol. Rep. 9, 374–382. doi: 10.1111/1758-2229.12539
Yu, T., Wu, W., Liang, W., Lever, M. A., Hinrichs, K. U., Wang, F., et al. (2018). Growth of sedimentary Bathyarchaeota on lignin as an energy source. Proc. Natl. Acad. Sci. U S A. 115, 6022–6027. doi: 10.1073/pnas.1718854115
Zhang, W., Ding, W., Yang, B., Tian, R., Gu, S., Luo, H., et al. (2016). Genomic and transcriptomic evidence for carbohydrate consumption among microorganisms in a cold seep brine pool. Front. Microbiol. 7, 1825. doi: 10.3389/fmicb.2016.01825
Zhilina, T. N., Zavarzina, D. G., Kevbrin, V. V., and Kolganova, T. V. (2013). Methanocalculus natronophilus sp. nov., a new alkaliphilic hydrogenotrophic methanogenic archaeon from a soda lake, and proposal of the new family Methanocalculaceae. Mikrobiologiia 82, 681–690. Russian.
Zhou, L., Zhou, Z. C., Lu, Y. W., Ma, L., Bai, Y., Li, X. X., et al. (2019). The newly proposed TACK and DPANN archaea detected in the production waters from a high-temperature petroleum reservoir. Int. Biodeterior. Biodegrad. 143. doi: 10.1016./j.ibiod.2019.104729
Zhou, Z., Pan, J., Wang, F., Gu, J. D., and Li, M. (2018). Bathyarchaeota: globally distributed metabolic generalists in anoxic environments. FEMS Microbiol. Rev. 42, 639–655. doi: 10.1093/femsre/fuy023
Keywords: uncultured microorganisms, archaea, anaerobic, methoxylated aromatic compounds, O-demethylase, Methanocalculus
Citation: Khomyakova MA, Merkel AY, Mamiy DD, Klyukina AA and Slobodkin AI (2023) Phenotypic and genomic characterization of Bathyarchaeum tardum gen. nov., sp. nov., a cultivated representative of the archaeal class Bathyarchaeia. Front. Microbiol. 14:1214631. doi: 10.3389/fmicb.2023.1214631
Received: 30 April 2023; Accepted: 31 July 2023;
Published: 22 August 2023.
Edited by:
Zhichao Zhou, University of Wisconsin-Madison, United StatesReviewed by:
Carolina A. Martinez-Gutierrez, Virginia Tech, United StatesCopyright © 2023 Khomyakova, Merkel, Mamiy, Klyukina and Slobodkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria A. Khomyakova, bWFyeV9rbGltb3ZhQG1haWwucnU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.