
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 19 June 2023
Sec. Microbial Physiology and Metabolism
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1213659
This article is part of the Research Topic Mechanisms of Prokaryotic Predation, Volume II View all 6 articles
 María José Soto1†
María José Soto1† Juana Pérez2†
Juana Pérez2† José Muñoz-Dorado2
José Muñoz-Dorado2 Francisco Javier Contreras-Moreno2
Francisco Javier Contreras-Moreno2 Aurelio Moraleda-Muñoz2*
Aurelio Moraleda-Muñoz2*Bacterial predation impacts microbial community structures, which can have both positive and negative effects on plant and animal health and on environmental sustainability. Myxococcus xanthus is an epibiotic soil predator with a broad range of prey, including Sinorhizobium meliloti, which establishes nitrogen-fixing symbiosis with legumes. During the M. xanthus-S. meliloti interaction, the predator must adapt its transcriptome to kill and lyse the target (predatosome), and the prey must orchestrate a transcriptional response (defensome) to protect itself against the biotic stress caused by the predatory attack. Here, we describe the transcriptional changes taking place in S. meliloti in response to myxobacterial predation. The results indicate that the predator induces massive changes in the prey transcriptome with up-regulation of protein synthesis and secretion, energy generation, and fatty acid (FA) synthesis, while down-regulating genes required for FA degradation and carbohydrate transport and metabolism. The reconstruction of up-regulated pathways suggests that S. meliloti modifies the cell envelop by increasing the production of different surface polysaccharides (SPSs) and membrane lipids. Besides the barrier role of SPSs, additional mechanisms involving the activity of efflux pumps and the peptide uptake transporter BacA, together with the production of H2O2 and formaldehyde have been unveiled. Also, the induction of the iron-uptake machinery in both predator and prey reflects a strong competition for this metal. With this research we complete the characterization of the complex transcriptional changes that occur during the M. xanthus-S. meliloti interaction, which can impact the establishment of beneficial symbiosis with legumes.
Bacteria interact with co-habiting microbes in different multispecies communities. The ecological and evolutionary success of microorganisms in a particular environment is not only governed by their capacity to adapt to external abiotic stresses, but also depends on their ability to detect and respond to competition with the neighboring cells. Consequently, the metabolic processes of one strain are influenced by the metabolic functions of the other members of the community. The relationships between microorganisms range from cooperative symbiotic associations to different competition strategies. In all cases, bacterial interactions involve complex processes that are key determinants that strongly shape the structure of bacterial communities (Bauer et al., 2018; Granato et al., 2019). In the last decades many studies have revealed how bacterial interactions that occur in small communities have consequences that affect in many cases human, animal, and plant health (Pérez et al., 2011; Stubbendieck et al., 2016; Niehaus et al., 2019; Molina-Santiago et al., 2021; Martins et al., 2022).
A particular type of interaction is represented by predatory bacteria, which are species that kill and lyse susceptible cells in order to consume the cellular materials as carbon and energy sources (Pérez et al., 2016; Whitworth et al., 2020). Most bacterial predators use two major approaches to kill prey: (i) the endobiotic strategy represented by Bdellovibrio and like organisms (BALOs) that mainly prey on diderm bacteria in the planktonic phase as well as in biofilms (Mookherjee and Jurkevitch, 2022), and (ii) the epibiotic predation exemplified by myxobacteria that can kill and externally lyse a great variety of microorganisms (Muñoz-Dorado et al., 2016; Pérez et al., 2016).
The capacity of bacterial predators to kill other bacteria, including multidrug-resistant pathogens, has attracted the attention of researchers as a feasible alternative to antibiotics in the actual crisis (Pérez et al., 2020). As the use of technologies such as next generation sequencing and meta-transcriptomics are increasingly being applied to the study of soil microbiota, bacterial predation is gaining relevant importance as a shaper of microbial communities. Although myxobacteria have been traditionally considered minority components of soil bacterial communities, several studies have revealed that this does not seem to be the case, and that the myxobacterial community is a predominant and highly diverse group within soil niches (Zhou et al., 2014). Until recently, protists have been considered the dominant group preying on bacteria. However, the results of recent studies strongly suggest the importance, and possibly even dominance, of myxobacteria as soil predators. In fact, an analysis of 28 European soils showed that in most of these soils myxobacteria comprise 1.5–9.7% of all obtained SSU rRNA transcripts and more than 60% of all identified potential bacterivores (Petters et al., 2021).
Myxococcus xanthus is a soil myxobacterium which has been extensively studied because of its unique complex lifecycle. This lifecycle consists of two stages: a vegetative growth stage in the presence of nutrients and/or prey (it is a facultative predator); and a developmental stage (with the formation of macroscopic fruiting bodies filled of myxospores) when nutrients are depleted (Muñoz-Dorado et al., 2016). It has a large genome which encodes all the genes that participate in the complex social and multicellular lifestyle exhibited during both growth and development (Goldman et al., 2006).
Myxococcus xanthus predation requires the participation of many weapons to kill and consume the prey, including a variety of hydrolytic enzymes, outer membrane vesicles, contact-dependent and independent elements, and the production of secondary metabolites such as antibiotics (Pérez et al., 2016; Thiery and Kaimer, 2020; Seef et al., 2021). In addition, this arsenal differs from one prey to another (Thiery et al., 2022).
In our laboratory we deciphered the transcriptomic changes that take place in M. xanthus during a complete lifecycle. During development, 1,415 genes were sequentially and differentially expressed in 10 discrete groups (Muñoz-Dorado et al., 2019). Moreover, we have also analyzed the predatosome of this myxobacterium when preying on Sinorhizobium meliloti. The results obtained revealed that the number of genes differentially expressed during predation is lower than during development. Among the transcripts that are up-regulated during predation, the most noteworthy are genes involved in the biosynthesis of secondary metabolites, in the synthesis and degradation of lipids, those encoding both extracellular and outer membrane hydrolytic enzymes, and genes related to social motility and Tad-like apparatuses (Pérez et al., 2022).
In predatory interactions, the prey will need to modify the network of genes and pathways required to mount an orchestrated defense against the biotic stress caused by the predatory attack. We will use the term “defensome” to refer to the whole set of genes that vary their transcription in response to the predatory bacterium, either to resist predation or to compete for resources. Nevertheless, some specific defense mechanisms and adaptations of different prey against M. xanthus attack have been reported, so far. For example, galactoglucan and melanin protect S. meliloti from predation by this myxobacterium (Pérez et al., 2014; Contreras-Moreno et al., 2020). The presence of this predator also induces the transcriptional activation of silenced genes coding for antibiotics in Streptomyces coelicolor (Pérez et al., 2011; Lee et al., 2020). Bacillus subtilis induces bacillaene synthesis and forms spore-filled megastructures against the attack of M. xanthus (Müller et al., 2014, 2015). On the other hand, studies of the prey response revealed a novel antibiotic resistance mechanism consisting of the glucosylation of the antibiotic myxovirescin TA, which was discovered in Bacillus licheniformis (Wang et al., 2019). Also, the transcriptome of Escherichia coli against M. xanthus in liquid media has been analyzed, and the results showed that the presence of the predator caused widespread induction in gene expression and enrichment of several pathways including ribosome production, lipopolysaccharide (LPS) biosynthesis, oxidative phosphorylation, production of antibiotics and secondary metabolites, energy and carbon metabolism, and vitamin and amino acid metabolism. However, only the pathway involved in glycerophospholipid metabolism was down-regulated (Livingstone et al., 2018).
In the current study, we have analyzed the defensome of the prey S. meliloti against attack by M. xanthus. S. meliloti is an alphaproteobacterium that establishes nitrogen-fixing symbiosis with legumes, thereby contributing to the fertility of soils. This soil bacterium can also exist as a free-living organism in natural environments, where it must adapt to diverse nutrient availability conditions, and compete with other neighboring microbes, including predators such as M. xanthus. These competitive interactions will affect not only the structure of the soils, but also, ultimately, will affect their fertility.
Our results reveal that the contact with M. xanthus induces in S. meliloti defense and/or adaptation genes that affect central pathways, such as protein biosynthesis and secretion. The reconstruction of other up-regulated pathways seems to indicate that S. meliloti not only protects itself against predator attack in a passive way by modifying the cell envelope, but also reacts actively by producing H2O2 or formaldehyde. The induction of genes related to iron uptake indicate ion competition. This research completes the study on transcriptomic changes undergone in both partners during the M. xanthus-S. meliloti interaction and draws a panoramic view of the mechanisms and pathways involved in attack, defense, and competition.
Sinorhizobium meliloti Rm1021 (Meade and Signer, 1977) was used as prey, whereas M. xanthus DK1622 (Kaiser, 1979) was used as predator. Tryptone yeast (TY) solid and liquid media (Beringer, 1974) were used for maintenance and growth of S. meliloti. CTT solid and liquid media (Hodgkin and Kaiser, 1977) were used to grow M. xanthus, and for the predation experiments. Solid media contained 1.5% Bacto-Agar (Difco, Le Pont de Claix, France), and liquid cultures were incubated with vigorous shaking at 30°C.
Sinorhizobium meliloti was grown in TY broth to an optical density at 600 nm (OD600) of 1 and then diluted using the same broth to a final OD600 of 0.2. Twenty 10-μl drops of the diluted culture were deposited on the surface of CTT agar plates for each replicate and incubated at 30°C for 24 h. After that time, S. meliloti cells from two replicates were harvested from plates to obtain t = 0 prey samples (samples Sm_t0). Then, to obtain samples of predatory and prey interacting cells, 10-μl drops of M. xanthus, grown in CTT liquid media to an OD600 of 1 and concentrated in TM buffer [10 mM Tris-HCl (pH 7.6), 1 mM MgSO4] to a final OD600 of 15, were deposited on top of each of the rhizobial colonies of a subset of the plates (samples Mx_Sm). Another subset of samples of S. meliloti was kept growing alone (samples Sm). Two replicates from each of the two conditions (predator/prey co-culture and pure culture of S. meliloti) were harvested from plates after 2 and 6 h of incubation. Pellets from each sample were resuspended in 0.5 ml of RNA Protect Bacteria Reagent (Qiagen, Hilden, Germany), incubated at room temperature for 5 min, and centrifuged at 5,000 g for 10 min (4°C). Next, pellets were stored at −80°C.
To purify RNA, cells were lysed for 10 min at room temperature with lysozyme and proteinase K [250 μl of 3 mg/ml lysozyme (Roche Diagnostic, Mannheim, Germany) and 0.4 mg/ml proteinase K (Ambion, Carlsbad, CA, United States)] prepared in TE buffer [10mMTris-HCl; 1mMethylenediaminetetraacetic acid (EDTA), pH 8.0]. RNeasy Mini Kit (Qiagen, Hilden, Germany) was used for RNA extraction [carrying out on-column DNase digestion with the RNAse-free DNase set (Qiagen, Hilden, Germany)], eluting each sample in 50 μl of RNase-free water.
Total RNA samples were processed by Novogene [Novogene Europe, Cambridge, United Kingdom], including rRNA depletion from total RNA samples with the Illumina Ribo-Zero Plus rRNA Reduction Kit (Illumina, Inc.). Remaining RNA was processed according to the procedures described in Pérez et al. (2022).
On average, 18.81 million raw reads and a coverage of 421.75x were obtained. After removing reads containing adapters, low quality reads and/or reads with more than 10 percent uncertain nucleotides, the genome coverage varied from 355.53x to 494.52x (median of 415.18x), which provide an excellent coverage of the mRNA fraction.
To facilitate comparison analyses with other S. meliloti transcriptomes we used the old nomenclature (SMa_, SMb_ or SMc_), since these identifiers are commonly used in the literature. However, the corresponding new localizers SM_RS are also indicated in the tables. To decipher the defensome, we have considered all the up- and down-regulated transcripts with | Log2 Fold Change| > 0 and padj < 0.05. It must be taken into account that predation is a multifaceted process, and any change could be of interest as we have demonstrated in the case of M. xanthus predatosome (Pérez et al., 2022). The adaptation and defense mechanisms expected may not be very drastic and, for this reason, our research has been focused on those routes in which there is gene enrichment or in those pathways in which many of the genes involved are up or down regulated in the presence of the predator. In all figures and tables the Log2 Fold Change is specified and those Log2 Fold Change > 1 or Log2 Fold Change < −1 are highlighted. For researchers interested in this transcriptomics, all data including reads, fragments per kilobase of transcript per million fragments mapped (FPKMs) and Log2 Fold Change are attached as Supplementary material. However, for improved confidence, | Log2 Fold Change| > 1 has been used in comparisons with other transcriptomes, and this threshold is also indicated in all the figures and tables.
Transcriptional changes in S. meliloti in response to the predatory attack by M. xanthus was investigated by using the RNA-seq technology. The M. xanthus-S. meliloti co-culture conditions and the preparation of libraries have been previously described in detail (Pérez et al., 2022). We focused our study on two points in time after contact: at 2 and 6 h, a period of time during which the prey has to adapt its metabolism and structures to the biotic stress that represents the predatory attack. Following the same procedure used to elucidate the M. xanthus predatosome (Pérez et al., 2022), five cDNA libraries were constructed to analyze the S. meliloti defensome: Sm_t0: S. meliloti alone at time 0 h; Sm_t2: S. meliloti alone collected after 2 h on solid CTT medium; Sm_t6: S. meliloti alone grown for 6 h on solid CTT medium; Mx_Smt2: cells collected after 2 h of interaction between M. xanthus and S. meliloti; and Mx_Smt6: cells harvested after 6 h of the M. xanthus-S. meliloti interaction. From now on, the terms t2 and t6 will be used to refer to results obtained at 2 and 6 h of the co-cultures of M. xanthus and S. meliloti (Mx_Smt2 and Mx_Smt6, respectively) compared to their respective controls (prey cells in pure culture at 2 or 6 h: Sm_t2 and Sm_t6, respectively).
The total number of raw reads from each sample, the clean reads (obtained after removal of raw reads containing adapters and/or of low quality reads), the errors Q20 and Q30, and the GC content of the clean reads are compiled in Supplementary Table 1A.
The Pearson correlation coefficients (R2) between replicates were satisfactory in all samples (≥ 0.91) (Supplementary Figure 1A). The principal component analysis (PCA) showed that genes obtained from the same condition cluster together and genes from different nutritional stages and times cluster separately as expected (Supplementary Figure 1B). The RNA mean reads were normalized to FPKM values (Supplementary Table 1B). The FPKM density distributions and the violin diagrams showing similar gene expression levels are depicted in Supplementary Figures 1C, D, respectively.
The volcano plots were constructed by using the transcripts of co-cultures at 2 and 6 h versus their respective controls filtered by their fold changes (| Log2 Fold Change| > 0) and padj < 0.05 (Figures 1A, B).
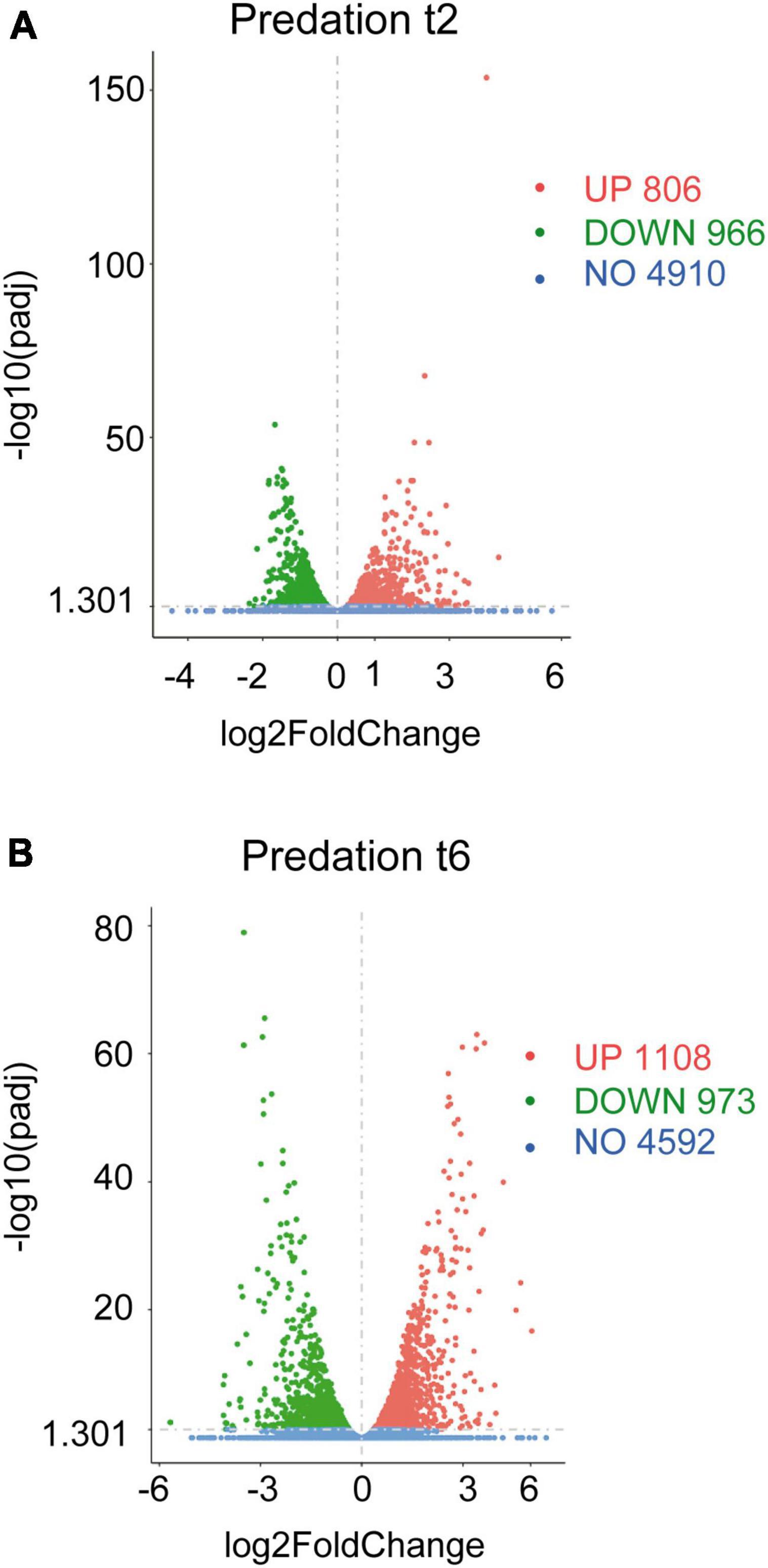
Figure 1. Differential gene expression of Sinorhizobium meliloti in response to Myxococcus xanthus predation. Volcano plots of up-regulated and down-regulated genes during the predatory interaction at (A) t2 and (B) t6 (2 and 6 h of contact). The estimated fold changes (x-axis) versus the minus log10 of the adjusted p-values (y-axis) from DESeq analysis are shown in the volcano plots. The significant genes with absolute values of | Log2 Fold Change| > 0 and padj < 0.05 are depicted in red (up-regulated) or in green (down-regulated). Blue dots indicate non-regulated genes (NO). Gray vertical dotted lines indicate zero-fold change.
Including novel genes and sRNAs (i.e., non-coding RNAs of 50–500 nt length), 1,772 and 2,081 transcripts were differentially regulated in S. meliloti in response to predator attack at 2 and 6 h, respectively (Figures 1A, B and Supplementary Tables 1C, D). Concerning the direction of transcript regulation, 806 transcripts (45.5%) were up-regulated and 966 (54.5%) down-regulated at 2 h, while 1,108 (53.2%), and 973 (46.8%) transcripts were up- and down-regulated, respectively, at 6 h. Overall, these data indicate that 26.5% (2 h) and 31.2% (6 h) of the S. meliloti transcriptome responded to predator attack. This represents a change in transcriptional activity notably greater than that observed in the predator under the same experimental conditions (Pérez et al., 2022). These results are in agreement with those obtained during predation on E. coli, where it was also found that predation caused a much more pronounced response in the prey than in the predator (Livingstone et al., 2018), indicating a strong adaptive, competitive and/or defensive response triggered by the presence of the predator. For further analyses of the defensome in this study, novel genes and sRNAs were not considered, leaving 1,361 and 1,818 transcripts as differentially regulated at 2 and 6 h, respectively. These genes have been organized in 973 up-regulated and 1,139 down-regulated genes identified at 2 h and/or 6 h in Supplementary Tables 2A, B.
We investigated whether predation alters gene expression in S. meliloti in a replicon-biased fashion. The S. meliloti Rm1021 genome (6.69 Mb in size) is composed of three large replicons: a chromosome (3.65 Mb) and two megaplasmids, pSymA (1.35 Mb), and pSymB (1.68 Mb) that contain 54, 21, and 25% of the total annotated genes (Galibert et al., 2001). We found that genes differentially regulated during predation were not proportionally distributed among the replicons. Instead, they were biased toward the chromosome (70.6%), whereas only 9.6 and 19.8% of the differentially expressed genes were associated to pSymA and pSymB, respectively (Supplementary Figure 2A). This chromosomal bias was mainly caused by genes whose expression was increased during predation since 83.2% were located on the chromosome with only 5.6% on pSymA and 11.2% on pSymB. In contrast, down-regulated genes were distributed more evenly, with 59.8% on the chromosome, 13% on pSymA and 27.2% on pSymB (Supplementary Figure 2B and Supplementary Table 3). These results indicate that in response to predator attack, S. meliloti activates chromosomal-encoded functions, whereas the symbiotic plasmids, especially pSymA, have a minor contribution.
To identify the main biological processes affected in S. meliloti during predation, two different approaches were used. In one of them, enrichment analyses were carried out using the associated pathways in the KEGG database (Kyoto Encyclopedia of Genes and Genomes; Kanehisa et al., 2021). The up-regulated pathways involved those related to ribosome production, oxidative phosphorylation, and biosynthesis of amino acids, secondary metabolites, and cofactors (Supplementary Figure 3A and Supplementary Tables 1E, F). The main down-regulated pathways during predation were related to valine, leucine, and isoleucine degradation, microbial metabolism in diverse environments, carbon metabolism, quorum sensing and ABC transporters (Supplementary Figure 3B and Supplementary Tables 1E, F). As a complementary approach, functional categories of the 973 up-regulated (Supplementary Table 2A) and the 1,139 down-regulated (Supplementary Table 2B) genes were determined using clusters of orthologous groups (COGs) according to the genome sequence annotations of the S. meliloti Genome Project1 (Table 1).
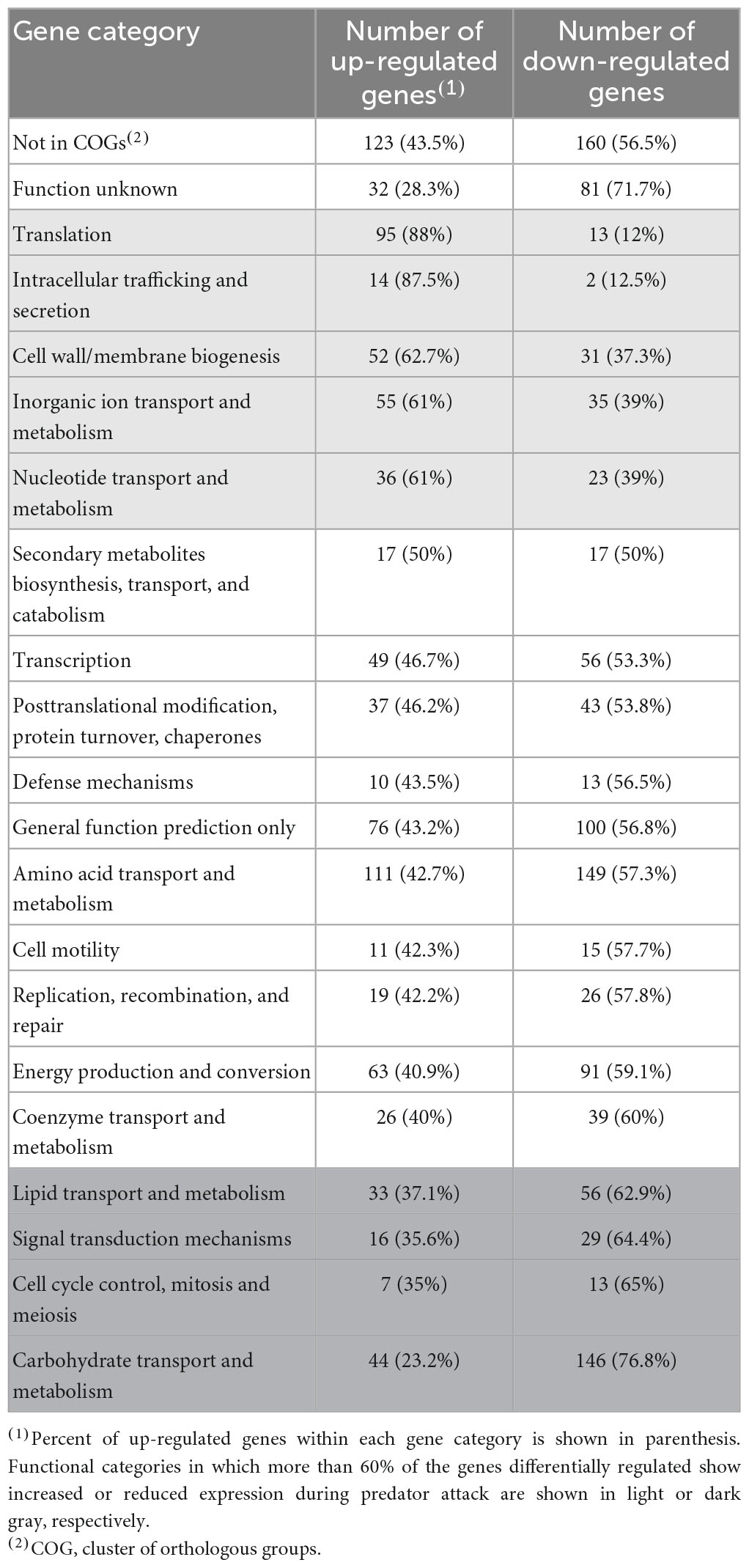
Table 1. Functional categories of genes differentially expressed in Sinorhizobium meliloti in response to predation by Myxococcus xanthus.
Many differentially regulated genes are annotated as either exhibiting partial or global homology to genes deposited in the databases (Not in COGs) or having unknown functions. Nevertheless, several functional categories could be identified associated to up-regulated genes (highlighted in light gray in Table 1), or to down-regulated genes (highlighted in dark gray in Table 1). These analyses suggest that the S. meliloti response to the predatory attack by M. xanthus involves the activation of protein synthesis and secretion, increased energy generation, changes in the cell envelope and membranes, and the stimulation of the transport and metabolism of inorganic ions and nucleotides. At the same time, carbohydrate and lipid transport and metabolism, as well as signal transduction mechanisms and cell division-related functions are repressed during predation. Comparison of the S. meliloti defensome obtained in the present study with that of E. coli (Livingstone et al., 2018) reveals some similarities but also some differences. Thus, increased protein production and energy generation, and the biosynthesis of secondary metabolites were detected during predation on the two preys. However, whereas carbon metabolism and glycerophospholipid metabolism were up- and down-regulated, respectively, in E. coli during predation, the opposite regulation was found for the same functional categories in S. meliloti. If these differences reflect different evasion/defense strategies of the prey or if they are the result of different experimental approaches remains unknown.
A large fraction (88%) of the genes involved in translation which were identified as differentially regulated in the transcriptome profile exhibited increased expression differentially expressed genes involved in translation identified in our transcriptome exhibited increased expression during predation compared with control conditions (Table 1). Of the 95 up-regulated genes related to translation, 28 code for ribosomal proteins and proteins involved in ribosome maturation and modification, and 33 for aminoacyl-tRNA synthetases and proteins related with tRNA modification. In addition, genes putatively coding for translation initiation (infA, infB, infC) and elongation (tsf, efp) factors, as well as probable peptide chain release factors (prfA, prfB, prfC) were also up-regulated, strongly suggesting the activation of protein synthesis during predation. Hence, translation is a biological process activated during predation in different prey (Livingstone et al., 2018), but also in the predator (Pérez et al., 2022).
In agreement with increased protein synthesis and the consequent greater demand for amino acids, 111 genes involved in amino acid transport and metabolism were also up-regulated. Moreover, genes coding for the ATP-dependent chaperone folding system DnaK/DnaJ/GrpE exhibited increased expression. This system plays a crucial role in microbial proteostasis under both normal and stress conditions by assisting the folding of newly synthesized polypeptides, and of misfolded or aggregated proteins (Winter and Jakob, 2004; Barriot et al., 2020). Recently, S. meliloti DnaJ has been shown to participate in tolerance to different stresses (Brito-Santana et al., 2023). Besides assisting protein folding, the DnaK chaperone also facilitates protein targeting to membranes and protein translocation (Barriot et al., 2020). Interestingly, we found that the majority of genes involved in intracellular trafficking and secretion identified as differentially expressed during predation were also up-regulated (Table 1), suggesting increased protein secretion. This was the case for several genes related to the Sec system, which is responsible for the insertion, translocation, and secretion across the membrane of unfolded polypeptides, which carry a removable N-terminal signal sequence. Thus, genes coding for the membrane-embedded SecYEG translocon, the SecA ATPase and piloting factors such as the signal recognition particle (SRP) Ffh or the SecB chaperone, which maintain newly synthesized proteins in an unfolded conformation and drive them to the membrane (Papanikou et al., 2007), exhibited increased expression during predation. In contrast to the Sec translocon, the Tat system is responsible for exporting previously folded proteins which have a particular signal peptide containing a recognizable twin-arginine motif (Pickering and Oresnik, 2010; Palmer and Berks, 2012). These proteins seem also to be actively exported in S. meliloti during predation, as suggested by the up-regulation of the tatA gene coding for a TatA/E translocase homolog. The up-regulation of protein secretion systems has also been reported in myxobacteria, where it has been associated to the secretion of factors required for predation of bacteria and fungi (Li et al., 2019; Pérez et al., 2022). In the case of the prey, increased protein secretion could be required to maintain bacterial cell envelope protein complexes, whose integrity might be damaged during predatory attack.
Many genes coding for the type II fatty acid synthase (FAS II) system, which is responsible for FA synthesis, were found to be up-regulated in S. meliloti during predation by M. xanthus (Figures 2A, 2C). These include genes coding for most of the enzymes involved in the initiation phase and elongation cycles of FA chains (López-Lara and Soto, 2018), as well as some paralogous genes. Activation of FA synthesis is also suggested by the increased expression of acpS (smc02654), the holo-acyl-carrier protein (ACP) synthase, which transfers the 4’-phosphopantetheine from coenzyme A (CoA) to apo-ACP, thereby converting it to the functional holo-ACP to which acyl intermediates can be bound. The up-regulation of fabA and fabB required for unsaturated FA synthesis, together with that of fabI and fabF, suggests that the synthesis of both saturated and unsaturated FAs is increased during predation (López-Lara and Soto, 2018). In contrast, FA degradation decreased during predatory attack, as indicated by the down-regulation of several genes required for this process. Among them, genes coding for proteins involved in the uptake of long-chain FAs (fadL), genes involved in the activation of different FAs with CoA (fadD, matB, smc00261, smb20650), as well as genes of the smc02229-fadA-fadB operon, which most likely code for the enzymes required for the different steps in the β-oxidation cycle, are found (Figures 2B, 2D). Decreased FA degradation, together with increased FA biosynthesis, could indicate that S. meliloti requires FAs for a function other than energy production, perhaps for building and remodeling the cell membrane in response to predatory attack (see section “3.4. Drastic changes in the S. meliloti cell envelope during predation”). Lipid biosynthesis is also activated in M. xanthus during predation, most likely to change the lipid composition of the cell envelope and to synthesize new secondary metabolites that will contribute to kill prey. However, and in contrast to its prey, genes coding for enzymes involved in the β-oxidation cycle are up-regulated indicating an increased energy demand during predatory attack (Pérez et al., 2022).
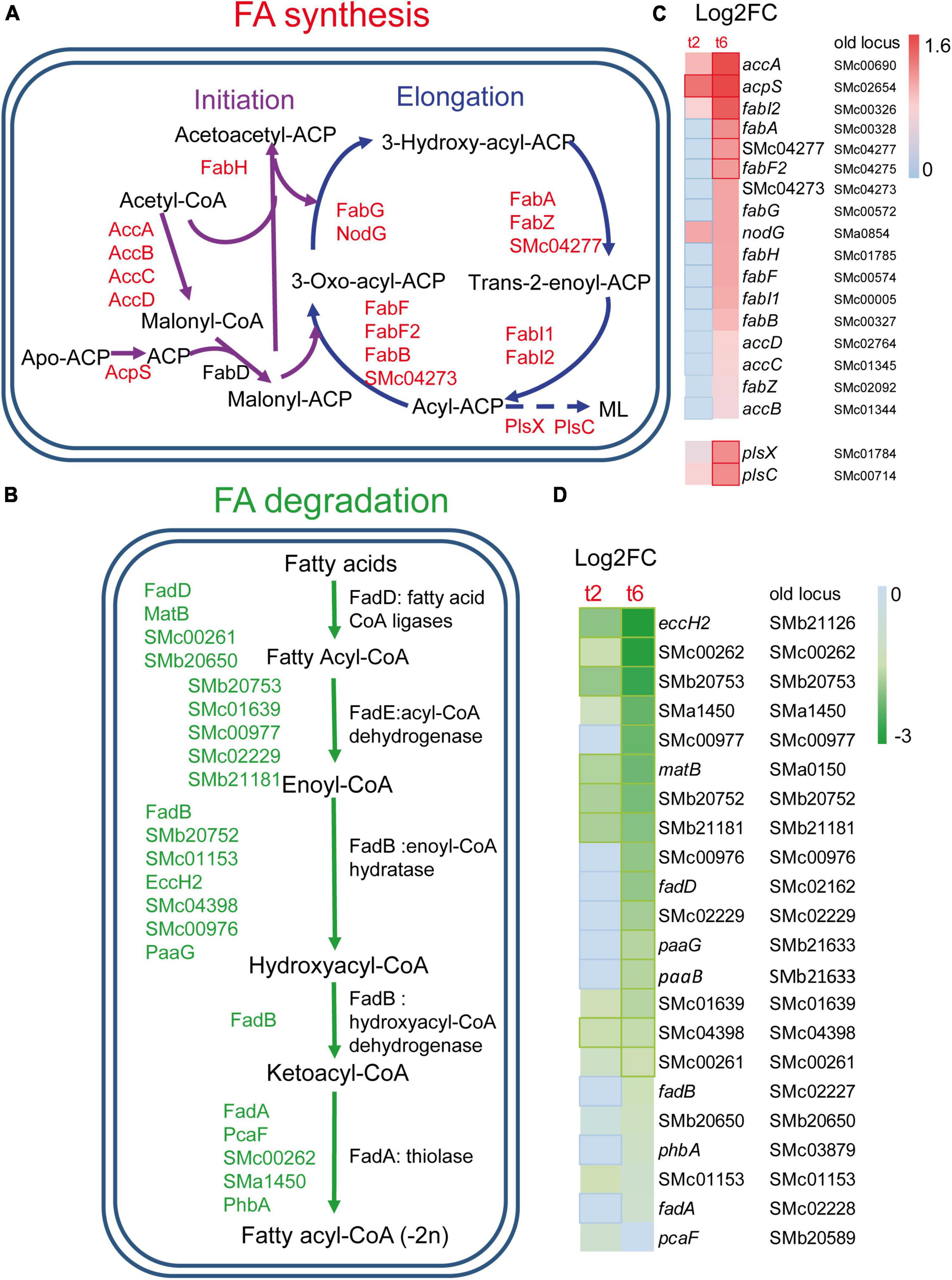
Figure 2. Changes in fatty acids (FA) metabolism in S. meliloti in co-cultures with Myxococcus xanthus after 2 and 6 h of contact (t2 and t6). (A) Up-regulation of genes involved in FA biosynthesis. ML, membrane lipids (see Figure 4 for more information). (B) Down-regulation of the genes responsible for the β-oxidation degradative pathway. Those genes (up or down-regulated) with demonstrated activity in the literature are indicated by their names (see text for details), while paralogous genes found in the KEGG database and that are also differentially expressed are represented by their gene identifiers. (C,D) Heatmaps of the genes involved in FA biosynthesis and FA β-oxidation, respectively. Red or green edges indicate genes with | Log2 Fold Change| > 1, and dotted edges indicate no differentially expressed genes at the indicated time.
Consistent with increased synthesis of proteins and FA, which are metabolically demanding processes, the up-regulation of up to 63 genes involved in energy production and conversion was detected. These include genes coding for different complexes of the respiratory chain and associated functions, such as the chromosomal nuo genes that code for the proton-pumping NADH: ubiquinone oxidoreductase (nuoA1B1C1D1E1F1HIJK1LMN), genes for cytochrome o ubiquinol oxidase (cyoABC), cytochrome c oxidase (ctaCD), cytochrome b (fbcB), and a putative ATP synthase (atpBCF2). Increased expression of genes that code for different enzymes of the tricarboxylic acid cycle was also detected (pdhABC, lpdA1, sdhABCD, fumC, sucA, icd, acnA, mdh).
Similar to genes involved in FA degradation, the majority of the genes belonging to the category of carbohydrate transport and metabolism (76.8%) were down-regulated in the transcriptome. Genes coding for different sugar ABC transporters (ugpABC, frcAK, rhaST), glucolytic enzymes (pgm, pgi, pgiA1, cbbA, cbbA2, tpiA1, gap, pgk, pykA), enzymes of the pentose phosphate pathway (cbbT, gnd, eda2, rbsK, ttuD1), and enzymes involved in glycogen (glgC, glgA1) and poly-3-hydroxybutyrate (phbABC) synthesis, showed reduced expression compared with control conditions. Disruption of the glycogen synthase gene glgA1 resulted in decreased polyhydroxybutyrate (PHB) levels and increased EPS levels compared to the wild type (D’Alessio et al., 2017). In starving conditions, the use of glycogen could be an alternative energy source. In favor of this hypothesis is the fact that gene glgX1, involved in glycogen-debranching, is up-regulated.
Inorganic transport and metabolism was a functional category that showed enrichment in up-regulated genes identified in the response to predatory attack (Table 1). Within this category, it was remarkable the up-regulation of many genes involved in iron uptake and metabolism, including genes involved in the synthesis (rhbABDEF and sma2339) and uptake (rhtA, rhtX) of the siderophore rhizobactin 1021 (Lynch et al., 2001). Additional genes related with iron and with increased expression are those involved in the uptake of heme and hydroxamate siderophores [shmR, hmuSTV, fhuA2/foxA, fhuA3, fhuP, sma1742 fepG (sma1742), fepB (sma1746)], the exbD gene encoding one of the components of the TonB-ExbB-ExbD complex, which provides the energy for the transport of heme and siderophore-mediated iron transport across the outer membrane, the fhuF gene coding for ferrioxamine B reductase, or genes for putative Fe3+ ABC transporters [irp6C (smb21429) and irp6B (smb21430)] (Fabiano and O’Brian, 2012; O’Brian, 2015) (Figures 3A–C). Genes coding for regulatory proteins involved in iron homeostasis (Supplementary Figures 4A, B and Supplementary Tables 4A, B) were also differentially expressed during predatory attack. Genes coding for HmuP, which controls hemin acquisition, RhrA, which controls both the synthesis and the uptake of rhizobactin 1021, and the iron regulator Irr were up-regulated, whereas the gene coding for the rhizobial iron regulator A (RirA) was down-regulated (Amarelle et al., 2010; O’Brian, 2015). Irr senses iron indirectly through the status of heme biosynthesis (Figure 3D). Under iron-limiting conditions, Irr represses genes encoding proteins that function under iron-sufficient conditions, including rirA (O’Brian, 2015). RirA is an iron-sulfur protein that acts as a repressor of iron-uptake under iron-replete conditions (Chao et al., 2005; Pellicer Martinez et al., 2017; Figure 3B). Therefore, data obtained here indicate that S. meliloti is experiencing iron-limiting conditions during co-culture with the myxobacterial predator, similar to the situation previously reported for S. coelicolor (Lee et al., 2020) and Pseudomonas putida (Akbar and Stevens, 2021). Up-regulation of the iron-uptake machinery was also observed in M. xanthus during co-culture with S. meliloti (Pérez et al., 2022), suggesting that both predator and prey are competing for iron. It would be interesting to test whether increased rhizobactin 1021 production in S. meliloti Rm1021 enhances rhizobial resistance to myxobacterial predation as shown for P. putida survivors that overproduce the siderophore pyoverdine (Akbar and Stevens, 2021).
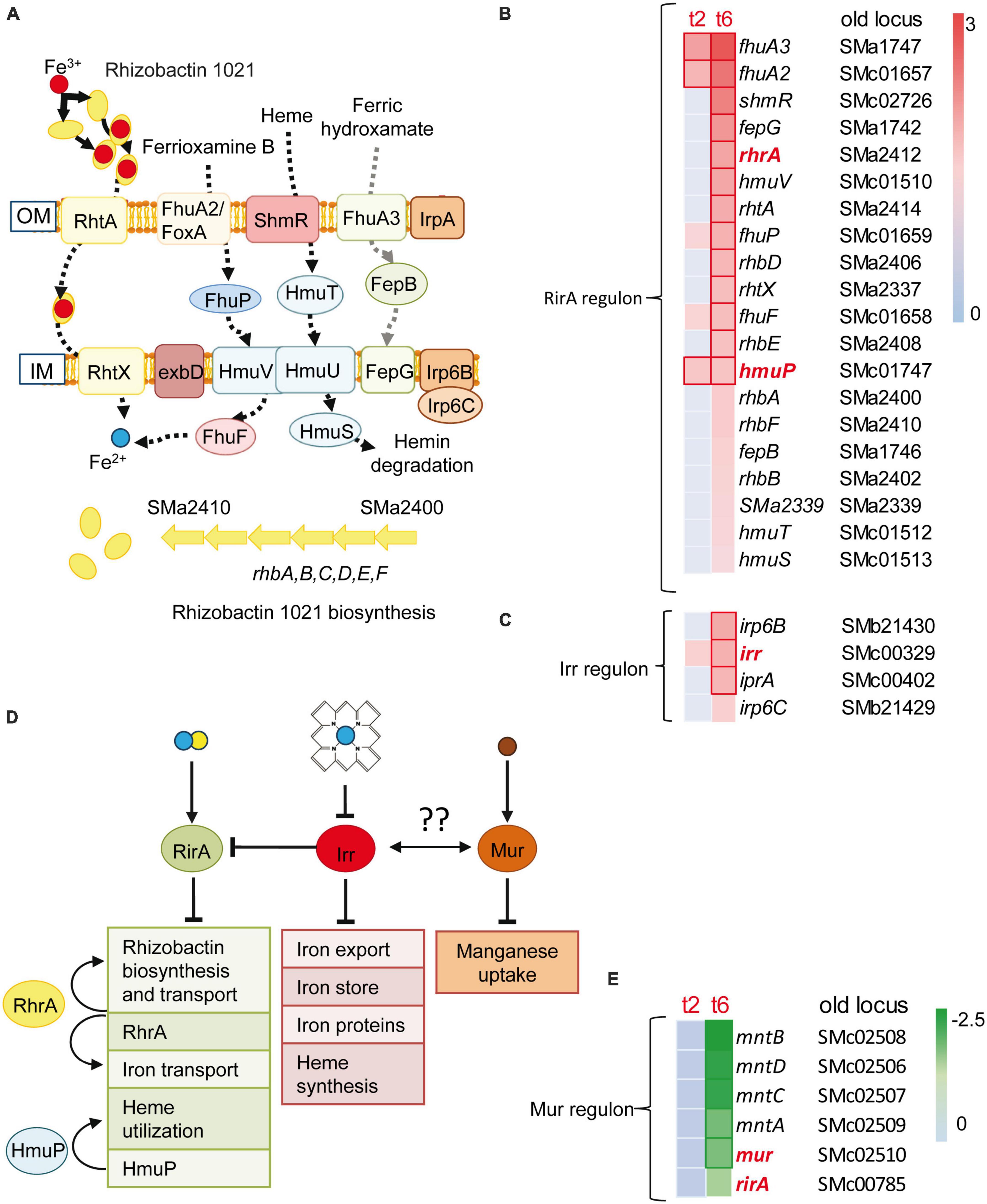
Figure 3. Iron uptake and rhizobactin 1021 biosynthesis are induced during competition. (A) Sinorhizobium meliloti genes involved in siderophore synthesis and iron-uptake regulation that are induced at t2 or/and t6. Red and blue circles represent Fe3+ and Fe2+, respectively. (B,C) Heatmaps of the RirA and Irr dependent genes which are depicted in panel (A). (D) Control of iron homeostasis by the regulators: RirA, RhrA, HmuP and Irr. RirA is a (4Fe–4S) cluster containing protein which represses many genes involved in iron uptake under iron-replete conditions. The manganese responsive Fur-like repressor, Mur, controls manganese uptake. Both global regulatory proteins are down-regulated during predation, indicating a mechanism for the control of iron homeostasis by manganese as has been suggested for other bacteria (see text for details). (Fe–S) clusters are depicted as blue and yellow circles. Brown circles represent Mn2+. Arrows and truncated lines indicate positive and negative regulation, respectively. OM, outer membrane; IM, inner membrane. (E) Down-regulation of rirA, mur and Mur-dependent genes (see text, and Supplementary Tables 4A, B for details).
It is known that the metabolism of iron and manganese are interrelated (O’Brian, 2015). Manganese can replace iron in many enzymes. However, and despite experiencing low iron concentrations, the expression of the mntABCD (formerly sitABCD) genes coding for a manganese ABC transporter (Platero et al., 2003) and that of its regulator Mur were down-regulated during predation (Figure 3E).
Several genes related with phosphorous uptake and metabolism were up-regulated in the defensome of S. meliloti, such as those coding for phosphate ABC-type transporters (phoCDE, pstA), phosphonate metabolism (phnGHIJ), alkaline phosphatase (phoX), polyphosphate kinase (ppk), as well as genes coding for regulatory proteins that are crucial for the maintenance of cellular phosphate homeostasis (phoB and phoU). Intriguingly, up to 29 of the up-regulated genes in the defensome were also found to be up-regulated in the phosphate starvation response of S. meliloti, with the majority of them (25) belonging to the cluster I of PhoB-dependent genes induced by phosphate stress (Supplementary Figure 4B and Supplementary Tables 4C, D; Krol and Becker, 2004). PhoB is the response regulator of the PhoR-PhoB two-component system that controls a set of genes known as the Pho regulon, which is involved in cell adaptation to phosphate starvation. In addition to genes related to phosphorus uptake and metabolism, the Pho regulon includes other genes such as those related to phosphorus-free membrane lipid biosynthesis (sqdB, btaAB, olsA, olsB), which were also up-regulated during predatory attack. Moreover, 29 genes up-regulated in the defensome increased their expression levels under phosphate starvation in a PhoB-independent manner. This group includes several genes involved in the synthesis of the exopolysaccharide I (EPS I) or succinoglycan (exoA, exoW, exoV, exoK, exoYF1, exoX) (see also Section “3.4. Drastic changes in the S. meliloti cell envelope during predation”). The transcriptional activation of low phosphate-responsive genes, including members of the Pho regulon, could be interpreted as S. meliloti cells facing phosphate-limiting conditions. However, this is difficult to believe considering that the medium used in the experimental setup is a phosphate-rich medium and that phoU is transcriptionally up-regulated. It has been suggested that the regulatory protein PhoU responds to elevated phosphate levels by significantly decreasing the phosphate transport of PstSCAB to prevent phosphate toxicity (diCenzo et al., 2017). Moreover, the activation of EPS I production is known to occur under high-phosphate conditions (Mendrygal and González, 2000; Acosta-Jurado et al., 2021). Therefore, and most likely, the up-regulation of low phosphate-responsive genes in the defensome cannot be the direct result of a phosphorous deficiency. In E. coli, PhoB is not only activated by low phosphate, but also by cell envelope stress (Choudhary et al., 2020). It is not unreasonable to think that compounds and hydrolytic enzymes secreted by M. xanthus during predatory attack disrupt different components of the S. meliloti cell envelope, causing cell envelope stress. Activation of the PhoB regulator by the cell envelope stress caused during predatory attack and/or any other mechanism, together with the known overlap and interaction of the Pho regulon with several other control circuits (Yuan et al., 2006), could explain the differential expression of low phosphate-induced genes during myxobacterial predation.
The bacterial cell envelope is a complex structure that provides structural integrity and protects the cytoplasm from changes in the surrounding environment. As a diderm bacterium, the rhizobial cell envelope consists of three layers: the cytoplasmic or inner membrane (IM), a thin peptidoglycan cell wall, and the outer membrane (OM) containing LPS in the outer leaflet. In addition, rhizobia produce different surface polysaccharides (SPSs) that serve different functions during the free-living and symbiotic lifestyles of these bacteria (Acosta-Jurado et al., 2021). Maintenance of cell envelope integrity is essential for viability, and bacteria have developed regulatory mechanisms to defend from envelope disturbance. Considering the epibiotic predatory strategy employed by M. xanthus, it is not surprising that many S. meliloti genes related with the different cell envelope structures are transcriptionally modulated in response to predator attack. Below, we provide interpretation for the transcriptional changes detected in genes associated to different cell envelope structures.
In S. meliloti, the exo/exs and wgx (formerly exp) genes are responsible for the production of two different kinds of acidic exopolysaccharides (EPSs): EPS I or succinoglycan, and EPS II or galactoglucan (Becker et al., 2002). The regulation of these EPSs is very complex, and several environmental conditions and regulatory proteins have been involved in their control (Barnett and Long, 2018). As previously mentioned, several exo/exs genes involved in the synthesis of EPS I (exoA, exoB, exoQ, exoF1, exoH, exoK, exoX, exoV, exoY, exoN, exoP, exoW, exoK, exsA, exsI) (Glucksmann et al., 1993) were found to be up-regulated during predation. Concerning EPS II, the S. meliloti strain Rm1021 used in this study lacks a functional ExpR, a LuxR-type regulator that is required for the quorum sensing-dependent production of high amounts of EPS II (Pellock et al., 2002). Nevertheless, the increased expression of wgcA (expC), wgdA (expE1) and that of the wggR (expG) gene that codes for the transcriptional activator of EPS II-related genes, suggest stimulation of EPS II synthesis. Transcriptional activation of EPS II-related genes could be mediated by PhoB, which activates expression of wggR (Bahlawane et al., 2008). As discussed in section “3.3. Increased expression of genes related to iron and phosphorus starvation responses,” the up-regulation of EPS I-related genes is independent of PhoB. In this case, the transcriptional activation of the exo/exs genes could be mediated by regulatory proteins and circuits known to influence EPS I production and whose expression was also induced during predation. This is the case of mucR (Bahlawane et al., 2008), the exoS/chvG gene of the ExoR-ExoS-ChvI system (Yao et al., 2004; Wells et al., 2007). and the ntrY and ntrX genes of the NtrY-NtrX regulatory system (Calatrava-Morales et al., 2017). Recently, the activation of the ChvG/ExoS-ChvI regulon in response to cell wall stress has been suggested for Alphaproteobacteria (Williams et al., 2022), as well as the co-ordinated work of the ExoR-ExoS-ChvI and NtrY-NtrX systems to control different functions, including regulation of the bacterial cell envelope (Stein et al., 2021).
In addition to EPS I and EPS II, the production of other SPSs seems to be activated in S. meliloti during predation. This is the case for the low-molecular mass K polysaccharide (KPS) (Fraysse et al., 2005), and an as-yet-uncharacterized SPS that confers resistance to the antibiotic phazolicin (PHZ) and provisionally named PPP (PHZ-protecting polysaccharide) (Travin et al., 2023). Several genes related with the production of KPS (rkpG, rkpI, rkpZ2, kpsF3, rkpU, rkpT2, rkpS) and seven out of the nine genes located between smb21252 and smb21271 that are necessary for the synthesis of PPP are up-regulated during predatory attack, suggesting that these two SPSs could also contribute to the defense against predation. KPS biosynthetic genes were also found to contribute to PHZ resistance in S. meliloti, although to a lesser extent than genes involved in PPP synthesis (Travin et al., 2023).
In rhizobia, SPSs contribute to protect cells against abiotic factors and defense products produced by the plant and other microbes (Soto et al., 2006; Lehman and Long, 2013; Arnold et al., 2017; Travin et al., 2023). Production of increased levels of different SPSs during predation might be a defense strategy of the prey, in which SPSs play a barrier role by sequestering and/or repulsing hydrolytic enzymes and toxic compounds released by the predator. Indeed, earlier findings showed that the production of high levels of EPS II, characteristic of S. meliloti ExpR-plus strains, contributes to rhizobial resistance to myxobacterial predation (Pérez et al., 2014). In support of an important role of SPSs for defense against predators, a recent study associated increased alginate production and mucoidy as a strategy in P. putida to survive predation by another myxobacterial predator, Cystobacter ferrugineus (Akbar and Stevens, 2021).
Besides the activation of SPS-related genes, many loci coding for enzymes that affect the peptidoglycan and structural components of the OM and IM were differentially expressed during co-culture with the myxobacterial predator, suggesting modifications of the cell envelope. Some genes related with peptidoglycan synthesis and remodeling were up-regulated, such as mltB2, murA, murC, murE, whereas murD, murF, murG, or pbpC, were down-regulated. The expression of a set of S. meliloti genes related with the synthesis of the lipid A, the core polysaccharide and the O-antigen of LPS (acpXL, smc04277-fabF2-smc04273, lpxXL, lpxK, ddhAB, kdsB, kdtA, lpsB, lpsCE, lpsS) (Lagares et al., 2001; Sharypova et al., 2003), as well as the lsrB gene that codes for a transcriptional regulator that activates the expression of LPS biosynthetic genes (Tang et al., 2014), were found to be up-regulated during predation. Likewise, the periplasmic TolB1 and the OM-anchored Pal lipoprotein, which are elements of the Tol/Pal system known to contribute to cell envelope integrity (Krol et al., 2020; Szczepaniak et al., 2020), showed increased expression. It was also remarkable that several genes involved in membrane lipid synthesis were up-regulated (Figures 4A, B). Under phosphate-sufficient conditions, S. meliloti produces the glycerophospholipids phosphatidylglycerol (PG), phosphatidylethanolamine (PE), phosphatidylcholine (PC) and cardiolipin (CL) as its major membrane lipids (Sohlenkamp and Geiger, 2016; López-Lara and Geiger, 2017). However, when phosphate levels are limiting, S. meliloti replaces its phospholipids with membrane lipids that do not contain any phosphorus, namely sulphoquinovosyl diacyl-glycerols (SQDG), diacylglyceryl-N,N,N-trimethylhomoserines (DGTS), and ornithine-containing lipids (OL) (López-Lara and Geiger, 2017). Intriguingly, in response to myxobacterial predation, S. meliloti increased the expression of genes required for the synthesis of both phospholipids (glpK, plsX, plsC, pgsA, pssA, psd, pmtA) and phosphorous-free membrane lipids (sqdB, btaAB, olsB, olsA) (Figures 4A, B). This contrasts with the down-regulation of glycerophospholipid metabolism observed in the defensome of E. coli against M. xanthus in liquid media (Livingstone et al., 2018). In the defensome of S. meliloti, the up-regulation of the plcP gene was also detected. This gene codes for a phospholipase C that degrades PC to release diacylglycerol (DAG) that can be used for DGTS synthesis (Zavaleta-Pastor et al., 2010). As with the genes coding for the enzymes participating in the synthesis of phosphorous-free membrane lipids (see section “3.3. Increased expression of genes related to iron and phosphorus starvation responses”), plcP is also regulated by PhoB under phosphorus limitation (Krol and Becker, 2004; Yuan et al., 2006; Zavaleta-Pastor et al., 2010). However, under our experimental conditions and as discussed in the previous section, it is very unlikely that low levels of phosphate are responsible for the induction of these genes during myxobacterial predation. Bacteria change their membrane lipid composition in response to environmental changes and abiotic stresses (Sohlenkamp and Geiger, 2016). Our transcriptomic data suggest that during predation, S. meliloti undergoes membrane remodeling in which the formation of several membrane lipids, including those devoid of phosphorous, is activated under phosphorous-sufficient conditions. If this is simply a consequence of predation stress or represents an adaptation strategy that provides a fitness advantage by allowing the prey to conserve phosphate for other cellular processes and/or by stabilizing their membranes, requires experimental investigation.
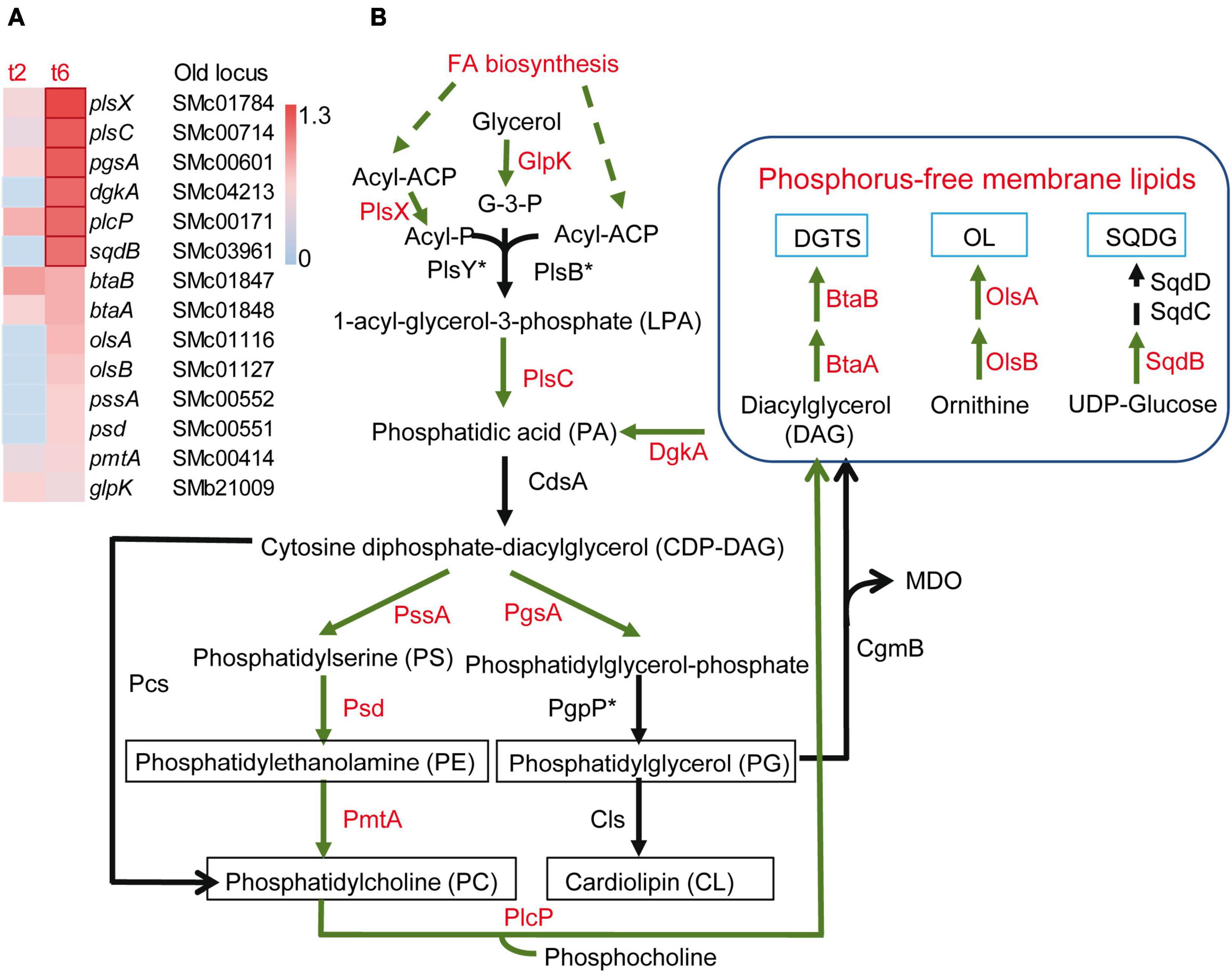
Figure 4. Increased expression of genes involved in membrane lipid formation in Sinorhizobium meliloti in co-culture with Myxococcus xanthus after 2 and/or 6 h of contact (t2 and/or t6). (A) Heatmap of up-regulated genes encoding different enzymes involved in membrane lipid formation. Red edges indicate genes with Log2 Fold Change > 1. (B) Metabolic routes for the synthesis of phospholipids and phosphorus-free membrane lipids. Up-regulated processes (FA biosynthesis) and enzymes exhibiting increased expression in S. meliloti during predation are shown in red. The asterisk indicates that the corresponding orthologous gene in S. meliloti has not been identified. DGTS, diacylglyceryl-N,N,N-trimethylhomoserine; OL, ornithine-containing lipids; SQDG, sulphoquinovosyl diacyl-glycerol; GlpK, glycerol kinase; PlsX/PlsY/PlsC and PlsB/PlsC are two different acyltransferase systems for the formation of PA, with the former most likely operating in S. meliloti; CdsA, CDP-DAG synthase; PssA, PS synthase; Psd, PS decarboxylase; PmtA, PE methyltransferase; Pcs, PC synthase; PgsA, PG-phosphate synthase; PgpP, PG-phosphate phosphatase; Cls, cardiolipin synthase; PlcP, phospholipase C; CgmB, cyclic glucan-modifying phosphoglycerol transferase; BtaA, S-adenosylmethionine: DAG 3-amino-3-carboxypropyl transferase; BtaB, diacylglyceryl homoserine N-methyltransferase; OlsA, O-acyltransferase; OlsB, lyso-ornithine lipid synthase; SqdB, UDP-sulfoquinovose synthase; SqdC, epimerase; SqdD, glycosyltransferase. Asterisks indicate that the corresponding genes have not been annotated in the S. meliloti Rm1021 genome.
Some features already described in previous sections, such as the unexpected activation of the PhoB regulon under phosphorous-sufficient conditions, the activation of the ExoS-ChvI and NtrY-NtrX regulatory systems, and the dramatic changes that the S. meliloti cell envelope is experiencing during co-culture with M. xanthus, are indicative of predatory stress responses. The up-regulation of rsiB1 and relA also detected in the transcriptome are compatible with this notion. RsiB1 is one of the components of the partner switching mechanism that controls the activity of the general stress response (GSR) σ factor RpoE2. RsiB1 is the anti-anti-σ factor, which binds the anti-σ RsiA1, thereby releasing the σ factor RpoE2, which alters the transcription of a large set of genes in response to several stresses (Bastiat et al., 2010). In S. meliloti, another σ factor, RpoH1, that regulates the expression of genes encoding the cell wall biosynthetic machinery and membrane biogenesis proteins (de Lucena et al., 2010), has been implicated in the regulation of membrane stress (Nicoud et al., 2021). Intriguingly, rpoH1 was found to be down-regulated in the defensome. Therefore, the contribution of RpoE2 and RpoH1 in the defense of S. meliloti requires further investigation. In addition, the up-regulation of relA suggests the activation of the stringent response, a complex bacterial regulatory mechanism that aids the adaptation to stressful conditions (Irving et al., 2021). In S. meliloti, RelA is involved in the synthesis and hydrolysis of the bacterial second messenger (p)ppGpp that plays a crucial role in the stringent response. S. meliloti relA mutants have pleiotropic phenotypes, including the overproduction of EPS I (Krol and Becker, 2011; Wippel and Long, 2019).
Together with the passive protective role of increased SPS production (see section “3.4. Drastic changes in the S. meliloti cell envelope during predation”), the overexpression of several efflux and transport proteins detected in this transcriptome could contribute to defend S. meliloti cells from predatory attack (Figure 5). Genes coding for the multidrug efflux systems EmrAB (Santos et al., 2014) and SmeABR (Eda et al., 2011), as well as the gene coding for the putative multidrug resistance EmrE protein, were found to be up-regulated during the co-culture. Multidrug resistance efflux pumps (MDR) enable bacteria to avoid the effects of a wide array of toxic compounds. The expression of the emrAB genes, which code for a putative major facilitator superfamily-type efflux pump, has been shown to increase in response to different abiotic stresses (e.g., heat shock, acidic pH) and a role for this efflux pump in the response to stresses by acting on the cell envelope has been suggested (Santos et al., 2014). In the case of SmeAB, this pump has been shown to confer resistance to different toxic compounds, including antibiotics, dyes, detergents, and antimicrobials produced by the host plant (Eda et al., 2011). Increased expression of the TetR-like regulator SmeR, which negatively regulates expression of SmeAB (Eda et al., 2011), suggests that the effector that releases the repression of SmeAB mediated by SmeR is being produced in the co-culture of S. meliloti with M. xanthus. Like S. meliloti, P. putida survivors to a different myxobacteria predation also exhibited increased expression of several efflux pumps (Akbar and Stevens, 2021), suggesting that this could be a common response used by prey to defend against predatory attack.
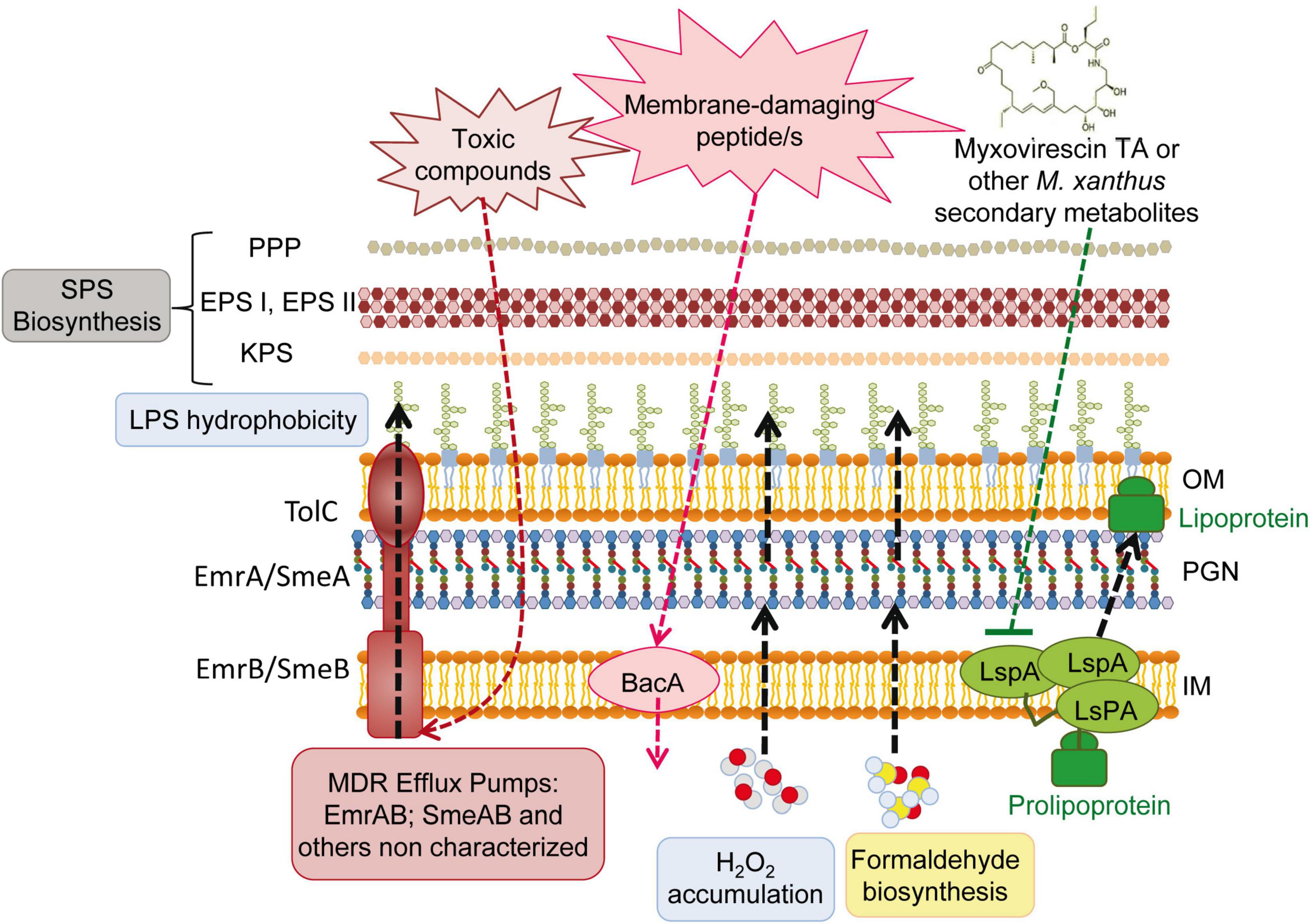
Figure 5. Overview of the deduced Sinorhizobium meliloti active and passive responses during the interaction with the predator Myxococcus xanthus. SPS, surface polysaccharides; PPP, PHZ-protecting polysaccharide; EPS, exopolysaccharides; EPS I, succinoglycan; EPS II, galactoglucan; KPS, K polysaccharide; LPS, lipopolysaccharide; OM, outer membrane; IM, inner membrane; PGN, peptidoglycan.
One of the transporters whose expression was up-regulated during M. xanthus predation was the broad-specificity peptide uptake transporter BacA (Figure 5). BacA is a SLiPT (SbmA-like peptide transporter) (Ghilarov et al., 2021), which was identified thirty years ago as an essential determinant in S. meliloti to establish a chronic infection inside the nodules of its host plant (Glazebrook et al., 1993). Years of investigation revealed that BacA is responsible for the uptake of nodule-specific cysteine-rich peptides (NCRs), defensin-like antimicrobial peptides that are produced inside the legume nodules and drive terminal bacteroid differentiation (Nicoud et al., 2021). With the internalization of NCRs, BacA protects rhizobial cells from the membrane-damaging activity of the host-produced peptides (Haag et al., 2011). Activation of bacA expression in response to predation could be indicative of the presence of as yet unknown membrane-damaging peptides released directly or indirectly by the predator.
The resistance to NCRs and other antimicrobial peptides is multifactorial (Arnold et al., 2017; Nicoud et al., 2021). In addition to BacA and the production of SPSs, modifications of the S. meliloti LPS mediated by LpsB and LpxXL have also been shown to contribute to resistance to NCR peptides (Nicoud et al., 2021). These genes code for a glycosyltransferase participating in the synthesis of the LPS core and a very-long-chain fatty acid (VLCFA; C ≥ 28) acyltransferase involved in the biosynthesis of lipid A, respectively (Figure 5). The unusual LpxXL-dependent acylation of the LPS is expected to increase the thickness of the OM and to increase lipid A hydrophobicity, which in turn reduces membrane fluidity. By increasing lipid A hydrophobicity, bacteria hamper the damaging effects of antimicrobial peptides on the membrane and contribute to increase resistance (Dalebroux and Miller, 2014). Interestingly, and as already mentioned in section “3.4. Drastic changes in the S. meliloti cell envelope during predation,” up-regulation of the lpsB and lpxXL genes was detected in response to predation. This, together with additional common transcriptional responses detected in the defensome and in the S. meliloti response to sublethal levels of NCR peptides (Penterman et al., 2014; see also next section “3.6. Transcriptional regulatory proteins that are differentially regulated”), supports the hypothesis of a rhizobial defense against putative predator-derived antimicrobial peptides.
Increased expression of the smc01129 gene, encoding a probable lipoprotein signal peptidase, LspA, could be another mechanism of predation resistance, more specifically against the antibiotic myxovirescin TA produced by M. xanthus, which targets bacterial type II signal peptidase to inhibit pro-lipoprotein processing (Figure 5; Wang et al., 2019). The overexpression of the lspA gene has been shown to confer TA resistance to E. coli cells (Xiao et al., 2011). Although the myxovirescin genes are not up-regulated in the M. xanthus predatosome against S. meliloti, the role of this antibiotic in the interaction should not be excluded, since its overproduction could be regulated by a post-transcriptional mechanism (Pérez et al., 2022).
Formaldehyde secretion has been described as a potential predator-resistance feature of Pseudomonas species (Sutton et al., 2019; Akbar and Stevens, 2021). Interestingly, in the defensome of S. meliloti during myxobacterial predation, the up-regulation of different subunits of sarcosine oxidase (soxA2, soxB2, and soxD2), together with the down-regulation of the gfa (smb20186) gene, were observed. Considering that sarcosine oxidase catalyzes the oxidative demethylation of sarcosine to yield formaldehyde, and that Gfa is a putative glutathione-dependent formaldehyde-activating enzyme that participates in the formaldehyde detoxification pathway, the observed transcriptional changes could lead to the increased production and accumulation of formaldehyde in S. meliloti cells. Sarcosine oxidase activity also leads to glycine and H2O2 production. This, together with the observed up-regulation of the superoxide dismutase-encoding gene sodB that generates H2O2, and the down-regulation of the katB gene that codes for a catalase that inactivates H2O2, suggests a possible accumulation of this reactive oxygen species as an additional defense mechanism used by S. meliloti to fight against myxobacterial predators (Figure 5).
Regulatory proteins whose expression levels change in response to the predator should, directly or indirectly, be involved in controlling the expression of those genes and pathways involved in adaptation, competition, or defense that have been discussed above. We have found 49 regulators that increase their expression during predation, out of which, 11 are two-component systems (TCSs), and the rest are one-component regulators (including 5 GntR, 3 TetR, 3 MarR, and 3 LysR) (Supplementary Table 5A). In contrast, 76 regulatory elements have been found that are down-regulated (including 9 FCD, 6 LacI, 4 LysR, and 17 forming part of TCSs) (Supplementary Table 5B). Eight of them are related to symbiosis and/or induced in bacteroids. As discussed in section “3.3. Increased expression of genes related to iron and phosphorus starvation responses” (Figure 3), the Fur type manganese-responsive regulator, Mur, and the transcriptional regulator of the iron limitation response, RirA, are down regulated. Finally, SoxR, could be regulating a global response against oxidative-generating agents and might provide broad antibiotic resistance as has been described in E. coli (Hidalgo and Demple, 1997).
Elucidation of the role of these regulatory elements during predation will help not only to draw a global picture of the response of S. meliloti to predation, but also will shed light on adaptation processes caused by bacterial interactions.
As already mentioned above, significant transcriptional changes occurring during co-culture with M. xanthus indicate that S. meliloti is experiencing stress when interacting with the predator. To identify putative common and/or differential mechanisms used by S. meliloti to cope with predatory stress and unfavorable conditions, we decided to compare the profile of the defensome with the transcriptome profile from cells grown under different abiotic stresses (osmotic, acid, or oxidative stress) (Domínguez-Ferreras et al., 2006; Hellweg et al., 2009; Lehman and Long, 2018), or treated with sublethal doses of NCR peptides (Penterman et al., 2014). To avoid noise, for all these comparisons, only differentially expressed transcripts in the defensome showing a | Log2 Fold Change| > 1 were considered.
Significant overlap was found between differentially expressed genes in the defensome and in response to abiotic stresses, especially under osmotic and oxidative stress. Intriguingly, the majority of the overlapping genes exhibited opposite regulation, suggesting different physiological adaptations of cells to abiotic or predatory stress (Supplementary Figures 5A–C and Supplementary Tables 6A–F). Among these genes, it was remarkable the differential regulation of many housekeeping genes involved in translation, and energy production and conversion, which were down-regulated under osmotic and oxidative stress conditions, but up-regulated during predation. Thus, whereas adaptation to abiotic stress involves a general slowing down of metabolism, the bacterial response to predatory attack requires activation of anabolism, especially protein and fatty acid synthesis.
A large overlap was also found between the defensome and genes differentially expressed in NCR-treated cells. Indeed, out of the 902 genes significantly altered in S. meliloti by NCR peptide treatment (Penterman et al., 2014), 180 genes were also found to be differentially expressed in response to myxobacterial predation (Supplementary Figure 5D and Supplementary Tables 6G, H). Approximately, 50% of the overlapping genes (91 genes) exhibited opposite regulation in both transcriptomes, but in this case, this group comprises genes down-regulated in the defensome and up-regulated in NCR-treated cells. Genes in this list include many genes involved in amino acid and carbohydrate transport and metabolism, energy production and conversion, and genes encoding proteins of unknown function. Interestingly, the catalase-encoding gene katB was included in this group, suggesting a putative H2O2 accumulation as a specific response to myxobacterial predation. Intriguingly, and contrasting with the results obtained comparing the defensome with the transcriptome profile of cells grown under abiotic stress, a greater number of genes (42) were found to be up-regulated in response to both NCR treatment and myxobacterial predation. This group comprises several genes involved in EPS synthesis and inorganic ion uptake (iron and potassium). Exposure of S. meliloti cells to cationic peptides leads to strong induction of the ExoS/ChvI, FeuP/FeuQ, and RirA regulons (Penterman et al., 2014). The transcriptomic data indicate that predatory attack activates ExoS/ChvI- and RirA-regulated genes (see sections “3.3. Increased expression of genes related to iron and phosphorus starvation responses” and “3.4. Drastic changes in the S. meliloti cell envelope during predation”). However, no significant overlap was found with genes regulated by the FeuP/FeuQ system that controls cyclic glucan production (Griffitts et al., 2008), suggesting that the Feu regulon is not relevant in the S. meliloti response to predatory attack, at least during the early stages of the interaction.
In an earlier transcriptome profile of S. meliloti cells exposed to 2 cationic NCR peptides, the down-regulation of genes involved in basic cellular functions, including translation, energy production, and fatty acid synthesis, was detected few minutes after treatment (Tiricz et al., 2013). This contrasts with the up-regulation of the same functional categories in response to predation. Nevertheless, some overlapping genes have been found between the study by Tiricz et al. (2013) and the defensome, including genes involved in stress responses (dnaJ, ipbA), iron uptake and metabolism (exbD, hmuVST, fhuA2), and genes encoding MDR efflux systems (emrAB).
The different comparisons performed in this study allowed the identification of 480 S. meliloti genes (205 up-regulated and 275 down-regulated) whose expression is altered specifically in response to myxobacterial predation (Supplementary Tables 7A, B). These defensome-specific genes belong to different functional categories, including many hypothetical genes. Nevertheless, the specific induction of several genes involved in cell wall and membrane biogenesis, as well as genes like bacA, sodB, or irr, suggests a role for cell envelope remodeling, transport of peptides, production of H2O2 and iron homeostasis in the adaptation/resistance response of S. meliloti cells against predation that awaits further experimentation.
This work completes the study at the transcriptional level of the predator-prey interaction established between M. xanthus and S. meliloti, which was previously initiated with the analyses of the M. xanthus predatosome. Here, we have analyzed the defensome of S. meliloti, that is, the transcriptomic changes that the prey undergoes when facing the predator. The main transcriptomic changes observed in the predator (Pérez et al., 2022) and in the prey have been compiled in Figure 6. This analysis has revealed that the interaction with M. xanthus causes a massive remodeling of the S. meliloti transcriptome that affects more than a quarter of the genome, indicating a strong adaptation/defense response of the prey. However, it is difficult to know if the observed transcriptional changes are triggered upon the detection of M. xanthus cells or secreted factors, by lysed S. meliloti cells, or both. Further investigations might give clues to answer the question.
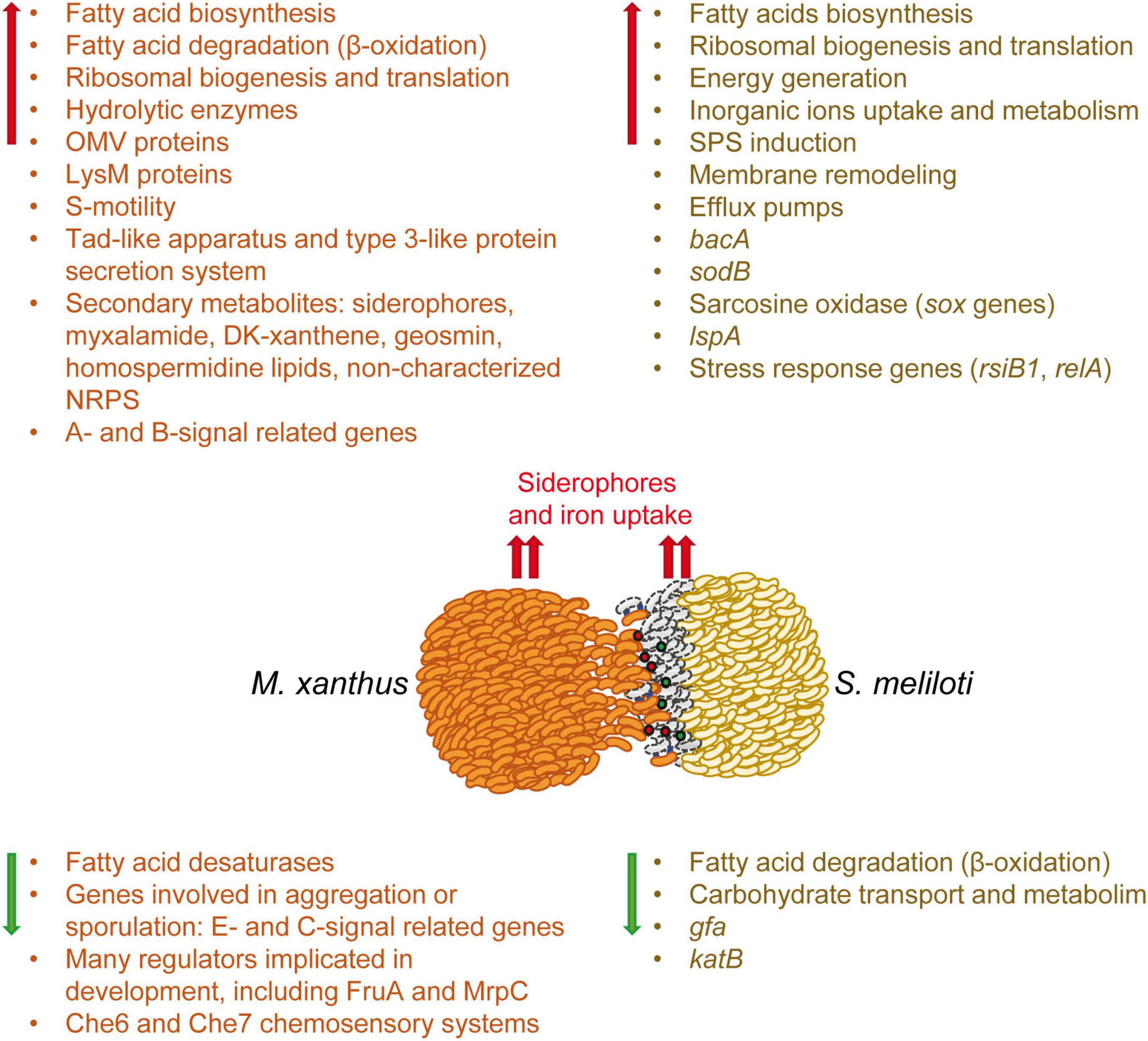
Figure 6. Schematic representation of the weapons used by the predator and the transcriptomic changes which occur in both the predator (predatosome) and the prey (defensome). Predatosome data have been compiled from Pérez et al. (2022). Red and green arrows represent up-regulation and down-regulation, respectively. Red and green balls represent extracellular weapons used by Myxococcus xanthus, such as hydrolytic enzymes and secondary metabolites, while blue lines represent contact dependent mechanisms. The dead prey cells at the interface are drawn as gray bacilli.
Sinorhizobium meliloti response involves the activation of mostly chromosomal-encoded functions, whereas the symbiotic plasmids have a minor contribution. During myxobacterial predation, S. meliloti up-regulates the expression of genes involved in protein synthesis and secretion, energy generation, fatty acid synthesis, and inorganic ion uptake and metabolism, while down-regulating genes required for FA degradation and carbohydrate transport and metabolism. This study highlights several significant changes that take place in the cell envelope of S. meliloti during myxobacterial predation. The up-regulation of many genes involved in the synthesis of different SPSs (EPS I, EPS II, KPS, PPP) suggest an increase in SPS production that could protect the cell by acting as a barrier against the action of hydrolytic enzymes and other antimicrobial compounds released by the predator. In addition to increased SPS production, transcriptomic changes detected in response to myxobacterial predation indicate the existence of modifications related to peptidoglycan and LPS, as well as significant membrane remodeling with the increased synthesis of different phospholipids and phosphorous-free membrane lipids. As previously observed in the predator, a remarkable up-regulation of iron-uptake mechanisms also takes place in S. meliloti, indicating that during the interaction, both the predator and the prey are experiencing iron starvation and need to compete for this essential nutrient.
Importantly, our findings have unveiled defensive mechanisms potentially used by S. meliloti during predatory attack. In addition to increased SPS production and modifications of the LPS that could protect rhizobial cells against membrane-damaging compounds produced by the predator, the activation of MDR efflux pumps and the broad-specificity peptide uptake transporter BacA could also contribute to defend S. meliloti from toxic compounds and membrane-damaging peptides potentially produced by M. xanthus. Additional defensive mechanisms include the increased expression of LspA to counteract the production of the antibiotic myxovirescin, and the production of formaldehyde and hydrogen peroxide that could have detrimental effects on the predator.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI–PRJNA937699.
MS and JP: substantial contributions to conception, design, analysis, interpretation of data, draft the manuscript and revising it critically for important intellectual content, and funding acquisition. JM-D: substantial contributions to conception, design and interpretation of the data, revising the manuscript critically for important intellectual content, and funding acquisition. FC-M: acquisition of data and revising the manuscript critically for important intellectual content. AM-M: acquisition of data, editing and revising the manuscript critically for important intellectual content, and funding acquisition. All authors contributed to the article and approved the submitted version.
This work has been supported by grant PID2020-112634GB-I00 funded by MCIN/AEI/10.13039/501100011033, grant PID20 21-123540NB-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe,” and grants A-BIO-126-UGR20 and P20_00225 funded by Consejería de Universidad, Investigación e Innovación de la Junta de Andalucía and by “ERDF A way of making Europe.”
We are grateful to I. M. López-Lara for her helpful comments to prepare Figures 2, 4 and to P. van Dillewijn for critical review of the manuscript.
JP was one of the topic editors for the research topic this manuscript is submitted to: “Mechanisms of Prokaryotic Predation, Volume II.”
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1213659/full#supplementary-material
Acosta-Jurado, S., Fuentes-Romero, F., Ruiz-Sainz, J. E., Janczarek, M., and Vinardell, J. M. (2021). Rhizobial exopolysaccharides: Genetic regulation of their synthesis and relevance in symbiosis with legumes. Int. J. Mol. Sci. 22:6233. doi: 10.3390/ijms22126233
Akbar, S., and Stevens, D. C. (2021). Functional genomics study of Pseudomonas putida to determine traits associated with avoidance of a myxobacterial predator. Sci. Rep. 11:16445. doi: 10.1038/s41598-021-96046-8
Amarelle, V., Koziol, U., Rosconi, F., Noya, F., O’Brian, M. R., and Fabiano, E. (2010). A new small regulatory protein, HmuP, modulates haemin acquisition in Sinorhizobium meliloti. Microbiology 156, 1873–1882. doi: 10.1099/mic.0.037713-0
Arnold, M. F. F., Shabab, M., Penterman, J., Boehme, K. L., Griffitts, J. S., and Walker, G. C. (2017). Genome-wide sensitivity analysis of the microsymbiont Sinorhizobium meliloti to symbiotically important, defensin-like host peptides. mBio 8:e01060-17. doi: 10.1128/mBio.01060-17
Bahlawane, C., Baumgarth, B., Serrania, J., Rüberg, S., and Becker, A. (2008). Fine-Tuning of galactoglucan biosynthesis in Sinorhizobium meliloti by differential WggR (ExpG)-, PhoB-, and MucR-dependent regulation of two promoters. J. Bacteriol. 190, 3456–3466. doi: 10.1128/JB.00062-08
Barnett, M. J., and Long, S. R. (2018). Novel genes and regulators that influence production of cell surface exopolysaccharides in Sinorhizobium meliloti. J. Bacteriol. 200:e00501-17. doi: 10.1128/JB.00501-17
Barriot, R., Latour, J., Castanie-Cornet, M. P., Fichant, G., and Genevaux, P. (2020). J-domain proteins in bacteria and their viruses. J. Mol. Biol. 432, 3771–3789. doi: 10.1016/j.jmb.2020.04.014
Bastiat, B., Sauviac, L., and Bruand, C. (2010). Dual control of Sinorhizobium meliloti RpoE2 sigma factor activity by two PhyR-type two-component response regulators. J. Bacteriol. 192, 2255–2265. doi: 10.1128/JB.01666-09
Bauer, M. A., Kainz, K., Carmona-Gutierrez, D., and Madeo, F. (2018). Microbial wars: Competition in ecological niches and within the microbiome. Microb. Cell 5, 215–219. doi: 10.15698/mic2018.05.628
Becker, A., Rüberg, S., Baumgarth, B., Bertram-Drogatz, P. A., Quester, I., and Pühler, A. (2002). Regulation of succinoglycan and galactoglucan biosynthesis in Sinorhizobium meliloti. J. Mol. Microbiol. Biotechnol. 4, 187–190.
Beringer, J. E. (1974). R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84, 188–198. doi: 10.1099/00221287-84-1-188
Brito-Santana, P., Duque-Pedraza, J. J., Bernabéu-Roda, L. M., Carvia-Hermoso, C., Cuéllar, V., Fuentes-Romero, F., et al. (2023). Sinorhizobium meliloti DnaJ is required for surface motility, stress tolerance, and for efficient nodulation and symbiotic nitrogen fixation. Int. J. Mol. Sci. 24:5848. doi: 10.3390/ijms24065848
Calatrava-Morales, N., Nogales, J., Ameztoy, K., van Steenbergen, B., and Soto, M. J. (2017). The NtrY/NtrX system of Sinorhizobium meliloti GR4 regulates motility, EPS I production, and nitrogen metabolism but is dispensable for symbiotic nitrogen fixation. Mol. Plant Microbe Interact. 30, 566–577. doi: 10.1094/MPMI-01-17-0021-R
Chao, T. C., Buhrmester, J., Hansmeier, N., Pühler, A., and Weidner, S. (2005). Role of the regulatory gene rirA in the transcriptional response of Sinorhizobium meliloti to iron limitation. Appl. Environ. Microbiol 71, 5969–5982. doi: 10.1128/AEM.71.10.5969-5982.2005
Choudhary, K. S., Kleinmanns, J. A., Decker, K., Sastry, A. V., Gao, Y., Szubin, R., et al. (2020). Elucidation of regulatory modes for five two-component systems in Escherichia coli reveals novel relationships. mSystems 5:e00980-20. doi: 10.1128/mSystems.00980-20
Contreras-Moreno, F. J., Muñoz-Dorado, J., García-Tomsig, N. I., Martínez-Navajas, G., Pérez, J., and Moraleda-Muñoz, A. (2020). Copper and melanin play a role in Myxococcus xanthus predation on Sinorhizobium meliloti. Front. Microbiol. 11:94. doi: 10.3389/fmicb.2020.00094
Dalebroux, Z. D., and Miller, S. I. (2014). Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr. Opin. Microbiol. 17, 106–113. doi: 10.1016/j.mib.2013.12.005
D’Alessio, M., Nordeste, R., Doxey, A. C., and Charles, T. C. (2017). Transcriptome analysis of polyhydroxybutyrate cycle mutants reveals discrete loci connecting nitrogen utilization and carbon storage in Sinorhizobium meliloti. mSystems 2:e00035-17. doi: 10.1128/mSystems.00035-17
de Lucena, D. K., Pühler, A., and Weidner, S. (2010). The role of sigma factor RpoH1 in the pH stress response of Sinorhizobium meliloti. BMC Microbiol. 10:265. doi: 10.1186/1471-2180-10-265
diCenzo, G. C., Sharthiya, H., Nanda, A., Zamani, M., and Finan, T. M. (2017). PhoU allows rapid adaptation to high phosphate concentrations by modulating pstscab transport rate in Sinorhizobium meliloti. J. Bacteriol. 199:e00143-17. doi: 10.1128/JB.00143-17
Domínguez-Ferreras, A., Pérez-Arnedo, R., Becker, A., Olivares, J., Soto, M. J., and Sanjuán, J. (2006). Transcriptome profiling reveals the importance of plasmid pSymB for osmoadaptation of Sinorhizobium meliloti. J. Bacteriol. 188, 7617–7625. doi: 10.1128/JB.00719-06
Eda, S., Mitsui, H., and Minamisawa, K. (2011). Involvement of the smeAB multidrug efflux pump in resistance to plant antimicrobials and contribution to nodulation competitiveness in Sinorhizobium meliloti. Appl. Environ. Microbiol. 77, 2855–2862. doi: 10.1128/AEM.02858-10
Fabiano, E., and O’Brian, M. R. (2012). “Mechanisms and regulation of iron homeostasis in the rhizobia,” in Molecular Aspects of Iron Metabolism in Pathogenic and Symbiotic Plant-Microbe Associations, eds D. Expert and M. R. O’Brian (Berlin: Springer), 41–86.
Fraysse, N., Lindner, B., Kaczynski, Z., Sharypova, L., Holst, O., Niehaus, K., et al. (2005). Sinorhizobium meliloti strain 1021 produces a low-molecular weight capsular polysaccharide that is a Kdo homopolymer harboring a phospholipidic anchor. Glycobiology 15, 101–108. doi: 10.1093/glycob/cwh142
Galibert, F., Finan, T. M., Long, S. R., Puhler, A., Abola, P., Ampe, F., et al. (2001). The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293, 668–672. doi: 10.1126/science.1060966
Ghilarov, D., Inaba-Inoue, S., Stepien, P., Qu, F., Michalczyk, E., Pakosz, Z., et al. (2021). Molecular mechanism of SbmA, a promiscuous transporter exploited by antimicrobial peptides. Sci. Adv. 7:eabj5363. doi: 10.1126/sciadv.abj5363
Glazebrook, J., Ichige, A., and Walker, G. C. (1993). A Rhizobium meliloti homolog of the Escherichia coli peptide-antibiotic transport protein SbmA is essential for bacteroid development. Genes Dev. 7, 1485–1497. doi: 10.1101/gad.7.8.1485
Glucksmann, M. A., Reuber, T. L., and Walker, G. C. (1993). Genes needed for the modification, polymerization, export, and processing of succinoglycan by Rhizobium meliloti: A model for succinoglycan biosynthesis. J. Bacteriol. 175, 7045–7055. doi: 10.1128/jb.175.21.7045-7055.1993
Goldman, B. S., Nierman, W. C., Kaiser, D., Slater, S. C., Durkin, A. S., Eisen, J. A., et al. (2006). Evolution of sensory complexity recorded in a myxobacterial genome. Proc. Natl. Acad. Sci. U. S. A. 103, 15200–15205. doi: 10.1073/pnas.0607335103
Granato, E. T., Meiller-Legrand, T. A., and Foster, K. R. (2019). The evolution and ecology of bacterial warfare. Curr. Biol. 29, R521–R537. doi: 10.1016/j.cub.2019.04.024
Griffitts, J. S., Carlyon, R. E., Erickson, J. H., Moulton, J. L., Barnett, M. J., Toman, C. J., et al. (2008). A Sinorhizobium meliloti osmosensory two-component system required for cyclic glucan export and symbiosis. Mol. Microbiol. 69, 479–490. doi: 10.1111/j.1365-2958.2008.06304.x
Haag, A. F., Baloban, M., Sani, M., Kerscher, B., Pierre, O., Farkas, A., et al. (2011). Protection of Sinorhizobium against host cysteine-rich antimicrobial peptides is critical for symbiosis. PLoS. Biol. 9:e1001169. doi: 10.1371/journal.pbio.1001169
Hellweg, C., Puhler, A., and Weidner, S. (2009). The time course of the transcriptomic response of Sinorhizobium meliloti 1021 following a shift to acidic pH. BMC Microbiol. 9:37. doi: 10.1186/1471-2180-9-37
Hidalgo, E., and Demple, B. (1997). Spacing of promoter elements regulates the basal expression of the soxS gene and converts SoxR from a transcriptional activator into a repressor. EMBO J. 16, 1056–1065. doi: 10.1093/emboj/16.5.1056
Hodgkin, J., and Kaiser, D. (1977). Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc. Natl. Acad. Sci. U. S. A. 74, 2938–2942. doi: 10.1073/pnas.74.7.2938
Irving, S. E., Choudhury, N. R., and Corrigan, R. M. (2021). The stringent response and physiological roles of (pp)pGpp in bacteria. Nat. Rev. Microbiol. 19, 256–271. doi: 10.1038/s41579-020-00470-y
Kaiser, D. (1979). Social gliding is correlated with the presence of pili in Myxococcus xanthus. Proc. Natl. Acad. Sci. U. S. A. 76, 5952–5956. doi: 10.1073/pnas.76.11.5952
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M., and Tanabe, M. (2021). KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551. doi: 10.1093/nar/gkaa970
Krol, E., and Becker, A. (2004). Global transcriptional analysis of the phosphate starvation response in Sinorhizobium meliloti strains 1021 and 2011. Mol. Genet. Genomics 272, 1–17. doi: 10.1007/s00438-004-1030-8
Krol, E., and Becker, A. (2011). ppGpp in Sinorhizobium meliloti: Biosynthesis in response to sudden nutritional downshifts and modulation of the transcriptome. Mol. Microbiol. 81, 1233–1254. doi: 10.1111/j.1365-2958.2011.07752.x
Krol, E., Yau, H. C. L., Lechner, M., Schaper, S., Bange, G., Vollmer, W., et al. (2020). Tol-Pal system and Rgs proteins interact to promote unipolar growth and cell division in Sinorhizobium meliloti. mBio 11:e00306-20. doi: 10.1128/mBio.00306-20
Lagares, A., Hozbor, D. F., Niehaus, K., Otero, A. J., Lorenzen, J., Arnold, W., et al. (2001). Genetic characterization of a Sinorhizobium meliloti chromosomal region in lipopolysaccharide biosynthesis. J. Bacteriol. 183, 1248–1258. doi: 10.1128/JB.183.4.1248-1258.2001
Lee, N., Kim, W., Chung, J., Lee, Y., Cho, S., Jang, K. S., et al. (2020). Iron competition triggers antibiotic biosynthesis in Streptomyces coelicolor during coculture with Myxococcus xanthus. ISME J. 14, 1111–1124. doi: 10.1038/s41396-020-0594-6
Lehman, A. P., and Long, S. R. (2013). Exopolysaccharides from Sinorhizobium meliloti can protect against H2O2-dependent damage. J. Bacteriol. 195, 5362–5369. doi: 10.1128/JB.00681-13
Lehman, A. P., and Long, S. R. (2018). OxyR-dependent transcription response of Sinorhizobium meliloti to oxidative stress. J. Bacteriol. 200, e622–e617. doi: 10.1128/JB.00622-17
Li, Z., Ye, X., Liu, M., Xia, C., Zhang, L., Luo, X., et al. (2019). A novel outer membrane β-1,6-glucanase is deployed in the predation of fungi by myxobacteria. ISME J. 13, 2223–2235. doi: 10.1038/s41396-019-0424-x
Livingstone, P. G., Millard, A. D., Swain, M. T., and Whitworth, D. E. (2018). Transcriptional changes when Myxococcus xanthus preys on Escherichia coli suggest myxobacterial predators are constitutively toxic but regulate their feeding. Microb. Genom. 4:e000152. doi: 10.1099/mgen.0.000152
López-Lara, I. M., and Geiger, O. (2017). Bacterial lipid diversity. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1862, 1287–1299. doi: 10.1016/j.bbalip.2016.10.007
López-Lara, I. M., and Soto, M. J. (2018). “Fatty acid synthesis and regulation,” in Biogenesis of Fatty Acids, Lipids and Membranes. Handbook of Hydrocarbon and Lipid Microbiology, ed. O. Geiger (Cham: Springer), doi: 10.1007/978-3-319-43676-0_26-1
Lynch, D., O’Brien, J., Welch, T., Clarke, P., Cuiv, P. O., Crosa, J. H., et al. (2001). Genetic organization of the region encoding regulation, biosynthesis, and transport of rhizobactin 1021, a siderophore produced by Sinorhizobium meliloti. J. Bacteriol. 183, 2576–2585. doi: 10.1128/JB.183.8.2576-2585.2001
Martins, S. J., Taerum, S. J., Triplett, L., Emerson, J. B., Zasada, I., de Toledo, B. F., et al. (2022). Predators of soil bacteria in plant and human health. Phytobiome 6, 184–200. doi: 10.1094/PBIOMES-11-21-0073-RVW
Meade, H. M., and Signer, E. R. (1977). Genetic mapping of Rhizobium meliloti. Proc. Natl. Acad. Sci. U. S. A. 74, 2076–2078. doi: 10.1073/pnas.74.5.2076
Mendrygal, K. E., and González, J. E. (2000). Environmental regulation of exopolysaccharide production in Sinorhizobium meliloti. J. Bacteriol. 182, 599–606. doi: 10.1128/JB.182.3.599-606.2000
Molina-Santiago, C., Vela-Corcía, D., Petras, D., Díaz-Martínez, L., Pérez-Lorente, A. I., Sopeña-Torres, S., et al. (2021). Chemical interplay and complementary adaptative strategies toggle bacterial antagonism and co-existence. Cell Rep. 36:109449. doi: 10.1016/j.celrep.2021.109449
Mookherjee, A., and Jurkevitch, E. (2022). Interactions between Bdellovibrio and like organisms and bacteria in biofilms: Beyond predator-prey dynamics. Environ. Microbiol. 24, 998–1011. doi: 10.1111/1462-2920.15844
Müller, S., Strack, S., Hoefler, B., Straight, P., Kearns, D., and Kirby, J. (2014). Bacillaene and sporulation protect Bacillus subtilis from predation by Myxococcus xanthus. Appl. Environ. Microbiol. 80, 5603–5610. doi: 10.1128/AEM.01621-14
Müller, S., Strack, S. N., Ryan, S. E., Kearns, D. B., and Kirby, J. R. (2015). Predation by Myxococcus xanthus induces Bacillus subtilis to form spore-filled megastructures. Appl. Environ. Microbiol. 81, 203–210. doi: 10.1128/AEM.02448-14
Muñoz-Dorado, J., Marcos-Torres, F. J., García-Bravo, E., Moraleda-Muñoz, A., and Pérez, J. (2016). Myxobacteria: Moving, killing, feeding, and surviving together. Front. Microbiol. 7:781. doi: 10.3389/fmicb.2016.00781
Muñoz-Dorado, J., Moraleda-Muñoz, A., Marcos-Torres, F. J., Contreras-Moreno, F. J., Martin-Cuadrado, A. B., Schrader, J. M., et al. (2019). Transcriptome dynamics of the Myxococcus xanthus multicellular developmental program. Elife 8:e50374. doi: 10.7554/eLife.50374
Nicoud, Q., Barrière, Q., Busset, N., Dendene, S., Travin, D., Bourge, M., et al. (2021). Sinorhizobium meliloti functions required for resistance to antimicrobial NCR peptides and bacteroid differentiation. mBio 12:e00895-21. doi: 10.1128/mBio.00895-21
Niehaus, L., Boland, I., Liu, M., Chen, K., Fu, D., Henckel, C., et al. (2019). Microbial coexistence through chemical-mediated interactions. Nat. Commun. 10:2052. doi: 10.1038/s41467-019-10062-x
O’Brian, M. R. (2015). Perception and homeostatic control of iron in the rhizobia and related bacteria. Annu. Rev. Microbiol. 69, 229–245. doi: 10.1146/annurev-micro-091014-104432
Palmer, T., and Berks, B. C. (2012). The twin- arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 10, 483–496. doi: 10.1038/nrmicro2814
Papanikou, E., Karamanou, S., and Economou, A. (2007). Bacterial protein secretion through the translocase nanomachine. Nat. Rev. Microbiol. 5, 839–851. doi: 10.1038/nrmicro1771
Pellicer Martinez, M., Martinez, A. B., Crack, J., Holmes, J., Svistunenko, D., Johnston, A., et al. (2017). Sensing iron availability via the fragile [4Fe-4S] cluster of the bacterial transcriptional repressor RirA. Chem. Sci. 8, 8451–8463. doi: 10.1039/c7sc02801f
Pellock, B. J., Teplitski, M., Boinay, R. P., Bauer, W. D., and Walker, G. C. (2002). A LuxR homolog controls production of symbiotically active extracellular polysaccharide II by Sinorhizobium meliloti. J. Bacteriol. 184, 5067–5076. doi: 10.1128/JB.184.18.5067-5076.2002
Penterman, J., Abo, R. P., De Nisco, N. J., Arnold, M. F. F., Longhi, R., Zanda, M., et al. (2014). Host plant peptides elicit a transcriptional response to control the Sinorhizobium meliloti cell cycle during symbiosis. Proc. Natl. Acad. Sci. U. S. A. 111, 3561–3566. doi: 10.1073/pnas.1400450111
Pérez, J., Contreras-Moreno, F. J., Marcos-Torres, F. J., Moraleda-Muñoz, A., and Muñoz-Dorado, J. (2020). The antibiotic crisis: How bacterial predators can help. Comput. Struct. Biotechnol. J. 18, 2547–2555. doi: 10.1016/j.csbj.2020.09.010
Pérez, J., Contreras-Moreno, F. J., Muñoz-Dorado, J., and Moraleda-Muñoz, A. (2022). Development versus predation: Transcriptomic changes during the lifecycle of Myxococcus xanthus. Front. Microbiol. 13:1004476. doi: 10.3389/fmicb.2022.1004476
Pérez, J., Jiménez-Zurdo, J. I., Martínez-Abarca, F., Millán, V., Shimkets, L. J., and Muñoz-Dorado, J. (2014). Rhizobial galactoglucan determines the predatory pattern of Myxococcus xanthus and protects Sinorhizobium meliloti from predation. Environ. Microbiol. 16, 2341–2350. doi: 10.1111/1462-2920.12477
Pérez, J., Moraleda-Muñoz, A., Marcos-Torres, F. J., and Muñoz-Dorado, J. (2016). Bacterial predation: 75 years and counting! Environ. Microbiol. 18, 766–779. doi: 10.1111/1462-2920.13171
Pérez, J., Muñoz-Dorado, J., Braña, A. F., Shimkets, L. J., Sevillano, L., and Santamaría, R. I. (2011). Myxococcus xanthus induces actinorhodin overproduction and aerial mycelium formation by Streptomyces coelicolor. Microb. Biotechnol. 4, 175–183. doi: 10.1111/j.1751-7915.2010.00208.x
Petters, S., Groß, V., Söllinger, A., Pichler, M., Reinhard, A., Bengtsson, M. M., et al. (2021). The soil microbial food web revisited: Predatory myxobacteria as keystone taxa? ISME J. 15, 2665–2675. doi: 10.1038/s41396-021-00958-2
Pickering, B. S., and Oresnik, I. J. (2010). The twin arginine transport system appears to be essential for viability in Sinorhizobium meliloti. J. Bacteriol. 193, 6409–6418. doi: 10.1128/JB.00206-10
Platero, R. A., Jaureguy, M., Battistoni, F. J., and Fabiano, E. R. (2003). Mutations in sitB and sitD genes affect manganese-growth requirements in Sinorhizobium meliloti. FEMS Microbiol. Lett. 218, 65–70. doi: 10.1111/j.1574-6968.2003.tb11499.x
Santos, M. íR., Marques, A. T., Becker, J. ÂD., and Moreira, L. M. (2014). The Sinorhizobium meliloti EmrR regulator is required for efficient colonization of Medicago sativa Root Nodules. Mol. Plant-Microbe Interact. 27, 388–399. doi: 10.1094/MPMI-09-13-0284-R
Seef, S., Herrou, J., de Boissier, P., My, L., Brasseur, G., Robert, D., et al. (2021). A tad-like apparatus is required for contact-dependent prey killing in predatory social bacteria. Elife 10:e72409. doi: 10.7554/elife.72409
Sharypova, L. A., Niehaus, K., Scheidle, H., Holst, O., and Becker, A. (2003). Sinorhizobium meliloti acpXL mutant lacks the C28 hydroxylated fatty acid moiety of lipid A and does not express a slow migrating form of lipopolysaccharide. J. Biol. Chem. 278, 12946–12954. doi: 10.1074/jbc.M209389200
Sohlenkamp, C., and Geiger, O. (2016). Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 40:133. doi: 10.1093/femsre/fuv008
Soto, M. J., Sanjuan, J., and Olivares, J. (2006). Rhizobia and plant-pathogenic bacteria: Common infection weapons. Microbiology 152, 3167–3174. doi: 10.1099/mic.0.29112-0
Stein, B. J., Fiebig, A., and Crosson, S. (2021). The ChvG-ChvI and NtrY-NtrX two-component systems coordinately regulate growth of Caulobacter crescentus. J. Bacteriol. 203:e0019921. doi: 10.1128/JB.00199-21
Stubbendieck, R. M., Vargas-Bautista, C., and Straight, P. D. (2016). Bacterial communities: Interactions to scale. Front. Microbiol. 7:1234. doi: 10.3389/fmicb.2016.01234
Sutton, D., Livingstone, P. G., Furness, E., Swain, M. T., and Whitworth, D. E. (2109). Genome-wide identification of myxobacterial predation genes and demonstration of formaldehyde secretion as a potentially predation-resistant trait of Pseudomonas aeruginosa. Front. Microbiol. 10:2650. doi: 10.3389/fmicb.2019.02650
Szczepaniak, J., Press, C., and Kleanthous, C. (2020). The multifarious roles of Tol-Pal in Gram-negative bacteria. FEMS Microbiol. Rev. 44, 490–506. doi: 10.1093/femsre/fuaa018
Tang, G., Wang, Y., and Luo, L. (2014). Transcriptional regulator LsrB of Sinorhizobium meliloti positively regulates the expression of genes involved in lipopolysaccharide biosynthesis. Appl. Environ. Microbiol. 80, 5265–5273. doi: 10.1128/AEM.01393-14
Thiery, S., and Kaimer, C. (2020). The predation strategy of Myxococcus xanthus. Front. Microbiol. 11:2. doi: 10.3389/fmicb.2020.00002
Thiery, S., Turowski, P., Berleman, J. E., and Kaimer, C. (2022). The predatory soil bacterium Myxococcus xanthus combines a Tad- and an atypical type 3-like protein secretion system to kill bacterial cells. Cell Rep. 40:111340. doi: 10.1016/j.celrep.2022.111340
Tiricz, H., Szucs, A., Farkas, A., Pap, B., Lima, R. M., Maroti, G., et al. (2013). Antimicrobial nodule-specific cysteine-rich peptides induce membrane depolarization-associated changes in the transcriptome of Sinorhizobium meliloti. Appl. Environ. Microbiol. 79, 6737–6746. doi: 10.1128/AEM.01791-13
Travin, D. Y., Jouan, R., Vigouroux, A., Inaba-Inoue, S., Lachat, J., Haq, F., et al. (2023). Dual-uptake mode of the antibiotic phazolicin prevents resistance acquisition by Gram-begative bacteria. mBio 14:e0021723. doi: 10.1128/mbio.00217-23
Wang, C., Liu, X., Zhang, P., Wang, Y., Li, Z., He, H., et al. (2019). Bacillus licheniformis escapes from Myxococcus xanthus predation by deactivating myxovirescin a through enzymatic glucosylation. Environ. Microbiol. 21, 4755–4772. doi: 10.1111/1462-2920.14817
Wells, D. H., Chen, E. J., Fisher, R. F., and Long, S. R. (2007). ExoR is genetically coupled to the ExoS-ChvI two-component system and located in the periplasm of Sinorhizobium meliloti. Mol. Microbiol. 64, 647–664. doi: 10.1111/j.1365-2958.2007.05680.x
Whitworth, D. E., Jurkevitch, E., Pérez, J., Fuhrmann, G., and Koval, S. F. (2020). Editorial: Mechanisms of prokaryotic predation. Front. Microbiol. 11:2071. doi: 10.3389/fmicb.2020.02071
Williams, M. A., Bouchier, J. M., Mason, A. K., and Brown, P. J. B. (2022). Activation of ChvG-ChvI regulon by cell wall stress confers resistance to β-lactam antibiotics and initiates surface spreading in Agrobacterium tumefaciens. PLoS Genetics 18:e1010274. doi: 10.1371/journal.pgen.1010274
Winter, J., and Jakob, U. (2004). Beyond transcription–new mechanisms for the regulation of molecular chaperones. Crit. Rev. Biochem. Mol. Biol. 39, 297–317. doi: 10.1080/10409230490900658
Wippel, K., and Long, S. R. (2019). Symbiotic performance of Sinorhizobium meliloti lacking ppGpp depends on the Medicago host species. Mol. Plant Microbe Interact. 32, 717–728. doi: 10.1094/MPMI-11-18-0306-R
Xiao, Y., Wei, X., Ebright, R., and Wall, D. (2011). Antibiotic production by myxobacteria plays a role in predation. J. Bacteriol. 193, 4626–4633. doi: 10.1128/jb.05052-11
Yao, S. Y., Luo, L., Har, K. J., Becker, A., Ruberg, S., Yu, G. Q., et al. (2004). Sinorhizobium meliloti ExoR and ExoS proteins regulate both succinoglycan and flagellum production. J. Bacteriol. 186, 6042–6049. doi: 10.1128/JB.186.18.6042-6049.2004
Yuan, Z. C., Zaheer, R., Morton, R., and Finan, T. M. (2006). Genome prediction of PhoB regulated promoters in Sinorhizobium meliloti and twelve proteobacteria. Nucleic Acids Res. 34, 2686–2697. doi: 10.1093/nar/gkl365
Zavaleta-Pastor, M., Sohlenkamp, C., Gao, J. L., Guan, Z., Zaheer, R., Finan, T. M., et al. (2010). Sinorhizobium meliloti phospholipase C required for lipid remodeling during phosphorus limitation. Proc. Natl. Acad. Sci. U. S. A. 107, 302–307. doi: 10.1073/pnas.0912930107
Keywords: bacterial predation, myxobacteria, Sinorhizobium meliloti, defensome, bacterial interactions
Citation: Soto MJ, Pérez J, Muñoz-Dorado J, Contreras-Moreno FJ and Moraleda-Muñoz A (2023) Transcriptomic response of Sinorhizobium meliloti to the predatory attack of Myxococcus xanthus. Front. Microbiol. 14:1213659. doi: 10.3389/fmicb.2023.1213659
Received: 28 April 2023; Accepted: 01 June 2023;
Published: 19 June 2023.
Edited by:
Christian Sohlenkamp, National Autonomous University of Mexico, MexicoReviewed by:
David Cole Stevens, University of Mississippi, United StatesCopyright © 2023 Soto, Pérez, Muñoz-Dorado, Contreras-Moreno and Moraleda-Muñoz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aurelio Moraleda-Muñoz, YXVyZWxpb21AdWdyLmVz
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.