 Liangzhi Li
Liangzhi Li Delong Meng
Delong Meng Huaqun Yin
Huaqun Yin Teng Zhang1,2,3*
Teng Zhang1,2,3*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 July 2023
Sec. Terrestrial Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1203164
This article is part of the Research Topic Microbial Mechanisms for the Behavior of Toxic Metals in Soil View all 7 articles
Microorganisms that exhibit resistance to environmental stressors, particularly heavy metals, have the potential to be used in bioremediation strategies. This study aimed to explore and identify microorganisms that are resistant to heavy metals in soil environments as potential candidates for bioremediation. Metagenomic analysis was conducted using microbiome metagenomes obtained from the rhizosphere of soil contaminated with heavy metals and mineral-affected soil. The analysis resulted in the recovery of a total of 175 metagenome-assembled genomes (MAGs), 73 of which were potentially representing novel taxonomic levels beyond the genus level. The constructed ecological network revealed the presence of keystone taxa, including Rhizobiaceae, Xanthobacteraceae, Burkholderiaceae, and Actinomycetia. Among the recovered MAGs, 50 were associated with these keystone taxa. Notably, these MAGs displayed an abundance of genes conferring resistance to heavy metals and other abiotic stresses, particularly those affiliated with the keystone taxa. These genes were found to combat excessive accumulation of zinc/manganese, arsenate/arsenite, chromate, nickel/cobalt, copper, and tellurite. Furthermore, the keystone taxa were found to utilize both organic and inorganic energy sources, such as sulfur, arsenic, and carbon dioxide. Additionally, these keystone taxa exhibited the ability to promote vegetation development in re-vegetated mining areas through phosphorus solubilization and metabolite secretion. In summary, our study highlights the metabolic adaptability and ecological significance of microbial keystone taxa in mineral-affected soils. The MAGs associated with keystone taxa exhibited a markedly higher number of genes related to abiotic stress resistance and plant growth promotion compared to non-keystone taxa MAGs.
Nowadays, the security of the soil’s ecosystem and utility has been significantly affected by the accumulation of heavy metals, which has emerged as a major issue in soil contamination. While some metals are essential for plant growth and development, e.g., copper, zinc, nickel, cobalt, but they can be cytotoxic at high concentrations. The toxicity of iron (Fe) and manganese (Mn) is strongly affected by the properties of soils (i.e oxygen content, pH). On the other hand, metals like chromium, mercury, arsenic, and lead are not essential for plants and can be severely toxic even at low concentrations (Tang et al., 2023). The primary cause of toxic metal pollution in the environment is mining operations. Toxic mineral deposits build up around mining and smelting plants may cause significant risks to the ecosystem and public health. An approximately seventy billion tons of wastes are generated by mining each year, of which 14 billion tons are fine particulate tailing matters (Jones and Boger, 2012). Because of the enormous amount of refuse produced, mining wastes are regarded as a significant contributor of environmental pollution through sub-terrestrial leaking, storm-water overflow, and atmospheric deposition (Tordoff et al., 2000). Owing to improper administration, derelict mining deposits have attracted special notice (Dold, 2008). Through stabilizing the mining dumps and lowering subsurface discharge and the aerial dispersion of toxic elements, re-vegetation of mining waste integrated with plant growth-promoting bacteria (PGPB) arbitrarily or naturally—presents a viable option to reduce the ecological effect of mining wastes (Ojuederie and Babalola, 2017; Ashraf et al., 2019).
Microbial community, a significant component of soil, plays a crucial role in the immobility and natural circulation of elements. It is also an important indicator for assessing the ecological effects of toxic metal contamination in the soils (Kumar et al., 2008). Previous studies have shown that the stress of contaminants like heavy metals can decrease the biomass and biodiversity of ecosystems and impact their composition and functionality (Kamal et al., 2010; Zhang et al., 2022). A potential ecologically sound and feasible solution for toxic metal decontamination is bio-remediation. Native microbes are also readily available bio-remediators around polluted locations. These microbes, naturally present in the soil, are capable of removing or transforming heavy metal contaminants into less harmful forms (He et al., 2009; Emenike et al., 2018; Liu et al., 2019; Ma et al., 2019; Li M. et al., 2020). Recent advancements in sequencing technologies have greatly contributed to the study of microbial populations in different habitats. Consequently, there is considerable interest in understanding the changes in microbial community composition and performance caused by heavy metal pollution (Feng et al., 2018; Akash et al., 2022).
Molecular transport mechanisms that facilitate the removal of hazardous metalloids from the cells, metabolic processes that transform metallic ions into less toxicforms, the development of more resilient cell walls and membranes as protection against metalloid toxicity—microbes have employed these intricate strategies to combat the detrimental effects of heavy metals, which can be acquired through the horizontal gene transfer (HGT) process (Li et al., 2019). Microbes are also important for regulating the fates of metalloid, such as speciation (Zhu et al., 2018; Park et al., 2021), solubilizing (Li et al., 2021), and migratory abilities (Lan et al., 2021), in addition to mineral weathering. Studies have determined that the predominant microbial species in areas subjected to long-term thallium pollution are Fe/Mn reducers and Fe/S oxidizers (She et al., 2022). Similarly, in antimony-contaminated streams, Geobacter and Acinetobacter are the most prevalent microbial species (Sun et al., 2016).
Studies on the spatiotemporal characteristics and successions of native microbial communities in regions with heavy metal contamination have contributed to the selection of potential treatment methods for mining and smelting-intensive areas. One study, for instance, found that Proteobacteria was the dominant phylum in all depth layers of heavy metal contaminated soils (Li S. et al., 2020). Understanding the response of microbial communities to metal pressure necessitates a comprehensive description of the ecological processes that regulate the assembly of these communities in environments contaminated with metalloids. Two ecological processes, determinism and stochasticism, are well-known principles that regulate the assembly of microbial community (Shi et al., 2018). Additionally, our previous study also found that the presence of heavy metals significantly impacts the diversity and composition of the microbial communities. Moreover, the community assembly of these communities process becomes more deterministic as the concentration of heavy metals increases. Notably, the proportions of heavy metal-tolerant microbial species, such as Thiobacillus, Euryarchaeota, and Crenarchaeota, have shown an increase (Kamal et al., 2010; Zhang et al., 2022).
Although the dominant groups and their geographic distributions in circumstances related to metal extraction and smelting have been extensively investigated, there are still gaps in our understanding of the ecological and regulatory mechanisms controlling microbial communities in sites with varying levels or types of metallic ions contamination. The use of genome-resolved metagenomics has increased our understanding of the genetic capabilities of microflora. This study retrieved public sequencing datasets of metagenome soil samples from sites contaminated with heavy metals or rich in metals from the National Center for Biotechnology Information (NCBI) database. Radionuclide (137Cs, 210Pb, 226Ra, 228Ra, 60Co, 241Am, 238U, 228Th, and 232Th) of high levels ranging from 30 to 3,750 Bq/kg and toxic metal (Co, Ni, Cu, Zn, As, Cd, Hg, Pb and Cr) of high levels ranging from 60 to 2,500 mg/kg soil are present in the tested soil samples (Rogiers et al., 2021). To explore microorganisms in metal-contaminated soils and identify potential candidates for bioremediation, the metabolism capacities of the microbial population residing in the metal-rich location were examined using metagenomic profiling. Additionally, metagenomic binning was employed to generate metagenome-assembled genomes (MAGs) and analyze the genetic profiles of metal resistance and transformation in the native microbes, particularly the keystone species. Our findings highlight the important role of the keystone species residing in the re-vegetated deposits, and deliberate managements of such microbiota may contribute to tailing restoration techniques. By examining the metabolic potency of significant microbiota, specifically keystone taxa, in understanding key natural processes involved in mining waste restoration, this study offered a novel perspective. Our research suggests that microbes such as Burkholderiaceae, Pseudomonadaceae and Xanthobacteraceae can transform different metals by modifying their transportation and cytotoxicity, potentially providing significant ecological benefits such as promote vegetation.
To investigate the metabolisms of key microbial community members that may be involved in the primary succession of the mineral-affected terrestrial areas, metagenome analysis was conducted on soil samples of metal-rich areas (n = 58, see Supplementary Table S1 at https://doi.org/10.6084/m9.figshare.22579942.v1). The taxonomic annotation revealed a predominance of phyla Actinobacteria, Proteobacteria and Acidobacteria in the tested contaminated soil metagenomes (Supplementary Figure S1), which is consistent with previous studies (Lin et al., 2022; Ma et al., 2022). Principal component analysis (PCA) based on the numerical distributions of microbial taxa in tested metagenomes showed that the metagenomes were significantly differentiated among samples. The three main axes explained a total of 76.8% variation (Figure 1A, left, analysis of similarity [ANOSIM] R = 0.854, p = 0.001). Metagenomes of bioproject PRJNA616017 (group A) from America have relatively lower alpha diversity indexes (i.e., Shannon, Simpson, Pielou, and invsimpson) compared toother samples (Figure 1A, right). The neutral community model (NCM) predicts approximately 55% of the variability in microbial community taxonomic occurrence frequency can be attributed to relative abundance, indicating a slight dominance of stochastic processes in community assembly (Figure 1B). Different enriched taxa were also identified (Supplementary Figure S2). For instance, plant-associated bacteria such as Phyllobacteriaceae, Burkholderiales, Pseudomonadaceae, Rhodobacteraceae, Rhizobiaceae, Sphingomonadaceae, Methylobacteriaceae, and Actinomycetia were found to be more abundant in the rhizosphere soil type, specifically in PRJNA766619 samples (Geng et al., 2022). Conversely, soil samples from the river basin, PRJNA630593 (Rogiers et al., 2021) and PRJNA616017 (Thomas et al., 2020), exhibited a higher abundance of nitrogen-transforming microbial taxa, such as Nitrobacteraceae, Nitrosomonadales and Thaumarchaeota. Additionally, redundancy analysis (RDA) indicated that the soil microorganisms displayed different responses to the types of metals. For example, Nitrobacteraceae demonstrated a positive correlation with the concentrations of arsenic and lead, but a negative correlation with other metals. Conversely, Burkholderiales exhibited the opposite trend.
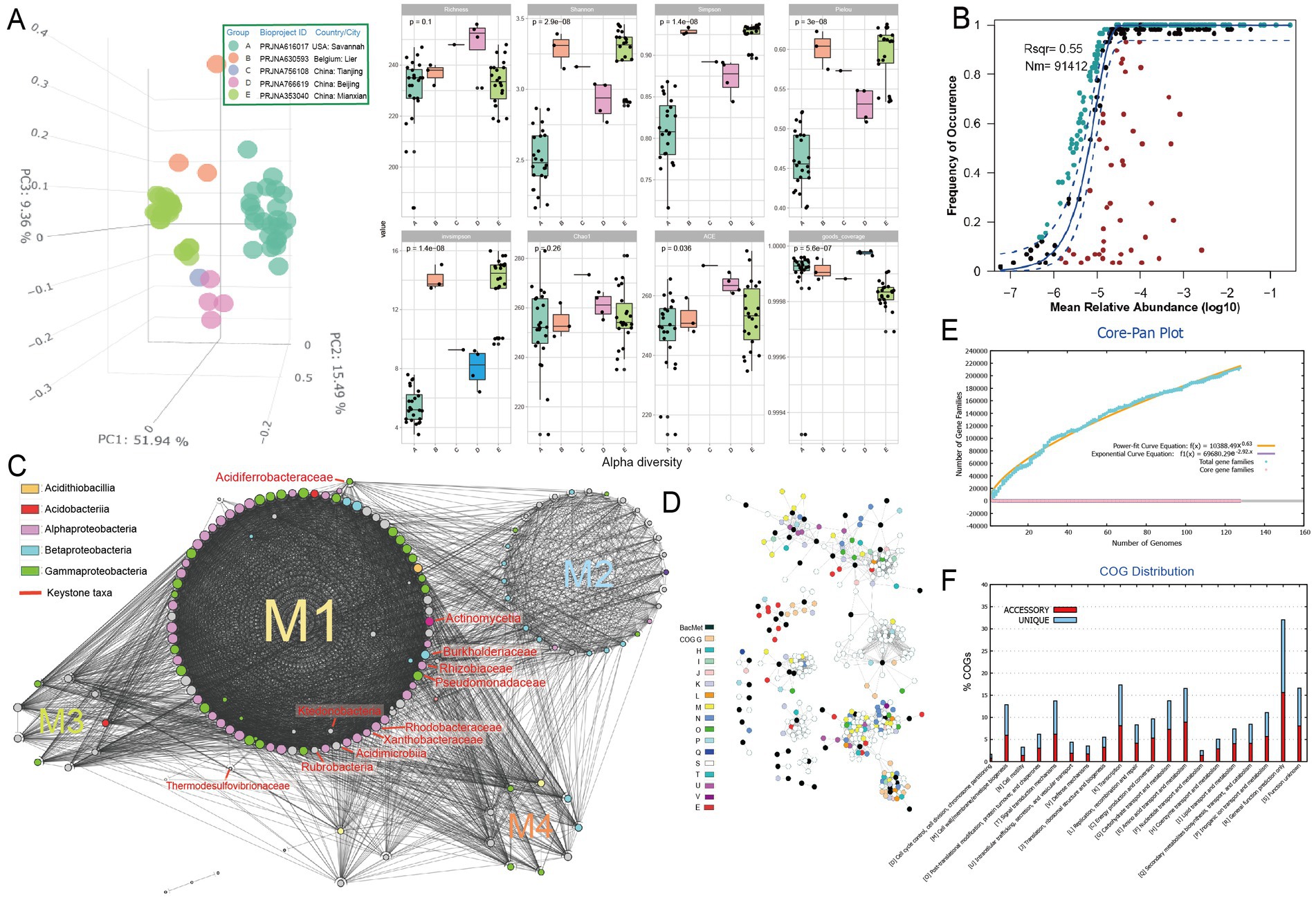
Figure 1. Microbial community analysis based on metagenome data of heavy metal-affected soils. (A) Left, Principal component analysis (PCA) of Bray–Curtis dissimilarity matrices showing clustering status of community structure based on the numerical distributions of microbial taxa in tested metagenomes [group (A–E)]; right, alpha diversity indexes including Richness, Shannon, Simpson, Pielou, invsimpson, Chao1, ACE, and goods coverage of microbial communities in each group (A–E). (B) Fit of the neutral community model (NCM) of community assembly. The OTUs more frequently present than predicted are in cyan, whereas those less frequently are in red. The blue dashed lines represent 95% confidence intervals around the model prediction and the OTUs fallen into the confidence intervals are regarded as neutrally distributed. Nm indicates the meta-community size times immigration, and Rsqr indicates the fit to the neutral model. Neutral processes are the part within 95% confidence interval (red) while non-neutral are the parts including above and below prediction (dark green). (C) Molecular ecological networks of microbial community visualized with circular layout in Cytoscape software. Modules (abbreviated as “M”) are separated by different circles. The size of each node is proportional to the degree (number of connections) and the colors of nodes represent different class. The links between the nodes indicate strong (cor > 0.8) and significant (p < 0.01) correlations. The identified keystone hubs by cytoHubba (Chin et al., 2014) are marked with red-color labels. (D) The sub-network containing genes annotated as BacMet (database of antibacterial biocide- and metal-resistance genes) entries and their first neighbors extracted from the gene co-occurrence network constructed across MAGs. The nodes are colored by their annotated function classes. Links between the nodes indicate strong (cor > 0.8) and significant (p < 0.01) gene co-occurrence in MAGs. (E) Pangenome analyzes and mathematical modeling of the size succession of the pangenome. (F) Bar chart showing functional proportions (based on COG categories) of different parts of the pangenome (i.e., accessory, unique).
The constructed microbial ecological network revealed four main modules, with the identification of Burkholderiaceae, Rhizobiaceae, Xanthobacteraceae, Pseudomonadaceae and other taxa as keystone hubs (Figure 1C, with labels marked with red color) by the software cytoHubba (Chin et al., 2014). After metagenome binning, a total of 175 metagenome-assembled genomes (MAGs) of medium quality or above (completeness >50% and contamination <10%) are recovered, in which 31 MAGs are of relatively high quality (completeness >80% and contamination <10%), and 73 MAGs were assigned to putative novel taxonomic level above genus (see Supplementary Table S2 at https://doi.org/10.6084/m9.figshare.22579942.v1). Pangenome mathematical modeling of the retrieved 175 MAGs revealed an “open” pangenome fitted into a power-law regression function [Ps (x) = 10388.49x0.63], while the core genome was fitted into an exponential regression [Fc (x) = 69680.29 e−2.92x] (Figure 1E). These results indicate vast genetic diversity within the microbiota from heavy metal contaminated soils and that the currently characterized features of the soil-borne microbial MAGs are still far from saturation. Functional annotation based on clusters of orthologous group (COG) revealed that the accessory genome of tested MAGs had a higher proportion of genes classified in COG categories such as K (Transcription), M (Cell wall/membrane/envelope biogenesis) and L (replication, recombination, and repair) (Figure 1F), which were probably related to environmental adaptions. Within the retrieved MAGs, 50 MAGs are related to the keystone taxa, such as Burkholderiaceae (n = 9), Rhizobiaceae (n = 8), Xanthobacteraceae (n = 4) and Pseudomonadaceae (n = 1). Still, we failed to retrieve MAGs of some keystone taxa, such as Acidiferrobacteraceae. We further constructed the gene co-occurrence network across the high-quality MAGs (Supplementary Figure S3) and extracted the sub-network containing genes annotated as BacMet entries (database of antibacterial biocide- and metal-resistance genes) (Pal et al., 2014) and their first neighbors (Figure 1D). We observed that toxic metal resistance genes (annotated as BacMet entries) co-occurred with the genes of COG classes E (Amino acid transport and metabolism), G (Carbohydrate transport and metabolism), U (Intracellular trafficking, secretion, and vesicular transport), V (Defense mechanisms), M (Cell wall/membrane/envelope biogenesis) and others. This finding is consistent with the consensus that microbial heavy metal stress response involves not only specialized metal transporters but also various cellular processes associated with systems-level maintenance (Pal et al., 2022). Accordingly, multiple gene families associated with heavy metal resilience, environmental adaption and plant growth promotion were identified from the MAGs (see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1), which will be discussed in the following sessions. Additionally, we performed selection pressure and codon adaption index (CAI) analyzes, indicating that nearly all metal resistance related genes that MAGs encoded exhibited a Ka/Ks ratio below the cutoff value 1 and relatively high CAI values (with average 0.70) (Figure 2).
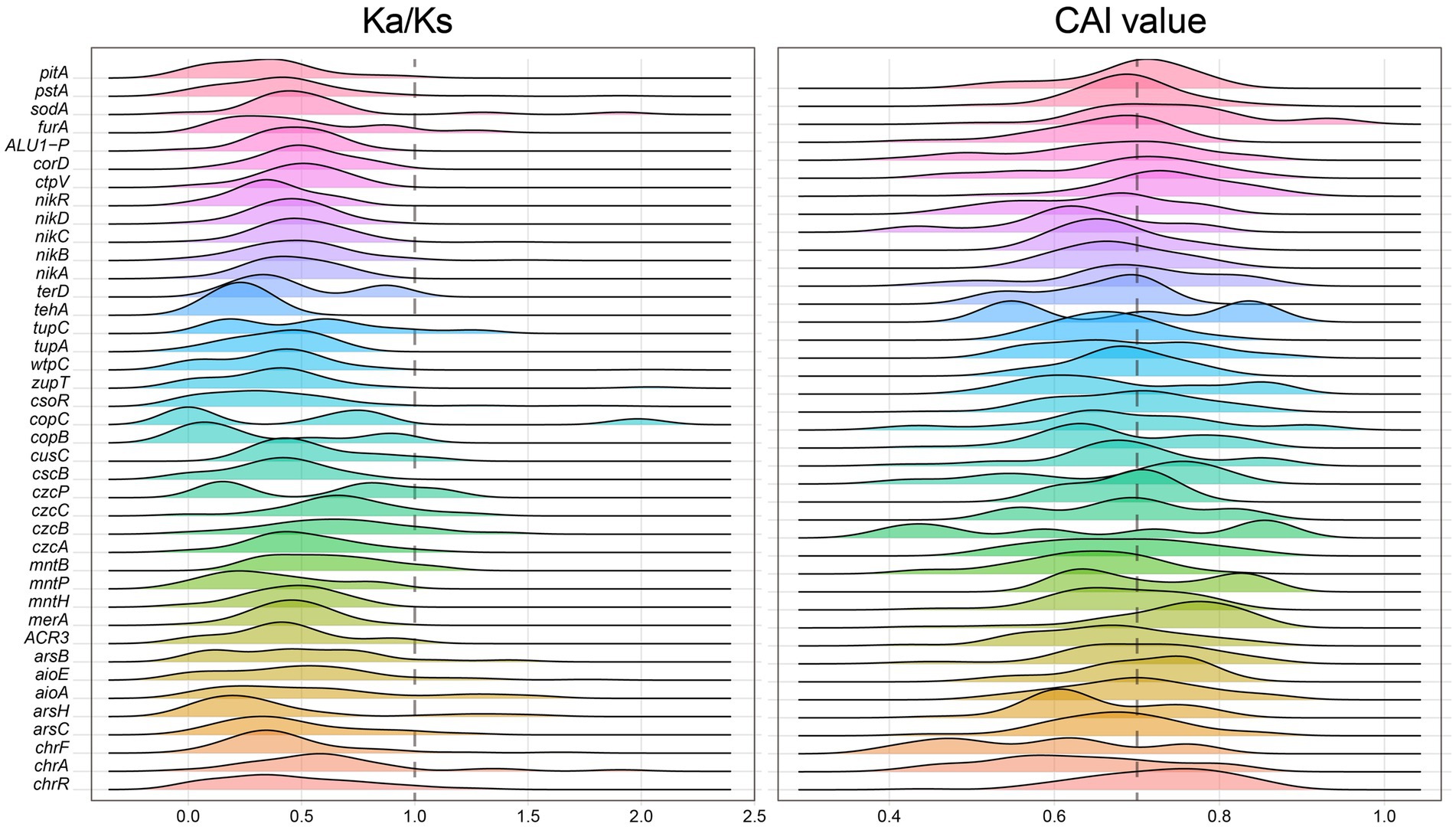
Figure 2. Distribution of Ka/Ks ratio (left) and CAI values (right) measuring the natural selection pressure and synonymous codon usage bias (the higher the CAI value, the higher the expression) for the heavy metal resistance genes classified by BacMet annotation, respectively.
The majority of the Proteobacteria and several Acidobacteriota-associated MAGs (n = 93, 56%) contained the Cr(VI) transporter-related genes (chrAH), and the best-studied Cr(VI) reductase encoding gene, chrR, was observed in almost all reconstructed MAGs (n = 126, 73.0%). This includes the MAGs classified into archaea Nitrososphaeraceae. Other genes that may be involved in Cr(VI) reduction, including the FMN reductase ssuE (n = 49, 31% MAGs), FMN-dependent NADH-azoreductase acpD/azoR (n = 64, 39% MAGs), 3-hydroxypropanoate dehydrogenase rutE (n = 64, 39% MAGs), nitroreductase nfsA (n = 64, 39% MAGs), riboflavin kinase/FMN adenylyltransferase ribF, and FMN reductase nrfA (Kwak et al., 2003; Mugerfeld et al., 2009; He et al., 2010; Zheng et al., 2015; Pradhan et al., 2016), were also annotated (Figure 3).

Figure 3. Heatmap showing distribution of gene families related with metal resistance, environmental adaption and metabolic function among tested MAGs. The taxonomic details are shown in the label. Branches leading to keystone taxa MAGs are marked in red color. The detailed description for gene abbreviations is provided in Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1.
The majority of the MAGs contained two putative genes arsC, which encodes the enzyme arsenate reductase, and arsR, which encodes the - transcriptional repressor responsive to arsenate, arsenite and antimonite. However, these genes were not found in MAGs belonging to Patescibacteria. The ACR3 gene, responsible for transporting As(III), was found in approximately 23.8% of the MAGs (Figure 4). The arsB gene, encoding the arsenite transporter, was also prevalent, present in 58% of the recovered MAGs. The arrB gene, which encodes a putative respiratory As(V) reductase involved in As detoxification, was detected in 29 MAGs (21%), mainly found in Acidobacteriota. In contrast, the aioE gene encoding putative As(III) oxidases (Wang Q. et al., 2017), was only found in one MAGs. Another arsenic resistance protein, ArsH, which acts as a methylarsenite oxidase, was also found encoded by 32 MAGs (17%), predominantly in Gammaproteobacteria and Alphaproteobacteria. This protein is an organoarsenical oxidase enzyme responsible for conferring resistance to methyl As(III) derivatives in both P. putida and S. meliloti (Páez-Espino et al., 2015).
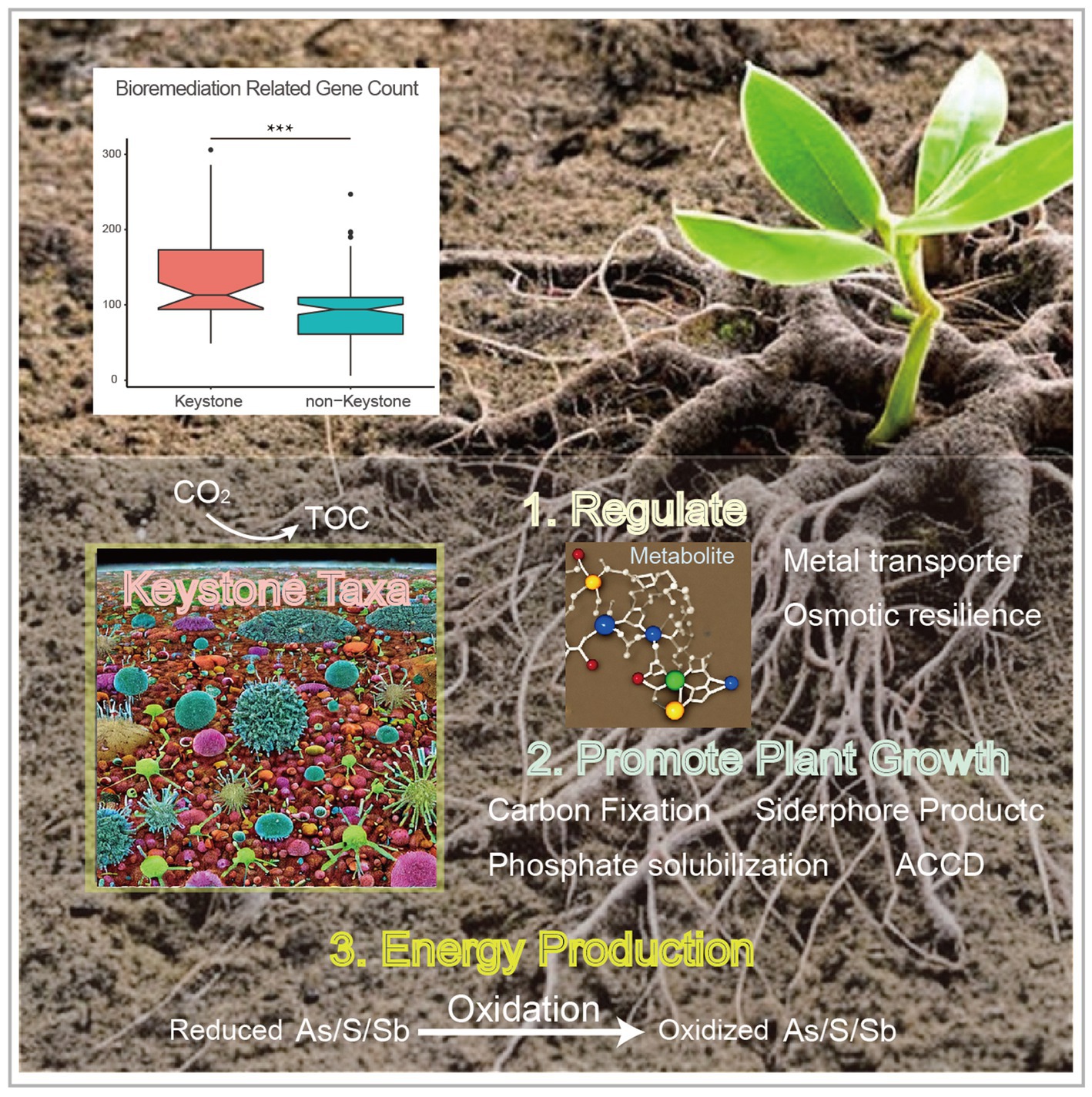
Figure 4. Comparison of the bioremediation-related gene amount between the keystone taxa MAGs and non-keystone MAGs (top, with the asterisks indicate significance; ***, p < 0.001), and the conceptual model of the ecological roles conducted by the keystone taxa within heavy metal contaminated soils (bottom).
A considerable number of putative genes encoding proteins implicated in diverse toxic ion transport and detoxification were found. These included the copper resistance system encoded by copABCDM/cutC/cusC, which was present in approximately 43% of the MAGs. Additionally, the cobalt-zinc-cadmium efflux system encoded by czcABCP was found in around 16% of the MAGs, and the zinc/manganese transport system encoded by mntABHP/znuB was identified in approximately 67% of the MAGs. The tellurite resistance system encoded by tehAB/terADZ was detected in about 10% of the MAGs. The fluoride exporter crcB, which was present in 68% of the MAGs, the ALU1-P gene for aluminum tolerance, found in 53% of the MAGs, and the peptide/nickel transport system encoded by nikBCDR, detected in around 10% of the MAGs, were also observed. Moreover, the nickel/cobalt transporter nixA/rcnA, the molybdate transport system modE, the ferrous iron transport system feoAB, and the storage bacterioferritin bfr were present in 29% of the MAGs. The tungstate transport system proteins tupAC were found in 6.9% of the MAGs, while the magnesium transporter corSAD was present in approximately 84% of the MAGs. Four MAGs contained the mercuric ion transport gene merT. Additionally, genes involved in biofilm formation, including rfbACD, pelAFG, and chitinase, were identified in 60.7, 13, and 13% of the MAGs, respectively (see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1).
Besides, components of the microbial adaptive immunity systems against exogenous DNA insertion, the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas (CRISPR-associated proteins) systems, including genes encoding CRISPR-associated exonuclease Cas2, Cas3, Cas4 and Cas5d, together with accessory proteins, such as Cmr6, Csd1, Csd2, Csm1, Csm3, Csm5 and Cst2 were detected in several MAGs belonging to Acidobacteriota (see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1), which might be further employed for gene editing in microbes or plants (Fasani et al., 2018).
A number of genes responsible to resist other abiotic environmental stresses (e.g., osmotic, acidic) were also observed in most MAGs (Figure 3, see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1). These genes encoded for the potassium transporting system (i.e., kdpBD, 96% MAGs; kup, 55% MAGs; kch, 37% MAGs; pch, 36% MAGs; trkA, 29% MAGs), phosphate transport system pst/pit (83% MAGs). Besides, genes encoded for organic acids degradation (e.g., acetyl-CoA synthetase, 87% MAGs), and Na+:H+ antiporter nhaA (82% MAGs) and pathways for arginine synthesis (e.g., arginine deiminase arcA, 39% MAGs, argininosuccinate lyase argH and argininosuccinate synthase argG, 82% MAGs) which produce ammonia as a by-product for acidic pressure resistance (Shek et al., 2017; Suryaletha et al., 2019) were also found.
In addition, a list of genes involved in the energy metabolism, biosynthesis of diverse secondary metabolites and xenobiotics biodegradation and metabolism was identified in the MAGs (Figure 3, see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1). Sulfur metabolism genes were widely identified, including those encoding sulfur-oxidizing protein (soxYZ, 14% MAGs), sulfide:quinone oxidoreductasewere (sqr, 24% MAGs), sulfate adenylyltransferase (sat, 19% MAGs), DMSO reductase (dmsC, 9% MAGs), alkanesulfonate monooxygenase (ssuD, 39% MAGs), thiosulfate dehydrogenase (doxD, 39% MAGs), taurine dioxygenase (tauD, 38% MAGs) and finally, sulfate transporter and reductase (cysCHJKP, 73% MAGs). Likewise, nitrogen fixation (i.e., nifD, 7% MAGs, mostly in the genus Pararhizobium) and nitrogen metabolism pathways (e.g., nirABD, narGIK, ncd2, cynS, arc) were also annotated. Ribulose-bisphosphate carboxylase (cbbLS), carbonic anhydrase (cah) and phosphoenolpyruvate carboxykinase (pckA) for carbon fixation were encoded by ~40% MAGs (mostly Actinomycetia, Gammaproteobacteria and Alphaproteobacteria). Also, five Beijerinckiaceae MAGs (5%) harbor genes encoding photosynthetic reaction center and light-harvesting complex components, including puhA, pufA, pufB, pufL and pufM. Xenobiotics biodegradation and metabolism genes, including those involved in the degradation of benzoate (i.e., pcaBCDHIJ, bcrBCD, boxC, ligABJ, catCE, galB, badAFH, xylEH), fluorobenzoate (i.e., dienelactone hydrolase family), chloroalkane and chloroalkene (i.e., haloacid/ haloalkane dehalogenase), steroid (i.e., hsaAC, choD, cyp125), nitrotoluene (i.e., hyaAB, nfsA), and aminobenzoate (i.e., nagH, mdlC, desB). Other genes related to secondary metabolism were also identified. For example, bacteriocin-related genes (e.g., ydeI) are detected in 74% MAGs; metabolism genes for compatible solutes such as trehalose (e.g., ostAB, treA) and ectoine (e.g., ectAC) are detected in 71 and 5% MAGs, respectively; genes for phosphate solubilization (e.g., phoND, ppa) are detected in 66% MAGs; genes for siderphore biosynthesis (e.g., frgA) are detected in 25% MAGs; and the gene encoding 1-amino cyclopropane-1-carboxylic acid (ACC) deaminase was detected in 20% MAGs. Detailed information of these metabolism genes could be found in the Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1.
In this study, we analyzed metagenome datasets from soils contaminated with heavy metals that were sampled from plant rhizosphere or naturally mineral areas. The main objectives were to investigate the microbial diversity, metabolic potential,and distribution. Additionally, the microbial correlation network assisted in identifying and gaining a deeper understanding of the diverse key microbes that likely participate in mining waste restoration. Comparing the metagenome-assembled genomes (MAGs) of the keystone taxa to the non-keystone taxa, we observed that the former had more genes related to abiotic stress resistance and promotion of plant growth, as confirmed by an unpaired t-test (p < 0.05). Based on these findings, we propose a conceptual model to illustrate the ecological role of the keystone taxa in mineral-affected soils (Figure 4).
Restoration and re-vegetation of heavy-metal polluted deposits are essential procedures for mitigating ecological risks (Bai et al., 2023). Significant efforts have been made to remediate and re-vegetate mineral areas such as tailing waste through microbe inoculation, which has shown certain effectiveness. Soil microorganisms play a crucial role in the initial accumulation of necessary ingredients, including hydrocarbon and accessible nitrogen, for other species (Sun et al., 2018). However, the harsh conditions in mineral-affected areas often limit bacterial growth, which negatively impacts remediation and re-vegetation efforts (Bai et al., 2023). Therefore, understanding the capabilities of soil microbes in terms of environmental adaptation and vegetation development is necessary for effective remediation strategies. Native microflora residing in heavy metal-rich soils or tailings is of considerable interest due to their potential ecological significance. The density of keystone species within the ecosystem may influence the composition of the microbial population (Agler et al., 2016). Their absence from the community can lead to significant disruptions in the ecological functions of the environment (van der Heijden and Hartmann, 2016). Given their importance in microbial populations and the natural “small world” phenomenon, the identification of keystone taxa is crucial (Dunne et al., 2002). Within the microbial ecological network, certain members such as Actinomycetia, Rhizobiaceae, Pseudonocardiaceae, Acidobacteria (Ventura et al., 2007) and Burkholderiaceae play a vital role and are considered keystone species in the community (Figure 1C). The keystone taxa in this study were chosen from the microbial ecological network according to the recently proposed standards of strong connectivity (closeness centrality >0.475) and small betweeness (betweenness centrality <0.025) (Berry and Widder, 2014; Banerjee et al., 2018a,b). These principles have commonly been employed in the identification of the keystone taxa in terrestrial ecosystems (Liang et al., 2016; Li et al., 2017; Banerjee et al., 2018a,b). The genetic composition of these species was analyzed using metagenomic binning analysis to identify species that are important to the remediation and vegetation. Due to their ability to perform crucial ecological functions, keystone taxa could contribute to re-vegetation efforts in areas affected by toxic waste. Actinobacteria, a phylum that encompasses members regarded as an ancient bacterial lineage with diverse environmental habitats, were specifically recognized as keystone taxa in extremely oligotrophic conditions such as Antarctic rock and soils (Le Roes-Hill et al., 2009; Hill et al., 2016), so do Acidobacteria (Xu et al., 2022). Members of the Rhizobiaceae, Burkholderiaceae and Pseudomonadaceae families are frequently found as keystone taxa in plant-associated environments (Chiarini et al., 1998). This finding aligns with the discovery of these microbes as keystone species in metal-rich deposits in our study. These taxa possess versatile and flexible metabolic pathways that can aid in plant growth promotion and resistance against pathogens (Jiang et al., 2017; Wang H. et al., 2017; Yan et al., 2017). Further investigation into the metabolic capabilities of such microbes will reveal the ecological significance of these keystone species and their potential in restoring harsh environments, such as heavy metal-contaminated soils or tailings. Our research also showed that MAGs associated with the keystone taxa have the ability to remediate heavy metals and improve nutrient availability for plants, highlighting their importance in ecological restoration. However, more research is required to fully comprehend the underlying mechanisms and develop effective strategies for utilizing these capabilities in environmental remediation.
Various metals can be transformed by microbes to modify their mobility and toxicity, potentially yielding ecological benefits (Giller et al., 2009). Multiple heavy metal resistance genes have been identified in the MAGs. Their functionality were highlighted by high CAI values, indicating ahigh predicted expression level, and low Ka/Ks ratio, indicating strong negative selection (Figure 2). Arsenic (As) is toxic to microbes and exerts significant selection pressure on the soil microbiota (Chen et al., 2020). Potential arsenic-resistance genes were found in recovered MAGs, particularly in the keystone taxa. Notably, genes involved in As detoxification (e.g., arsR) and those encoding the As(III) efflux pump (e.g., ACR3) were frequently detected, especially in keystone taxa like Rhizobiaceae and Burkholderiaceae (Figure 3). This suggests that soil-borne microbes have played a role in resisting the harmful effects of arsenic. Furthermore, the As(V) respiratory reductase encoded by arrB gene and arsenate reductase encoded by gene arsC are responsible for As(V) reduction. Microbes can generate energy during the redox processes of arsenic. As(V) reduction is likely common among the microbiome in metal-rich soil, as microbe-mediated As recycling could alter the transportation and cytotoxicity of As.
Furthermore, several genes associated with the reduction of Cr(VI) have been identified. The chromate ion [Cr(VI)] is highly toxic to microbes. Microbes have evolved two primary mechanisms to counteract Cr(VI): ion transport (efflux) and Cr(VI) reduction. The protein ChrA, which acts as a chromate transporter, has been discovered in various microbes, and it plays a role in moderating the extravasation of Cr(VI) (Ouertani et al., 2020; Kusumawardhani et al., 2022). Potential chrA genes were extensively discovered in the MAGs of Deinococcus, Ktedonobacteraceae and keystone taxa Burkholderiaceae, Pseudomonadaceae, and Xanthobacteraceae (Figure 3, see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1). This finding suggests that these microorganisms may be involved in the transport of Cr(VI) ions across cell membranes. Deinococcus, for instance, is a kind of radio-resistant bacterium that can detoxify Cr(VI), U(VI) and Tc(VII) from soil (Fredrickson et al., 2000). However, the chromate resistance of Ktedonobacteraceae has been rarely reported. The reduction of toxic Cr(VI) to Cr(III) by microorganisms has significant microbiological implications since Cr(III) is less bioactive and hazardous as compared to Cr(VI), although it is a thermodynamically stable compound. Several microorganisms, including Pseudomonas (Rajkumar et al., 2005), Bacillus (Das et al., 2014) and Arthrobacter (Mauricio et al., 2010), have been found to reduce Cr(VI). The best-characterized Cr(VI) reducing gene chrR, which encodes the hydrophilic, homodimer, NADH-dependent flavoprotein chromate reductase (Ackerley et al., 2004), was also widely found in MAGs. Additionally, diverse metal-resistant genes, such as cobalt-zinc-cadmium efflux systems czc, cus, and cop, have been detected in the MAGs (Figure 3, see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1). This suggests that the efflux of metal ions may serve as an important strategy for resisting metal stress. Therefore, metal content may be an important factor influencing the abundances of microbes, in addition to pH. Indeed, many keystone taxa were rich with various metal resistance genes against As, Cr, and Cu in this study. Numerous proteins implicated in diverse toxic ion tolerance and metal transformations were also present in the MAGs. For instance, gene crcB that encodes fluoride exporter was detected in 68% of MAGs (Figure 3, see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1), which may be utilized to counteract the presence of toxic fluoride compounds ubiquitous in the environment (Sonne et al., 2023). Collectively, the detection of genes associated with metal resistance and transformation in these MAGs supports the hypothesis that that the native microbes may alleviate the metal stress in the metal-rich soils.
Moreover, we observed a significant cross-linking of phylogenetically distant taxa in the sequence similarity network (SSN) of heavy metal resistance proteins retrieved from tested metagenome bins (Figure 5). This finding indicates that adaptive gene-sharing via horizontal gene transfer (HGT), as previously observed in mine drainage environments (Li et al., 2019), is also taking place in our study. Additionally, we have identified heavy metal resistance genes (e.g., arsC, terCD, copC) encoded by viral scaffolds recovered from the tested contaminated soil metagenomes (Figure 5, marked with black arrow, see Supplementary data at https://doi.org/10.6084/m9.figshare.22226998.v1). This suggests that the temperate viruses, such as lysogenic bacteriophage, may serve as the vectors for HGT, leading to the accelerated spread of adaptive genetic materials and the increased stress resilience within the microbial community as a whole. Consistent with our previous research, viruses appear to play a crucial role in regulating microbial community assembly (Liu et al., 2023).
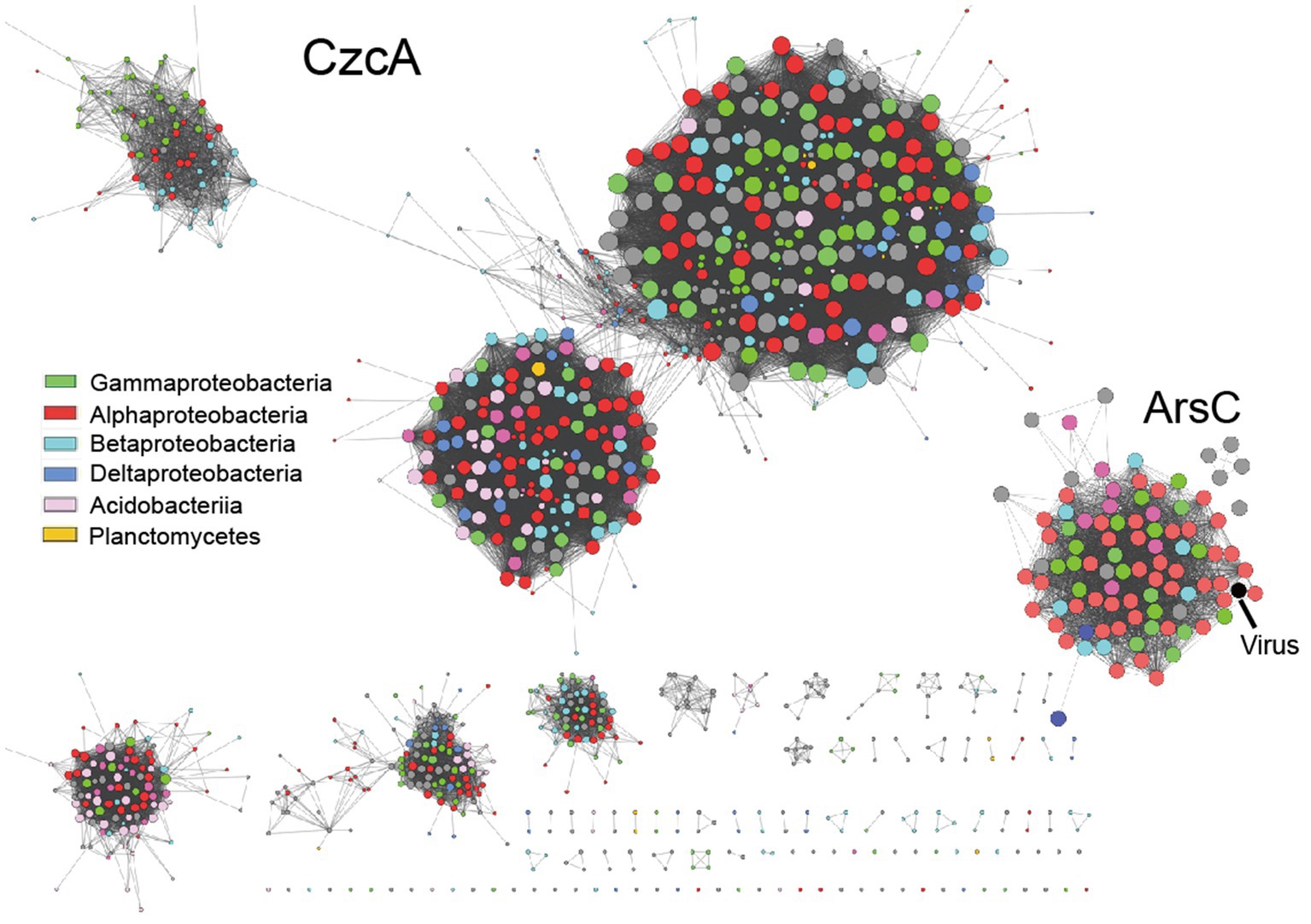
Figure 5. The representative sequence similarity network (SSN) displays heavy metal resistance proteins obtained from metagenome bins that have been tested.
The survival of microbiota in polluted soils requires resistance to a variety of adverse geochemical conditions, in addition to heavy metals. To maintain a balanced and near-neutral cytoplasm under osmotic pressure, proton buffer mechanisms, such as phosphate-specific transporter (Pst), potassium transporting ATPase system (Kdp), and arginine deiminase (ArcA), Na+:H+ antiporter (NhaA) are responsible to maintain an equilibrated and near-neutral cytoplasm (Tan et al., 2019). The identification of these associated genes in most of the keystone MAGs (especially Xanthobacteraceae, Burkholderiaceae and Pseudomonadaceae, as shown in Figure 3) suggests that they facilitate cytoplasmic homeostasis through buffering mechanisms. Specifically, 96% of the microbial genomes were found to possess kdpBD genes, which encode the K+ transporting ATPase system, indicating the capacity of soil-borne microbiota to relieve osmotic pressure by stimulating K+ transporter (Hua et al., 2015). Moreover, the extensive identification of genes encoding acetyl-CoA synthetase (ACSS), argininosuccinate lyase (argH) and argininosuccinate synthase (argG), dTDP 4-dehydrorhamnose reductase (rfbD) and others (Figure 3, see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1) suggests that organic degradation, polyamine production, formation of biofilm and impermeable cell membrane against harmful ions are additional survival strategies for the resistance to osmotic and pH perturbation (Baker-Austin and Dopson, 2007; Suryaletha et al., 2019). These genes may also contribute to the microbial community’s response to heavy metal stress, in line with the consensus that microbial heavy metal stress response involves specialized metal transporters and various cellular processes associated with systems-level maintenance (Pal et al., 2022).
The verdant site is hypothesized to be aided in tailing replanting by the microflora. Therefore, their metabolic activities are of great interest. Autotrophic organisms play a vital role in environmental restoration as they are the primary producers of organic matter in the environment. They are often present in high numbers during the early stages of restoration. It has been demonstrated that organic matter promotes vegetation development during soil remediation procedures in deposits. In addition to biological carbon, the oxidation of inorganic elements can also impact other geological conditions. For example, oxidation can significantly reduce the movement and cytotoxicity of As. Therefore, the presence of chemolitho-autotrophs in the deposits is essential for environmental recovery. In our study, many keystone MAGs showed the ability to generate energy from inorganic electron donors. For instance, almost all MAGs of keystone Burkholderiaceae and Xanthobacteraceae contained the sulfur-oxidizing sox genes (Figure 3). This suggests that sulfur oxidation may be important for keystone species. This study confirmed previous findings that sulfur oxidation genes and microbial species were highly enriched in the mining-impacted region (Sun et al., 2020). Since metal ores are commonly found in the form of sulfide minerals, tailings often contain high amounts of reduced sulfur compounds. Therefore, energetic production through the oxidation of reduced sulfur molecules may fuel the keystone taxa responsible for carbon fixation in the microbial community. On the other hand, the oxidation of large amounts of arsenic (As) and antimony (Sb) pollutants can also contribute to the flourishing of keystone taxa in metal-rich areas like tailings (Zhang et al., 2020), since the keystone taxa Rhizobiaceae, Xanthobacteraceae, Burkholderiaceae and Actinomycetia harbor the gene arsH or aioE (Figure 3).
The energy efficiency, imperative for surviving in nutrient-poor tailing environments, may be influenced by the abundance of carbon fixation genes (Ryu et al., 2003). Previous studies have investigated the functional genes involved in carbon fixation within bacterial life in mineral areas (Sun et al., 2018, 2020). These keystone MAGs may play a crucial role in supplying organic carbon in the re-vegetated mining areas (Schmidt et al., 2008). MAGs of keystone taxa such as Rhizobiaceae, Burkholderiaceae and Actinomycetia harbor the potency of fixing carbon (Figure 3). Moreover, the microbial ability to degrade xenobiotics, complex organic compounds, and synthesize secondary metabolites aids in their ecological roles within the microflora. The metabolism of essential metabolites can significantly alter the variety and interactions of soil microbes (Coyte et al., 2015; Banerjee et al., 2016). Secondary metabolites refer to a wide range of organic chemicals that interact with other species in soil ecosystems or vegetation, even though they are not necessary for the development or propagation of the microbial species (Coyte et al., 2015; Banerjee et al., 2016; Tyc et al., 2017). For instance, the competition and niche development within the soil’s microbiota may depend on the production bacteriocins (Dobson et al., 2012).
Further investigation was conducted on the capacity of keystone taxa to promote plant growth (PGP), which has potential applications in phytoremediation procedures and supporting vegetation development (Grandlic et al., 2008, 2009). Most of the keystone MAGs such as Burkholderiaceae, encoded PGP genes related to siderophore production, phosphate solubilization, and 1-amino cyclopropane-1-carboxylic acid (ACC) deaminase pathways (Figure 3, see Supplementary Table S3 at https://doi.org/10.6084/m9.figshare.22579942.v1). The development of siderophore production and strong iron uptake and retention abilities can provide strategic advantages to microbes during colonization of ecological niches, such as the rhizosphere, and promote vegetation development (Compant et al., 2005). Additionally, the enzymatic hydrolysis of phosphorus and other nutritional compounds promotes vegetation development by converting refractory nutrients like polyphosphate into an ionic form (Rodríguez et al., 2006). Plant growth may be supported by ACC deaminase, which reduces ethylene production in plants (Contesto et al., 2008). The identification of PGP genes in the keystone taxa reinforces previous research findings. For instance, it has been demonstrated that the keystone taxa, Burkholderiaceae and Pseudomonadaceae produce various auxiliary compounds. Burkholderiaceae species, resistant to toxic metals, significantly promote vegetation development by enhancing iron uptake, ACC deaminase activity, and polyphosphate solubility (Jiang et al., 2008). Similarly, metal-resistant Pseudomonas strains exhibit these PGP traits in growing vegetation (Loaces et al., 2011; Oves et al., 2013; Hsu and Micallef, 2017). The effective competitiveness and niche maintenance of keystone taxa within soil ecosystems may depend on the production of secondary metabolites like bacteriocins. Consequently, in-depth research on the physiological characteristics of secondary metabolites produced by keystone species may facilitate replanting efforts by regulating the rhizosphere microbiota (Vetsigian et al., 2011). The number of shotgun metagenomes related to metal contaminated soils is still limited compared with the amplicon sequencing data. As sequencing technology advances, future studies will include a wider range of soil samples with metal contamination to confirm the ecological roles of the identified keystone taxa in this study.
We first queried and retrieved all the items in the National Center for Biotechnology Information (NCBI) database (Benson et al., 2018) with the keywords “soil” and “metal” within the “biosample” regions. Only “shotgun metagenome” with the total size of clean data over 5GB were retained. This process has filtered the amplicon sequence data that do not provide functional information. We then downloaded from database the out-coming 58 soil metagenomes of microbiome as listed in Supplementary Table S1 at https://doi.org/10.6084/m9.figshare.22579942.v1 for downstream analyzes. Metagenome reads were assembled with MEGAHIT version 1.1.1 with default k-mer options (Li et al., 2015), followed by routine analyzes with the pipeline metawrap v.1.0 (Uritskiy et al., 2018). A combination of metabat2 (Kang et al., 2019), maxbin2 (Wu et al., 2016), concoct (Alneberg et al., 2014) were used for metagenome binning. GTDB-TK (Chaumeil et al., 2022) was applied for taxonomic assignment. Metagenome-assembled genomes (MAGs) quality assessment was conducted by CheckM (Parks et al., 2015) and we categorized the bins into high quality (completeness >80% and contamination <10%) and medium quality (completeness >50 to <80% and contamination <10%) according to a previous study (Tandon et al., 2022). This was followed by protein sequence clustering and analysis through software BPGA v.1.0 (Chaudhari et al., 2016) by default procedures, as well as functional annotation against the eggNOG database version 5.0 (Huerta-Cepas et al., 2019) and BacMet (database of antibacterial biocide- and metal-resistance genes) (Pal et al., 2014). The recovered MAGs in “fasta” format are available at https://doi.org/10.6084/m9.figshare.22579957.v1. A combination of VIBRANT v.2.0 (Kieft et al., 2020) and DeepVirFinder v.1.0 (Ren et al., 2020) were applied for viral scaffold recovery and analyzes under default parameters. The retrieved viral sequences in “gbk” format are available at https://doi.org/10.6084/m9.figshare.22226998.v1.
Codon adaption index (CAI) was used as a numerical estimator of gene expression level (Hiraoka et al., 2009; Zhou et al., 2013), and correspondingly, the webserver CAIcal (Puigbò et al., 2008a,b)1 was applied to calculate the CAI values for respective genes. A higher CAI value indicates a higher level of gene expression (Puigbò et al., 2008a,b; Li et al., 2019). We used Ka/Ks Calculation tool2 to calculate the ratio of nonsynonymous (Ka) to synonymous (Ks) nucleotide substitution rates is an indicator of selective pressures on metal resistance genes.
To construct the microbe or gene orthologue association network, correlations between pairwise operational taxonomic or gene orthologues that were present in more than half of the samples were calculated using the SparCC (Friedman and Alm, 2012) or CoNet (Faust and Raes, 2016) methods. Only edges with a significant correlation higher than 0.8 (p < 0.01) were retained for network construction. Cytoscape v.3.9.13 was used for network visualization. Network topological characteristics were calculated using NetworkAnalyzer tool in Cytoscape. Modular structure of highly interconnected nodes was analyzed using the MCODE application with default parameters. Cytoscape plugin cytoHubba (Chin et al., 2014) with “ClusteringCoefficient” and “BottleNeck” methods was used to predict keystone nodes in the network with the criteria: closeness centrality >0.475 and betweenness centrality <0.025 (Banerjee et al., 2018a,b). To determine the potential importance of stochastic processes on community assembly, the neutral community model (NCM) was used to predict the relationship between microbial taxa detection frequencies and their relative abundance across the wider meta-community, performed using R (version 3.6.3). The NCM is a validated method for deducing stochastic processes related to community assembly, which has been useful in deciphering ecological phenomena (Roguet et al., 2015). This model can quantify the significance of undetectable processes that might have a great impact on microbial communities (i.e., dispersal and ecological drift).
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
LL, DM, and HY conceived and designed the research. LL, TZ, and YL analyzed the data. LL wrote the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the Key Projects of Science and Technology of Hunan Branch of China National Tobacco Corporation (grants nos. 2021539200340244, 2020530000242025, xj202106, and 202104) and Fundamental Research Funds for the Central Universities of Central South University (no. 2022ZZTS0420). This study received funding from the Key Research and Development Program of Hunan Province and Natural Science Foundation of Changsha. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
We are grateful for resources from the High-Performance Computing Center of Central South University.
TZ was employed by Hunan Urban and Rural Environmental Construction Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1203164/full#supplementary-material
Supplementary Figure S1 | Bar chart showing the taxonomic compositions of tested metagenome of mineral-affected soil samples.
Supplementary Figure S2 | (A) Kruskal-Wallis analysis was performed to assess the distribution of taxa among the sample groups. (B) Redundancy analysis (RDA) was conducted to investigate the interaction between metal type and microorganism abundance.
Supplementary Figure S3 | The gene co-occurrence network across MAGs was constructed based on strong Spearman correlation (correlation coefficient > 0.8, p-value < 0.05). Nodes in the network were colored according to COG class.
Ackerley, D. F., Gonzalez, C. F., Park, C. H., Blake, R. N., Keyhan, M., and Matin, A. (2004). Chromate-reducing properties of soluble flavoproteins from pseudomonas putida and Escherichia coli. Appl. Environ. Microbiol. 70, 873–882. doi: 10.1128/AEM.70.2.873-882.2004
Agler, M. T., Ruhe, J., Kroll, S., Morhenn, C., Kim, S. T., Weigel, D., et al. (2016). Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 14:e1002352. doi: 10.1371/journal.pbio.1002352
Akash, S., Sivaprakash, B., Raja, V. C. V., Rajamohan, N., and Muthusamy, G. (2022). Remediation techniques for uranium removal from polluted environment 鈥?Review on methods, mechanism and toxicology. Environ. Pollut. 302:119068. doi: 10.1016/j.envpol.2022.119068
Alneberg, J., Bjarnason, B. S., de Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Ashraf, S., Ali, Q., Zahir, Z. A., Ashraf, S., and Asghar, H. N. (2019). Phytoremediation: environmentally sustainable way for reclamation of heavy metal polluted soils. Ecotoxicol. Environ. Saf. 174, 714–727. doi: 10.1016/j.ecoenv.2019.02.068
Bai, S., Han, X., and Feng, D. (2023). Shoot-root signal circuit: phytoremediation of heavy metal contaminated soil. Front. Plant Sci. 14:1139744. doi: 10.3389/fpls.2023.1139744
Baker-Austin, C., and Dopson, M. (2007). Life in acid: PH homeostasis in acidophiles. Trends Microbiol. 15, 165–171. doi: 10.1016/j.tim.2007.02.005
Banerjee, S., Kirkby, C. A., Schmutter, D., Bissett, A., Kirkegaard, J. A., and Richardson, A. E. (2016). Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 97, 188–198. doi: 10.1016/j.soilbio.2016.03.017
Banerjee, S., Schlaeppi, K., and van der Heijden, M. G. A. (2018a). Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576. doi: 10.1038/s41579-018-0024-1
Banerjee, S., Thrall, P. H., Bissett, A., van der Heijden, M., and Richardson, A. E. (2018b). Linking microbial co-occurrences to soil ecological processes across a woodland-grassland ecotone. Ecol. Evol. 8, 8217–8230. doi: 10.1002/ece3.4346
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Ostell, J., Pruitt, K. D., et al. (2018). GenBank. Nucleic Acids Res. 46, D41–D47. doi: 10.1093/nar/gkx1094
Berry, D., and Widder, S. (2014). Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 5:219. doi: 10.3389/fmicb.2014.00219
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2022). GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316. doi: 10.1093/bioinformatics/btac672
Chen, S., Sun, G., Yan, Y., Konstantinidis, K. T., Zhang, S., Deng, Y., et al. (2020). The great oxidation event expanded the genetic repertoire of arsenic metabolism and cycling. Proc. Natl. Acad. Sci. 117, 10414–10421. doi: 10.1073/pnas.2001063117
Chiarini, L., Bevivino, A., Tabacchioni, S., and Dalmastri, C. (1998). Inoculation of Burkholderia cepacia, Pseudomonas fluorescens and Enterobacter sp. on Sorghum bicolor: root colonization and plant growth promotion of dual strain inocula. Soil Biol. Biochem. 30, 81–87. doi: 10.1016/S0038-0717(97)00096-5
Chin, C. H., Chen, S. H., Wu, H. H., Ho, C. W., Ko, M. T., and Lin, C. Y. (2014). CytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8:S11. doi: 10.1186/1752-0509-8-S4-S11
Compant, S., Duffy, B., Nowak, J., Clément, C., and Barka, E. A. (2005). Use of plant growth-promoting bacteria for biocontrol of plant diseases: principles, mechanisms of action, and future prospects. Appl. Environ. Microbiol. 71, 4951–4959. doi: 10.1128/AEM.71.9.4951-4959.2005
Contesto, C., Desbrosses, G., Lefoulon, C., Béna, G., Borel, F., Galland, M., et al. (2008). Effects of rhizobacterial ACC deaminase activity on Arabidopsis indicate that ethylene mediates local root responses to plant growth-promoting rhizobacteria. Plant Sci. 175, 178–189. doi: 10.1016/j.plantsci.2008.01.020
Coyte, K. Z., Schluter, J., and Foster, K. R. (2015). The ecology of the microbiome: networks, competition, and stability. Science 350, 663–666. doi: 10.1126/science.aad2602
Das, S., Mishra, J., Das, S. K., Pandey, S., Rao, D. S., Chakraborty, A., et al. (2014). Investigation on mechanism of Cr(VI) reduction and removal by Bacillus amyloliquefaciens, a novel chromate tolerant bacterium isolated from chromite mine soil. Chemosphere 96, 112–121. doi: 10.1016/j.chemosphere.2013.08.080
Dobson, A., Cotter, P. D., Ross, R. P., and Hill, C. (2012). Bacteriocin production: a probiotic trait? Appl. Environ. Microbiol. 78, 1–6. doi: 10.1128/AEM.05576-11
Dold, B. (2008). Sustainability in metal mining: from exploration, over processing to mine waste management. Rev. Environ. Sci. Biotechnol. 7, 275–285. doi: 10.1007/s11157-008-9142-y
Dunne, J. A., Williams, R. J., and Martinez, N. D. (2002). Network structure and biodiversity loss in food webs: robustness increases with connectance. Ecol. Lett. 5, 558–567. doi: 10.1046/j.1461-0248.2002.00354.x
Emenike, C. U., Jayanthi, B., Agamuthu, P., and Fauziah, S. H. (2018). Biotransformation and removal of heavy metals: a review of phytoremediation and microbial remediation assessment on contaminated soil. Environ. Rev. 26, 156–168. doi: 10.1139/er-2017-0045
Fasani, E., Manara, A., Martini, F., Furini, A., and DalCorso, G. (2018). The potential of genetic engineering of plants for the remediation of soils contaminated with heavy metals. Plant Cell Environ. 41, 1201–1232. doi: 10.1111/pce.12963
Faust, K., and Raes, J. (2016). CoNet app: inference of biological association networks using Cytoscape. F1000Res 5:1519. doi: 10.12688/f1000research.9050.2
Feng, G., Xie, T., Wang, X., Bai, J., Tang, L., and Zhao, H. (2018). Metagenomic analysis of microbial community and function involved in cd-contaminated soil. BMC Microbiol. 18:11. doi: 10.1186/s12866-018-1152-5
Fredrickson, J. K., Kostandarithes, H. M., Li, S. W., Plymale, A. E., and Daly, M. J. (2000). Reduction of Fe(III), Cr(VI), U(VI), and Tc(VII) by deinococcus radiodurans r1. Appl. Environ. Microbiol. 66, 2006–2011. doi: 10.1128/AEM.66.5.2006-2011.2000
Friedman, J., and Alm, E. J. (2012). Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8:e1002687. doi: 10.1371/journal.pcbi.1002687
Geng, H., Wang, F., Yan, C., Ma, S., Zhang, Y., Qin, Q., et al. (2022). Rhizosphere microbial community composition and survival strategies in oligotrophic and metal(loid) contaminated iron tailings areas. J. Hazard. Mater. 436:129045. doi: 10.1016/j.jhazmat.2022.129045
Giller, K. E., Witter, E., and McGrath, S. P. (2009). Heavy metals and soil microbes. Soil Biol. Biochem. 41, 2031–2037. doi: 10.1016/j.soilbio.2009.04.026
Grandlic, C. J., Mendez, M. O., Chorover, J., Machado, B., and Maier, R. M. (2008). Plant growth-promoting bacteria for phytostabilization of mine tailings. Environ. Sci. Technol. 42, 2079–2084. doi: 10.1021/es072013j
Grandlic, C. J., Palmer, M. W., and Maier, R. M. (2009). Optimization of plant growth-promoting bacteria-assisted phytostabilization of mine tailings. Soil Biol. Biochem. 41, 1734–1740. doi: 10.1016/j.soilbio.2009.05.017
He, Z., Gao, F., Sha, T., Hu, Y., and He, C. (2009). Isolation and characterization of a Cr(VI)-reduction Ochrobactrum sp. strain CSCr-3 from chromium landfill. J. Hazard. Mater. 163, 869–873. doi: 10.1016/j.jhazmat.2008.07.041
He, M., Li, X., Guo, L., Miller, S. J., Rensing, C., and Wang, G. (2010). Characterization and genomic analysis of chromate resistant and reducing Bacillus cereus strain SJ1. BMC Microbiol. 10:221. doi: 10.1186/1471-2180-10-221
Hill, R., Saetnan, E. R., Scullion, J., Gwynn-Jones, D., Ostle, N., and Edwards, A. (2016). Temporal and spatial influences incur reconfiguration of Arctic heathland soil bacterial community structure. Environ. Microbiol. 18, 1942–1953. doi: 10.1111/1462-2920.13017
Hiraoka, Y., Kawamata, K., Haraguchi, T., and Chikashige, Y. (2009). Codon usage bias is correlated with gene expression levels in the fission yeast Schizosaccharomyces pombe. Genes Cells 14, 499–509. doi: 10.1111/j.1365-2443.2009.01284.x
Hsu, C. K., and Micallef, S. A. (2017). Plant-mediated restriction of Salmonella enterica on tomato and spinach leaves colonized with pseudomonas plant growth-promoting rhizobacteria. Int. J. Food Microbiol. 259, 1–6. doi: 10.1016/j.ijfoodmicro.2017.07.012
Hua, Z. S., Han, Y. J., Chen, L. X., Liu, J., Hu, M., Li, S. J., et al. (2015). Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 9, 1280–1294. doi: 10.1038/ismej.2014.212
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). EggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Jiang, Y., Li, S., Li, R., Zhang, J., Liu, Y., Lv, L., et al. (2017). Plant cultivars imprint the rhizosphere bacterial community composition and association networks. Soil Biol. Biochem. 109, 145–155. doi: 10.1016/j.soilbio.2017.02.010
Jiang, C. Y., Sheng, X. F., Qian, M., and Wang, Q. Y. (2008). Isolation and characterization of a heavy metal-resistant Burkholderia sp. from heavy metal-contaminated paddy field soil and its potential in promoting plant growth and heavy metal accumulation in metal-polluted soil. Chemosphere 72, 157–164. doi: 10.1016/j.chemosphere.2008.02.006
Jones, H., and Boger, D. V. (2012). Sustainability and waste management in the resource industries. Ind. Eng. Chem. Res. 51, 10057–10065. doi: 10.1021/ie202963z
Kamal, S., Prasad, R., and Varma, A. (2010). “Soil microbial diversity in relation to heavy metals” in in (Berlin, Heidelberg: Springer), 31–63.
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 7:e7359. doi: 10.7717/peerj.7359
Kieft, K., Zhou, Z., and Anantharaman, K. (2020). VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 8:90. doi: 10.1186/s40168-020-00867-0
Kumar, K. V., Singh, N., Behl, H. M., and Srivastava, S. (2008). Influence of plant growth promoting bacteria and its mutant on heavy metal toxicity in Brassica juncea grown in fly ash amended soil. Chemosphere 72, 678–683. doi: 10.1016/j.chemosphere.2008.03.025
Kusumawardhani, H., Hosseini, R., Verschoor, J. A., and de Winde, J. H. (2022). Comparative analysis reveals the modular functional structure of conjugative megaplasmid pTTS12 of Pseudomonas putida S12: a paradigm for transferable traits, plasmid stability, and inheritance? Front. Microbiol. 13:1001472. doi: 10.3389/fmicb.2022.1001472
Kwak, Y. H., Lee, D. S., and Kim, H. B. (2003). Vibrio harveyi nitroreductase is also a chromate reductase. Appl. Environ. Microbiol. 69, 4390–4395. doi: 10.1128/AEM.69.8.4390-4395.2003
Lan, J., Zhang, S., Dong, Y., Li, J., Li, S., Feng, L., et al. (2021). Stabilization and passivation of multiple heavy metals in soil facilitating by pinecone-based biochar: mechanisms and microbial community evolution. J. Hazard. Mater. 420:126588. doi: 10.1016/j.jhazmat.2021.126588
Le Roes-Hill, M., Rohland, J., Meyers, P. R., Cowan, D. A., and Burton, S. G. (2009). Streptomyces hypolithicus sp. Nov., isolated from an Antarctic hypolith community. Int. J. Syst. Evol. Microbiol. 59, 2032–2035. doi: 10.1099/ijs.0.007971-0
Li, F., Chen, L., Zhang, J., Yin, J., and Huang, S. (2017). Bacterial community structure after long-term organic and inorganic fertilization reveals important associations between soil nutrients and specific taxa involved in nutrient transformations. Front. Microbiol. 8:187. doi: 10.3389/fmicb.2017.00187
Li, M., Huang, Y., Yang, Y., Wang, H., Hu, L., Zhong, H., et al. (2020). Heavy metal ions removed from imitating acid mine drainages with a thermoacidophilic archaea: Acidianus manzaensis YN25. Ecotoxicol. Environ. Saf. 190:110084. doi: 10.1016/j.ecoenv.2019.110084
Li, D., Li, G., and Zhang, D. (2021). Field-scale studies on the change of soil microbial community structure and functions after stabilization at a chromium-contaminated site. J. Hazard. Mater. 415:125727. doi: 10.1016/j.jhazmat.2021.125727
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, L., Liu, Z., Meng, D., Liu, X., Li, X., Zhang, M., et al. (2019). Comparative genomic analysis reveals the distribution, organization, and evolution of metal resistance genes in the genus acidithiobacillus. Appl. Environ. Microbiol. 85. doi: 10.1128/AEM.02153-18
Li, S., Zhao, B., Jin, M., Hu, L., Zhong, H., and He, Z. (2020). A comprehensive survey on the horizontal and vertical distribution of heavy metals and microorganisms in soils of a pb/Zn smelter. J. Hazard. Mater. 400:123255. doi: 10.1016/j.jhazmat.2020.123255
Liang, Y., Zhao, H., Deng, Y., Zhou, J., Li, G., and Sun, B. (2016). Long-term oil contamination alters the molecular ecological networks of soil microbial functional genes. Front. Microbiol. 7:60. doi: 10.3389/fmicb.2016.00060
Lin, H., Tang, Y., Dong, Y., Wei, Z., and Liu, C. (2022). Characterization of heavy metal migration, the microbial community, and potential bioremediating genera in a waste-rock pile field of the largest copper mine in Asia. J. Clean. Prod. 351:131569. doi: 10.1016/j.jclepro.2022.131569
Liu, Z., Huang, Y., Zhang, T., Meng, D., Jiang, Z., Yang, Z., et al. (2023). Viruses regulate microbial community assembly together with environmental factors in acid mine drainage. Appl. Environ. Microbiol. 89, e0197322–e0191973. doi: 10.1128/aem.01973-22
Liu, B., Su, G., Yang, Y., Yao, Y., Huang, Y., Hu, L., et al. (2019). Vertical distribution of microbial communities in chromium-contaminated soil and isolation of Cr(vi)-reducing strains. Ecotoxicol. Environ. Saf. 180, 242–251. doi: 10.1016/j.ecoenv.2019.05.023
Loaces, I., Ferrando, L., and Scavino, A. F. (2011). Dynamics, diversity and function of endophytic siderophore-producing bacteria in rice. Microb. Ecol. 61, 606–618. doi: 10.1007/s00248-010-9780-9
Ma, S., Qiao, L., Liu, X., Zhang, S., Zhang, L., Qiu, Z., et al. (2022). Microbial community succession in soils under long-term heavy metal stress from community diversity-structure to KEGG function pathways. Environ. Res. 214:113822. doi: 10.1016/j.envres.2022.113822
Ma, Y., Zhong, H., and He, Z. (2019). Cr(VI) reductase activity locates in the cytoplasm of Aeribacillus pallidus BK1, a novel Cr(VI)-reducing thermophile isolated from Tengchong geothermal region, China. Chem. Eng. J. 371, 524–534. doi: 10.1016/j.cej.2019.04.085
Mauricio, G. A., Peña, C. J., and Maldonado, V. M. (2010). Isolation and characterization of hexavalent chromium-reducing rhizospheric bacteria from a wetland. Int. J. Phytoremediation 12, 317–334. doi: 10.1080/15226510902968118
Mugerfeld, I., Law, B. A., Wickham, G. S., and Thompson, D. K. (2009). A putative azoreductase gene is involved in the Shewanella oneidensis response to heavy metal stress. Appl. Microbiol. Biotechnol. 82, 1131–1141. doi: 10.1007/s00253-009-1911-1
Ojuederie, O. B., and Babalola, O. O. (2017). Microbial and plant-assisted bioremediation of heavy metal polluted environments: a review. Int. J. Environ. Res. Public Health 14:1504. doi: 10.3390/ijerph14121504
Ouertani, R., Ouertani, A., Mahjoubi, M., Bousselmi, Y., Najjari, A., Cherif, H., et al. (2020). New plant growth-promoting, chromium-detoxifying microbacterium species isolated from a tannery wastewater: performance and genomic insights. Front. Bioeng. Biotechnol. 8:521. doi: 10.3389/fbioe.2020.00521
Oves, M., Khan, M. S., and Zaidi, A. (2013). Chromium reducing and plant growth promoting novel strain Pseudomonas aeruginosa OSG41 enhance chickpea growth in chromium amended soils. Eur. J. Soil Biol. 56, 72–83. doi: 10.1016/j.ejsobi.2013.02.002
Páez-Espino, A. D., Durante-Rodríguez, G., and de Lorenzo, V. (2015). Functional coexistence of twin arsenic resistance systems in Pseudomonas putida KT2440. Environ. Microbiol. 17, 229–238. doi: 10.1111/1462-2920.12464
Pal, C., Bengtsson-Palme, J., Rensing, C., Kristiansson, E., and Larsson, D. G. (2014). BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 42, D737–D743. doi: 10.1093/nar/gkt1252
Pal, A., Bhattacharjee, S., Saha, J., Sarkar, M., and Mandal, P. (2022). Bacterial survival strategies and responses under heavy metal stress: a comprehensive overview. Crit. Rev. Microbiol. 48, 327–355. doi: 10.1080/1040841X.2021.1970512
Park, S., Boyanov, M. I., Kemner, K. M., Loughlin, O., and Kwon, M. J. (2021). Distribution and speciation of Sb and toxic metal(loid)s near an antimony refinery and their effects on indigenous microorganisms. J. Hazard. Mater. 403:123625. doi: 10.1016/j.jhazmat.2020.123625
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pradhan, S. K., Singh, N. R., Rath, B. P., and Thatoi, H. (2016). Bacterial chromate reduction: a review of important genomic, proteomic, and bioinformatic analysis. Crit. Rev. Environ. Sci. Technol. 46, 1659–1703. doi: 10.1080/10643389.2016.1258912
Puigbò, P., Bravo, I. G., and Garcia-Vallve, S. (2008a). CAIcal: a combined set of tools to assess codon usage adaptation. Biol. Direct 3:38. doi: 10.1186/1745-6150-3-38
Puigbò, P., Romeu, A., and Garcia-Vallvé, S. (2008b). HEG-DB: a database of predicted highly expressed genes in prokaryotic complete genomes under translational selection. Nucleic Acids Res. 36, D524–D527. doi: 10.1093/nar/gkm831
Rajkumar, M., Nagendran, R., Lee, K. J., and Lee, W. H. (2005). Characterization of a novel Cr6+ reducing pseudomonas sp. with plant growth-promoting potential. Curr. Microbiol. 50, 266–271. doi: 10.1007/s00284-005-4470-4
Ren, J., Song, K., Deng, C., Ahlgren, N. A., Fuhrman, J. A., Li, Y., et al. (2020). Identifying viruses from metagenomic data using deep learning. Quant. Biol. 8, 64–77. doi: 10.1007/s40484-019-0187-4
Rodríguez, H., Fraga, R., Gonzalez, T., and Bashan, Y. (2006). Genetics of phosphate solubilization and its potential applications for improving plant growth-promoting bacteria. Plant Soil 287, 15–21. doi: 10.1007/s11104-006-9056-9
Rogiers, T., Claesen, J., Van Gompel, A., Vanhoudt, N., Mysara, M., Williamson, A., et al. (2021). Soil microbial community structure and functionality changes in response to long-term metal and radionuclide pollution. Environ. Microbiol. 23, 1670–1683. doi: 10.1111/1462-2920.15394
Roguet, A., Laigle, G. S., Therial, C., Bressy, A., Soulignac, F., Catherine, A., et al. (2015). Neutral community model explains the bacterial community assembly in freshwater lakes. FEMS Microbiol. Ecol. 91:fiv125. doi: 10.1093/femsec/fiv125
Ryu, C. M., Farag, M. A., Hu, C. H., Reddy, M. S., Wei, H. X., Paré, P. W., et al. (2003). Bacterial volatiles promote growth in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 100, 4927–4932. doi: 10.1073/pnas.0730845100
Schmidt, S. K., Reed, S. C., Nemergut, D. R., Grandy, A. S., Cleveland, C. C., Weintraub, M. N., et al. (2008). The earliest stages of ecosystem succession in high-elevation (5000 metres above sea level), recently deglaciated soils. Proc. Biol. Sci. 275, 2793–2802. doi: 10.1098/rspb.2008.0808
She, J., Liu, J., He, H., Zhang, Q., Lin, Y., Wang, J., et al. (2022). Microbial response and adaption to thallium contamination in soil profiles. J. Hazard. Mater. 423:127080. doi: 10.1016/j.jhazmat.2021.127080
Shek, R., Dattmore, D. A., Stives, D. P., Jackson, A. L., Chatfield, C. H., Hicks, K. A., et al. (2017). Structural and functional basis for targeting campylobacter jejuni agmatine deiminase to overcome antibiotic resistance. Biochemistry 56, 6734–6742. doi: 10.1021/acs.biochem.7b00982
Shi, Y., Li, Y., Xiang, X., Sun, R., Yang, T., He, D., et al. (2018). Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China plain. Microbiome 6:27. doi: 10.1186/s40168-018-0409-4
Sonne, C., Bank, M. S., Jenssen, B. M., Cieseielski, T. M., Rinklebe, J., Lam, S. S., et al. (2023). PFAS pollution threatens ecosystems worldwide. Science 379, 887–888. doi: 10.1126/science.adh0934
Sun, W., Sun, X., Li, B., Xu, R., Young, L. Y., Dong, Y., et al. (2020). Bacterial response to sharp geochemical gradients caused by acid mine drainage intrusion in a terrace: relevance of C, N, and S cycling and metal resistance. Environ. Int. 138:105601. doi: 10.1016/j.envint.2020.105601
Sun, W., Xiao, E., Dong, Y., Tang, S., Krumins, V., Ning, Z., et al. (2016). Profiling microbial community in a watershed heavily contaminated by an active antimony (Sb) mine in Southwest China. Sci. Total Environ. 550, 297–308. doi: 10.1016/j.scitotenv.2016.01.090
Sun, W., Xiao, E., Häggblom, M., Krumins, V., Dong, Y., Sun, X., et al. (2018). Bacterial survival strategies in an alkaline tailing site and the physiological mechanisms of dominant phylotypes as revealed by metagenomic analyses. Environ. Sci. Technol. 52, 13370–13380. doi: 10.1021/acs.est.8b03853
Suryaletha, K., Narendrakumar, L., John, J., Radhakrishnan, M. P., George, S., and Thomas, S. (2019). Decoding the proteomic changes involved in the biofilm formation of Enterococcus faecalis SK460 to elucidate potential biofilm determinants. BMC Microbiol. 19:146. doi: 10.1186/s12866-019-1527-2
Tan, S., Liu, J., Fang, Y., Hedlund, B. P., Lian, Z. H., Huang, L. Y., et al. (2019). Insights into ecological role of a new deltaproteobacterial order Candidatus Acidulodesulfobacterales by metagenomics and metatranscriptomics. ISME J. 13, 2044–2057. doi: 10.1038/s41396-019-0415-y
Tandon, K., Ricci, F., Costa, J., Medina, M., Kühl, M., Blackall, L. L., et al. (2022). Genomic view of the diversity and functional role of archaea and bacteria in the skeleton of the reef-building corals Porites lutea and Isopora palifera. Gigascience 12. doi: 10.1093/gigascience/giac127
Tang, Z., Wang, H. Q., Chen, J., Chang, J. D., and Zhao, F. J. (2023). Molecular mechanisms underlying the toxicity and detoxification of trace metals and metalloids in plants. J. Integr. Plant Biol. 65, 570–593. doi: 10.1111/jipb.13440
Thomas, J. T., Oladeinde, A., Kieran, T. J., Finger, J. J., Bayona-Vásquez, N. J., Cartee, J. C., et al. (2020). Co-occurrence of antibiotic, biocide, and heavy metal resistance genes in bacteria from metal and radionuclide contaminated soils at the Savannah River site. Microb. Biotechnol. 13, 1179–1200. doi: 10.1111/1751-7915.13578
Tordoff, G. M., Baker, A. J., and Willis, A. J. (2000). Current approaches to the revegetation and reclamation of metalliferous mine wastes. Chemosphere 41, 219–228. doi: 10.1016/s0045-6535(99)00414-2
Tyc, O., Song, C., Dickschat, J. S., Vos, M., and Garbeva, P. (2017). The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol. 25, 280–292. doi: 10.1016/j.tim.2016.12.002
Uritskiy, G. V., DiRuggiero, J., and Taylor, J. (2018). MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6:158. doi: 10.1186/s40168-018-0541-1
van der Heijden, M. G., and Hartmann, M. (2016). Networking in the plant microbiome. PLoS Biol. 14:e1002378. doi: 10.1371/journal.pbio.1002378
Ventura, M., Canchaya, C., Tauch, A., Chandra, G., Fitzgerald, G. F., Chater, K. F., et al. (2007). Genomics of actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 71, 495–548. doi: 10.1128/MMBR.00005-07
Vetsigian, K., Jajoo, R., and Kishony, R. (2011). Structure and evolution of streptomyces interaction networks in soil and in silico. PLoS Biol. 9:e1001184. doi: 10.1371/journal.pbio.1001184
Wang, Q., Han, Y., Shi, K., Fan, X., Wang, L., Li, M., et al. (2017). An oxidoreductase AioE is responsible for bacterial arsenite oxidation and resistance. Sci. Rep. 7:41536. doi: 10.1038/srep41536
Wang, H., Wei, Z., Mei, L., Gu, J., Yin, S., Faust, K., et al. (2017). Combined use of network inference tools identifies ecologically meaningful bacterial associations in a paddy soil. Soil Biol. Biochem. 105, 227–235. doi: 10.1016/j.soilbio.2016.11.029
Wu, Y., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Xu, R., Sun, X., Häggblom, M. M., Dong, Y., Zhang, M., Yang, Z., et al. (2022). Metabolic potentials of members of the class Acidobacteriia in metal-contaminated soils revealed by metagenomic analysis. Environ. Microbiol. 24, 803–818. doi: 10.1111/1462-2920.15612
Yan, Y., Kuramae, E. E., de Hollander, M., Klinkhamer, P. G., and van Veen, J. A. (2017). Functional traits dominate the diversity-related selection of bacterial communities in the rhizosphere. ISME J. 11, 56–66. doi: 10.1038/ismej.2016.108
Zhang, M., Li, Z., Häggblom, M. M., Young, L., He, Z., Li, F., et al. (2020). Characterization of nitrate-dependent as(III)-oxidizing communities in arsenic-contaminated soil and investigation of their metabolic potentials by the combination of DNA-stable isotope probing and metagenomics. Environ. Sci. Technol. 54, 7366–7377. doi: 10.1021/acs.est.0c01601
Zhang, M., Zhang, T., Zhou, L., Lou, W., Zeng, W., Liu, T., et al. (2022). Soil microbial community assembly model in response to heavy metal pollution. Environ. Res. 213:113576. doi: 10.1016/j.envres.2022.113576
Zheng, Z., Li, Y., Zhang, X., Liu, P., Ren, J., Wu, G., et al. (2015). A Bacillus subtilis strain can reduce hexavalent chromium to trivalent and an nfrA gene is involved. Int. Biodeterior. Biodegradation 97, 90–96. doi: 10.1016/j.ibiod.2014.10.017
Zhou, M., Guo, J., Cha, J., Chae, M., Chen, S., Barral, J. M., et al. (2013). Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495, 111–115. doi: 10.1038/nature11833
Zhu, X., Yao, J., Wang, F., Yuan, Z., Liu, J., Jordan, G., et al. (2018). Combined effects of antimony and sodium diethyldithiocarbamate on soil microbial activity and speciation change of heavy metals. Implications for contaminated lands hazardous material pollution in nonferrous metal mining areas. J. Hazard. Mater. 349, 160–167. doi: 10.1016/j.jhazmat.2018.01.044
Keywords: heavy metal resistance, metagenome-assembled genomes, contaminated soils, keystone taxa, metagenomics
Citation: Li L, Meng D, Yin H, Zhang T and Liu Y (2023) Genome-resolved metagenomics provides insights into the ecological roles of the keystone taxa in heavy-metal-contaminated soils. Front. Microbiol. 14:1203164. doi: 10.3389/fmicb.2023.1203164
Edited by:
Zhimin Xu, Zhongkai University of Agriculture and Engineering, ChinaReviewed by:
Shihong Guo, Fujian Provinical Academy Environmental Sciences, ChinaCopyright © 2023 Li, Meng, Yin, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teng Zhang, Njc1MTA0Nzc4QHFxLmNvbQ==; Yongjun Liu, VmluY2VudGxpdTIwMjBAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.