 Lingli Min1
Lingli Min1 Aharon Oren
Aharon Oren Qiliang Lai
Qiliang Lai Zhaobin Huang
Zhaobin Huang- 1School of Resources and Environmental Sciences, Quanzhou Normal University, Quanzhou, China
- 2College of Oceanology and Food Science, Quanzhou Normal University, Quanzhou, China
- 3The Institute of Life Sciences, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Jerusalem, Israel
- 4Key Laboratory of Marine Genetic Resources, Third Institute of Oceanography, Ministry of Natural Resources, Xiamen, China
- 5Fujian Province Key Laboratory for the Development of Bioactive Material from Marine Algae, Quanzhou, China
Members of the class Opitutae are widely distributed in various environments such as rice paddy soil, freshwater lakes, seawater, marine sediment, and invertebrate digestive tracts. The class currently consists of two orders, Opitutales and Puniceicoccales, represented by the families Opitutaceae and Puniceicoccaceae, respectively, which are primarily delineated on the basis of 16S rRNA gene sequences and limited phenotypic characterizations of a few type strains. The scarcity of 16S rRNA gene and genome sequences generated from the type strains of the class Opitutae constrained our understanding of the ecological distribution and adequate resolution of its taxonomy. Here, an Opitutae strain designated WMMB3T, isolated from a mangrove sediment, was subjected to taxonomic characterization. The 16S rRNA gene of strain WMMB3T shared high sequence similarities with Coraliomargarita akajimensis DSM 45221T and C. sinensis WN38T of 96.1 and 95.9%, respectively. Phylogenetic analysis suggested that strain WMMB3T formed a monophyletic branch affiliated to the genus Coraliomargarita. The average nucleotide identity (ANI) values, digital DNA–DNA hybridization (dDDH) values and average amino acid identity (AAI) values of strain WMMB3T compared between Coraliomargarita members were 71.8–72.5, 20.7, and 68.2–68.7%, respectively, indicating that strain WMMB3T represented a novel species of Coraliomargarita. The genome of strain WMMB3T was 4.5 Mbp with a DNA G + C content of 56.0%. The respiratory quinone was menaquinone-7. The major fatty acids were iso-C14:0, and C18:1ω9c. Based on genomic, phenotypic, and chemotaxonomic characterizations, strain WMMB3T represents a novel species, and Coraliomargarita parva sp. nov. is proposed. Additionally, the phylogenomic analysis of more than 500 genomes of the class Opitutae, encompassing a majority of uncultivated bacteria and a few type strains, was performed using the Genome Taxonomic Database toolkit (GTDB-Tk) to present adequate resolution of the taxonomy. Combined with 16S rRNA gene sequence phylogeny and genomic relatedness, five novel families retrieved mainly from marine habitats were proposed: Coraliomargaritaceae fam. nov., Pelagicoccaceae fam. nov., Cerasicoccaeae fam. nov., Oceanipulchritudinaceae fam. nov., and Alterococcaeae fam. nov. AAI values of 58–60% could be considered as the boundary to delineate families of the class Opitutae. This study provided a new taxonomic framework of the class Opitutae based on the genomic data.
1. Introduction
The class Opitutae within the phylum Verrucomicrobiota is widely distributed in various environments, including rice paddy soil, freshwater lakes, seawater, marine sediment, and digestive tracts of invertebrate hosts, such as marine clamworms, ciliates, and sea cucumbers (Choo et al., 2007). The class currently comprises two orders, Opitutales and Puniceicoccales, represented by the families Opitutaceae and Puniceicoccaceae, respectively, primarily classified based on the phylogeny of 16S rRNA gene sequences. Members of the family Opitutaceae were mainly retrieved from soil and terrestrial habitats, while those of the family Puniceicoccaceae were derived from marine environments (Choo et al., 2007). Nearly 20 species with validly published names were described in the class Opitutae (Parte et al., 2020). The scarcity of the 16S rRNA gene sequences and genome sequences generated from the type strains of the class Opitutae constrains our understanding of its taxonomy.
Coraliomargarita, a genus of the family Puniceicoccaceae, order Puniceicoccales, is described on the basis of 16S rRNA gene phylogeny and physiological and chemotaxonomic characteristics (Yoon et al., 2007b). Until now, this genus includes two species with validly published names, Coraliomargarita akajimensis (Yoon et al., 2007b) and Coraliomargarita sinensis (Zhou et al., 2019). Cells of Coraliomargarita are Gram-stain-negative, obligately aerobic, coccus-shaped, non-motile, and oxidase- and catalase- positive (Yoon et al., 2007b). The major respiratory quinone is menaquinone-7 (MK-7). The genomic DNA G + C contents calculated from genome sequences are 53.6–54.7%. The predominant cellular fatty acids are C14:0, C18:1 ω9c, and C18:0 (Yoon et al., 2007b; Zhou et al., 2019). The 16S rRNA gene sequences of Coraliomargarita showed low sequence similarities of 88.2–89.2% with Puniceicoccus vermicola IMCC1545T, which made us question the taxonomic placement of Coraliomargarita into the family Puniceicoccaceae.
In this study, a novel strain designated WMMB3T, isolated from a mangrove sediment, was found to represent a novel species of the genus Coraliomargarita. This study aimed to determine the taxonomic status of strain WMMB3T using a polyphasic taxonomic approach. Additionally, the phylogeny of the class Opitutae members was investigated based on the currently available genomes including a majority of uncultivated bacteria obtained using metagenomic assembled genomes (MAGs) and single cell genomes (SAGs) and a few type strains. This study provided new insights into the taxonomy of the class Opitutae.
2. Materials and methods
2.1. Strain isolation and cultivation
Strain WMMB3T was isolated from a mangrove sediment, collected from a mangrove preservation area (E 118.699o, N 24.937o) in Quanzhou on Sep. 15, 2022. About 1 g of sediment was added to 9 mL sterile natural seawater and vigorously shaken to make a suspension. Serial 10-fold dilutions were spread on 2216E culture plates (5 g/L peptone, 1 g/L yeast extract, 15 g/L agar, 1 L natural seawater). The plates were maintained at 30°C for 14 days. Strain WMMB3T was picked and streaked twice on Marine Broth 2216 (MB, BD) agar plates to obtain a pure culture. The strain was routinely cultured in MB and on MB agar plates at 30°C, and stored at -80°C in 20% glycerol (v/v). Strain WMMB3T was deposited in the Marine Culture Collection of China (MCCC 1K08426T) and the Korean Collection for Type Cultures (KCTC 92914T).
For comparative purpose, the two type strains of species of the genus Coraliomargarita, C. akajimensis DSM 45221T (=04OKA010-24T = MCCC 1A12044T) and C. sinensis WN38T (=KCTC 62602T = MCCC 1H00313T) were used as reference strains.
2.2. Phylogenetic analysis of 16S rRNA gene sequences
The genomic DNA of strain WMMB3T was extracted using a bacterial genomic DNA extraction kit (Saibaisheng, Shanghai, China). The 16S rRNA gene of strain WMMB3T was PCR-amplified in a 50 μL amplification system (Ex Taq, TaKaRa) using primers Eub27F and 1492R (DeLong, 1992). The PCR product of ~1.5 kb was detected using 1.5% agarose gel electrophoresis, and the sequence was determined by using an ABI 3730 sequencer (Sanger sequencing).
The closest type strains of strain WMMB3T were identified by using the EzBioCloud (Yoon et al., 2017a), nr database in GenBank1 and SILVA 132 database (Yilmaz et al., 2014). The 16S rRNA gene sequences were aligned using ClustalW, and neighbor-joining and maximum-likelihood phylogenetic trees were reconstructed using MEGA 7.0 (Kumar et al., 2016). The models used in neighbor-joining (NJ) and maximum-likelihood (ML) were maximum composite likelihood (MCL) and K2 + G + I, respectively. The topology was evaluated based on 1,000 bootstrap replicates for the NJ and ML methods.
2.3. Genome sequencing and annotation
The draft genome sequence of strain WMMB3T was determined using the Illumina Hiseq platform following the manufacturer’s instruction (Shanghai Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China). Paired-end reads (PE reads) of 2 × 151 bp were quality checked (q > 20 and length > 50 bp) using sickle,2 and assembled into contigs using SPAdes v. 3.8.0 (Bankevich et al., 2012) with a serial of k values (21, 33, 55, 77, 99, 127) and -careful flag. The assembled contigs with <1 kbp were removed from the following analysis. The prediction of functional genes was performed using prodigal (Hyatt et al., 2010), and also annotated using the RAST server (Aziz et al., 2008).
2.4. Phylogenomic analysis
The genomes of type strains affiliated to the class Opitutae were used to reconstruct a phylogenomic tree using GTDB-Tk v1.3.0 (Parks et al., 2018) with 120 conserved concatenated proteins referred to as the Bac120 set. The tree was visualized using the Interactive Tree of Life (iTOL) online (Letunic and Bork, 2007).
2.5. Genomic relatedness
The average nucleotide identity (ANI) values were calculated using ANI Calculator (Yoon et al., 2017b). The digital DNA–DNA hybridization (dDDH) values were calculated using the online Genome-to-Genome Distance Calculator (GGDC, version 3.0) (Meier-Kolthoff et al., 2013). The average amino acid identity (AAI) values were calculated using CompareM v0.1.2.3
2.6. Characterization of cell morphology and physiology
Gram staining of strain WMMB3T was conducted by using a Gram staining kit (Solarbio Co, Beijing). Catalase activity was tested by using 3% (v/v) H2O2 solution. Oxidase activity was tested by using an oxidase reagent (N,N,N,N-tetramethyl-p-phenylenediamine dihydrochloride, bioMérieux, France). To observe the growth temperature range, strains were streaked on MB agar plates and maintained at various temperatures (10, 15, 20, 25, 30, 35, 40, and 45°C) for 7 days. The ability of degrading substrates, including soluble starch (1%, w/v), skimmed milk (1%, w/v), cellulose (1%, w/v), Tween 20 (1%, v/v), Tween 40 (1%, v/v), Tween 60 (1%, v/v), and Tween 80 (1%, v/v) were tested by streaking the strains onto the MB agar plates with each substrate (Huang et al., 2021). Additional physiological and biochemical characteristics were tested by using three API strips, including API ZYM, API 20NE and API 20E (bioMérieux product, France) according to the manufacturer’s instructions.
2.7. Chemotaxonomic characteristics
For cellular fatty acids composition analysis, strain WMMB3T and reference strains were cultured in MB at 30°C for 5 days and cells were harvested by centrifugation at 8,000 rpm. The cellular fatty acids were extracted and identified by gas chromatography following the standard MIDI protocol (Sherlock Microbial Identification System, version 6B). The respiratory quinone of strain WMMB3T was extracted using chloroform/methanol (2:1, v/v), and identified and quantified using reversed phase high-performance liquid chromatography as described previously (Komagata and Suzuki, 1987).
2.8. Phylogenomic analysis of the class Opitutae
The genomes affiliated to the class Opitutae (until Nov. 17. 2022) were downloaded from the genome portal in GenBank.4 The quality of these genomes was evaluated using CheckM v1.0.12 (Parks et al., 2015), and the genomes with > = 90% completeness and < =5% contamination were maintained for the subsequent phylogenomic study (Supplementary Table S1). A phylogenomic tree of the high-quality genomes was inferred using a concatenated alignment of Bac120 with GTDB-Tk v. 1.3.0 by using FastTree (Parks et al., 2018). The tree was visualized using the iTOL online (Letunic and Bork, 2007).
2.9. Phylogeny of 16S rRNA gene sequences of the class Opitutae
The 16S rRNA gene sequences were identified and extracted from all downloaded Opitutae genomes using RNAmmer with default parameters (Lagesen et al., 2007). The 16S sequences exhibiting >1,200 bp in length, < 8 polymers and no ambiguous bases were selected by using screen.seqs command in mothur (Schloss et al., 2009). The representative sequences were identified by using RDP Classifier (Wang et al., 2007) and aligned using ClustalW. Phylogenetic trees of 16S rRNA gene sequences were constructed using NJ and ML in MEGA 7.0 (Kumar et al., 2016). The trees were finally visualized using the Interactive Tree of Life (iTOL) online (Letunic and Bork, 2007).
3. Results and discussion
3.1. Phylogeny of the 16S rRNA gene
Phylogeny of the 16S rRNA gene sequences showed that strain WMMB3T belonged to the genus Coraliomargarita of the class Opitutae, and formed a monophyletic branch with C. akajimensis DSM 45221T and C. sinensis WN38T (Figure 1; Supplementary Figure S1), sharing sequence similarities of 96.1 and 95.9%, respectively. The 16S rRNA gene of strain WMMB3T had <88.3% sequence similarity with Puniceicoccus vermicola IMCC1545T and the other species with validly published names within the class Opitutae. The low sequence similarity of its 16S rRNA gene with its closest relatives demonstrated that strain WMMB3T may represent a novel species.
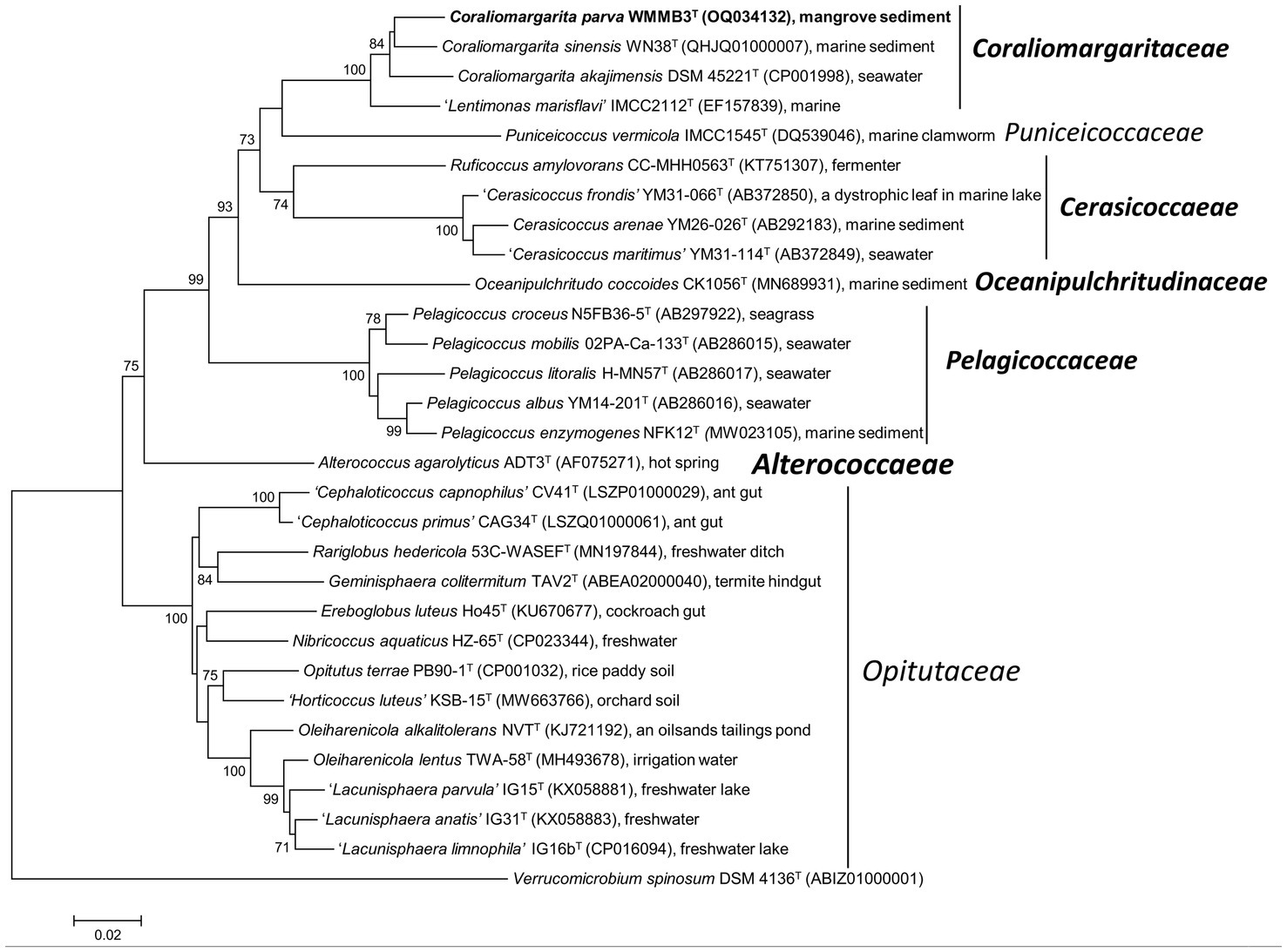
Figure 1. Neighbor joining phylogenetic tree of strain WMMB3T and its close relatives constructed based on 16S rRNA gene sequences. Coraliomargarita parva WMMB3T is marked bold. Bootstrapping was carried out with 1,000 replicates. Node values below 70% are not shown. Verrucomicrobium spinosum DSM 4136T was selected as the outgroup. Bar, 0.02 means the nucleotide substitution per position. The newly described families are marked bold.
Phylogeny of 16S rRNA gene of the type strains also revealed that nine genus-level members formed a highly supported clade (bootstrap values of 99–100%), belonging to the family Opitutaceae (Figure 1; Supplementary Figure S1). They were Opitutus (type genus), ‘Cephaloticoccus’, Rariglobus, Geminisphaera, Ereboglobus, Nibricoccus, “Horticoccus”, Oleiharenicola, and “Lacunisphaera”. These members are mainly terrestrial in origin, for instance, soil, freshwater lakes and the guts of terrestrial invertebrates (Figure 1). Alterococcus agarolyticus ADT3T, currently classified in the family Opitutaceae (Yoon et al., 2017a; Parte et al., 2020), formed a distant branch with Optitutus and other members of the family Opitutaceae, and could therefore be assigned to a novel family. Thus, a new family Alterococcaeae fam. nov. is proposed to accommodate this genus Alterococcus. A. agarolyticus ADT3T is a halophilic thermophilic bacterium, isolated from an intertidal hot spring (Shieh and Jean, 1998), a habitat different from that of the other members of the family Opitutaceae. The 16S rRNA gene of A. agarolyticus ADT3T shared 88.3 and 84.5% sequence similarity with Opitutus terrae PB90-1T and P. vermicola IMCC1545T, respectively.
Seven other genera, including Pelagicoccus, Oceanipulchritudo, Cerasicoccus, Ruficoccus, Puniceicoccus (type genus), Coraliomargarita, and “Lentimonas” grouped into a coherent clade. Members of these seven genera are primarily marine in origin (Figure 1). However, these groups are need to be re-classified into different families based on phylogenomic analysis of the genome sequence and genomic relatedness described below.
3.2. Genome-based analysis
The draft genome size of strain WMMB3T was 4,497,538 bp on 27 assembled contigs (>1 kb in length), which was larger than that of C. akajimensis DSM 45221T with 3,750,771 bp and that of C. sinensis WN38T with 3,569,655 bp. The genomic DNA G + C content of strain WMMB3T was 56.0% (Table 1), which was also higher than C. akajimensis DSM 45221T (53.6%) and C. sinensis WN38T (54.7%).
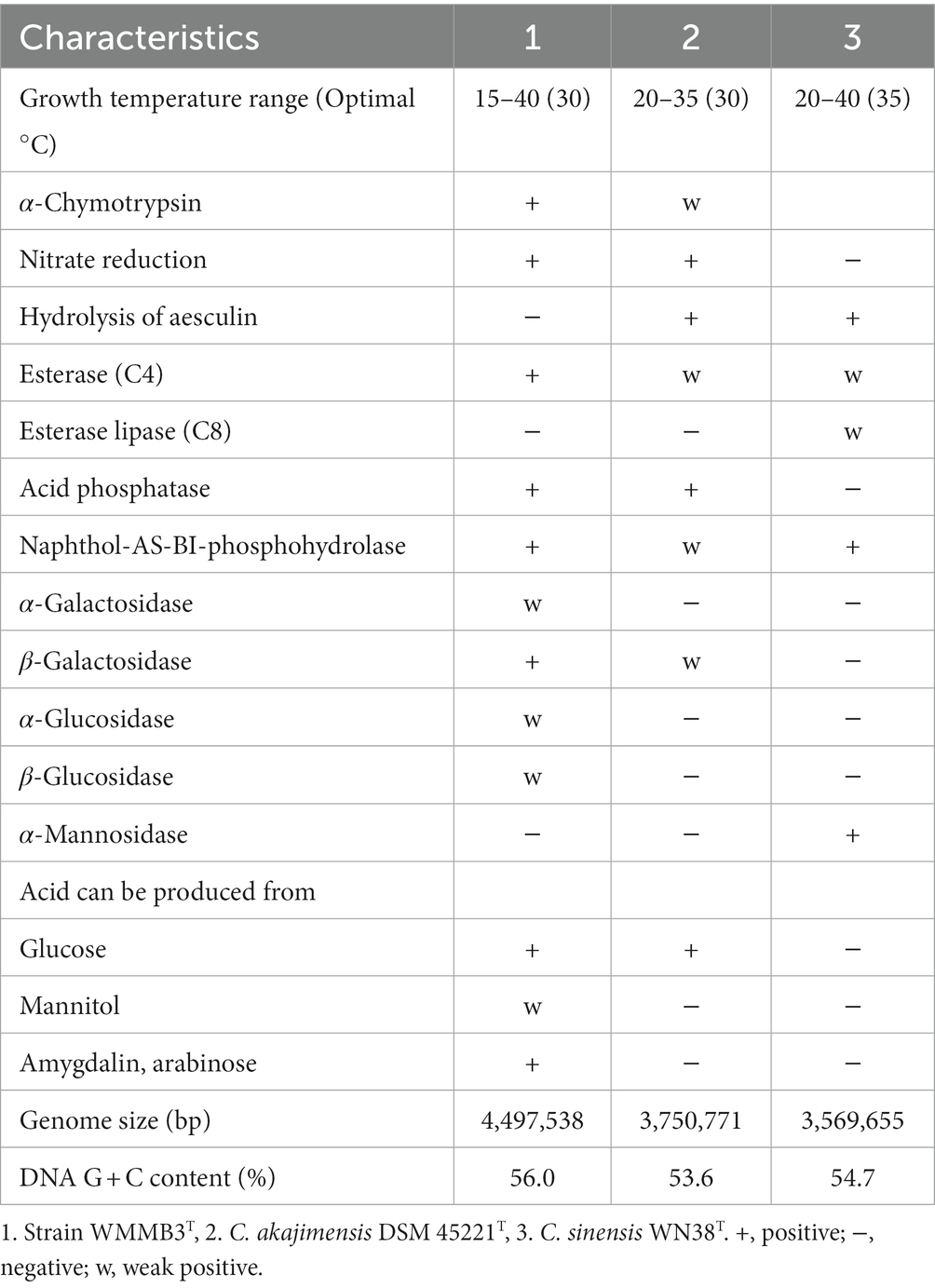
Table 1. Differential characteristics of strain WMMB3T compared to close relatives of the genus Coraliomargarita.
The average nucleotide identity (ANI) values of WMMB3T compared against C. akajimensis DSM 45221T and C. sinensis WN38T were 71.8 and 72.5%, respectively. The digital DNA–DNA hybridization (dDDH) values of WMMB3T against C. akajimensis DSM 45221T and C. sinensis WN38T were both 20.7%. These values were below the threshold values of prokaryotic species definition (95–96% ANI and 70% DDH) (Richter and Rosselló-Móra, 2009), which strongly supported that strain WMMB3T belonged to a novel species. The average amino acid identity (AAI) values of strain WMMB3T against C. akajimensis DSM 45221T and C. sinensis WN38T were 68.2 and 68.7%, respectively, which exceeded the cutoff of the same genus (65–95% AAI) (Konstantinidis et al., 2017), suggesting that WMMB3T belonged to the genus Coraliomargarita.
The AAI values calculated among the 10 species of the family Opitutaceae were 59.9–79.1% (Figure 2). The AAI values of the Pelagicoccus species and the Coraliomargarita species were 73.2–74.5% and 67.5–69.0%, respectively, and were clearly separated from other members of the class Opitutae. The two genera could be considered two families, as also supported by phylogenetic analysis. Cerasicoccus arenae KCTC 12870T had 60.1% AAI with Ruficoccus amylovorans JCM 31066T, which could be united into the same family. The above four genera had <60% AAI with P. vermicola JCM14086T and Oceanipulchritudo coccoides CK1056T. Thus, AAI value of 60% could be regarded the threshold to differentiate these genera. The boundary of AAI values to divide these species into two orders Opitutales and Puniceicoccales is not distinct. To be congruent with GTDB taxonomy (Chaumeil et al., 2019), we suggested to unite the two orders.
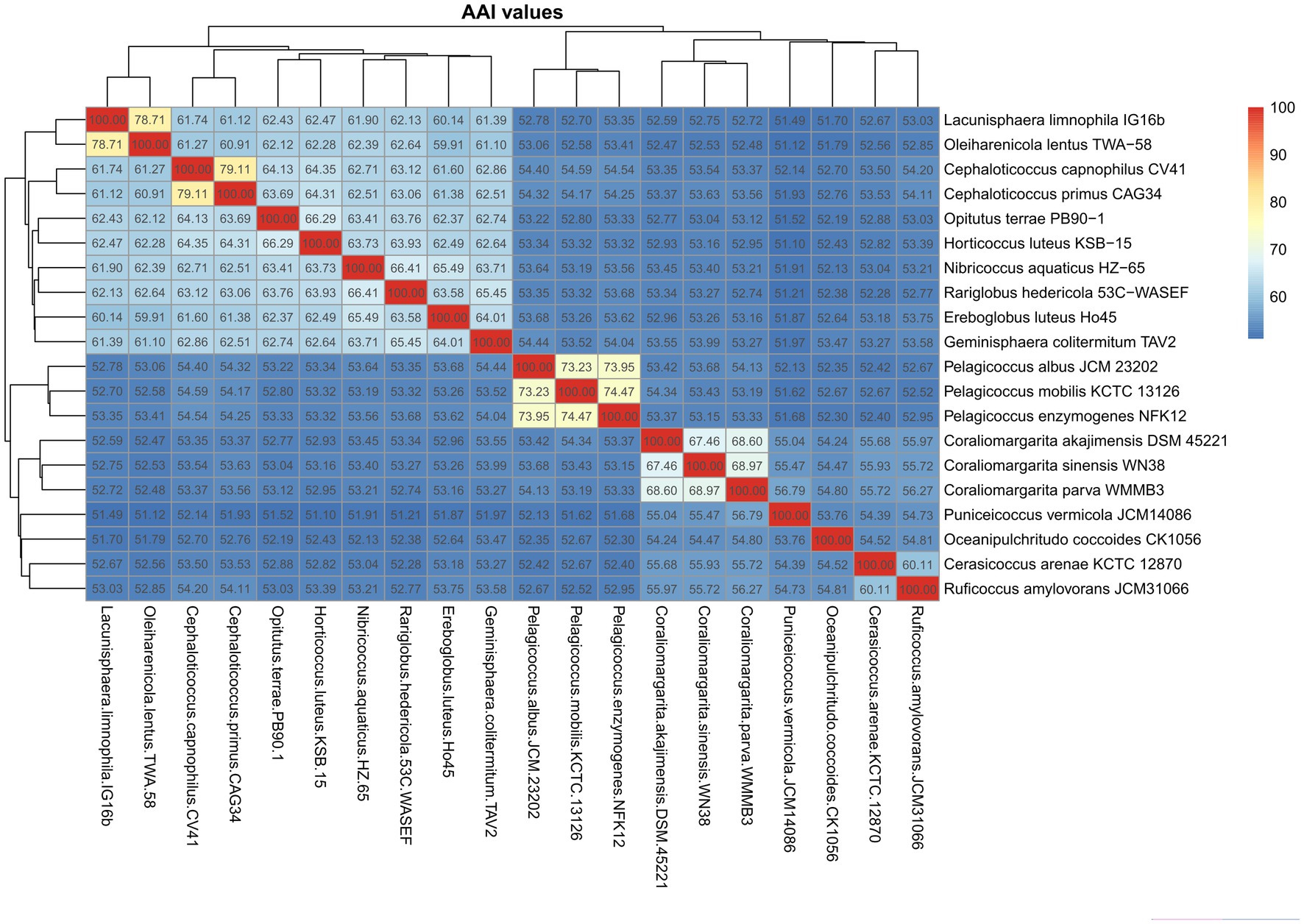
Figure 2. Heatmap showing the AAI values calculated among the species of the class Opitutae.
3.3. Phylogenomic analysis
A phylogenomic tree including the type strains affiliated to the class Opitutae was constructed using GTDB-Tk v.1.3.0 (Figure 3), a powerful tool to resolve the taxonomic position of the bacterial and archaeal genomes based on the Bac120 set (Chaumeil et al., 2019). The phylogenomic tree showed that strain WMMB3T formed a highly supported and monophyletic clade with C. akajimensis DSM 45221T and C. sinensis WN38T, supporting that strain WMMB3T represented a novel species within the genus Coraliomargarita (Figure 3). Moreover, we found that Pelagicoccus members formed a distinct group, which should be regarded as a novel family, but not as a member in the family Puniceicoccaceae (Yoon et al., 2017a; Parte et al., 2020). Thus, Pelagicoccaceae fam. nov. was proposed to accommodate the genus Pelagicoccus. Members of Pelagicoccus were found in seawater, marine sediment and seagrass-associated environments (Figure 1).
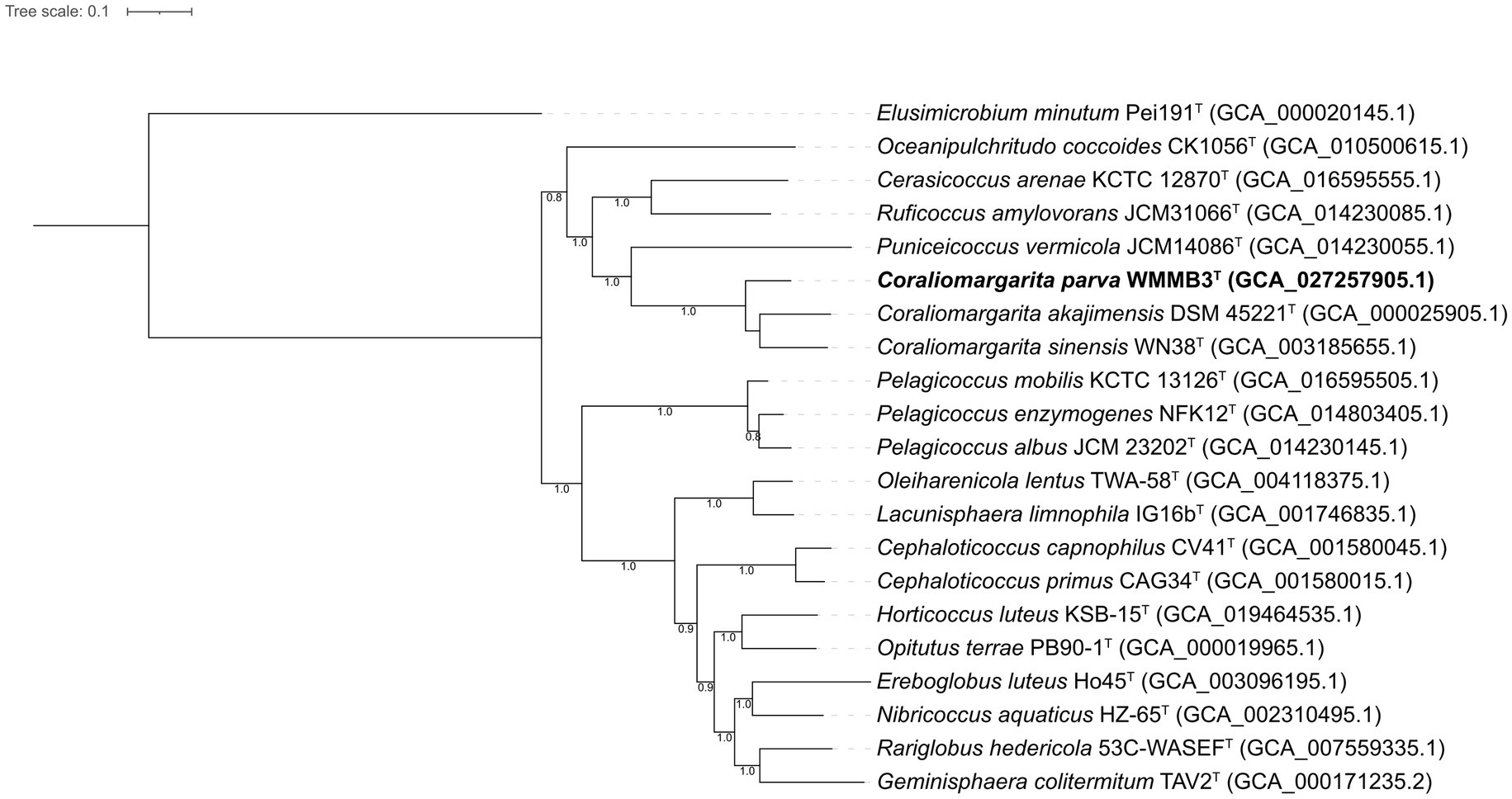
Figure 3. Phylogenomic tree constructed based on 120 bacterial conserved concatenated proteins showing the relationship between strain WMMB3T and the type strains of the class Opitutae. The tree was rooted by Elusimicrobium minutum Pei191T. Coraliomargarita parva WMMB3T is marked bold. The bootstrap values of nodes greater than 0.7 are displayed. Bar, 0.1 means the amino acid substitutions per position.
3.4. Morphological and physiological characteristics
Colonies of strain WMMB3T on agar plates cultured at 30°C for 5 days were small and round. Cells were Gram-stain-negative, aerobic, and coccus-shaped. Oxidase and catalase activities were positive, as for C. akajimensis DSM 45221T and C. sinensis WN38T. The growth temperature range of strain WMMB3T was 15–40°C with optimal temperature of 30°C (Table 1). Growth was not observed at 10 and 45°C.
Hydrolysis of Tween 20, Tween 40, Tween 60 and Tween 80 was not observed for strain WMMB3T, as for C. akajimensis DSM 45221T and C. sinensis WN38T. Strain WMMB3T cannot degrade soluble starch, CMC, and skimmed milk, similar to C. akajimensis DSM 45221T and C. sinensis WN38T. Nitrate can be reduced to nitrite for strain WMMB3T and C. akajimensis DSM 45221T, but different from C. sinensis WN38T (Table 1 and species description). Production of indole is positive and hydrolysis of gelatin is negative. The other phenotypic characteristics are summarized in the species description.
3.5. Chemotaxonomic characteristics
The sole respiratory quinone of strain WMMB3T was menaquinone 7 (MK-7), similar to C. akajimensis DSM 45221T (Yoon et al., 2007b) and C. sinensis WN38T (Zhou et al., 2019). The major fatty acids (>10%) of strain WMMB3T were iso-C14:0 (23.1%), and C18:1 ω9c (19.0%), similar to C. sinensis WN38T and C. akajimensis DSM 45221T. However, there were differences in the relative abundance of some fatty acids (Table 2).
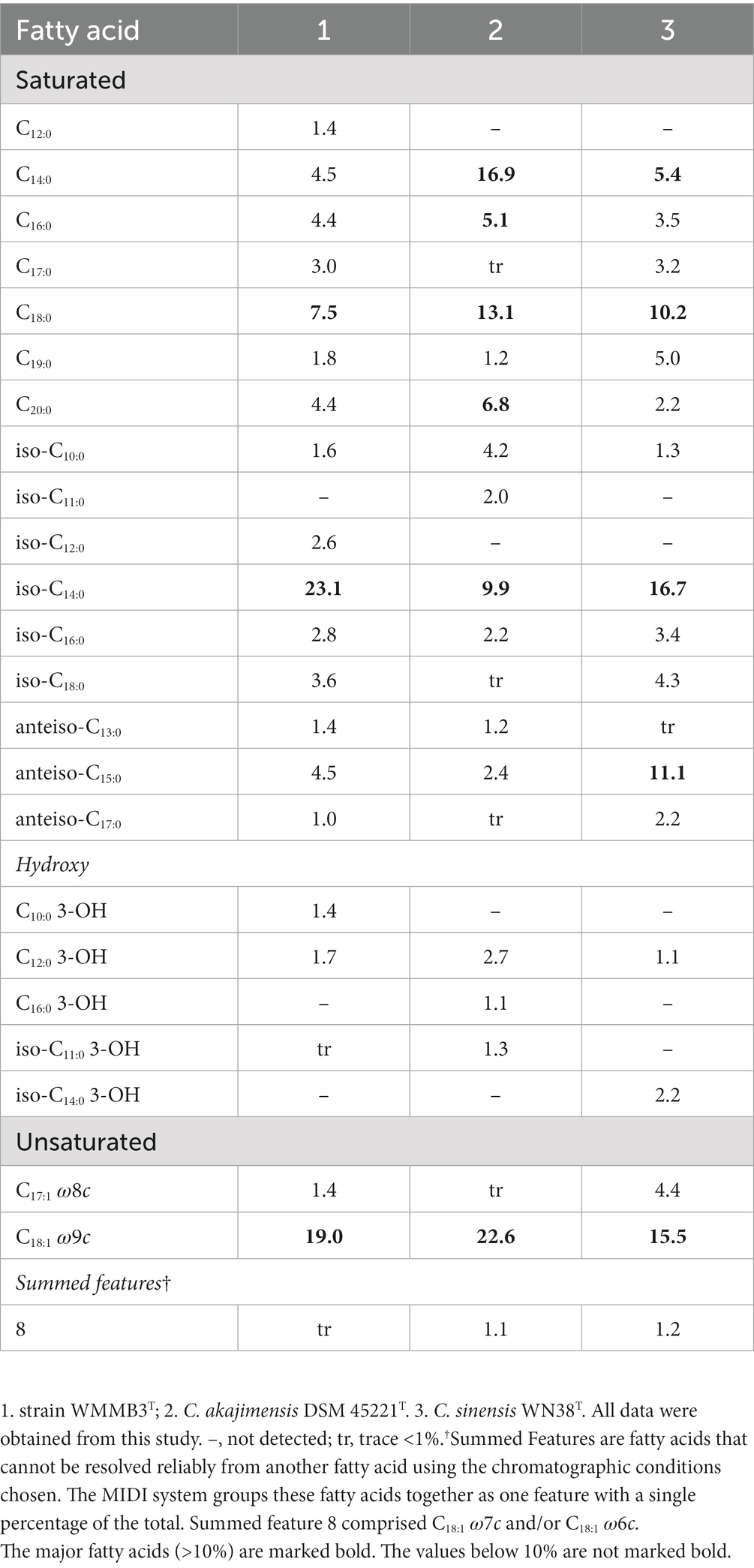
Table 2. Cellular fatty acid composition of strain WMMB3T compared to its close relatives of the genus Coraliomargarita.
3.6. Phylogenomic analysis of Opitutae genomes
A total of 1,105 genomes affiliated to the class Opitutae were downloaded from the Genome portal of GenBank (Supplementary Table S1). Genomes with ≥90% completeness and ≤ 5% contamination were used, which were verified to perform accurate phylogenetic analysis. The phylogenomic tree including a total of 546 genomes that met the above standards was inferred by using GTDB-Tk (Bowers et al., 2017; Figure 4; Supplementary Figure S2).
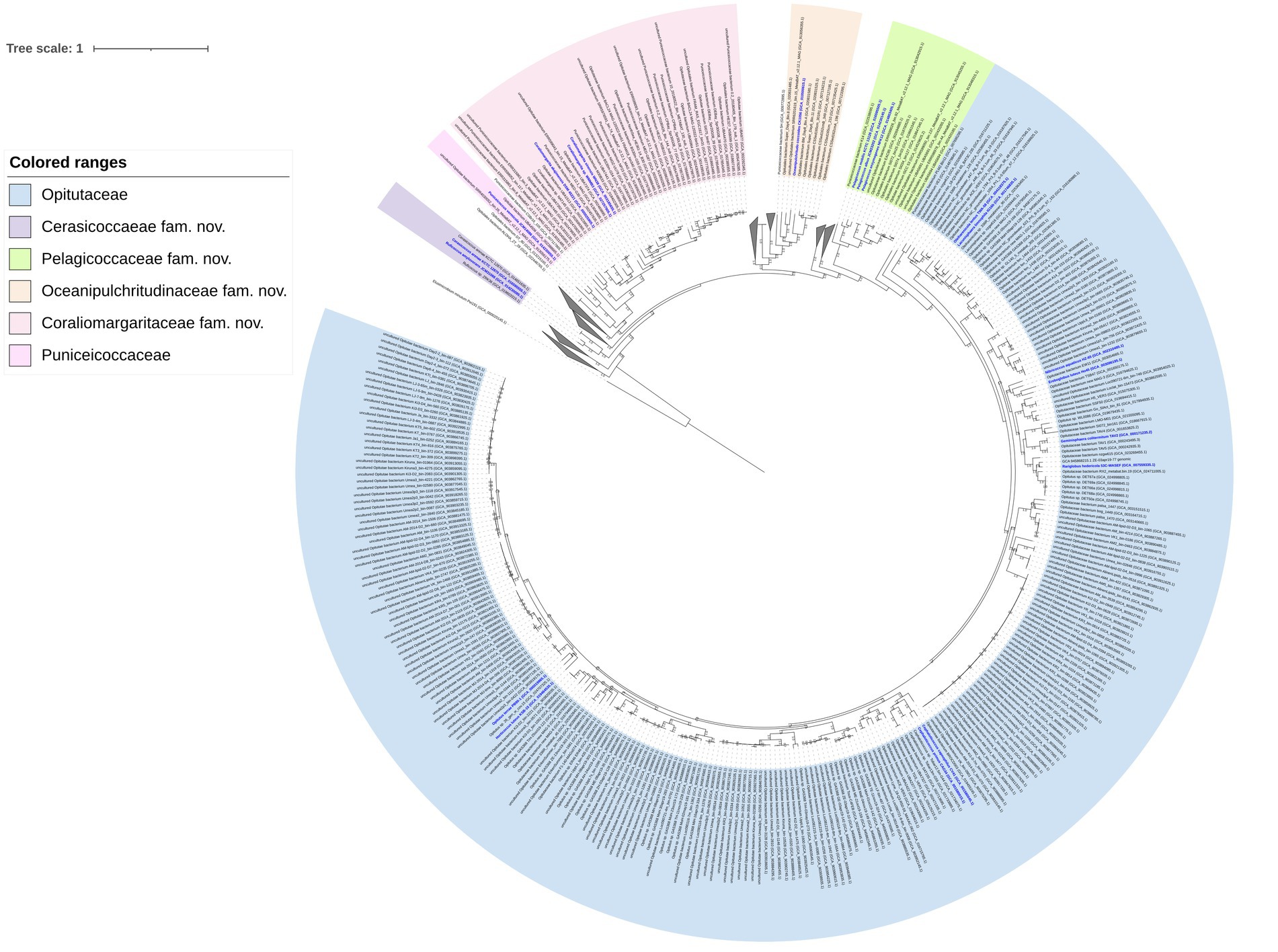
Figure 4. Phylogenomic tree constructed based on 120 bacterial conserved concatenated proteins showing the relationship of the 546 genomes affiliated to the class Opitutae. The tree was rooted by Elusimicrobium minutum Pei191T. The bootstrap values of the node greater than 0.7 are displayed. Bar, 1 means the amino acid substitutions per position. Type strains are marked blue. The clades without cultivated representatives were collapsed.
Strain WMMB3 was placed in the lineage including C. sinensis WN38T and C. akajimensis DSM 45221T. This lineage containing Coraliomargarita should be considered as a novel family of the class Opitutae. Thus, Coraliomargaritaceae fam. nov. was proposed. The AAI values of the Coraliomargaritaceae members were 59.7–100%. The genome size of Coraliomargaritaceae members was 1.2–4.7 Mbp with a genomic G + C content of 38.5–56.4%.
Cerasicoccus arenae KCTC 12870T, Ruficoccus amylovorans JCM 31066T and Ruficoccus sp. ZRK36 formed a deeply branched clade, which should be recognized as a novel family (Figure 3). The AAI values among the three strains were 59.3–78.9%, demonstrating they belonged to two different genera of the same family (Konstantinidis et al., 2017). Thus, a novel family named Cerasicoccaceae fam. nov. was proposed to accommodate the genera Cerasicoccus and Ruficoccus. The genome size was 3.8–4.5 Mbp with genomic G + C content of 52.6–60.3%. Members were found in marine sediments, seawater, deep-sea cold seeps and liquid fertilizer of a fermenter in a greenhouse facility (Figure 1).
Puniceicoccus vermicola JCM14086T and two uncultivated bacterial genomes formed a tight clade, constituting the members of the family Puniceicoccaceae. The genome size of this family was 2.6–5.2 Mbp with genomic G + C content of 54.3–68.6%. The AAI values computed with the three genomes were 61.2–75.1%.
Oceanipulchritudo coccoides CK1506T grouping with eight uncultivated bacterial genomes formed a monophyletic clade, which was distant from the family Puniceicoccaceae. Oceanipulchritudo coccoides should be classified in a novel family, and thus, Oceanipulchritudinaceae fam. nov. was proposed. The AAI values computed with the uncultivated genomes were 58.4–99.3%. The genome size of members of the Oceanipulchritudinaceae was 3.5–5.3 Mbp with genomic G + C content of 54.0–63.8%.
Pelagicoccus, currently including five species with validly published names (Parte et al., 2020), also represented a novel family, which was confirmed by phylogenomic analysis based on a few type strains (Figure 3). The genome size was 3.1–7.5 Mbp with genomic G + C content of 46.1–56.8%. The AAI values of Pelagicoccaceae genomes including uncultivated bacteria were 60.5–99.9%.
The differential characteristics of the novel families compared to Opitutaceae and Puniceicoccaceae were summarized in Table 3. A large number of clades (collapsed lineages) lacking species with validly published names or cultivated strains may represent novel families of the class Opitutae. The bacteria in these clades are waiting cultivation, taxonomic characterization and naming.
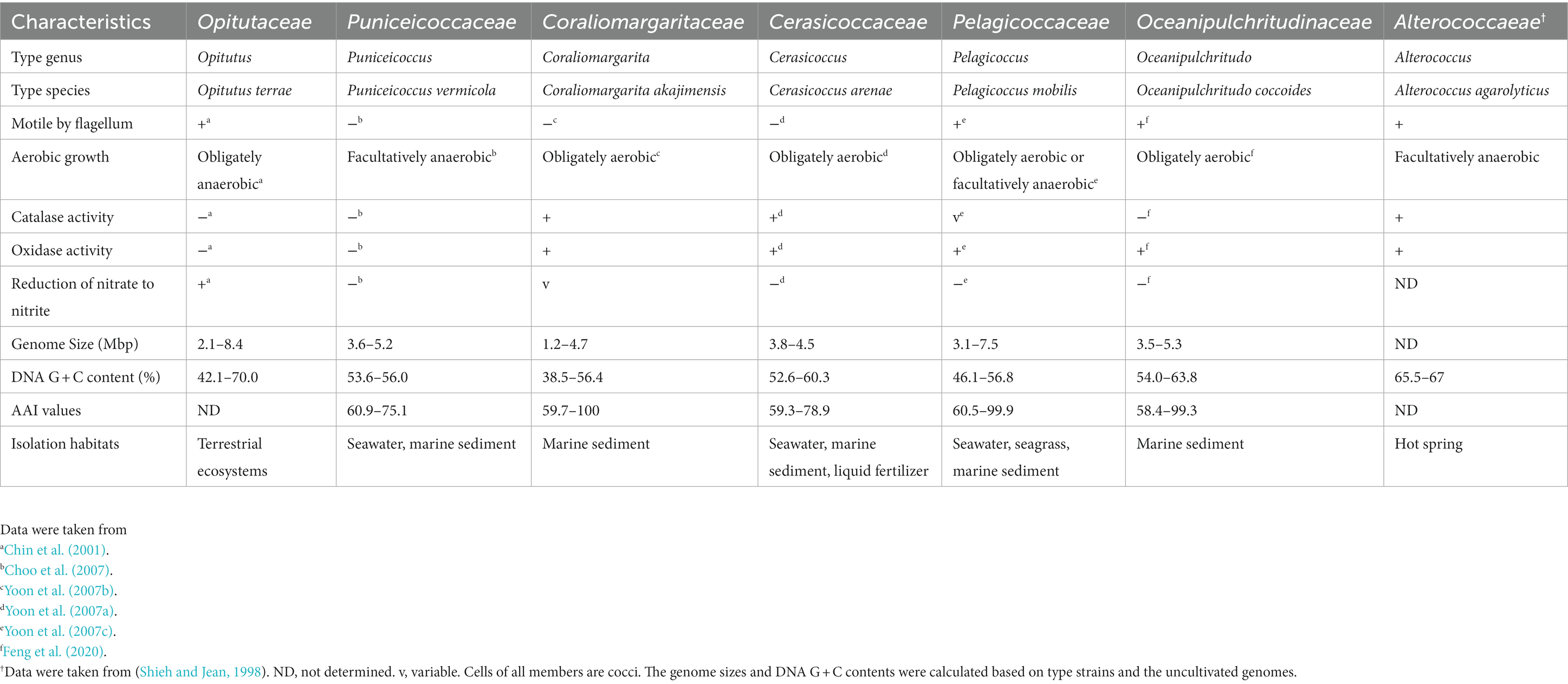
Table 3. Differential characteristics compared among the members in the class Opitutae.
3.7. Phylogenetic analysis based on the 16S rRNA gene of Opitutae
A total of 156 high-quality 16S rRNA gene sequences with > = 1,200 bp in length were retrieved from 1,105 Opitutae genomes. The 16S rRNA sequences were not frequently extracted from MAGs or SAGs, resulting in a smaller dataset than the genomic data used in phylogenomic tree. The phylogeny of 16S rRNA gene sequences revealed similar topology (Figure 5) with the phylogenomic analysis based on a few type strains (Figure 3) and uncultivated bacteria (Figure 4). Pelagicoccus is a phylogenetically distant group from the members of the family Puniceicoccaceae and represented a novel family (Figure 5). Similar to the genus Pelagicoccus, O. coccoides is phylogenetically distant from other members of the class Opitutae, and should be classified in a novel family. C. arenae KCTC 12870T and R. amylovorans JCM 31066T formed a deeply branched clade. They are phylogenetically related with the members of the family Puniceicoccaceae, and phylogenomic analysis could warrant the creation of a novel family (Figures 3, 4). The majority of clades presented in Figure 4 lacked cultivated representatives.

Figure 5. Phylogenetic analysis of high-quality 16S rRNA gene sequences of the class Opitutae. The tree was rooted by Elusimicrobium minutum Pei191T. Type strains are marked blue. The bootstrap values of nodes greater than 0.7 are displayed. Bar, 0.1 means the nucleotide substitution per position.
4. Conclusion
Based on the genomic, phenotypic, and chemotaxonomic characteristics, strain WMMB3T represented a novel species of the genus Coraliomargarita, for which the name Coraliomargarita parva sp. nov. was proposed. The type strain is WMMB3T (= MCCC 1K04284T = KCTC 92914T). Based on the phylogenetic analysis of whole genome sequences and 16S rRNA genes affiliated to the class Opitutae, five novel families Coraliomargaritaceae fam. nov., Pelagicoccaceae fam. nov., Cerasicoccaeae fam. nov., Oceanipulchritudinaceae fam. nov., and Alterococcaeae fam. nov., were proposed. AAI values of 58–60% could be considered the threshold for delineating families of the class Opitutae. This study provided a new taxonomic framework of the class Opitutae based on genomic data.
4.1. Description of Coraliomargarita parva sp. nov.
4.1.1. Coraliomargarita parva (par’va. L. fem. adj. parva, small)
Colonies on MB agar plates cultured for 5 days at 30°C are less than 1 mm, small and round. Cells are Gram-stain-negative and coccus-shaped. Catalase-positive and oxidase-positive. Growth occurs between 15 and 40°C with an optimum at 30°C. Nitrate can be reduced to nitrite. Production of indole is positive, and hydrolysis of aesculin and gelatin is negative. Positive for alkaline phosphatase, acid phosphatase, esterase (C4), naphthol-AS-BI-phosphohydrolase, and β-galactosidase; weakly positive for α-galactosidase, α-glucosidase, and β-glucosidase. Acid can be produced from glucose, amygdalin, mannitol and arabinose. The genome size of the type strain is 4.5 Mbp with a DNA G + C content of 56.0%. The respiratory quinone is menaquinone-7 (MK-7). The major fatty acids are iso-C14:0 and C18:1 ω9c.
The type strain is WMMB3T (= MCCC 1K08426T = KCTC 92914T), isolated from a mangrove sediment collected from a mangrove preservation area in Quanzhou Bay.
The GenBank/EMBL/DDBJ accession number of the 16S rRNA gene sequence and draft genome sequence of strain WMMB3T are Q034132 and JAPZEI000000000, respectively.
4.2. Taxonomic consequences: new families
4.2.1. Description of Coraliomargaritaceae fam. nov.
Coraliomargaritaceae (Co.ra.li.o.mar.ga.ri.ta.ce’ae. N.L. fem. n. Coraliomargarita, a bacterial genus; −aceae, ending to denote a family; N.L. fem. pl. n. Coraliomargaritaceae, the Coraliomargarita family).
The description is the same as for the genus Coraliomargarita (Yoon et al., 2007b), the type genus and the sole genus in this family. Members are all found in marine sediment and seawater. Delineation of the family is mainly determined by phylogeny of 16S rRNA gene, genomic relatedness and phylogenomic analysis. The genome size is 1.2–4.7 Mbp with genomic G + C content of 38.5–56.4%.
4.2.2. Description of Pelagicoccaceae fam. nov.
Pelagicoccaceae (Pe.la.gi.coc.ca.ce’ae. N.L. masc. n. Pelagicoccus, a bacterial genus; −aceae, ending to denote a family; N.L. fem. pl. n. Pelagicoccaceae, the Pelagicoccus family).
The description is the same as for the genus Pelagicoccus (Yoon et al., 2007c), the type and the sole genus in this family. Delineation of the family is mainly determined by phylogeny of 16S rRNA gene, genomic relatedness and phylogenomic analysis. The genome size is 3.1–7.5 Mbp with genomic G + C content of 46.1–56.8%.
4.2.3. Description of Oceanipulchritudinaceae fam. nov.
Oceanipulchritudinaceae (O.ce.a.ni.pul.chri.tu.di.na.ce’ae. N.L. fem. n. Oceanipulchritudo, a bacterial genus; −aceae, ending to denote a family; N.L. fem. pl. n. Oceanipulchritudinaceae, the Oceanipulchritudo family).
The description is the same as for the genus Oceanipulchritudo (Feng et al., 2020), the type and the sole genus in this family. Delineation of the family is mainly determined by phylogeny of 16S rRNA gene, genomic relatedness and phylogenomic analysis. The genome size is 3.5–5.3 Mbp with genomic G + C content of 54.0–63.8%.
4.2.4. Description of Cerasicoccaceae fam. nov.
Cerasicoccaceae (Ce.ra.si.coc.ca.ce’ae. N.L. masc. n. Cerasicoccus, a bacterial genus; −aceae, ending to denote a family; N.L. fem. pl. n. Cerasicoccaceae, the Cerasicoccus family).
Cells are Gram-stain negative, cocci, and obligately aerobic. Cells lack flagella and are non-motile. Spores are not formed. Catalase- and oxidase-positive. The major respiratory quinone is MK-7. Predominant cellular fatty acids are C14:0 and C18:1 ω9c. It includes two genera Cerasicoccus and Ruficoccus. Phylogeny of 16S rRNA gene, genomic relatedness and phylogenomic analysis indicate that Cerasicoccusaceae should be separated from the family Puniceicoccaceae. The genome size was 3.8–4.5 Mbp with genomic G + C content of 52.6–60.3%. The type genus is Cerasicoccus.
4.2.5. Description of Alterococcaeae fam. nov.
Alterococcaeae (Al.te.ro.coc.ca.ce’ae. N.L. masc. n. Alterococcus, a bacterial genus; −aceae, ending to denote a family; N.L. fem. pl. n. Alterococcaeae, the Alterococcus family).
The description is the same as for the genus Alterococcus (Shieh and Jean, 1998), the type and sole genus in this family. Delineation of the family is mainly determined by phylogeny of 16S rRNA gene.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
ZH and QL conceived the study. LM, WW, and QL conducted the experiments. AO proposed names, wrote, and checked etymologies. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Quanzhou City Science & Technology Program of China (No. 2022C018R), National Infrastructure of Natural Resources for Science and Technology Program of China (NIMR-2021-9), and Natural Science Foundation of Fujian Province (2022 J011106).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1202141/full#supplementary-material
Abbreviations
MCCC, Marine Culture Collection of China, KCTC, Korean Collection for Type Cultures, MCL, maximum composite likelihood, NJ, neighbor-joining, ML, maximum-likelihood, iTOL, Interactive Tree of Life, dDDH, digital DNA–DNA hybridization, ANI, average nucleotide identity, AAI, average amino acid identity,
Footnotes
1. ^https://blast.ncbi.nlm.nih.gov/Blast.cgi
2. ^https://github.com/najoshi/sickle
References
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731. doi: 10.1038/nbt.3893
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chin, K. J., Liesack, W., and Janssen, P. H. (2001). Opitutus terrae gen. nov., sp. nov., to accommodate novel strains of the division 'Verrucomicrobia' isolated from rice paddy soil. Int. J. Syst. Evol. Microbiol. 51, 1965–1968. doi: 10.1099/00207713-51-6-1965
Choo, Y. J., Lee, K., Song, J., and Cho, J. C. (2007). Puniceicoccus vermicola gen. nov., sp. nov., a novel marine bacterium, and description of Puniceicoccaceae fam. nov., Puniceicoccales Ord. nov., Opitutaceae fam. nov., Opitutales Ord. nov. and Opitutae classis nov. in the phylum 'Verrucomicrobia'. Int. J. Syst. Evol. Microbiol. 57, 532–537. doi: 10.1099/ijs.0.64616-0
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U. S. A. 89, 5685–5689. doi: 10.1073/pnas.89.12.5685
Feng, X., Zou, Q. H., Zhang, X. Y., Ye, M. Q., and Du, Z. J. (2020). Oceanipulchritudo coccoides gen. Nov., sp. nov., isolated from marine sediment within the family Puniceicoccaceae. Int. J. Syst. Evol. Microbiol. 70, 5654–5664. doi: 10.1099/ijsem.0.004458
Huang, Z., Su, P., and Lai, Q. (2021). Proposal of Zooshikellaceae fam. nov. to accommodate the genera Zooshikella and Spartinivicinus and reclassification of Zooshikella marina as a later heterotypic synonym of Zooshikella ganghwensis based on whole genome sequence analysis. Int. J. Syst. Evol. Microbiol. 71:005055. doi: 10.1099/ijsem.0.005055
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Komagata, K., and Suzuki, K. (1987). Lipid and cell-wall analysis in bacterial systematics. Meth. Microbiol. 19, 161–207. doi: 10.1016/S0580-9517(08)70410-0
Konstantinidis, K. T., Rosselló-Móra, R., and Amann, R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. doi: 10.1038/ismej.2017.113
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Letunic, I., and Bork, P. (2007). Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14, 1–14. doi: 10.1186/1471-2105-14-60
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). Check M: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parte, A. C., Sarda Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C., and Göker, M. (2020). List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Shieh, W. Y., and Jean, W. D. (1998). Alterococcus agarolyticus, gen. nov., sp.nov., a halophilic thermophilic bacterium capable of agar degradation. Can. J. Microbiol. 44, 637–645. doi: 10.1139/cjm-44-7-637
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017a). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., and Chun, J. (2017b). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Yoon, J., Matsuo, Y., Matsuda, S., Adachi, K., Kasai, H., and Yokota, A. (2007a). Cerasicoccus arenae gen. nov., sp. nov., a carotenoid-producing marine representative of the family Puniceicoccaceae within the phylum 'Verrucomicrobia', isolated from marine sand. Int. J. Syst. Evol. Microbiol. 57, 2067–2072. doi: 10.1099/ijs.0.65102-0
Yoon, J., Yasumoto-Hirose, M., Katsuta, A., Sekiguchi, H., Matsuda, S., Kasai, H., et al. (2007b). Coraliomargarita akajimensis gen. nov., sp. nov., a novel member of the phylum 'Verrucomicrobia' isolated from seawater in Japan. Int. J. Syst. Evol. Microbiol. 57, 959–963. doi: 10.1099/ijs.0.64755-0
Yoon, J., Yasumoto-Hirose, M., Matsuo, Y., Nozawa, M., Matsuda, S., Kasai, H., et al. (2007c). Pelagicoccus mobilis gen. nov., sp. nov., Pelagicoccus albus sp. nov. and Pelagicoccus litoralis sp. nov., three novel members of subdivision 4 within the phylum 'Verrucomicrobia', isolated from seawater by in situ cultivation. Int. J. Syst. Evol. Microbiol. 57, 1377–1385. doi: 10.1099/ijs.0.64970-0
Keywords: Coraliomargarita , Opitutae , genome-based analysis, phylogeny, polyphasic taxonomy, Puniceicoccaceae , new families
Citation: Min L, Wang W, Oren A, Lai Q and Huang Z (2023) Coraliomargarita parva sp. nov., isolated from mangrove sediment and genome-based analysis of the class Opitutae revealed five novel families: Coraliomargaritaceae fam. nov., Pelagicoccaceae fam. nov., Cerasicoccaeae fam. nov., Oceanipulchritudinaceae fam. nov., and Alterococcaeae fam. nov. Front. Microbiol. 14:1202141. doi: 10.3389/fmicb.2023.1202141
Edited by:
Vasco Ariston De Carvalho Azevedo, Federal University of Minas Gerais, BrazilCopyright © 2023 Min, Wang, Oren, Lai and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiliang Lai, bGFpNzE5QDE2My5jb20=; Zhaobin Huang, emJodWFuZ2VtYWlsQGdtYWlsLmNvbQ==