 Yan Lin
Yan Lin Meili Sun2†
Meili Sun2† Junjie Zhang
Junjie Zhang Keli Yang
Keli Yang Hasan Zulfiqar
Hasan Zulfiqar Hongyan Lai
Hongyan Lai- 1Key Laboratory for Animal Disease-Resistance Nutrition of the Ministry of Agriculture, Animal Nutrition Institute, Sichuan Agricultural University, Chengdu, China
- 2Beidahuang Industry Group General Hospital, Harbin, China
- 3Chifeng Product Quality Inspection and Testing Centre, Chifeng, China
- 4Nonlinear Research Institute, Baoji University of Arts and Sciences, Baoji, China
- 5Baotou Teacher’s College, Inner Mongolia University of Science and Technology, Baotou, China
- 6Yangtze Delta Region Institute (Huzhou), University of Electronic Science and Technology of China, Huzhou, Zhejiang, China
- 7Chongqing Key Laboratory of Big Data for Bio Intelligence, Chongqing University of Posts and Telecommunications, Chongqing, China
Promoters are the basic functional cis-elements to which RNA polymerase binds to initiate the process of gene transcription. Comprehensive understanding gene expression and regulation depends on the precise identification of promoters, as they are the most important component of gene expression. This study aimed to develop a machine learning-based model to predict promoters in Klebsiella aerogenes (K. aerogenes). In the prediction model, the promoter sequences in K. aerogenes genome were encoded by pseudo k-tuple nucleotide composition (PseKNC) and position-correlation scoring function (PCSF). Numerical features were obtained and then optimized using mRMR by combining with support vector machine (SVM) and 5-fold cross-validation (CV). Subsequently, these optimized features were inputted into SVM-based classifier to discriminate promoter sequences from non-promoter sequences in K. aerogenes. Results of 10-fold CV showed that the model could yield the overall accuracy of 96.0% and the area under the ROC curve (AUC) of 0.990. We hope that this model will provide help for the study of promoter and gene regulation in K. aerogenes.
1. Introduction
Klebsiella aerogenes (K. aerogenes) is a ubiquitous Gram-negative bacterium found in a variety of environments, such as soil, sewage, mammalian gastrointestinal tract et al. The K. aerogenes can also colonize in human gut and most community-or hospital-acquired bloodstream infections are caused by this common multi-drug resistant pathogen, which is a source of opportunistic infections. Although most of these bacteria are sensitive to the antibiotics targeting them, the drug resistance still exists, and the induced resistance mechanisms are complex (Price and Sleigh, 1970). Promoters are the genomic regions upstream of genes, where RNA polymerase and other transcription factors bind together to initiate genes transcription (Sawadogo and Roeder, 1985). Thus, promoter identification is the first step to understand gene expression mechanism. Thus, a precise identification of promoter sequence could generate dynamic signs for understanding its mechanism of regulation (Zuo and Li, 2010).
In fact, several experimental methods, such as mass spectrometry (Flusberg et al., 2010), reduced-representation bisulfite sequencing (Doherty and Couldrey, 2014), and single-molecule real-time sequencing (Boch and Bonas, 2010), have been developed to recognize promoters. Although these methods are relatively helpful in the identification of promoters, they are exorbitant when implemented to large sequencing data (Hu et al., 2022a). Therefore, a bioinformatics tool to identify promoter sequence is instantly needed.
At present, some machine learning-based methods have been presented to predict promoters in multiple species (Ao et al., 2022). Li and Lin have ever designed a position weight matrix (PWM) method to identify sigma70 promoters in Escherichia coli (E. coli) (Li and Lin, 2006). Subsequently, they developed a hybrid approach (called IPMD) to identify eukaryotic and prokaryotic promoters (Lin and Li, 2011). PePPER is another webserver for recognizing prokaryote promoter elements and regulons (de Jong et al., 2012). In 2014, Lin et al. proposed a first model called iPro54-PseKNC to predict sigma54 promoters in prokaryotes (Lin et al., 2014). Liu et al. established a friendly tool called iPromoter-2 l for the prediction of bacterial promotors. These works mainly used sequence composition to perform prediction. By using Z-curve theory, the bacterial promoters could also be formulated and predicted (Song, 2012; Lin et al., 2019). Combining various of sequence information, Lai et al. built a powerful model named iProEP for the identification of promoters in three kinds of eukaryotes and two kinds of bacteria (Lai et al., 2019). Chevez-Guardado designed a general tool (Promotech) for bacterial promoter recognition (Chevez-Guardado and Peña-Castillo, 2021). Recently, the promoters in two prokaryotes: Corynebacterium glutamicum and Agrobacterium Tumefaciens Strain C58 were studied by using machine learning based models (Zulfiqar et al., 1011; Li et al., 2023). Among them, the sigma70 promoter is the most extensively studied in prokaryotes (Patiyal et al., 2022). iProm-phage is a two-layer model for phage promoters and their types prediction (Shujaat et al., 2022).
Although there are already many prediction models for prokaryotic promoters, due to species specificity and prediction performance limitations, there is a need for trainning more specific promoter prediction models for K. aerogenes (Hu et al., 2022b). Thus, in this paper, we designed a SVM-based model to predict the promoters of K. aerogenes. The Figure 1 illustrates the workflow of this project, mainly including the core content and key steps. Thereinto, two feature extraction methods, namely PseKNC and PCSF, were employed to convert DNA sequences into numerical features. And then these features were optimized by using mRMR feature selection algorithm based on SVM machine learning model and 5-fold CV. Moreover, the selected optimal feature subset was applied to train a SVM classifier for identifying K. aerogenes promoter sequences on the basis of 10-fold CV. As a result, an ideal model with prediction accuracy and AUC of 96.0% and 0.990 was attained.
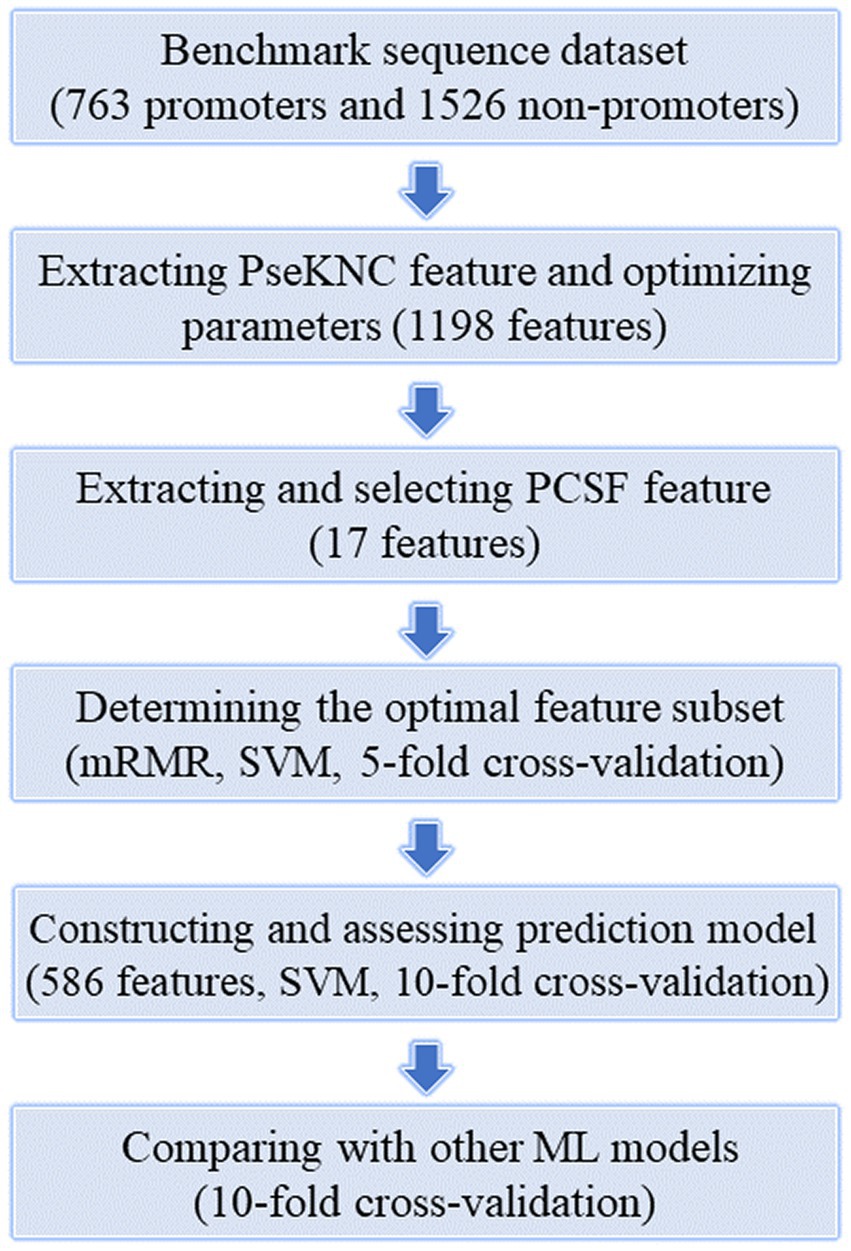
Figure 1. The overall workflow of this study.
2. Materials and methods
2.1. Data collection and preprocessing
The construction of a prokaryotic promoter dataset is crucial for obtaining a good promoter model. Prokaryotic Promoter Database (PDD, http://lin-group.cn/database/ppd/) developed by Lin et al. contains comprehensive information on experimentally verified promoters of numerous prokaryotic species and can be freely accessed (Su et al., 2021). The sequence data of 763 K. aerogenes promoters were downloaded from the database and defined as positive dataset. Each promoter sequence was composed of 81 nucleotides, including transcription start site (TSS) (namely the 0-th site), upstream 20 bp and downstream 60 bp regions of TSS. In order to generate a reliable negative dataset, we firstly extracted the convergent intergenic (length greater than 81 bp) and coding (length greater than 2000 bp) regions from K. aerogenes genome. Secondly, sliding window method with step of 1 bp was applied to generate convergent intergenic and coding sequences, with length of 81 bp. Then, we used CD-HIT program to estimate the sequence similarity of convergent intergenic and coding sequences, and filtered highly similar sequences by setting cutoff value as 0.8. Finally, 763 convergent intergenic sequences and 763 coding sequences were randomly picked out and regarded as negative dataset.
2.2. Feature extraction
Referring to the well-designed eukaryotic and prokaryotic promoter identification tool, iProEP,1 we also adopted two algorithms, including pseudo k-tuple nucleotide composition (PseKNC) and position-correlation scoring function (PCSF), to transform raw promoter/non-promoter sequence data into suitable numeric features for modeling.
In this study, the type II PseKNC method was used to transform each nucleotide sequence into a feature vector of dimensions (Tang et al., 2021),
where means -tuple nucleotide component, is an integer less than ( denotes the length of a DNA sequence). And is the number of physicochemical properties, the value of which is 6 corresponding to the six types of DNA local structural properties included in this work. Each element in is defines as:
The former elements are nucleotide composition features, which can reflect local or short-range sequence-order information. The latter factors are pseudo nucleotide composition features corresponding to global or long-range effect. In equation (2), represents the normalized frequency of occurrence of the -th -tuple nucleotides in the sample sequence. The weight factor can adjust the effects of nucleotide composition and local structural properties of DNA. And indicates the -tier correlation factor and is formulated with the form of equation (3), the value of which corresponds to the sequence-order correlation between all the -tier contiguous -tuple nucleotide component along a promoter/non-promoter sequence.
where
where is the standardized value of the -th DNA local structural properties for the dinucleotide at position . The original values of these physicochemical properties are provided by Goñi et al. (2008) and the standardization approach are the same as previously described in iProEP. In addition, the processes of Position-Correlation Scoring Matrix (PCSM) construction and PCSF feature transformation and selection are directly referring to the E. coli model in iProEP.
2.3. mRMR
mRMR is a well-known feature selection method and has been used in many computational and biological applications (Zulfiqar et al., 2021; Su et al., 2023). The density functions are described as ‘i’ and ‘y’ and their corresponding probabilities are and . The common information between these two functions can be demarcated as
If the target is then calculating the mutual information in relation to the target and can be defined as
2.4. Machine learning classifiers
SVM is a well-known classifier and has been utilized in many bioinformatics and computational biology related tools (Basith et al., 2021; Arif et al., 2022; Basith et al., 2022; Bupi et al., 2023; Dao et al., 2023). It is typically used to perform binary classification. Ada boost (AB) is another famous classifier (Wang et al., 2021). The main idea of AB is to set the classifiers weights and trained the data in each and every iteration. Naïve Bayes (NB) classifier has been widely used in bioinformatics due to its simplicity (Naseer et al., 2022; Zulfiqar et al., 2022). This classification method totally depends on the Bayes theorems. Random Forest (RF) is a collective knowledge algorithm and broadly used in bioinformatics (Zhu et al., 2022; Zhang et al., 2023). The main idea of this is to unite multiple weak classifiers and outcome generated on the basis of voting (Zulfiqar et al., 2023). The brief description is clearly described in (Zulfiqar et al., 2021). The k-nearest neighbor (KNN) is a non-parametric and supervised learning classifier, which uses vicinity to make classifications about the grouping of an individual data point. Logistic Regression (LR) is a classification algorithm and used when the value of the target variable is categorical in nature (Yang et al., 2021). We have executed these algorithms in Weka version 3.8.4. by using the default values.
2.5. Evaluation metrics
Accuracy, sensitivity, specificity (Cao et al., 2017; Tang et al., 2022; Yang et al., 2022; Zhang et al., 2022; Chen et al., 2023) were utilized to evaluate the performance of the prediction model and termed as
where ‘tp’ represents the correctly predicted promoter sequences and ‘fp’ shows the non-promoter sequences classified as promoter sequence. And the other hand, ‘tn’ characterizes the correctly recognized non-promotor sequences and ‘fn’ exhibit the promoter sequences which were classified as non-promoter sequence.
3. Results and discussion
In the fields of statistical analysis and machine learning (ML) prediction, cross-validation (CV) strategy has been widely utilized to evaluate the prediction performance of ML models (Hasan et al., 2022; Shoombuatong et al., 2022; Xiao et al., 2022; Yu et al., 2022; Zhang et al., 2022). In this work, 5-fold CV technique was used in the processes of PseKNC parameter optimization and optimal feature subset selection and 10-fold CV technique was used to assess the performance of the six machine learning methods. In n-fold CV, the benchmark dataset was randomly divided into n groups with equal size. Each group was individually tested on the model which was trained with the remaining n-1 groups. According to this, the n-fold CV method was performed n times, and the final evaluation result was the average prediction performance of the n models.
We constructed a computational model on the basis of sequence features to recognize promoter sequences in K. aerogenes. Based on the definition of pseudo nucleotide characteristics, we debugged the parameters , , and according to the following range to determine the optimal combination of k-mer nucleotide composition information and long-range sequence order information.,
Based on the feature set generated by each combination and the LIBSVM algorithm, we can construct promoter prediction models and evaluate their accuracies using a 5-fold CV method. The final determined values of k, λ, and ω were 5, 29, and 0.1, respective. The original vector contains 1,198 features which could produce the prediction accuracy of 88.0%. Then, 17 positional correlation scoring features were calculated based on the most conserved sites in the promoter sequence of the 3-mer nucleotide fragment. After integrating two types of features, the mRMR algorithm was applied to sort all features, and an incremental feature selection (IFS) method was applied to eliminate redundant information to obtain the optimal feature subset for improving the accuracy of the promoter classifier. In the process of IFS, we also used a 5-fold CV method to evaluate the promoter prediction accuracy of each classifier, as shown in Figure 2A. As shown in the figure, the model constructed based on the first 586 features has the highest prediction accuracy of 95.9%.
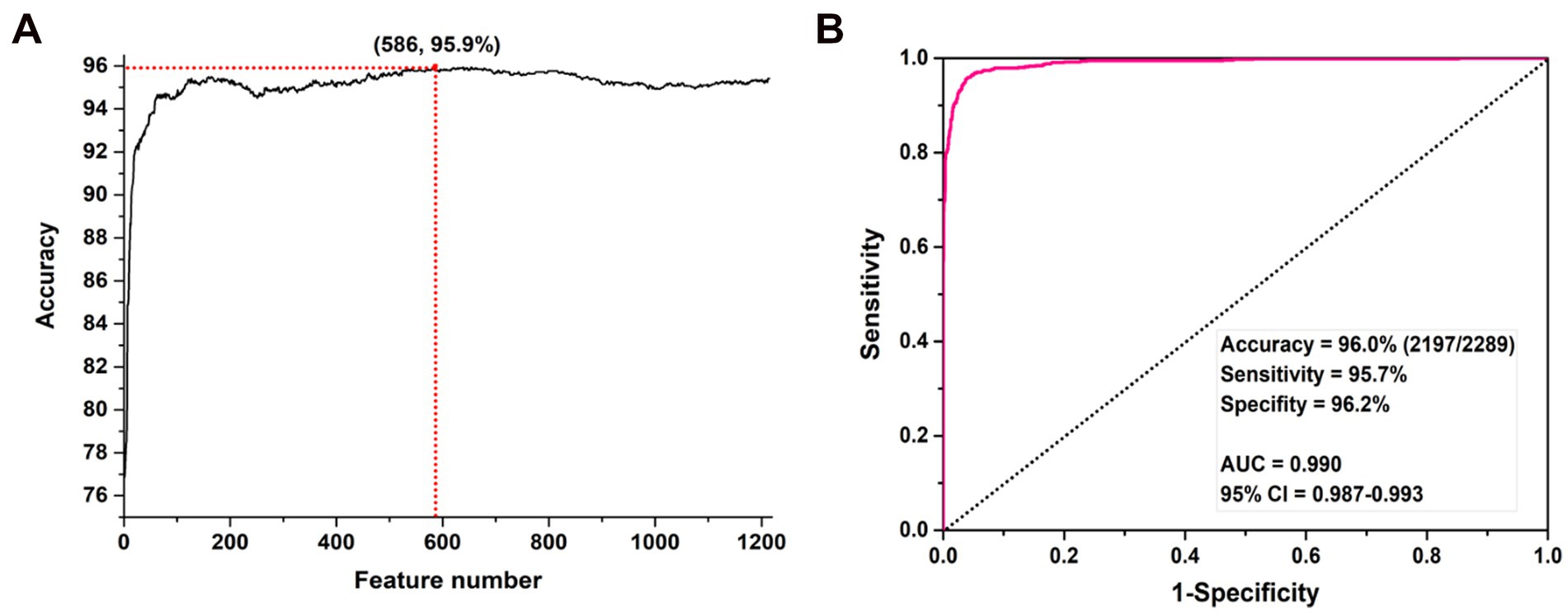
Figure 2. The prediction accuracies of SVM models constructed with different numbers of features. (A) IFS process for feature selection and (B) ROC curve based on the optimal features.
After determining the optimal subset of features, we further evaluated its promoter prediction ability using a 10-fold CV method for determining the parameters c and γ in SVM, where c ∈ [2−5, 215] with a step of 2, γ∈ [23,2−15] with a step size of 2−1. The final optimal values of c and γ are 2 and 2−3, respectively. The optimal SVM model could produce the best performance with the accuracy of 96.0%, sensitivity of 95.7%, and specificity of 96.2%. The area under the ROC curve (AUC) was 0.990 with 95% confidence interval (CI): 0.987–0.993 (as shown in Figure 2B).
In order to evaluate the performance of this SVM prediction model, we also constructed five models based on LR, KNN, RF, AB and NB for K. aerogenes promoter recognition by using the same optimal features. The 10-fold CV results showed that the AUC values of the LR, KNN, RF, and AB models were 0.960, 0.941, 0.939, and 0.959, respectively, as shown in Figure 3. We observed that the sensitivity of the RF model was poor (68.8%), while the overall predictive performance of the NB model was the weakest, with accuracy and AUC values of 81.3% and 0.882 (Table 1). The accuracy of SVM-based model was 96.0% which was 5.6–14.7% higher than the other five classifiers. Overall, identifying K. aerogenes promoter sequences based on optimal pseudo nucleotide features and positional correlation scoring features is effective, and the model constructed based on SVM algorithm has the best predictive performance.
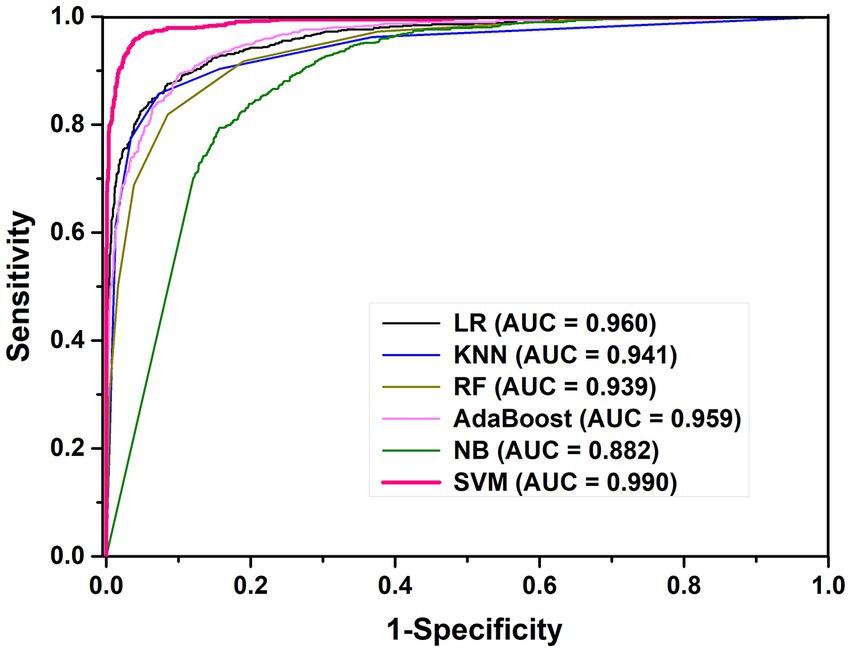
Figure 3. The ROC curves for different machine learning models.
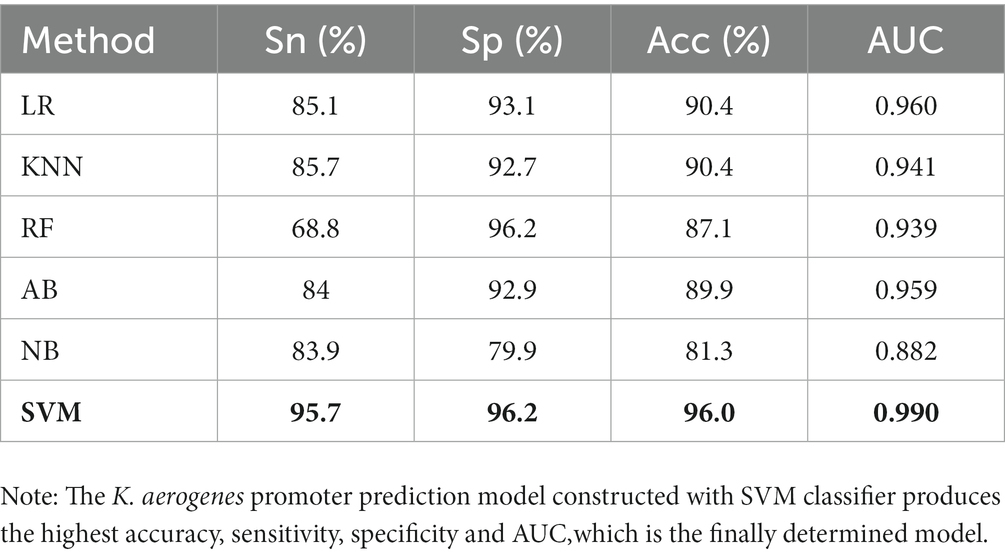
Table 1. The prediction performance of different machine learning models based on 10-fold cross-validation.
4. Conclusion
Promoters play an important role in the initiation of transcription, because they are located upstream of genes. RNA polymerase and a quantity of transcription factors bind to promoter to start the transcription. Therefore, studying promoters is crucial for studying gene expression regulation. In this study, we proposed an SVM-based model to identify promoter sequences in K. aerogenes. In the proposed model, sequences were encoded using PseKNC and PCSF and then optimized with mRMR and SVM-based algorithm on 5-fold CV. Then, these optimized features were inputted into SVM-based classifier using 10-fold CV and achieved the best model. The results show that our model can predict promoters accurately, suggesting that our feature extraction and selection methods are able to capture the important sequence features. In the future, we will develop more suitable and robust models for more prokaryotic species.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found at: http://lin-group.cn/database/ppd/.
Author contributions
YL, HZ, and HL project design and oversight, and manuscript writing and revision. MS and HZ sample collection and curation. YL, JZ, HZ, ML, and KY experiment conduction and data analysis. YL and ML table preparation. YL, MS, and CW result interpretation and discussion. YL and JZ funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the grant from the National Natural Science Foundation of Sichuan Province (no. 2022NSFSC0058), the Sichuan Key Science and Technology Project (no. 2021ZDZX0009), the Research Program of Science and Technology at Universities of Inner Mongolia Autonomous Region under Grant NJZZ18381 and the National Natural Science Foundation of China (62262049).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Ao, C., Jiao, S., Wang, Y., Yu, L., and Zou, Q. (2022). Biological sequence classification: a review on data and general methods. Research 2022:0011. doi: 10.34133/research.0011
Arif, M., Ahmed, S., Ge, F., Kabir, M., Khan, Y. D., Yu, D. J., et al. (2022). StackACPred: prediction of anticancer peptides by integrating optimized multiple feature descriptors with stacked ensemble approach. Chemom. Intell. Lab. Syst. 220:104458. doi: 10.1016/j.chemolab.2021.104458
Basith, S., Hasan, M. M., Lee, G., Wei, L., and Manavalan, B. (2021). Integrative machine learning framework for the identification of cell-specific enhancers from the human genome. Brief. Bioinform. 22:bbab252. doi: 10.1093/bib/bbab252
Basith, S., Lee, G., and Manavalan, B. (2022). STALLION: a stacking-based ensemble learning framework for prokaryotic lysine acetylation site prediction. Brief. Bioinform. 23:bbab376. doi: 10.1093/bib/bbab376
Boch, J., and Bonas, U. (2010). Xanthomonas Avr Bs3 family-type III effectors: discovery and function. Annu. Rev. Phytopathol. 48, 419–436. doi: 10.1146/annurev-phyto-080508-081936
Bupi, N., Sangaraju, V. K., Phan, L. T., Lal, A., Vo, T. T. B., Ho, P. T., et al. (2023). An effective integrated machine learning framework for identifying severity of tomato yellow leaf curl virus and their experimental validation. Research 6:0016. doi: 10.34133/research.0016
Cao, R., Freitas, C., Chan, L., Sun, M., Jiang, H., and Chen, Z. (2017). Pro Lan GO: protein function prediction using neural machine translation based on a recurrent neural network. Molecules 22:1732. doi: 10.3390/molecules22101732
Chen, L., Yu, L., and Gao, L. (2023). Potent antibiotic design via guided search from antibacterial activity evaluations. Bioinformatics 39:btad059. doi: 10.1093/bioinformatics/btad059
Chevez-Guardado, R., and Peña-Castillo, L. (2021). Promotech: a general tool for bacterial promoter recognition. Genome Biol. 22:318. doi: 10.1186/s13059-021-02514-9
Dao, F. Y., Liu, M. L., Su, W., Lv, H., Zhang, Z. Y., Lin, H., et al. (2023). AcrPred: a hybrid optimization with enumerated machine learning algorithm to predict anti-CRISPR proteins. Int. J. Biol. Macromol. 228, 706–714. doi: 10.1016/j.ijbiomac.2022.12.250
de Jong, A., Pietersma, H., Cordes, M., Kuipers, O. P., and Kok, J. (2012). PePPER: a webserver for prediction of prokaryote promoter elements and regulons. BMC Genomics 13:299. doi: 10.1186/1471-2164-13-299
Doherty, R., and Couldrey, C. (2014). Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: a technical assessment. Front. Genet. 5:126. doi: 10.3389/fgene.2014.00126
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465. doi: 10.1038/nmeth.1459
Goñi, J. R., Fenollosa, C., Pérez, A., Torrents, D., and Orozco, M. (2008). DNAlive: a tool for the physical analysis of DNA at the genomic scale. Bioinformatics 24, 1731–1732. doi: 10.1093/bioinformatics/btn259
Hasan, M. M., Tsukiyama, S., Cho, J. Y., Kurata, H., Alam, M. A., Liu, X., et al. (2022). Deepm 5C: a deep-learning-based hybrid framework for identifying human RNA N5-methylcytosine sites using a stacking strategy. Mol. Ther. 30, 2856–2867. doi: 10.1016/j.ymthe.2022.05.001
Hu, Y., Zhang, Y., Zhang, H., Gao, S., Wang, L., Wang, T., et al. (2022a). Mendelian randomization highlights causal association between genetically increased C-reactive protein levels and reduced Alzheimer’s disease risk. Alzheimers Dement. 18, 2003–2006. doi: 10.1002/alz.12687
Hu, Y., Zhang, Y., Zhang, H., Gao, S., Wang, L., Wang, T., et al. (2022b). Cognitive performance protects against Alzheimer’s disease independently of educational attainment and intelligence. Mol. Psychiatry 27, 4297–4306. doi: 10.1038/s41380-022-01695-4
Lai, H.-Y., Zhang, Z.-Y., Su, Z.-D., Su, W., Ding, H., Chen, W., et al. (2019). iProEP: a computational predictor for predicting promoter. Mol. Ther. Nucleic Acids 17, 337–346. doi: 10.1016/j.omtn.2019.05.028
Li, Q. Z., and Lin, H. (2006). The recognition and prediction of sigma (70) promoters in Escherichia coli K-12. J. Theor. Biol. 242, 135–141. doi: 10.1016/j.jtbi.2006.02.007
Li, H., Zhang, J., Zhao, Y., and Yang, W. (2023). Predicting Corynebacterium glutamicum promoters based on novel feature descriptor and feature selection technique. Front. Microbiol. 14:1141227. doi: 10.3389/fmicb.2023.1141227
Lin, H., Deng, E. Z., Ding, H., Chen, W., and Chou, K. C. (2014). iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 42, 12961–12972. doi: 10.1093/nar/gku1019
Lin, H., and Li, Q. Z. (2011). Eukaryotic and prokaryotic promoter prediction using hybrid approach. Theory Biosci. 130, 91–100. doi: 10.1007/s12064-010-0114-8
Lin, H., Liang, Z. Y., Tang, H., and Chen, W. (2019). Identifying Sigma70 promoters with novel Pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 1316–1321. doi: 10.1109/TCBB.2017.2666141
Naseer, S., Ali, R. F., Khan, Y. D., and Dominic, P. D. D. (2022). iGluK-deep: computational identification of lysine glutarylation sites using deep neural networks with general pseudo amino acid compositions. J. Biomol. Struct. Dyn. 40, 11691–11704. doi: 10.1080/07391102.2021.1962738
Patiyal, S., Singh, N., Ali, M. Z., Pundir, D. S., and Raghava, G. P. (2022). Sigma70Pred: a highly accurate method for predicting sigma70 promoter in Escherichia coli K-12 strains. Front. Microbiol. 13:1042127. doi: 10.3389/fmicb.2022.1042127
Price, D., and Sleigh, J. (1970). Control of infection due to Klebsiella aerogenes in a neurosurgical unit by withdrawal of all antibiotics. Lancet 296, 1213–1215. doi: 10.1016/S0140-6736(70)92179-3
Sawadogo, M., and Roeder, R. G. (1985). Interaction of a gene-specific transcription factor with the adenovirus major late promoter upstream of the TATA box region. Cells 43, 165–175. doi: 10.1016/0092-8674(85)90021-2
Shoombuatong, W., Basith, S., Pitti, T., Lee, G., and Manavalan, B. (2022). THRONE: a new approach for accurate prediction of human RNA N7-Methylguanosine sites. J. Mol. Biol. 434:167549. doi: 10.1016/j.jmb.2022.167549
Shujaat, M., Jin, J. S., Tayara, H., and Chong, K. T. (2022). iProm-phage: a two-layer model to identify phage promoters and their types using a convolutional neural network. Front. Microbiol. 13:1061122. doi: 10.3389/fmicb.2022.1061122
Song, K. (2012). Recognition of prokaryotic promoters based on a novel variable-window Z-curve method. Nucleic Acids Res. 40, 963–971. doi: 10.1093/nar/gkr795
Su, W., Liu, M. L., Yang, Y. H., Wang, J. S., Li, S. H., Lv, H., et al. (2021). PPD: a manually curated database for experimentally verified prokaryotic promoters. J. Mol. Biol. 433:166860. doi: 10.1016/j.jmb.2021.166860
Su, W., Xie, X. Q., Liu, X. W., Gao, D., Ma, C. Y., Zulfiqar, H., et al. (2023). iRNA-ac4C: a novel computational method for effectively detecting N4-acetylcytidine sites in human mRNA. Int. J. Biol. Macromol. 227, 1174–1181. doi: 10.1016/j.ijbiomac.2022.11.299
Tang, Q., Nie, F., Kang, J., and Chen, W. (2021). mRNALocater: enhance the prediction accuracy of eukaryotic mRNA subcellular localization by using model fusion strategy. Mol. Ther. 29, 2617–2623. doi: 10.1016/j.ymthe.2021.04.004
Tang, Q., Nie, F., Zhao, Q., and Chen, W. (2022). A merged molecular representation deep learning method for blood-brain barrier permeability prediction. Brief. Bioinform. 23:bbac357. doi: 10.1093/bib/bbac357
Wang, H., Liang, P. F., Zheng, L., Long, C. S., Li, H. S., and Zuo, Y. (2021). eHSCPr discriminating the cell identity involved in endothelial to hematopoietic transition. Bioinformatics 37, 2157–2164. doi: 10.1093/bioinformatics/btab071
Xiao, J., Liu, M., Huang, Q., Sun, Z., Ning, L., Duan, J., et al. (2022). Analysis and modeling of myopia-related factors based on questionnaire survey. Comput. Biol. Med. 150:106162. doi: 10.1016/j.compbiomed.2022.106162
Yang, Y., Gao, D., Xie, X., Qin, J., Li, J., Lin, H., et al. (2022). DeepIDC: a prediction framework of injectable drug combination based on heterogeneous information and deep learning. Clin. Pharmacokinet. 61, 1749–1759. doi: 10.1007/s40262-022-01180-9
Yang, H., Luo, Y., Ren, X., Wu, M., He, X., Peng, B., et al. (2021). Risk prediction of diabetes: big data mining with fusion of multifarious physical examination indicators. Inf. Fusion 75, 140–149. doi: 10.1016/j.inffus.2021.02.015
Yu, L., Zheng, Y. J., and Gao, L. (2022). MiRNA-disease association prediction based on meta-paths. Brief. Bioinform. 23:bbab571. doi: 10.1093/bib/bbab571
Zhang, Q., Li, H., Liu, Y., Li, J., Wu, C., and Tang, H. (2022). Exosomal non-coding RNAs: new insights into the biology of hepatocellular carcinoma. Curr. Oncol. 29, 5383–5406. doi: 10.3390/curroncol29080427
Zhang, Z. Y., Ning, L., Ye, X., Yang, Y. H., Futamura, Y., Sakurai, T., et al. (2022). iLoc-miRNA: extracellular/intracellular miRNA prediction using deep BiLSTM with attention mechanism. Brief. Bioinform. 23:bbac395. doi: 10.1093/bib/bbac395
Zhang, Y. F., Wang, Y. H., Gu, Z. F., Pan, X., Li, J., Ding, H., et al. (2023). Bitter-RF: a random forest machine model for recognizing bitter peptides. Front. Med. 10:1052923. doi: 10.3389/fmed.2023.1052923
Zhu, H., Ao, C. Y., Ding, Y. J., Hao, H. X., and Yu, L. (2022). Identification of D modification sites using a random Forest model based on nucleotide chemical properties. Int. J. Mol. Sci. 23:3044. doi: 10.3390/ijms23063044
Zulfiqar, H., Guo, Z., Grace-Mercure, B. K., Zhang, Z. Y., Gao, H., Lin, H., et al. (2023). Empirical comparison and recent advances of computational prediction of hormone binding proteins using machine learning methods. Comput. Struct. Biotechnol. J. 21, 2253–2261. doi: 10.1016/j.csbj.2023.03.024
Zulfiqar, H., Huang, Q.-L., Lv, H., Sun, Z. J., Dao, F. Y., and Lin, H. (2022). Deep-4mCGP: a deep learning approach to predict 4mC sites in Geobacter pickeringii by using correlation-based feature selection technique. Int. J. Mol. Sci. 23:1251. doi: 10.3390/ijms23031251
Zulfiqar, H., Khan, R. S., Hassan, F., Hippe, K., Hunt, C., Ding, H., et al. (2021). Computational identification of N4-methylcytosine sites in the mouse genome with machine-learning method. Math. Biosci. Eng. 18, 3348–3363. doi: 10.3934/mbe.2021167
Zulfiqar, H., Yuan, S.-S., Huang, Q.-L., Sun, Z. J., Dao, F. Y., Yu, X. L., et al. (2021). Identification of cyclin protein using gradient boost decision tree algorithm. Comput. Struct. Biotechnol. J. 19, 4123–4131. doi: 10.1016/j.csbj.2021.07.013
Zulfiqar, H., Zahoor, A., Kissanga Grace-Mercure, B., Hassan, F., Zhang, Z. Y., and Liu, F. (1011). Computational prediction of promotors in Agrobacterium Tumefaciens strain C58 by using machine learning technique. Front. Microbiol. 14
Keywords: promoter, pseudo k-tuple nucleotide composition, position-correlation scoring function, feature selection, support vector machine
Citation: Lin Y, Sun M, Zhang J, Li M, Yang K, Wu C, Zulfiqar H and Lai H (2023) Computational identification of promoters in Klebsiella aerogenes by using support vector machine. Front. Microbiol. 14:1200678. doi: 10.3389/fmicb.2023.1200678
Edited by:
Yongchun Zuo, Inner Mongolia University, ChinaReviewed by:
Yaser Daanial Khan, University of Management and Technology, Lahore, PakistanWei Chen, Chengdu University of Traditional Chinese Medicine, China
Copyright © 2023 Lin, Sun, Zhang, Li, Yang, Wu, Zulfiqar and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Lin, TGlueWFuOTM2QDE2My5jb20=; Hasan Zulfiqar, aGFzYW56dWxmaXFhckB1ZXN0Yy5lZHUuY24=; Hongyan Lai, bGFpaHlAY3F1cHQuZWR1LmNu
†These authors have contributed equally to this work