 Marcelina M. Jaworska1Paulina Pecyna1Katarzyna Jaskiewicz2Małgorzata Rydzanicz3Malgorzata Kaluzna4
Marcelina M. Jaworska1Paulina Pecyna1Katarzyna Jaskiewicz2Małgorzata Rydzanicz3Malgorzata Kaluzna4 Krzysztof Pawlaczyk5Rafal Ploski3Dorota M. Nowak-Malczewska1
Krzysztof Pawlaczyk5Rafal Ploski3Dorota M. Nowak-Malczewska1 Justyna A. Karolak1
Justyna A. Karolak1 Marzena Gajecka1,2*
Marzena Gajecka1,2*- 1Chair and Department of Genetics and Pharmaceutical Microbiology, Poznan University of Medical Sciences, Poznan, Poland
- 2Institute of Human Genetics, Polish Academy of Sciences, Poznan, Poland
- 3Department of Medical Genetics, Medical University of Warsaw, Warsaw, Poland
- 4Chair and Department of Endocrinology, Metabolism and Internal Diseases, Poznan University of Medical Sciences, Poznan, Poland
- 5Department of Nephrology, Transplantology and Internal Medicine, Poznan University of Medical Sciences, Poznań, Poland
Introduction: The development of molecular biology methods and their application in microbial research allowed the detection of many new pathogens that cause urinary tract infections (UTIs). Despite the advances of using new research techniques, the etiopathogenesis of UTIs, especially in patients undergoing dialysis and patients after kidney transplantation, is still not fully understood.
Methods: This study aimed to characterize and compare the composition of the bacterial element of the urinary tract microbiome between the groups of patients undergoing dialysis (n = 50) and patients after kidney transplantation (n = 50), with positive or negative urine culture, compared to healthy individuals (n = 50).
Results: Asymptomatic bacteriuria was observed in 30% of the urine cultures of patients undergoing dialysis and patients after kidney transplantation, with Escherichia coli as the most dominant microorganism (73%) detected with the use of classical microbiology techniques. However, differences in the bacterial composition of the urine samples between the evaluated patient groups were demonstrated using the amplicon sequencing. Finegoldia, Leptotrichia, and Corynebacterium were found to be discriminative bacteria genera in patients after dialysis and kidney transplantation compared to the control group. In addition, in all of urine samples, including those without bacteriuria in classical urine culture, many types of bacteria have been identified using 16S rRNA sequencing.
Discussion: The revealed microbial characteristics may form the basis in searching for new diagnostic markers in treatment of patients undergoing dialysis and patients after kidney transplantation.
1. Introduction
While historically urine has been considered sterile in healthy individuals, recent studies have revealed the presence of a wide variety of bacteria in the urinary tract that are usually unidentifiable by conventional microbiological techniques, but can be recognized using molecular methods, including the 16S amplicon sequencing (Wolfe et al., 2012; Lewis et al., 2013; Johnson et al., 2019).
Since the discovery of the human urobiome, which is defined as a group of microorganisms and their genomes occurring in the urinary system, some research has been done to characterize its composition and understand its relationship to human health and disease (Wolfe and Brubaker, 2019). Disturbances in the functioning of the bacterial element of the urinary tract microbiome can cause diseases of the urinary system such as overactive bladder, interstitial cystitis, or neurogenic bladder dysfunction (Pearce et al., 2014; Whiteside et al., 2015).
Urinary tract infection (UTI) is a serious problem in patients with chronic kidney disease (CKD), especially in individuals undergoing dialysis or kidney transplant (Grabe et al., 2015; Coussement et al., 2020). UTI is associated with increased risks of acute rejection, allograft dysfunction, graft loss, increased hospital stays, and mortality (Ariza-Heredia et al., 2014; Koga et al., 2022). Furthermore, a recurrent UTI, which occurs in 7% of the patients after kidney transplantation, is one of the leading causes of allograft loss and death (Britt et al., 2017). Therefore, prediction, early detection, and prevention of UTI are essential. In patients with CKD, UTI can be caused by both, opportunistic bacteria and uropathogens responsible for mixed infections (Naumnik and Myśliwiec, 2013). For many decades, knowledge about uropathogens was considered to be well-established and fairly consistent. Currently, the development of molecular biology methods and their application in research have allowed the identification of many new pathogens, also involved in the etiology of UTI (Price et al., 2018).
Knowledge about the bacterial component of the urinary tract in patients undergoing dialysis or in patients after kidney transplantation remains limited. Thus far, scarce information, including the differentiation of the bacterial element of the urinary tract microbiome depending on the gender of patients after kidney transplantation (Fricke et al., 2014) and differences in the microbiota composition between urine samples of patients after kidney transplantation and healthy individuals (Modena et al., 2017; Rani et al., 2017) have been reported. However, there is no data about differences in the urinary tract microbiome between patients undergoing dialysis and patients after kidney transplantation.
The detection of bacteria in urine samples using culturing methods is the main diagnostic criterion for UTI (Hryniewicz and Holecki, 2015). While culture-dependent techniques allow the identification of typical uropathogens, the isolation and identification of highly demanding and non-cultured microorganisms are not routinely performed in microbiological diagnostics of UTI Consequently, the role of highly demanding and non-cultured bacteria remains elusive in the etiology of UTIs (Magistro and Stief, 2019). Therefore, the molecular biology-based approach could be essential in the identification of bacteria in patients with negative urine cultures (Thomas-White et al., 2018; Wolfe and Brubaker, 2019).
Due to limited data on the bacterial component of the urinary tract microbiome in patients with kidney diseases, the aim of this research was to characterize and compare the composition of the bacterial element of the urinary tract microbiome between the groups of patients with positive or negative urine culture in classical microbiology undergoing dialysis or kidney transplant, compared to control individuals without UTI.
2. Materials and methods
2.1. Patient ascertainment and sample collection
Study individuals were recruited in the outpatient clinic and inpatient ward in the Chair and Department of Endocrinology, Metabolism, and Internal Diseases at the Poznan University of Medical Sciences (PUMS). Patients with end-stage-renal diseases (ESRD), including patients after renal transplantation and patients undergoing dialysis, as well as individuals without urinary tract dysfunction (controls), were enrolled in this study. Patients were assigned to the adequate study group based on the ESRD-treatment. The inclusion criterion for the control group was lack of active UTI. Antibiotic/antifungal treatment 3 weeks before the urine sample collection was the exclusion criterion in the subgroups of patients and controls.
A urine sample was obtained once from each study participant by a sterile mid-stream collection. Derived samples were divided into two aliquots—the first part was immediately used for the assessment by techniques of classical microbiology, while the second part was centrifuged at 800 rcf for 10 min and the pellet was stored at −80°C for the genetic testing.
The study was carried out in accordance with the research protocol approved by the Bioethics Committee at the PUMS (no. 942/14 (2014.12.04), 191/15 (2015.02.05) and 1170/19 (2019.12.05)). The possible consequences of the study were explained, and informed consent was obtained from all participants, according to the Declaration of Helsinki.
2.2. Assessment of bacterial cultures of urine samples
Urine samples were inoculated by a Hoeprich quantitative method on the chromogenic medium Chrom ID® CPS Elite (Biomerieux), then used for isolation, quantification, and presumptive identification of Enterococcus, Proteeae (Proteus, Providencia, Morganella), KESC group (Klebsiella, Enterobacter, Serratia, Citrobacter), Staphylococcus saprophyticus, and Streptococcus agalactiae. After incubation at 35–37°C for 24–48 h, the grown colonies were counted and reported as the number of colony forming units (CFUs) per 1 mL of urine sample.
2.3. DNA isolation
Genomic microbial DNA was extracted from urine pellets using Genomic Mini AX Body Fluids extraction kit (A&A Biotechnology), according to the manufacturer’s protocol with one modification, in a form of adding 10 μL of each of mutanolysin (activity >10,000 U/mg), lysostaphin (concentration: 15 U/μL), lysozyme (concentration: 10 mg/mL), and lyticase (concentration: l0 U/μL) to ensure cell lysis.
2.4. 16S rRNA amplicon sequencing
All steps were performed according to the 16S Metagenomic Sequencing Library Preparation—Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System protocol (Illumina). Briefly, V3 and V4 hypervariable regions of the bacterial 16S rRNA gene were amplified using 515F and 806R primers. Libraries were prepared with Nextera XT Index Kit (Illumina) followed by paired-end sequencing (2×300 bp) using MiSeq System (Illumina, San Diego, CA, United States). The sequencing run was performed with 10% of reference PhiX Control v3 Library (Illumina) spike-in to improve sequencing quality of 16S rRNA amplicon low diversity libraries. The sequencing was performed at the Medical University of Warsaw in Poland.
2.5. Bioinformatic analysis and taxonomic assignment
Bioinformatic analysis of obtained 16S rRNA amplicon sequence reads was performed by the bioinformatics company, ideas4biology Sp. z o.o. in Poznan, Poland, in accordance with the previously described protocol (Katoh et al., 2002; McDonald et al., 2012; Callahan et al., 2016; Bokulich et al., 2018; Bolyen et al., 2019). Analyzes of 16Sr RNA amplicon sequencing data were performed using QIIME 2 version 2019.7 (Bolyen et al., 2019). The control and improvement of the quality of the readings (including the removal of phiX and chimeras) was based on the q2-dada2 function implementing the DADA2 algorithm (Callahan et al., 2016). The obtained amplicon sequence variants (ASVs) were aligned using the mafft algorithm (Katoh et al., 2002) implemented as part of the q2-alignment function. ASVs are generated by a de novo process, in which biological sequences are distinguished on the basis of more frequent repetition than sequence error, and are applied to individual DNA sequences recovered from high-throughput sequencing. The taxonomic affiliation was assigned to the ASV with the help of a naive Bayesian class fitter trained using the q2-feature-class plugin (Bokulich et al., 2018). The Greengenes 13_8 99% OTUs reference sequences database (McDonald et al., 2012) was used to teach the fi cator class, taken from the ftp address: http://qiime.org/home_static/dataFiles.html Alpha-diversity (Shannon’s diversity index) and beta-diversity (Bray-Curtis distance) analyzes were carried out. The overall structural similarity and variation (beta diversity between samples) between the bacterial element of urine microbiomes from the patients undergoing dialysis, patients after kidney transplantation, and controls were then examined using the Bray–Curtis dissimilarity distance analysis with Principal Coordinates Analysis (PCoA).
2.6. Statistical analyzes
Statistical analyzes were performed using Statistica, version 13.4 (Dell Inc.). The compliance of the empirical data distributions with the normal distribution was verified with the Shapiro–Wilk W test. Due to the fact that the distribution of data was not consistent with the normal distribution, the analysis of differences between the study groups was assessed using the Kruskal-Wallis test with the post-hoc multiple comparison test (Dunn’s test).
2.7. Comparison of the bacterial element of the urinary tract microbiome between patients undergoing dialysis and patients after kidney transplantation
In order to differentiate bacterial species identified in the 16S rRNA amplicon sequencing between the samples in the groups of patients undergoing dialysis and patients after kidney transplantation, with the positive or negative results of urine culture, statistical analyzes were performed using the chi square test. The analyzes were carried out in the following scheme: (i) patients undergoing dialysis with a positive urine culture vs. patients undergoing dialysis with a negative urine culture, (ii) patients after kidney transplantation with a positive urine culture vs. patients after kidney transplantation with a negative urine culture, and (iii) patients undergoing dialysis and patients after kidney transplantation with a positive urine culture vs. patients undergoing dialysis and patients after kidney transplantation with a negative urine culture.
3. Results
3.1. Characteristics of patients and controls
In total, 100 patients and 50 control individuals, with the age range of 18–80 years participated in the study. In the group of patients undergoing dialysis, 15 and 35 individuals were treated with peritoneal dialysis (DO) or hemodialysis (HD), respectively. In Table 1 demographic characteristics of study participants are compiled.
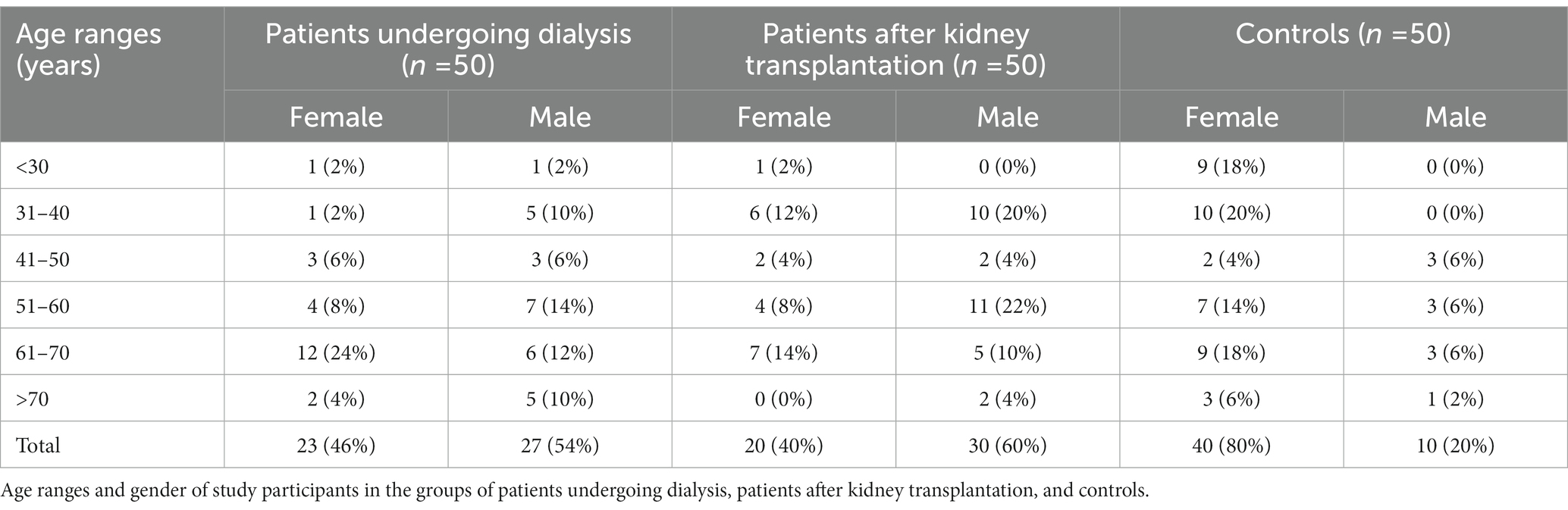
Table 1. Demographic characteristics of the studied groups.
Polycystic kidney disease and glomerulonephritis were the most common indications for treatment with DO and HD, respectively. In 6% of patients, the cause of the renal dysfunction leading to the decision to apply dialysis therapy was undetermined nephropathy.
Glomerulonephritis was the most common indication for kidney transplantation. In 22% patients, the cause of the renal dysfunction remained undetermined nephropathy. Diseases identified in patients undergoing dialysis or patients after kidney transplantation are listed in Table 2.
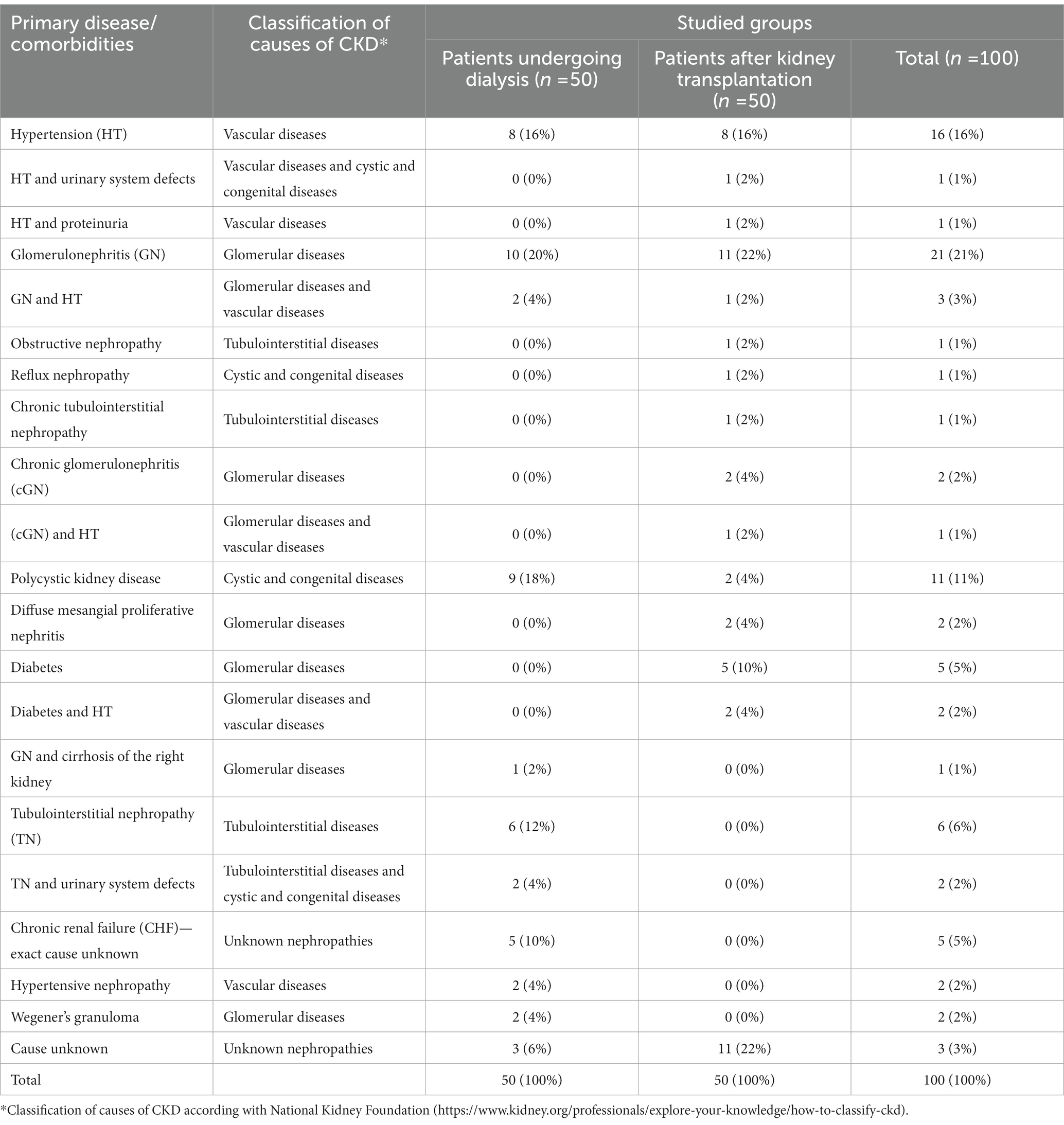
Table 2. Primary disease or urinary system dysfunction, treated with dialysis or resulting in a kidney transplant in studied patients.
3.2. Characteristics of microorganisms identified in urine cultures
The etiological factor responsible for UTI was identified (bacteriuria ≥105 CFU/mL) in 30% of the samples obtained from patients undergoing dialysis and patients after kidney transplantation. Asymptomatic bacteriuria was found to be more common in women (18/43 = 42%) than in men (13/57 = 23%). In the group of patients undergoing dialysis, asymptomatic bacteriuria was recognized in 8/23 women (35%) and 7/27 men (26%), while in patients after kidney transplantation, asymptomatic bacteriuria was recognized in 10/20 women (50%) and 5/30 men (17%) Escherichia coli was the most common etiological factor of asymptomatic bacteriuria and was detected in 80% of patients undergoing dialysis and 67% of patients after kidney transplantation. Staphylococcus spp., Enterococcus spp., Kocuria spp., Morganella morganii, and Enterobacter cloacae complex were also recognized as etiological factors of asymptomatic bacteriuria in these study groups. Detailed information about bacteria identified in urine cultures is presented in Supplementary Tables S1–S3.
3.3. The composition of the bacterial element of urinary tract microbiome varies between patients undergoing dialysis and patients after renal transplantation
The bacterial DNA was extracted from all of the collected 150 urine samples. Out of them, 148 samples met the required qualitative and quantitative criteria and were subjected to the 16S rRNA amplicon sequencing.
Without taking into account the division into study groups, Actinobacteria (100%), Firmicutes (97%), Proteobacteria (91%), Fusobacteria (50%), Cyanobacteria (40%), Acidobacteria (22%), Bacteroidetes (16%), and Thermi (4%) were found as the most common types of bacteria detected in the assessed samples. The substantial difference in bacteria abundance in urine samples was detected for Proteobacteria between patients undergoing dialysis and controls (p = 0.047), and between patients undergoing dialysis and patients after renal transplantation (p = 0.037). The superior abundance was found in a group of patients undergoing dialysis.
Overall, at the genus level, 76 different genera were detected in the samples derived from patients. The highest number of genera were identified in patients undergoing dialysis (n = 60), followed by patients after renal transplantation (n = 58). In urine samples obtained from controls, 50 different genera were detected. The number and variability of identified bacteria genera of the bacterial element of the urinary tract microbiome between groups are presented in Figure 1.
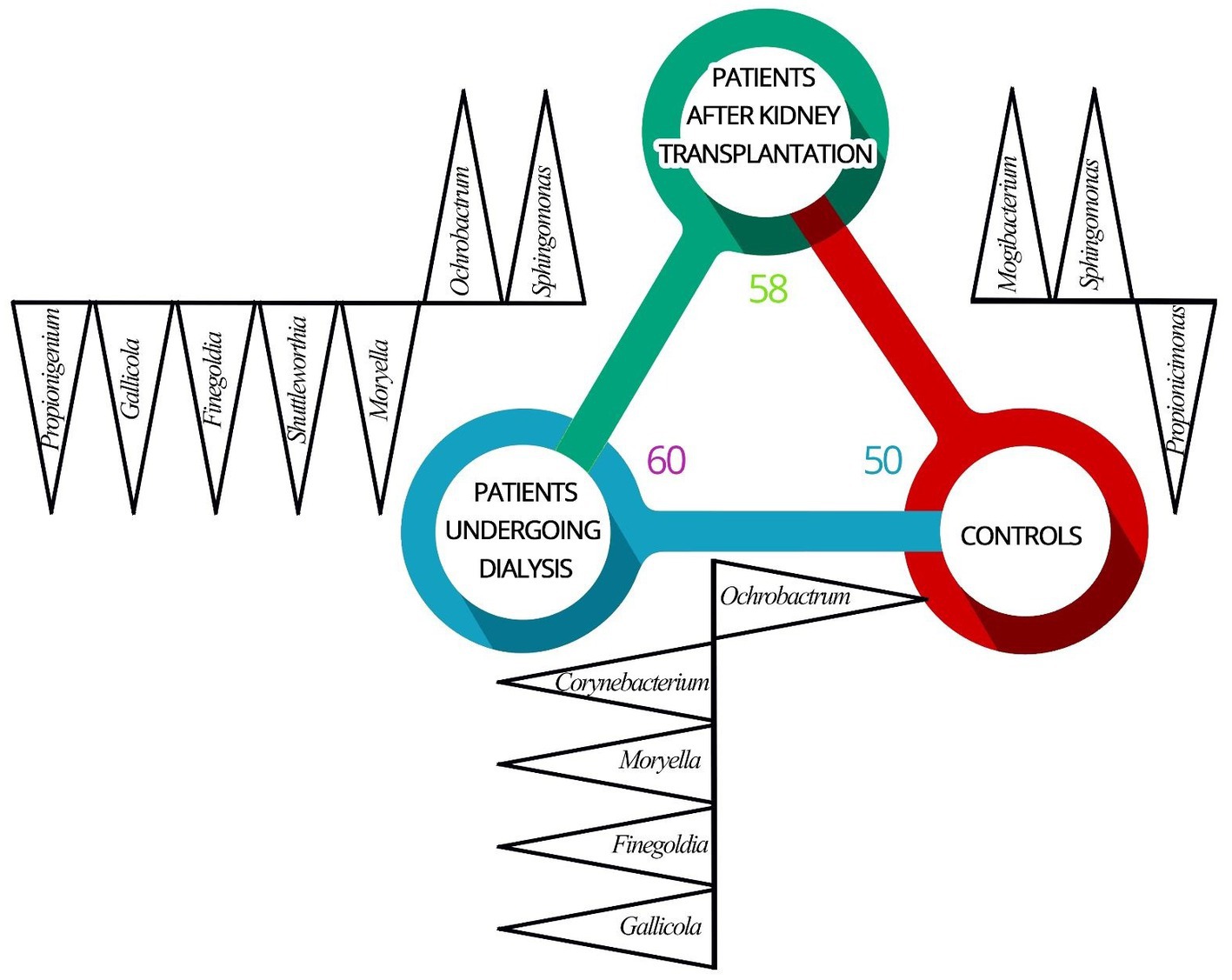
Figure 1. Differences in the composition of the bacterial element of urinary tract microbiome between patients undergoing dialysis (D), patients after renal transplantation (T), and control individuals (C). Identified genera (black triangles) of the bacterial element of the urinary tract microbiome varies between patients undergoing dialysis (blue circle), patients after kidney transplantation (green circle), and control individuals (red circle). Increased abundance in (D) vs. (C) were found for Corynebacterium, Moryella, Finegoldia, and Gallicola. Ochrobactrum was identified more often in (C) comparing to (D). Mogibacterium and Sphingomonas were more often identified in (T) comparing to (C). Propionicimonas was identified with a higher abundance in (C) compared to (T). Increased abundance of genera Moryella, Shuttleworthia, Finegoldia, Gallicola, and Propionigenium was recognized in (D) and Ochrobactrum and Sphingomonas were detected with higher abundance in (T) compared to (D).
The genera Corynebacterium (p = 0.002), Moryella (p = 0.033), Finegoldia (p < 0.001), and Gallicola (p = 0.002) were detected with increased abundance in urine samples of patients undergoing dialysis comparing to urine samples derived from controls. Representatives of the genus Ochrobactrum were identified more often in controls compared to patients undergoing dialysis (p < 0.0001).
Bacteria from genera Mogibacterium (p = 0.049) and Sphingomonas (p < 0.0001) were more often identified in the urine samples from patients after renal transplantation compared to controls. The sequences corresponding to the genus Propionicimonas were identified with a higher abundance in urine samples derived from controls (p = 0.041).
Differences in relative abundances of selected genera were also observed between urine samples obtained from patients undergoing dialysis and patients after kidney transplantation. Increased abundance of genera Moryella (p = 0.033), Shuttleworthia (p = 0.003), Finegoldia (p < 0.001), Gallicola (p = 0.002), and Propionigenium (p = 0.001) was recognized in patients undergoing dialysis. On the other hand, the genera Ochrobactrum (p < 0.0001) and Sphingomonas (p < 0.0001) were detected with higher abundance in urine samples from renal transplant patients.
The obtained values of alpha-diversity indicate heterogeneity of the samples (Figure 2). Bacterial relative abundance in samples derived from patients undergoing dialysis and patients after kidney transplantation was found to be larger as compared to control samples (p = 0.003 and p = 0.004, respectively). There was no significant difference in the relative abundance of bacteria in patients after renal transplantation compared to patients undergoing dialysis (p = 0.68).
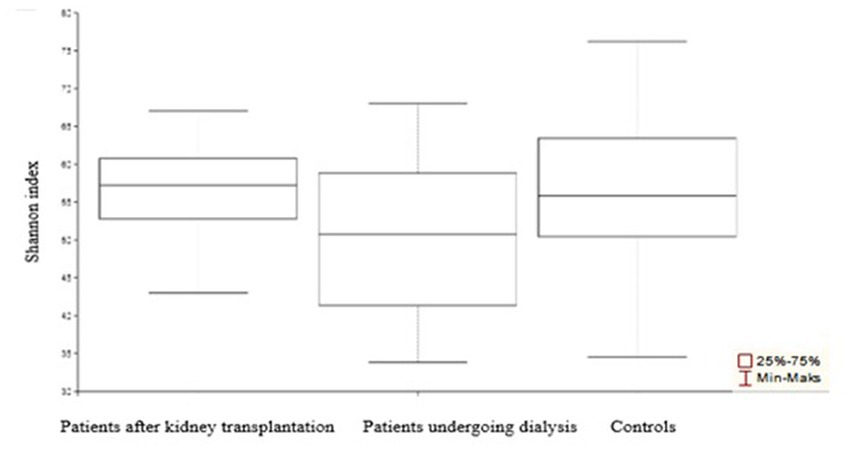
Figure 2. α-Diversity of the urinary tract microbiome bacterial element among patients after transplantation, undergoing dialysis, and control individuals. Compared to control samples, the most diverse was the urine microbiota composition among patients undergoing dialysis.
The analyses of bacterial structural similarity and variation revealed substantial differences between samples were detected and the results are presented in Figure 3. Bacterial element of the urinary tract microbiome in samples from patients undergoing dialysis was the most diverse in terms of composition. On the other hand, urine samples from controls were characterized by the smallest diversity.
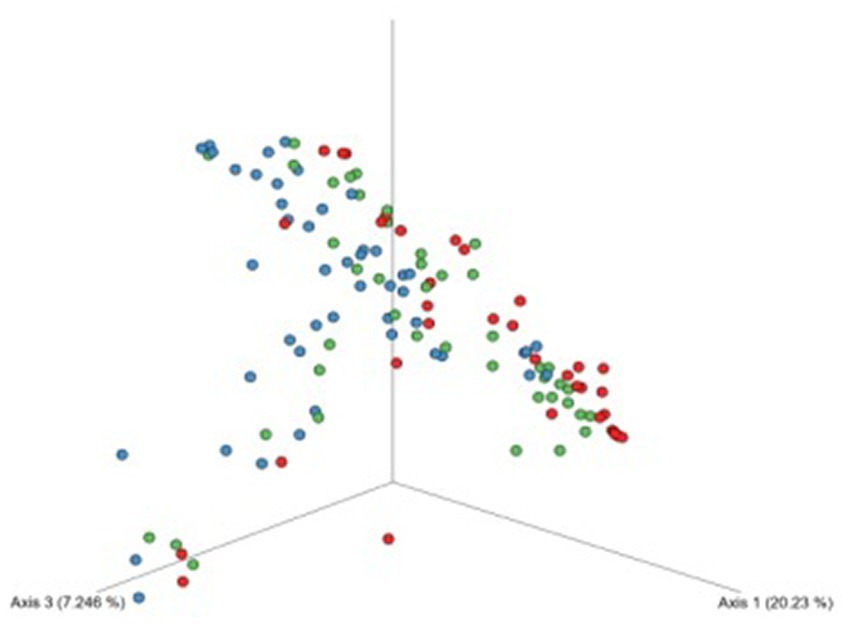
Figure 3. Beta-diversity assessed by the Bray–Curtis dissimilarity tests between patients undergoing dialysis (blue dots), patients after kidney transplantation (green dots), and controls (red dots) based on ASVs. The bacterial element of urine microbiota of patients undergoing dialysis was more variable in composition of microorganisms.
3.4. Composition of the urinary tract microbiome in patients with positive vs. negative urine cultures
Bacterial element of the urinary tract microbiome in patients with positive and negative urine cultures was compared (Figure 4). In urine samples derived from patients with positive urine culture in classical microbiology, lower diversity of bacterial genera comparing to samples from patients with negative urine culture was found in the 16S amplicon sequencing. In addition, bacteria from Finegoldia, Leptotrichia, and Corynebacterium genera were found to be discriminative in patients undergoing dialysis and patients after kidney transplantation compared to individuals without urinary tract dysfunction.
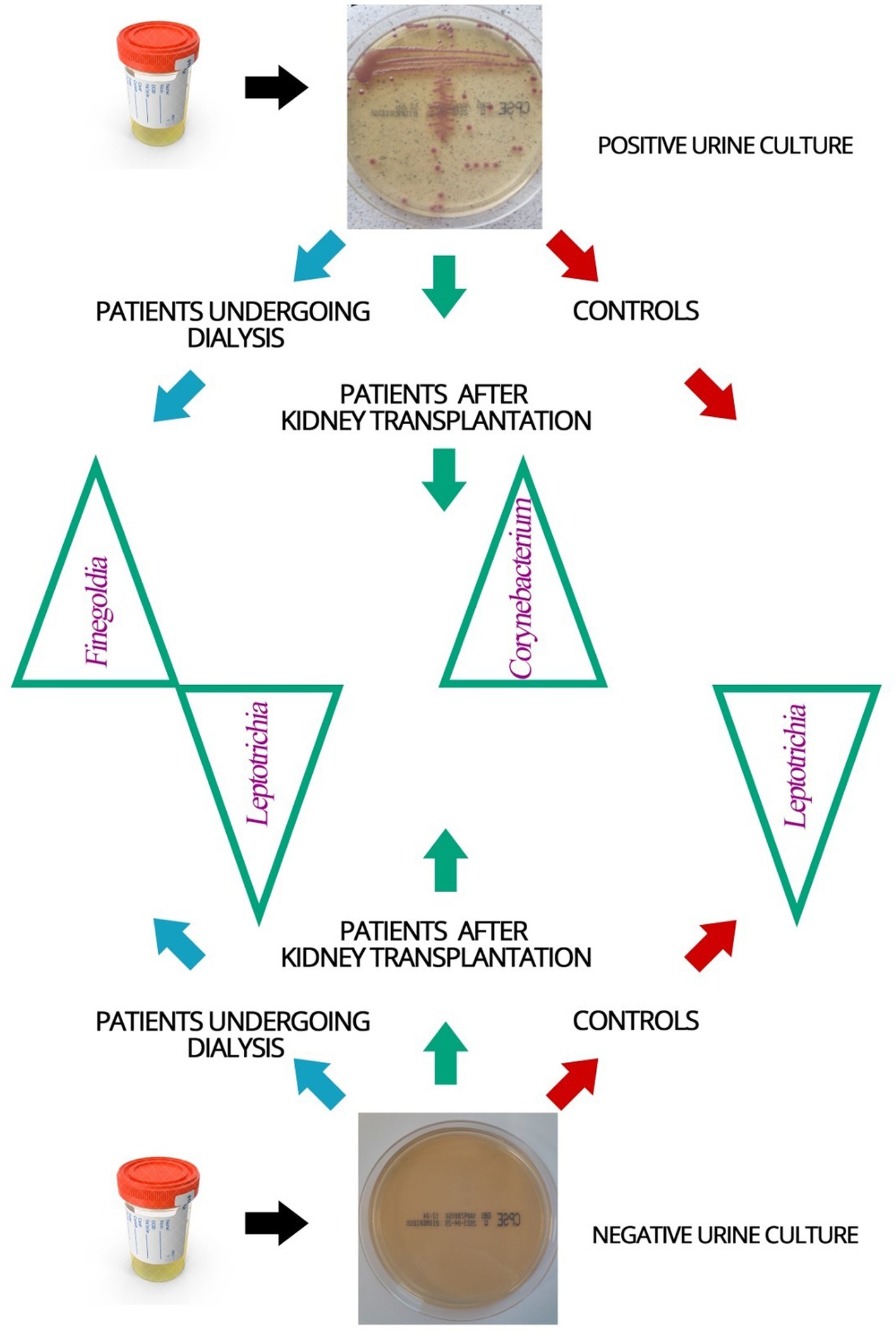
Figure 4. Schematic representation of the bacterial element of the urinary tract microbiome identified in the urine samples of patients and control individuals with positive and negative urine cultures. Blue arrow—dialysis patients, green arrow—patients after kidney transplantation, red arrow—controls; green triangles—names of identified bacteria genera of the bacterial element of the urinary tract microbiome that were found to be discriminative in the studied groups of patients.
In urine samples from patients undergoing dialysis, with a negative urine culture, bacteria Leptotrichia were identified more frequently (p = 0.003), and those from the genus Finegoldia less often (p = 0.038) than in the group of patients undergoing dialysis with a positive urine culture.
Corynebacterium bacteria were recognized less often in the urine samples of kidney transplant patients with negative urine culture than in positive urine culture samples from this group (p = 0.016).
In patients undergoing dialysis and kidney transplant patients with a negative urine culture, Leptotrichia was identified in the larger number of samples (p = 0.037) than in patients undergoing dialysis and kidney transplant patients with a culture-positive urine.
These results point to Leptotrichia, Finegoldia and Corynebacterium as potential diagnostics indicators of UTI.
4. Discussion
Chronic renal failure is a global health problem (Qiu et al., 2017) with the overall incidence of 242 people per million and an annual increase of 8% (Kim, 2019). In Poland, about 4.2 million people suffer from CKD (Gellert et al., 2020). Since the prevalence of CKD is calculated based on the number of patients with the end-stage disease only (Włodarczyk and Felcenloben, 2017–2018), the actual prevalence of CKD could be underestimated, both in Poland and worldwide (Włodarczyk and Felcenloben, 2017–2018).
UTI complication in patients undergoing dialysis is a significant problem in the process of renal replacement therapy that increases the frequency of hospitalization and the risk of death (Gołębiewska et al., 2011) Also, in the group of kidney recipients, UTI is a leading cause of post-transplant infectious complications in both men and women (Abbott et al., 2004; Tenke et al., 2008; Naber et al., 2010). In this study, asymptomatic bacteriuria, defined in accordance with the current recommendations (Hryniewicz and Holecki, 2015), was detected in 30% of patients undergoing dialysis or kidney transplantation.
Typical pathogens of the urinary tract are also most often responsible for asymptomatic bacteriuria in patients undergoing dialysis and patients after kidney transplantation. Thus, the results of our study are in line with previous reports showing the presence of Escherichia coli in 70% (Willner et al., 2014), 72.9% (Michno et al., 2018), or 82% (Olenski et al., 2019) of the urine samples with a positive culture result.
In order to extend the knowledge about urobiome, we evaluated groups of patients rarely included in microbiome studies, namely patients undergoing dialysis and patients after kidney transplantation. Assessment of the bacterial composition using 16S amplicon sequencing revealed 60 and 58 genera in urine samples from patients undergoing dialysis and patients after kidney transplantation, respectively. In controls, 50 different genera were recognized.
Culture-based methods, compared to the 16S rRNA amplicon sequencing, are less effective in detecting bacteria in urine samples and identify a limited number of microorganisms, mainly aerobic and fast-growing bacteria such as Escherichia coli (García and Isenberg, 2010; Wolfe and Brubaker, 2015). Using classical microbiology techniques, four genera of microorganisms were detected in urine samples from individuals without urinary tract dysfunction, 12 genera of bacteria in urine samples from patients undergoing dialysis, and 16 genera of microbes in urine samples from patients after kidney transplantation. In contrast, using 16S rRNA sequencing, we recognized higher bacterial heterogeneity, 58 genera were recorded in urine samples from patients after kidney transplantation and 60 genera of bacteria in the group of patients undergoing dialysis. Also, 37 detected genera were found to be common to all study groups.
Since as many as 80% of the phylotypes detected by 16S rRNA sequencing correspond to bacterial species that cannot be cultured in vitro (Jach and Sajnaga, 2017), the differences in the number of recognized microbes identified in our study are coherent. Such large disproportion in the number of bacterial genera obtained due to classical microbiology and 16S rRNA sequencing, as well as the identification of non-cultivated microorganisms, gave the opportunity to get a more complete picture of the microbiome and highlighted the role of molecular biology methods in the clinical examination of urine samples. Therefore, a combination of 16S rRNA gene sequencing along with quantitative urine culture would provide a reliable characterization of microbial communities in the urinary tract, since culture-based approaches would allow an approximate bacterial quantification and would therefore be useful as clinically relevant indicators (Perez-Carrasco et al., 2021).
Bacteria of the genera Corynebacterium, Moryella, Finegoldia, and Gallicola were more often identified in urine samples from patients undergoing dialysis than in control individuals, in which the dominant genus was Ochrobactrum. It has been reported that the midstream urinary microbiomes varied in adult patients with CKD and that variability was lower in more advanced CKD (Kramer et al., 2018). In recent years, there has been an interest in rare or infrequently described Corynebacterium species, which are emerging opportunistic pathogens considered to be potential uropathogens due to their detection in patients with UTIs (Barberis et al., 2021). Corynebacterium phoceense has been isolated from the urine of a kidney transplant recipient (Cresci et al., 2016). Ochrobactrum anthropi is considered atypical for dialysis peritonitis, a major complication of peritoneal dialysis therapy, as well as for other human infections (Gałgowska et al., 2020). However, this bacterium is an increasingly recognized cause of infection in immunocompromised hosts such as patients with kidney failure treated by dialysis (Abbassi-Nik et al., 2022).
Also, the composition of the urobiome in patients after kidney transplantation has not been sufficiently evaluated. Rani et al. indicated that urine microbiota in kidney transplant recipients was less diverse and dominated by potentially pathogenic Gram-negative bacteria of the genus Escherichia or Enterobacter, while urine microbiome in controls showed greater diversity and a higher incidence of non-pathogenic Gram-positive organisms such as Propionibacterium, Corynebacterium, and Mobiluncus (Rani et al., 2017). The study showed significant differences in the presence of bacteria of the genera Mogibacterium and Sphingomonas, which were more often identified in urine samples from transplant patients than in controls (Rani et al., 2017). The results of these studies may suggest that under the multiple stressors associated with kidney transplantation (including antibiotics or immunosuppression), the urinary microbiota of kidney-transplant recipients can decrease in diversity compared to healthy controls, while the abundance of opportunistic pathogens may increase, which may favor the promotion of antibiotic resistance (Rani et al., 2017).
Mogibacterium are Gram-positive, strictly anaerobic bacilli present in the oral environment and involved in oral infections (Casarin et al., 2012). Previously, the role of these microorganisms as the cause of urolithiasis was evaluated and the study concluded that differences and interactions between gut bacteria might predict certain types of urolithiasis (Zhao et al., 2020). The genus Sphingomonas is comprised of more than 30 species including human pathogen, Sphingomonas paucimobilis (Ryan and Adley, 2010). The most Sphingomonas infections are nosocomial and usually occur in immunocompromised individuals, including the kidney transplant patients (Ryan and Adley, 2010; Yu et al., 2015).
In a pilot study on the urinary microbiome of 21 renal transplant recipients and nine patients with acute kidney injury, rich and differentiated microbiome was found (Gerges-Knafl et al., 2020). Also, the same study demonstrated significant differences in the certain bacterial genera profiles between renal transplant recipients and patients with acute kidney injury, but the overall diversity did not differ between the two groups (Gerges-Knafl et al., 2020). In our study, comparing the urinary microbiota of patients undergoing dialysis and patients after transplantation, genera of Sphingomonas, Ochrobactrum, and Actinocorallia were found with a higher frequency in patients after kidney transplantation. On the other hand, genera Moryella, Shuttleworthia, Finegoldia, Gallicola, and Propionigenium were identified in urine samples from patients undergoing dialysis with a significantly higher frequency than in urine samples from patients after kidney transplantation. In the future research, numerous and longitudinal sampling of the patient’ urinary microbiome should be implemented to detect deviations from microbiome stability. If these changes indicate a risk of organ damage or loss, analysis of microbiome could be useful as a non-invasive method for early detection of transplanted organ failure (Campbell et al., 2020).
The genus Ochrobactrum includes nine species, with Ochrobactrum anthropi, Gram-negative oxygen-rod, increasingly recognized as a potentially problematic, opportunistic, and nosocomial pathogen (Hagiya et al., 2013). Asymptomatic bacteriuria associated with the administration of anti-thymocytic globulin contaminated with Ochrobactrum anthropi have been described in organ recipients (Gałgowska et al., 2020). Other complications caused by Ochrobactrum anthropi were associated with a superinfection of tissues implanted from deceased donors or contamination of the preservative fluid (Gałgowska et al., 2020).
Moryella indoligenes is a strictly anaerobic bacterium from the Lachnospiraceae family of the order Clostridiales (Carlier et al., 2007). Bacteria belonging to the genus Moryella were also detected in urine samples (Sabat et al., 2017) and the first case of bacteriemia involving this microorganism has been described in a patient with a history of prostatic hyperplasia complicated by recurrent UTI (Hansen et al., 2020).
Shuttleworthia is another discriminative type of bacteria found in our study. Previously Shuttleworthia has been suggested as a potential biomarker of inflammation in the urinary tract (Ling et al., 2017).
Here, the genus Finegoldia (formerly Peptostreptococcus) was identified in a high percentage in all three study groups with the highest number detected in the group of patients undergoing dialysis. Finegoldia are Gram-negative anaerobic opportunistic microorganisms that colonize the skin and mucous membranes (Song and Finegold, 2007; Murphy and Frick, 2013). The representative of this genus, Peptostreptococcus prevotii was often identified in men with non-gonococcal urethritis, while Peptostreptococcus magnus was the most common anaerobic species detected in patients without urethritis (Tungland, 2018). Finegoldia is also present in the urine of both sexually active adult males and sexually inactive adolescents (Tungland, 2018). In another study, two specific species of Finegoldia found in the urine of the bladder, including Finegoldia magna, were associated with preoperative worsening of urinary symptoms in women undergoing stress urinary incontinence and pelvic prolapse (Fok et al., 2018).
A comparison of 16S rRNA amplicon sequencing results in mid-stream urine samples from renal transplant patients with confirmed interstitial fibrosis and tubular atrophy (IFTA) and healthy controls showed that in IFTA patients, Streptococcus and Lactobacillus were more commonly identified than in controls (Modena et al., 2017). In patients with IFTA an increase in the number of pathogenic bacteria, Propionibacterium acnes (formerly Cutibacterium acnes), Prevotella disiens, Gardnerella vaginalis, and Finegoldia magna was found (Modena et al., 2017). It has been suggested that as the enrichment of uropathogens increases, the immune response in IFTA patients may be enhanced. In turn, it might result in an increased risk of transplant rejection in these individuals (Modena et al., 2017).
In another study, Colas et al. identified major changes in the urinary microbiota of patients following kidney transplantation with a significant impact on recipient status (spontaneously tolerant patient—TOL; minimal immunosuppressed patient—MIS; stable patient—STA). Their results highlighted a unique and specific urinary microbiota associated with spontaneous tolerance characterized by a high diversity and a clear Proteobacteria profile. In addition, TOL recipients were characterized by a specific and unique increase in Proteobacteria phylum (Oxalobacteraceae [genus Janthinobacterium], Caulobacteraceae, Comamonadaceae, Moraxellaceae [genus Acinetobacter], Xanthomonadaceae, Achromobacter, Yersinia) (Colas et al., 2020).
In another study performed to identify differences in the urinary microbiome associated with chronic allograft dysfunction (CAD), Corynebacterium was more prevalent in female and male patients with CAD compared to non-CAD female patients. On the other hand, analysis of male CAD and female CAD patients showed greater abundance of phylum Proteobacteria in males (Wu et al., 2018).
In our study, 30% of patients undergoing dialysis and also 30% of patients after kidney transplantation had a positive urine culture with identification of an etiological factor indicative of UTI in classical microbiology. The impact of UTI on distant graft function is not fully understood. The predominance of pathogenic Gram-negative bacteria in urobiome of patients after kidney transplantation, indicated in some reports, highlights the problem of UTIs in recipients (Sabé et al., 2022) and a negative impact of early or late UTIs on the functioning of the transplanted kidney has been confirmed (Rani et al., 2017; Fiorentino et al., 2019; Pesce et al., 2019).
While the V3-V4 hypervariable regions of 16S rRNA are frequently used for human microbiome profiling, sequencing only two fragments of the 16S rRNA gene does not allow to narrow down the classified the bacteria to the species level due to the homology between sequences of phylogenetically related bacteria and/or issues related to bacterial nomenclature and taxonomy (Vetrovsky and Baldrian, 2013; Bukin et al., 2019; Winand et al., 2020). Many investigators have found resolution problems at the genus and/or species level with 16S rRNA gene sequencing data (Janda and Abbott, 2007; Liu et al., 2020), especially for the family Enterobacteriaceae (in particular, Enterobacter and Pantoea), Acinetobacter baumannii—A. calcoaceticus complex, Achromobacter, Stenotrophomonas, and Actinomyces (Chang et al., 2005; Janda and Abbott, 2007). In addition, 16S rRNA gene sequencing cannot distinguish between recently diverged species (Peker et al., 2019). The difference between the closest and next closest match to the unknown strain is < 0.5% divergence (>99.5% similarity) (Petti, 2007; Paul et al., 2019). In these circumstances, such small differences cannot justify choosing the closest match as a definitive identification. Since classification to the species level based on V3-V4 hypervariable regions of 16S rRNA is debatable and usually not achievable (Hiergeist et al., 2023), we performed data analysis at the bacterial genus level. Also, due to the inability to quantify microorganisms in amplicon sequencing, only qualitative assessment of microorganisms identified by this method was performed. Thus, in this study, data analysis at the level of bacterial genus has been performed. Also, due to the inability to quantify microorganisms in amplicon sequencing, only qualitative analyzes of microorganisms identified by this method were performed.
Summarizing, our study provides evidence of the diversity of bacterial composition in urine samples in patients undergoing dialysis and patients after kidney transplantation compared to unaffected individuals. The applied amplicon sequencing allowed a better characterization of the composition of urobiome than classical microbiology methods. Leptotrichia, Finegoldia, and Corynebacterium were revealed as discriminative bacteria in patients undergoing dialysis and after kidney transplantation with UTI. This latest finding may form the basis of a future diagnostic process involving these bacteria as diagnostic biomarkers or in future treatment strategies for the diseases discussed here.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: BioProject: PRJNA949101.
Ethics statement
The study protocol was approved by the Bioethics Committee at Poznan University of Medical Sciences (nr 942/14 (04.12.2014), nr 191/15 (05.02.2015) and 1170/19 (05.12.2019)). The possible consequences of the study were explained, and informed consent was obtained from all participants, according to the Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study.
Author contributions
MJ and MG designed the experiment. MJ and PP collected the urine samples and prepared bacterial genetic material to 16S rRNA sequencing. KJ and MR performed the amplicon sequencing, supervised by RP. MK and KP performed medical examination and enrolled patients in this study. MJ conducted experiments using classical microbiology. DN-M performed the statistical analysis. MJ, JK, and MG participated in the data analysis. MJ and JK wrote the manuscript. MG revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Science Centre in Poland, grant no. 2015/19/N/NZ6/01780 (to MJ).
Acknowledgments
Special thanks to colleagues at the Chair and Department of Genetics and Pharmaceutical Microbiology, PUMS, for technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1187625/full#supplementary-material
References
Abbassi-Nik, A., Sprenger-Mähr, H., Zitt, E., Hartmann, G., Dirschmid, H., and Lhotta, K. (2022). Ochrobactrum anthropi infection in dialysis patients: a case report and literature summary. Clin. Nephrol. 97, 188–192. doi: 10.5414/CN110692
Abbott, K. C., Swanson, S. J., Richter, E. R., Bohen, E. M., Agodoa, L. Y., Peters, T. G., et al. (2004). Late urinary tract infection after renal transplantation in the United States. Am. J. Kidney Dis. 44, 353–362. doi: 10.1053/j.ajkd.2004.04.040
Ariza-Heredia, E. J., Beam, E. N., Lesnick, T. G., Cosio, F. G., Kremers, W. K., and Razonable, R. R. (2014). Impact of urinary tract infection on allograft function after kidney transplantation. Clin. Transpl. 28, 683–690. doi: 10.1111/ctr.12366
Barberis, C. M., Traglia, G. M., Almuzara, M. N., Rocha, D. J. P. G., Santos, C. S., Aguiar, E. R. G. R., et al. (2021). Corynebacterium phoceense – a rare Corynebacterium species isolated from a urine sample. Access Microbiol. 3:000197. doi: 10.1099/acmi.0.000197
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Britt, N. S., Hagopian, J. C., Brennan, D. C., Pottebaum, A. A., Santos, C. A. Q., Gharabagi, A., et al. (2017). Effects of recurrent urinary tract infections on graft and patient outcomes after kidney transplantation. Nephrol. Dial. Transpl. 32, 1758–1766. doi: 10.1093/ndt/gfx237
Bukin, Y. S., Galachyants, Y. P., Morozov, I. V., Bukin, S. V., Zakharenko, A. S., and Zemskaya, T. I. (2019). The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci. Data. 6:190007. doi: 10.1038/sdata.2019.7
Callahan, B., McMurdie, P., Rosen, M., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Campbell, P. M., Humphreys, G. J., Summers, A. M., Konkel, J. E., Knight, C. G., Augustine, T., et al. (2020). Does the microbiome affect the outcome of renal transplantation? Front. Cell. Infect. Microbiol. 10:558644. doi: 10.3389/fcimb.2020.558644
Carlier, J. P., K’Ouas, G., and Han, X. Y. (2007). Moryella indoligenes gen. nov., sp. nov., an anaerobic bacterium isolated from clinical specimens. Int. J. Syst. Evol. Microbiol. 57, 725–729. doi: 10.1099/ijs.0.64705-0
Casarin, R. C., Saito, D., Santos, V. R., Pimentel, S. P., Duarte, P. M., Casati, M. Z., et al. (2012). Detection of Mogibacterium timidum in subgingival biofilm of aggressive and non-diabetic and diabetic chronic periodontitis patients. Braz. J. Microbiol. 43, 931–937. doi: 10.1590/S1517-83822012000300012
Chang, H. C., Wei, Y. F., Dijkshoorn, L., Vaneechoutte, M., Tang, C. T., and Chang, T. C. (2005). Species-level identification of isolates of the Acinetobacter calcoaceticus – Acinetobacter baumannii complex by sequence analysis of the 16S-23S rRNA gene spacer region. J. Clin. Microbiol. 43, 1632–1639. doi: 10.1128/JCM.43.4.1632-1639.2005
Colas, L., Mongodin, E. F., Montassier, E., Chesneau, M., Guerif, P., Hittle, L., et al. (2020). Unique and specific proteobacteria diversity in urinary microbiota of tolerant kidney transplanted recipients. Am. J. Transplant. 20, 145–158. doi: 10.1111/ajt.15549
Coussement, J., Kaminski, H., Scemla, A., and Manuel, O. (2020). Asymptomatic bacteriuria and urinary tract infections in kidney transplant recipients. Curr. Opin. Infect. Dis. 33, 419–425. doi: 10.1097/QCO.0000000000000678
Cresci, M., Ibrahima Lo, C., Khelaifia, S., Mouelhi, D., Delerce, J., di Pinto, F., et al. (2016). Corynebacterium phoceense sp. nov., strain MC1T a new bacterial species isolated from human urine. NMNI 14, 73–82. doi: 10.1016/j.nmni.2016.09.001
Fiorentino, M., Pesce, F., Schena, A., Simone, S., Castellano, G., and Gesualdo, L. (2019). Updates on urinary tract infections in kidney transplantation. J. Nephrol. 32, 751–761. doi: 10.1007/s40620-019-00585-3
Fok, C. S., Gao, X., Lin, H., Thomas-White, K. J., Mueller, E. R., Wolfe, A. J., et al. (2018). Urinary symptoms are associated with certain urinary microbes in urogynecologic surgical patients. Int. Urogynecol. J. 29, 1765–1771. doi: 10.1007/s00192-018-3732-1
Fricke, W. F., Maddox, C., Song, Y., and Bromberg, J. S. (2014). Human microbiota characterization in the course of renal transplantation. Am. J. Transplant. 14, 416–427. doi: 10.1111/ajt.12588
Gałgowska, J., Ruszkowski, J., Chmielewski, M., and Lichodziejewska-Niemierko, M. (2020). Dialysis peritonitis caused by an atypical pathogen Ochrobactrum anthropi. Ochrobactrum anthropi as an unusual cause of dialysis-associated peritonitis. Nephrol. Forum 13, 138–141.
García, L. S., and Isenberg, H. D., (2010) Clinical microbiology procedures handbook. 3rd edition, Vol. 6 (Washington, DC: ASM Press)
Gellert, R., Durlik, M., and Małgorzewicz, S., (2020) Raport 2019. National Study of Nephrological Patients, Kidney Disease: a population-based polish Nationwide study, Hope for Health Foundation, nephrology forum, 13, nr 3, 149–163.
Gerges-Knafl, D., Pichler, P., Zimprich, A., Hotzy, C., Barousch, W., Lang, R. M., et al. (2020). The urinary microbiome shows different bacterial genera in renal transplant recipients and non-transplant patients at time of acute kidney injury – a pilot study. BMC Nephrol. 21:117. doi: 10.1186/s12882-020-01773-1
Gołębiewska, J., Dębska-Ślizień, A., and Rutkowski, B. (2011). Urinary tract infections in patients after kidney transplantation. Nephrol. Forum 4, 110–118.
Grabe, M., Bartoletti, R., Bjerklund Johansen, T. E., Cai, T., Çek, M., Köves, B., et al. (2015) Guideliness on urological infections. European Association of Urology 2015. Available at: http://uroweb.org/wp-content/uploads/19-Urological-infectionsLR2.pdf, 1–86.
Hagiya, H., Ohnishi, K., Maki, M., Watanabe, N., and Murase, T. (2013). Clinical characteristics of Ochrobactrum anthropi bacteremia. J. Clin. Microbiol. 51, 1330–1333. doi: 10.1128/JCM.03238-12
Hansen, S., Lřfberg, S. V., Nielsen, D. K., Kobberø, H., and Justesen, U. S. (2020). Bacteraemia with Moryella indoligenes and Fastidiosipila sanguinis: a case report. Access Microbiol. 2:acmi000108. doi: 10.1099/acmi.0.000108
Hiergeist, A., Ruelle, J., Emler, S., and Gessner, A. (2023). Reliability of species detection in 16S microbiome analysis: Comparison of five widely used pipelines and recommendations for a more standardized approach. PLoS One 18:e0280870.
Hryniewicz, W., and Holecki, M. (2015) Recommendations for diagnosis, therapy and prevention of urinary tract infections in adults. Publishing house financed from the funds at the disposal of the minister of health under the health program entitled: "National Program for the Protection of Antibiotics for 2011-2015", Warsaw.
Jach, M. E., and Sajnaga, E., (2017) Composition and function of the intestinal microbiome based on metagenomic studiem. Abstract-book-Metagenomy-2017.Pdf.
Janda, J. M., and Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Johnson, J. S., Spakowicz, D. J., Hong, B.-Y., Petersen, L. M., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10:5029. doi: 10.1038/s41467-019-13036-1
Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kim, G. H. (2019). Pharmacologic treatment of chronic hyperkalemia in patients with chronic kidney disease. Electrolyte Blood Press. 17, 1–6. doi: 10.5049/EBP.2019.17.1.1
Koga, S., Yamanaga, S., Hidaka, Y., Tanaka, K., Kaba, A., Toyoda, M., et al. (2022). Influence of graft ureter length, a donor-related factor, on urinary tract infections after living-donor kidney transplantation: a single-center analysis of 211 cases. Transpl. Int. 35:10754. doi: 10.3389/ti.2022.10754
Kramer, H., Kuffel, G., Thomas-White, K., Wolfe, A. J., Vellanki, K., Leehey, D. J., et al. (2018). Diversity of the midstream urine microbiome in adults with chronic kidney disease. Int. Urol. Nephrol. 50, 1123–1130. doi: 10.1007/s11255-018-1860-7
Lewis, D. A., Brown, R., Williams, J., White, P., Jacobson, S. K., Marchesi, J. R., et al. (2013). The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front. Cell. Infect. Microbiol. 3:41. doi: 10.3389/fcimb.2013.00041
Ling, Z., Liu, F., Shao, L., Cheng, Y., and Li, L. (2017). Dysbiosis of the urinary microbiota associated with urine levels of proinflammatory chemokine interleukin-8 in female type 2 diabetic patients. Front. Immunol. 8:1032. doi: 10.3389/fimmu.2017.01032
Liu, P.-Y., Wu, W.-K., Chen, C.-C., Panyod, S., Sheen, L. Y., and Wu, M. S. (2020) Evaluation of compatibility of 16S rRNA V3V4 and V4 amplicon libraries for clinical microbiome profiling. bioRxiv. [Epub ahead of preprint]. doi: 10.1101/2020.08.18.256818
Magistro, G., and Stief, C. G. (2019). The urinary tract microbiome: the answer to all our open questions? Eur. Urol. Focus 5, 36–38. doi: 10.1016/j.euf.2018.06.011
McDonald, D., Price, M., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Michno, M., Sydor, A., Wałaszek, M., and Sułowicz, W. (2018). Microbiology and drug resistance of pathogens in patients hospitalized at the nephrology department in the South of Poland. Pol. J. Microbiol. 67, 517–524. doi: 10.21307/pjm-2018-061
Modena, B. D., Milam, R., Harrison, F., Cheeseman, J. A., Abecassis, M. M., Friedewald, J. J., et al. (2017). Changes in urinary microbiome populations correlate in kidney transplants with interstitial fibrosis and tubular atrophy documented in early surveillance biopsies. Am. J. Transplant. 17, 712–723. doi: 10.1111/ajt.14038
Murphy, E. C., and Frick, I. M. (2013). Gram-positive anaerobic cocci – commensals and opportunistic pathogens. FEMS Microbiol. Rev. 37, 520–553. doi: 10.1111/1574-6976.12005
Naber, K. G., Schaeffer, A. J., Hynes, C. F., Matsumoto, T., Shoskes, D. A., and Bjerklund Johansen, T. E.. (2010) EAU/International Consultation on Urological Infections. European Association of Urology. Arnhem.
Naumnik, B., and Myśliwiec, M. (2013). Dialysis: heparin-free haemodialysis-use and outcomes. Nat. Rev. Nephrol. 9, 381–382. doi: 10.1038/nrneph.2013.106
Olenski, S., Scuderi, C., Choo, A., Bhagat Singh, A. K., Way, M., and Jeyaseelan, L., et.al. (2019) Urinary tract infections in renal transplant recipients at a quaternary care Centre in Australia. BMC Nephrol., 20,:479. doi: 10.1186/s12882-019-1666-6
Paul, B., Dixit, G., Murali, T. S., and Satyamoorthy, K. (2019). Genome-based taxonomic classification. Genome 62, 45–52. doi: 10.1139/gen-2018-0072
Pearce, M. M., Hilt, E. E., Rosenfeld, A. B., Zilliox, M. J., Thomas-White, K., Fok, C., et al. (2014). The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. MBio 5, e01283–e01214. doi: 10.1128/mBio.01283-14
Peker, N., Garcia-Croes, S., Dijkhuizen, B., Wiersma, H. H., van Zanten, E., Wisselink, G., et al. (2019). A comparison of three different bioinformatics analyses of the 16S-23S rRNA encoding region for bacterial identification. Front. Microbiol. 10:620. doi: 10.3389/fmicb.2019.00620
Perez-Carrasco, V., Soriano-Lerma, A., Soriano, M., Gutiérrez-Fernández, J., and Garcia-Salcedo, J. A. (2021). Urinary microbiome: Yin and Yang of the urinary tract. Front. Cell. Infect. Microbiol. 11:617002. doi: 10.3389/fcimb.2021.617002
Pesce, F., Martino, M., Fiorentino, M., Rollo, T., Simone, S., Gallo, P., et al. (2019). Recurrent urinary tract infections in kidney transplant recipients during the first-year influence long-term graft function: a single-center retrospective cohort study. J. Nephrol. 32, 661–668. doi: 10.1007/s40620-019-00591-5
Petti, C. A. (2007). Detection and identification of microorganisms by gene amplification and sequencing. Clin. Infect. Dis. 44, 1108–1114. doi: 10.1086/512818
Price, T. K., Hilt, E. E., Dune, T. J., Mueller, E. R., Wolfe, A. J., and Brubaker, L. (2018). Urine trouble: should we think differently about UTI? Int. Urogynecol. J. Pelvic Floor Dysfunct. 29, 205–210. doi: 10.1007/s00192-017-3528-8
Qiu, Z., Zheng, K., Zhang, H., Feng, J., Wang, L., and Zhou, H. (2017). Physical exercise and patients with chronic renal failure: a meta-analysis. Biomed. Res. Int. 2017, 1–8. doi: 10.1155/2017/7191826
Rani, A., Ranjan, R., McGee, H. S., Andropolis, K. E., Panchal, D. V., Hajjiri, Z., et al. (2017). Urinary microbiome of kidney transplant patients reveals dysbiosis with potential for antibiotic resistance. Transl. Res. 181, 59–70. doi: 10.1016/j.trsl.2016.08.008
Ryan, M. P., and Adley, C. C. (2010). Sphingomonas paucimobilis: a persistent gram-negative nosocomial infectious organism. J. Hosp. Infect. 75, 153–157. doi: 10.1016/j.jhin.2010.03.007
Sabat, A. J., van Zanten, E., Akkerboom, V., Wisselink, G., van Slochteren, K., de Boer, R. F., et al. (2017). Targeted next-generation sequencing of the 16S-23S rRNA region for culture-independent bacterial identification – increased discrimination of closely related species. Sci. Rep. 7:3434. doi: 10.1038/s41598-017-03458-6
Sabé, N., Maristany, M., Tuells, M., Favà, A., Melilli, E., Tubau, F., et al. (2022). Risk factors and outcomes of acute graft pyelonephritis with bacteremia due to multidrug-resistant gram-negative Bacilli among kidney transplant recipients. J. Clin. Med. 11:3165. doi: 10.3390/jcm11113165
Song, Y., and Finegold, S. M. (2007). “Peptostreptococus, Finegoldia, Anaerococcus, Peptoniphilus, Veillonella, and other anaerobic cocci” in Manual of clinical microbiology. eds. P. R. Murray, E. J. Baron, J. H. Jorgensen, M. L. Landry, and M. A. Pfaller. 9th ed (Washington, DC: ASM Press), 862–871.
Tenke, P., Kovacs, B., Bjerklund Johansen, T. E., Matsumoto, T., Tambyah, P. A., and Naber, K. G. (2008). European and Asian guidelines on management and prevention of catheter-associated urinary tract infections. Int. J. Antimicrob. Agents 31, 68–78. doi: 10.1016/j.ijantimicag.2007.07.033
Thomas-White, K., Forster, S. C., Kumar, N., van Kuiken, M., Putonti, C., Stares, M. D., et al. (2018). Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat. Commun. 9:1557. doi: 10.1038/s41467-018-03968-5
Tungland, B. (2018). Microbiota of the male urogenital tract. Human microbiota in health and disease. From pathogenesis to therapy. Chapter 14 Microbiota and the urogenital tract, pathogenesis, and therapies in human microbiota in health and disease, 14.2.1, 605–647.
Vetrovsky, T., and Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923
Whiteside, S. A., Razvi, H., Dave, S., Reid, G., and Burton, J. P. (2015). The microbiome of the urinary tract–a role beyond infection. Nat. Rev. Urol. 12, 81–90. doi: 10.1038/nrurol.2014.361
Willner, D., Low, S., Steen, J. A., George, N., Nimmo, G. R., Schembri, M. A., et al. (2014). Single clinical isolates from acute uncomplicated urinary tract infections are representative of dominant in situ populations. MBio 5, e01064–e01013. doi: 10.1128/mBio.01064-13
Winand, R., Bogaerts, B., Hoffman, S., Lefevre, L., Delvoye, M., Braekel, J. V., et al. (2020). Targeting the 16S rRNA gene for bacterial identification in complex mixed samples: comparative evaluation of second (Illumina) and third (Oxford Nanopore technologies) generation sequencing technologies. Int. J. Mol. Sci. 21:298. doi: 10.3390/ijms21010298
Włodarczyk, E., and Felcenloben, J. (2017–2018) in cooperation with Department of the European Social Fund. Program for early detection of chronic kidney disease for residents of the Łódź Voivodeship. Available at: https://rpo.lodzkie.pl/images/2016/354-poddzialanie-x32/Zalacznik_12_Program.doc
Wolfe, A. J., and Brubaker, L. (2015). “Sterile urine” and the presence of Bacteria. Eur. Urol. 68, 173–174. doi: 10.1016/j.eururo.2015.02.041
Wolfe, A. J., and Brubaker, L. (2019). Urobiome updates: advances in urinary microbiome research. Nat. Clin. Pract. Urol. 16, 73–74. doi: 10.1038/s41585-018-0127-5
Wolfe, A. J., Toh, E., Shibata, N., Rong, R., Kenton, K., FitzGerald, M. P., et al. (2012). Evidence of uncultivated bacteria in the adult female bladder. J. Clin. Microbiol. 50, 1376–1383. doi: 10.1128/JCM.05852-11
Wu, J. F., Muthusamy, A., al-Ghalith, G. A., Knights, D., Guo, B., Wu, B., et al. (2018). Urinary microbiome associated with chronic allograft dysfunction in kidney transplant recipients. Clin. Transpl. 32:e13436. doi: 10.1111/ctr.13436
Yu, H., Meng, H., Zhou, F., Ni, X., Shen, S., and das, U. N. (2015). Urinary microbiota in patients with prostate cancer and benign prostatic hyperplasia. Arch. Med. Sci. 2, 385–394. doi: 10.5114/aoms.2015.50970
Keywords: urobiome, urinary tract infection, dialysis, renal transplantation, urine culture
Citation: Jaworska MM, Pecyna P, Jaskiewicz K, Rydzanicz M, Kaluzna M, Pawlaczyk K, Ploski R, Nowak-Malczewska DM, Karolak JA and Gajecka M (2023) Differences in the composition of the bacterial element of the urinary tract microbiome in patients undergoing dialysis and patients after kidney transplantation. Front. Microbiol. 14:1187625. doi: 10.3389/fmicb.2023.1187625
Edited by:
Arabella Touati, Centre Hospitalier Universitaire de Bordeaux, FranceReviewed by:
Mangesh Vasant Suryavanshi, Cleveland Clinic, United StatesBeata Krawczyk, Gdansk University of Technology, Poland
Copyright © 2023 Jaworska, Pecyna, Jaskiewicz, Rydzanicz, Kaluzna, Pawlaczyk, Ploski, Nowak-Malczewska, Karolak and Gajecka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marzena Gajecka, Z2FtYXJAbWFuLnBvem5hbi5wbA==