 Qing Su1
Qing Su1 Chen Jin2
Chen Jin2 Zhiyuan Bo3
Zhiyuan Bo3 Yi Yang1
Yi Yang1 Jingxian Wang1
Jingxian Wang1 Juejin Wang1Junxi Zhou1Yaqing Chen1Hao Zeng1
Juejin Wang1Junxi Zhou1Yaqing Chen1Hao Zeng1 Gang Chen3*
Gang Chen3* Yi Wang1*
Yi Wang1*- 1Department of Epidemiology and Biostatistics, School of Public Health and Management, Wenzhou Medical University, Wenzhou, China
- 2Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing, China
- 3Department of Hepatobiliary Surgery, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
Background: The gut microbiome is closely related to gastrointestinal (GI) cancer, but the causality of gut microbiome with GI cancer has yet to be fully established. We conducted this two-sample Mendelian randomization (MR) study to reveal the potential causal effect of gut microbiota on GI cancer.
Materials and methods: Summary-level genetic data of gut microbiome were derived from the MiBioGen consortium and the Dutch Microbiome Project. Summary statistics of six GI cancers were drawn from United Kingdom Biobank. Inverse-variance-weighted (IVW), MR-robust adjusted profile score (MR-RAPS), and weighted-median (WM) methods were used to evaluate the potential causal link between gut microbiota and GI cancer. In addition, we performed sensitivity analyses and reverse MR analyses.
Results: We identified potential causal associations between 21 bacterial taxa and GI cancers (values of p < 0.05 in all three MR methods). Among them, phylum Verrucomicrobia (OR: 0.17, 95% CI: 0.05–0.59, p = 0.005) retained a strong negative association with intrahepatic cholangiocarcinoma after the Bonferroni correction, whereas order Bacillales (OR: 1.67, 95% CI: 1.23–2.26, p = 0.001) retained a strong positive association with pancreatic cancer. Reverse MR analyses indicated that GI cancer was associated with 17 microbial taxa in all three MR methods, among them, a strong inverse association between colorectal cancer and family Clostridiaceae1 (OR: 0.91, 95% CI: 0.86–0.96, p = 0.001) was identified by Bonferroni correction.
Conclusion: Our study implicates the potential causal effects of specific microbial taxa on GI cancer, potentially providing new insights into the prevention and treatment of GI cancer through specific gut bacteria.
1. Introduction
In 2020, the five major types of gastrointestinal (GI) cancer (including esophageal, gastric, colorectal, liver, and pancreatic cancers) account for 25.8% of the global cancer incidence and 35.4% of the global cancer-related deaths (Sung et al., 2021). GI cancers are significant contributors to the global burden of cancer and pose a serious challenge to public health (Arnold et al., 2020), so finding the etiology and applying suitable preventive measures are urgent.
There is increasing evidence that intestinal microbiota is closely related to GI cancer (Tong et al., 2021). Intestinal flora is involved in the occurrence and progression of colorectal cancer (CRC) by affecting the inflammatory process in the intestine and producing metabolites (Brennan and Garrett, 2016; Wong and Yu, 2019). Epidemiological studies have shown that the microbial composition differs between CRC patients and healthy controls, and may serve as biomarkers for CRC screening and prognosis (Wong and Yu, 2019). The intestinal microbiota not only influences the occurrence of CRC via local effects but also has long-distance effects on other cancers, for example, affecting the development of liver cancer through the gut–liver axis (Schwabe and Jobin, 2013; Yu and Schwabe, 2017). Intestinal microbial metabolites and microbial components can be transferred to the liver through the gut–liver axis (Ohtani and Hara, 2021). In addition, abundance differences in gut microbes between patients with other GI cancers (e.g., pancreatic, esophageal, and gastric cancers) and healthy controls were also found (Yu et al., 2021; Cheung et al., 2022; Kartal et al., 2022). However, the causality of gut microbiota with GI cancer has not been fully established due to the potential effects of residual confounding and reverse causality.
Mendelian randomization (MR), a common method for examining causal relationships between exposures and outcomes, has been used to explore potential causal associations between gut microbiota and multiple diseases (Li et al., 2022, 2023). Recently, using two-sample MR analysis, two studies reported the potential causal association between intestinal microbiota and CRC and one study reported the potential causality of gut microbiota with gastric cancer (Ni et al., 2022; Long et al., 2023). In addition, there was an MR analysis exploring the relationship of 57 bacterial taxa (including four phyla, eight classes, six orders, 10 families, and 29 genera) with liver cancer (Ma et al., 2023). However, potential causal associations of many other gut microbial taxa with liver cancer are unknown, and the potential causal relationship of gut microbiome with pancreatic and esophageal cancer is not well established. Therefore, this study performed two-sample MR analysis using the genome-wide association study (GWAS) datasets containing 211 bacterial taxa at the phylum to genus level from the MiBioGen consortium (Kurilshikov et al., 2021) and 105 bacterial taxa at the species level from the Dutch Microbiome Project (Lopera-Maya et al., 2022) to reveal the potential causal relationships of gut microbiota with esophageal, gastric, colorectal, liver, and pancreatic cancers.
2. Materials and methods
2.1. Study design
The study design of the present two-sample MR analysis is shown in Figure 1. To reliably infer the potential causality of gut microbiome with GI cancer risk using MR approach, we tried to meet three key assumptions of MR analysis. First, the instrumental variables (IVs) are correlated with gut microbiome. Second, IVs are unrelated to confounders influencing this association. Third, IVs influence the GI cancer risk only through gut microbiota (Davies et al., 2018).
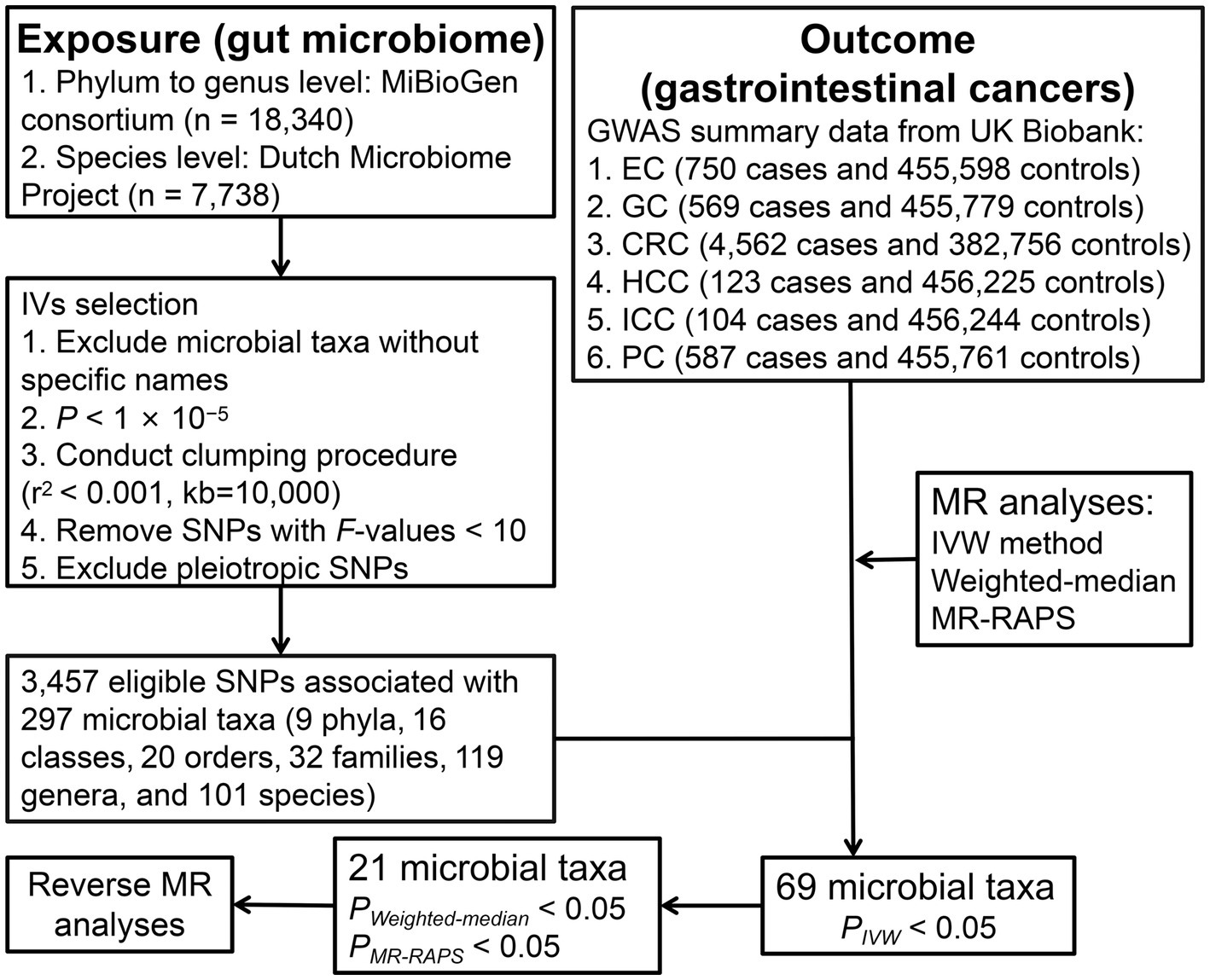
Figure 1. The study design and workflow of the present MR study. EC, esophageal cancer; GC, gastric cancer; CRC, colorectal cancer; HCC, hepatocellular carcinoma; ICC, intrahepatic cholangiocarcinoma; PC, pancreatic cancer; IVs, instrumental variables; MR, Mendelian randomization; SNP, single-nucleotide polymorphism; IVW, Inverse-variance-weighted; and MR-RAPS, MR-robust adjusted profile score.
2.2. Data sources and instruments selection
The genetic data of human gut microbiome at the phylum to genus level were obtained from the multi-ethnic MiBioGen consortium comprising 24 population-based cohorts with 18,340 participants. A total of 211 gut microbial taxa were included in this GWAS dataset, of which 15 were unknown families or genera and were excluded, leaving 196 microbial taxa for MR analysis. The summary statistics on the species level of gut microbiota were derived from the Dutch Microbiome Project including a total of 105 species with 7,738 participants of European ancestry. To obtain more comprehensive results, IVs that attained locus-wide significance (p < 1 × 10−5) were selected. In parallel, single-nucleotide polymorphisms (SNPs) in linkage disequilibrium were excluded by the PLINK clumping method (r2 < 0.001, kb = 10,000). Then, SNPs with F-statistics [formula: R2/K × (N − K − 1)/(1 − R2)] < 10 were removed (Palmer et al., 2012). Finally, we searched the PhenoScanner website for additional phenotypes associated with gut microbiota-related SNPs and removed SNPs associated with confounders (body mass index, waist circumference, smoking, alcohol intake, blood pressure, blood lipids, coronary artery disease, weight, hip circumference, fat percentage, diabetes, worrier or anxious feelings, nervous feelings, chronotype, birth weight, hypothyroidism, gout, Gamma glutamyl transferase, skin cancer, ovarian cancer, malabsorption or coeliac disease, primary biliary cholangitis, and Hodgkin’s disease). Four species with less than three available SNPs were excluded. A total of 297 bacterial taxa were included in the MR analysis.
Genome-wide association study summary statistics for esophageal cancer (750 cases and 455,598 controls), gastric cancer (569 cases and 455,779 controls), CRC (4,562 cases and 382,756 controls), hepatocellular carcinoma (HCC, 123 cases and 456,225 controls), intrahepatic cholangiocarcinoma (ICC, 104 cases and 456,244 controls), and pancreatic cancer (587 cases and 455,761 controls) were obtained from United Kingdom Biobank, with details described elsewhere (Zhou et al., 2018; Jiang et al., 2021). No additional ethics approval or informed consent was required due to our study was based on public databases.
2.3. Statistical analysis
The potential causality of gut microbiota and GI cancer risk was primarily calculated by inverse-variance-weighted (IVW) method. Cochran’s Q test was used for assessment of heterogeneity, using random-effects IVW when heterogeneity was significant (p < 0.05) and, conversely, fixed-effects IVW. The consistency of results was examined by two additional approaches: MR-robust adjusted profile score (MR-RAPS) method and weighted-median (WM) method. The condition for WM method to obtain consistent estimates of causal effects is that half of SNPs are valid IVs (Bowden et al., 2016). MR-RAPS can make robust inferences when it contains weak IVs (Zhao et al., 2019). To assess pleiotropy, we performed MR-Egger and MR-Pleiotropy RESidual Sum and Outlier (MR-PRESSO) tests (Bowden et al., 2015; Verbanck et al., 2018), the latter of which could also detect outliers and test for differences in results before and after eliminating outliers (Verbanck et al., 2018). In addition, leave-one-out analysis was conducted to assess the impact on overall estimates by a single SNP.
To obtain a more rigorous explanation of causality, we used Bonferroni method to establish multiple testing significance thresholds at different taxonomic levels separately based on the number of bacteria under each taxonomic level [5.6 × 10−3 (0.05/9) for phylum, 3.1 × 10−3 (0.05/16) for class, 2.5 × 10−3 (0.05/20) for order, 1.6 × 10−3 (0.05/32) for family, 4.2 × 10−4 (0.05/119) for genus, and 5.0 × 10−4 (0.05/101) for species]. P values reaching nominal significance (p < 0.05) were considered to have nominal potential causal effects. Finally, reverse MR analyses utilizing SNPs correlated with GI cancer (p < 5 × 10−6) as IVs were performed to examine whether GI cancer had a causal impact on gut microbial taxa. “TwoSampleMR” and “MR-PRESSO” packages were used for analyses in R program (version 4.2.2).
3. Results
3.1. Overview
After a series of IV screening steps, a total of 3,457 eligible SNPs from 297 microbial taxa were finally included in this analysis. Details of IVs are listed in Supplementary Table S1. The IVW method identified 69 microbial taxa associated with esophageal cancer, gastric cancer, CRC, HCC, ICC, or pancreatic cancer (Figure 2). However, only 21 microbial taxa remained stable in both WM and MR-RAPS methods (Table 1). The scatter plots of the associations of these 21 microbial taxa with the corresponding GI cancers are shown in Supplementary Figures S1–S6. And the statistical power of these microbial taxa calculated by an online tool1 is presented in Table 1.

Figure 2. The potential causal relationship between gut microbiome and gastrointestinal (GI) cancer risk in the IVW method (p < 0.05). Red represents the risk taxa for GI cancer, blue represents the protective taxa for GI cancer, and white represents no causal taxa for GI cancer. EC, esophageal cancer; GC, gastric cancer; CRC, colorectal cancer; HCC, hepatocellular carcinoma; ICC, intrahepatic cholangiocarcinoma; PC, pancreatic cancer; and NS, no significant association.
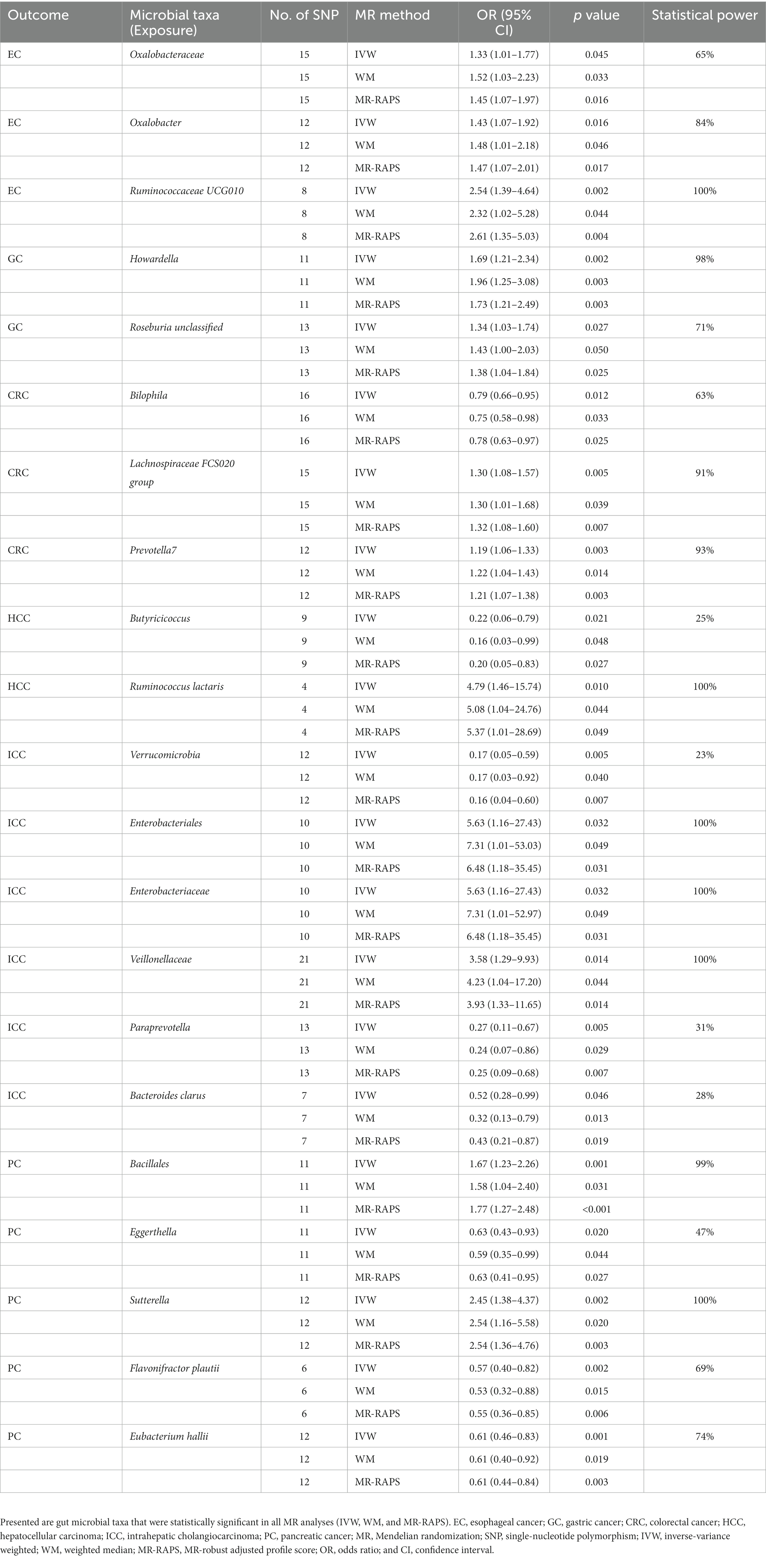
Table 1. Causal associations between genetically predicted 21 microbial taxa and gastrointestinal cancer risk.
3.2. Esophageal cancer
The IVW analysis indicated that Oxalobacteraceae (OR: 1.33, 95% CI: 1.01–1.77), Coprobacter (OR: 1.60, 95% CI: 1.15–2.21), Oxalobacter (OR: 1.43, 95% CI: 1.07–1.92), Ruminococcaceae UCG010 (OR: 2.54, 95% CI: 1.39–4.64), Sellimonas (OR: 1.46, 95% CI: 1.07–1.99), Akkermansia muciniphila (OR: 1.57, 95% CI: 1.13–2.17), Bacteroides salyersiae (OR: 1.24, 95% CI: 1.01–1.52), and Roseburia unclassified (OR: 1.33, 95% CI: 1.06–1.68) were correlated with increased esophageal cancer risk (p < 0.05), whereas Butyricicoccus (OR: 0.56, 95% CI: 0.33–0.94), Lachnospira (OR: 0.44, 95% CI: 0.21–0.93), and Bacteroides plebeius (OR: 0.72, 95% CI: 0.57–0.92) were related to a reduced risk of esophageal cancer (p < 0.05; Figure 2). However, only Oxalobacteraceae, Oxalobacter, and Ruminococcaceae UCG010 maintained consistent results in both WM and MR-RAPS methods (Table 1). Leave-one-out analysis of these three bacterial taxa found that some SNPs of Oxalobacteraceae and Oxalobacter might dominate the positive results (Supplementary Figure S7).
3.3. Gastric cancer
According to IVW method (Figure 2), Bacteroidales S24 7group (OR: 0.56, 95% CI: 0.34–0.90) and Bacteroides eggerthii (OR: 0.69, 95% CI: 0.49–0.97) were associated with a lower risk of gastric cancer (p < 0.05), while Clostridium innocuum group (OR: 1.48, 95% CI: 1.04–2.10), Dialister (OR: 1.85, 95% CI: 1.06–3.25), Howardella (OR: 1.69, 95% CI: 1.21–2.34), Lachnospiraceae NC2004 group (OR: 1.72, 95% CI: 1.10–2.70), Paraprevotella clara (OR: 1.48, 95% CI: 1.10–2.00), Lactobacillus delbrueckii (OR: 1.18, 95% CI: 1.01–1.38), Phascolarctobacterium succinatutens (OR: 1.33, 95% CI: 1.01–1.75), Desulfovibrio piger (OR: 1.55, 95% CI: 1.02–2.35), and Roseburia unclassified (OR: 1.34, 95% CI: 1.03–1.74) were correlated with higher gastric cancer risk (p < 0.05). Of these, only Howardella and Roseburia unclassified remained stable in WM and MR-RAPS methods (Table 1), and SNPs with significant effects were not identified in Howardella but in Roseburia unclassified by leave-one-out analysis (Supplementary Figure S8).
3.4. Colorectal cancer
Bifidobacteriales (OR: 0.75, 95% CI: 0.62–0.91), Bifidobacteriaceae (OR: 0.75, 95% CI: 0.62–0.91), Bilophila (OR: 0.79, 95% CI: 0.66–0.95), Coprococcus2 (OR: 0.80, 95% CI: 0.65–0.99), Fusicatenibacter (OR: 0.82, 95% CI: 0.68–0.99), Odoribacter (OR: 0.73, 95% CI: 0.57–0.94), Ruminococcaceae UCG005 (OR: 0.81, 95% CI: 0.67–0.98), Alistipes finegoldii (OR: 0.84, 95% CI: 0.71–0.99), Bacteroides faecis (OR: 0.93, 95% CI: 0.88–1.00), Lachnospiraceae bacterium 3_1_46FAA (OR: 0.83, 95% CI: 0.70–0.98), Lachnospiraceae bacterium 7_1_58FAA (OR: 0.85, 95% CI: 0.73–1.00), and Roseburia intestinalis (OR: 0.84, 95% CI: 0.72–0.98) were negatively correlated with CRC risk in the IVW approach (p < 0.05; Figure 2). As for Cyanobacteria (OR: 1.19, 95% CI: 1.01–1.40), Eubacterium brachy group (OR: 1.17, 95% CI: 1.03–1.31), Coprobacter (OR: 1.16, 95% CI: 1.02–1.32), Family XIII UCG001 (OR: 1.24, 95% CI: 1.00–1.54), Lachnospiraceae FCS020 group (OR: 1.30, 95% CI: 1.08–1.57), Prevotella7 (OR: 1.19, 95% CI: 1.06–1.33), and Bifidobacterium longum (OR: 1.17, 95% CI: 1.00–1.36), we observed positive associations with CRC in IVW analysis (p < 0.05; Figure 2). However, only Bilophila, Lachnospiraceae FCS020 group, and Prevotella7 obtained similar estimates in WM and MR-RAPS methods (Table 1). Leave-one-out analysis detected that the results of Lachnospiraceae FCS020 group and Prevotella7 remained stable (Supplementary Figure S9).
3.5. Liver cancer
We noticed 11 and 13 bacterial taxa associated with HCC and ICC in IVW test, respectively (Figure 2). Six taxa, namely, Clostridia (OR: 3.21, 95% CI: 1.01–10.22), Enterobacteriales (OR: 4.52, 95% CI: 1.05–19.40), Enterobacteriaceae (OR: 4.52, 95% CI: 1.05–19.40), Ruminococcus torques group (OR: 4.79, 95% CI: 1.09–21.10), Ruminococcus lactaris (OR: 4.79, 95% CI: 1.46–15.74), and Bacteroides clarus (OR: 1.96, 95% CI: 1.09–3.53) were positively associated with HCC (p < 0.05). Of these, Enterobacteriales (OR: 5.63, 95% CI: 1.16–27.43) and Enterobacteriaceae (OR: 5.63, 95% CI: 1.16–27.43) were also positively correlated with ICC risk (p < 0.05). Additionally, Veillonellaceae (OR: 3.58, 95% CI: 1.29–9.93), Alistipes (OR: 5.65, 95% CI: 1.32–24.24), Oscillospira (OR: 6.38, 95% CI: 1.44–28.30), and Slackia (OR: 3.34, 95% CI: 1.06–10.52) were also related to a higher risk of ICC (p < 0.05).
On the contrary, we found negative associations of Christensenellaceae (OR: 0.13, 95% CI: 0.03–0.57), Christensenellaceae R 7group (OR: 0.16, 95% CI: 0.04–0.77), Butyricicoccus (OR: 0.22, 95% CI: 0.06–0.79), Parabacteroides goldsteinii (OR: 0.43, 95% CI: 0.23–0.83), and Lachnospiraceae bacterium 7_1_58FAA (OR: 0.25, 95% CI: 0.10–0.63) with HCC risk (p < 0.05), and Verrucomicrobia (OR: 0.17, 95% CI: 0.05–0.59), Anaerostipes (OR: 0.14, 95% CI: 0.03–0.64), Paraprevotella (OR: 0.27, 95% CI: 0.11–0.67), Parasutterella (OR: 0.32, 95% CI: 0.11–0.99), Bifidobacterium longum (OR: 0.31, 95% CI: 0.11–0.88), Bacteroides clarus (OR: 0.52, 95% CI: 0.28–0.99), as well as Bacteroides fragilis (OR: 0.48, 95% CI: 0.28–0.83) were negatively associated with ICC risk (p < 0.05).
The WM and MR-RAPS analyses indicated that the associations of Butyricicoccus and Ruminococcus lactaris with HCC risk remained, and the associations of Verrucomicrobia, Enterobacteriales, Enterobacteriaceae, Veillonellaceae, Paraprevotella, and Bacteroides clarus with ICC risk remained (Table 1). Finally, stable results were achieved in Verrucomicrobia, Veillonellaceae, and Paraprevotella by leave-one-out analysis, whereas there were some SNPs in Butyricicoccus, Ruminococcus lactaris, Enterobacteriales, Enterobacteriaceae, and Bacteroides clarus with dominant effects on the causal estimations (Supplementary Figures S10, S11).
3.6. Pancreatic cancer
Genetically predicted Bacillales (OR: 1.67, 95% CI: 1.23–2.26), Defluviitaleaceae UCG011 (OR: 1.63, 95% CI: 1.01–2.64), Sutterella (OR: 2.45, 95% CI: 1.38–4.37), and Bacteroides plebeius (OR: 1.41, 95% CI: 1.07–1.85) were associated with higher pancreatic cancer risk in IVW method (p < 0.05; Figure 2). Differently, Ruminococcaceae (OR: 0.49, 95% CI: 0.26–0.93), Eggerthella (OR: 0.63, 95% CI: 0.43–0.93), Lachnospiraceae UCG004 (OR: 0.52, 95% CI: 0.29–0.92), Alistipes putredinis (OR: 0.38, 95% CI: 0.18–0.82), Flavonifractor plautii (OR: 0.57, 95% CI: 0.40–0.82), Eubacterium hallii (OR: 0.61, 95% CI: 0.46–0.83), Dialister invisus (OR: 0.51, 95% CI: 0.33–0.79), Parasutterella excrementihominis (OR: 0.70, 95% CI: 0.51–0.96), and Bacteroides stercoris (OR: 0.62, 95% CI: 0.40–0.97) were related to lower pancreatic cancer risk (p < 0.05; Figure 2). As shown in Table 1, the WM and MR-RAPS methods revealed similar results in the potential causal association analysis of Bacillales, Eggerthella, Sutterella, Flavonifractor plautii, and Eubacterium hallii with pancreatic cancer. Furthermore, leave-one-out test indicated that except for Eggerthella, no SNPs with dominant effects were identified in other genetic predictions (Supplementary Figure S12).
3.7. Bonferroni correction and sensitivity analysis
The Bonferroni correction indicated that phylum Verrucomicrobia retained a strong negative association with ICC (OR: 0.17, 95% CI: 0.05–0.59, p = 0.005), whereas order Bacillales retained a strong positive association with pancreatic cancer (OR: 1.67, 95% CI: 1.23–2.26, p = 0.001). The p values of the intercept terms of MR-Egger regression were all greater than 0.05, revealing no notable pleiotropy (Supplementary Table S2). Cochran’s Q test and MR-PRESSO detected no evidence of heterogeneity and outliers (p > 0.05; Supplementary Table S2).
3.8. Reverse MR analysis
After a series of IV screening steps, 11 esophageal cancer associated-SNPs, eight gastric cancer-associated SNPs, 24 CRC-associated SNPs, five HCC-associated SNPs, three ICC-associated SNPs, and 13 pancreatic cancer-associated SNPs were eligible IVs. For HCC and ICC, reverse MR analysis was not performed because the number of SNPs available in the outcome (gut microbiome) was less than 3 and no proxy SNPs (R2 > 0.8) were found or proxy SNPs were also unavailable in the outcome. Details of IVs for reverse MR are listed in Supplementary Table S3.
We found that esophageal cancer, gastric cancer, CRC, and pancreatic cancer were associated with 9, 12, 23, and 20 microbial taxa, respectively, by the IVW method (Supplementary Table S4). Among them, the associations of esophageal cancer with two microbial taxa, gastric cancer with four microbial taxa, CRC with six microbial taxa, and pancreatic cancer with five microbial taxa remained stable in WM and MR-RAPS methods (Supplementary Table S5). In particular, esophageal cancer was negatively correlated with Ruminococcaceae UCG004 and Dorea longicatena, CRC was negatively correlated with Lentisphaerae, Lentisphaeria, Victivallales, Clostridiaceae1, Victivallaceae and Bacteroides ovatus, as well as pancreatic cancer was negatively correlated with Lachnospiraceae and Eubacterium eligens group. Conversely, gastric cancer was positively associated with Candidatus Soleaferrea, Barnesiella intestinihominis, Parabacteroides unclassified, and Clostridium leptum, as well as pancreatic cancer was positively associated with Alistipes finegoldii, Pseudoflavonifractor capillosus, and Bacteroides fragilis. The Bonferroni correction revealed that CRC retained a strong negative association with family Clostridiaceae1 (OR: 0.91, 95% CI: 0.86–0.96, p = 0.001).
4. Discussion
Through MR analysis of 297 microbial taxa and six GI cancers (esophageal cancer, gastric cancer, CRC, HCC, ICC, and pancreatic cancer), we identified potential causal associations between 21 bacterial taxa and GI cancers (values of p < 0.05 in all three MR methods), and a strong potential causality was identified in two of them by Bonferroni correction (phylum Verrucomicrobia and order Bacillales). Reverse MR indicated that GI cancer was associated with 17 microbial taxa in all three MR methods, among them, a strong inverse association between CRC and family Clostridiaceae1 was identified by Bonferroni correction. To our knowledge, this work comprehensively reveals the potential causality between gut microbiota and GI cancer using MR analysis for the first time.
We discovered that Howardella and Roseburia unclassified were positively related to gastric cancer, and Lachnospiraceae FCS020 group was positively associated with CRC in all three MR methods. Howardella, Roseburia unclassified, and Lachnospiraceae FCS020 group belong to the family Lachnospiraceae, which can produce short-chain fatty acids with an inhibitory effect on inflammation, such as acetate and butyrate, and is generally considered a beneficial bacterium (Sivaprakasam et al., 2016; Sorbara et al., 2020). However, some studies have found that the gut microbiota of patients with metabolic diseases, non-alcoholic fatty liver disease, primary sclerosing cholangitis with inflammatory bowel disease, and chronic kidney disease is enriched with Lachnospiraceae (Vacca et al., 2020). Results regarding the association of Lachnospiraceae with gastric cancer and CRC are also inconsistent (Aviles-Jimenez et al., 2014; Yang et al., 2019; Clos-Garcia et al., 2020; Yu et al., 2021). This discrepancy may be due to the different genera and species of family Lachnospiraceae playing different roles in diseases, and the relationship of Lachnospiraceae with gastric cancer and CRC requires further exploration at more nuanced levels (i.e., genus and species levels). The exact mechanism underlying the effect of Howardella and Roseburia unclassified on gastric cancer as well as Lachnospiraceae FCS020 group on CRC deserves validation studies. At present, there are many studies on the association between gastric microbiome and gastric cancer, while there are few studies on the association between gut microbiome and gastric cancer, especially the research using metagenomic sequencing is more limited. Therefore, the positive associations between gastric cancer and four bacterial taxa (one genus and three species) identified in our reverse MR analysis were also not found in previous observational studies.
The three MR analysis methods also identified that Bilophila and Prevotella7 were negatively and positively correlated with CRC risk, respectively. Bilophila wadsworthia, one species of genus Bilophila, can produce hydrogen sulfide, which has a promoting effect on CRC (Yazici et al., 2017). However, the association between Bilophila and CRC has obtained conflicting results in observational studies (Feng et al., 2015; Yang et al., 2021). The function and mechanism of Bilophila on CRC warrant verification studies. Previous studies have indicated that the increase of Prevotella abundance is related to the deficiency of the anti-inflammatory cytokine IL-10 and that inflammation is a recognized driver of colorectal carcinogenesis (Janney et al., 2020). Ternes et al. (2022) also found that Prevotella7 was enriched in fecal samples from CRC patients. The comparison of the results of our study with those of previous studies on gut microbiota and CRC is shown in Supplementary Table S6. Furthermore, reverse MR demonstrated that CRC was negatively related to Lentisphaerae, Lentisphaeria, Victivallales, Clostridiaceae1, Victivallaceae, and Bacteroides ovatus. Only two microbial taxa, Clostridiaceae1 and Bacteroides ovatus, each had an observational study consistent with our findings (Supplementary Table S6).
Butyricicoccus, a butyrate producer, was detected to be inversely associated with HCC in our study, which remained stable across the three MR methods. Consistently, several observational studies also found that Butyricicoccus abundance was reduced in HCC patients (Ren et al., 2019; Lapidot et al., 2020). Supplementary Table S7 showed the comparison of our results with previous studies on gut microbiota and HCC. We also observed negative associations of Verrucomicrobia and Paraprevotella with ICC, and positive correlations of Enterobacteriales, Enterobacteriaceae, and Veillonellaceae with ICC. There are currently few studies concentrating on the relationship between intestinal microbiota and ICC, but some prior studies indicated that Verrucomicrobia and Paraprevotella were decreased in HCC patients, while Enterobacteriales, Enterobacteriaceae, and Veillonellaceae were enriched (Ponziani et al., 2019; Huang et al., 2020; Lapidot et al., 2020). Similarly, our study also found that Enterobacteriales and Enterobacteriaceae were associated with increased HCC risk in IVW method. In addition, we found a positive association between Ruminococcus lactaris and HCC and a negative association between Bacteroides clarus and ICC, which have not been reported in previous studies. We could not yet prove the mechanisms of the effects of these bacterial taxa on liver cancer, as our study mainly focused on correlation analysis. Future mechanism explanation studies are needed.
Our study also suggested that Bacillales and Sutterella were potentially causally related to increased pancreatic cancer risk, while Eggerthella, Flavonifractor plautii, and Eubacterium hallii were associated with decreased pancreatic cancer risk. The comparison of our results with previous studies on gut microbiota and pancreatic cancer is presented in Supplementary Table S8. Sutterella is a pro-inflammatory bacterium (Hiippala et al., 2016), and accumulating studies have suggested that gut microbiota may influence pancreatic carcinogenesis by modulating inflammatory and immune responses (Meng et al., 2018). Eggerthella is also considered a pro-inflammatory genus (Nikolova et al., 2021), but one study found that obese individuals had a lower relative proportion of Eggerthella compared to non-obese individuals (Pinart et al., 2022), and obesity significantly increased the risk of pancreatic cancer. Similarly, another study found that Flavonifractor plautii was more abundant in non-obese compared with obese individuals (Kasai et al., 2015). This seems to suggest the beneficial roles of Eggerthella and Flavonifractor plautii. The mechanism underlying the influence of Eggerthella and Flavonifractor plautii on pancreatic cancer deserves to be studied. Eubacterium hallii is one of the major producers of butyrate in the human gut with health-promoting effects (Hiippala et al., 2018). Observational studies also found that the abundance of Eubacterium hallii was higher in healthy controls than in pancreatic cancer patients (Matsukawa et al., 2021; Zhou et al., 2021). As for order Bacillales, further studies are warranted to clarify the functional significance of specific species and strains for pancreatic cancer. Besides, among the five microbial taxa found to be affected by pancreatic cancer in reverse MR, only the inverse associations of pancreatic cancer with Lachnospiraceae and Eubacterium eligens group were also found in previous observational studies (Supplementary Table S8).
In addition, some gut bacteria including Oxalobacteraceae, Oxalobacter, and Ruminococcaceae UCG010 were found to be positively correlated with esophageal cancer in all three MR analysis methods. Reverse MR indicated that esophageal cancer was negatively correlated with Ruminococcaceae UCG004 and Dorea longicatena. At present, little is known about the relevance between intestinal microbiota and esophageal cancer, and the association of Oxalobacteraceae, Oxalobacter, Ruminococcaceae UCG010, Ruminococcaceae UCG004, and Dorea longicatena with esophageal cancer has not been reported. Therefore, our study provided a new direction to unravel the role of gut microflora in esophageal cancer, and the mechanism of these bacteria in esophageal cancer requires further exploration.
The major advantage of our study is that we comprehensively analyzed the potential causalities of 297 microbial taxa and six GI cancers using two-sample MR method. Using MR method to investigate the association between gut microbiota and GI cancer has the following advantages. First, according to Mendel’s Laws of Inheritance, alleles are randomly allocated among the descendants, similar to randomization in randomized controlled trials (Zheng et al., 2017). In addition, genotypes are fixed at conception and cannot be modified by diseases (Zheng et al., 2017). Thus, causal inference is unlikely to be influenced by reverse causality and confounders. Second, the two-sample MR is based on publicly available large-scale GWAS summary-level data without additional experimental costs. However, our study has several limitations. First, since SNPs with p < 5 × 10−8 were too limited, we selected SNPs with p < 1 × 10−5 as IVs. To obtain reliable IVs, we performed a series of IV screening steps, including excluding SNPs with F < 10 to avoid weak IVs bias and searching all SNPs in PhenoScanner to avoid confounding effects. Second, although 297 microbial taxa were included in our analysis, the potential causal associations of many other microbial taxa with GI cancers were not explored. Especially at the species level, only 101 species were included in our study. Third, this MR is a correlation analysis of gut microbiota and GI cancer without explaining the mechanism. Fourth, the MR analysis may be affected by potential pleiotropy. Of note, all exposures in our MR analysis had 3 or more IVs, which may mitigate the impact of potential pleiotropy to some extent, because pleiotropy is unlikely to generate the same association for different IVs (Davey Smith and Hemani, 2014). Fifth, the variances of some microbial taxa explained by the genetic IVs were small, so estimates of the associations might be hampered by limited statistical power. Sixth, the participants in present study were mostly of European ancestries and only a small number of intestinal microbiome data were drawn from other ethnicities, which were less affected by ethnic bias. However, this may limit the applicability of the results to other populations.
In conclusion, this MR study demonstrates that gut microbiota has potential causal impacts on GI cancer. Our results probably offer useful biomarkers for non-invasive early diagnosis of GI cancer. In addition, our results imply that modulation of intestinal microbiome may be a potential intervention target for GI cancer prevention.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
QS, GC, and YW designed the research. QS, CJ, ZB, YY, JiW, JuW, JZ, YC, and HZ collected and analyzed the data. QS drafted the manuscript. GC and YW supervised the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was jointly supported by the National Natural Science Foundation of China (No. 81703310, 81772628, and 82072685) and the Science and Technology Plan Project of Wenzhou (No. Y2020153).
Acknowledgments
The authors gratefully acknowledge all researchers and participants who provided publicly available GWAS summary data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1181328/full#supplementary-material
Footnotes
References
Arnold, M., Abnet, C. C., Neale, R. E., Vignat, J., Giovannucci, E. L., McGlynn, K. A., et al. (2020). Global burden of 5 major types of gastrointestinal Cancer. Gastroenterology 159, 335–349.e15. doi: 10.1053/j.gastro.2020.02.068
Aviles-Jimenez, F., Vazquez-Jimenez, F., Medrano-Guzman, R., Mantilla, A., and Torres, J. (2014). Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci. Rep. 4:4202. doi: 10.1038/srep04202
Bowden, J., Davey Smith, G., and Burgess, S. (2015). Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int. J. Epidemiol. 44, 512–525. doi: 10.1093/ije/dyv080
Bowden, J., Davey Smith, G., Haycock, P. C., and Burgess, S. (2016). Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314. doi: 10.1002/gepi.21965
Brennan, C. A., and Garrett, W. S. (2016). Gut microbiota, inflammation, and colorectal Cancer. Annu. Rev. Microbiol. 70, 395–411. doi: 10.1146/annurev-micro-102215-095513
Cheung, M. K., Yue, G. G. L., Lauw, S., Li, C. S. Y., Yung, M. Y., Ng, S. C., et al. (2022). Alterations in gut microbiota of esophageal squamous cell carcinoma patients. J. Gastroenterol. Hepatol. 37, 1919–1927. doi: 10.1111/jgh.15941
Clos-Garcia, M., Garcia, K., Alonso, C., Iruarrizaga-Lejarreta, M., D’Amato, M., Crespo, A., et al. (2020). Integrative analysis of fecal metagenomics and metabolomics in colorectal Cancer. Cancers 12:1142. doi: 10.3390/cancers12051142
Davey Smith, G., and Hemani, G. (2014). Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–R98. doi: 10.1093/hmg/ddu328
Davies, N. M., Holmes, M. V., and Davey Smith, G. (2018). Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362:k601. doi: 10.1136/bmj.k601
Feng, Q., Liang, S., Jia, H., Stadlmayr, A., Tang, L., Lan, Z., et al. (2015). Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 6:6528. doi: 10.1038/ncomms7528
Hiippala, K., Jouhten, H., Ronkainen, A., Hartikainen, A., Kainulainen, V., Jalanka, J., et al. (2018). The potential of gut commensals in reinforcing intestinal barrier function and alleviating inflammation. Nutrients 10:988. doi: 10.3390/nu10080988
Hiippala, K., Kainulainen, V., Kalliomaki, M., Arkkila, P., and Satokari, R. (2016). Mucosal prevalence and interactions with the epithelium indicate commensalism of Sutterella spp. Front. Microbiol. 7:1706. doi: 10.3389/fmicb.2016.01706
Huang, H., Ren, Z., Gao, X., Hu, X., Zhou, Y., Jiang, J., et al. (2020). Integrated analysis of microbiome and host transcriptome reveals correlations between gut microbiota and clinical outcomes in HBV-related hepatocellular carcinoma. Genome Med. 12:102. doi: 10.1186/s13073-020-00796-5
Janney, A., Powrie, F., and Mann, E. H. (2020). Host-microbiota maladaptation in colorectal cancer. Nature 585, 509–517. doi: 10.1038/s41586-020-2729-3
Jiang, L., Zheng, Z., Fang, H., and Yang, J. (2021). A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 53, 1616–1621. doi: 10.1038/s41588-021-00954-4
Kartal, E., Schmidt, T. S. B., Molina-Montes, E., Rodriguez-Perales, S., Wirbel, J., Maistrenko, O. M., et al. (2022). A faecal microbiota signature with high specificity for pancreatic cancer. Gut 71, 1359–1372. doi: 10.1136/gutjnl-2021-324755
Kasai, C., Sugimoto, K., Moritani, I., Tanaka, J., Oya, Y., Inoue, H., et al. (2015). Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 15:100. doi: 10.1186/s12876-015-0330-2
Kurilshikov, A., Medina-Gomez, C., Bacigalupe, R., Radjabzadeh, D., Wang, J., Demirkan, A., et al. (2021). Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165. doi: 10.1038/s41588-020-00763-1
Lapidot, Y., Amir, A., Nosenko, R., Uzan-Yulzari, A., Veitsman, E., Cohen-Ezra, O., et al. (2020). Alterations in the gut microbiome in the progression of cirrhosis to hepatocellular carcinoma. mSystems 5:e00153-20. doi: 10.1128/mSystems.00153-20
Li, P., Wang, H., Guo, L., Gou, X., Chen, G., Lin, D., et al. (2022). Association between gut microbiota and preeclampsia-eclampsia: a two-sample Mendelian randomization study. BMC Med. 20:443. doi: 10.1186/s12916-022-02657-x
Li, N., Wang, Y., Wei, P., Min, Y., Yu, M., Zhou, G., et al. (2023). Causal effects of specific gut microbiota on chronic kidney diseases and renal function-a two-sample Mendelian randomization study. Nutrients 15:360. doi: 10.3390/nu15020360
Long, Y., Tang, L., Zhou, Y., Zhao, S., and Zhu, H. (2023). Causal relationship between gut microbiota and cancers: a two-sample Mendelian randomisation study. BMC Med. 21:66. doi: 10.1186/s12916-023-02761-6
Lopera-Maya, E. A., Kurilshikov, A., van der Graaf, A., Hu, S., Andreu-Sánchez, S., Chen, L., et al. (2022). Effect of host genetics on the gut microbiome in 7,738 participants of the Dutch microbiome project. Nat. Genet. 54, 143–151. doi: 10.1038/s41588-021-00992-y
Ma, J., Li, J., Jin, C., Yang, J., Zheng, C., Chen, K., et al. (2023). Association of gut microbiome and primary liver cancer: a two-sample Mendelian randomization and case-control study. Liver Int. 43, 221–233. doi: 10.1111/liv.15466
Matsukawa, H., Iida, N., Kitamura, K., Terashima, T., Seishima, J., Makino, I., et al. (2021). Dysbiotic gut microbiota in pancreatic cancer patients form correlation networks with the oral microbiota and prognostic factors. Am. J. Cancer Res. 11, 3163–3175.
Meng, C., Bai, C., Brown, T. D., Hood, L. E., and Tian, Q. (2018). Human gut microbiota and gastrointestinal Cancer. Genom. Proteom. Bioinform 16, 33–49. doi: 10.1016/j.gpb.2017.06.002
Ni, J. J., Li, X. S., Zhang, H., Xu, Q., Wei, X. T., Feng, G. J., et al. (2022). Mendelian randomization study of causal link from gut microbiota to colorectal cancer. BMC Cancer 22:1371. doi: 10.1186/s12885-022-10483-w
Nikolova, V. L., Smith, M. R. B., Hall, L. J., Cleare, A. J., Stone, J. M., and Young, A. H. (2021). Perturbations in gut microbiota composition in psychiatric disorders: a review and Meta-analysis. JAMA Psychiat. 78, 1343–1354. doi: 10.1001/jamapsychiatry.2021.2573
Ohtani, N., and Hara, E. (2021). Gut-liver axis-mediated mechanism of liver cancer: a special focus on the role of gut microbiota. Cancer Sci. 112, 4433–4443. doi: 10.1111/cas.15142
Palmer, T. M., Lawlor, D. A., Harbord, R. M., Sheehan, N. A., Tobias, J. H., Timpson, N. J., et al. (2012). Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat. Methods Med. Res. 21, 223–242. doi: 10.1177/0962280210394459
Pinart, M., Dötsch, A., Schlicht, K., Laudes, M., Bouwman, J., Forslund, S. K., et al. (2022). Gut microbiome composition in obese and non-obese persons: a systematic review and Meta-analysis. Nutrients 14:12. doi: 10.3390/nu14010012
Ponziani, F. R., Bhoori, S., Castelli, C., Putignani, L., Rivoltini, L., del Chierico, F., et al. (2019). Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 69, 107–120. doi: 10.1002/hep.30036
Ren, Z., Li, A., Jiang, J., Zhou, L., Yu, Z., Lu, H., et al. (2019). Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 68, 1014–1023. doi: 10.1136/gutjnl-2017-315084
Schwabe, R. F., and Jobin, C. (2013). The microbiome and cancer. Nat. Rev. Cancer 13, 800–812. doi: 10.1038/nrc3610
Sivaprakasam, S., Prasad, P. D., and Singh, N. (2016). Benefits of short-chain fatty acids and their receptors in inflammation and carcinogenesis. Pharmacol. Ther. 164, 144–151. doi: 10.1016/j.pharmthera.2016.04.007
Sorbara, M. T., Littmann, E. R., Fontana, E., Moody, T. U., Kohout, C. E., Gjonbalaj, M., et al. (2020). Functional and genomic variation between human-derived isolates of Lachnospiraceae reveals inter- and intra-species diversity. Cell Host Microbe 28, 134–146.e4. doi: 10.1016/j.chom.2020.05.005
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. doi: 10.3322/caac.21660
Ternes, D., Tsenkova, M., Pozdeev, V. I., Meyers, M., Koncina, E., Atatri, S., et al. (2022). The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat. Metab. 4, 458–475. doi: 10.1038/s42255-022-00558-0
Tong, Y., Gao, H., Qi, Q., Liu, X., Li, J., Gao, J., et al. (2021). High fat diet, gut microbiome and gastrointestinal cancer. Theranostics 11, 5889–5910. doi: 10.7150/thno.56157
Vacca, M., Celano, G., Calabrese, F. M., Portincasa, P., Gobbetti, M., and De Angelis, M. (2020). The controversial role of human gut Lachnospiraceae. Microorganisms 8:573. doi: 10.3390/microorganisms8040573
Verbanck, M., Chen, C. Y., Neale, B., and Do, R. (2018). Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698. doi: 10.1038/s41588-018-0099-7
Wong, S. H., and Yu, J. (2019). Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 16, 690–704. doi: 10.1038/s41575-019-0209-8
Yang, Y., du, L., Shi, D., Kong, C., Liu, J., Liu, G., et al. (2021). Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat. Commun. 12:6757. doi: 10.1038/s41467-021-27112-y
Yang, J., McDowell, A., Kim, E. K., Seo, H., Lee, W. H., Moon, C. M., et al. (2019). Development of a colorectal cancer diagnostic model and dietary risk assessment through gut microbiome analysis. Exp. Mol. Med. 51, 1–15. doi: 10.1038/s12276-019-0313-4
Yazici, C., Wolf, P. G., Kim, H., Cross, T. L., Vermillion, K., Carroll, T., et al. (2017). Race-dependent association of sulfidogenic bacteria with colorectal cancer. Gut 66, 1983–1994. doi: 10.1136/gutjnl-2016-313321
Yu, L. X., and Schwabe, R. F. (2017). The gut microbiome and liver cancer: mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 14, 527–539. doi: 10.1038/nrgastro.2017.72
Yu, D., Yang, J., Jin, M., Zhou, B., Shi, L., Zhao, L., et al. (2021). Fecal Streptococcus alteration is associated with gastric Cancer occurrence and liver metastasis. MBio 12:e0299421. doi: 10.1128/mBio.02994-21
Zhao, Q., Chen, Y., Wang, J., and Small, D. S. (2019). Powerful three-sample genome-wide design and robust statistical inference in summary-data Mendelian randomization. Int. J. Epidemiol. 48, 1478–1492. doi: 10.1093/ije/dyz142
Zheng, J., Baird, D., Borges, M. C., Bowden, J., Hemani, G., Haycock, P., et al. (2017). Recent developments in Mendelian randomization studies. Curr Epidemiol Rep 4, 330–345. doi: 10.1007/s40471-017-0128-6
Zhou, W., Nielsen, J. B., Fritsche, L. G., Dey, R., Gabrielsen, M. E., Wolford, B. N., et al. (2018). Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341. doi: 10.1038/s41588-018-0184-y
Keywords: gut microbiota, gastrointestinal cancer, Mendelian randomization, instrumental variable, causal relationship
Citation: Su Q, Jin C, Bo Z, Yang Y, Wang J, Wang J, Zhou J, Chen Y, Zeng H, Chen G and Wang Y (2023) Association between gut microbiota and gastrointestinal cancer: a two-sample bi-directional Mendelian randomization study. Front. Microbiol. 14:1181328. doi: 10.3389/fmicb.2023.1181328
Edited by:
Jian Li, Tulane University, United StatesReviewed by:
Igor Łoniewski, Pomeranian Medical University, PolandZhendong Mei, Brigham and Women’s Hospital, United States
Copyright © 2023 Su, Jin, Bo, Yang, Wang, Wang, Zhou, Chen, Zeng, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Chen, Y2hlbi5nYW5nQHdtdS5lZHUuY24=; Yi Wang, d2FuZy55aUB3bXUuZWR1LmNu