
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 June 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1180474
This article is part of the Research Topic Insights on Plant-Associated Microorganisms: Diversity, Systematics and Genomics View all 23 articles
 Linyan Xie1
Linyan Xie1 Lufeng Liu1,2,3Yanju Luo1Xibing Rao1Yining Di1,2,3Han Liu1Zhenfeng Qian1
Lufeng Liu1,2,3Yanju Luo1Xibing Rao1Yining Di1,2,3Han Liu1Zhenfeng Qian1 Qingqing Shen1Lilian He1,3*Fusheng Li1,3*
Qingqing Shen1Lilian He1,3*Fusheng Li1,3*Introduction: Sugarcane is one of the most important sugar crops worldwide, however, sugarcane production is seriously limited by sugarcane red rot, a soil-borne disease caused by Colletotrichum falcatum. Bacillus velezensis YC89 was isolated from sugarcane leaves and can significantly inhibited red rot disease caused by C. falcatum.
Methods: In this study, the genome of YC89 strain was sequenced, its genome structure and function were analyzed using various bioinformatics software, and its genome was compared with those of other homologous strains. In addition, the effectiveness of YC89 against sugarcane red rot and the evaluation of sugarcane plant growth promotion were also investigated by pot experiments.
Results: Here, we present the complete genome sequence of YC89, which consists of a 3.95 Mb circular chromosome with an average GC content of 46.62%. The phylogenetic tree indicated that YC89 is closely related to B. velezensis GS-1. Comparative genome analysis of YC89 with other published strains (B. velezensis FZB42, B. velezensis CC09, B. velezensis SQR9, B. velezensis GS-1, and B. amyloliquefaciens DSM7) revealed that the strains had a part common coding sequences (CDS) in whereas 42 coding were unique of strain YC89. Whole-genome sequencing revealed 547 carbohydrate-active enzymes and identified 12 gene clusters encoding secondary metabolites. Additionally, functional analysis of the genome revealed numerous gene/gene clusters involved in plant growth promotion, antibiotic resistance, and resistance inducer synthesis. In vitro pot tests indicated that YC89 strain controlled sugarcane red rot and promoted the growth of sugarcane plants. Additionally, it increased the activity of enzymes involved in plant defense, such as superoxide dismutase, peroxidase, polyphenol oxidase, chitinase, and β-1,3-glucanase.
Discussion: These findings will be helpful for further studies on the mechanisms of plant growth promotion and biocontrol by B. velezensis and provide an effective strategy for controlling red rot in sugarcane plants.
Sugarcane (Saccharum officinarum L.) is one of the most important sugar crops in the world. In China, the sugarcane planting area is >85% of the sugar crop planting area, and > 92% of the total sugar is obtained from sugarcane. China is a major sugarcane planting and sugar production and marketing country (Wu et al., 2017). In 2021, the sugarcane planting area exceeded 1.301 million hm2, and sugarcane sugar output reached 104.4043 million tons (National Bureau of Statistics). However, the frequent occurrence of sugarcane diseases affects yield and quality, and soil-borne diseases are particularly difficult to control due to their persistence and widespread. Sugarcane red rot is a fungal disease caused by Colletotrichum falcatum Went.; its sexual stage is Physalospora tucumanensis Speg. (Glomerella tucumanensis) (Nithya et al., 2012). Red rot was first reported in Java, Indonesia, in 1893, representing one of the first known sugarcane diseases, leading to outbreaks in many sugarcane-producing countries (Viswanathan et al., 2009). The pathogen overwinters as hyphae, conidia, and chlamydospores in sugarcane seeds and sugarcane plants, which is the primary infection source of the disease. Pathogenic spores can be transmitted by rain, airflow, insects, and mechanical damage. In addition, they can invade plants through borer holes and growth cracks (Peñaflor and Bento, 2019). Sugarcane red rot primarily damages the stem and leaf midrib of sugarcane, which can occur from seedling stage to adult plant stage and late harvest stage. The disease in the seedling stage prevents seedlings from germinating normally, which often causes serious loss of sugarcane plants. Meanwhile, disease in the adult stage not only affects the normal growth of sugarcane plants, but also causes decay and even death (Costa et al., 2021). Indeed, red rot disease is one of the most critical sugarcane diseases in many sugarcane-growing countries, including India, Pakistan, the United States, and Bangladesh, as it can result in 25–50% sugarcane yield loss (Saksena et al., 2013) and can reduce the sugar content and purity of sugarcane juice, thus, significantly impacting the productivity of the sugarcane industry (Bukhari et al., 2012).
The main prevention and control methods for sugarcane red rot disease include disease resistance breeding, chemical control, and biological control measures. The pathogen of sugarcane red rot is prone to mutation, with more than six known variants; a disease-resistant plant becomes susceptible within 4–5 years (Sumana and Singh, 2012). Currently, the prevention and control of sugarcane red rot mainly rely on chemical fungicides. However, excessive use of chemical fungicides can lead to fungicide resistance, environmental pollution, and pose a threat to human health (Mishra and Behera, 2009). Therefore, cost-effective, chemical-free, and eco-friendly strategies for controlling sugarcane red rot are urgently required. Using bioactive agents to control plant diseases is an attractive alternative and has significant potential in the future. In fact, microorganisms with antifungal activity have been used in many crop systems. For example, Pseudomonas spp. (Shair et al., 2021), Bacillus spp. (Amna et al., 2020), and Trichoderma spp. (Singh et al., 2014) have been used to control sugarcane red rot in the field. Among the microbial candidates used for biocontrol, Bacillus species are the most useful as they can produce spores and a series of bioactive compounds that allow them to survive in adverse environmental conditions. Therefore, Bacillus has become one of the most studied biocontrol bacteria (Li et al., 2021; Pang et al., 2021).
Biocontrol boasts certain advantages, including environmental friendliness, safety, and the lack of induction of pesticide resistance. Microorganisms, including Bacillus, Pseudomonas, and Paenibacillus spp., have been used to suppress soil-borne pathogens. The main mechanisms of biological control of plant diseases include the secretion of antimicrobial substances and competition for nutrients and niches with pathogens (Backer et al., 2018; Rodriguez et al., 2019). In this way, they can induce disease resistance by promoting plant growth. In addition to these effects, many biocontrol strains increase plant resistance to pathogens via induced systemic resistance (ISR), which is triggered by a range of secondary metabolites known as elicitors (Shi et al., 2017; Gilardi et al., 2021). Different signaling pathways, such as the jasmonic acid and ethylene pathways, are activated to induce plant resistance (Ryu et al., 2004). In addition, some phenolic substances and pathogenesis-related proteins, such as chitinase, β-l,3-glucanase (β-1,3-GA), peroxidase (POD), polyphenol oxidase (PPO), superoxide dismutase (SOD), and phenylalanine aminolysis, are involved in plant disease resistance.
Bacillus velezensis has been widely used in agricultural production owing to its environmental safety, straightforward industrial production, and good biocontrol efficacy (Sun et al., 2022). B. velezensis YC89 is a sugarcane endophyte isolated from sugarcane leaves and can inhibit 78% of the etiological pathogens of sugarcane red rot (Xie et al., 2020). However, the relative efficacy of B. velezensis YC89 against sugarcane red rot and its biological control mechanism remains unknown. The current study sought to investigate the mechanisms underlying the biocontrol ability of B. velezensis YC89 and determine its complete genome sequence. To this end, a greenhouse experiment was performed. Results revealed B. velezensis YC89 could serve as a potential biocontrol agent of sugarcane red rot and promote sugarcane plant growth. To understand the molecular mechanism involved in the plant–microbe interaction, high-quality genome assembly and annotation of B. velezensis YC89 was performed. Collectively, the results of the current study support the application of B. velezensis to sugarcane plants in the field to protect against red rot disease.
The fungus C. falcatum Went., antagonistic bacterium B. velezensis YC89, and an unknown strain X22 used in this study were isolated from the leaves of infected sugarcane plants and healthy plants, respectively. B. velezensis YC89 was deposited as a reference strain (strain N°60,902) at the China General Microbiological Culture Collection Center. All bacteria were cultured on Luria-Bertani agar media and incubated at 28°C for 24 h. C. falcatum Went. was cultivated on a potato dextrose agar (PDA) at 28°C for 7 d.
The genomic DNA of B. velezensis YC89 was extracted using the HiPure Bacterial DNA kit (Magen, Guangzhou, China) according to the manufacturer’s instructions. The quality of the extracted DNA was determined using Qubit (Thermo Fisher Scientific, Waltham, MA) and Nanodrop (Thermo Fisher Scientific, Waltham, MA) for the corresponding assays.
The whole genome was sequenced using the Pacific Biosciences platform (PacBio, Menlo Park, CA) and Illumina Miseq platform at Genedenovo Biotechnology (Guangzhou, China). Qualified genomic DNA was fragmented with G-tubes (Covaris, Woburn, MA, United States) and end-repaired to prepare SMRTbell DNA template libraries (with fragment size of >10 Kb selected by blue pippin system) according to the manufacturer’s specification (PacBio, Menlo Park, CA). Liraray quality was detected by Qubit® 2.0 Flurometer (Life Technologies, CA, United States) and average fragment size was estimated on a Bioanalyzer 2,100 (Agilent, Santa Clara, CA). SMRT sequencing was performed on the Pacific Biosciences Sequel (PacBio, Menlo Park, CA) according to standard protocols. The PacBio reads were de novo assembled using Microbial Assembly (smrtlink8), HGAP4, and Canu (v.1.6) software. The depth of genome coverage was analyzed using the pbalign (BLASR, v.0.4.1) tool. The complete genome sequence of B. velezensis YC89 was deposited in GenBank under the accession number CP0392499.
The genes were annotated by alignment with those deposited in diverse protein databases, including the NCBI non-redundant (NR) protein sequence database, UniProt/Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), gene ontology (GO), and Cluster of Orthologous Groups of proteins (COG). Protein family annotation was performed using Pfam_Scan (version 1.6) based on the Pfam database (version 32.0) (Finn et al., 2014). Gene sequences annotated from the YC89 genome were basic local alignment search tool (BLAST)ed. using the carbohydrate-active enzyme (CAZyme) database.
To determine the taxonomic position of YC89 in the genus Bacillus, sequence analysis of YC89 and 30 other Bacillus strains was performed based on the 16S rDNA gene, and an evolutionary tree was constructed. Genome-wide comparative analysis was then performed for YC89 and B. velezensis FZB42, B. velezensis CC09, B. velezensis SQR9, B. velezensis GS-1, and B. amyloliquefaciens DSM7, and genes that were single copies in the whole genome were selected. The evolutionary tree was constructed using iqtree (version 1.6.3) (Lam-Tung et al., 2015).
All genomes used in this study were downloaded in the FASTA format from the NCBI database. Basic information on the reference strains is provided in Supplementary Table S1. For comparative analyses of the orthologous and exclusive genes between YC89 and the other closest genomes, the amino acid (or nucleotide) sequences of B. velezensis FZB42, B. velezensis CC09, B. velezensis SQR9, B. velezensis GS-1, and B. amyloliquefaciens DSM7 were filtered in FASTA format to remove low-quality sequences based on the length and percentage of stop codons. Diamond (version 2.0.7) was used to compare the amino acid (or nucleotide) sequences of all species investigated in the study (Buchfink et al., 2021). OrthoMCL (version 1.4) was used for similarity clustering, a list of homologous genes grouped into clusters was obtained, and the species distribution of each protein cluster was determined (Li et al., 2003). Subsequently, the Mauve software (version 2.3.1) was employed to analyze the genome collinearity of six Bacillus strains (FZB42, CC09, SQR9, GS-1, DSM7, YC89) (Darling et al., 2004).
Based on collinearity, Pyani was used to calculate the average nucleotide identity (ANI) of the pairwise genome alignment region between the target genome and closely related reference genome (Pritchard et al., 2016). The ANI value is an index for comparing two genomic relatives at the nucleotide level and is defined as the average base similarity between homologous segments of two microbial genomes, which is characterized by a high degree of discrimination between closely related species. Usually, an ANI value of 95% is set as the classification threshold to distinguish species.
The genome of B. velezensis YC89 was analyzed using antiSMASH 4.0 (version 4.1.0) with web server1 to predict putative secondary metabolites (Blin et al., 2017). Functional genes involved in plant growth promotion, such as those responsible for indole acetic acid (IAA) production, phosphate solubilization, nitrogen fixation, and genes related to the induction of resistance substance synthesis, were searched in the KEGG database according to the methods described by Kumar et al. (2019) and Shi et al. (2017), and gene similarity was compared using BLAST.
The ability of the YC89 strain to produce siderophores was determined according to the method of Schwyn and Neilands (1987). The activated strain was inoculated on Chrome Azurol S (CAS) medium and incubated at 28°C for 7 d to observe whether a yellow transparent aperture was produced. All experiments were performed in triplicate.
Carboxymethyl cellulose agar (Zheng et al., 2011) was used to detect cellulase activity. The strains were inoculated in carboxymethyl cellulase identification medium and incubated at 28°C for 3 d. The plates were then dyed with Congo red (1 mg/mL) for 15 min and rinsed with NaCl (1 mol/L) for several minutes (Guo et al., 2016). The plates were then examined to determine whether cellulose hydrolysis circle was produced around the colonies; the presence of a cellulose hydrolysis circle indicates that the strain can produce carboxymethyl cellulase. All experiments were performed in triplicate.
The strains were inoculated on a solid phosphate solubilization medium with tricalcium phosphate and calcium phytate as substrates (Kumar et al., 2013). The formation of transparent halo zones around the bacterial colonies after 7 d of incubation at 28°C was considered an indication of phosphate solubilizing activity. All experiments were performed in triplicate.
One single colony on the medium was picked and inoculated into Luria-Bertani broth and incubated at 28°C for 24 h under shaking conditions (180 rpm). The fermented liquid was centrifuged at 10,000 rpm for 10 min, and the supernatant was discarded, suspended in sterile water, and adjusted to a concentration of 1 × 108 CFU/mL for further experiments.
Colletotrichum falcatum Went. was inoculated onto solid PDA medium and incubated at 28°C for 7 d. A sterile punch (d = 5 mm) was used to remove a plug from the edge of the pathogen colony and transfer it to the potato liquid culture solution. The culture was then incubated for 7 d at 28°C with shaking at 180 rpm, and filtered through a double layer of sterile gauze to obtain a suspension of pathogen spores. The spores were enumerated under a microscope with a hemocytometer plate, and diluted with sterile water to a concentration of 1 × 107 conidia/mL.
For the greenhouse experiment, the sugarcane cultivar (ROC 22) was the test plant. Two sugarcane endophytes, YC89 and X22, were used for the pot experiments. Five treatments were applied: (1) water (control), (2) carbendazim (1 mg/mL, positive control), (3) YC89, (4) X22, and (5) YC89 + X22. All experiments had six replicates, with 12 seedlings per treatment. From seedling emergence to six leaves, the plants in treatment groups 3, 4, and 5 were treated with three applications of bacterial suspension, while group 2 was treated twice with carbendazim. Water was applied as a control. At 15 d after the final bacterial treatment, the seedlings in each treatment group were inoculated with 1 L of C. falcatum spore suspension (1 × 107 conidia/mL).
After inoculation with C. falcatum, sugarcane leaves were collected on Days 0, 1, 3, 5, and 7 to detect the changes in related defense enzyme activities in sugarcane at different time points. The leaves were stored in a deep freezer at −80°C for further biochemical analysis.
Disease incidence and severity were recorded within 7–10 d of the appearance of the first symptoms. Disease severity was rated on a scale from 0 to 5 according to the symptoms of plant disease in each plant: 0, no symptoms; 1, appearance of small red spots; 2, enlargement of red spots; 3, spots extend along the midrib to form strips of red to brown parallel to the vein; 4, breakage of blade; 5, spear leaf and growth point rot to death. The disease index and disease control effects were calculated as follows:
Disease index = [∑(The number of diseased plants in this grade × Disease grade)/(Total number of plants investigated × the highest disease grade)] × 100.
Control efficacy (%) = [(Disease index of control – Disease index of treated group)/Disease index of control] × 100.
The activities of plant defense-related enzymes (SOD, PPO, chitinase, and β-1,3-GA) were assessed during the process of B. velezensis YC89-triggered ISR against C. falcatum in sugarcane. The activities of SOD, PPO, chitinase, and β-1,3-GA in plant leaves were measured using the corresponding test kits as described in the study by Jiang et al. (2019) (Nanjing Jiancheng Biological Engineering Institute, Nanjing, China), according to the manufacturer’s instructions.
All data were analyzed by analysis of variance in IBM SPSS Statistics 25 (IBM Corp., Armonk, NY, United States). Differences between means were compared using the LSD test (Fisher’s protected least significant difference test) at p = 0.05. p < 0.05 was considered statistically significant.
The complete genome sequence of B. velezensis YC89 was composed of a 3.95 Mb circular chromosome that included 3,711 coding sequences (CDS) with an average length of 922.05 bp and an average G + C content of 46.62% (Figure 1). The chromosome contained 27 rRNAs, 86 tRNAs, and two genomic islands, with an average length of 39.39 kb. Among the 3,711 CDS, 3698 were annotated functionally, and the remaining 13 were hypothetical. Of the predicted genes, 3,696 (99.60%), 3,380 (91.08%), 2,785 (75.05%), and 2,278 (61.38%) matched in the NR, Swiss-Prot, COG, and KEGG databases, respectively. The features of the YC89 genome and a comparison with other closely related Bacillus genomes are presented in Table 1. In comparison, the entire genome size of the five B. velezensis strains ranged from 3.92–4.16 Mb, the G + C content ranged from 46.1–47.06%, and the predicted coding genes ranged from 3,711–4,187. The YC89 strain and the other five strains contained one circular chromosome without a plasmid. The genome size and protein-coding genes of strains YC89 and FZB42 were smaller than those of the other strains.

Figure 1. Circular genome map of Bacillus velezensis YC89. From the innermost to the outermost: ring 1 GC skew positive (Orange) and negative (purple); ring 2 for GC content, Orange is greater than the average, blue is less than the average; ring 3 for distribution of rRNAs (red) and tRNAs (black); ring 4 COG classifications of protein-coding genes on the reverse strand; ring 5 COG classifications of protein-coding genes on the forward strand; ring 6 for genome size (black line).

Table 1. Genomic features of Bacillus velezensis YC89 and its close-related strains.
According to GO annotation, the genes were classified into 59 functional groups, and the genes involved in biological processes were the most abundant (Supplementary Figure S1). Among the biological process groups, the number of genes related to cellular processes was the highest (2708) (Supplementary Table S2). Based on the COG database, 3,380 genes were assigned to 21 COG categories (Figure 2). The general function prediction-only category represented the largest group (443 genes, 11.94% of all CDS), followed by amino acid transport and metabolism (346 genes, 9.32% of all CDS). According to KEGG annotation, 2,172 genes (58.52% of all CDS) were assigned to 23 KEGG pathways, and the largest number of identified genes were classified into metabolic pathways. Among these pathways, the most represented pathways included carbohydrate metabolism (242 genes, 6.52% of all CDS), followed by amino acid metabolism, and metabolism of cofactors and vitamin pathways (Supplementary Figure S2).
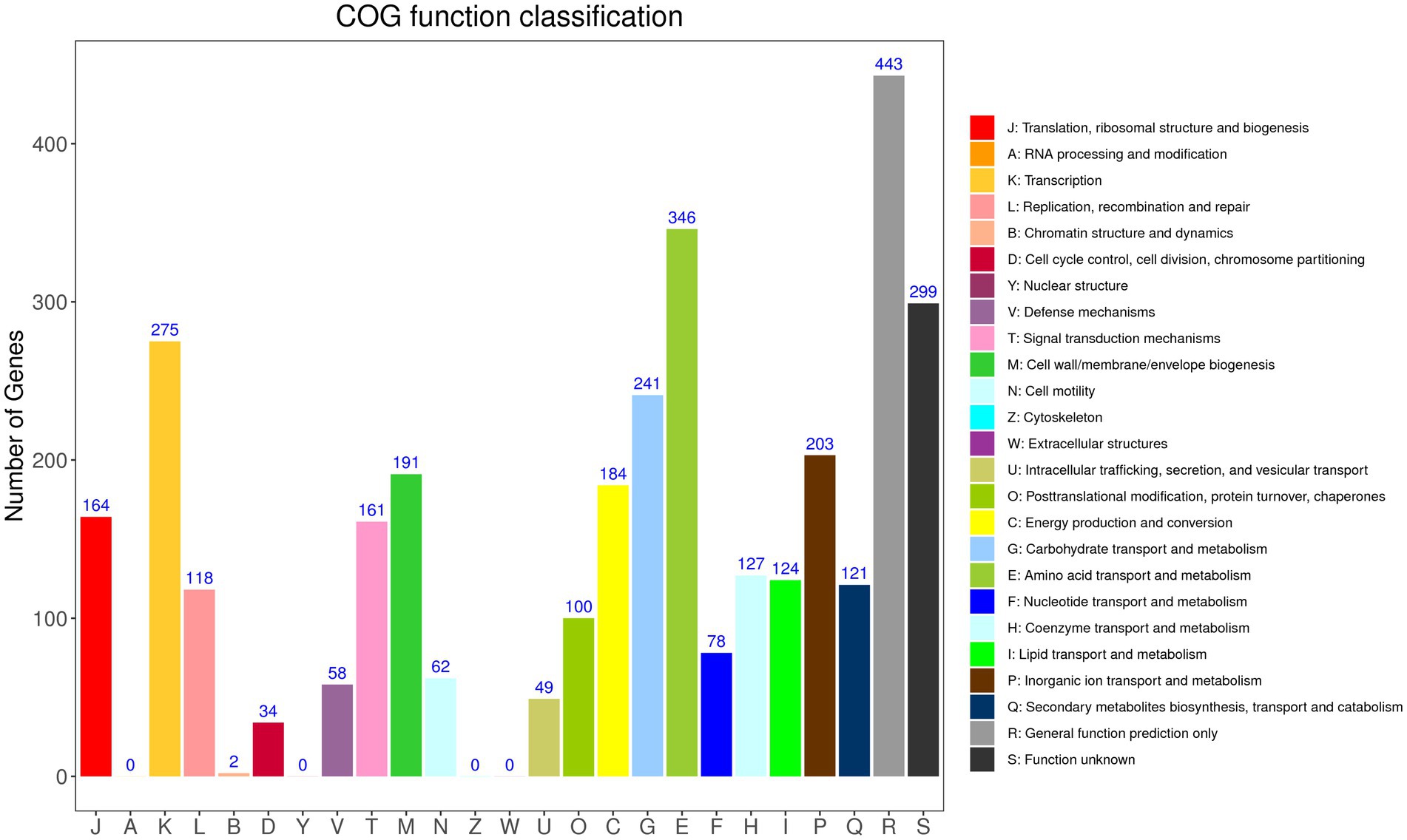
Figure 2. Distribution of genes across COG functional categories in the chromosome of YC89.
These results indicate that YC89 is closely related to B. velezensis FZB42 and B. amyloliquefaciens 6B (Supplementary Figure S3A). For an in-depth analysis of the taxonomic status of YC89 strain, a genome-wide phylogenomic tree was constructed. The results revealed that strain YC89 formed a close genetic relationship with strain B. velezensis GS-1 (Supplementary Figure S3B).
ANI is a powerful approach for assessing the evolutionary distance among bacterial species based on digital whole-genome comparisons, with values closer to 1 indicating higher similarity. Based on ANI values, the genome sequence of YC89 displayed the highest similarity with B. velezensis with ANI values >97%; however, the ANI values between YC89 and B. amyloliquefaciens were < 95% (Figure 3). Strains being compared having ANI values >96% are typically regarded as the same species. Therefore, YC89 does not belong to B. amyloliquefaciens.
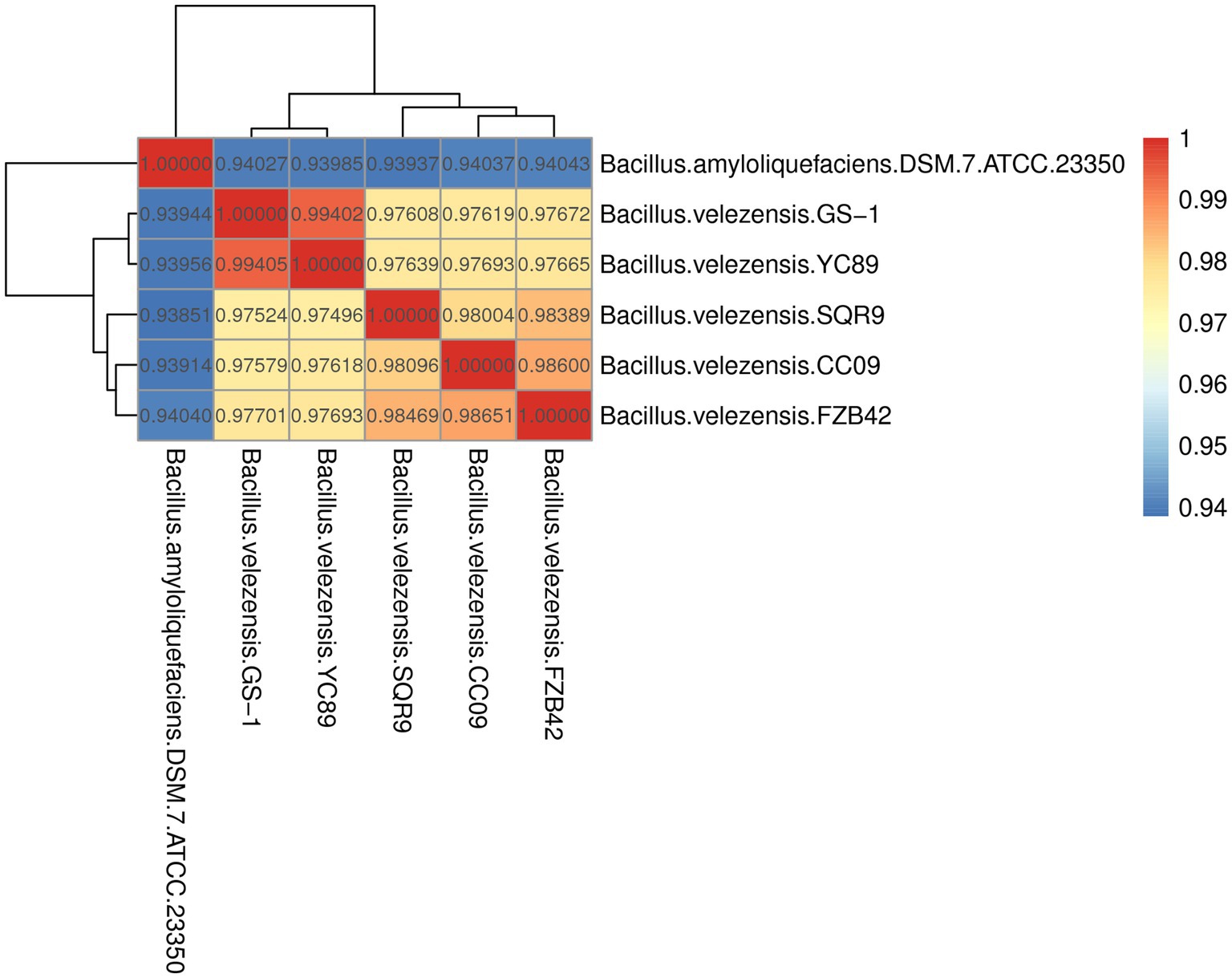
Figure 3. Heatmap of pairwise average nucleotide identity (ANI) values for whole genomes of YC89 and five other Bacillus species.
To evaluate the evolutionary distance among these sequenced strains in relation to several Bacillus strains, the genome sequence of YC89 was compared with that of four sequenced B. velezensis strains (GS-1, FZB42, CC09, and SQR9) and one B. amyloliquefaciens strain (DSM7). Alignments of Bacillus strains are presented in Figure 4A. Eight LCBs representing chromosomal similarity regions among the six strains were identified. Except for B. velezensis CC09, the collinearity level among the genomes of other strains was high, with only a few insertions, deletions, and inversions. The nucleotide similarity between B. velezensis YC89 and B. velezensis GS-1 was significantly higher than that between the other strains. Additionally, a pangenome analysis to compare YC89 with five other strains was performed. As presented in Figure 4B, 3,157 orthologous protein-coding genes constituted the core genome; 42 genes were unique to strain YC89. Annotation revealed that these specific genes encode a large number of transcriptional regulators, helicase domain proteins, hypothetical proteins, aminotransferases, transposases, drug resistance transporters, and chloramphenicol resistance proteins (Supplementary Table S3).
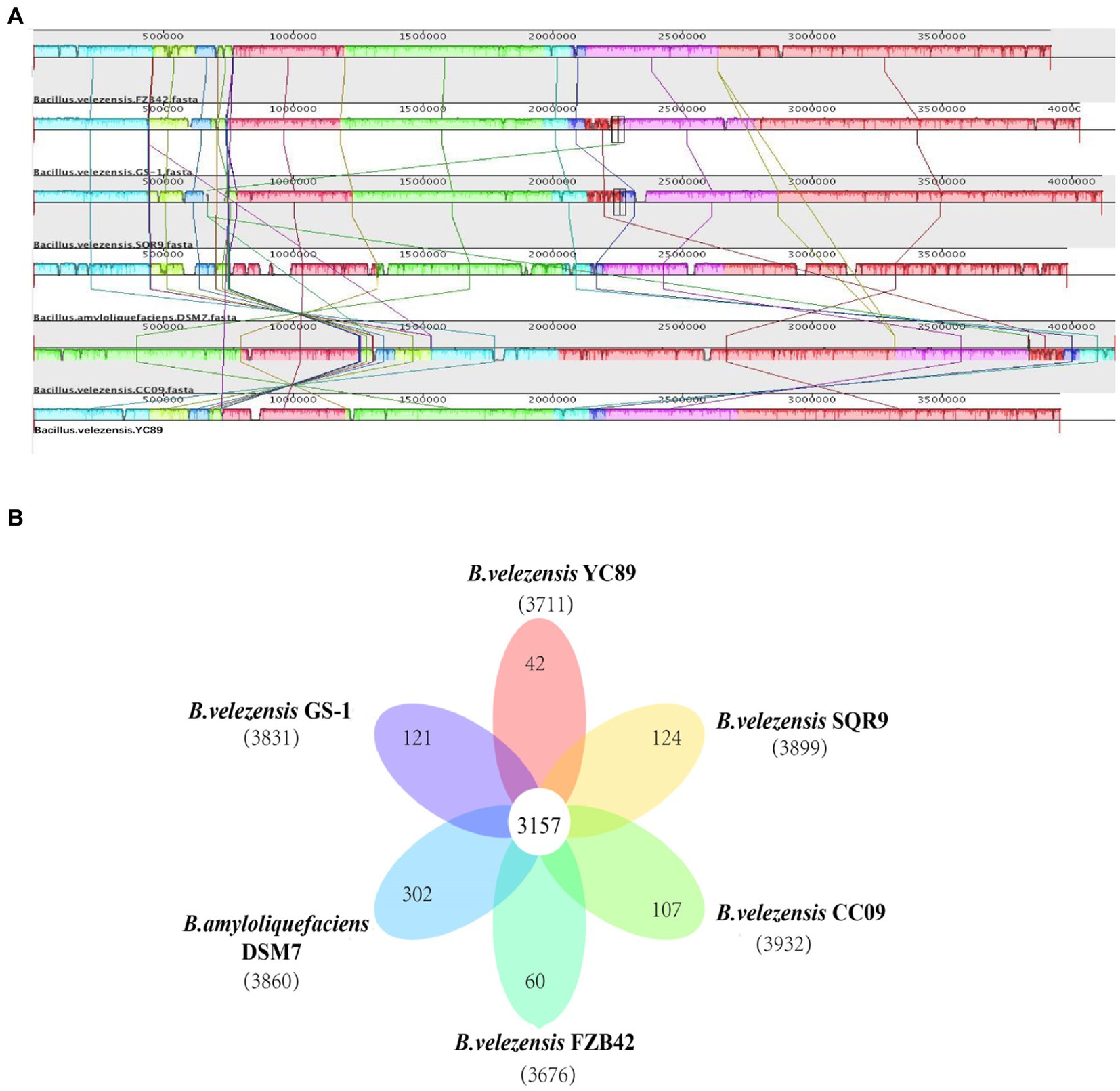
Figure 4. Comparison of B. velezensis YC89 genome sequences against five other Bacillus species genome sequences. (A) Synteny analysis of B. velezensis YC89, B. velezensis FZB42, B. velezensis CC09, B. velezensis SQR9, B. velezensis GS-1 and B. amyloliquefaciens DSM7 genomes. Pairwise alignments of the genomes were generated using MAUVE. The genome of strain YC89 was used as the reference genome. Boxes with the same color indicate syntenic regions. Boxes below the horizontal strain line indicate inverted regions. Rearrangements are shown and by colored lines. Scale is in nucleotides. (B) Venn diagram showing the number of clusters of orthologous genes shared and unique genes.
CAZyme prediction of the B. velezensis YC89 strain revealed that 547 CAZyme genes; 5 had auxiliary activity, 87 were carbohydrate-binding modules (CBM), 46 carbohydrate esterases, 156 glycoside hydrolases, 250 glycosyl transferases, and three polysaccharide lyase genes (Figure 5). After a detailed classification of these six enzyme groups, 547 genes in the B. velezensis YC89 strain were assigned to 118 CAZyme families; the most abundant of these families was GT2 (108 members), which is responsible for transferring nucleotide bisphosphate sugars to substrates, such as polysaccharides and lipids. The CBM50 family contains 47 members that promote the antifungal activity of the enzyme by binding to the chitinous component of the fungal cell wall (Supplementary Table S4). Glucosidases (GH4 and GH13_29), chitinases (GH23 and GH18), and glucanases (GH5_2 and GH16) exhibited potential antifungal activities. These results suggested that CAZymes in YC89 played a significant role in antifungal activity.
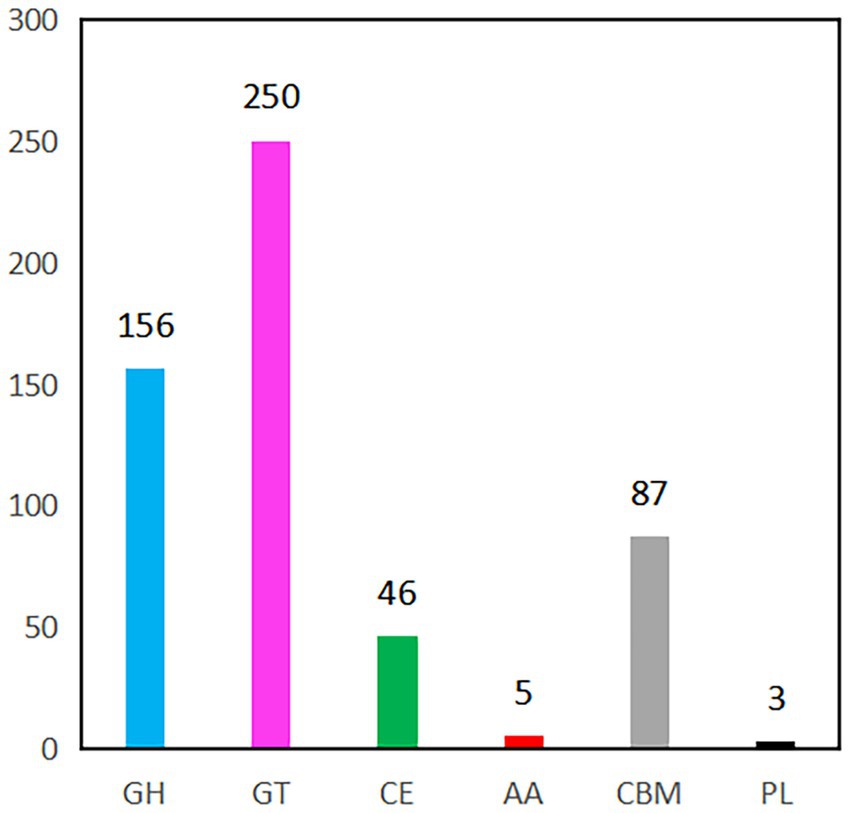
Figure 5. CAZymes gene classification in YC89 genome. GH, Glycoside Hydrolases; GT, Glycosyl Transferases, CE, Carbohydrate Esterases, AA, Auxiliary Activities, CBM, Carbohydrate-Binding Modules; PL, Polysaccharide Lyase.
The YC89 genome was predicted using the online secondary metabolite gene cluster prediction tool antiSMASH; 12 secondary metabolite clusters were predicted, accounting for 18.62% of the whole genome (Table 2). These included two Transatpks gene clusters, two Transatpks-NRPS heterozygous gene clusters, two Terpene gene clusters, one NRPS gene cluster, one OtherKS gene cluster, one Lantipeptide gene cluster, one T3pks gene cluster, one Bacteriocin-NRPS gene cluster, and one Other gene cluster. Cluster 1 had 82% similarity to the surfactin synthesis gene cluster from BGC0000433; Cluster 2 had 7% similarity to the butirosin A/butirosin B synthesis gene cluster from BGC0000693; Cluster 5 had 7% similarity to the Macroprotein A/butirosin B synthesis gene cluster from BGC0000181; Cluster 6 had 100% similarity with the bacillaene synthetic gene cluster from BGC0001089; Cluster 7 had 100% similarity with the fengycin synthetic gene cluster from BGC0001095; Cluster 10 had 100% similarity with the bacillibactin synthetic gene cluster from BGC0000176; Cluster 11 had 100% similarity with the bacillibactin synthetic gene cluster from BGC0000309. Clusters 3, 4, 8, and 9 were functionally unknown, with no homologous sequences matched in the MIBiG accession database, suggesting that these secondary metabolic gene clusters may encode secondary metabolites that generate novel structures and warrant further isolation and identification.
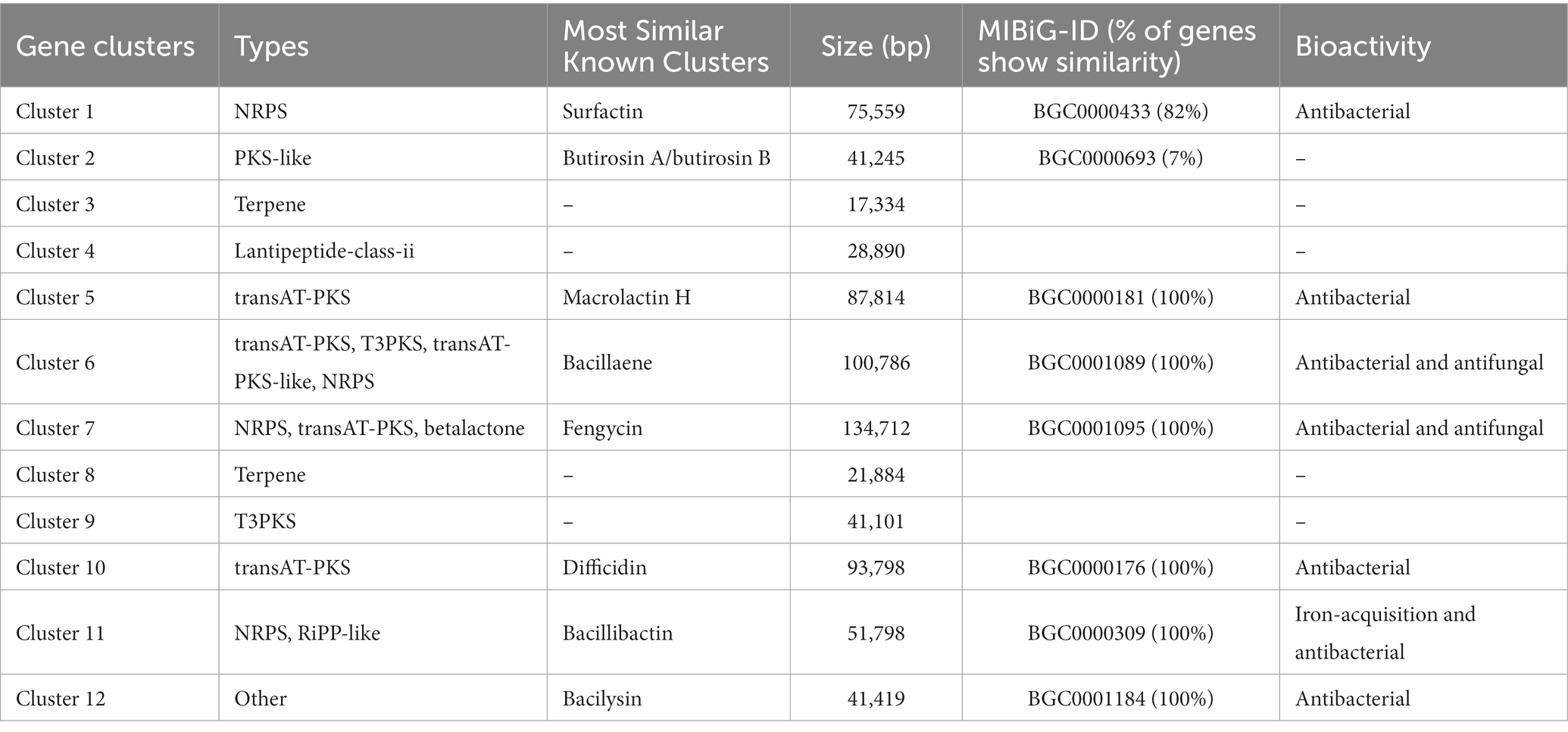
Table 2. Putative gene clusters that encoded for secondary metabolites in YC89.
Based on numerous reported examples of elicitors, genes encoding resistance inducers were selected for comparison among strains YC89, FZB42, and GS-1 using BLAST. As presented in Table 3, the genes encoding several elicitors, such as 2, 3-butanediol, acetoin, peptidoglycan, and EF-Tu, were detected in YC89, FZB42, and GS-1. However, the sequence identity between YC89 and GS-1 was higher than that between YC89 and GS-1. This finding indicates that although all three strains exhibited similar effects on inducing resistance, they may have certain differences in their efficiency to induce plant disease resistance owing to different gene similarities.
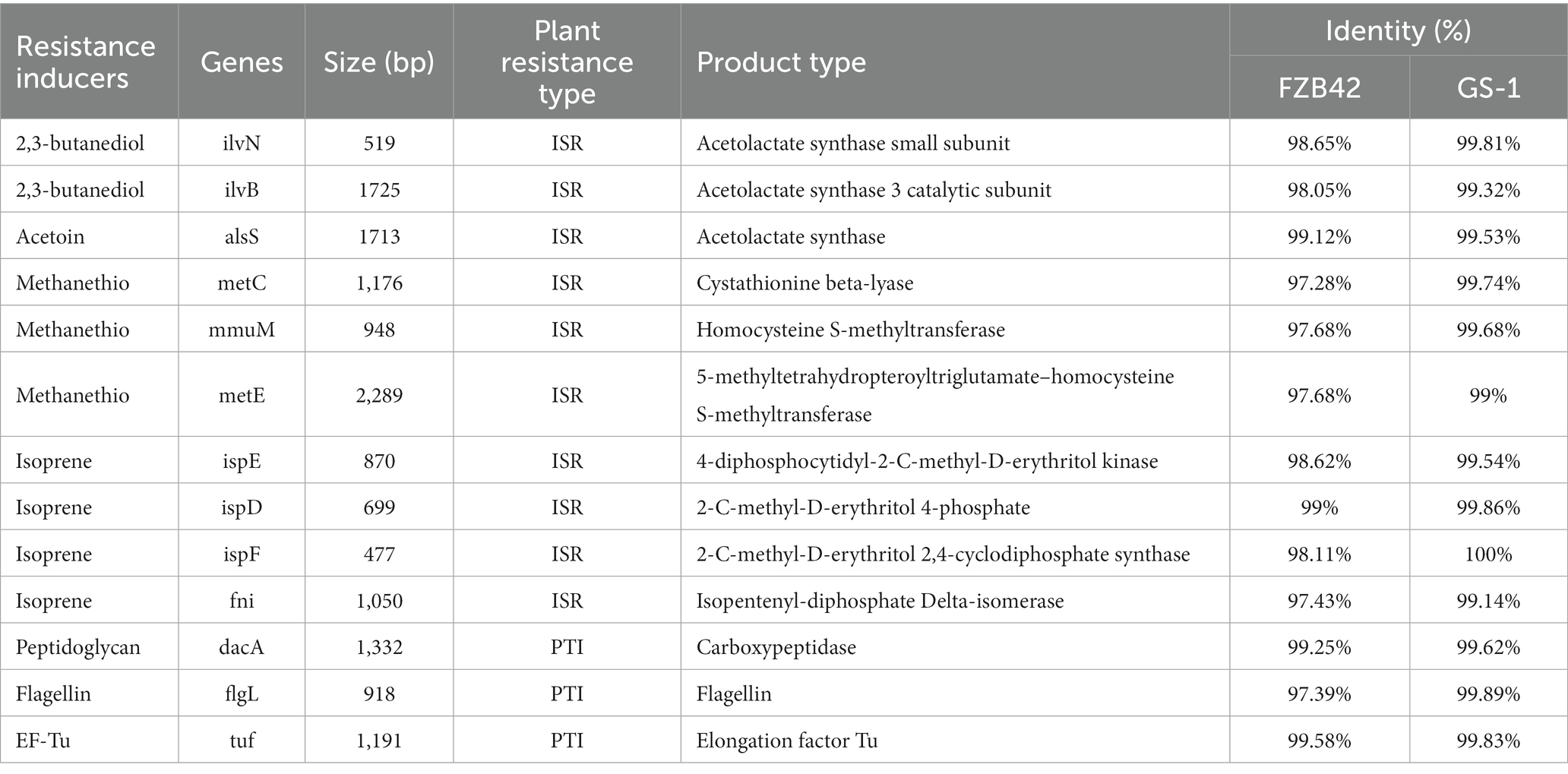
Table 3. Comparison of genes involved in synthesis of resistance inducers in strain YC89, FZB42, and GS-1.
Beneficial bacteria promote plant growth by influencing nutrient uptake, most genes related to plant growth promotion and protection were detected in B. velezensis YC89 (Table 4). IAA is an important phytohormone that controls cell enlargement and tissue differentiation in plants; eight genes related to IAA biosynthesis have been identified in strain YC89. Nitrogen, phosphorus, and potassium are essential for plant growth and development. One gene cluster, nar (H–K), nasD, and nirD, was predicted to be involved in nitrate transport and reduction. A gene cluster consisting of three genes, pst (ABC), was found in the YC89 genome and was predicted to be involved in phosphate solubilization. KtrC and yugO were found in the YC89 genome and predicted to be involved in potassium uptake. In addition, two genes, mgtE and corA, were predicted to be involved in Mg transport.
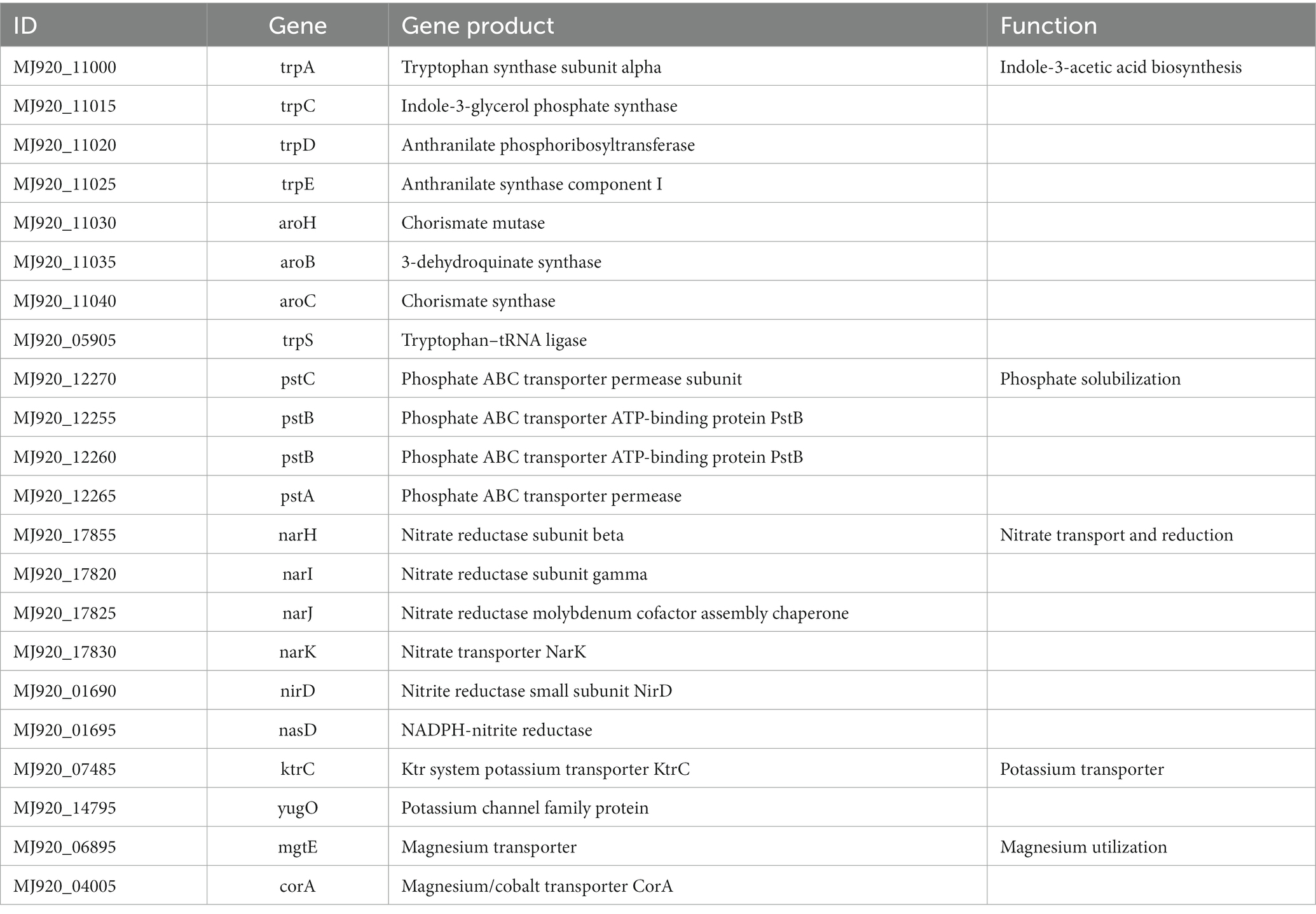
Table 4. Genes related to plant growth promotion in the B. velezensis YC89 genome.
To investigate the biocontrol mechanism of YC89, siderophore production, phosphate solubilization, and cellulase production of the YC89 strain were determined. The results revealed that the strain inoculated on the CAS plate produced a yellow transparent aperture; however, the strain inoculated on a solid phosphate solubilization medium with tricalcium phosphate and calcium phytate as substrate media produced transparent apertures, and a cellulose hydrolysis circle was formed in the carboxymethyl cellulase identification medium. This indicated that YC89 could produce siderophores, dissolve phosphorus, and produce cellulase (Figure 6).
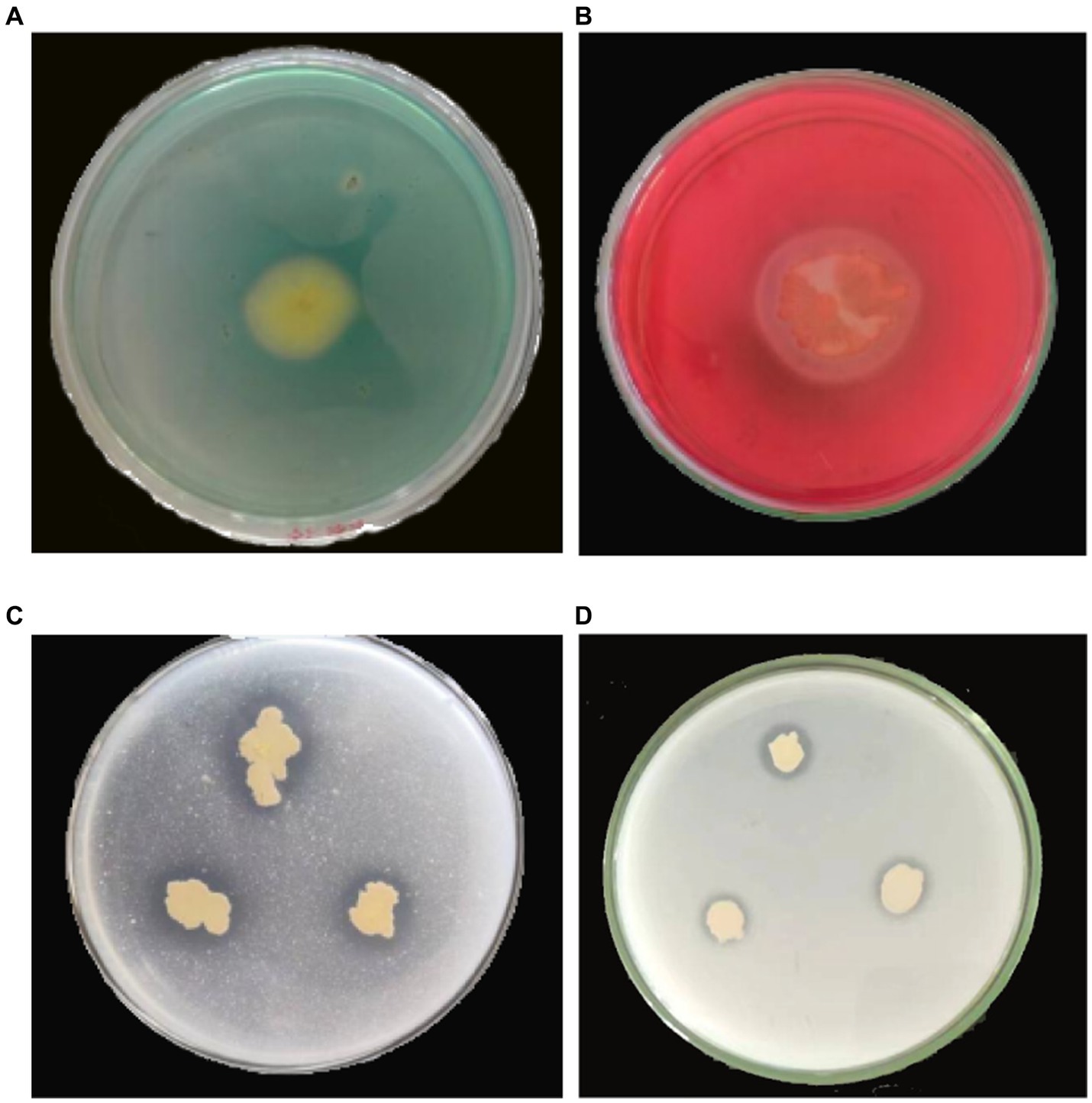
Figure 6. Determination of biological activity of B. velezensis YC89. (A): siderophore, (B): cellulase, (C): Monkina, (D): NBRIP.
In order to determine the effect of biocontrol bacteria on the activity of disease resistance-related enzymes, C. falcatum was inoculated after inoculation of biocontrol bacteria, and the activities of SOD, β-1,3-GA, chitinase and PPO were measured at different times after inoculation of C. falcatum. The results are shown in Figure 7. Compared with the control treatment group, the biocontrol bacteria treatment group could increase the activities of SOD, β-1,3-GA, chitinase and PPO in sugarcane leaves and maximize their activities to cope with the invasion of C. falcatum. The SOD enzyme activity of sugarcane leaves reached the highest value 5 days after inoculation of C. falcatum, at this time, the enzyme activity difference was the largest. The enzyme activity of the biocontrol bacteria treatment group was significantly higher than the CK, in which the enzyme activity of YC89 treatment group was 41.90% higher than the CK, and 16.32% higher than the carbendazim treatment group (Figure 7A). The β-1,3-GA enzyme activity peaked on day 1 and 5 after inoculation of the C. falcatum, and the difference in enzyme activity between treatments was not significant on day 1, but was significantly higher than the CK. On the fifth day, the enzyme activity of biocontrol bacteria treatment group was significantly higher than the carbendazim treatment group and CK treatment group (Figure 7B). The effect of each treatment group on the chitinase activity was not significant from 0 to 3 days after inoculation with C. falcatum, and the difference between treatments was not significant. However, after the fifth day, the difference in chitinase activity was significant, with the YC89 treatment group and YC89 + X22 treatment group having significantly higher enzyme activity than the CK and carbendazim treatment groups (Figure 7C). PPO enzyme activity reached its peak three days after inoculation of C. falcatum, at this time, the enzyme activity difference was the largest, and the enzyme activity of YC89 treatment group was significantly higher than the CK and carbendazim treatment group (Figure 7D).
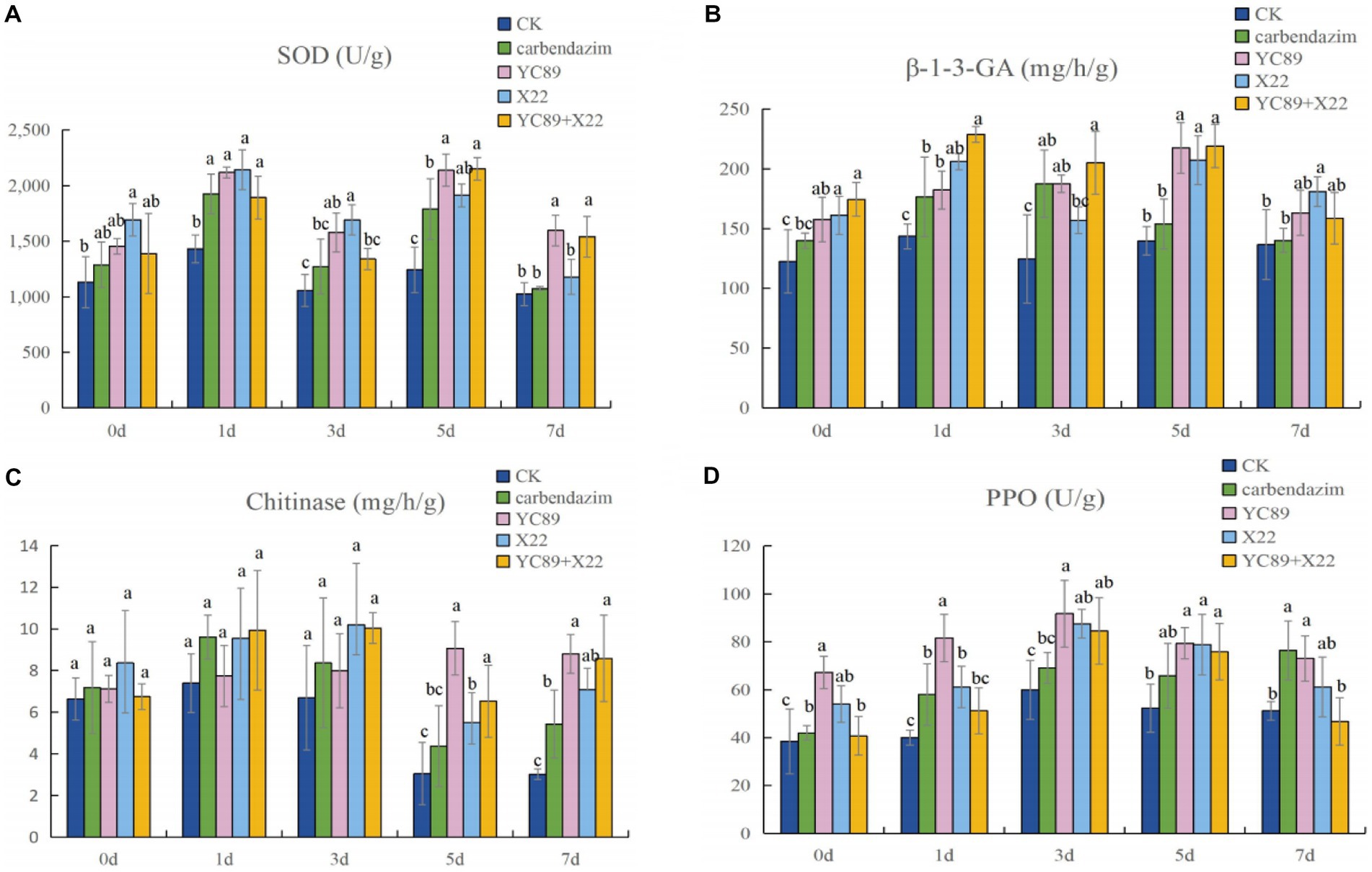
Figure 7. Plant Defense-Related Enzyme Analysis. (A) Superoxide dismutase activity (SOD), (B) β-1-3- glucase activity, (C) chitinase activity, (D) Polyphenol oxidase activity (PPO).
The pot-control effects of the biocontrol strains were also determined (Table 5). The results revealed that strains YC89 and X22 had certain control effects on sugarcane red rot and that the control effect was greater than that of carbendazim. The control effect of strain YC89 on sugarcane red rot disease was 61.91%. The control effect of the X22 strain on sugarcane red rot was worse than the YC89; however, the relative control effect of the mixed inoculation of the two strains was slightly different from that of YC89 alone. In addition, the plant growth-promoting capabilities of YC89 were evaluated using greenhouse experiments. As presented in Table 5, the growth parameters (plant height, stem diameter, and root length) of sugarcane subjected to the YC89 treatment were significantly higher than the control treatment. Collectively, the potted results indicated that the YC89 strain could control the red rot disease of sugarcane and promote the growth of sugarcane plants (Supplementary Figure S4).
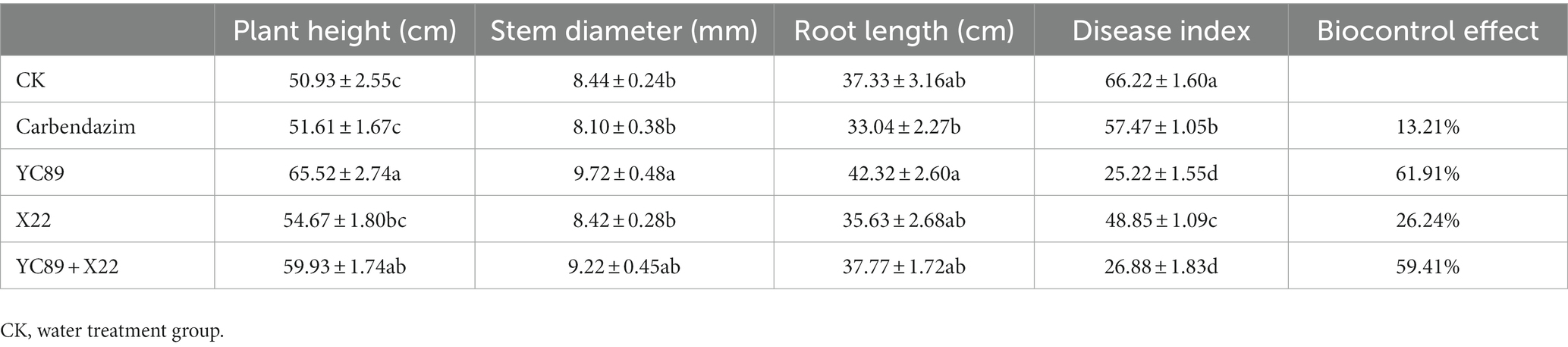
Table 5. Evaluation of biocontrol efficacy of B. velezensis YC89 and its effect on plant growth parameters in sugarcane in greenhouse.
The genus Bacillus covers a large number of species with a high degree of similarity among many Bacillus species (in terms of the current classification), making it difficult to effectively determine the taxonomic status of strains using traditional classification methods (Hamdache et al., 2013). Several B. amyloliquefaciens are now reported as B. velezensis, such as type strain B. amyloliquefaciens FZB42 (Schilling et al., 2018), B. amyloliquefaciensvb7 (Saravanan et al., 2021), B. amyloliquefaciens SQR9 (Feng et al., 2018) so on. To determine the relationship between YC89 and Bacillus spp. strains, phylogenetic trees based on 16S rRNA gene sequences were constructed. These results indicate that YC89 is closely related to B. velezensis FZB42 and B. amyloliquefaciens. In order to be more accurate, we constructed a genome-wide phylogenomic tree, the phylogenetic analysis showed that the YC89 strain and B. velezensis GS-1 were in the same branch with the highest similarity.
Biological control, which has become an effective measure, is one of the safest and most environmentally friendly methods of plant disease control. Among biological control bacteria, Bacillus spp. are believed to be effective. The primary mechanisms used by Bacillus to control plant diseases include antagonism, competition, and induction of disease resistance in plants, however, they can also enhance their resistance by promoting plant growth, which is usually synergistic (Backer et al., 2018; Rodriguez et al., 2019). However, endophytic bacteria can promote the uptake and utilization of nutrients, such as nitrogen, phosphorus, and potassium, by host plants, promote plant growth and enhance their resistance, and produce certain plant growth hormones, such as IAA and indoleacetonitrile, which can directly promote plant growth (Pii et al., 2015). Pot experiments using YC89 to treat sugarcane plants revealed an increase in shoot height and root length. In this study, the YC89 genome annotation demonstrated that a large number of YC89 genes are involved in plant growth by enhancing nutrient uptake and availability. In the YC89 genome, the nitrate transporter nark and the nitrate reductases narH-narI-narJ, nirD, and nasD were found. These gene clusters were predicted to be involved in nitrate transport and reduction (Wray et al., 1994). Additionally, genes involved in potassium transport were identified in the YC89 genome; two genes, ktrC and yugO, that have been identified and characterized as potassium transporters in Bacillus spp., were identified in the YC89 genome. In a recent full-genome analysis of B. velezensis FZB42, mgtE, and corA were predicted to be involved in Mg uptake and detoxification of heavy metal ions in host plants (Schilling et al., 2018). Hence, MgtE and corA likely have similar roles in YC89. Biocontrol bacteria can compete with pathogens for iron to inhibit pathogen growth; in this study, strain YC89 was able to produce siderophores. In addition, a cluster (bacillibactin) was found in the YC89 genome that predicted putative iron availability.
In recent years, research has focused on investigating the ability of B. velezensis to produce various secondary metabolites and enzymes with broad-spectrum resistance to plant pathogens (Wang et al., 2021). Antimicrobial cyclic lipopeptides synthesized by the NRPS and polyketides synthesized by the polyketide synthesis pathway form the basis of their broad-spectrum antibacterial activity. Cyclic lipopeptides primarily include the three families of surfactin, iturin, and fengycin, all of which can induce systemic resistance to enhance plant defense against pathogens (Jourdan et al., 2009; Wu et al., 2018). However, the gene cluster encoding iturin was not found in the YC89 genome. Polyketides, including macrolactin, bacillaene, and diffidin, can be used as antibiotics, antifungals, or natural insecticides. The main polyketides identified in Bacillus are derived from B. subtilis and B. velezensis, among which B. velezensis is particularly abundant in polyketides (Baek, 2020). Using whole genome sequencing, Zhang et al. (2022) found that B. velezensis GS-1 was able to produce the lipopeptide compounds, surfactin, fengycin, and plantazolicin, and confirmed the inhibitory effect of lipopeptide compounds on M. oryzae. Meanwhile, eight secondary metabolite-related gene clusters were identified in the Bacillus velezensis VB7 genome, the Bacillus velezensis VB7 was a potential antagonist for managing carnation infection by Sclerotinia sclerotiorum, cotton infection by tobacco streak virus, and groundnut bud necrosis infection of tomato (Saravanan et al., 2021). In the current study, the YC89 genome was predicted using the online secondary metabolite gene cluster prediction tool antiSMASH, which predicted 12 gene clusters, including four of unknown function and seven antibiotic synthesis gene clusters with high similarity (surfactin, macrolactin H, bacillaene, fengycin, difficidin, bacillibactin, and bacilysin). One antibiotic gene cluster (butirosin) showed only 7% similarity. Butirosin is an aminoglycoside antibiotic that is particularly effective against gram-negative bacteria (Kudo and Eguchi, 2009). Although 12 secondary metabolite gene clusters were identified by whole-genome sequencing, the isolation and extraction of secondary metabolites and their inhibitory effects requires further investigation.
The main component of the cell wall in most pathogenic fungi is chitin. Chitinase catalyzes chitin hydrolysis to produce N-acetylglucosamine, which destroys the structural integrity of the fungal cell wall (Zhang et al., 2018). Endophytic bacteria can also produce hydrolases, such as chitinase, cellulase, and glucanase. These hydrolases can destroy fungal cell structure and dissolve the cell wall, thereby inhibiting fungal growth (Verma and Gange, 2014). Indeed, plant cell walls are composed of a complex network of carbohydrates, such as cellulose, hemicellulose, pectin, proteins, and glycoproteins. CAZymes cleave polysaccharides and other structural compounds associated with similar microorganisms into oligomers and simple monomers for nutrient uptake by microorganisms (Stafford and Satur, 2017). CAZyme analysis showed that the B. velezensis YC89 genome contained 547 CAZyme genes, including those associated with cell wall degrading enzymes, such as β-1,3-glucanase, and chitinase; it was hypothesized that degradation of the cell wall of pathogenic fungi serves as an inhibitory mechanism of B. velezensis YC89. Thus, the CAZymes in B. velezensis YC89 strain likely have an important role in spatial competition and nutrient acquisition and play an integral part in suppressing the virulence of the sugarcane red rot pathogen.
Additionally, endophytes can induce systemic resistance in plants to resist pathogen infestation and reduce disease incidence. Plant-induced resistance is synthesized by a combination of physiological and biochemical factors, such as defense enzyme activity, lignin, disease-course-associated proteins, and multi-post compounds. SOD, PPO, gibberellinase, and β-1,3 glucanase (β-1,3-GA) resistance enzymes in plants play an important role in the defense against foreign pathogens, and their enzyme activity level is often used to indicate the strength of disease resistance in plants (Song et al., 2006). To understand the mechanism of B. velezensis F21-mediated ISR in Fusarium wilt, Jiang et al. (2019) determined the expression levels of defense-related genes and the activities of defense-related enzymes (CAT, POD, and SOD) in the roots of watermelon inoculated with Fusarium oxysporum f. sp. niveum (Fon) only and in those pretreated with B. velezensis F21 before inoculation with Fon. The results indicated that compared with the control treatment, B. velezensis F21 could increase the activities and prepare the defense-related enzymes for maximum activity in advance of Fon invasion. In this study, we measured the activity and relative efficacy of sugarcane leaf resistance enzymes in a pot test and identified that YC89 and X22 in the biocontrol solution increased SOD, PPO, gibberellinase, and β-1,3-GA activities. In fact, the relative efficacy of the treatment groups with a biocontrol solution was higher than that of the water and carbendazim groups. In addition, ten genes related to ISR were identified in B. velezensis YC89. These results demonstrate that B. velezensis YC89 induces similar systemic resistance in plants.
Although the optimal method for controlling sugarcane red rot includes the application of chemical fungicides, long-term use can lead to the development of resistance by the pathogenic bacteria, resulting in a decrease in treatment efficacy. Moreover, the frequent use of high-concentration chemical fungicides can cause the formation of pesticide residues, which pollute the environment and pose a severe threat to human health. As an alternative, various biocontrol agents have been implemented. In particular, Pseudomonas spp., Trichoderma spp., Bacillus spp., and Streptomyces spp. are the most effective biocontrol agents against sugarcane red rot (Wu et al., 2015). In this study, the relative efficacy of B. velezensis YC89 against sugarcane red rot was determined using a pot test, and the results showed that B. velezensis YC89 had a relative efficacy of 61.91% against sugarcane red rot, in addition to promoting the growth of sugarcane plants. Although the greenhouse pot experiment simulates the field environment to a large extent, compared to the field environment, greenhouse pots have controlled characteristics and less interference than plants in the field, so that the effects of the strain on the plant would be more pronounced in the greenhouse pots. The induction of biocontrol agents is easily affected by various factors in the field, including environment, temperature, climate, and terrain; therefore, further research is required in field trials of strain YC89.
The colonization of a single biocontrol bacterium in the soil is unknown and has a limited effect on the control of soil pathogens, thus affecting the promotion of their use. However, the synergistic effects among different biocontrol agents and strains are difficult to predict and may reduce the control effect (Xu et al., 2011; Pertot et al., 2017). In this study, strain YC89 was mixed with strain X22 to control sugarcane red rot; although the effect did not differ significantly from that of YC89 alone, it was superior to that of X22 alone, indicating that the biological control ability of strain YC89 was not inhibited by strain X22 after combined inoculation. Hence, given that many biocontrol agents are being used to control plant diseases, the effects of combining B. velezensis YC89 with other beneficial microorganisms warrants further investigation.
Our study findings indicate that B. velezensis YC89 could serve as a potential biocontrol agent for sugarcane red rot and promote sugarcane plant growth. Whole-genome and phylogenetic analyses revealed that YC89 belongs to B. velezensis and is closely related to B. velezensis GS-1. Genome analysis demonstrated that YC89 harbors multiple genes related to IAA production, nitrogen fixation, phosphate solubilization, antifungal activity, and resistance inducer biosynthesis. The underlying biocontrol mechanisms can be inferred as follows: (1) production of active antibacterial substances, such as antibiotics and hydrolases; (2) significant promotion of plant growth through nitrogen fixation, phytohormone synthesis, and phosphorus augmentation; and (3) induction of ISR in plants for defense against pathogenic bacterial infection. Overall, the features of B. velezensis YC89 make it a potential biocontrol agent and biofertilizer. These results will contribute to in-depth research on sugarcane plant growth promotion and biocontrol mechanisms.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
FL, LH, and LX designed the experiments. LX conducted most of the experiments. LL, YL, and XR performed a small number of experiments and provided experimental methods. YD, HL, ZQ, and QS carried out the statistical analyses and organized the data. LX wrote the manuscript. FL and LH revised the manuscript. All authors contributed to the article and approved the submitted version.
This project was supported by the Major Science and Technology Special Project of Yunnan Province (202202AE090021), Yunnan Provincial Key Laboratory of Crop Production and Smart Agriculture Special Project (202105AG070007), Yunnan Provincial Education Department Scientific Research Fund Project (2022 J0286) and Yunnan Province Modern Agricultural Sugarcane Industry Technical System Construction Project (2022–2023).
The authors would like to thank all the project funds for supporting the experiment, Guangzhou Genedenovo Biotechnology Co., Ltd. for assisting in sequencing and/or bioinformatics analysis, and Editage (www.editage.cn) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1180474/full#supplementary-material
β-1,3-GA, β-l,3-glucanase, POD, peroxidase; PPO, polyphenol oxidase; SOD, superoxide dismutase; ISR, induced systemic resistance; CDS, coding sequences; NCBI, National Center for Biotechnology Information; LCBs, locally collinear blocks; ANI, average nucleotide identity; PDA, potato dextrose agar; NR, non-redundant; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, gene ontology; COG, Cluster of Orthologous Groups; CAZyme, carbohydrate-active enzyme; BLAST, basic local alignment search tool; IAA, indole acetic acid; CAS, Chrome Azurol S; NRPS, non-ribosomal peptide synthetase; CK, water treatment group.
Amna, Y., Xia, Y., Farooq, M. A., Javed, M. T., Kamran, M. A., Mukhtar, T., et al. (2020). Multi-stress tolerant pgpr bacillus xiamenensis pm14 activating sugarcane (saccharum officinarum L.) red rot disease resistance. Plant Physiol. Biochem. 151, 640–649. doi: 10.1016/j.plaphy.2020.04.016
Backer, R., Rokem, J. S., Ilangumaran, G., Lamont, J., Praslickova, D., Ricci, E., et al. (2018). Plant growth-promoting rhizobacteria: context, mechanisms of action, and roadmap to commercialization of biostimulants for sustainable agriculture. Front. Plant Sci. 9, 1473–1489. doi: 10.3389/fpls.2018.01473
Baek, K. H. (2020). Antimicrobial activities of lipopeptides and polyketides of Bacillus velezensis for agricultural applications. Molecules 25:4973. doi: 10.3390/molecules25214973
Blin, K., Wolf, T., Chevrette, M. G., Lu, X., Schwalen, C. J., Kautsar, S. A., et al. (2017). Antismash 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41. doi: 10.1093/nar/gkx319
Buchfink, B., Reuter, K., and Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using Diamond. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Bukhari, K., Nithya, K., Valluvaparidasan, V., Paranidharan, V., and Velazhahan, R. (2012). Detection of Colletotrichum falcatum causing red rot of sugarcane by enzyme-linked immunosorbent assay. Arch. Phytopathol. Plant Protect. 45, 823–830. doi: 10.1080/03235408.2011.597959
Costa, M. M., Silva, B., Moreira, G. M., and Pfenning, L. H. (2021). Colletotrichum falcatum and fusarium species induce symptoms of red rot in sugarcane in Brazil. Plant Pathol. 70, 1807–1818. doi: 10.1111/ppa.13423
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Feng, H. C., Zhang, N., Fu, R. X., Liu, Y. P., and Krell, T. (2018). Recognition of dominant attractants by key chemoreceptors mediates recruitment of plant growth-promoting rhizobacteria. Environ. Microbiol. 21, 402–415. doi: 10.1111/1462-2920.14472
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Gilardi, G., Vasileiadou, A., Garibaldi, A., and Gullino, M. L. (2021). Biocontrol agents and resistance inducers reduce phytophthora crown rot (phytophthora capsici) of sweet pepper in closed soilless culture. Phytopathol. Mediterr. 1:60. doi: 10.36253/phyto-11978
Guo, X., Chen, D. D., Peng, K. S., Cui, Z. W., Zhang, X. J., Li, S., et al. (2016). Identification and characterization of Bacillus subtilis from grass carp (Ctenopharynodon idellus) for use as probiotic additives in aquatic feed. Fish Shellfish Immunol. 52, 74–84. doi: 10.1016/j.fsi.2016.03.017
Hamdache, A., Azarken, R., Lamarti, A., Aleu, J., and Collado, I. G. (2013). Comparative genome analysis of Bacillus spp. and its relationship with bioactive nonribosomal peptide production. Phytochem. Rev. 12, 685–716. doi: 10.1007/s11101-013-9278-4
Jiang, C. H., Yao, X. F., Mi, D. D., Li, Z. J., Yang, B. Y., Zheng, Y., et al. (2019). Comparative transcriptome analysis reveals the biocontrol mechanism of Bacillus velezensis F21 against fusarium wilt on watermelon. Front. Microbiol. 10:652. doi: 10.3389/fmicb.2019.00652
Jourdan, E., Henry, G., Duby, F., Dommes, J., Barthélemy, J. P., Thonart, P., et al. (2009). Insights into the defense-related events occurring in plant cells following perception of surfactin-type lipopeptide from bacillus subtilis. MolPlant-Microbe Interact 22, 456–468. doi: 10.1094/MPMI-22-4-0456
Kudo, F., and Eguchi, T. (2009). Biosynthetic enzymes for the aminoglycosides butirosin and neomycin. Methods Enzymol. 459:493. doi: 10.1016/S0076-6879(09)04620-5
Kumar, R., Acharya, V., Mukhia, S., Singh, D., and Kumar, S. (2019). Complete genome sequence of Pseudomonas frederiksbergensis ERDD5: 01 revealed genetic bases for survivability at high altitude ecosystem and bioprospection potential. Genomics 111, 492–499. doi: 10.1016/j.ygeno.2018.03.008
Kumar, V., Singh, P., Jorquera, M. A., Sangwan, P., Kumar, P., Verma, A. K., et al. (2013). Isolation of phytase-producing bacteria from Himalayan soils and their efect on growth and phosphorus uptake of Indian mustard (Brassica juncea). World J. Microb. Biol. 29, 1361–1369. doi: 10.1007/s11274-013-1299-z
Lam-Tung, N., Schmidt, H. A., Arndt, V. H., and Quang, M. B. (2015). Iq-tree: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 1, 268–274. doi: 10.1093/molbev/msu300
Li, C., Cheng, P., Zheng, L., Li, Y., and Yu, G. (2021). Comparative genomics analysis of two banana fusarium wilt biocontrol endophytes Bacillus subtilis r31 and tr21 provides insights into their differences on phytobeneficial trait. Genomics 113, 900–909. doi: 10.1016/j.ygeno.2021.02.006
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Mishra, M. K., and Behera, B. (2009). Pathogenic and molecular variability of Colletotrichum falcatum went. Isolates from sugarcane with red rot disease symptoms. J. Crop. Sci. Biotechnol. 12, 31–35. doi: 10.1007/s12892-008-0066-4
Nithya, K., Bukhari, K. A. I. M., Valluvaparidasan, V., Paranidharan, V., and Velazhahan, R. (2012). Molecular detection of Colletotrichum falcatum causing red rot disease of sugarcane (Saccharum officinarum) using a SCAR marker. Ann. Appl. Biol. 160, 168–173. doi: 10.1111/j.1744-7348.2011.00529.x
Pang, Y., Yang, J., Chen, X., Jia, Y., Li, T., Jin, J., et al. (2021). An antifungal chitosanase from Bacillus subtilis SH21. Molecules 26:1863. doi: 10.3390/MOLECULES26071863
Peñaflor, M. F. G. V., and Bento, J. M. S. (2019). Red-rot infection in sugarcane attenuates the attractiveness of sugarcane borer-induced plant volatiles to parasitoid. Arthropod Plant Interact. 13, 117–125. doi: 10.1007/s11829-018-9629-6
Pertot, I., Giovannini, O., Benanchi, M., Caffi, T., Rossi, V., and Mugnai, L. (2017). Combining biocontrol agents with different mechanisms of action in a strategy to control botrytis cinerea on grapevine. Crop Prot. 97, 85–93. doi: 10.1016/j.cropro.2017.01.010
Pii, Y., Mimmo, T., Tomasi, N., Terzano, R., Cesco, S., and Crecchio, C. (2015). Microbial interactions in the rhizosphere: beneficial influences of plant growth-promoting rhizobacteria on nutrient acquisition process. A review. Biol. Fertil. Soils. 51, 403–415. doi: 10.1007/s00374-015-0996-1
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Rodriguez, P. A., Rothballer, M., Chowdhury, S. P., Nussbaumer, T., and Falter-Braun, P. (2019). Systems biology of plant-microbiome interactions. Mol. Plant 12, 804–821. doi: 10.1016/j.molp.2019.05.006
Ryu, C. M., Farag, M. A., Hu, C. H., Reddy, M. S., and Pare, K. P. W. (2004). Bacterial volatiles induce systemic resistance in arabidopsis. Plant Physiol. 134, 1017–1026. doi: 10.2307/4281635
Saksena, P., Vishwakarma, S. K., Tiwari, A. K., Singh, A., and Kumar, A. (2013). Pathological and molecular variation in colletotrichum falcatum went isolates causing red rot of sugarcane in the northwest zone of India. J. Plant Protect. Res. 53, 37–51. doi: 10.2478/jppr-2013-0005
Saravanan, R., Nakkeeran, S., Saranya, N., Senthilraja, C., Renukadevi, P., and Krishnamoorthy, A. S. (2021). Mining the genome of bacillus velezensis vb7 (cp047587) for mamp genes and non-ribosomal peptide synthetase gene clusters conferring antiviral and antifungal activity. Microorganisms 9:2511. doi: 10.3390/microorganisms9122511
Schilling, T., Hoppert, M., Daniel, R., and Hertel, R. (2018). Complete genome sequence of vb_bvep-goe6, a virus infecting bacillus velezensis fzb42. Genome Announc. 6, e00008–e00018. doi: 10.1128/genomeA.00008-18
Schwyn, B., and Neilands, J. B. (1987). Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160, 47–56. doi: 10.1016/0003-2697(87)90612-9
Shair, F., Yasmin, H., Hassan, M. N., Alzahrani, O. M., and Noureldeen, A. (2021). Pseudomonas spp. mediate defense response in sugarcane through differential exudation of root phenolics. Saudi J. Biol. Sci. 28, 7528–7538. doi: 10.1016/j.sjbs.2021.09.030
Shi, Y., Id Niu, K., Huang, B., and Liu, W. (2017). Transcriptional responses of creeping bentgrass to 2,3-butanediol, a bacterial volatile compound (bvc) analogue. Molecules 22:1318. doi: 10.3390/molecules22081318
Singh, R. K., Kumar, P., Tiwari, N. N., Singh, S. P., Tiwari, A. K., Vishwakarma, S. K., et al. (2014). Role of endochitinase gene and efficacy of trichoderma against colletotrichum falcatum went. Causing red rot disease in sugarcane. Sugar Tech. 16, 180–188. doi: 10.1007/s12355-013-0254-5
Song, Y. Y., Ping, L. I., Jin, L. S., Zheng, A. P., and Wang, L. X. (2006). Studies on promoting ability and induced resistance of antifungal bacterial strain lm-3 in rice. Southwest China J. Agric. Sci. 3, 438–441. doi: 10.1016/S1872-2032(06)60050-4
Stafford, F., and Satur,. (2017). Periodontal pathogen CaZymes: host-pathogen biology, biochemistry and biotechnological exploitation. J. Oral Microbiol. 9:1325208. doi: 10.1080/20002297.2017.1325208
Sumana, A., and Singh, B. P. (2012). Evaluation of epiphytic microflora as antagonists of red rot pathogen, colletotrichum falcatum in sugarcane under subtropical conditions. J. Biol. Control. 4. doi: 10.18641/JBC/26/4/46782
Sun, X., Xu, Z., Xie, J., Viktor, H. T., Tan, T., Zheng, D., et al. (2022). Bacillus velezensis stimulates resident rhizosphere Pseudomonas stutzeri for plant health through metabolic interactions. ISME J. Multidiscip. J. Microb. Ecol. 3:16. doi: 10.1101/2021.06.02.446779
Verma, V. C., and Gange, A. C. (2014). Advances in endophytic research || biological control of insect-pest and diseases by endophytes, (Chapter13), 231–256. doi: 10.1007/978-81-322-1575-2
Viswanathan, R., Ramesh Sundar, A., Malathi, P., Rahul, P. R., Ganesh Kumar, V., Banumathy, R., et al. (2009). Interaction between sugarcane and Colletotrichum falcatum causing red rot: understanding disease resistance at transcription level. Sugar Tech. 11, 44–50. doi: 10.1007/s12355-009-0008-6
Wang, C., Ye, X., Ng, T. B., and Zhang, W. (2021). Study on the biocontrol potential of antifungal peptides produced by Bacillus velezensis against fusarium solani that infects the passion fruit passiflora edulis. J. Agric. Food Chem. 69, 2051–2061. doi: 10.1021/acs.jafc.0c06106
Wray, L. V., Atkinson, M. R., and Fisher, S. H. (1994). The nitrogen-regulated Bacillus subtilis nrgAB operon encodes a membrane protein and a protein highly similar to the Escherichia coli glnB-encoded PII protein. J. Bacteriol. 176, 108–114. doi: 10.1128/jb.176.1.108-114.1994
Wu, G. W., Liu, Y. P., Xu, Y., Zhang, G. S., Shen, Q., and Zhang, R. F. (2018). Exploring elicitors of the beneficial Rhizobacterium Bacillus amyloliquefaciens SQR9 to induce plant systemic resistance and their interactions with plant signaling pathways. Molecular plant-microbe interactions. MPMI 31, 560–567. doi: 10.1094/MPMI-11-17-0273-R
Wu, X. M., Shen, W. K., Luo, M. Z., Rao, D. H., and Chen, P. S. (2015). Progress on sugarcane red rot disease and its causal fungus Colletotrichum falcatum. Sugar Crops China 37, 66–70. doi: 10.13570/j.cnki.scc.2015.06.024
Wu, J. T., Xie, J., Wang, Q. N., and Qiu, Y. S. (2017). Research on the development status of sugarcane industry in China[J]. Guangdong Agric. Sci. 44, 154–160. doi: 10.16768/j.issn.1004-874X.2017.07.024
Xie, L. Y., Di, Y. N., He, L. L., Liu, L. F., Shen, X. Y., and Li, F. S. (2020). Screening and identification of endophytic antagonistic Bacteria YC89 in sugarcane red rot. Acta Microbiol Sin. 60, 739–748. doi: 10.13343/j.cnki.wsxb.20190292
Xu, X. M., Jeffries, P., Pautasso, M., and Jeger, M. J. (2011). A numerical study of combined use of two biocontrol agents with different biocontrol mechanisms in controlling foliar pathogens. Phytopathology 101, 1032–1044. doi: 10.1094/PHYTO-10-10-0267
Zhang, Y., Zhao, M., Chen, W., Yu, H., Jia, W., Pan, H., et al. (2022). Multi-omics techniques for analysis antifungal mechanisms of Lipopeptides produced by Bacillus velezensis GS-1 against Magnaporthe oryzae in vitro. Int. J. Mol. Sci. 23:3762. doi: 10.3390/ijms23073762
Zhang, B. Y., Zhu, T. H., Han, S., Wang, Y., Li, S. J., and Qiao, T. M. (2018). Cloning, expression and antibacterial functions of ChiKJ40, a chitinase gene from Streptomyces sampsonii. Microbiol. China 45, 1016–1026. doi: 10.13344/j.microbiol.china.170572
Keywords: Bacillus velezensis , biocontrol, sugarcane red rot, genome sequencing, biocontrol mechanism
Citation: Xie L, Liu L, Luo Y, Rao X, Di Y, Liu H, Qian Z, Shen Q, He L and Li F (2023) Complete genome sequence of biocontrol strain Bacillus velezensis YC89 and its biocontrol potential against sugarcane red rot. Front. Microbiol. 14:1180474. doi: 10.3389/fmicb.2023.1180474
Edited by:
Mamadou L. Fall, Agriculture and Agri-Food Canada (AAFC), CanadaReviewed by:
Giorgia Pertile, Polish Academy of Sciences, PolandCopyright © 2023 Xie, Liu, Luo, Rao, Di, Liu, Qian, Shen, He and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fusheng Li, bGZzODEwQHNpbmEuY29t; Lilian He, aGVsaWxpYW45MDVAc29odS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.