 Ying Jiang
Ying Jiang Jie Tang
Jie Tang Xiangjian Liu
Xiangjian Liu Maurycy Daroch
Maurycy Daroch- 1School of Environment and Energy, Peking University Shenzhen Graduate School, Shenzhen, China
- 2School of Food and Bioengineering, Chengdu University, Chengdu, Sichuan, China
Thermophilic cyanobacteria play a crucial role as primary producers in hot spring ecosystems, yet their microbiological, taxonomic, and ecological characteristics are not extensively studied. This study aimed to characterize a novel strain of thermophilic cyanobacteria, PKUAC-SCTA174 (A174), using a combination of traditional polyphasic methods and modern genomic-based approaches. The study included 16S rRNA-based phylogeny, ITS secondary structure prediction, morphological and habitat analyses, as well as high-quality genome sequencing with corresponding phylogenomic analyses. The results of the 16S rRNA, 16S-23S ITS secondary structure, morphology, and habitat analyses supported the classification of the strain as a member of a novel genus within the family Oculatellaceae, closely related to Albertania and Trichotorquatus. Genomic analysis revealed the presence of a sophisticated carbon-concentrating mechanism (CCM) in the strain, involving two CO2 uptake systems NDH-I3, and NDH-I4, three types of bicarbonate transporters (BCT1, bicA, sbtA,) and two distinct putative carboxysomal carbonic anhydrases (ccaA1 and ccaA2). The expression of CCM genes was investigated with a CO2 shift experiment, indicating varying transcript abundance among different carbon uptake systems. Based on the comprehensive characterization, the strain was delineated as Thermocoleostomius sinensis, based on the botanical code. The study of the complete genome of strain A174 contributes valuable insights into the genetic characteristics of the genus Thermocoleostomius and related organisms and provides a systematic understanding of thermophilic cyanobacteria. The findings presented here offer valuable data that can be utilized for future research in taxogenomics, ecogenomics, and geogenomics.
Introduction
Thermophilic cyanobacteria are essential primary producers of geothermal ecosystems that profoundly impact the ecology and biological productivity of these sites (Esteves-Ferreira et al., 2018). In recent years, there has been increased interest in these extremophilic organisms with a focus on their ecology, taxonomy, genomics, and biotechnology as donors of useful enzymes and metabolites (Liang et al., 2018; Patel et al., 2019) and microbial cell factories for carbon photo valorization (Liang et al., 2019). Despite an observable increase in interest in this group of photosynthetic extremophiles, many organisms remain relatively poorly described compared to their mesophilic counterparts. In recent years, low costs and increased availability of next-generation sequencing has led to increases in metagenomic studies of hot spring microbial communities (Alcorta et al., 2020). Numerous Metagenome-Assembled Genomes (MAGs) of thermophilic cyanobacteria have been recovered from these sequencing projects highlighting genomic features of new thermophilic microorganisms and potential mechanisms of adaptations to the environment (Chen et al., 2020; Kees et al., 2022). Until recently, it was difficult to conduct fundamental and applied studies on thermophilic strains due to the lack of well-described thermophilic isolates that contain complete genomic, taxonomic, physiological, and morphological data. However, as more studies characterizing thermophilic strains have been published, the new genera have been proposed (Perona et al., 2022). Simultaneously, thermophilic cyanobacteria share many of the same challenges regarding their taxonomy to their mesophilic counterparts, most notably simple morphology (Komárek and Anagnostidis, 2005). Those features, often difficult to identify and plastic, like colony and filament morphology, types and sizes of cells, and characteristic features of sheaths, aerotopes, or branches make their assignment particularly challenging. The absence of sufficient genetic, physiological, or morphological data exacerbates this problem. To effectively identify cyanobacterial isolates and resolve their precise taxonomic status, a polyphasic approach combining multiple datasets is necessary (Raabova et al., 2019; Shalygin et al., 2020).
In the past few years, there has been a significant increase in the discovery of new species of cyanobacteria (Casamatta, 2023; Strunecký et al., 2023). This can be also observed among thermophilic strains where simple unicellular organisms such as Thermosynechococcus, Synechococcus, and Thermostichus (Komárek et al., 2020; Tang et al., 2022c) and filamentous strains exhibiting Leptolyngbya-like morphology underwent a series of revisions in recent years (Sciuto and Moro, 2016; Mai et al., 2018). Meanwhile, extensive revisions have also been made in the family Oculatellaceae, and numerous clades and new genera have been delineated from the strains previously assigned to Leptolyngbya, e.g., Drouetiella (Mai et al., 2018), Thermoleptolyngbya (Sciuto and Moro, 2016), Timaviella (Mai et al., 2018), and Trichotorquatus (Pietrasiak et al., 2021).
However, despite the wide and rapid deployment of next-generation sequencing technology (NGS) in almost all areas of microbiology, cyanobacterial taxonomy often lacks sufficient genomic components. This can lead to taxonomic revisions of a well-established cyanobacterial taxon based exclusively on single-gene sequencing and microscopic observations (Nowicka-Krawczyk et al., 2019). To enhance the credibility of taxonomic revisions, it is crucial, in our opinion, to incorporate comparative genomic analyses to strain descriptions that include morphological and ecological data. Examples of revisions that have been backed by genomic data include the delineation of Thermoleptolynbya (Sciuto and Moro, 2016) and Trichothermofontia (Tang et al., 2023). While many of the delineations that originally did not include comparative genomics were already confirmed and expanded thanks to those datasets (Strunecký et al., 2023), in many other instances it was not possible due to sequence unavailability. With the renewed interest in cyanobacterial taxonomy in recent years, it is important that those consolidated community efforts based on a polyphasic approach also include genomic and physiological data, to better guide taxonomic revisions in future.
In this study, an isolate PKUAC-SCTA174 (A174 thereafter), isolated from the Erdaoqiao Hot Spring in Ganzi Prefecture, western Sichuan Province of China, previously suggested as a potential new genus (Tang et al., 2018a) is described. The strain is the third potential novel organism from the hot spring after Thermoleptolyngbya sichuanensis (Tang et al., 2021) and Leptodesmis sichuanensis (Tang et al., 2022a) described earlier. A complete dataset containing genomic, morphological, and physiological data of the isolated thermophilic filamentous strain is presented. On the basis of the data and utilizing a polyphasic approach, a new genus is proposed Thermocoleostomius sinensis gen. et sp. nov., based on the botanical code. Finally, the genes responsible for the strain's sophisticated carbon concentration mechanism (CCM) were studied in response to changes in inorganic carbon availability.
Materials and methods
Origins, cultivation, deposition, and basic physiological assessment
The strain A174 was initially isolated from light green biofilm deposited on the surface of calcareous sinter in the Erdaoqiao Hot Spring in the Ganzi Prefecture, western Sichuan Province, China. Original sampling was performed on 12 May 2016 under environmental conditions including an ambient temperature of 15°C, a relative humidity of 71%, and a light intensity of ~1,000 lux. The temperature and pH of the thermal spring were 40.8°C and 6.32, respectively. The total dissolved solids in the spring were 447 mmol L−1. The hot spring sampling site description and preliminary taxonomic identification have been reported earlier earlier (Tang et al., 2018a,b). The uni-cyanobacterial culture was routinely cryopreserved as 10% DMSO in BG11 stocks at −80°C. Unless stated otherwise, the pre-cultures for the experiments were established as 150 ml BG-11 (Stanier et al., 1971) medium in 500 ml Erlenmeyer flasks agitated at 100 rpm in an illuminated shaking incubator (MGC-450BP-2, Yiheng, Shanghai, China) for 14 days. Unless stated otherwise, the light parameters were set at 45 μmol m−2 s−1 provided by cool white light fluorescent tubes, temperature 45°C, and photoperiod 12L: 12D. The strain was deposited in the Freshwater Algae Culture Collection at the Institute of Hydrobiology (FACHB-collection) with an accession number FACHB-3572. Simultaneously, the dried inactive holotype was deposited in the Herbarium of North Minzu University with the voucher number: NMU00174 along with other strains collected in Sichuan Ganzi Prefecture, which belonged to other genera and were described earlier and deposited in the same herbarium with voucher numbers: NMU00183 (A183 strain; Tang et al., 2021), NMU00121 (A121 strain; Tang et al., 2022a), NMU00231 (B231 strain), and NMU00412 (E412 strain; Tang et al., 2022b). All the herbarium specimens were prepared by harvesting and gently drying on the filter paper their biomass before deposition. Strain capacity to utilize various nitrogen and carbon sources was tested as follows: the strain was cultured in a series of concentrations of sodium bicarbonate (0, 0.1, 0.3, 0.5, 0.7, and 1 M) and nitrogen sources, including sodium nitrate (0, 0.075, 0.5, 1.5, 5, and 7.5 g L−1) and sodium nitrite (1.218 g L−1). Batch experiments were performed in 250 ml flasks and 50 ml breathable capped tubes containing 100 and 25 ml culture medium, respectively. The control groups were cultured in a standard BG11 medium. Similar sizes and numbers of cell pellets containing trichoid granules were inoculated in the experimental and control groups. The temperature was maintained at 45°C, and the experiment was performed continuously for 21 days, with adjustment for the evaporative losses.
Genome sequencing, assembly, and annotation of strain A174
Genomic DNA was extracted as previously described (Tang et al., 2021) and tested for integrity using agarose gel electrophoresis. The whole-genome sequencing was conducted using a combination of long-read Oxford Nanopore Technologies (ONT) performed with the PromethION sequencer, and Illumina short-read PE150 approach using the NovaSeq 6000 sequencer. For ONT sequencing, the libraries were generated using SQK-LSK109 Kit according to the manufacturer's guidelines. ONT sequencing yielded 1,387,280,929 bp of an average read length of 10,405 bp. These long-reads were used to assemble the draft circular contig of the genome using Flye v2.7 (Kolmogorov et al., 2019) plugin to the commercially available Geneious Prime 2022.2 package (Kearse et al., 2012). Subsequently, the draft contig was error-corrected with 6,098,395 filtered PE150 reads (clean data) generated with short-read technology, using Geneious mapper on default settings. Finally, the final genome was annotated using a customized pipeline, as described before (Tang et al., 2019). In short, the gene prediction and annotation were automatically performed using the NCBI prokaryotic genome annotation pipeline (O'Leary et al., 2016) and polished utilizing the RAST annotation system (Brettin et al., 2015) to minimize poor calls. The genome annotation of strain A174 was summarized in Supplementary Table 1. The complete genome was deposited in GenBank under accession number CP113797.
Phylogenetic reconstruction of 16S rRNA
The sequences of the 16S rRNA gene were extracted from the complete genome sequence of the A174 strain. The reference sequences with high similarity to the A174 were selected based on the BLAST algorithm from the GenBank database or by identifying with the genera in the family Oculatellaceae. Each sequence was trimmed to a similar length of ~1,100 bp, aligned with ClustalW, refined by adjustment of poorly aligned regions, and used to generate a phylogenetic tree representing the taxonomic assignment of the A174 strain. Meanwhile, the 16S rRNA gene sequence similarities between species were calculated based on pairwise alignment using an online calculator from enveomics collection toolbox (Rodriguez-R and Konstantinidis, 2016). Independent phylogenetic relationships of cyanobacteria species within the order Synechococcales (including family Oculatellaceae and Leptolyngbyaceae) were simultaneously conducted using PhyML V3.0 (Guindon et al., 2010) and Bayesian analysis. The nexus file generated from the alignment was run in MrBayes 3.2.7a (Ronquist et al., 2012), available on CIPRES Science Gateway (Miller et al., 2010). The best-fit substitution evolutionary model was chosen by jModelTest based on the Bayesian Information Criterion. Bayesian analysis was run using the TVM + invariant + gamma (TVM+I+R) substitution model with the parameters: NST = 6, Rates = invgamma, and MCMC Ngen = 10,000,000. An initial 25% of samples were discarded as the burn-in fraction. The potential scale reduction factor (PSRF) for parameter values was 1.00, which realized the purpose of convergent statistics. Maximum likelihood was conducted using PhyML, employing the GTR model, with 1,000 bootstrap replications. Bayesian inference results showed the best result to classify the family and the genus and were used to map correct phylogenetic distances and evaluate the relative support of branches. All phylogenies were visualized using FigTree (Rambaut, 2007) and subsequently annotated in Adobe Illustrator.
Prediction of secondary structures
The 16S−23S intergenic spacer (ITS) region was extracted from the annotated genome of the strain and analyzed for the presence of conserved domains. The conserved D1–D1′ domain and variable domains V2 and boxB of the 16S-23S ITS region were identified as described before (Iteman et al., 2000), and their secondary structures were computed with RNAstructure and visualized with Structure Editor 1.0 (Mathews, 2014). The tRNA sequences were identified using tRNA scan-SE v.1.3.1 (Lowe and Eddy, 1997).
Genome-based analyses
A series of pairwise genomic comparisons were performed to further investigate the taxonomic relationships between A174 and related strains. The nucleotide and amino acid sequences were retrieved from GenBank databases. Pairwise average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated for the A174 genome against both closely related strains and focus taxa. ANI/AAI parameters were calculated using a publicly available algorithm provided through the Environmental Microbial Genomics Laboratory at Georgia Tech (http://enve-omics.ce.gatech.edu/index) and visualized as a matrix table. Further genome similarity was compared using digital DNA–DNA hybridization (dDDH) between pairs of genomes. The estimated DDH distances were calculated using an online tool (Meier-Kolthoff et al., 2022) based on a generalized linear model (GLM) by submitting and comparing two genomes using the BLAT program to obtain HSPs/MUMs (high-scoring segment pair/ maximal matches that are unique in both sequences) (Chatterjee et al., 2015) and infer distances using formula most suitable for genomes.
Phylogenomic reconstruction
Concatenated protein sequences generated with the shared single-copy genes among all focus taxa were used to ascertain the phylogenomic position of strain A174 essentially as described before (Tang et al., 2022a). In short, the homologous gene clusters were identified with OrthoMCL (Li et al., 2003) and concatenated. The resultant multisequence alignment was generated using MAFFT v7453 (Katoh and Standley, 2013) and analyzed for phylogenomic inference using IQ-TREE v2.1.3 (Minh et al., 2020). Subsequently, 546 protein models were used to create an optimal substitution model using a ModelFinder module of IQ-TREE. The final assessment of the generated tree topology was performed with UltraFast Bootstrap (Thi Hoang et al., 2017), employing 1000 bootstrap tests (replicates).
Microscopic analysis
The light micrographs of the A174 strain were obtained from ~5 μl of a healthy 2-week-old culture grown at standard conditions at 400 × magnification using light microscopy (LM, DP72, OLYMPUS, Japan) equipped with an image acquisition system (U-TV0.63XC, OLYMPUS, Japan). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) micrographs were generated using the protocols described earlier (Tang et al., 2021). In short, the cells for SEM were washed in phosphate-buffered saline (PBS), fixed for 2 h in fixation solution (Servicebio, G1102), post-fixed with 1% OsO4, and dehydrated with ethanol and isoamyl acetate before taking micrographs with Hitachi, Tokyo, Japan, SU8100. Cells for TEM were additionally embedded in agarose and cut to 60–80 nm thin layers with ultra-microtome Leica EM UC7 (Leica, Wetzlar, Germany), stained with 2% uranium acetate saturated alcohol solution and lead citrate for 8 min, and examined using TEM (Hitachi, HT7800).
Investigation of the carbon concentration mechanism in strain A174
The carbon concentration mechanism (CCM) components were identified as previously described (Tang et al., 2022c). In short, the 28 well-described protein sequences involved in the CCM of a model strain Synechocystis sp. PCC 6803 were retrieved as a reference. The orthologous gene set of the A174 strain was obtained using bidirectional best hit methodology employing E-value cutoff of 1E-6, ≥30% identity, and 70% coverage with BLASTP and subsequently manually curated for accuracy using annotated genomes and summarized in Supplementary Table 2. Nucleotide sequences of Rubisco large subunit (rbcL), carboxysome shell proteins ccmK1, ccmK2, and two variants of each of the four genes of the putative high-affinity bicarbonate transporter BCT1, i.e., cmpA, cmpB, cmpC, and cmpD were extracted from the genomic sequence. All nucleotide sequences were translated into protein sequences and subjected to maximum-likelihood (ML) analysis as previously described (Tang et al., 2022c).
Gene transcription analysis during CO2 shift experiment
A CO2 shift experiment was performed to analyze the expression profiles of 10 putative genes responsible for carbon uptake in the A174 strain. As described earlier, the cells of A174 were cultivated in a shaking incubator to induce the formation of biomass pellets of ~1 mm in diameter. The pellets were harvested by filtration, and ~6,000 pellets corresponding to the dry weight of nearly 0.2 g were used for the CO2 shift experiment. The strain pellets have been pre-cultured in BG-11 liquid medium under a continuous supply of ambient air for 24 h to adjust to the cultivation mode. All the cell pellets have been harvested by centrifugation at 1,500 × g, washed, and subsequently resuspended in fresh BG-11 medium and grown for 24 h in HC [4% CO2 in air (v/v)]. At the end of this cultivation period, ~200 pellets have been collected, flash-frozen in liquid nitrogen, and used for RNA extraction (HC). The remaining cells have been shifted to lower, ambient, carbon environment by replacing the CO2-enriched air with ambient air (0.042% CO2, defined as low CO2, LC). The cultivation in LC lasted for 1 h, and an equal amount of biomass was used for RNA extraction (LC1). The remaining cells were grown for another 23 h, and another batch of cells was used for RNA extraction (LC24). The total RNA from each of the three biological replicate samples was extracted using RNAiso Plus reagent (Takara, Dalian, China) and treated with DNAseI (Thermo Fisher Scientific, Waltham, USA) according to the manufacturer's instructions at the following three time point: HC, LC1, and LC24. RNA concentration and integrity have been assessed with Nanophotometer (Implen, Germany) and agarose gel electrophoresis, respectively.
Real-time quantitative polymerase chain reaction (RT-pPCR) was used to assess 10 genes representing predicted distinct components of the A174 CCM system. The following genes have been selected for the analysis: ndhF3, ndhF4, cmpA1, cmpA2, bicA, sbtA, rbcL, ccmK1, ccaA1, and ccaA2. The rpoB was used as an internal control among other genes based on the expression stability vs. target genes and relatively high Ct value. The oligonucleotides used in the qPCR reaction are summarized in Supplementary Table 3. Reverse transcription was performed with PrimeScript RT reagent Kit (Perfect Real-Time kit, Takara, China). The resultant cDNA was a template for the quantitative estimation of gene expression levels. The quantitative PCR reaction was done with TB Green Premix Ex Taq (Tli RnaseH Plus) (TaKaRa, Dalian, China) in an Applied Biosystems QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific, Waltham, America) using the following conditions: denaturation at 95°C for 30 s, annealing at 95°C for 5 s, and then 40 cycles of 60°C for 30 s for extension, followed by a melt cycle of 95°C for 15 s, 65°C for 60 s, and 95°C for 15 s. Data were analyzed by the 2−ΔΔCT method, which directly uses the threshold cycle (CT) value to calculate the relative quantification of gene expression.
Taxonomic evaluation
The taxonomic description followed the recommendations of the Botanical Code, International Code of Nomenclature for Algae, Fungi, and Plants (Shenzhen code) (Turland et al., 2018) and was based on the classification system developed by Komárek et al. (2014).
Results and discussion
General genomic characteristics of strain A174
The complete genome of the A174 strain was obtained using a combination of Oxford Nanopore and Illumina sequencing platforms, resulting in a genome coverage of 239 × and 157 × , respectively. No plasmids were identified in the sequencing data by the analysis of closed circular sequences assembled from long-read sequencing. The strain's final genome comprised a single circular chromosome of 5,809,202 bp and exhibited a GC content of 48.6%. The overall analysis of the A174 genome revealed the existence of three identical ribosomal RNA operons, 48 tRNA genes, 1 tmRNA, 4,864 protein-coding sequences, and 4 repeat regions. In total 2,068 protein-coding sequences have been predicted to be hypothetical proteins, consistent with previous findings on thermophilic cyanobacteria (Cheng et al., 2020; Kono et al., 2022).
Phylogeny reconstruction using 16S rRNA gene
The genome of the A174 strain has three identical copies of the ribosomal RNA operon. The sequence of 16S rRNA gene was extracted and aligned with other focus taxa. According to the results of the alignment, 91 cyanobacterial strains' sequences were selected to construct 16S rRNA phylogenetic trees (Figure 1, Supplementary Figures 1, 2). The Bayesian tree inferred from nucleotide sequences in NCBI clearly separated new strains from other well-described genera. The tree was categorized into 22 genera, belonging to the order Synechococcales and including families of Oculatellaceae and Leptolyngbyaceae. The branch containing members of the family Leptolyngbyaceae included Stenomitos (Shalygin et al., 2020), Kovacikia (Miscoe et al., 2016), Leptothermofonsia (Tang et al., 2022b), Phormidesmis (Komárek et al., 2009), Leptodesmis (Raabova et al., 2019), Alkalinema (Vaz et al., 2015), Myxacorys (Pietrasiak et al., 2019; Soares et al., 2019), and Pantanalinema (Vaz et al., 2015). Family Oculatellaceae included Drouetiella (Mai et al., 2018), Pegethrix (Mai et al., 2018), Cartusia (Mai et al., 2018), Siamcapillus (Tawong et al., 2022), Elainella (Jahodárová et al., 2018), Albertania (Zammit, 2018), Trichotorquatus (Pietrasiak et al., 2021), Timaviella (Sciuto et al., 2017), Kaiparowitsia (Mai et al., 2018), Tildeniella (Mai et al., 2018; Strunecky et al., 2019), Shackletoniella (Strunecky et al., 2019), Thermoleptolyngbya (Sciuto and Moro, 2016), and Oculatella (Zammit et al., 2012). In addition, Gloeobacter genus which has a distinct, independent basal position, was used as an out-group to root the resultant phylogenetic tree accurately.
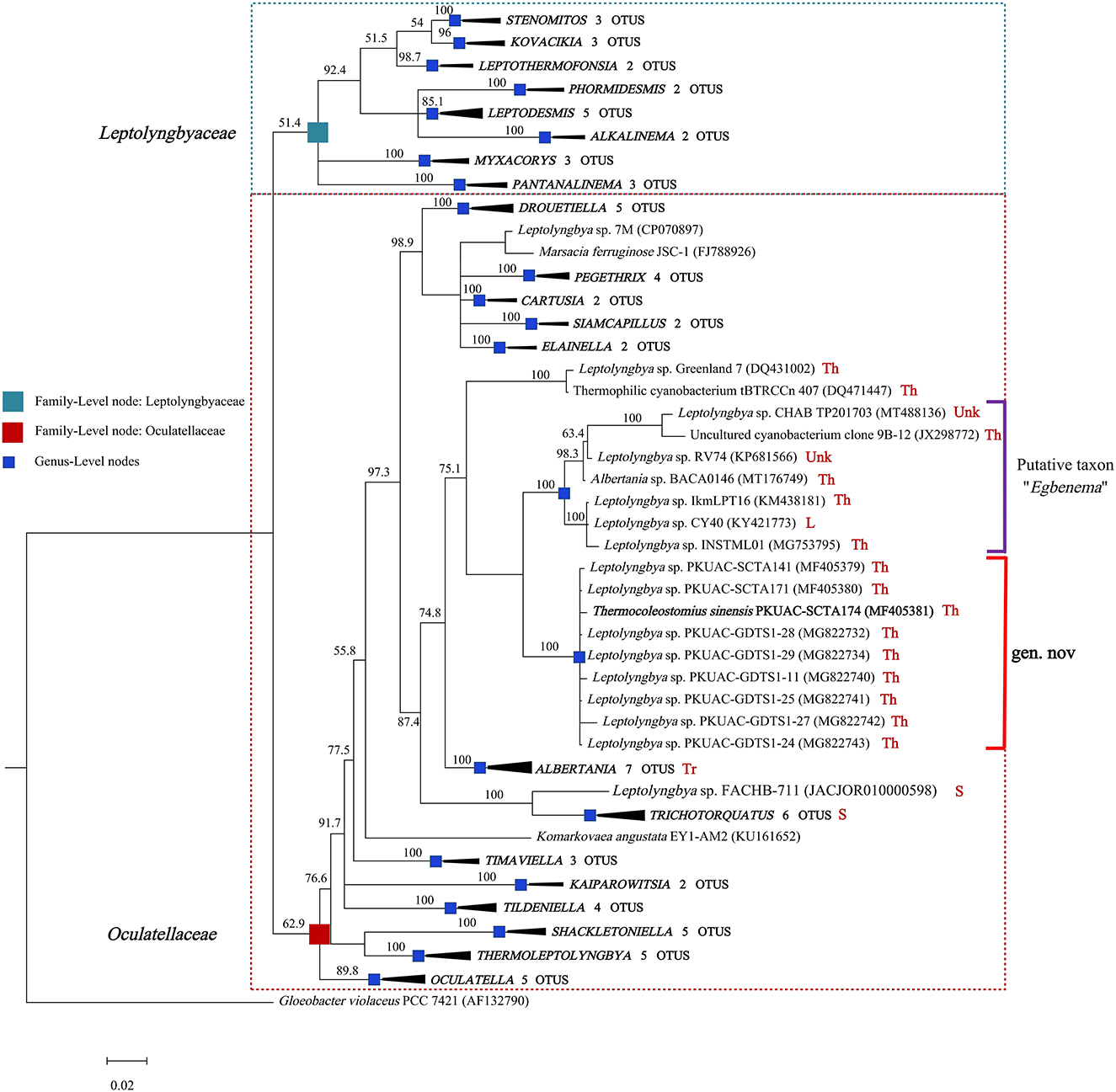
Figure 1. Bayesian inference of 16S rRNA gene sequences representing 91 cyanobacterial strains. Collapsed genera are indicated by black polygons, with a length corresponding to the distance from the most basal sequence to the most diverged sequence of the genus. The complete phylogram refers to Supplementary Figure 1. Posterior probabilities (%) are given above the nodes. The origin (red) of the sequences in the focus clade is provided using the following key: Th, thermal spring; L, lake; S, soil; Tr, terrestrial; Unk, unknown.
The position of A174, another two sequences retrieved from the isolates of this spring A141 and A171 and clones isolated from the thermal spring of Huizhou region in China (GDTS1-11, GDTS1-24, GDTS1-25, GDTS1-27, GDTS1-28, GDTS1-29) (Zhang et al., 2019), formed a well-defined clade with high posterior probabilities and 98.96% 16S rRNA sequence identity (Supplementary Table 4) that was clearly separated from other related cyanobacteria. All strains in this clade shared 16S rRNA molecular signature, morphological, and ecological traits. Phylogenetic analysis indicated that the novel clade containing A174-like species is closely related to Albertania (93.86%−94.90% 16S rRNA sequence identity) and Trichotorquatus (91.59%−92.27% identity) genera already described (Zammit, 2018; Pietrasiak et al., 2021). The closest related clades were occupied by the strains exhibiting Leptolyngbya-like (sensu lato) morphology such as Leptolyngbya sp. IkmLPT16, Leptolyngbya sp. CY40, Leptolyngbya sp. INSTML01, Leptolyngbya sp. RV74, and Leptolyngbya sp. CHAB TP201703. The 16S rRNA sequence identity between A174 and the neighboring strains are as follows: Leptolyngbya sp. IkmLPT16 (95.36%), Leptolyngbya sp. CY40 (95.36%), Leptolyngbya sp. INSTML01 (94.9%), Leptolyngbya sp. RV74 (95.48%), Albertania sp. BACA0146 (95.36%), Leptolyngbya sp. CHAB TP201703 (95.36%), and Uncultured cyanobacterium clone 9B-12 (95.24%). All these strains are likely to belong to a recently proposed novel taxon “Egbenema” (Akagha, 2022). Meanwhile, there are insufficient data to classify the two more distant strains, a benthic isolate from Artic hot springs of Kap Tobin (Greenland_7) (Roeselers et al., 2007) (94.22% sequence identity) and an unidentified filamentous strain from thermal springs in Jordan (tBTRCCn407) (Bruno et al., 2009) (94.11% sequence identity). This level of sequence identity between the representatives of the two clades is at the border of differentiation of a novel genus (Rodriguez-R et al., 2018) and should be supported with additional analyses. To further classify these strains to their respective taxonomic groups, additional analyses were performed using a polyphasic approach.
Genome-based analyses
Since 16S rRNA-based taxonomy, based on a single evolutionary marker, has some limitations (Johnson et al., 2019), it is important to supplement it with other methods, especially when borderline values of genus or species delineation are observed. To make the most of the complete genome sequence of the strain and to ascertain the taxonomic position of the clade containing A174, genome-based analyses were employed. In total, three genome-based coefficients were applied to aid the demarcation of the strain: average nucleotide identity (ANI), average amino acid identity (AAI), and digital DNA–DNA hybridization (dDDH). These parameters are presented in Table 1 and Supplementary Table 6. Methods based on ANI and AAI have proven to be useful for identifying and classifying different genera and are widely employed in taxogenomics. The ANI values (< 83%) (Walter et al., 2017) and AAI values (<65%–72%) (Konstantinidis and Tiedje, 2007) are considered accepted values for genus-level discrimination. Meanwhile, two genomes are considered to belong to the same species if both ANI and AAI values between them are equal to or greater than 95% (Jain and Rodriguez, 2018).
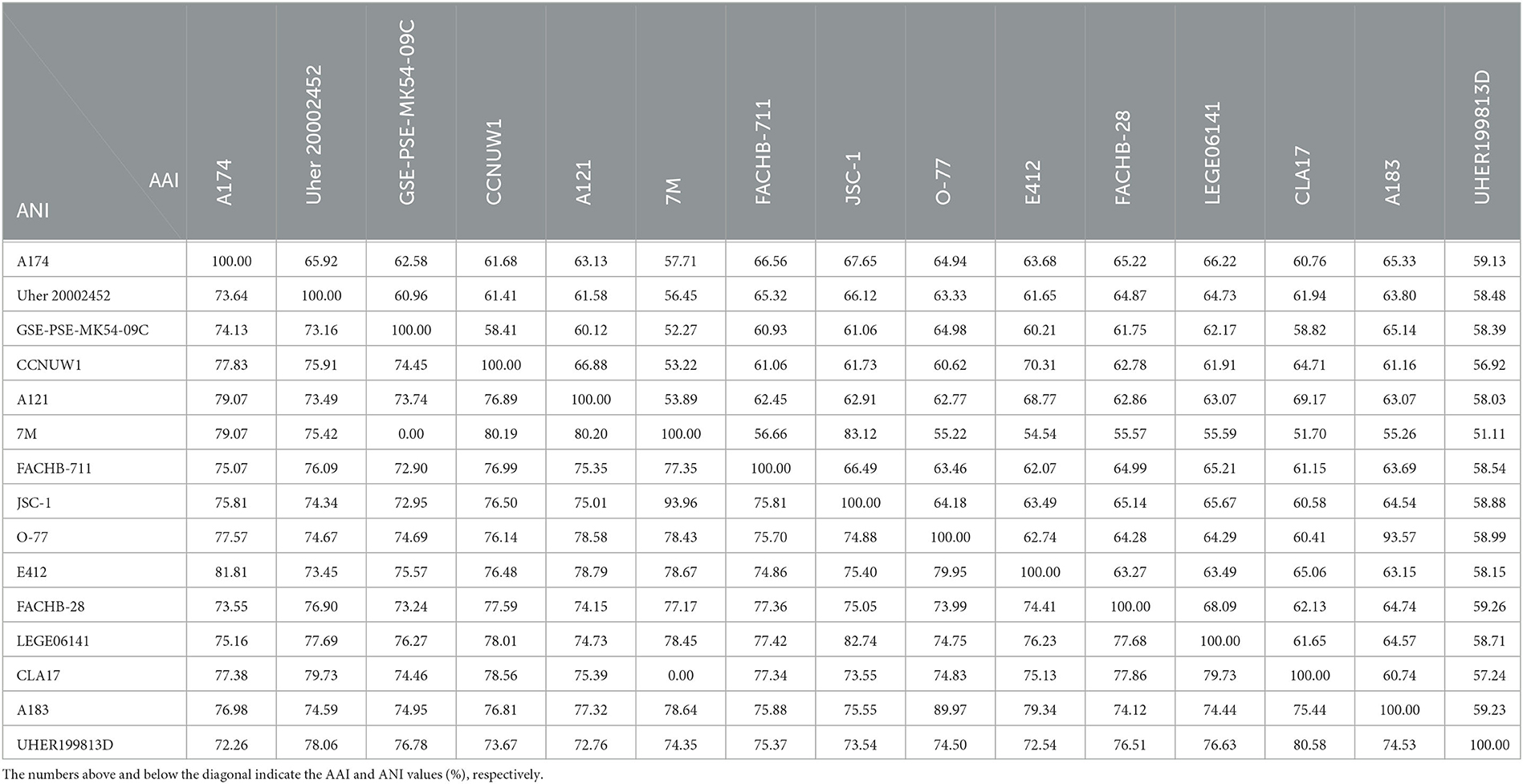
Table 1. Values of average nucleotide identity (ANI) and average amino acid identity (AAI) between genomes studied.
The two coefficients can be used to verify the taxonomic position of the strains on the phylogenetic trees. The strain A174 has the closest ANI of 81.81%, to the recently sequenced L. sichuanensis E412. These parameters are below the level of both species and genus demarcation. The 16S rRNA taxonomy-based analysis revealed that among the strains with whole genomes sequenced, the strain most closely related to A174 was Leptolyngbya sp. FACHB-711. The ANI between the two strains was 75.07%, markedly lower than that of E412. Currently, there is a lack of conclusive evidence to determine the exact cause of this phenomenon. There are two non-mutually exclusive hypotheses. One possibility is the convergent evolution of A174 and E412. The two strains could independently evolve similar traits as a result of adapting to similar environments. This hypothesis is supported by the fact that the two strains were found in hot springs of similar temperatures in Kangding County of Ganzi Prefecture, and they both exhibit tolerance to elevated bicarbonate concentrations and display similar nitrogen metabolism. Another possible explanation is horizontal gene transfer. Almost a quarter of the protein-coding genes from the E412 strain was found to have been potentially acquired through this mechanism (Tang et al., 2022b). This finding suggests that the ANI of E412 may have been influenced by the transfer of genetic material from another organism resulting in an aberration of the ANI relationship resulting from the evolution. It should be noted that these explanations are not mutually exclusive, and further research is needed to fully comprehend the complex relationship between the ANIs of these two strains. Meanwhile, the AAI coefficient between the A174 and FACHB-711 was 66.56%. This parameter is at the lower boundary for genus demarcation, indicating slightly closer proximity than that of the L. sichuanensis E412 (63.68%).
This combined with the results of 16S rRNA phylogeny suggests that the FACHB-711 strain should be reclassified to the genus Trichotorquatus, closely related to the clade containing the A174 strain. ANI and AAI values between A174 and other reference genomes were in the 72–82 and 57–68% range, respectively. Those significant genome-level differences support the delineation of the clade at the genus level. The dDDH parameters further support these findings. The calculated dDDH between the A174 and other focus taxa ranged between 16.80 and 31.70%, significantly below the 70% hybridization threshold for prokaryotic species demarcation.
To further analyze the taxogenomic relationships between the strains, a phylogenomic reconstruction based on single-copy genes shared among the focus taxa with complete genome sequences was performed. A total of 642 single-copy genes were identified to be shared by all the surveyed genomes, generating concatenated alignment with a length of 199,585 aligned amino acid sites. The ML phylogram of the supergene alignment was inferred using the optimal substitution model (LG+F+R5). Each branch of the genomic phylogram is supported by strong bootstrap values (Figure 2), defining tremendous divergence among representative species from each genus. The topology of the resultant tree largely reflects that of the 16S rRNA tree (Figure 1 and Supplementary Figures 1, 2). The unique position of strain A174 confirms its classification to a novel genus. Strain FACHB-711, the closest to the A174 strain with a known genome sequence, is located in a sister clade to strain A174. The genetic distance between those two strains supports their classification to different genera, in line with the 16S rRNA phylogram and the results of genome comparisons, i.e., ANI, AAI, and dDDH. Taken together, Leptolyngbya sp. FACHB-711 could be reclassified to the genus Trichotorquatus, closely related to the novel genus containing the A174 strain if further genomic studies of well-described Trichotorquatus support such findings.
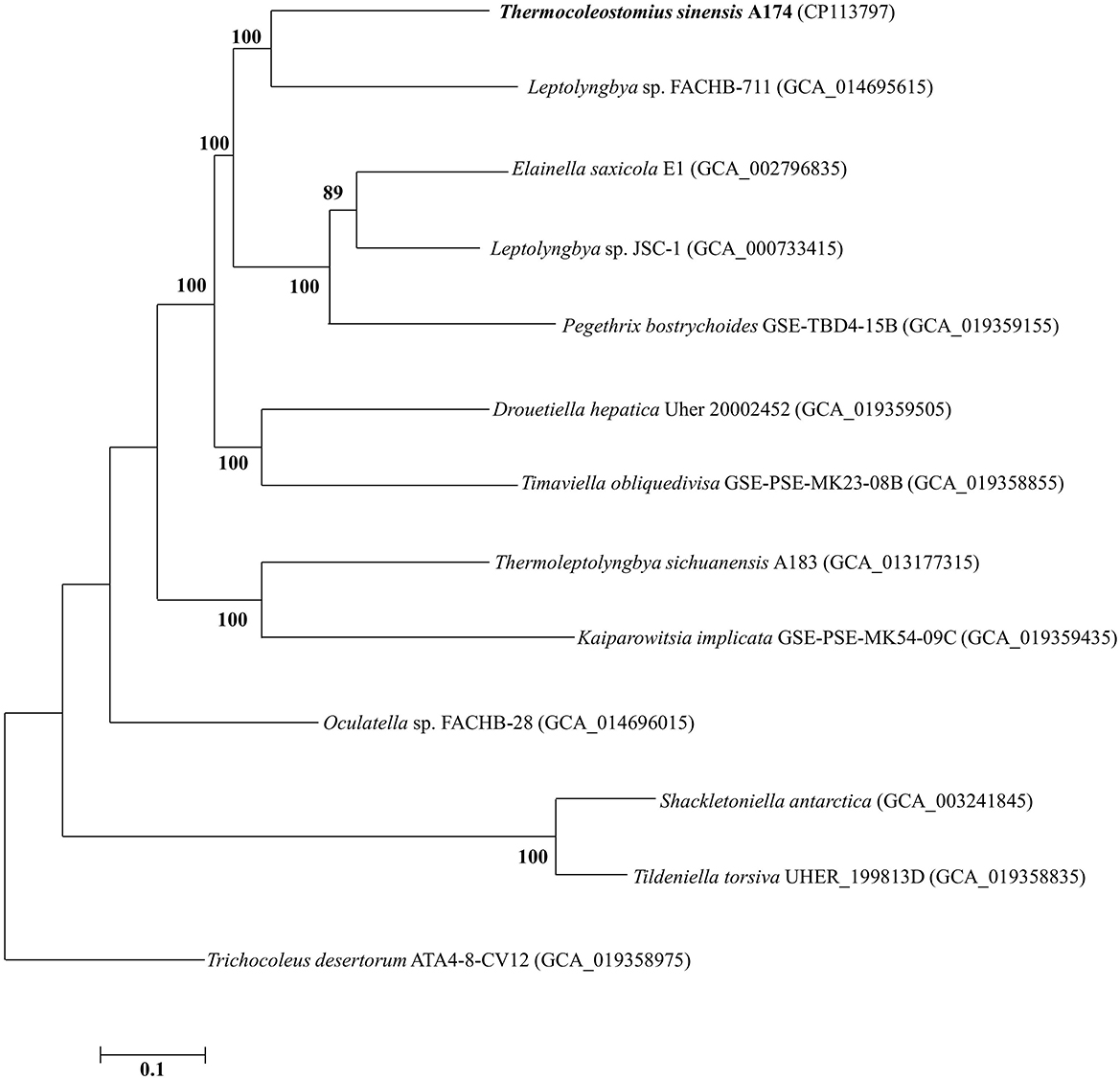
Figure 2. ML phylogenomic tree of a concatenated alignment of 642 single-copy genes shared by all genomes. Strain no. in bold represents the strain identified in this study. Bootstrap values (1,000 replications) are indicated at nodes. Scale bar = 5% substitutions per site.
Analyzing all genome-based parameters and phylogenomic reconstruction based on single-copy genes, it can be concluded that the clade containing the A174 strain can be delineated from other taxa as a separate genus. Unfortunately, the scarcity of genomic sequences in the close proximity of the novel clade made its further description using genome-based parameters impossible and more traditional polyphasic methods encompassing detailed analysis of variable regions of ITS, in addition to morphological and habitat characterization need to be employed.
16S−23S ITS region and secondary structures
The ITS regions of all three ribosomal operons of strain A174 were extracted from the genome sequence and analyzed along with eight closely related strains regarding sequences and predicted structures of their ITS. Sequence alignment has shown that the three ITS regions of ribosomal operons in A174 were identical. Consequently, only one sequence was analyzed along with the related strains. The highly conserved tRNA regions were removed, and the resulting ITS sequences were processed. When selecting the corresponding operons in related organisms, the specificity of Trichotorquatus genus and its operon variability were also considered. Two operons (operons 2 and 3, sensu Pietrasiak et al., 2021) of 16S-23S ITS regions were included and compared with the target strains. The length of the resultant sequences varied from 145 to 353 bp (Supplementary Table 5). The variation could be ascribed to the significant difference in variable V2 and V3 helix structure regions. Strains in Trichotorquatus genus universally have V3 helixes of ~110 nucleotides. Meanwhile, in other strains, those sequences were either very short (20 nt) or absent.
The focus of the secondary structure analyses was placed on D1–D1′ and boxB regions (Supplementary Figures 3A, B). The evaluation of the predicted structures of the D1–D1′ helix revealed two structural variants that significantly differ in length. The shorter one, encompassing ~56–65 nucleotides sharing a similar overall fold, and a longer one of 80 nucleotides are characteristic for ITS operon 2 of the three main representatives of subclades of Trichotorquatus: Trichotorquatus andrei CMT-3FSIN-NPC37, Trichotorquatus ladouxae WJT66-NPBG9, and Trichotorquatus maritimus SMER-A. Additionally, some Trichotorquatus species had shorter ITS sequences present in their ITS operon 3. Trichotorquatus maritimus had a two nucleotide unilateral bulge when compared to other strains. Meanwhile, Trichotorquatus species 5 WJT32-NPBGA lacked bilateral bulge and had three unilateral bulges, which distinguishes it from other structures. It is the most similar in structure to FACHB-711, but the terminal loop of 711 had 4 nucleotides, whereas WJT32-NPBGA had seven nucleotides. One strain, Albertania sp. MAR67, had a unique D1–D1′ helix of 111 nucleotides. In addition, except for the similar basal clamp of five base pairs of the D1–D1′ structure, the relatively short LPT16, FACHB-711, and WJT32-NPBGA helices do not have distinct asymmetric loops of multiple nucleotides characteristic for A174 or SA373. The terminal loops of A174 and SA373 were composed of five nucleotides, while that of IkmLPT16 had 12 nucleotides. On the basis of the current, limited description of “Egbenema,” “Egbenema gypsiphila” species possessed an enlarged terminal loop of 16 nucleotides, which was absent in other strains (Akagha, 2022). The length of the boxB region varied from 35 to 65 bp. Analysis of the boxB region's calculated structures shows similarity of A174 and LPT16 structures. The difference was that the end of the A174 hairpin was a 5-base asymmetric loop, while LPT16 was a 6-base symmetric loop. Meanwhile, T. maritimus, T. andrei in operon 2, and Albertania sp. MAR67 have shorter helices than helices in other strains, but the first two are more symmetrical. Albertania sp. BACA0713 has one more unilateral bulge in helices of similar length. Moreover, comparing the target strain with the putative Egebenema taxon, A174 has three asymmetrical additional bilateral bulge structures in BoxB helix, which all putative “Egbenema” strains do not have. The BoxB helix in T. maritimus in operon 3 has a unique and large unilateral bulge, which is obviously longer than in other strains. In addition, FACHB-711 is identical in structure and sequence to Trichotorquatus species 5 WJT32-NPBGA in BoxB helix, reinforcing that FACHB-711 most likely belongs to the Trichotorquatus genus.
The combined analysis of the predicted secondary structures of the D1–D1′ (Supplementary Figure 3A) and boxB (Supplementary Figure 3B) regions in the context of 16S rRNA phylogeny (Figure 1, Supplementary Figures 1, 2) and genomic analyses (Figure 2) suggests that the strain FACHB-711 should be reclassified as Trichotorquatus. The genus itself is composed of at least five distinctive species including Trichotorquatus species 5 (WJT32-NPBGA), T. maritimus, Trichotorquatus coquimbo, T. andrei, and T. ladouxae. All these strains were isolated from dryland soils in North and South America indicating that molecular evidence is consistent with their habitat. In addition, there is considerable diversity among the strains containing Albertania, and a currently limited amount of molecular data suggests several divergent species within this genus. Finally, the A174 strain belongs to a novel genus different from the putative taxon “Egbenema,” Trichotorquatus, and Albertania.
Morphological and physiological characteristics of strain A174
The strain A174, when grown in the shaking flask cultures, formed very dense, isolated, small trichoid pellets of ~1 mm in diameter containing many entangled filaments. Cells aggregated when reaching a stationary phase in a liquid medium, forming bridges between granules and resulting in a dense mat. Meanwhile, the strain formed dispersed mats or thin laminae on a solid medium. The isolated cyanobacterium was investigated by light microscopy, SEM, and TEM. The image taken by light microscopy (Figures 3A, D) reveals flexuous or bent, sometimes straight trichomes. The strain was phenotypically simple and was composed of elongated cylindrical-shaped cells (Figures 3A–F) surrounded by colorless sheaths that were frequently open at the end, with the terminal cells rounded. False branching was absent. Filaments were solitary with a single trichome per sheath, blue-green in color, accompanied by some minor changes depending on the growth condition or the number of cells. TEM (Figures 3C, F) micrographs indicated that trichomes were cylindrical, composed of isodiametric cells, and the cross-walls of cells had small constrictions. The sheath was thin and colorless, 0.20–0.60 μm thick, usually longer than the trichome, and could be easily observed. Cells are circular in cross section and rectangular in the longitudinal section. Cells of the strain were isodiametric, longer than they were wide (1.5–2.5 μm wide, 2.0–5.8 μm long), with a length-to-width ratio ranging from 1.0 to 2.5 under light microscopy. The number of thylakoids varied from 4 to 6 layers. Thylakoids were arrayed in parallel lines in order at the inner edges of cells (Figures 3C, F). Sheath, septum, phycobilisome, carboxysome, and polyphosphate bodies can also be identified in the cytoplasm (Figures 3C, E, F). The morphological characteristics of other isolates from the same spring, the A141 and A171, were near-identical to the strain A174 (Supplementary Figure 13) and isolates of Huizhou region, i.e., GDTS1–24 and GDTS1–29 (Zhang et al., 2019) confirming the similar morphology of the entire clade.
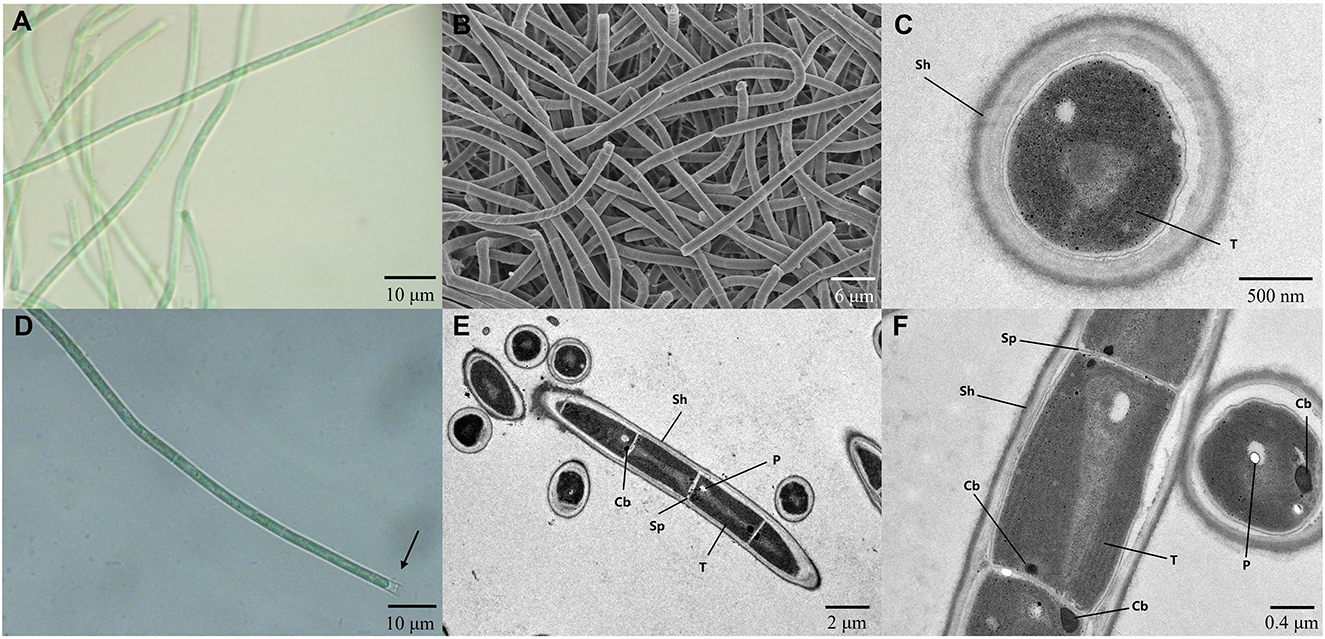
Figure 3. (A, D) Light micrographs of Thermocoleostomius sinensis sp. A174. Sheath are open at the end (arrow). Scales = 10 μm. (B) SEM image of the A174 strain. Scales = 6 μm. (C, E, F) TEM micrographs of A174. Short filament showing the round apical cell. A174 cells with carboxysome (Cb), polyphosphate body (P), sheath (Sh), septum (Sp), and thylakoid (T) in the cytoplasm. Magnifications: 400× (A, D), 2,000× (B), 8,000× (C), 1,200× (E), and 5,000× (F).
The overall morphology of the described clade was similar to the previously described cyanobacterium Albertania skiophila (Zammit, 2018), with some notable exceptions. Light microscope images revealed that A. skiophila frequently exhibited a spiral shape, a feature, which was not observed in A174 and closely related strains. Moreover, A. skiophila grown at its physiological conditions exhibited cylindrical trichomes widened at their apices; meanwhile, the A174 was typically thinner than wide. Other strains belonging to Albertania, e.g., freshwater A. alaskaenisis, were characterized by apparent false branching and sometimes formed characteristic mushroom-like calyptra. These features were not observed in A174. Comparison of strain A174 to Trichotorquatus is problematic due to the absence of good-quality electron micrographs, but several distinguishing features can be inferred from light microscopy. Trichotorquatus strains described vary in their sheath description, ranging from thick to absent (Table 2). This may be attributed to the different life cycle stages and environmental conditions that these strains exhibited during taking the micrographs. Meanwhile, A174 had a defined colorless sheath under the conditions tested. Cell size, shape, and spacing were also distinguishing factors between these two genera. Moreover, the individual cells of Trichotorquatus were easily distinguished on light micrographs, while the cells were not well distinguishable in the A174. The cells of the representatives of a putative taxon “Egbenema” were often shorter than the wide based on the available light micrographs (Akagha, 2022). The average length and width were 1.9 μm and 2.2 μm in “Egbenema aeruginosa,” which is significantly shorter than the long, cylindrical cells of A174. All “Egbenema” strains had visible false branches, and the width and length of each cell were very comparable. Trichomes of “E. gypsiphila” were mostly constricted at cross-walls.
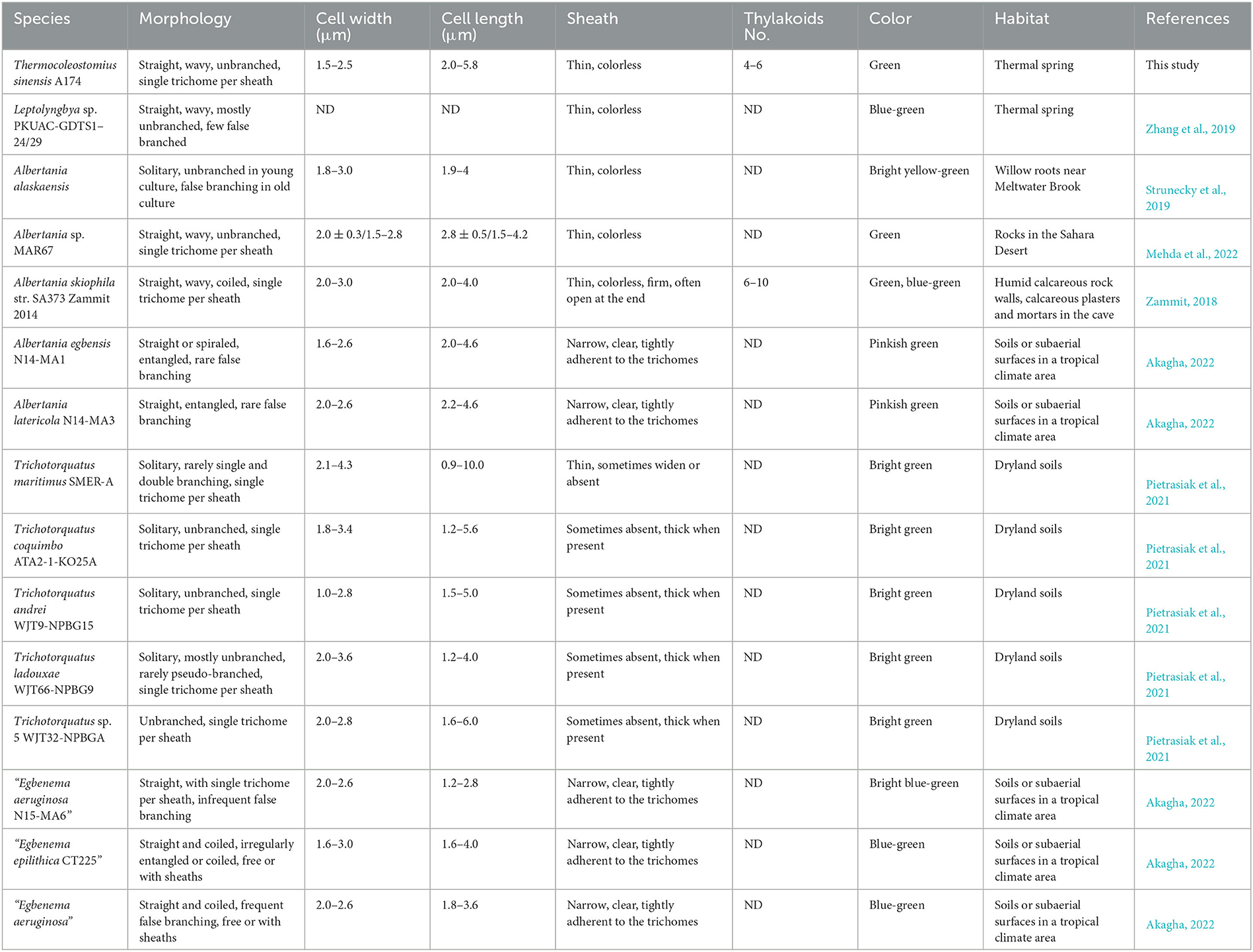
Table 2. Comparison of morphological features and habitats of Thermocoleostomius sinensis and closely related strains.
The A174 strain was subjected to physiological tests using varying concentrations of bicarbonate and nitrate in different modifications of the BG11 medium (Table 3). It exhibited growth in most nitrogen conditions except for nitrite, and optimal growth was observed at 1.5 g/L nitrate concentration. The strain demonstrated a high tolerance for bicarbonate, with a maximum concentration of 0.7 M. When treated for 21 days with 1 M bicarbonate, the cells turned yellow-green and exhibited poor growth. The strain's tolerance to bicarbonate was attributed to its possession of bicarbonate transporters (BCT1, bicA, and sbtA). Furthermore, the strain displayed phototaxis when grown under directional light (Supplementary Figure 11).
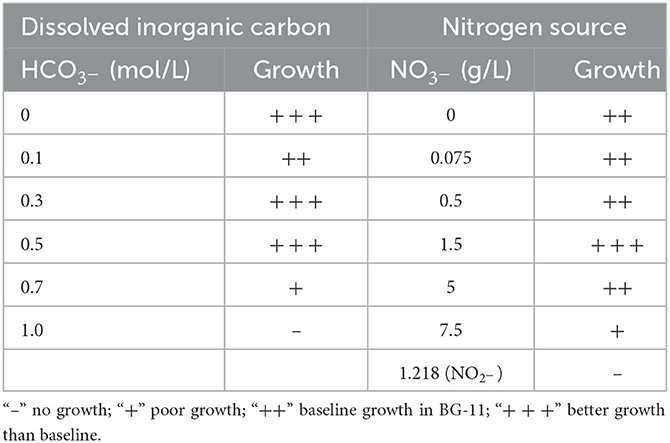
Table 3. Physiological characteristic of Thermocoleostomius sinensis A174 grown in 45°C 150 μmol m−2 s−1.
Taxonomic position of A174 strain using polyphasic approach
In accordance with contemporary cyanobacterial taxonomy standards, Komárek et al. (2014) have defined the concept of a cyanobacterial genus based on three criteria. These criteria include the following: (i) a distinct taxonomic position with a discernible divergence (of 95% or less 16S rRNA gene sequence similarity) from the nearest sister clade, (ii) unique morphological traits or biological specificity (for example, type of cell division, formation of heterocyte or akinete, etc.) that distinguish it from other genera, and (iii) clear ecological niches that are relevant to the genus. The molecular 16S rRNA gene sequence similarities between the clade of interest and related clades of Trichotorquatus and Albertania were lower than 95% similarity. Delineation of A174 as a new genus was additionally supported with genomic similarity coefficients, phylogenomics, and secondary structures of D1–D1′ and boxB regions. Analysis of the habitat and morphological features reinforces the molecular evidence. Strains in Trichotorquatus genus were isolated from dryland soils in the desert or coastal scrub, which exhibit the sheath with a distinctive collar-like fraying and widening mid-filament. Albertania skiophila filaments formed biofilms on the rock surface at the crypt, and MAR67 was collected from gypsum blocks in the hyper-arid Sahara Desert. Meanwhile, Alaskan strains of Albertania isolated from willow roots appear unique morphologically with undulating trichomes with perpendicular cells and false branching in old cultures. Both related genera Trichotorquatus and Albertania have morphological and ecological niche differences that can support the delineation of the thermal clade containing the A174 strain as a new genus.
The exact separation between the clade and adjacent clade, putatively described as “Egbenema,” is more problematic due to the lack of sufficient data on the strains that are not a part of this study. Analysis of the three aspects of genus-level delineation: molecular, morphological or physiological characteristics and habitat data, allows for drawing the following conclusions. Based on the 16S rRNA sequence identity between the Chinese strains (A174, A171, GDTS1–24, and GDTS1–29) and sister clade containing “Egbenema” (Figure 1), which ranges from 94.9 to 95.48%, indicating that based on the 16S rRNA sequence alone, it is uncertain if strains belong to the same genus. Analysis of the secondary structure of ITS helices D1–D1′ and BoxB reveals that the A174 strain has a more diverse asymmetrical additional bilateral bulge structure. The habitat data (Figure 1 and Table 2) show that strains in the focus clade originate from thermal environments supporting the ecological niche requirement for the genus delineation. Meanwhile, the strains proposed as “Egbenema” show diverse habitats. “Egbenema” strains were isolated from soil or subaerial samples in a tropical climate area and can survive in perennially wet habitat. There is also a clear morphological difference between A174 clade and putative “Egbenema” taxon. The members of A174 clade, contrary to strains described as “Egbenema,” had no false branching, and their overall cell shape was different. Cells of the A174 clade are longer than wide, while those of “Egbenema” representatives had similar length and width. The terminal cells of the trichomes have also shown different morphologies. The characteristic pellet-forming feature of A174 was not observed in “Egbenema” according to the available literature.
To summarize, at least at the level of the strains with morphological data available, the focus clade of this study is consistent in morphology, habitat, and molecular data. A monophyletic cluster, comprising three strains (A171, A174, and A141) isolated from two hot springs in Erdaoqiao (Sichuan Province) and two closely related strains isolated from a single hot spring in Guangdong Province (Zhang et al., 2019), has been included in this clade. These strains exhibit similar morphology and 16S rRNA sequence identity of over 98.9% and have been found to originate from hot springs. This allows us to conclude that they belong to the same species. Consequently, the different morphology combined with very distinct environmental niches and some limited molecular evidence suggests that the clade containing A174 and the putative “Egbenema” clade belong to two different genera.
Genomic outlook on the carbon uptake and concentration in A174 strain
Due to the low solubility of gaseous CO2 at elevated temperatures and higher ionic strengths, many thermophilic cyanobacteria deal with higher limitations of inorganic carbon than their mesophilic counterparts (Tang et al., 2022c). In response to this limitation, cyanobacteria frequently form microbial mats in hot springs where the microorganisms are stratified. The top layer is occupied by cyanobacteria that perform photosynthesis and release O2, the subsequent layer contains heterotrophs that utilize oxygen for respiration and release CO2 that is acquired by the top layer cyanobacteria (Ferris et al., 1997). In the absence of cohabitating organisms, cyanobacteria can also utilize carbon concentration mechanisms (CCM) that employ active transport to increase the amount of carbon dioxide in the carboxysome.
Like their mesophilic counterparts, thermophilic cyanobacteria perform photosynthesis in specialized protein-based structures called carboxysomes, where cytoplasm-stored bicarbonate is transported and converted into gaseous CO2 by a carboxysomal carbonic anhydrase to generate high concentration of CO2 around the main carboxylating enzyme, Rubisco (Figure 4). The large subunit of this protein, rbcL, has been used to discriminate between α- and β-cyanobacteria (Badger and Price, 2003). The rbcL gene of the A174 strain was extracted from the genome sequence and analyzed with other focus taxa using an ML phylogram (Supplementary Figure 4). The results have confirmed that the strain possesses Rubisco 1B form and thus belongs to β-cyanobacteria similarly to terrestrial, freshwater and other thermal strains in accordance with previous studies (Tang et al., 2022c). The gene itself is positioned within an operon containing genes sequentially encoding the Rubisco large subunit (rbcL), Rubisco chaperonin (rbcX), and Rubisco small subunit (rbcS) (Figure 5).
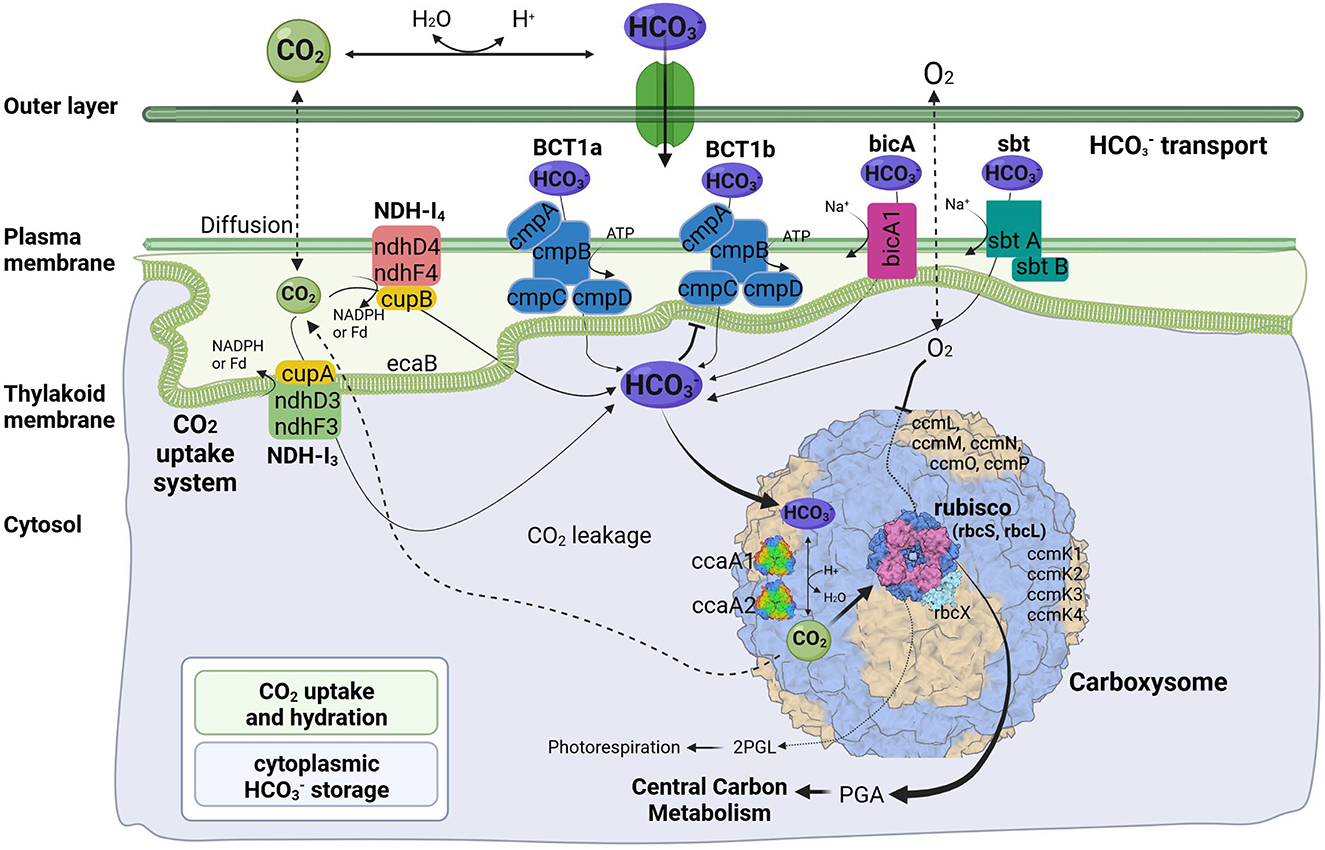
Figure 4. Predicted molecular components of CCM for Thermocoleostomius sinensis A174. 2PGL, 2-phosphoglycolate; PGA, phosphoglyceric acid. Three-dimensional structures of proteins visualized using related structures of 2YBV, 6OWF, 5SWC, 5BS1, 6KI1. Figure created with BioRender.com.

Figure 5. Genomic organization of CCM-related genes for Thermocoleostomius sinensis A174. Solid arrow boxes refer to genes and the direction of transcription.
Cyanobacterial CCMs are primarily composed of two main components: inorganic carbon transport system responsible for carbon uptake and carboxysomes responsible for its fixation. Two complexes NDH-I3 and NDH-I4, responsible for CO2 uptake, are ubiquitous in β-cyanobacteria, but there exists a significant diversity in the bicarbonate uptake systems (Price et al., 2008). Analysis of genomic components of the A174 strain reveals a similar pattern. The strain possesses a full complement of genes encoding NDH-I3 (low-CO2 inducible, high-affinity CO2 uptake system) and NDH-I4 (constitutive, low-affinity CO2 uptake system). The amino acid sequence identity to the orthologous proteins from a mesophilic reference strain Synechocystis PCC 6803 of the constitutive ndhD4, ndhF4, cupB, and inducible ndhD3, and ndhF3 components varies from 55.8% (ndhF4) to 62.1% (cupB). Meanwhile, the carbonic anhydrase cupA associated with low-CO2 inducible ndhD3 and ndhF3 is more conserved and exhibits an identity of 83.4%. These findings are in accordance with our previous findings on thermophilic strains (Tang et al., 2022c).
The genomic makeup of A174 bicarbonate transport system is diverse compared to other thermophilic strains and most similar to Leptolyngbya JSC-1 and Thermoleptolyngbya O-77 (Tang et al., 2022c). The strain shows a repertoire of known bicarbonate transporters: BCT1, bicA, and sbtA. Based on the genomic information alone, there is uncertainty about the number of ATP-dependent inducible high-affinity transporters, BCT1. There are two regions of high homology to the BCT1 transporters of model strains Synechocystis PCC6803 and Synechococcus PCC7942 positioned in forms discrete operons at 2.77 and 5.37 Mbp (Figure 5). Both comprise four polypeptides cmpA-D, consistent with other thermophilic cyanobacteria (Tang et al., 2022c). The two putative bicarbonate transporters are the members of two distinct clades (Supplementary Figures 6–9), and their transcription profiles are further analyzed in the CO2 shift experiment. In addition to BCT1, the strain possesses two genes of the sbt, the least abundant bicarbonate transporter in thermophilic cyanobacteria, sbtA and sbtB. The two copies are dispersed in distant loci of the genome. Finally, the A174 strain possesses a single bicA transporter (bicA1), similar to most thermophilic cyanobacteria except for Thermoleptolyngbya A183 and Leptothermofonsia E412, which have two gene copies of such transporter, bicA1 and bicA2.
Carboxysomes are evolutionarily conserved protein structures that facilitate efficient carbon fixation in cyanobacteria. Carboxysome structures comprise a proteinaceous shell and two encapsulated enzymes, the main carboxylating protein Rubisco and carboxysomal carbonic anhydrase that transforms the soluble bicarbonate into gaseous CO2 (Figure 4). Among β-cyanobacteria, the typical structure of shell proteins comprises ccmK-P proteins. Typically, ccmKLMNO genes form a discrete operon, and ccmP is found in a separate genomic locus. This arrangement is conserved in A174. The strain contains four ccmK genes, namely ccmK1, K2, K3, and K4. The genes coding for two main structural proteins ccmK1, K2 (Cai et al., 2016), are conserved with other cyanobacteria but do not always cluster with their other thermophilic orthologs on the phylogenetic tree (Supplementary Figure 5). Meanwhile, the sequence of the remaining auxiliary shell proteins ccmK3 and K4 shows the highest similarity to the orthologous thermophilic proteins of Leptolyngbya JSC-1. When it comes to encapsulated enzymes, rbcL is clearly positioned within the thermophilic cluster of Rubisco but exhibits a relatively diverse sequence compared to other thermophilic cyanobacteria (Supplementary Figure 4) and 96.8% sequence identity to the enzyme of Leptothermofonsia E412. Interestingly, there appear to be two copies of carboxysomal carbonic anhydrase ccaA dispersed in two loci of the A174 genome, again highlighting higher diversity of strain's CCM than most other thermophilic cyanobacteria. Alignment of the two sequences reveals that the second copy of the gene is ~30% longer than the first (Supplementary Figure 10). More detailed studies will be required to validate the biological function of both carbonic anhydrases in vivo.
Gene expression profiles of the A174 strain during CO2 shift experiment
The CO2 shift experiment was performed to check the relative transcription levels of the A174 strain in response to carbon limitation. The cells were initially cultured in a high-carbon environment for 24 h (4% CO2 in air) and were subsequently shifted to ambient air. The gene expressions of various bioinformatically identified genes involved in carbon uptake and conversion, including bicA, ccaA1, ccaA2, cmpA1, cmpA2, ndhF3, ndhF4, rbcL, and sbtA, were subsequently analyzed at two different time point: 1 h and 24 h after the shift. The data were normalized first to the relative abundance of the rpoB and subsequently to the transcript abundance under a carbon-replete environment (HC) and are presented in Figure 6.
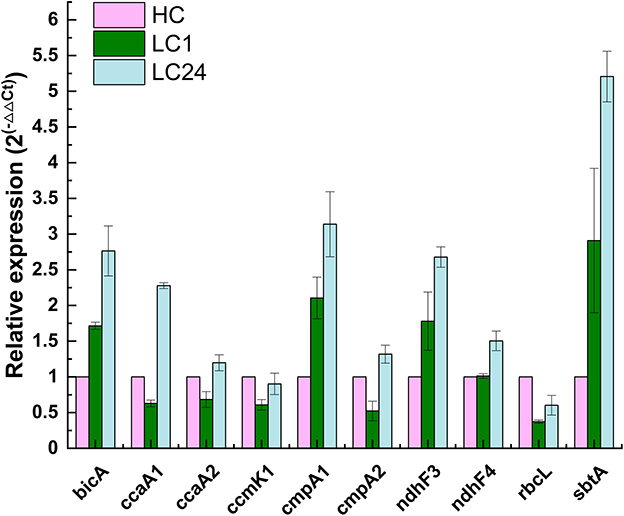
Figure 6. Relative changes in transcript abundance of ndhF3, ndhF4, cmpA1, cmpA2, bicA, sbtA, rbcL, ccmK1, ccaA1, and ccaA2 genes of Thermocoleostomius sinensis A174 cells during CO2 shift experiment assessed with RT-qPCR. HC indicates high carbon for 24 h (4% CO2), LC1 (1 h of carbon limitation), LC24 (24 h of carbon limitation). The data have been normalized to the reference gene rpoB and HC conditions.
The results showed that genes related to both low-affinity and high-affinity uptake systems responded positively to carbon limitation but in different magnitudes. NDH-I3, the high-affinity system for gaseous CO2 uptake, was found to be the dominant system in A174, with 2- to 3-fold increases in expression levels after 1 and 24 h of carbon limitation, respectively. These findings are in line with those for Synechocystis PCC6803 (McGinn et al., 2003), where the ndhF3 gene is undergoing significant upregulation, and ndhF4 is slightly downregulated during the CO2 shift experiment. Similarly, among bicarbonate uptake systems, the transcript levels of high-affinity systems were higher than those of low-affinity ones. The gene expression level of sbtA was found to have increased the most compared to other transcripts assessed. Interestingly, the two putative cmpA genes exhibited different behaviors in response to carbon limitation, highlighting the need for further function loss studies when appropriate molecular tools become available.
Carboxysome proteins such as shell protein ccmK1 and two encapsulated enzymes Rubisco (rbcL) and carbonic anhydrase ccaA2 all show a similar pattern of transcription, with a relative drop in gene expression levels after 1 h of carbon limitation, followed by a recovery to baseline levels after 24 h, likely due to the sufficient supply of carbon by the transport components of the CCM system. Interestingly, the second of the carbonic anhydrases, ccaA1, exhibited markedly higher expression levels, suggesting a possible different cellular localization than suggested by bioinformatic analyses. Further functional studies are required to validate the function of both putative carbonic anhydrases on the carbon fixation of the A174 strain.
Conclusion
In this manuscript, we have characterized, using a polyphasic approach, a novel hot-spring cyanobacterium strain A174 as a representative of a novel taxon of thermophilic cyanobacteria. The subject of this study was capable of growth at 50°C and 0.7M bicarbonate. The combination of phylogenetic, taxogenomic, morphological, habitat, and physiological data suggests that the strain could belong to a novel genus of thermophilic cyanobacteria. Based on the results of 16S rRNA phylogeny, comparative genomics, secondary structures of 16S-23S ITS, and strain's distinct morphology to Albertania, Trichotorquatus, and recently proposed “Egbenema” support the delineation of the clade. Based on those data, we proposed a new genus T. sinensis as a best-described representative of this taxon and proposed its delineation within the family Oculatellaceae. Additionally, the inorganic carbon uptake and concentration genes have been extracted from the assembled high-quality genome sequence and analyzed. The results have shown that the strain firmly belongs to the β-cyanobacteria and has a diverse array of bicarbonate uptake proteins encompassing bicA, sbtA, and at least one variant of BCT1, and two carbonic anhydrases ccaA. The results of gene expression studies under CO2 limitation revealed that all the bicarbonate transporters, bicA, sbtA, and BCT1 are likely to respond to carbon limitation but at different degrees, and the increase of transcript abundance in NDH-I3 is higher than that of NDH-I4, consistently with mesophilic counterparts.
Taxonomic treatment and description of Thermocoleostomius sinensis Daroch, Jiang, Tang et al. gen. et sp. nov.
The taxonomic classification methodology used for the strain proposed in this study follows the polyphasic approach as described by Komárek et al. (2014). The taxon description was consistent with the prescriptions of the International Code of Nomenclature for Algae, Fungi, and Plants (Shenzhen code) (Turland et al., 2018).
Phylum: Cyanobacteria
Order: Synechococcales
Family: Oculatellaceae
Genus: Thermocoleostomius, gen. nov.
Description: Filamentous cyanobacteria, forming small pellets and radiating outward, colonies intertwined, forming green and compact biofilms and new pellets. Filaments flexuous, wavy or bent, occasionally straight trichomes. Filaments with a single trichome per sheath, no false branching. Sheath frequently open at the trichome termini. Cylindrical trichomes, cross-walls of cells with minor constrictions, cells constricted toward the end. Cells variable in shape, mostly isodiametric to longer than wide, with parietal thylakoids. Apical cell rounded, shorter on average than regular vegetative cells, morphologically similar to regular cells.
Etymology: “Thermo” similar to thermophilic (high temperature tolerant), “coleo” genus epithet derived from the Greek word koleos meaning sheath or scabbard, “stomius” genus epithet derived from Greek word meaning mouth, outlet, indicating that the sheath of the strain is frequently open.
Type species: Thermocoleostomius sinensis.
Thermocoleostomius sinensis Daroch, Jiang, Tang et al. gen. et. sp. nov.
Diagnosis: Differing from other species of the genus based on the 16S rRNA sequence identity.
Description: Filamentous cyanobacteria (Figures 3A–F), in laboratory cultures, originating from small pellets and radiating outward in diverse directions (Supplementary Figure 14), tiny colonies intertwined and connected, forming green and compact biofilms, appearance of new pellets on solid agar plates (Supplementary Figure 11). Filaments flexuous, wavy or bent, occasionally straight trichomes (Figures 3A, D). Filaments with a single trichome per sheath, no false branching (Figures 3A–F). Sheath frequently open at the trichome termini (Figure 3D). Cylindrical trichomes, cross-walls of cells with minor constrictions, cells slightly constricted toward the end, bright green in healthy cultures, yellow in senescent cultures or in a high light environment. Cells variable in shape, mostly isodiametric to longer than wide (Figures 3A–F), length-to-width ratio 0.8:2.3. Cells size 1.5–2.5 μm long, 2.0–5.8 μm wide, parietal thylakoids arrayed in parallel lines (Figures 3C, E, F). Apical cell rounded, shorter on average than regular vegetative cells, morphologically similar to regular cells (Figure 3E). Strain capable of growth in standard BG11 medium to a maximum temperature of 50°C and medium with 0.7 M NaHCO3. Successful cryopreservation of the strain in 10% DMSO for over 2 years. Phototaxis observed (Supplementary Figure 11).
Etymology: “sinensis” species epithet derives from the fact that all representatives of the genus have been isolated from various provinces of China.
Type locality: Thermal spring. Erdaoqiao village in Ganzi Prefecture of Sichuan Province, China.
Ecology of type locality: The sample occurred as a light green biofilm deposited on the surface of calcareous sinter (Supplementary Figure 12).
Habitat: Thermal spring in Ganzi Prefecture of Sichuan Province, China (30°05′14″N, 101°56′55″E).
Holotype here designated: The dried inactive holotype was deposited in the Herbarium of North Minzu University with the voucher number: NMU00174 (contact: Lei Zhang, emhhbmdzYW5zaGktMDMxOUAxNjMuY29t).
Reference strain: The culture of Thermocoleostomius sinensis Daroch, Jiang, Tang et al. gen. et sp. nov. was initially denoted and deposited in Peking University Algae Collection as PKUAC-SCTA174, and it also has been deposited in the Freshwater Algae Culture Collection at the Institute of Hydrobiology (FACHB) as Oculatellaceae sp. FACHB-3572 after identification and authentication based on the full-length sequencing of the 16S rRNA gene along with folding of the secondary structures of the 16S−23S ITS region.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
YJ: formal analysis, software, investigation, data curation, visualization, writing—original draft, and writing—reviewing and editing. JT: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, writing—reviewing and editing, visualization, supervision, project administration, and funding acquisition. MD: conceptualization, methodology, resources, data curation, writing—original draft, writing—reviewing and editing, supervision, project administration, and funding acquisition.
Funding
This research was funded by the National Natural Science Foundation of China (31970092, 32071480, and 3221101094) and Tenure-Track Fund to MD. Funding bodies had no influence over the design and execution of this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1176500/full#supplementary-material
References
Akagha, M. (2022). “Albertania” and “Egbenema,” Panding Biodiversity in Oculatellaceae (Cyanobacteria). John Carroll University.
Alcorta, J., Alarcón-Schumacher, T., Salgado, O., and Díez, B. (2020). Taxonomic novelty and distinctive genomic features of hot spring cyanobacteria. Front. Genet. 11, 568223. doi: 10.3389/fgene.2020.568223
Badger, M. R., and Price, G. D. (2003). CO2 concentrating mechanisms in cyanobacteria: molecular components, their diversity and evolution. J. Exp. Bot. 54, 609–622. doi: 10.1093/jxb/erg076
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5, 8365. doi: 10.1038/srep08365
Bruno, L., Dan, I., Bellezza, S., and Albertano, P. (2009). Cytomorphological and genetic characterization of troglobitic Leptolyngbya strains isolated from Roman Hypogea. Appl. Environ. Microbiol. 75, 608–617. doi: 10.1128/AEM.01183-08
Cai, F., Bernstein, S. L., Wilson, S. C., and Kerfeld, C. A. (2016). Production and characterization of synthetic carboxysome shells with incorporated luminal proteins. Plant Physiol. 170, 1868–1877. doi: 10.1104/pp.15.01822
Casamatta, D. (2023). Giving form to the formless: an updated classification of cyanobacterial taxonomy. J. Phycol. 59, 9–11. doi: 10.1111/jpy.13313
Chatterjee, S., Alampalli, S. V., Nageshan, R. K., Chettiar, S. T., Joshi, S., Tatu, U. S., et al. (2015). Draft genome of a commonly misdiagnosed multidrug resistant pathogen Candida auris. BMC Genomics 16, 686. doi: 10.1186/s12864-015-1863-z
Chen, M.-Y. Y., Teng, W.-K. K., Zhao, L., Hu, C.-X. X., Zhou, Y.-K. K., Han, B.-P. P., et al. (2020). Comparative genomics reveals insights into cyanobacterial evolution and habitat adaptation. ISME J. 15, 211–227. doi: 10.1038/s41396-020-00775-z
Cheng, Y. I., Chou, L., Chiu, Y. F., Hsueh, H. T., Kuo, C. H., Chu, H. A., et al. (2020). Comparative genomic analysis of a novel strain of taiwan hot-spring cyanobacterium Thermosynechococcus sp. CL-1. Front. Microbiol. 11, 1–13. doi: 10.3389/fmicb.2020.00082
Esteves-Ferreira, A. A., Inaba, M., Fort, A., Araújo, W. L., and Sulpice, R. (2018). Nitrogen metabolism in cyanobacteria: metabolic and molecular control, growth consequences and biotechnological applications. Crit. Rev. Microbiol. 44, 541–560. doi: 10.1080/1040841X.2018.1446902
Ferris, M. J., Nold, S. C., Revsbech, N. P., and Ward, D. M. (1997). Population structure and physiological changes within a hot spring microbial mat community following disturbance. Appl. Environ. Microbiol. 63, 1367–1374. doi: 10.1128/aem.63.4.1367-1374.1997
Guindon, S., Dufayard, J.-F. F. F., Lefort, V., Anisimova, M., Hordijk, W., Gascuel, O., et al. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Iteman, I., Rippka, R., Marsac, d. e., Herdman, N. T., Tandeau, M., Herdman, D. M. N. M., et al. (2000). Comparison of conserved structural and regulatory domains within divergent 16S rRNA-23S rRNA spacer sequences of cyanobacteria. Microbiology 146, 1275–1286. doi: 10.1099/00221287-146-6-1275
Jahodárová, E., Dvorák, P., Hašler, P., Holušová, K., and Poulíčková, A. (2018). Elainella gen. nov.: a new tropical cyanobacterium characterized using a complex genomic approach. Eur. J. Phycol. 53, 39–51. doi: 10.1080/09670262.2017.1362591
Jain, C., and Rodriguez, R. L. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114. doi: 10.1038/s41467-018-07641-9
Johnson, J. S., Spakowicz, D. J., Hong, B. Y., Petersen, L. M., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10, 5029. doi: 10.1038/s41467-019-13036-1
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kees, E. D., Murugapiran, S. K., Bennett, A. C., and Hamilton, T. L. (2022). Distribution and genomic variation of thermophilic cyanobacteria in diverse microbial mats at the upper temperature limits of photosynthesis. mSystems 7, e0031722. doi: 10.1128/msystems.00317-22
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Komárek, J., and Anagnostidis, K. (2005). Cyanoprokaryota 2. Teil/2ndPart : Oscillatoriales. München: SpringerSpektrum.
Komárek, J., Johansen, J. R., Šmarda, J., and Strunecký, O. (2020). Phylogeny and taxonomy of Synechococcus-like cyanobacteria. Fottea. 20, 171–191. doi: 10.5507/fot.2020.006
Komárek, J., Kaštovský, J., Mares, J., and Johansen, J. (2014). Taxonomic classification of cyanoprokaryotes (cyanobacterial genera) 2014, using a polyphasic approach. Preslia 86, 295–335.
Komárek, J., Kaštovský, J., Ventura, S., Turicchia, S., and Šmarda, J. (2009). The cyanobacterial genus Phormidesmis. Arch. Hydrobiol. Suppl. Algol. Stud. 129, 41–59. doi: 10.1127/1864-1318/2009/0129-0041
Kono, M., Martinez, J., Sato, T., and Haruta, S. (2022). Draft genome sequence of the thermophilic unicellular cyanobacterium Synechococcus sp. strain C9. Microbiol. Resour. Announc. 11, e0029422. doi: 10.1128/mra.00294-22
Konstantinidis, K. T., and Tiedje, J. M. (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr. Opin. Microbiol. 10, 504–509. doi: 10.1016/j.mib.2007.08.006
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Liang, Y., Kaczmarek, M. B., Kasprzak, A. K., Tang, J., Shah, M. M. R., Jin, P., et al. (2018). Thermosynechococcaceae as a source of thermostable C-phycocyanins : properties and molecular insights. Algal Res. 35, 223–235. doi: 10.1016/j.algal.2018.08.037
Liang, Y., Tang, J., Luo, Y., Kaczmarek, M. B., Li, X., Daroch, M., et al. (2019). Thermosynechococcus as a thermophilic photosynthetic microbial cell factory for CO 2 utilisation. Bioresour. Technol. 278, 255–265. doi: 10.1016/j.biortech.2019.01.089
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for inproved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Mai, T., Johansen, J. R., Pietrasiak, N., Bohunicka, M., and Martin, M. P. (2018). Revision of the Synechococcales (cyanobacteria) through recognition of four families including Oculatellaceae fam. nov., Trichocoleaceae fam nov., seven new genera containing 14 species. Phytotaxa 365, 1–59. doi: 10.11646/phytotaxa.365.1.1
Mathews, D. H. (2014). RNA secondary structure analysis using RNAstructure. Curr. Protoc. Bioinformatics 46, 1–25. doi: 10.1002/0471250953.bi1206s46
McGinn, P. J., Price, G. D., Maleszka, R., and Badger, M. R. (2003). Inorganic carbon limitation and light control the expression of transcripts related to the CO2-concentrating mechanism in the cyanobacterium Synechocystis sp. strain PCC6803. Plant Physiol. 132, 218–229. doi: 10.1104/pp.019349
Mehda, S., Muñoz-Martín, M. Á., Oustani, M., Hamdi-Aïssa, B., Perona, E., Mateo, P., et al. (2022). Lithic cyanobacterial communities in the polyextreme Sahara Desert: implications for the search for the limits of life. Environ. Microbiol. 24, 451–474. doi: 10.1111/1462-2920.15850
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., and Göker, M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in 2010 Gateway Computing Environments Workshop, GCE 2010 (New Orleans, LA: IEEE). doi: 10.1109/GCE.2010.5676129
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Miscoe, L. H., Johansen, J. R., Kociolek, J. P., Lowe, R. L., Vaccarino, M. A., Pietrasiak, N., et al. (2016). The diatom flora and cyanobacteria from caves on Kauai, Hawaii. Acta Botanica Hungarica. 58, 3–4.
Nowicka-Krawczyk, P., Mühlsteinová, R., and Hauer, T. (2019). Detailed characterization of the Arthrospira type species separating commercially grown taxa into the new genus Limnospira (Cyanobacteria). Scient. Rep. 9, 694. doi: 10.1038/s41598-018-36831-0
O'Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, 733–745. doi: 10.1093/nar/gkv1189
Patel, A., Matsakas, L., Rova, U., and Christakopoulos, P. (2019). A perspective on biotechnological applications of thermophilic microalgae and cyanobacteria. Bioresour. Technol. 278, 424–434. doi: 10.1016/j.biortech.2019.01.063
Perona, E., Muñoz-Martín, M. Á., and Berrendero Gómez, E. (2022). “Recent trends of polyphasic approach in taxonomy and cyanobacterial diversity,” in Expanding Horizon of Cyanobacterial Biology, eds P. K. Singh, M. F. Fillat, and V. Sitther (Amsterdam: Elsevier), 1–49. doi: 10.1016/B978-0-323-91202-0.00008-7
Pietrasiak, N., Osorio-Santos, K., Shalygin, S., Martin, M. P., and Johansen, J. R. (2019). When is a lineage a species? A case study in Myxacorys gen. nov. (Synechococcales: Cyanobacteria) with the description of two new species from the Americas. J. Phycol. 55, 976–996. doi: 10.1111/jpy.12897
Pietrasiak, N., Reeve, S., Osorio-Santos, K., Lipson, D. A., and Johansen, J. R. (2021). Trichotorquatus gen. nov. - a new genus of soil cyanobacteria discovered from American drylands. J. Phycol. 57, 886–902. doi: 10.1111/jpy.13147
Price, G. D., Badger, M. R., Woodger, F. J., and Long, B. M. (2008). Advances in understanding the cyanobacterial CO2-concentrating- mechanism (CCM): functional components, Ci transporters, diversity, genetic regulation and prospects for engineering into plants. J. Exp. Bot. 59, 1441–1461. doi: 10.1093/jxb/erm112
Raabova, L., Kovacik, L., Elster, J., Strunecky, O., Raabová, L., Kovacik, L., et al. (2019). Review of the genus Phormidesmis (Cyanobacteria) based on environmental, morphological, and molecular data with description of a new genus Leptodesmis. Phytotaxa 395, 1–16. doi: 10.11646/phytotaxa.395.1.1
Rodriguez-R, L. M., Gunturu, S., Harvey, W. T., Rosselló-Mora, R., Tiedje, J. M., Cole, J. R., et al. (2018). The microbial genomes atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288. doi: 10.1093/nar/gky467
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 4, e1900v1 doi: 10.7287/peerj.preprints.1900v1
Roeselers, G., Norris, T. B., Castenholz, R. W., Rysgaard, S., Glud, R. N., Kühl, M., et al. (2007). Diversity of phototrophic bacteria in microbial mats from Arctic hot springs (Greenland). Environ. Microbiol. 9, 26–38. doi: 10.1111/j.1462-2920.2006.01103.x
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Sciuto, K., and Moro, I. (2016). Detection of the new cosmopolitan genus Thermoleptolyngbya (Cyanobacteria, Leptolyngbyaceae) using the 16S rRNA gene and 16S−23S ITS region. Mol. Phylogenet. Evol. 105, 15–35. doi: 10.1016/j.ympev.2016.08.010
Sciuto, K., Moschin, E., and Moro, I. (2017). Cryptic cyanobacterial diversity in the giant cave (Trieste, Italy): the new genus Timaviella (Leptolyngbyaceae). Cryptogam. Algol. 38, 285–323. doi: 10.7872/crya/v38.iss4.2017.285
Shalygin, S., Shalygina, R., Redkina, V., Gargas, C., and Johansen, J. (2020). Description of Stenomitos kolaenensis and S. hiloensis sp. nov. (Leptolyngbyaceae, Cyanobacteria) with an emendation of the genus. Phytotaxa 440, 108–128. doi: 10.11646/phytotaxa.440.2.3
Soares, F., Tiago, I., Trovo, J., Coelho, C., and Portugal, A. (2019). Description of Myxacorys almedinensis sp. nov. (Synechococcales, Cyanobacteria) isolated from the limestone walls of the Old Cathedral of Coimbra, Portugal (UNESCO World Heritage Site). Phytotaxa 419, 77–90. doi: 10.11646/phytotaxa.419.1.5
Stanier, R. Y., Kunisawa, R., Mandel, M., and Cohen-Bazire, G. (1971). Purification and properties of unicellular blue-green algae (order Chroococcales). Bacteriol. Rev. 35, 171–205. doi: 10.1128/br.35.2.171-205.1971
Strunecký, O., Ivanova, A. P., and Mareš, J. (2023). An updated classification of cyanobacterial orders and families based on phylogenomic and polyphasic analysis. J. Phycol. 59, 12–51. doi: 10.1111/jpy.13304
Strunecky, O., Raabova, L., Bernardova, A., Ivanova, A. P., Semanova, A., Crossley, J., et al. (2019). Diversity of cyanobacteria at the Alaska North Slope with description of two new genera: Gibliniella and Shackletoniella. FEMS Microbiol. Ecol. 96, iz189. doi: 10.1093/femsec/fiz189
Tang, J., Du, L.-M. L., Li, M., Yao, D., Waleron, M., Waleron, K. F., et al. (2022a). Characterization of a novel hot-spring cyanobacterium Leptodesmis sichuanensis sp. Nov., genomic insights of molecular adaptations into its habitat. Front. Microbiol. 12, 739625. doi: 10.3389/fmicb.2021.739625
Tang, J., Du, L.-M. L.-M., Liang, Y.-M. Y.-M., and Daroch, M. (2019). Complete genome sequence and comparative analysis of Synechococcus sp. CS-601 (SynAce01), a cold-adapted cyanobacterium from an oligotrophic antarctic habitat. Int. J. Mol. Sci. 20, 152. doi: 10.3390/ijms20010152
Tang, J., Jiang, D., Luo, Y., Liang, Y., Li, L., Shah, M. M. R., et al. (2018a). Potential new genera of cyanobacterial strains isolated from thermal springs of western Sichuan, China. Algal Res. 31, 14–20. doi: 10.1016/j.algal.2018.01.008
Tang, J., Li, L., Li, M., Du, L., Shah, M. R. M. M. R., Waleron, M. M. M., et al. (2021). Description, taxonomy, and comparative genomics of a novel species, Thermoleptolyngbya sichuanensis sp. nov., isolated from hot springs of Ganzi, Sichuan, China. Front. Microbiol. 12, 696102. doi: 10.3389/fmicb.2021.696102
Tang, J., Liang, Y., Jiang, D., Li, L., Luo, Y., Shah, M. M. R., et al. (2018b). Temperaturecontrolled thermophilic bacterial communities in hot springs of western Sichuan, China. BMC Microbiol. 18, 134. doi: 10.1186/s12866-018-1271-z
Tang, J., Shah, M. R., Yao, D., Du, L., Zhao, K., Li, L., et al. (2022b). Polyphasic identification and genomic insights of Leptothermofonsia sichuanensis gen. sp. nov., a novel thermophilic cyanobacteria within Leptolyngbyaceae. Front. Microbiol. 13, 765105. doi: 10.3389/fmicb.2022.765105
Tang, J., Zhou, H., Jiang, Y., Yao, D., Waleron, K. F., Du, L. M., et al. (2023). Characterization of a novel thermophilic cyanobacterium within Trichocoleusaceae, Trichothermofontia sichuanensis gen. et sp. nov., its CO2-concentrating mechanism. Front. Microbiol. 14, 1111809. doi: 10.3389/fmicb.2023.1111809
Tang, J., Zhou, H., Yao, D., Riaz, S., You, D., Klepacz-Smółka, A., et al. (2022c). Comparative genomic analysis revealed distinct molecular components and organization of CO2-concentrating mechanism in thermophilic cyanobacteria. Front. Microbiol. 13, 876272. doi: 10.3389/fmicb.2022.876272
Tawong, W., Pongcharoen, P., Nishimura, T., and Saijuntha, W. (2022). Siamcapillus rubidus gen. et sp. nov. (Oculatellaceae), a novel filamentous cyanobacterium from Thailand based on molecular and morphological analyses. Phytotaxa 558, 33–52. doi: 10.11646/phytotaxa.558.1.2
Thi Hoang, D., Chernomor, O., von Haeseler, A., Quang Minh, B., Sy Vinh, L., Rosenberg, M. S., et al. (2017). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol 35, 518–522. doi: 10.1093/molbev/msx281
Turland, N., Wiersema, J., Barrie, F. R., Greuter, W., and Smith, G. F. (2018). International Code of Nomenclature for Algae, Fungi, and Plants (Shenzhen Code) Adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017. Koeltz botanical Books.
Vaz, M. G. M. V., Genuário, D. B., Andreote, A. P. D., Malone, C. F. S., Sant'Anna, C. L., Barbiero, L., et al. (2015). Pantanalinema gen. nov., Alkalinema gen. nov.: novel pseudanabaenacean genera (Cyanobacteria) isolated from saline–alkaline lakes. Int. J. Syst. Evol. Microbiol. 65, 298–308. doi: 10.1099/ijs.0.070110-0
Walter, J. M., Coutinho, F. H., Dutilh, B. E., Swings, J., Thompson, F. L., Thompson, C. C., et al. (2017). Ecogenomics and taxonomy of cyanobacteria phylum. Front. Microbiol. 8, 2132. doi: 10.3389/fmicb.2017.02132
Zammit, G. (2018). Systematics and biogeography of sciophilous cyanobacteria; an ecological and molecular description of Albertania skiophila (Leptolyngbyaceae) gen. and sp. nov. Phycologia 57, 481–491. doi: 10.2216/17-125.1
Zammit, G., Billi, D., and Albertano, P. (2012). The subaerophytic cyanobacterium Oculatella subterranea (Oscillatoriales, Cyanophyceae) gen. et sp. nov.: a cytomorphological and molecular description. Eur. J. Phycol. 47, 341–354. doi: 10.1080/09670262.2012.717106
Keywords: Thermocoleostomius, 16S rRNA, 16S-23 ITS, CO2-concentrating mechanism, Thermophilic cyanobacterium, Oculatellaceae, Leptolyngbyaceae, Albertania
Citation: Jiang Y, Tang J, Liu X and Daroch M (2023) Polyphasic characterization of a novel hot-spring cyanobacterium Thermocoleostomius sinensis gen et sp. nov. and genomic insights into its carbon concentration mechanism. Front. Microbiol. 14:1176500. doi: 10.3389/fmicb.2023.1176500
Received: 28 February 2023; Accepted: 06 July 2023;
Published: 26 July 2023.
Edited by:
Brian P. Hedlund, University of Nevada, Las Vegas, United StatesReviewed by:
Wittaya Tawong, Naresuan University, ThailandSenthil Murugapiran, Diversigen, United States
Copyright © 2023 Jiang, Tang, Liu and Daroch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurycy Daroch, bS5kYXJvY2hAcGt1c3ouZWR1LmNu