 Weifeng Huang1†
Weifeng Huang1† Yuan Fang
Yuan Fang Yihan Lu
Yihan Lu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 30 May 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1157888
Chlamydia psittaci, a strictly intracellular bacterium, is an underestimated etiologic agent leading to infections in a broad range of animals and mild illness or pneumonia in humans. In this study, the metagenomes of bronchoalveolar lavage fluids from the patients with pneumonia were sequenced and highly abundant C. psittaci was found. The target-enriched metagenomic reads were recruited to reconstruct draft genomes with more than 99% completeness. Two C. psittaci strains from novel sequence types were detected and these were closely related to the animal-borne isolates derived from the lineages of ST43 and ST28, indicating the zoonotic transmissions of C. psittaci would benefit its prevalence worldwide. Comparative genomic analysis combined with public isolate genomes revealed that the pan-genome of C. psittaci possessed a more stable gene repertoire than those of other extracellular bacteria, with ~90% of the genes per genome being conserved core genes. Furthermore, the evidence for significantly positive selection was identified in 20 virulence-associated gene products, particularly bacterial membrane-embedded proteins and type three secretion machines, which may play important roles in the pathogen-host interactions. This survey uncovered novel strains of C. psittaci causing pneumonia and the evolutionary analysis characterized prominent gene candidates involved in bacterial adaptation to immune pressures. The metagenomic approach is of significance to the surveillance of difficult-to-culture intracellular pathogens and the research into molecular epidemiology and evolutionary biology of C. psittaci.
Chlamydia psittaci is a gram-negative, obligate intracellular pathogen belonging to the family Chlamydiaceae (Beeckman and Vanrompay, 2009). It is a zoonotic agent that can cause infections in humans and a broad range of animals, also known as psittacosis (Cong et al., 2013; Vorimore et al., 2015). In people, psittacosis generally presents mild symptoms, including fever, headache, dry cough, and muscle aches; while it can also lead to life-threatening pneumonia, especially in immunocompromised patients (Branley et al., 2016). The agent has been frequently detected in avian and non-avian animals, which has posed economic impacts on the poultry and livestock industry (Knittler and Sachse, 2014). Based on sequencing of the major outer membrane protein OmpA, C. psittaci has been divided into nine genotypes, of which genotypes A and E have been described as common etiologic agents causing respiratory illnesses in humans (Van Lent et al., 2012; Wolff et al., 2015).
Clinically, it is difficult to diagnose psittacosis and to detect the related pathogens directly from the specimens through traditional culture, due to Chlamydia spp. being strictly intracellular bacteria (Wu et al., 2021). The serological assay and polymerase chain reaction can be utilized to identify the pathogens in patients with suspected psittacosis (Nieuwenhuizen et al., 2018). However, it is impractical to foresee the culprit causing infection in prior in most cases. The application of metagenomic next-generation sequencing (mNGS) has effectively made up for this issue by summarizing the microbial profile in the specimen (Gu et al., 2019). For instance, Chen et al. (2020) have reported that nine cases of pneumonia have negative results for the routine etiological pathogen tests, and these are positive for C. psittaci DNA fragments by mNGS. In addition, mNGS enables providing comprehensive genetic information to capture abnormal changes in microbial compositions in vivo (Yin et al., 2021). A recent study has revealed that the abundance outliers of certain microbes could prompt their pathogenic risk, and further infer if opportunistic pathogens are harmful or commensal in the niches (Chen et al., 2022).
Decoding genome sequences of the target pathogens from microbiome data is meaningful for tracing microbial evolution and transmission, as well as genetic characteristics of clinically relevant genotypes. Lam et al. (2021) have presented a genomic surveillance framework to detect and type Klebsiella pneumoniae and virulence loci genotypes direct from gut metagenome data. Bacterial colonization and transmission patterns in a newly built hospital have been deciphered by metagenomic analysis, which uncovered persistent unique genotypes of Staphylococcus and Propionibacterium circulating in healthcare environments (Lax et al., 2017). To date, there have been few surveys on the utility of metagenomic surveillance for understanding the genomic dynamics and evolutionary biology of C. psittaci.
In this study, C. psittaci was detected with significant abundance in the bronchoalveolar lavage fluid (BALF) of two pneumonia human cases. Nearly-complete genomes of C. psittaci were reconstructed directly from the shotgun metagenomic sequencing on the specimens. Genomic analyses combined with the public isolates of C. psittaci were performed to characterize the molecular epidemiology and pan-genomic structure of the metagenome strains. Furthermore, evidence for positive Darwinian selection acting on virulence-associated genes was first revealed to explore the molecular mechanisms of adaptive evolution for this intracellular pathogen.
The two patients included in this study had an acute onset of pneumonia. A 72-year-old male patient (#1) with a history of pigeon breeding was admitted to Shanghai General Hospital, Shanghai JiaoTong University School of Medicine (Shanghai, China), in December 2020 due to cough, fever, and dyspnea for 2 weeks. A 66-year-old male patient (#2), a poultry breeder, was admitted to Shanghai Jiao Tong University Affiliated Sixth People’s Hospital (Shanghai, China) in June of 2021 due to fever and chills for 5 days, and chest distress and shortness of breath for 2 days. The patient had a maximum body temperature of 40°C, asthma, decreased appetite, and general fatigue. BALF was harvested in strict accordance with clinical practice. The samples were then sent for mNGS at Genoxor (Shanghai, China). Patient characteristics and clinical tests are given in Supplementary Table S1. Anti-infective therapy with third-generation cephalosporins was started according to the guidelines on the treatment of adult community-acquired pneumonia (Qu and Cao, 2016).
Samples were processed through 10,000 g centrifugation for 5 min, and the pellets were collected for nucleic acid extraction using DP710-T2A (TIANGEN, China). Approximately 10 ng DNA was used for library construction with the Hieff NGS OnePot Pro DNA Library Prep Kit (Yeasen Biotech., China). Using the mode of single-end 76-bp reads, the library was sequenced by the Illumina NextSeq 550 platform (Illumina Inc., United States). The raw sequencing reads were trimmed and filtered using the program Fastp v0.21.1 (Chen et al., 2018). Host-derived reads were further subtracted by aligning the sequences to the human reference genome GRCh38 using Bowtie v2.2.6 (Langmead and Salzberg, 2012). The remaining reads were used to compute the taxonomic abundances of individual species by Kraken v2.0.9 (Wood et al., 2019) and Bracken v2.2 (Lu et al., 2017).
Bacterial genomes (<200 contigs) of C. psittaci isolates were retrieved from the NCBI Assembly database in March 2022 (Kitts et al., 2016; Supplementary Table S2). For the specimens sequenced in this study, the metagenomic reads derived from C. psittaci were first extracted based on the reads mapping to the genomes using BBmap v38.18. The species-specific reads were assembled by Spades v3.15.4 with the options -t 24 -m 128 --cov-cutoff auto --isolate (Bankevich et al., 2012). Quality assessment of the resulting assemblies was conducted by both programs QUAST v5.0.2 (Gurevich et al., 2013) and CheckM v1.0.18 (Parks et al., 2015).
Average nucleotide identity (ANI) between genomes was calculated by using PYANI v0.2.11 with the option -m ANIb (Pritchard et al., 2016). The resulting matrix was visualized using the R package pheatmap v1.0.12. Multilocus sequence typing (MLST) was performed to detect bacterial sequence types (STs) by Fastmlst v0.0.15 (Guerrero-Araya et al., 2021) according to the scheme Chlamydiales with seven alleles: gatA, oppA, hflX, gidA, enoA, hemN, fumC. New STs in the genome assemblies generated above were submitted to the PubMLST website (Jolley et al., 2018). For the pan-genome analysis, all the genomes were annotated uniformly by Prokka v1.14.6 (Seemann, 2014). Gene clustering was performed using Roary v3.13.0 (Page et al., 2015) with the options -p 36 -i 80 -e -n -t 11 -s -cd 100 -a -v. A concatenated alignment of core genes was then generated to build a maximum-likelihood phylogenomic tree that was visualized with ggtree v3.2.1 (Yu et al., 2018). Genome-scale sequence conservation was investigated between the metagenome strains and reference strains of nine C. psittaci genotypes (Van Lent et al., 2012). A circular genome map was constructed by using BRIG v0.95 (Alikhan et al., 2011).
For protein functional classification, we employed Blastp v2.9.0+ (Shiryev et al., 2007) with the E-value threshold of 1e−20 searching against the COG database. Using the VFDB core database with 4,194 protein sequences (Liu et al., 2018), the genes encoding virulence factors (VFs) were detected by Blastp with the E-value threshold of 1e−20. Protein subcellular localization was predicted by PSORTb v3.0.3 (Yu et al., 2010). Secondary structure prediction of the proteins was performed using PROTEUS2 (Montgomerie et al., 2008). Protein structure modeling was carried out using the intensive mode of Phyre2 web portal (Kelley et al., 2015). Selection pressure acting on the virulence-associated genes (VAGs) was detected by the PAML v4.9 package (Yang, 2007), which can estimate the ratios (ω) of non-synonymous (dN) to synonymous (dS) substitutions among the aligned codons. Briefly, protein multiple sequence alignments were initially performed by MAFFT v7.505 (Katoh et al., 2019). Codon alignments were then generated using amino acid sequence alignment and nucleotide sequences per gene. The resulting alignment per gene was further improved to remove the unreliable aligned sites using Gblocks v0.91b (Talavera and Castresana, 2007). The maximum-likelihood algorithm was used to build gene trees by PhyMLv3.1 (Guindon et al., 2010). The codon alignments and the corresponding trees were used for positive selection scanning by the codeml program in PAML. Two site-specific models M1a (NearlyNeutral) and M2a (PositiveSelection) were applied herein. Likelihood ratio test (LRT) of positive selection was performed to infer if the data is a significantly better fit for the alternative model M2a, which allows some sites with ω > 1; whereas the null model M1a does not allow this. Posterior probabilities for each site were then calculated using Bayes empirical Bayes and the sites with ω > 1 and p > 95% were predicted to be under positive selection (Yang et al., 2005).
In this retrospective study, we investigated the metagenomic data of BALF from two cases of pneumonia. The numbers of species present in the lung microbiome were 69 in patient #1 and 48 in patient #2 (Supplementary Table S3). C. psittaci was observed to be the most predominant bacteria in the microbial community, whose abundance was 79.4% in patient #1 and 89.8% in patient #2. It implied that C. psittaci was the etiological agent closely related to the pulmonary infection in both cases. According to the results of mNGS, the patient #1 was treated with high-flow nasal oxygenation and the antibiotics doxycycline and moxifloxacin. Consequently, the body temperature of the patient gradually reached the normal level, and he was discharged 14 days later. Patient #2 received 16 days of anti-infection treatment with moxifloxacin and minocycline before discharge.
Next, we reconstructed bacterial genomes of two C. psittaci strains from the metagenomes of BALF: CPMBF01 and CPMBF02. The assembly metrics are summarized in Table 1. The genome sizes were 1,176,486 bp for CPMBF01 and 1,158,597 bp for CPMBF02, with >99% completeness for both assemblies. The numbers of protein-coding sequences (CDSs) were 978 for CPMBF01 and 975 for CPMBF02. The average GC content of two metagenome-assembled genomes (MAGs) was about 39%, which was consistent with the median GC content of nine C. psittaci genotype reference strains (Van Lent et al., 2012). Furthermore, Figure 1 displays the pairwise ANI values between two MAGs and 21 isolate genomes derived from eight species within the genus Chlamydia. CPMBF01 shared 97.3% (M56) ~ 99.0% (WC) ANI with three C. psittaci isolates (Supplementary Table S4). CPMBF02 shared 97.0% (M56) ~ 98.6% (WC) ANI with three C. psittaci isolates. On the other hand, the genomes of CPMBF01 and CPMBF02 possessed relatively low ANI values with the genomes from the other Chlamydia species, ranging from 71.3 to 93.5%. The results demonstrate that the strains CPMBF01 and CPMBF02 are derived from C. psittaci.
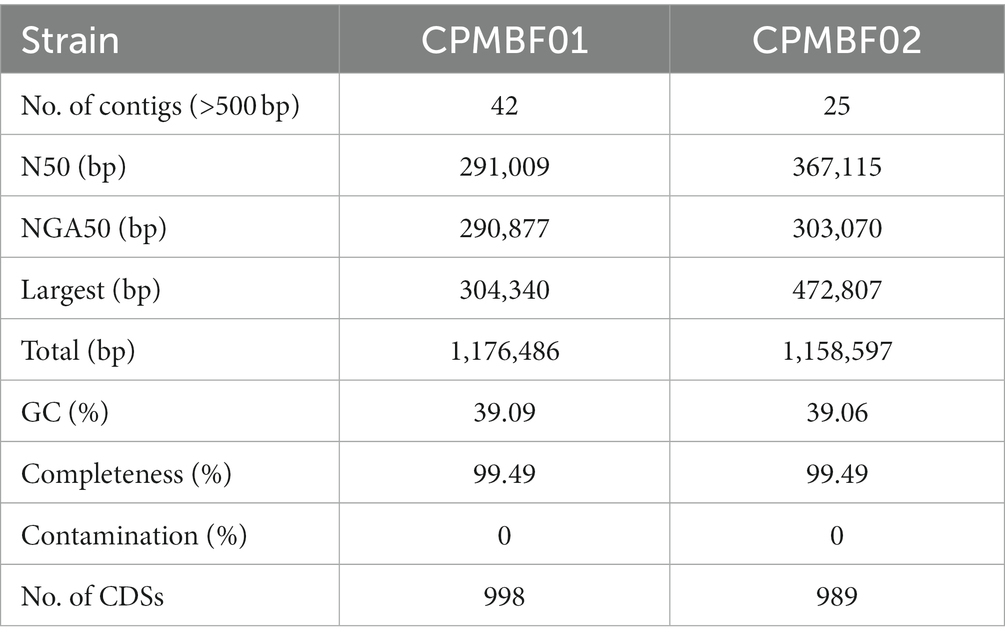
Table 1. Genome features of Chlamydia psittaci recovered from the metagenomic data of bronchoalveolar lavage fluid in patients with pneumonia.
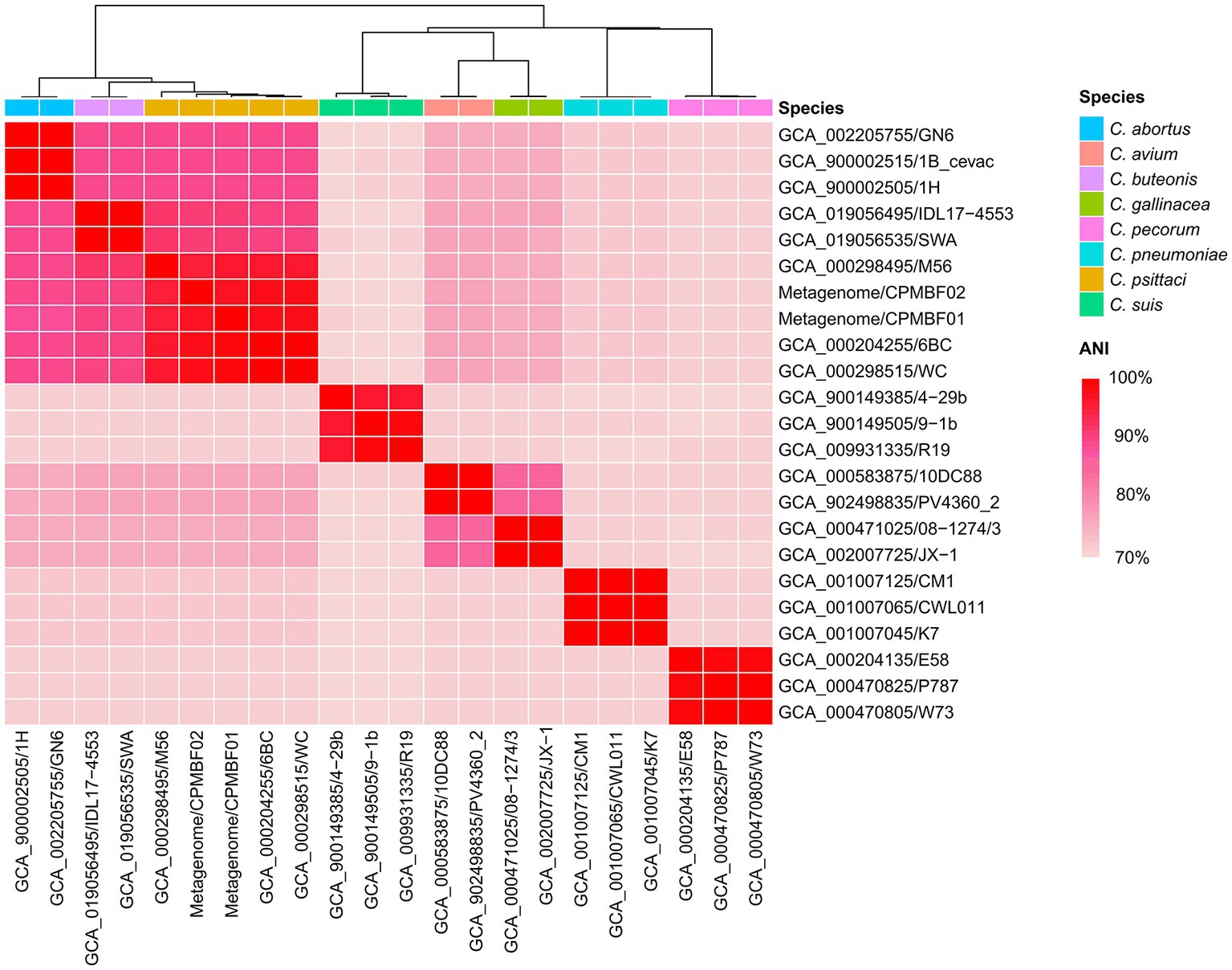
Figure 1. Species identification for the metagenomic strains CPMBF01 and CPMBF02 recovered from the BALF specimen. The heatmap shows the pairwise ANI values between metagenome-assembled genomes and the isolate genomes from eight species within the genus Chlamydia. The sequence accessions of the isolate genomes are listed in Supplementary Table S2.
Furthermore, genome sequence conservation of the metagenomic strains and representative isolates of nine C. psittaci genotypes was shown in Figure 2. The highly homologous regions (>95% identity) between the metagenomic strains and genotype E isolate MN accounted for 97.6% (1,140,068 bp; CPMBF01) and 95.0% (1,110,205 bp; CPMBF02) of the MN genome, respectively. Genomic regions (>1 kb) that are present in the MN genome but absent in the MAGs are summarized in Supplementary Table S5. It suggests that the majority of genomic sequences are very conserved across the C. psittaci strains.
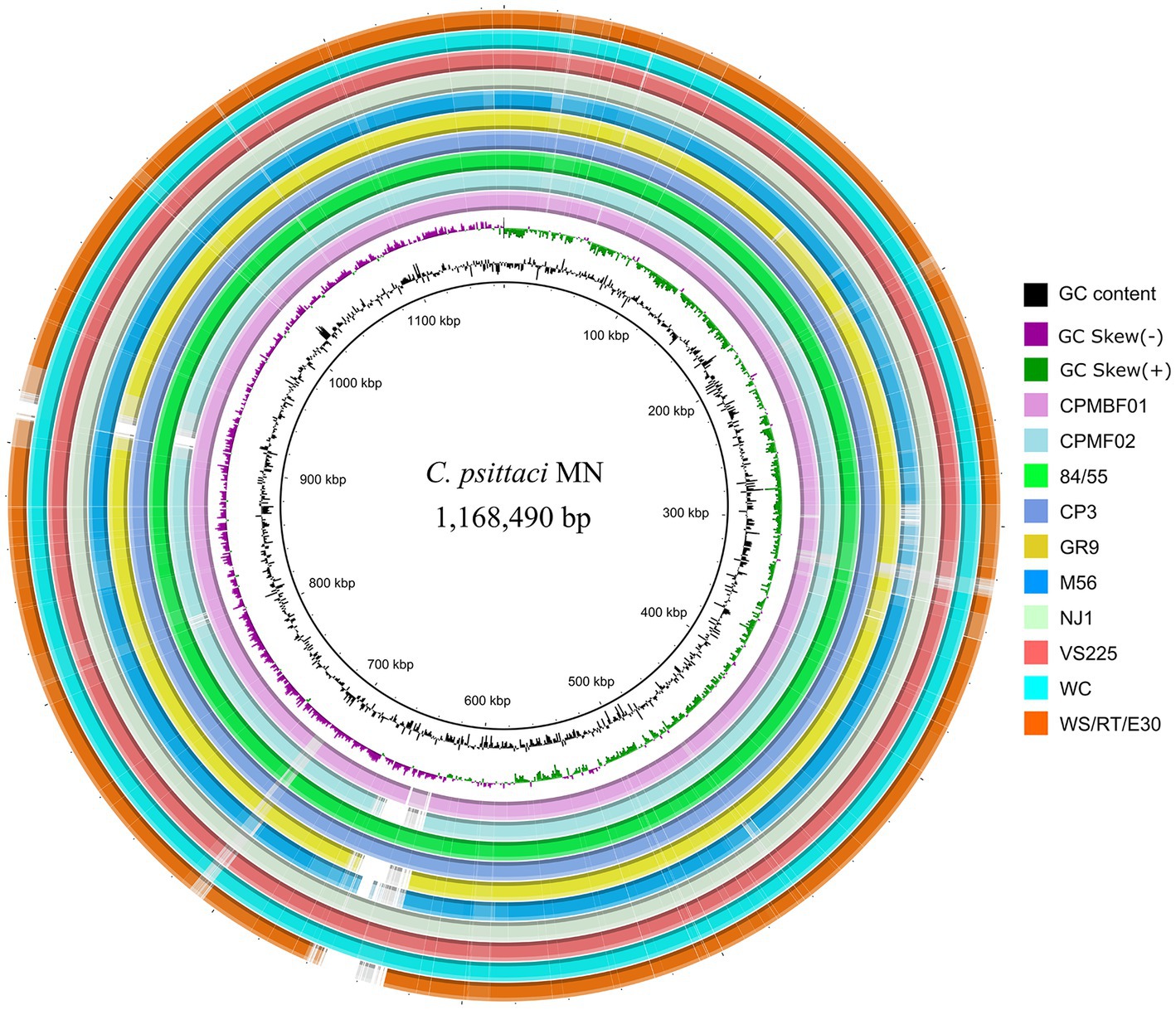
Figure 2. Circular genome representation of eleven C. psittaci strains. The innermost two rings denote the tick mark and GC skew plot of the MN genome of C. psittaci genotype E. The remaining rings from innermost to outermost represent the two metagenome strains and the isolates from the other eight genotypes, i.e., CPMBF01, CPMBF02, 84/55 of genotype A, CP3 of genotype B, GR9 of genotype C, M56 of genotype M56, NJ1 of genotype D, VS225 of genotype F, WC of genotype WC, and WS/RT/E30 of genotype E/B. The rings corresponding to individual genomes are color-coded based on the metrics of BLAST matches with a minimum sequence identity of 80% and an E-value threshold of 1e−10.
The relationships between the two metagenomic strains and 56 isolates of C. psittaci were analyzed according to a phylogenetic tree built with a core-gene alignment (560,571 nt). Figure 3 shows the strain phylogeny integrated with the bacterial metadata (Supplementary Table S2). Among 14 STs observed in the isolate genomes, ST24 (34 strains) was the most prevalent, followed by ST28 (5), ST47 (4), and ST35 (3). Novel STs were detected in both metagenomic strains, including 85-78-108-112-13-9-78 for CPMBF01 and 12-13-11-13-13-28-12 for CPMBF02. Both ST profiles have been deposited in PubMLST, with the identifiers ST335 assigned to CPMBF01 and ST334 to CPMBF02. As shown in the tree, the strains were distributed into eight clades. In clade VII, the strain CPMBF01 of ST335 was neighboring to the genotype D strain NJ1 (GCA_000298555; ST43), which was isolated from Meleagris gallapavo, a large ground-dwelling bird. The strain CPMBF02 of ST334 was clustered with five ST28 strains, two of which were isolated from birds in Germany and the others were from sheep, goats and vulpes in Russia. In terms of geographical distribution, most strains were sampled from Germany (16), Australia (12), the United States (6), China (5), and Russia (5). The frequency of the isolation countries by different hosts is illustrated in Supplementary Figure S1. It seemed that the phylogenetic topology of most strains was consistent with the groups of STs, which was irrelevant to the places of isolation or host preference. For instance, all the strains of ST24 clustered together in clade I were from multiple sources, most of which were isolated from humans (11), birds (7), cattle (7), and equine (5). Of these, the strains of human origin are closely related to the strains from mammals and birds. It indicates that infection caused by C. psittaci may be attributed to its wide zoonotic transmission all over the world.
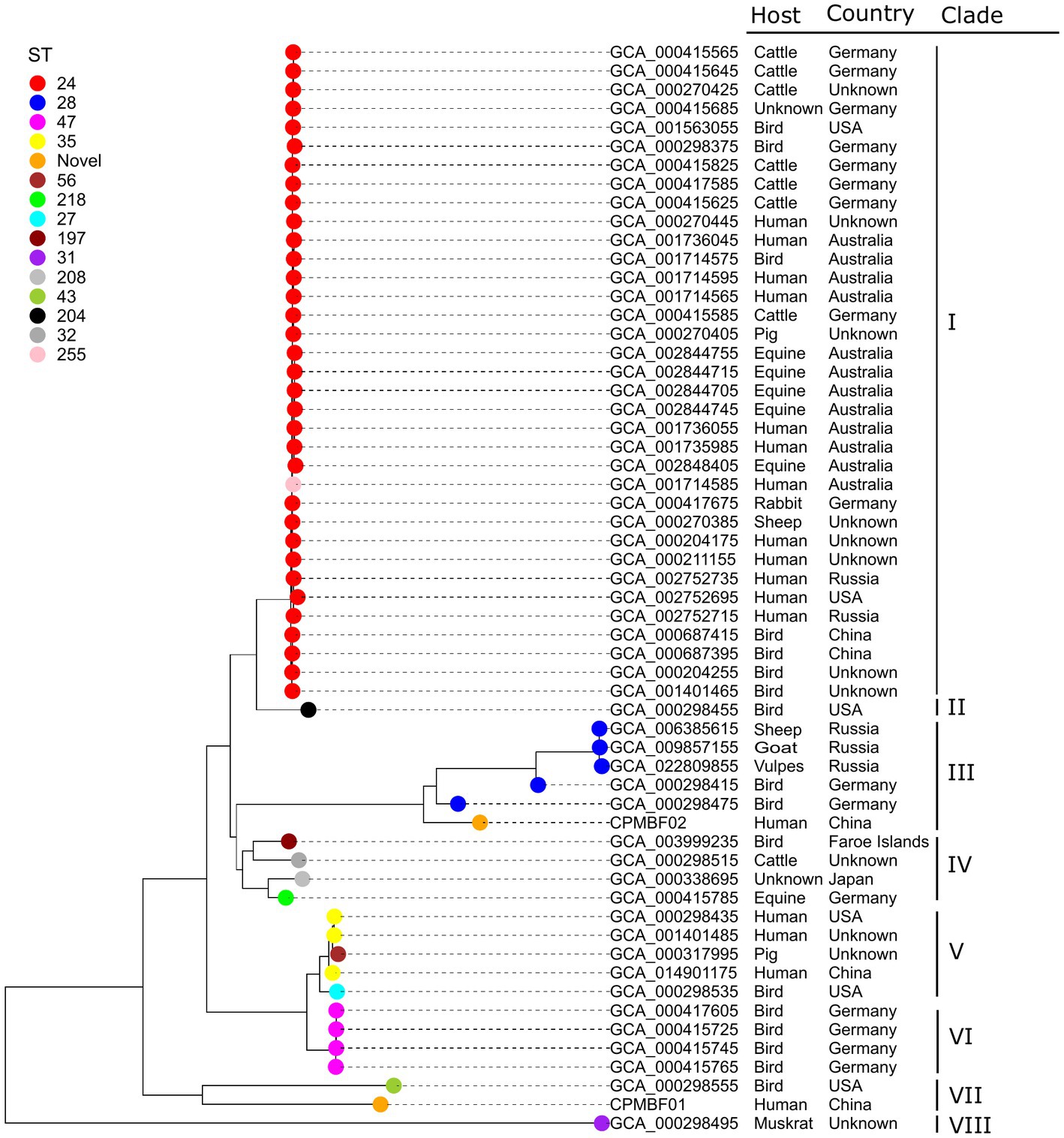
Figure 3. Maximum-likelihood phylogenetic tree of C. psittaci. The tip nodes are color-coded by different sequence types. Both metagenomic strains CPMBF01 and CPMBF02 with novel sequence types ST335 and ST334 are represented as orange nodes. The isolation countries, hosts, and evolutionary clades of individual strains are displayed in the right panel.
Based on the clustering of 58,326 CDSs across all the strains, 1,157 orthologous genes (OGs) were detected in the pan-genome of C. psittaci (Supplementary Table S6). Of these, over three-quarters (n = 890) were conserved core genes present in all the strains. Besides, 157 (14%) accessory genes and 110 (10%) strain-specific genes were found in the pan-genome. Using an exponential decay model on 58 genomes, the number of core genes in C. psittaci was estimated to be ~880 genes (Figure 4A), accounting for ~90% of the genes per genome. Notably, there are few strain-specific genes (~3 genes) per newly sequenced C. psittaci genome (Figure 4B). In contrast, the gram-negative model organism Escherichia coli encodes about 300 truly unique genes per genome and the percentage of the core genes per genome is ~47% (Rasko et al., 2008). It was apparent that C. psittaci possessed a stable gene repertoire and a weakening ability in the acquisition of genetic materials, perhaps due to the strictly intracellular properties of this bacterium. In addition, the proportion of three gene-sets distributed in the COG functional categories is shown in Figure 4C. Except for the genes encoding unknown functions, the most abundant category is “Translation and ribosomal structure,” with 131 genes. Several categories consisted of conserved core genes without the other dispensable genes, such as “Cell wall/membrane/envelope biogenesis,” “Defense mechanisms,” and “Inorganic ion transport and metabolism.” Generally, the genic components of C. psittaci tend to be more conserved than other extracellular bacterial pathogens whose horizontal gene transfers are vibrant (Arnold et al., 2022).
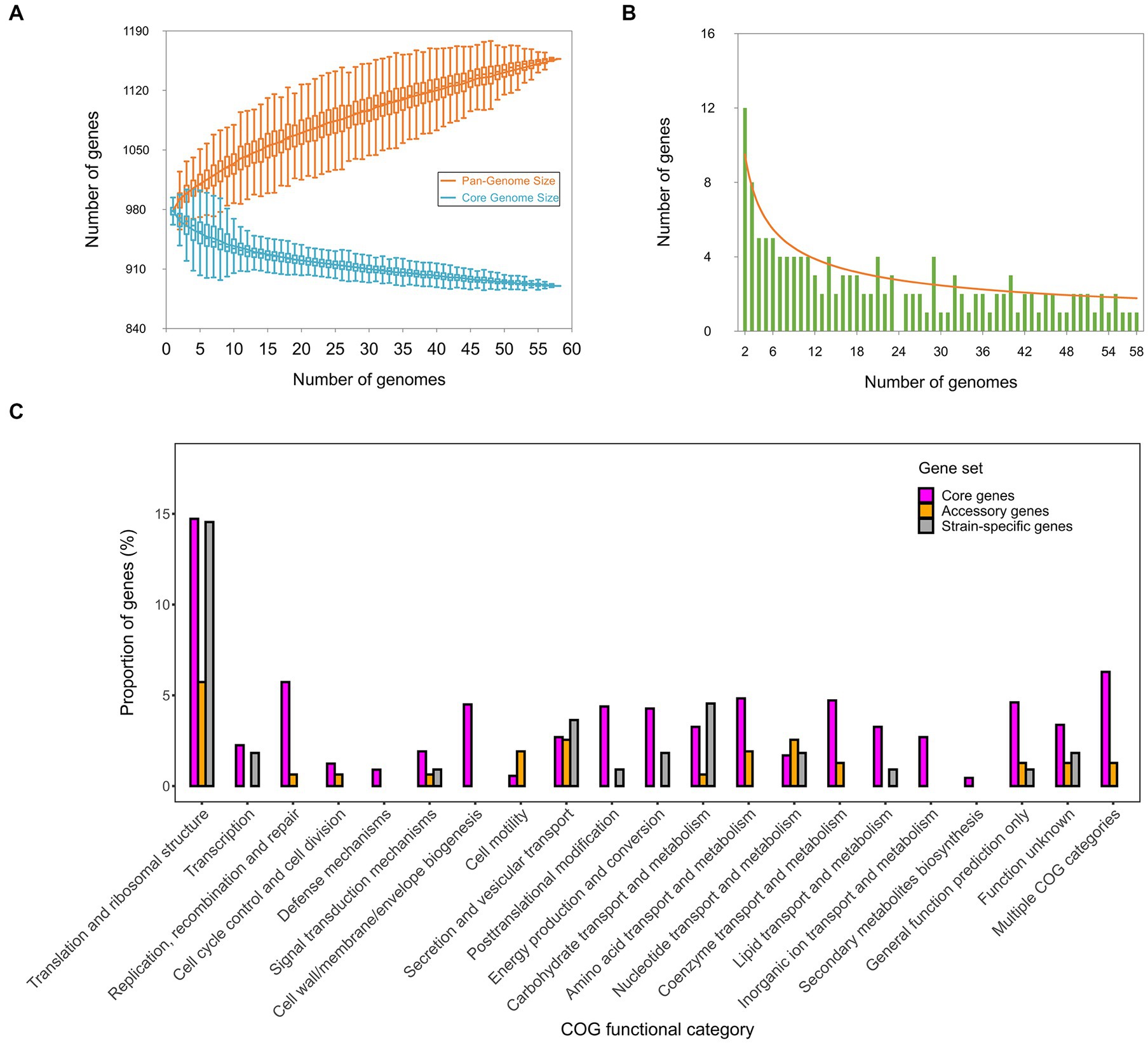
Figure 4. The pan-genomic structure and function of C. psittaci. (A) Estimation of the sizes of the pan-genome and core-genome is shown by (A), and estimation of the number of strain-specific genes per genome is shown by (B). (C) Distribution of the core genes, accessory genes, and strain-specific genes affiliated with individual COG functional categories. The COG assignments of all genes are summarized in Supplementary Table S5.
To depict genetic features of virulence factors possessed by C. psittaci, 168 VAGs were identified in the bacterial pan-genome. The majority of VAGs (n = 158) were core genes (Supplementary Table S5). Based on the classification of virulence factors, the VAGs were assigned to 12 functional groups. The largest group “Effector delivery system” included 85 genes, followed by “Adherence” with 23 genes, “Immune modulation” with 20 genes, “Nutritional/metabolic factor” with 15 genes, and “Stress survival” with eight genes. A full spectrum of genes coding for products from classical virulence factors in Chlamydia was identified. For instance, we detected 30 genes involved in the assembly of type III secretion system (T3SS) and 49 genes encoding effector proteins secreted by T3SS (Supplementary Table S6). A total of 16 genes encoding polymorphic membrane proteins (Pmps), which can be vital for mediating adhesion to host cells (Favaroni and Hegemann, 2021), were detected in the pan-genome of C. psittaci.
Next, we investigated the evidence for positive selection operating on the 135 single-copy core genes associated with bacterial virulence and cell surface components. According to LRT between the null model M1a and the selection model M2a, 20 C. psittaci genes were identified as undergoing strong positive selection (p-value < 0.05; Table 2 and Supplementary Table S7). Half of these genes were encoding membrane-associated proteins whose adaptive evolution might be actively induced by selective immune pressure. For instance, the products encoded by both genes OG0548/0590 were predicted to localize on the bacterial outer membrane. Figure 5 shows structural models of the outer membrane proteins and the relevant sites subject to positive selection.
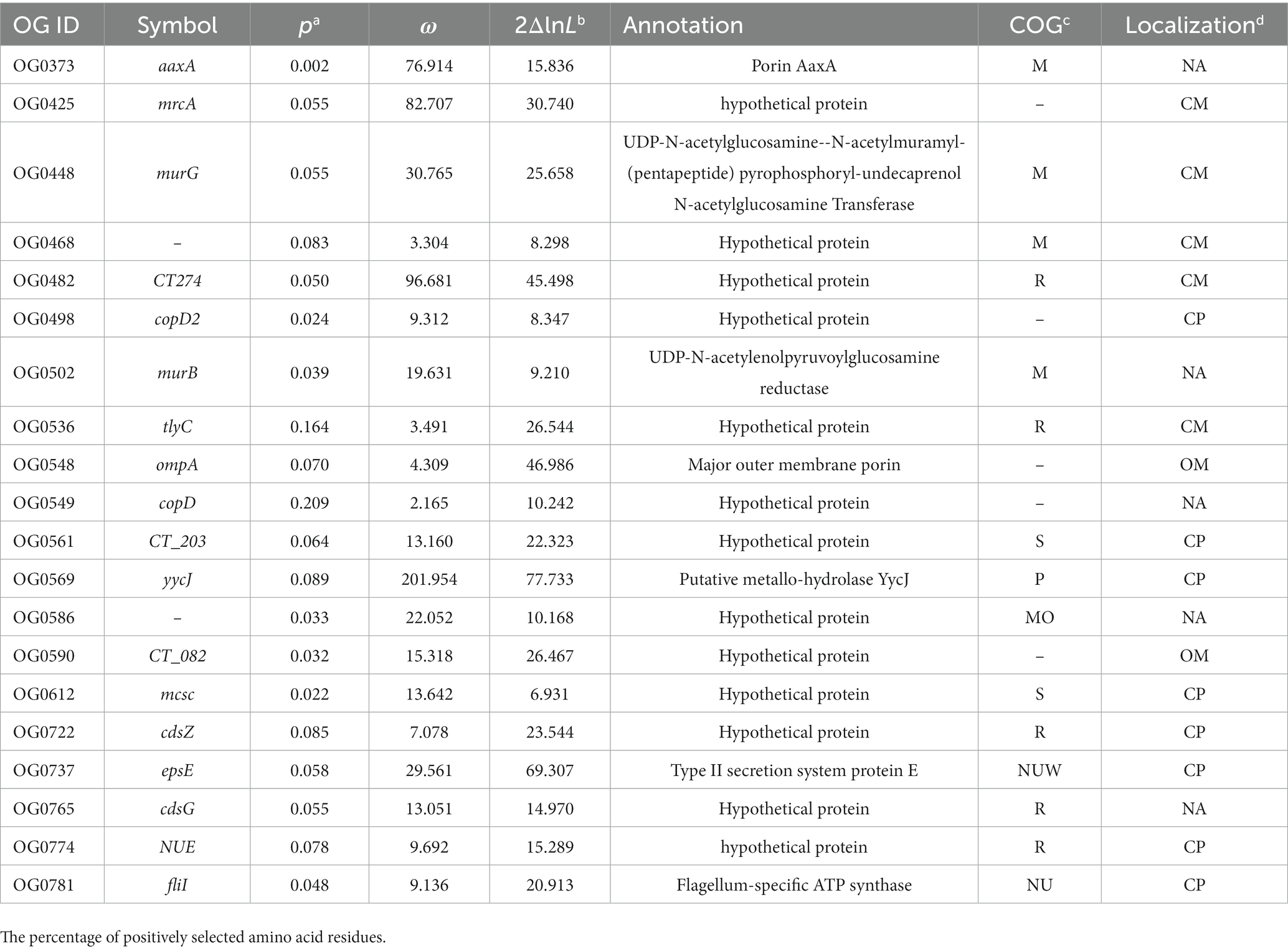
Table 2. Positively selected genes encoding proteins involved in the cell wall biogenesis and virulence of C. psittaci.
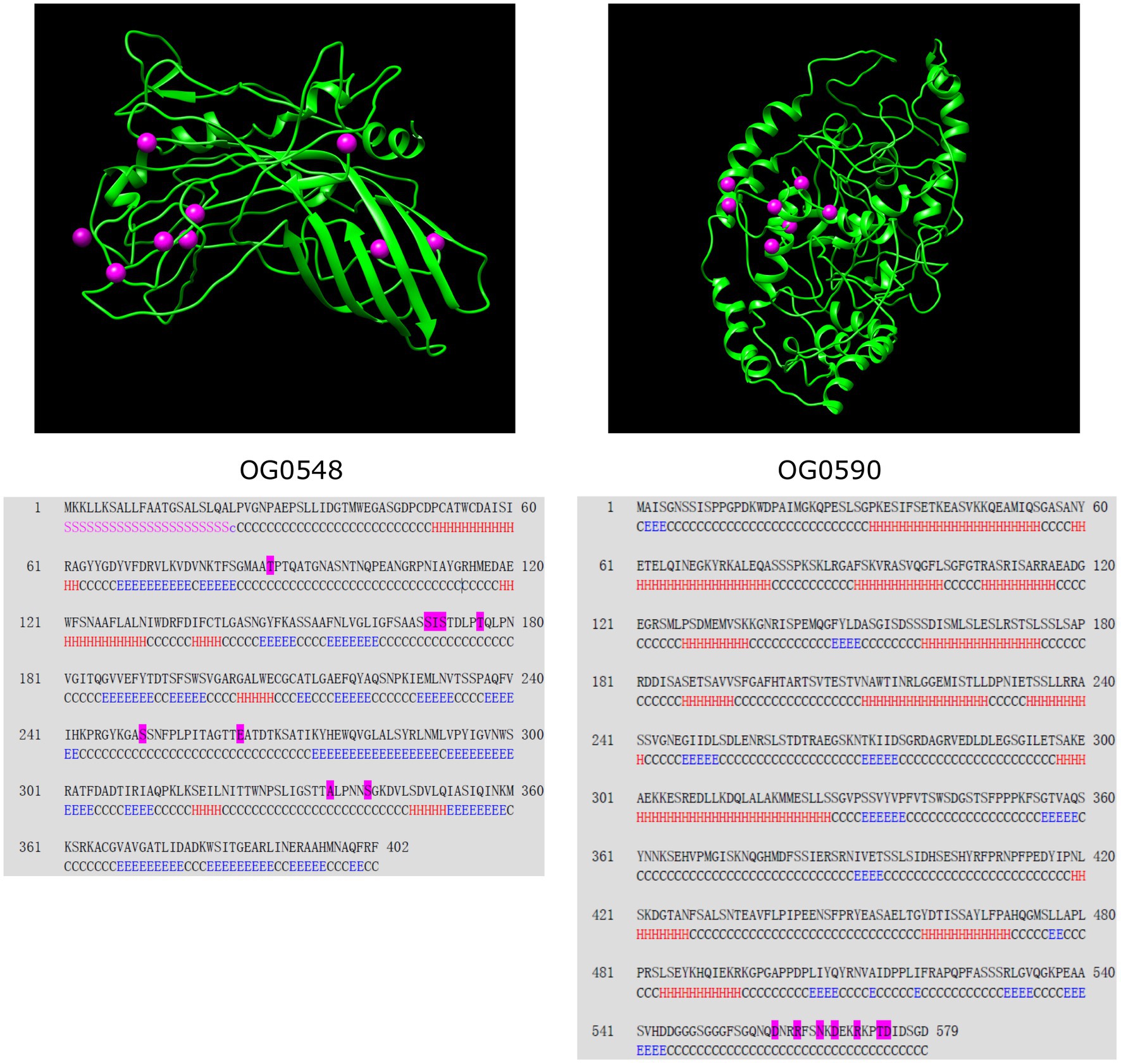
Figure 5. Secondary and tertiary structure of the outer membrane proteins encoded by two C. psittaci genes. Positively selected sites are illustrated by magenta spheres in the structural models (upper panel) and magenta residues in the corresponding sequences (lower panel). The segments of the secondary structure are marked by ‘H’ for helix, ‘E’ for β-strand, ‘C’ for coil, ‘S’ for signal peptide, and ‘c’ for cleavage site. Both proteins are identical to the following NCBI reference sequences: OG0548 to WP_006342723 (402 aa) and OG0590 to WP_006343129 (579 aa). The protein sequences displayed herein are provided in Supplementary Data Sheet 1.
The incidences of lung infection caused by C. psittaci are usually underestimated due to complicated clinical presentations varying from severe pneumonia to asymptomatic infection (De Gier et al., 2018). Diagnosis of psittacosis can be challenging through routine methods for detecting the infectious agent C. psittaci in the hospital setting (Nieuwenhuizen et al., 2018). Like C. trachomatis, C. psittaci also lacks genetic manipulation systems to resolve the function of genes (Kari et al., 2011). To better depict the molecular features of this difficult-to-culture intracellular pathogen, we reconstructed the draft genomes of C. psittaci, and further characterize the phylogeny of novel strains, pan-genomic structure and adaptive evolution of virulence genes under a metagenomic framework.
The obligate intracellular lifestyle of Chlamydia spp. may restrict genic acquisition via lateral transfer in the bacterial community (Stepkowski and Legocki, 2001). It was worth noting that the genes encoding structural proteins responsible for the biosynthesis of T4SS were absent in the pan-genome of C. psittaci (Supplementary Table S6). Meanwhile, C. psittaci possesses a high proportion (~90%) of core genes per genome, which is similar to that (~92%) of C. trachomatis (McInerney et al., 2017). Based on the metric, Chlamydia spp. have relatively low genomic plasticity in comparison to other intracellular bacteria, like Anaplasma phagocytophilum (~72%) and Coxiella burnetii (~60%) (Dugat et al., 2014; Abou Abdallah et al., 2022). In contrast, the proportion of conserved core genes is lower in the extracellular bacteria, such as E. coli (~47%), Pseudomonas aeruginosa (~41%), and Klebsiella pneumoniae (~32%) (Mosquera-Rendón et al., 2016; Wyres et al., 2020). It implies that natural selection or other forces driving the evolution of Chlamydia spp. may be reflected by small genetic variations in the coding regions rather than frequent gene gain or loss.
Chlamydial virulence factors, particularly surface appendages, play key roles in the interaction with the host immune system and are associated with prevalence and disease pathogenesis (Byrne, 2010). Herein, we focused on 20 C. psittaci VAGs showing a significant signal of positive selection. Half of these genes code for products localized on the cell surface/membrane, which are prone to natural selection pressures and other evolutionary forces (Xu et al., 2011). For instance, OmpA encoded by OG0548 (402 aa) shares 64.9% amino acid sequence identity with the homolog MOMP (393 aa) in C. trachomatis D/UW-3/CX, which acts as a cytoadhesin involved in the binding of chlamydial elementary bodies to heparan sulfate proteoglycans on the host cells (Su et al., 1996). Nine positively selected amino acid residues were detected in C. psittaci OmpA (Figure 5), the genetic changes of which may be associated with the diversity of bacterial serotypes (Knittler and Sachse, 2014). Another positively selected gene OG0373 (448 aa) encodes a putative porin belonging to the OprB family (PF04966) of carbohydrate-selective porins and shares 75% similarity with an integral outer membrane protein AaxA (438 aa, WP_014518085) in C. pneumoniae, which can enhance arginine uptake and decarboxylation activity of bacterial cells (Smith and Graham, 2008).
Besides, the roles of the effector delivery system in chlamydial infection have been extensively studied, such as attachment and invasion into host cells, regulation of intracellular trafficking and innate immune signaling (Mojica et al., 2015; Carpenter et al., 2017; Sixt et al., 2017). In C. psittaci, we detected 13 positively selected genes involved in the effector delivery system, including five genes of T3SS, one gene of T2SS, and seven effector genes (Supplementary Table S7). Of these, both genes OG0549/0498 (440 aa/494 aa) encoding translocators are homologous to the C. pneumoniae CopD (444 aa) and CopD2 (497 aa), of which CopD has been verified as a hydrophobic translocator with functions in bacterial infectivity (Bulir et al., 2014). The product of OG0590 (579 aa), another positively selected outer membrane protein of C. psittaci (Figure 5), shares 44.9% identity with C. trachomatis CT_082 (560 aa) coding for a T3SS secreted effector. Additionally, significant evidence for positive selection on yycJ (262 aa) coding for a metallo-hydrolase was identified (p < 0.001; Supplementary Table S7), which shares 55% similarity with a zinc metallo-beta-lactamase (264 aa) from Bacillus cereus (Carfi et al., 1995). YycJ is a pleiotropic protein with a domain (Lactamase_B, PF00753) catalyzing the hydrolysis of most beta-lactams that may facilitate enzymatic evolution for bacterial survival (Bebrone, 2007). Since genetic intractability for Chlamydia spp., decoding the function of their genes has remained challenging (McClure et al., 2017). Detection of genes subject to positive selection should provide evidence-based candidates for elucidating the molecular mechanisms of adaptation and immune evasion for C. psittaci.
In this study, we performed comparative genomic analysis of 56 C. psittaci isolates and two novel strains directly from the BALF specimens of pneumonia. New sequence types ST335 and ST334 were assigned to the two metagenomic strains that were closely related to the animal-borne isolates belonging to ST43 and ST28. The pan-genome analysis revealed this obligate intracellular pathogen exhibited a low genomic plasticity represented by a highly conserved core-genome. Furthermore, we identified 20 positively selected virulence genes and related amino acid sites showing evidence for adaptive evolution and pathogen-host interactions. Our findings indicate the metagenomic technique will be a new approach to clinical surveillance and research into the epidemiology and evolutionary biology of difficult-to-culture pathogens.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA895174.
The studies involving human participants were reviewed and approved by the Ethics Committee of Shanghai Sixth People’s Hospital. The patients/participants provided their written informed consent to participate in this study.
YL, YF, and RW conceived and designed the study. WH, SH, and XZ performed the experiments. WH, SH, YL, and RW contributed to materials. WH, YF, YL, SL, and YZ performed sequence analysis and data interpretation. WH, SH, YZ, and XZ wrote the manuscript. SL, YL, YF, and RW provided major revisions to the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Shanghai Municipal Science and Technology Major Project (grant no. ZD2021CY001).
XZ and YF were employed by Genoxor Medical Science and Technology Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1157888/full#supplementary-material
Abou Abdallah, R., Million, M., Delerce, J., Anani, H., Diop, A., Caputo, A., et al. (2022). Pangenomic analysis of Coxiella burnetii unveils new traits in genome architecture. Front. Microbiol. 13:1022356. doi: 10.3389/fmicb.2022.1022356
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). Blast ring image generator (Brig): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Arnold, B. J., Huang, I. T., and Hanage, W. P. (2022). Horizontal gene transfer and adaptive evolution in bacteria. Nat. Rev. Microbiol. 20, 206–218. doi: 10.1038/s41579-021-00650-4
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bebrone, C. (2007). Metallo-beta-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharmacol. 74, 1686–1701. doi: 10.1016/j.bcp.2007.05.021
Beeckman, D., and Vanrompay, D. (2009). Zoonotic Chlamydophila psittaci infections from a clinical perspective. Clin. Microbiol. Infect. 15, 11–17. doi: 10.1111/j.1469-0691.2008.02669.x
Branley, J., Bachmann, N. L., Jelocnik, M., Myers, G. S., and Polkinghorne, A. (2016). Australian human and parrot Chlamydia psittaci strains cluster within the highly virulent 6bc clade of this important zoonotic pathogen. Sci. Rep. 6:30019. doi: 10.1038/srep30019
Bulir, D. C., Waltho, D. A., Stone, C. B., Mwawasi, K. A., Nelson, J. C., and Mahony, J. B. (2014). Chlamydia pneumoniae CopD translocator protein plays a critical role in type iii secretion (T3S) and infection. PLoS One 9:e99315. doi: 10.1371/journal.pone.0099315
Byrne, G. I. (2010). Chlamydia trachomatis strains and virulence: rethinking links to infection prevalence and disease severity. J. Infect. Dis. 201, S126–S133. doi: 10.1086/652398
Carfi, A., Pares, S., Duée, E., Galleni, M., Duez, C., Frère, J. M., et al. (1995). The 3-D structure of a zinc metallo-beta-lactamase from Bacillus cereus reveals a new type of protein fold. EMBO J. 14, 4914–4921. doi: 10.1002/j.1460-2075.1995.tb00174.x
Carpenter, V., Chen, Y. S., Dolat, L., and Valdivia, R. H. (2017). The effector TepP mediates recruitment and activation of phosphoinositide 3-kinase on early Chlamydia trachomatis vacuoles. mSphere 2:e00207-17. doi: 10.1128/mSphere.00207-17
Chen, X., Cao, K., Wei, Y., Qian, Y., Liang, J., Dong, D., et al. (2020). Metagenomic next-generation sequencing in the diagnosis of severe pneumonias caused by Chlamydia psittaci. Infection 48, 535–542. doi: 10.1007/s15010-020-01429-0
Chen, J., Sun, L., Liu, X., Yu, Q., Qin, K., Cao, X., et al. (2022). Metagenomic assessment of the pathogenic risk of microorganisms in sputum of postoperative patients with pulmonary infection. Front. Cell. Infect. Microbiol. 12:855839. doi: 10.3389/fcimb.2022.855839
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one Fastq preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cong, W., Huang, S. Y., Zhang, X. Y., Zhou, D. H., Xu, M. J., Zhao, Q., et al. (2013). Seroprevalence of Chlamydia psittaci infection in market-sold adult chickens, ducks and pigeons in North-Western China. J. Med. Microbiol. 62, 1211–1214. doi: 10.1099/jmm.0.059287-0
De Gier, B., Hogerwerf, L., Dijkstra, F., and Van Der Hoek, W. (2018). Disease burden of psittacosis in the Netherlands. Epidemiol. Infect. 146, 303–305. doi: 10.1017/S0950268817003065
Dugat, T., Loux, V., Marthey, S., Moroldo, M., Lagrée, A. C., Boulouis, H. J., et al. (2014). Comparative genomics of first available bovine Anaplasma phagocytophilum genome obtained with targeted sequence capture. BMC Genomics 15:973. doi: 10.1186/1471-2164-15-973
Favaroni, A., and Hegemann, J. H. (2021). Chlamydia trachomatis polymorphic membrane proteins (Pmps) form functional Homomeric and heteromeric oligomers. Front. Microbiol. 12:709724. doi: 10.3389/fmicb.2021.709724
Gu, W., Miller, S., and Chiu, C. Y. (2019). Clinical metagenomic next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 14, 319–338. doi: 10.1146/annurev-pathmechdis-012418-012751
Guerrero-Araya, E., Muñoz, M., Rodríguez, C., and Paredes-Sabja, D. (2021). FastMLST: a multi-core tool for multilocus sequence typing of draft genome assemblies. Bioinform Biol Insights 15:11779322211059238. doi: 10.1177/11779322211059238
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of Phyml 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). Quast: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3:124. doi: 10.12688/wellcomeopenres.14826.1
Kari, L., Goheen, M. M., Randall, L. B., Taylor, L. D., Carlson, J. H., Whitmire, W. M., et al. (2011). Generation of targeted Chlamydia trachomatis null mutants. Proc. Natl. Acad. Sci. 108, 7189–7193. doi: 10.1073/pnas.1102229108
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). Mafft online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kitts, P. A., Church, D. M., Thibaud-Nissen, F., Choi, J., Hem, V., Sapojnikov, V., et al. (2016). Assembly: a resource for assembled genomes at Ncbi. Nucleic Acids Res. 44, D73–D80. doi: 10.1093/nar/gkv1226
Knittler, M. R., and Sachse, K. (2014). Chlamydia psittaci: update on an underestimated zoonotic agent. Pathog Dis 73, 1–15. doi: 10.1093/femspd/ftu007
Lam, M. M. C., Wick, R. R., Watts, S. C., Cerdeira, L. T., Wyres, K. L., and Holt, K. E. (2021). A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 12:4188. doi: 10.1038/s41467-021-24448-3
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lax, S., Sangwan, N., Smith, D., Larsen, P., Handley, K. M., Richardson, M., et al. (2017). Bacterial colonization and succession in a newly opened hospital. Sci. Transl. Med. 9:eaah6500. doi: 10.1126/scitranslmed.aah6500
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2018). Vfdb 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Lu, J., Breitwieser, F. P., Thielen, P., and Salzberg, S. L. (2017). Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci 3:e104. doi: 10.7717/peerj-cs.104
Mcclure, E. E., Chávez, A. S. O., Shaw, D. K., Carlyon, J. A., Ganta, R. R., Noh, S. M., et al. (2017). Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat. Rev. Microbiol. 15, 544–558. doi: 10.1038/nrmicro.2017.59
Mcinerney, J. O., Mcnally, A., and O'connell, M. J. (2017). Why prokaryotes have pangenomes. Nat Microbiol 2:17040. doi: 10.1038/nmicrobiol.2017.40
Mojica, S. A., Hovis, K. M., Frieman, M. B., Tran, B., Hsia, R. C., Ravel, J., et al. (2015). Sinc, a type iii secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol. Biol. Cell 26, 1918–1934. doi: 10.1091/mbc.E14-11-1530
Montgomerie, S., Cruz, J. A., Shrivastava, S., Arndt, D., Berjanskii, M., and Wishart, D. S. (2008). Proteus2: a web server for comprehensive protein structure prediction and structure-based annotation. Nucleic Acids Res. 36, W202–W209. doi: 10.1093/nar/gkn255
Mosquera-Rendón, J., Rada-Bravo, A. M., Cárdenas-Brito, S., Corredor, M., Restrepo-Pineda, E., and Benítez-Páez, A. (2016). Pangenome-wide and molecular evolution analyses of the Pseudomonas aeruginosa species. BMC Genomics 17:45. doi: 10.1186/s12864-016-2364-4
Nieuwenhuizen, A. A., Dijkstra, F., Notermans, D. W., and Van Der Hoek, W. (2018). Laboratory methods for case finding in human psittacosis outbreaks: a systematic review. BMC Infect. Dis. 18:442. doi: 10.1186/s12879-018-3317-0
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Qu, J. M., and Cao, B. (2016). Guidelines for the diagnosis and treatment of adult community acquired pneumonia in China (2016 Edition). Zhonghua Jie He He Hu Xi Za Zhi 39, 241–242. doi: 10.3760/cma.j.issn.1001-0939.2016.04.001
Rasko, D. A., Rosovitz, M. J., Myers, G. S., Mongodin, E. F., Fricke, W. F., Gajer, P., et al. (2008). The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 190, 6881–6893. doi: 10.1128/JB.00619-08
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shiryev, S. A., Papadopoulos, J. S., Schaffer, A. A., and Agarwala, R. (2007). Improved Blast searches using longer words for protein seeding. Bioinformatics 23, 2949–2951. doi: 10.1093/bioinformatics/btm479
Sixt, B. S., Bastidas, R. J., Finethy, R., Baxter, R. M., Carpenter, V. K., Kroemer, G., et al. (2017). The Chlamydia trachomatis inclusion membrane protein CpoS counteracts sting-mediated cellular surveillance and suicide programs. Cell Host Microbe 21, 113–121. doi: 10.1016/j.chom.2016.12.002
Smith, C. B., and Graham, D. E. (2008). Outer and inner membrane proteins compose an arginine-agmatine exchange system in Chlamydophila pneumoniae. J. Bacteriol. 190, 7431–7440. doi: 10.1128/JB.00652-08
Stepkowski, T., and Legocki, A. B. (2001). Reduction of bacterial genome size and expansion resulting from obligate intracellular lifestyle and adaptation to soil habitat. Acta Biochim. Pol. 48, 367–381. doi: 10.18388/abp.2001_3922
Su, H., Raymond, L., Rockey, D. D., Fischer, E., Hackstadt, T., and Caldwell, H. D. (1996). A recombinant Chlamydia trachomatis major outer membrane protein binds to heparan sulfate receptors on epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 93, 11143–11148. doi: 10.1073/pnas.93.20.11143
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Van Lent, S., Piet, J. R., Beeckman, D., Van Der Ende, A., Van Nieuwerburgh, F., Bavoil, P., et al. (2012). Full genome sequences of all nine Chlamydia psittaci genotype reference strains. J. Bacteriol. 194, 6930–6931. doi: 10.1128/JB.01828-12
Vorimore, F., Thebault, A., Poisson, S., Cleva, D., Robineau, J., De Barbeyrac, B., et al. (2015). Chlamydia psittaci in ducks: a hidden health risk for poultry workers. Pathog Dis 73, 1–9. doi: 10.1093/femspd/ftu016
Wolff, B. J., Morrison, S. S., Pesti, D., Ganakammal, S. R., Srinivasamoorthy, G., Changayil, S., et al. (2015). Chlamydia psittaci comparative genomics reveals intraspecies variations in the putative outer membrane and type iii secretion system genes. Microbiology 161, 1378–1391. doi: 10.1099/mic.0.000097
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
Wu, H. H., Feng, L. F., and Fang, S. Y. (2021). Application of metagenomic next-generation sequencing in the diagnosis of severe pneumonia caused by Chlamydia psittaci. BMC Pulm. Med. 21:300. doi: 10.1186/s12890-021-01673-6
Wyres, K. L., Lam, M. M. C., and Holt, K. E. (2020). Population genomics of Klebsiella pneumoniae. Nat. Rev. Microbiol. 18, 344–359. doi: 10.1038/s41579-019-0315-1
Xu, Z., Chen, H., and Zhou, R. (2011). Genome-wide evidence for positive selection and recombination in Actinobacillus pleuropneumoniae. BMC Evol. Biol. 11:203. doi: 10.1186/1471-2148-11-203
Yang, Z. (2007). Paml 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., Wong, W. S. W., and Nielsen, R. (2005). Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Yin, X. W., Mao, Z. D., Zhang, Q., Ou, Q. X., Liu, J., Shao, Y., et al. (2021). Clinical metagenomic sequencing for rapid diagnosis of pneumonia and meningitis caused by Chlamydia psittaci. World J. Clin. Cases 9, 7693–7703. doi: 10.12998/wjcc.v9.i26.7693
Yu, G., Lam, T. T.-Y., Zhu, H., and Guan, Y. (2018). Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol. Biol. Evol. 35, 3041–3043. doi: 10.1093/molbev/msy194
Yu, N. Y., Wagner, J. R., Laird, M. R., Melli, G., Rey, S., Lo, R., et al. (2010). Psortb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. doi: 10.1093/bioinformatics/btq249
Keywords: metagenomics, comparative genomics, Chlamydia psittaci, obligate intracellular pathogen, phylogeny, pangenome, positive selection
Citation: Huang W, Hu S, Zhu Y, Liu S, Zhou X, Fang Y, Lu Y and Wang R (2023) Metagenomic surveillance and comparative genomic analysis of Chlamydia psittaci in patients with pneumonia. Front. Microbiol. 14:1157888. doi: 10.3389/fmicb.2023.1157888
Edited by:
Debmalya Barh, Federal University of Minas Gerais, BrazilReviewed by:
Junping Peng, Institute of Pathogen Biology (CAMS), ChinaCopyright © 2023 Huang, Hu, Zhu, Liu, Zhou, Fang, Lu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Fang, eXVhbi5mYW5nQGdlbm94b3IuY29t; Yihan Lu, bHV5aWhhbkBmdWRhbi5lZHUuY24=; Ruilan Wang, d2FuZ3l1c3VuQGhvdG1haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.