 Jing Li1
Jing Li1 Fengjuan Tian2
Fengjuan Tian2 Sen Zhang1
Sen Zhang1 Shun-Shuai Liu1
Shun-Shuai Liu1 Xiao-Ping Kang1Ya-Dan Li1Jun-Qing Wei2Wei Lin2
Xiao-Ping Kang1Ya-Dan Li1Jun-Qing Wei2Wei Lin2 Zhongyi Lei2Ye Feng1Jia-Fu Jiang1*
Zhongyi Lei2Ye Feng1Jia-Fu Jiang1* Tao Jiang1*
Tao Jiang1* Yigang Tong2*
Yigang Tong2*- 1State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology, AMMS, Beijing, China
- 2Beijing Advanced Innovation Center for Soft Matter Science and Engineering (BAIC-SM), College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, China
Introduction: Coronaviruses (CoVs) are naturally found in bats and can occasionally cause infection and transmission in humans and other mammals. Our study aimed to build a deep learning (DL) method to predict the adaptation of bat CoVs to other mammals.
Methods: The CoV genome was represented with a method of dinucleotide composition representation (DCR) for the two main viral genes, ORF1ab and Spike. DCR features were first analyzed for their distribution among adaptive hosts and then trained with a DL classifier of convolutional neural networks (CNN) to predict the adaptation of bat CoVs.
Results and discussion: The results demonstrated inter-host separation and intra-host clustering of DCR-represented CoVs for six host types: Artiodactyla, Carnivora, Chiroptera, Primates, Rodentia/Lagomorpha, and Suiformes. The DCR-based CNN with five host labels (without Chiroptera) predicted a dominant adaptation of bat CoVs to Artiodactyla hosts, then to Carnivora and Rodentia/Lagomorpha mammals, and later to primates. Moreover, a linear asymptotic adaptation of all CoVs (except Suiformes) from Artiodactyla to Carnivora and Rodentia/Lagomorpha and then to Primates indicates an asymptotic bats-other mammals-human adaptation.
Conclusion: Genomic dinucleotides represented as DCR indicate a host-specific separation, and clustering predicts a linear asymptotic adaptation shift of bat CoVs from other mammals to humans via deep learning.
1. Introduction
RNA viruses from natural reservoir hosts continuously pose a threat to human health, such as coronaviruses (CoVs) from bats (Liu et al., 2021; Wang et al., 2022) and avian influenza viruses from birds (Liu et al., 2014; Sun et al., 2014; Deng et al., 2017). In particular, bat-originated CoVs either caused high-pathogenic but low-transmissible infections of Severe Acute Respiratory Syndrome (SARS) or Middle East Respiratory Syndrome (MERS) or launched a widespread pandemic of low-pathogenic human CoVs, such as HcoV-NL63, HcoV-229E, HcoV-OC43, and HKU1 (Su et al., 2016; Forni et al., 2017). The ongoing global spread of SARS-CoV-2 has not only caused huge damage to public health (WHO, 2022) but also radically changed social habits and lifestyles (West et al., 2020; El-Sayed and Kamel, 2021). Orthocoronavirinae, known as CoV, is one of the two subfamilies in Coronavirinae and consists of four genera of alpha-, beta-, gamma-, and delta-coronaviruses that infect mammalian or avian hosts, especially those specific to species (Woo et al., 2012). The two former CoV members only infect mammals, and the two latter CoVs dominantly infect birds, with some exceptions for mammalian infection (Woo et al., 2012; Ji et al., 2022). According to current CoV databases, almost all human CoVs, with HcoV-OC43 and HKU1 as the exceptional origins of rodents (Forni et al., 2017), have been indicated to have originated in bats (Cui et al., 2019; Ruiz-Aravena et al., 2022). SARS-CoV-2 likely originated from bats as well (Zhou et al., 2020). Additionally, approximately half of the 20 Alphacoronavirus or Betacoronavirus species were identified only in bats (Cui et al., 2019). Taken together, bats are most likely natural reservoirs of CoVs.
Bats are the second largest order of mammals after rodents, widely inhabiting all continents except Antarctica (Gentles et al., 2020), accounting for approximately one-third of CoV sequences before Coronavirus Disease 2019 (COVID-19) (Ruiz-Aravena et al., 2022). The natural reservoir role of bats for CoVs is attributed to host/virus co-existence in an equilibrium pattern, which is also interpreted as the virus adapting to the host, enabling effective bat infection and inter-bat transmission but with limited pathogenicity (Li et al., 2020, 2022) due to several factors. First, bats exhibit extraordinary immune tolerance, which maintains a moderate immune response to invading viruses such as CoVs, leading to limited viral replication and asymptomatic or mild CoV infections (Baker et al., 2013; Olival et al., 2017; Banerjee et al., 2018; Skirmuntt et al., 2020; Sia et al., 2022). Second, bats have a high body temperature, which resembles other mammals' febrile responses and infection immune responses, helping to keep virus infections at a tolerable level (O'Shea et al., 2014). Third, factors such as the large and closely aggregated population, sustained flight capability, and extreme roosting proximity of bats support the widespread and sustained existence of CoVs within the bat population (Maganga et al., 2014; Olival et al., 2017; Roes, 2020). Thus, the sustained infection and transmission in bats provide CoVs with a high probability of accumulating mutations, leading to variants with marginal adaptation to other mammalian hosts and causing spillover infections in humans and other mammals.
Numerous bat CoVs have been isolated and sequenced in recent years. A total of 78% (2,209/2,820) of the recorded CoV sequences in NCBI were uploaded since 2015, before the COVID-19 pandemic (https://www.ncbi.nlm.nih.gov/nuccore). However, it is challenging to assess the risk of new bat CoV isolates that cause infection or pandemics in human or other mammalian populations (Seyran et al., 2021). Traditional phylogenetic analysis can sufficiently evaluate the cross-species infection risk or any bat CoV (Lima et al., 2013; Seyran et al., 2021). More recently, machine learning or deep learning approaches based on big sequencing data have led to remarkable predictions of the host adaptation (Li et al., 2022; Nan et al., 2022), evolution (Hie et al., 2021), transmissibility (Fischhoff et al., 2021), virus–host interaction (Dey et al., 2020), and pathogenicity (Gussow et al., 2020) of SARS-CoV-2 and other viruses (Li et al., 2020). Host-specific compositional features in the virus genome have been indicated by the representation traits, such as dinucleotides (DNTs) (Li et al., 2020), DNT composition representation (DCR) (Li et al., 2022), and Uniform Manifold Approximation and Projection (UMAP) (Hie et al., 2021). Unfortunately, there is no pipeline or framework available to predict the adaptation of recorded or newly isolated bat CoVs to main mammalian hosts, such as Primates, Rodents, Artiodactyla, Suiformes, or Carnivora.
The present study aimed to represent the genome composition of the two main genes, i.e., Spike, the receptor binding glycoprotein, and ORF1ab, the RNA-dependent RNA polymerase complex, and then to predict the adaptive host of recorded or newly isolated CoVs. In this study, the viral genome representation and adaptive host prediction framework provide an intelligent approach to assessing the risk of cross-species infection and transmission for bat CoVs.
2. Methods
2.1. Data preprocessing and genomic compositional trait parsing of ssRNA viruses
Full genome sequences of coronaviruses were downloaded from the NCBI nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore) and cleaned by removing records with multiple imprecise nucleotides or filtering with sequence length thresholds of 27,000 and 32,000 bp. Six types of adaptive host labels, including Chiroptera (CHI), Artiodactyla (ART), Suiformes (SUI), Rodents/Lagomorphs (Rodents, ROD), Carnivora (CAR), and Primates (PRI), were extracted from the “FEATURES”-“source”-“host” of each sequence record in the genebank sequence files and were manually checked one by one according to the host family (genus for porcine CoVs). The coding sequences of the two main CoV genes, ORF1ab and Spike, were parsed with the Biopython Python package. Genomic nucleotide composition traits of mononucleotide (NT), dinucleotide (DNT), DNT composition representation (DCR), trinucleotide (codon), codon pair, and amino acid (AA) were counted as a frequency value for each ORF1ab or spike sequence sample with a nucleotide counting script. The traits of NT, DNT, and DCR were counted as codon nucleotide-dependent sequences (Li et al., 2022). In sum, 12, 48, 1,536, 64, 3,721, and 20 features of the aforementioned six types of compositional traits were counted and utilized for genome composition analysis.
2.2. Clustering in genomic composition traits of coronaviruses
To visualize data distribution and clustering, dimension reduction was performed using Principal Component Analysis (PCA) and t-Distributed Stochastic Neighbor Embedding (t-SNE) for the full-dimension features of 12 NTs, 48 DNTs, 1,536 DCRs, 64 codons, 3,721 codon pairs, or 20 Aas for ORF1ab or Spike. PCA and t-SNE were performed using sklearn.decomposition.PCA (Jolliffe and Cadima, 2016) and sklearn.manifold.TSNE (https://scikit-learn.org/stable/about.html#citing-scikit-learn), respectively. Two main components (PCA1 and PCA2, or t-SNE1 and t-SNE2) were plotted with a host label for each data point using the Python Seaborn package. An unsupervised machine learning approach based on hierarchical clustering was used to observe the clustering and homology of CoVs with various adaptation host labels based on full-dimension features of each compositional trait. Euclidean distance was utilized as a hierarchical clustering scalar, and hierarchical clustering was performed using the sns.clustermap package. Additionally, to balance the biased sample number of CoVs with the six host labels, random down- and up-sampling were performed using the imblearn.over_sampling.SMOTE package before dimension reduction and visualization.
2.3. Building and training of deep learning predictors for adaptive hosts
To predict the adaptation of bat CoVs to other mammalian hosts, a deep learning predictor of convolutional neural networks (CNN) (Li et al., 2022) was built based on the 1,536 DCR features and five host labels (ART, SUI, ROD, CAR, and PRI). Five adaptive hosts were labeled as {0: 'SUI,' 1:'ART', 2:'ROD', 3:'CAR',4: 'PRI'}, respectively. Two packages, pandas.DataFrame.sample and imblearn.over_sampling.SMOTE, were utilized to perform down- and up-sampling to maintain the sample number balance of various host-originated CoVs. Two CNN models were built, one for ORF1ab and another for Spike. sklearn.model_selection import train_test_split was utilized for random training/test data splitting with a test data size of 25%. All bat CoV samples, either for ORF1ab or Spike, were not included in either the training or test data sets to avoid data leaks and were only utilized for the adaptive host prediction with trained models. The 1,536 DCR features of ORF1ab or Spike sequences were reshaped into an array of (6, 16, 16) for a 3D-CNN model of three convolutional layers. Out-channels of (8, 16, 32), a stride of (1, 1, 1), a padding of (0,1,1), and a kernel_size of (1, 3, 3) were set for the three layers of CNN. ReLU activation and average pooling were followed for each CNN layer. Two linear transformations were performed into the 192- and 5-dimensions, respectively, from the 768- and 192-dimensions of a fully connected layer. The sigmoid activation function was utilized for the 192-dimensions of the full-connected layer after one time of linear transformation to output prediction, and the Softmax function was utilized to output the prediction probability. A learning rate of 0.001 and a training epoch of 50 were set uniformly for the ORF1ab or Spike 3D-CNN model.
2.4. Evaluating the deep learning predictor
To evaluate the predictor's performance, the prediction of adaptive hosts and the adaptation probability for each host label were the outputs for each model. The confusion matrix (Townsend, 1971) and micro-average receiver operating characteristic (ROC) (Fawcett, 2005) with AUCs were plotted. A pair plot of the PCA-reduced, fully connected layer data (768-dimension) was performed with the two components to visualize the separation or clustering of the CoVs from different or the same host(s). The PCA1 value was also plotted and compared between/among these CoVs. Statistical significance in the PCA1 value of the PCA-reduced fully connected data was analyzed using an unpaired, non-parametric Mann–Whitney test, based on the hypothesis of non-Gaussian data distribution using GraphPad Prism 9.
2.5. Predict adaptive hosts for bat coronaviruses via the deep learning predictor
To evaluate the adaptive host(s) of bat CoVs, each of the bat CoV samples was predicted using the trained ORF1ab or Spike 3D-CNN model based on 1,536 ORF1ab or Spike DCR data. The adaptation and adaptation probability were output for each of the five hosts (ART, SUI, ROD, CAR, and PRI). The probability vector (five probability values) of all bat CoVs and the CoVs from other mammalian hosts were reduced to two main values by PCA and plotted, with each data point labeled with its host type or virus name.
3. Results
3.1. The architecture of genomic parsing and adaptation for predicting bat CoVs
To represent viral genome composition, full-length CoV sequences were selected and labeled with each of the six adaptive hosts (ART, CHI, CAR, ROD, PRI, and SUI). Complete open reading frames (ORFs) of Spike and ORF1ab were parsed for adaptation analysis. Six types of codon-dependent compositional traits of mononucleotides (NTs, NNTdimension = 12), amino acids (AAs, NAAdimension = 20), DNTs (NDNTdimension = 48), codons (Ncodondimension = 64), DCR (NDCRdimension = 1,536), and codon pairs (codonpairs, Ncodonpairdimension = 3,721) were embedded for Spike and ORF1ab, respectively, with previously reported approaches (Li et al., 2022) (Figure 1A). Unsupervised machine learning methods such as t-SNE, PCA, and hierarchical clustering were performed to visualize the separation and clustering of CoVs based on their abovementioned traits (Figure 1B). A DCR-based CNN (Li et al., 2022) was utilized to classify the CoVs based on each of the five adaptive host labels (Figure 1C). Finally, the adaptive host was predicted for bat CoVs, which were recorded in the database (Figure 1D) or were newly isolated and sequenced CoV strains (Figure 1E).
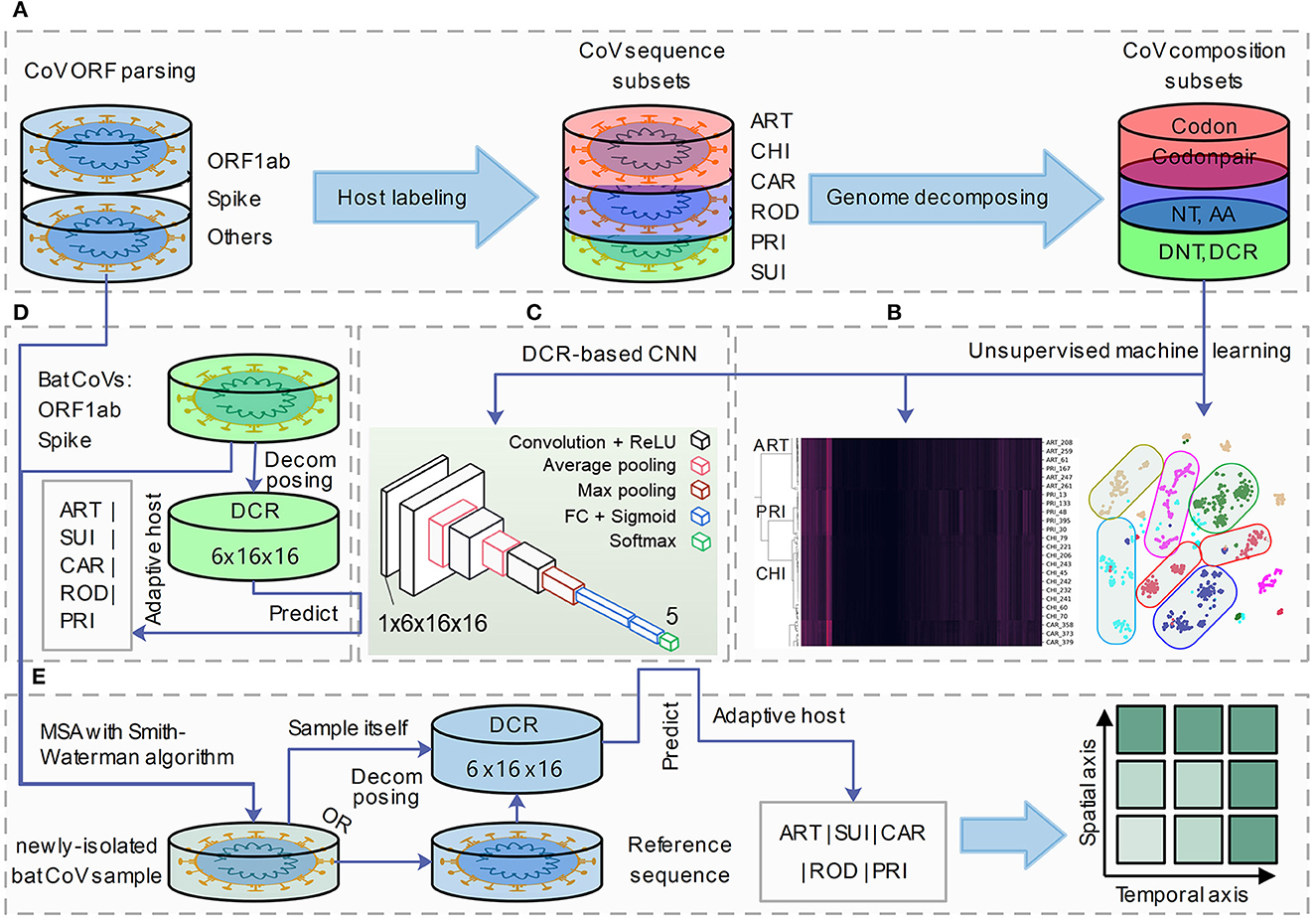
Figure 1. A pipeline for genome decomposition and adaptive host prediction of coronaviruses. The study was designed as the following pipeline of four sections. (A) Coronavirus (CoV) open reading frame (ORF) parsing (ORF1ab and Spike), adaptive host labeling (ART, Artiodactyla; CHI, Chiroptera; CAR, Carnivora; ROD, Rodents and Lagomorphs; PRI, Primates; SUI, Suiformes) and genome decomposition (into six types of NT, nucleotide; AA, amino acid; DNT, dinucleotide, codon; DCR, DNT compositional representation and codon pair); (B, C) unsupervised machine learning analysis (t-distributed stochastic neighbor embedding: t-SNE, Principal Component Analysis: PCA and hierarchical clustering) (B) and supervised deep learning (DCR-based Convolutional Neural Networks, CNN) prediction building of CoV genes; (D) field coronavirus sample collection, sequencing and Multiple Sequence Aligning (MSA); (E) host adaptation prediction of novel isolated CoV strains based on their decomposed DCR or based on their reference CoV sequences.
3.2. Representation and visualization of DCR and other compositional traits for CoVs
Dimension reduction was performed using t-SNE or PCA into two main components for each trait type of the CoVs. We then used Synthetic Minority Over-sampling Technique (SMOTE) to correct the data imbalance among host labels by up- and down-sampling. Given the high importance of SARS-CoV-2-related pangolin CoVs, we also added pangolin CoV data for unsupervised learning analysis. In the ORF1ab DNT trait, we observed a clear separation among CoVs with the five host labels in the two reduced t-SNE components (upper part, Figure 2A) and a much more diffuse distribution in the two reduced PCA components (lower part, Figure 2A). The intra-host clustering and the inter-host separation were also indicated using the hierarchical clustering of ORF1ab DNTs (Figure 2B). Similar clustering and separation of ORF1ab DCR were also observed post-t-SNE/PCA reduction and using hierarchical clustering (Figures 2C, D). The Spike in DNT and DCR also indicated intra-host clustering and inter-host separation in both DNT and DCR traits using the three types of unsupervised machine learning methods (Figures 2E–H). Interestingly, the pangolin CoVs were closely clustered with PRI CoVs, either for the reduced DNT or DCR features of ORF1ab (Figures 2A–D) of Spike. Moreover, the compositional traits of AAs and NTs for ORF1ab (Supplementary Figures S1a–f) and Spike (Supplementary Figures S1g–l) and the compositional traits of codons and codonpairs for ORF1ab (Supplementary Figures S2a–f) and Spike (Supplementary Figures S2g–l) were also observed. Additionally, some obvious disseminated distribution for ROD or CAR samples was mainly enlarged for abnormally disseminated samples using SMOTE sampling; the wide distribution of CHI samples had no association with data sampling and probably implied the wide host adaptation of CHI CoVs. Taking these results together, there was a host specificity in DCR and other compositional traits for the ORF1ab and Spike of CoVs.
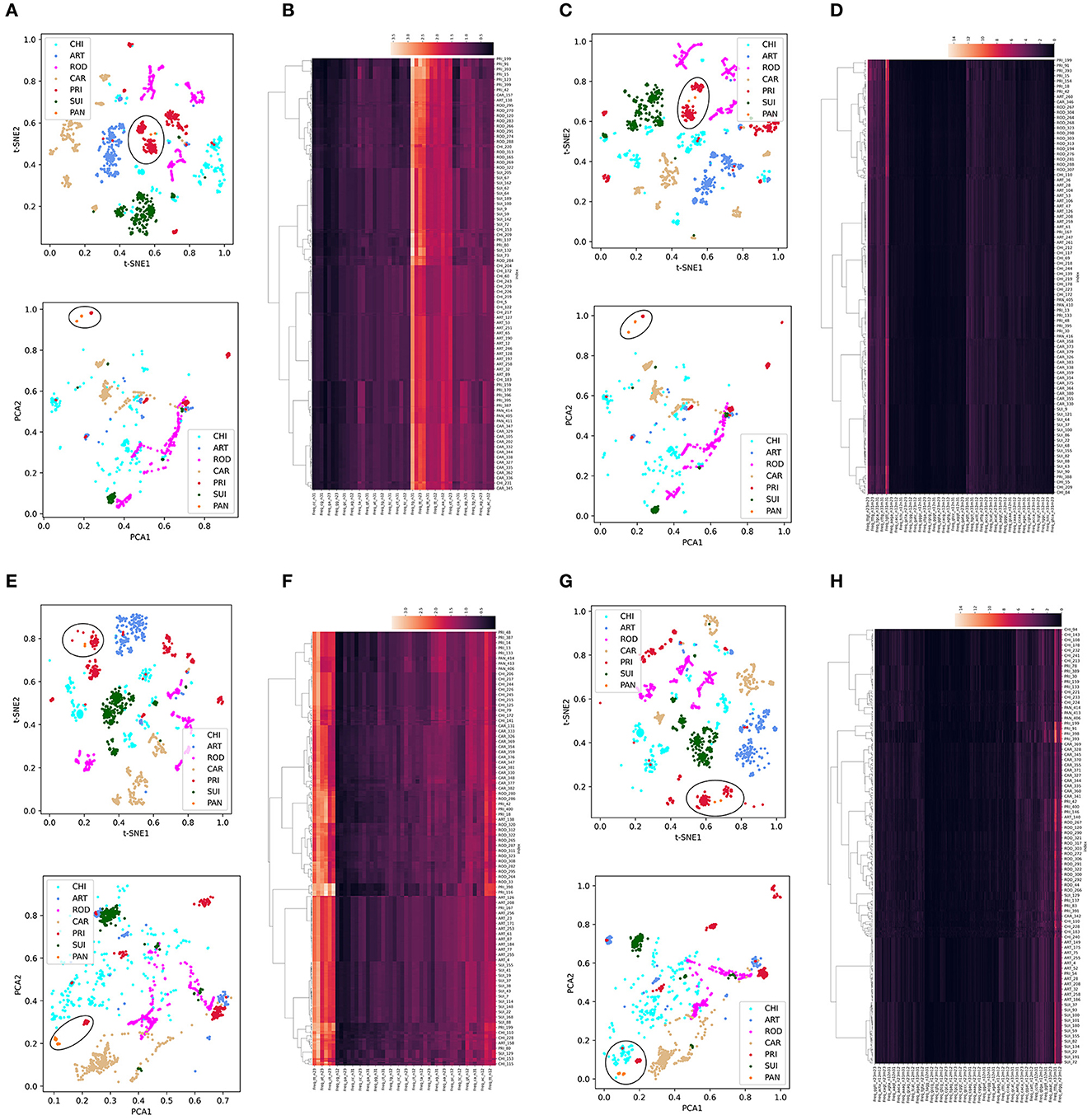
Figure 2. Distribution and clustering analysis of CoVs with host labels using unsupervised machine learning methods based on DNTs and DCR. Distribution of the two main components of compositional DNTs with t-SNE [(A) up] or PCA [(A) down] reduction and hierarchical clustering of 48 compositional DNTs of CoV ORF1ab (B); (C, D) distribution of the two main t-SNE [(C) up] and PCA [(C) down] DCR components of ORF1ab and hierarchical clustering of all 1,536 DCR features (D); (E–H) Similar unsupervised machine learning analysis of DNTs (E, F) and DCR features (G, H) of Spike.
Additionally, the other three genes, E, M, and N, were analyzed for the abovementioned six types of compositional traits. The severe mixture was observed for each type of trait in the two-dimensional space of t-SNE1 and t-SNE2 or of PCA1 and PCA2 (in order of amino acid, NTS, DNTS, DCR, codons, and codonpairs, respectively, for a-f, Supplementary Figures S3–S5).
3.3. Performance of the DCR-based CNN model to predict adaptive hosts CoVs
A deep learning model of CNN was built to predict the adaptation of bat CoVs to various types of mammalian hosts. The classification model with five labels (ART, CAR, ROD, PRI, and SUI) was trained using the 1,536-dimension DCR features of either ORF1ab or Spike. A training epoch-dependent performance elevation was observed for the classification of valid data based on DCR features of ORF1ab according to the confusion matrix (for epochs 10, 30, and 50, respectively, in Figures 3A–C; or for epochs 10–50, respectively, in Supplementary Figures S6a–e) or area under the receiver operating characteristic curve (ROC_AUC) (Figures 3D–F; Supplementary Figures S6f–j). Another model based on spike DCR features was also trained for the classification of CoV adaptive hosts. A high prediction accuracy was also obtained post-50-epoch training, as indicated by the confusion matrix (higher than 97% for epoch 50, Figures 3G–I; Supplementary Figures S6k–o) or ROC_AUC (Figures 3J–L; Supplementary Figures S6p–t). The training loss for either classifier descended quickly within the first 10 epochs and reached a plateau at approximately 20 epochs (respectively for ORF1ab and Spike Figures 3M, N; Supplementary Figures S6p–t).
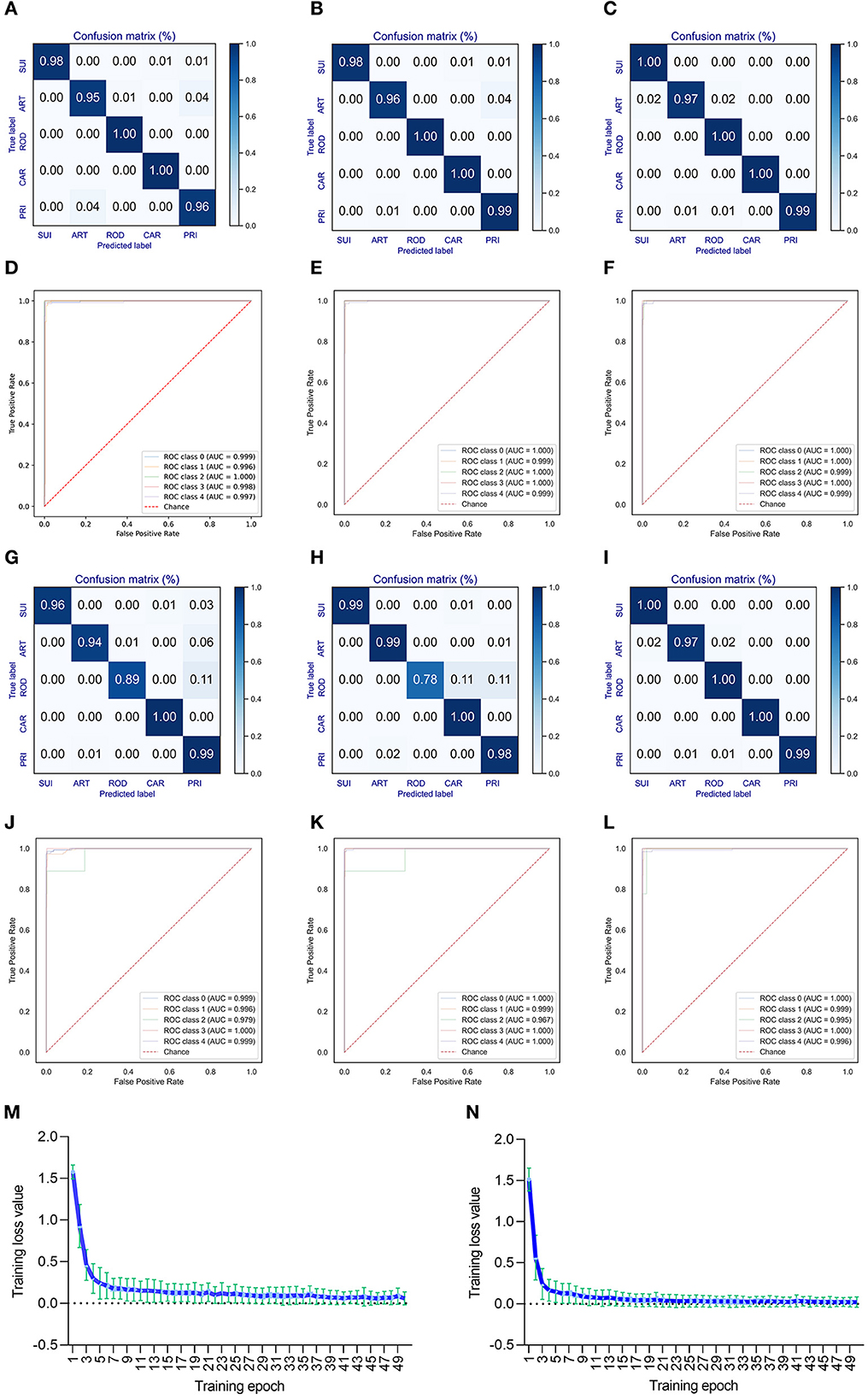
Figure 3. Performance of the DCR-based deep learning approach for predicting adaptive hosts of bat coronaviruses. The performance of the DCR-based deep learning predictor was evaluated with a confusion matrix [(A–C) for training epochs of 10, 30, and 50, respectively] and receiver operating characteristic ROC curve () [(D–F) for training epochs of 10, 30, and 50, respectively] for CoV ORF1ab; a similar evaluation was performed with a confusion matrix [(G–I), respectively] and ROC [(J–L), respectively] for CoV Spike. (M, N) Curve of the average training loss for validated data for the predictors for ORF1ab (M) and Spike (N). ART, Artiodactyla; SUI, Suiformes; ROD, Rodents and Lagomorphs; CAR, Carnivora; PRI, Primates.
To interpret the two trained classifiers, the reduction of the model's full-connected layer with PCA was visualized by plotting each pair of PCA1/PCA2 and PCA2/PCA1. The plotting results demonstrated that there was a sequential distribution of SUI, ART ROD, CAR, and PRI for the ORF1ab samples with five host labels for epochs 10, 30, and 50 (Figures 4A–C) or for 10–50 epochs (Supplementary Figures S7a–e). A significant separation of PRI (from other mammalian hosts) CoV samples was also observed from the distribution of the trained full-connected layer of spike DCR for epochs 10, 30, and 50 (Figures 4D–F) or for 10–50 epochs (Supplementary Figures S7f, g), with a different sequence of CAR, ART, SUI, and ROD for the other four host labels. The statistical analysis of the PCA1 values for each group indicated a significant difference between each neighboring pair of hosts in the ORF1ab samples (P < 0.01, except for ART vs. ROD with P > 0.5, Figure 4G). The difference was also significant for the neighboring ART/SUI or ROD/PRI ORF1ab samples (P < 0.01, Figure 4H).
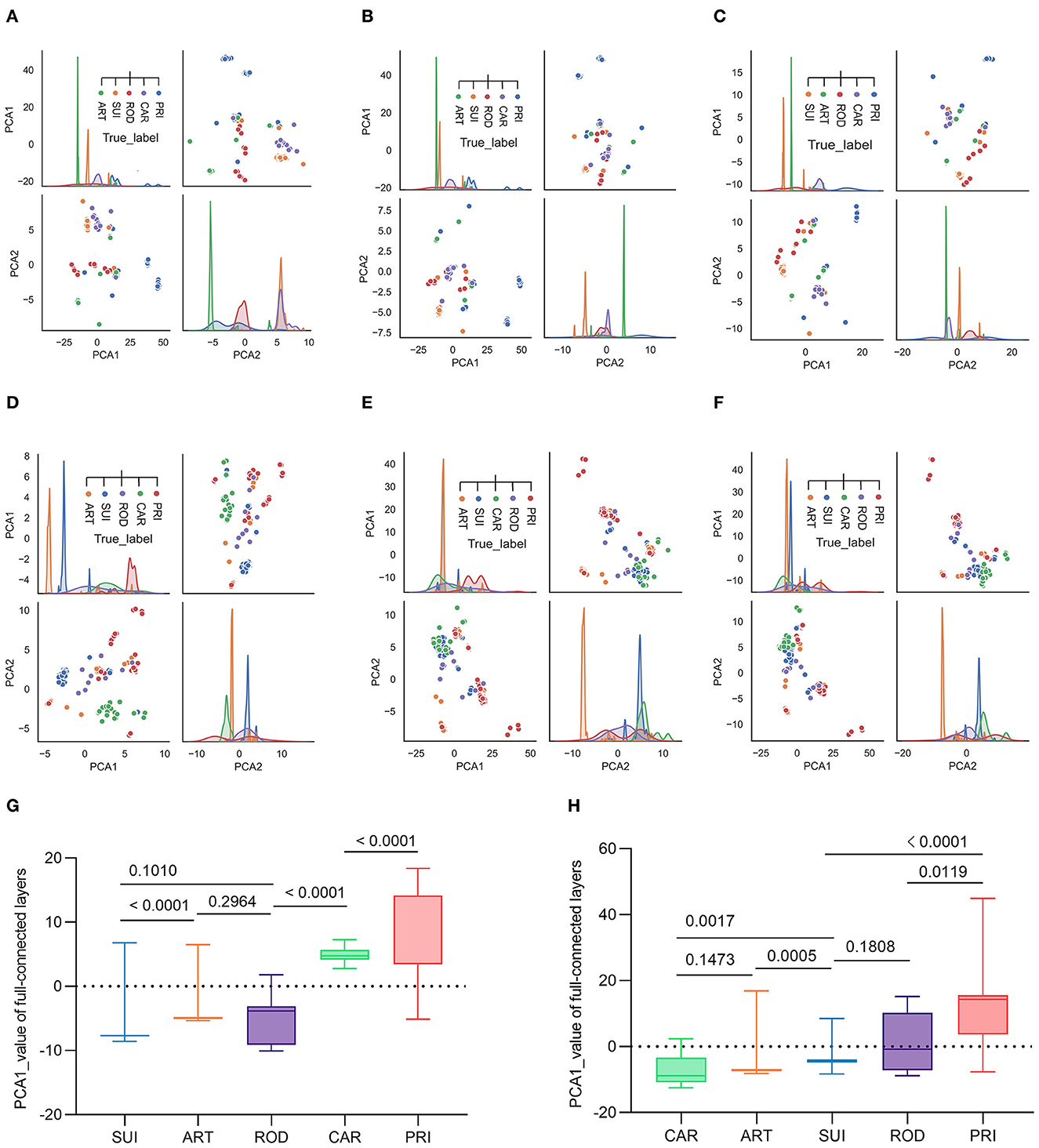
Figure 4. Visualization of the complete connection layer post-deep learning training of CoV ORF1ab and Spike. Full connection layers after three rounds of convolution were reduced by PCA into two principal components and plotted to visualize the distribution of CoV samples with the five host labels. Pair plots of reduced PCA1 and PCA2, post-training epochs of 10, 30, and 50, were plotted for ORF1ab (A–C) or Spike (D–F), respectively. (G, H) Statistics of the reduced PCA1 value post-training epoch of 50 [(G) for ORF1ab and (H) for Spike].
3.4. DCR-based CNN predicts asymptotic bat-to-human adaptation of bat CoVs
To assess the adaptation of bat CoVs to other mammalian hosts, bat CoV sequences were fed to the two trained classifiers for ORF1ab and Spike. The results showed that 53% of CoV ORF1ab sequences were predicted as ART adaptive, while the percentages of adaptive samples for SUI, PRI, CAR, and ROD were 26, 11, 5, and 4%, respectively (Figure 5A). The average standardized probability of the predicted five groups of ART, PRI, SUI, CAR, and ROD were 0.640, 0.477, 0.276, 0.085, and 0.042, respectively (Figure 5B). The second classifier predicted almost the same percentage of Spike-adapted CV for ART hosts (54%). The percentages of adaptive samples for the other four types of hosts were 7, 12, 22, and 5%, respectively (Figure 5C), with an average standardized probability of 0.623, 0.451, 0.081, 0.456, and 0.048 for the five groups (Figure 5D). To further assess the distribution of bat CoVs and other mammalian CoVs in the adaptation space of mammalian hosts, five probability values for the five hosts were taken as a vector for each sample and were reduced to two main components with PCA. Interestingly, except for CoVs with an SUI host label, other mammalian but bat ORF1ab samples were almost linearly distributed, with ART samples on the lower left, CAR, and ROD samples in the middle, and PRI samples mainly on the upper right (Figure 5E), indicating a linear asymptotic adaptation shift from ART to CAR/ROD and then to PRI. Particularly, there was a linear-like distribution of all human CoV or human CoV-related ORF1ab samples in the two-dimensional space. MERS/bat MERS-related CoVs, SARS/bat SARS-like CoVs, and human CoVs of OC43, 229E, and others were successively distributed from the lower left to the upper right (Figure 5E). Similar linear asymptotic adaptation shifts of CoV spike samples were also observed (Figure 5F). Additionally, bat CoVs were disseminated in the adaptation space, with varied distances in PCA1 or PCA2 values for each of the five groups of CoVs (Figures 5E, F). Taken together, the two adaptation classifiers predicted a unanimous linear asymptotic adaptation shift from the ART host to humans.
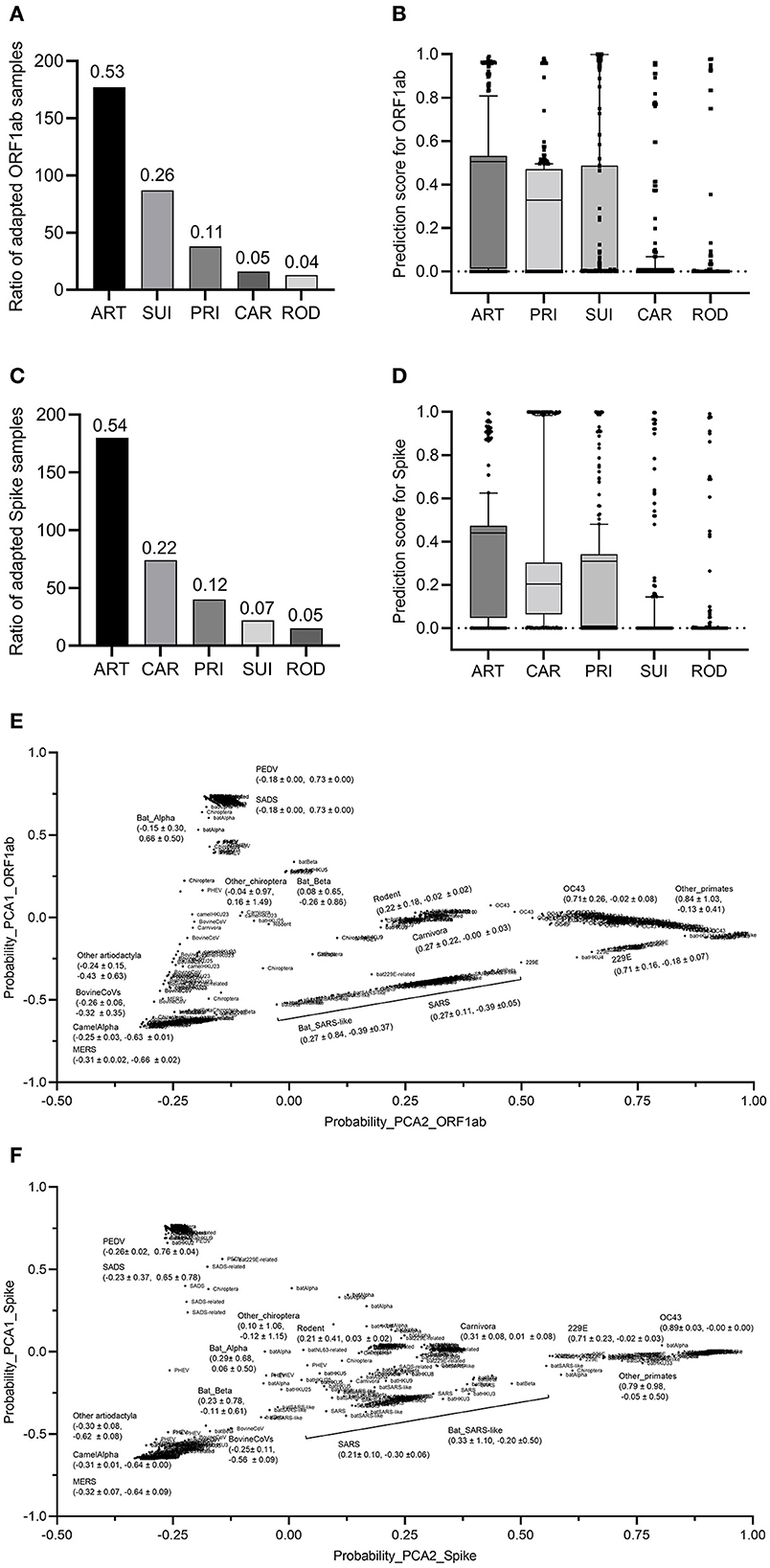
Figure 5. Prediction and prediction probabilities of bats and other coronaviruses. (A, B): Prediction (A) and prediction probability (B) of ORF1ab for the adaptation to the five mammalian hosts of bat coronaviruses; (C, D) prediction (C) and prediction probability (D) of Spike for the adaptation to the five mammalian hosts of bat coronaviruses; (E, F) visualization of the PCA-reduced prediction probability of ORF1ab (E) and Spike (F) for bat and other coronaviruses.
4. Discussion
The present study aimed to predict the potential for viruses, such as influenza A viruses and coronaviruses, to cause infection and transmission in the human population.
Thus, we defined it as “the capability to infect humans easily, to transmit among populations efficiently, and to be virulent to some degree to humans” previously (Li et al., 2020, 2022). Genomic traits for virus adaptation have been biologically interpreted as shaping viral mRNA decay (Contu et al., 2021), methylation (Upadhyay et al., 2013), translation (Chen et al., 2020), replication efficiency (Forsberg, 2003; Bahir et al., 2009; Li et al., 2011), and antagonizing host anti-virus immune response (Xia, 2020), all of which reflect viral adaptive phenotypic traits to their hosts. Moreover, such adaptive genotypes were distinguishable and predictable with machine learning or deep learning approaches. Adaptation phenotypes of viruses to bats and other mammals are supported by parallel viral genotypes. A coarse-grained representation of the viral genome as compositional traits, such as DNT and DCR, is host-specific and predictable with machine learning or deep learning approaches for CoVs (Pollock et al., 2020; Li et al., 2022; Nan et al., 2022), influenza viruses (Taubenberger and Kash, 2010; Li et al., 2020), and other viruses (Bahir et al., 2009; Babayan et al., 2018; Chen et al., 2020). Fine-tuned sequential representation has been indicated to be sensitive to predicting the adaptation of SARS-CoV-2 Omicron sublineages with deep learning (Nan et al., 2022). In the present study, representative compositional traits of DCR and others confirmed the intra-host clustering and inter-host separability of various host-specific CoVs. Interestingly, there was a disseminated distribution of bat (CHI) CoVs into the areas of the CoVs with other host labels, indicating multiple adaptations to other hosts of bat CoVs. Additionally, the dispersed distribution of ROD samples was mainly caused by SMOTE up-sampling. Pangolin has been shown to play an intermediate role in the cross-species infection of SARS-CoV-2 viruses (Lam et al., 2020; Xiao et al., 2020) or MERS-CoV (Chen et al., 2020). The compositional traits indicated a close clustering of these pangolin CoVs with human CoVs, either for ORF1ab or Spike genes, implying a human adaptation. However, we did not set pangolin as an independent host label for supervised learning due to the small sample size of the whole genome and also due to the too-close clustering of pangolin viruses to human CoVs. Multiple genes other than ORF1ab and Spike might mediate the adaptation of CoVs to human and other mammalian hosts. However, the three other genes, E, M, and N, were mixed for CoVs of various host labels, suggesting less host specificity.
In the present study, the deep learning classifier with five host labels (ART, CAR, ROD, SUI, and PRI) targeting either the ORF1ab or Spike gene, accurately predicted the host of the five groups of CoVs. A complete landscape of mammalian CoV samples in the predicted adaptation space constructed by the adaptation probability for the five hosts (Figure 5) unanimously showed a clearer intra-host clustering and inter-host separability of all CoV samples than the distribution of the original DCR features. Interestingly, a linear-like distribution of the CoV samples, except for the SUI CoVs, was observed in the adaptation space, suggesting CoV's asymptotic adaptation from ART to CAR/ROD and then to PRI hosts. Taking these results together, we proposed a possible niche distance-related landscape of host adaptation for bat CoVs (Figure 6): a dominant adaptation to the ART hosts, followed by a relatively less adaptation to CAR/ROD hosts, and finally to PRI hosts. Such asymptotic adaptation to the bat's close and far niche distances (Corman et al., 2018) reconfirmed the mediation of these natural hosts in the adaptation shift of bat CoVs to human beings. The ranked adaptation for bat CoVs provides more clues that CoVs might shift more probably from ART to a CAR/ROD host and then to humans than directly from CHI hosts, considering the closer niche distance between humans and these mediator hosts.
Figure 6. Summary diagram of adaptive hosts predicted by DCR-based deep learning. Adaption of Chiroptera CoVs to sequential ecological niches of ART/SUI, ROD/CAR, and then PRI are shown. ART, Artiodactyla; CAR, Carnivora; CHI, Chiroptera; PRI, Primates; ROD, Rodents and Lagomorphs; SUI, Suiformes.
Additionally, the domestic pig in the SUI host type is the key mediator for the adaptation shift to a human host for the other major respiratory infectious agent, influenza A viruses (Neumann et al., 2009); however, the results in the present study indicated a significantly independent distribution of SUI CoVs from the linear-like and asymptotic distribution of the CoVs from other mammals. CoVs have been reported to cause infection and transmission in domestic pigs worldwide, such as porcine transmissible gastroenteritis virus (TGEV) (Brian and Baric, 2005), porcine enteric diarrhea virus (PEDV) (Lin et al., 2016), and swine acute diarrhea syndrome (SADS) CoV (Zhou et al., 2018). SUI CoVs are not likely to cause transmission in the human population, although the porcine delta coronavirus has been reported to infect malnourished Haitian children (Lednicky et al., 2021). SUI CoVs did not cause cross-species transmission in humans, as they were not closely related to human CoVs in the adaptation space predicted in this study. Therefore, we speculate that the risk of SUI CoVs threatening human populations is lower. However, it is important to note that overfitting can occur in machine learning or deep learning models to varying degrees. Additionally, there is a significant bias, with a smaller number of ROD CoVs and a much larger number of SUI or ART CoVs. The use of up-sampling for ROD CoVs and down-sampling for SUI and ART CoVs may lead to overfitting of the model and potentially explain the wide range of predicted adaptation probabilities (Figure 4).
In summary, the genomic dinucleotides represented as DCR indicate a host-specific separation and clustering that can predict a linear and asymptotic adaptation shift of bat CoVs from other mammals to humans through deep learning techniques.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
JL, J-FJ, TJ, and YT conceived the study. JL, FT, and SZ contributed to the acquisition and interpretation of data. S-SL, X-PK, J-QW, and WL performed data cleaning and statistical analysis. JL, SZ, ZL, Y-DL, and YF performed genome parsing and unsupervised and supervised learning with the assistance of J-FJ, TJ, and YT. JL drafted the manuscript, coded all scripts for genome parsing, deep learning, and data visualization. All authors contributed to the critical revision of the manuscript for important intellectual content.
Funding
This study was supported by grants from the National Key Research and Development Program of China (Grant Nos. 2021YFC2302004, 2021YFC0863400, 2019YFC1200501, and 2018YFA0903000) and the National Natural Science Foundation of China (Grant No. 32070166).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1157608/full#supplementary-material
References
Babayan, S. A., Orton, R. J., and Streicker, D. G. (2018). Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science 362, 577–580. doi: 10.1126/science.aap9072
Bahir, I., Fromer, M., Prat, Y., and Linial, M. (2009). Viral adaptation to host: a proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 5, 311. doi: 10.1038/msb.2009.71
Baker, M. L., Schountz, T., and Wang, L. F. (2013). Antiviral immune responses of bats: a review. Zoonoses Public Health 60, 104–116. doi: 10.1111/j.1863-2378.2012.01528.x
Banerjee, A., Misra, V., Schountz, T., and Baker, M. L. (2018). Tools to study pathogen-host interactions in bats. Virus Res. 248, 5–12. doi: 10.1016/j.virusres.2018.02.013
Brian, D. A., and Baric, R. S. (2005). Coronavirus genome structure and replication. Curr. Top Microbiol. Immunol. 287, 1–30. doi: 10.1007/3-540-26765-4_1
Chen, F., Wu, P., Deng, S., Zhang, H., Hou, Y., Hu, Z., et al. (2020). Dissimilation of synonymous codon usage bias in virus-host coevolution due to translational selection. Nat. Ecol. Evol. 4, 589–600. doi: 10.1038/s41559-020-1124-7
Contu, L., Balistreri, G., Domanski, M., Uldry, A., and Mühlemann, O. (2021). Characterisation of the Semliki Forest Virus-host cell interactome reveals the viral capsid protein as an inhibitor of nonsense-mediated mRNA decay. PLoS Pathog. 17, e1009603. doi: 10.1371/journal.ppat.1009603
Corman, V. M., Muth, D., Niemeyer, D., and Drosten, C. (2018). Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 100, 163–188. doi: 10.1016/bs.aivir.2018.01.001
Cui, J., Li, F., and Shi, Z. (2019). Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192. doi: 10.1038/s41579-018-0118-9
Deng, Y., Li, C., Han, J., Wen, Y., Wang, J., Hong, W., et al. (2017). Phylogenetic and genetic characterization of a 2017 clinical isolate of H7N9 virus in Guangzhou, China during the fifth epidemic wave. Sci. China Life Sci. 60, 1331–1339. doi: 10.1007/s11427-017-9152-1
Dey, L., Chakraborty, S., and Mukhopadhyay, A. (2020). Machine learning techniques for sequence-based prediction of viral-host interactions between SARS-CoV-2 and human proteins. Biomed. J. 43, 438–450. doi: 10.1016/j.bj.2020.08.003
El-Sayed, A., and Kamel, M. (2021). Coronaviruses in humans and animals: the role of bats in viral evolution. Environ. Sci. Pollut. Res. Int. 28, 19589–19600. doi: 10.1007/s11356-021-12553-1
Fawcett, T. (2005). An introduction to ROC analysis. Patt. Recogn. Lett. 27, 861–874. doi: 10.1016/j.patrec.2005.10.010
Fischhoff, I. R., Castellanos, A. A., Rodrigues, J., Varsani, A., and Han, B. A. (2021). Predicting the zoonotic capacity of mammals to transmit SARS-CoV-2. Proc. Biol. Sci. 288, 20211651. doi: 10.1098/rspb.2021.1651
Forni, D., Cagliani, R., Clerici, M., and Sironi, M. (2017). Molecular evolution of human coronavirus genomes. Trends Microbiol. 25, 35–48. doi: 10.1016/j.tim.2016.09.001
Forsberg, R. (2003). A codon-based model of host-specific selection in parasites, with an application to the influenza a virus. Mol. Biol. Evol. 20, 1252–1259. doi: 10.1093/molbev/msg149
Gentles, A. D., Guth, S., Rozins, C., and Brook, C. E. (2020). A review of mechanistic models of viral dynamics in bat reservoirs for zoonotic disease. Pathog. Glob Health 114, 407–425. doi: 10.1080/20477724.2020.1833161
Gussow, A. B., Auslander, N., Faure, G., Wolf, Y. I., Zhang, F., and Koonin, E. V. (2020). Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc. Natl. Acad. Sci. U S A. 117, 15193–15199. doi: 10.1073/pnas.2008176117
Hie, B., Zhong, E. D., Berger, B., and Bryson, B. (2021). Learning the language of viral evolution and escape. Science 371, 284–288. doi: 10.1126/science.abd7331
Ji, W., Peng, Q., Fang, X., Li, Z., Li, Y., Xu, C., et al. (2022). Structures of a deltacoronavirus spike protein bound to porcine and human receptors. Nat. Commun. 13, 1467. doi: 10.1038/s41467-022-29062-5
Jolliffe, I. T., and Cadima, J. (2016). Principal component analysis: a review and recent developments. Philosop. Transac. R. Soc. A. 374, 20150202. doi: 10.1098/rsta.2015.0202
Lam, T. T., Jia, N., Zhang, Y. W., Shum, M. H., Jiang, J. F., Zhu, H. C., et al. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 583, 282–285. doi: 10.1038/s41586-020-2169-0
Lednicky, J. A., Tagliamonte, M. S., White, S. K., Elbadry, M. A., Alam, M. M., Stephenson, C. J., et al. (2021). Independent infections of porcine deltacoronavirus among Haitian children. Nature 600, 133–137. doi: 10.1038/s41586-021-04111-z
Li, J., Liu, B., Chang, G., Hu, Y., Zhan, D., Xia, Y., et al. (2011). Virulence of H5N1 virus in mice attenuates after in vitro serial passages. Virol. J. 8, 93. doi: 10.1186/1743-422X-8-93
Li, J., Wu, Y., Zhang, S., Kang, X., and Jiang, T. (2022). Deep learning based on biologically interpretable genome representation predicts two types of human adaptation of SARS-CoV-2 variants. Brief Bioinform. 23, bbac036. doi: 10.1093/bib/bbac036
Li, J., Zhang, S., Li, B., Hu, Y., Kang, X., Wu, X., et al. (2020). Machine learning methods for predicting human-adaptive influenza a viruses based on viral nucleotide compositions. Mol. Biol. Evol. 37, 1224–1236. doi: 10.1093/molbev/msz276
Lima, F. E., Campos, F. S., Kunert, F. H., Batista, H. B., Carnielli, P. J., Cibulski, S. P., et al. (2013). Detection of Alphacoronavirus in velvety free-tailed bats (Molossus molossus) and Brazilian free-tailed bats (Tadarida brasiliensis) from urban area of Southern Brazil. Virus Genes 47, 164–167. doi: 10.1007/s11262-013-0899-x
Lin, C. M., Saif, L. J., Marthaler, D., and Wang, Q. (2016). Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Res. 226, 20–39. doi: 10.1016/j.virusres.2016.05.023
Liu, K., Pan, X., Li, L., Yu, F., Zheng, A., Du, P., et al. (2021). Binding and molecular basis of the bat coronavirus RaTG13 virus to ACE2 in humans and other species. Cell 184, 3438–3451. doi: 10.1016/j.cell.2021.05.031
Liu, W., Fan, H., Raghwani, J., Lam, T. T., Li, J., Pybus, O. G., et al. (2014). Occurrence and reassortment of avian influenza A (H7N9) viruses derived from coinfected birds in China. J. Virol. 88, 13344–13351. doi: 10.1128/JVI.01777-14
Maganga, G. D., Bourgarel, M., Vallo, P., Dallo, T. D., Ngoagouni, C., Drexler, J. F., et al. (2014). Bat distribution size or shape as determinant of viral richness in african bats. PLoS ONE 9:e100172. doi: 10.1371/journal.pone.0100172
Nan, B. G., Zhang, S., Li, Y. C., Kang, X. P., Chen, Y. H., Li, L., et al. (2022). Convolutional neural networks based on sequential spike predict the high human adaptation of SARS-CoV-2 Omicron Variants. Viruses 14, 1072. doi: 10.3390/v14051072
Neumann, G., Noda, T., and Kawaoka, Y. (2009). Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459, 931–939. doi: 10.1038/nature08157
Olival, K. J., Hosseini, P. R., Zambrana-Torrelio, C., Ross, N., Bogich, T. L., and Daszak, P. (2017). Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650. doi: 10.1038/nature22975
O'Shea, T. J., Cryan, P. M., Cunningham, A. A., Fooks, A. R., Hayman, D. T., Luis, A. D., et al. (2014). Bat flight and zoonotic viruses. Emerg. Infect. Dis. 20, 741–745. doi: 10.3201/eid2005.130539
Pollock, D. D., Castoe, T. A., Perry, B. W., Lytras, S., Wade, K. J., Robertson, D. L., et al. (2020). Viral CpG deficiency provides no evidence that dogs were intermediate hosts for SARS-CoV-2. Mol Biol Evol 37, 2706–2710. doi: 10.1093/molbev/msaa178
Roes, F. L. (2020). On the evolution of virulent zoonotic viruses in bats. Biol. Theory 15, 223–225. doi: 10.1007/s13752-020-00363-6
Ruiz-Aravena, M., McKee, C., Gamble, A., Lunn, T., Morris, A., Snedden, C. E., et al. (2022). Ecology, evolution and spillover of coronaviruses from bats. Nat. Rev. Microbiol. 20, 299–314. doi: 10.1038/s41579-021-00652-2
Seyran, M., Hassan, S. S., Uversky, V. N., Pal, C. P., Uhal, B. D., Lundstrom, K., et al. (2021). Urgent Need for Field Surveys of Coronaviruses in Southeast Asia to Understand the SARS-CoV-2 Phylogeny and Risk Assessment for Future Outbreaks. Biomolecules 11, 398. doi: 10.3390/biom11030398
Sia, W. R., Zheng, Y., Han, F., Chen, S., Ma, S., Wang, L. F., et al. (2022). Exploring the role of innate lymphocytes in the immune system of bats and virus-host interactions. Viruses 14, 150. doi: 10.3390/v14010150
Skirmuntt, E. C., Escalera-Zamudio, M., Teeling, E. C., Smith, A., and Katzourakis, A. (2020). The potential role of endogenous viral elements in the evolution of bats as reservoirs for zoonotic viruses. Annu. Rev. Virol. 7, 103–119. doi: 10.1146/annurev-virology-092818-015613
Su, S., Wong, G., Shi, W., Liu, J., Lai, A., Zhou, J., et al. (2016). Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 24, 490–502. doi: 10.1016/j.tim.2016.03.003
Sun, H., Sun, Y., Pu, J., Zhang, Y., Zhu, Q., Li, J., et al. (2014). Comparative virus replication and host innate responses in human cells infected with three prevalent clades (2.3.4, 2.3.2, and 7) of highly pathogenic avian influenza H5N1 viruses. J. Virol. 88, 725–729. doi: 10.1128/JVI.02510-13
Taubenberger, J. K., and Kash, J. C. (2010). Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe. 7, 440–451. doi: 10.1016/j.chom.2010.05.009
Townsend, J. T. (1971). Theoretical analysis of an alphabetic confusion matrix. Percept. Psychophy. 9, 40–50. doi: 10.3758/BF03213026
Upadhyay, M., Samal, J., Kandpal, M., Vasaikar, S., Biswas, B., Gomes, J., et al. (2013). CpG dinucleotide frequencies reveal the role of host methylation capabilities in parvovirus evolution. J. Virol. 87, 13816–13824. doi: 10.1128/JVI.02515-13
Wang, Z., Huang, G., Huang, M., Dai, Q., Hu, Y., Zhou, J., et al. (2022). Global patterns of phylogenetic diversity and transmission of bat coronavirus. Sci. China Life Sci. 66, 861–874. doi: 10.1007/s11427-022-2221-5
West, R., Michie, S., Rubin, G. J., and Amlot, R. (2020). Applying principles of behaviour change to reduce SARS-CoV-2 transmission. Nat. Hum. Behav. 4, 451–459. doi: 10.1038/s41562-020-0887-9
WHO (2022). Coronavirus (COVID-19) Dashboard. Available online at: https://covid19.who.int/ (accessed June 30, 2022).
Woo, P. C., Lau, S. K., Lam, C. S., Lau, C. C., Tsang, A. K., Lau, J. H., et al. (2012). Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 86, 3995–4008. doi: 10.1128/JVI.06540-11
Xia, X. (2020). Extreme genomic CpG deficiency in SARS-CoV-2 and evasion of host antiviral defense. Mol. Biol. Evol. 37, 2699–2705. doi: 10.1093/molbev/msaa094
Xiao, K., Zhai, J., Feng, Y., Zhou, N., Zhang, X., Zou, J. J., et al. (2020). Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. 583, 286–289. doi: 10.1038/s41586-020-2313-x
Zhou, P., Fan, H., Lan, T., Yang, X. L., Shi, W. F., Zhang, W., et al. (2018). Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 556, 255–258. doi: 10.1038/s41586-018-0010-9
Keywords: bat coronavirus, asymptotic adaptation, deep learning, dinucleotide composition representation (DCR), convolutional neural networks
Citation: Li J, Tian F, Zhang S, Liu S-S, Kang X-P, Li Y-D, Wei J-Q, Lin W, Lei Z, Feng Y, Jiang J-F, Jiang T and Tong Y (2023) Genomic representation predicts an asymptotic host adaptation of bat coronaviruses using deep learning. Front. Microbiol. 14:1157608. doi: 10.3389/fmicb.2023.1157608
Received: 02 February 2023; Accepted: 03 April 2023;
Published: 05 May 2023.
Edited by:
Wenshi Wang, Xuzhou Medical University, ChinaReviewed by:
Yang Li, Chinese Academy of Sciences (CAS), ChinaZhijiang Miao, Erasmus Medical Center, Netherlands
Copyright © 2023 Li, Tian, Zhang, Liu, Kang, Li, Wei, Lin, Lei, Feng, Jiang, Jiang and Tong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yigang Tong, dG9uZ3lpZ2FuZ0BtYWlsLmJ1Y3QuZWR1LmNu; Tao Jiang, amlhbmd0YW9AYm1pLmFjLmNu; Jia-Fu Jiang, amlhbmdqZjIwMDhAZ21haWwuY29t