 Dong Hyun Kim
Dong Hyun Kim Yu-Min Choi
Yu-Min Choi Junghwa Jang1
Junghwa Jang1 Bum-Joon Kim
Bum-Joon Kim
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 09 March 2023
Sec. Virology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1137084
This article is part of the Research Topic Origin and Evolution of Hepatitis Viruses, Volume II View all 5 articles
Hepatitis B Virus (HBV) genotypes reflect geographic, ethical or clinical traits and are currently divided into 10 genotypes (A–J). Of these, genotype C is mainly distributed in Asia, is the largest group and comprises more than seven subgenotypes (C1–C7). Subgenotype C2 is divided into three phylogenetically distinct clades, C2(1), C2(2), and C2(3), and is responsible for most genotype C infections in three East Asian nations, including China, Japan, and South Korea, which are major HBV endemic areas. However, despite the significance of subgenotype C2 with regard to clinical or epidemiologic aspects, its global distribution and molecular characteristics remain largely unknown. Here, we analyze the global prevalence and molecular characteristics between 3 clades within subgenotype C2 using 1,315 full genome sequences of HBV genotype C retrieved from public databases. Our data show that almost all HBV strains from South Korean patients infected with genotype C belong to clade C2(3) within subgenotype C2 [96.3%] but that HBV strains from Chinese or Japanese patients belong to diverse subgenotypes or clades within genotype C, suggesting clonal expansion of a specific HBV type, C2(3), among the Korean population. Our genome sequence analysis indicated a total of 21 signature sequences specific to the respective clades C2(1), C2(2), and C2(3). Of note, two types of four nonsynonymous C2(3) signature sequences, sV184A in HBsAg and xT36P in the X region, were detected in 78.9 and 82.9% of HBV C2(3) strains, respectively. In particular, HBV strains C2(3) versus C2(1) and C2(2) show a higher frequency of reverse transcriptase mutations related to nucleot(s)ide analog (NA) resistance, including rtM204I and rtL180M, suggesting an increased possibility of C2(3) infection in those with NA treatment failure. In conclusion, our data show that HBV subgenotype C2(3) is extremely prevalent in Korean patients with chronic HBV infection, which is distinct from two other East Asian nations, China and Japan, where diverse subgenotypes or clades within genotype C coexist. This epidemiologic trait might affect distinct virological and clinical traits in chronic HBV patients in Korea, where exclusively C2(3) infection is predominant.
The discovery of hepatitis B virus (HBV) led to the first vaccine not prepared through tissue culture but that was initially directly prepared from plasma, with a later recombinant vaccine produced from yeast (da Fonseca, 2010). Despite an effective vaccine, HBV infection remains a severe global health issue, with more than 240 million people being chronic carriers and approximately 786,000 patient deaths annually worldwide due to HBV-related diseases, including liver cirrhosis (LC) and hepatocellular carcinoma (HCC; Schweitzer et al., 2015; Guvenir and Arikan, 2020). HBV is an endemic disease in South Korea (Korean National Health and Nutrition Survey of 2011); the prevalence of hepatitis B virus surface antigen (HBsAg) positivity is 3.4% in males and 2.6% in females (Yim and Kim, 2019).
HBV, which belongs to the Hepadnaviridae family, is an enveloped virus with a partially double-stranded DNA genome of approximately 3.2 kb in length that includes 4 overlapping open reading frames (ORFs) encoding the surface protein (S), core protein (C), polymerase (Pol), and HBx protein (X; Liang, 2009; Datta et al., 2012). HBV has been characterized into 10 genotypes, A to J (Sunbul, 2014), which are subdivided into more than 30 subgenotypes (Kurbanov et al., 2010; Shi et al., 2012). Each subgenotype has been reported to show distinct geographical patterns and clinical traits (Liu et al., 2021). Genotype C is the oldest and most common extant genotype and is endemic in the Asia-Pacific region (Velkov et al., 2018; Kyaw et al., 2020). Compared to genotype B, genotype C exhibits higher HBV replication capacity and tends to result in chronic infection, which may lead to the development of LC and/or HCC (Chan et al., 2003; Gao et al., 2007). In addition, incomplete response to IFN therapy and higher levels of mutations have been reported in genotype C HBV infections (Kao et al., 2000).
Genotype C is further divided into many subgenotypes, C1 to C11 (McNaughton et al., 2020). Subgenotype C2 is endemic in Far Eastern countries, including Korea, China and Japan, which are close geographically and similar in historical and social-economic aspects (Lin et al., 2019). Rather diverse genotypes are observed in China and Japan in addition to subgenotype C2, whereas an extremely high prevalence of subgenotype C2 has been reported in Korea (Kim et al., 2007; Cho et al., 2009). More recent studies have shown that subgenotype C2 can be further divided into three phylogenetically distinct clades, C2(1), C2(2), and C2(3) (Yin et al., 2019; McNaughton et al., 2020), but the correlation of these clades with geographic and clinical traits has not yet been analyzed.
Studies on the correlation between regional-specific genotypes and epidemiologic traits may provide an explanation for specific clinical or virological issues in local population (Lin et al., 2019). In Korea, there are higher rates of naturally occurring HBV variants related to clinical severity compared to other countries, and occult infection or potential antiviral drug resistance and a higher frequency of IFN therapy failure occur even with intensive medical care (Yim and Kim, 2019). In this study, we investigated the prevalence of three clades within subgenotype C2, i.e., C2(1), C2 (2), and C2 (3), in East Asian countries. A total of 683 whole-genome sequences of HBV subgenotype C2 of 1,315 HBV genotype C retrieved from public databases were used to analyze overall mutation rates and signature mutations specific for each clade.
A total of 1,315 HBV genome sequences of genotype C were used in this study. With 1,096 sequences from a previous research dataset (McNaughton et al., 2020), additional Korean and Japan sequences from NCBI1 databases with the keywords “HBV,” “complete genome,” “Korea,” or “Japan” were downloaded, and the final 219 sequences were genotyped using phylogenetic analysis. HBV genotype C and genotype A sequence (accession no. AB116076, AB116080, AB452979) alignments were fitted to 3,215 and 3,221 bp, respectively. The genotyped datasets used in this manuscript were constructed in FASTA format (Supplementary Table S1). A flowchart of the database configuration in each analysis is shown (Figure 1).
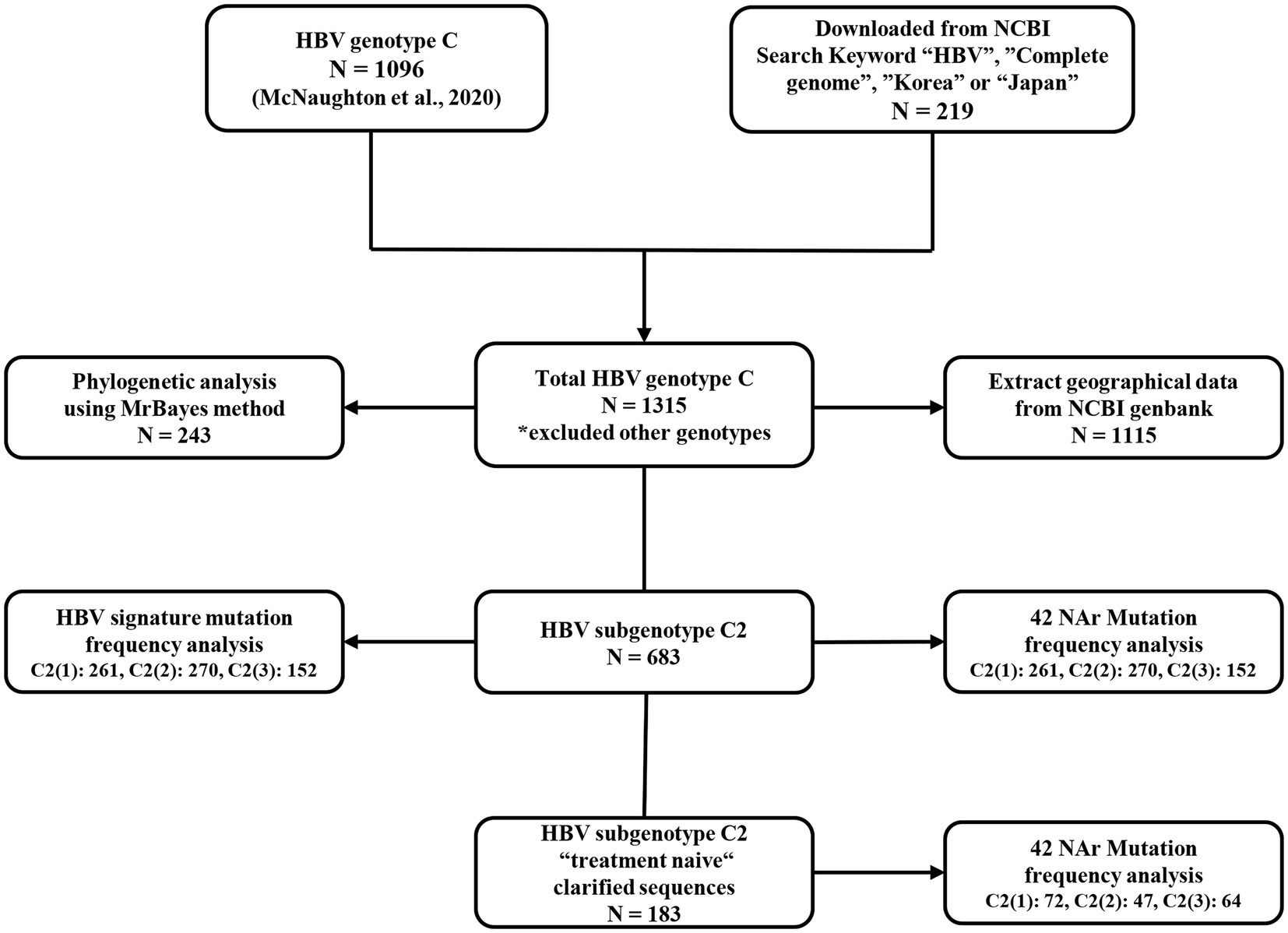
Figure 1. Flowchart of dataset preparation and data composition for each analysis.
Alignments were constructed using MAFFT with the FFT-NS-2 algorithm in Geneious Prime software (2022.1.1, Biomatters, Inc., New Zealand), and all indels in alignments were deleted. A total of 1,315 sequences were genotyped by phylogenetic analysis using the approximate maximum likelihood method with the FastTree program in Geneious Prime software (2022.1.1, Biomatters, Inc., New Zealand; Supplementary Figure S1). Then, 243 genotype C sequences, including 40, 34, and 36 well-branched sequences of each C2(1), C2(2), and C2(3) clade, were extracted. A phylogenetic tree with the 243 sequences was constructed by the MrBayes program (Huelsenbeck and Ronquist, 2001) using the MCMC method and GTR substitution model. The topology of the phylogenetic tree was confirmed by the maximum likelihood method with the Tamura-Nei model (Tamura and Nei, 1993) using the MEGA X program (Tamura et al., 2021). An accession number list organized by subgenotype, including geographical information, was constructed in Excel format (Supplementary Table S2).
A total of 683 subgenotype C2 sequences, 261, 270, and 152 sequences belonging to subgenotypes C2(1), C2(2), and C2(3), respectively, were used for genetic distance calculations. Consensus sequences were extracted using the majority rule from each subgenotype and aligned using Geneious Prime software (2022.1.1, Biomatters, Inc., New Zealand). Pairwise distance between sequences and the consensus in each subgenotype was calculated using the MEGA X program (Tamura et al., 2021).
The number of preC/Core and BCP mutations and polymorphism rates for the rt269 region were analyzed for each clade (Tong et al., 2007; Jeong et al., 2021). The amino acid composition of each clade was analyzed for 42 potential analog-resistant (NAr)-related amino acid mutations identified in a previous study (Liu et al., 2010). The total mutation rate for all 42 NAr sites was calculated by dividing the number of sequences in each clade multiplied by 42 by the number of mutations in each clade. By examining the source of the obtained accession number, we checked whether the patients with respective sequences experienced antiviral treatment before serum extraction. Then, the sequences were further classified into four categories: “treatment naïve,” “antiviral treatment,” “not mentioned,” and “unpublished” (Supplementary Table S3). The frequency of 42 potential NAr-related amino acid mutations with sequences from 183 “treatment-naïve” patients was further analyzed. A polymorphism that uniquely appeared with a frequency of ≥70% in each subgenotype was defined as a signature sequence. Signature sequences were found throughout the HBV genome, and the nucleotide composition ratio (A, T, G, C) with other clades was compared for each signature sequence. Due to the characteristics of the overlapping genome of HBV, nonsynonymous or synonymous mutations of two proteins can occur simultaneously within one signature sequence, and both were considered. Gaps in alignment or strains that were not properly sequenced were excluded.
Serum DNA samples from 127 Korean HBV patients who visited Konkuk University Hospital were extracted as previously described (Kim et al., 2017). For the frequency of xT36P in HBx, the HBx region of 127 DNA samples was newly sequenced using the following primers (FW: CTC TGC CGA TCC ATA CTG CGG AA, RV: TTA ACC TAA TCT CCT CCC CCA; Sugauchi et al., 2001). The frequency of sV184A in HBsAg was analyzed using the overlapping HBsAg region of reverse transcriptase sequences from the 131 Korean patients analyzed in our previous study (accession nos. KX264864~KX264922, KX264792~KX264863; Kim et al., 2017). As shown in Figure 2A, 65 patient DNA samples overlapped between the 131 samples with the reverse transcriptase sequenced in a previous study and the HBx sequences from 127 samples in this study. The frequency of the sV184A and xT36P sequences from the overlapping 65 patients was calculated. The 127 HBx sequences are shown in Supplementary Table S4. As previously extracted virion DNA from isolates was used in this study, informed consent and waiver of informed consent were not required by the IRB of the hospital (IRB1012-131-346).
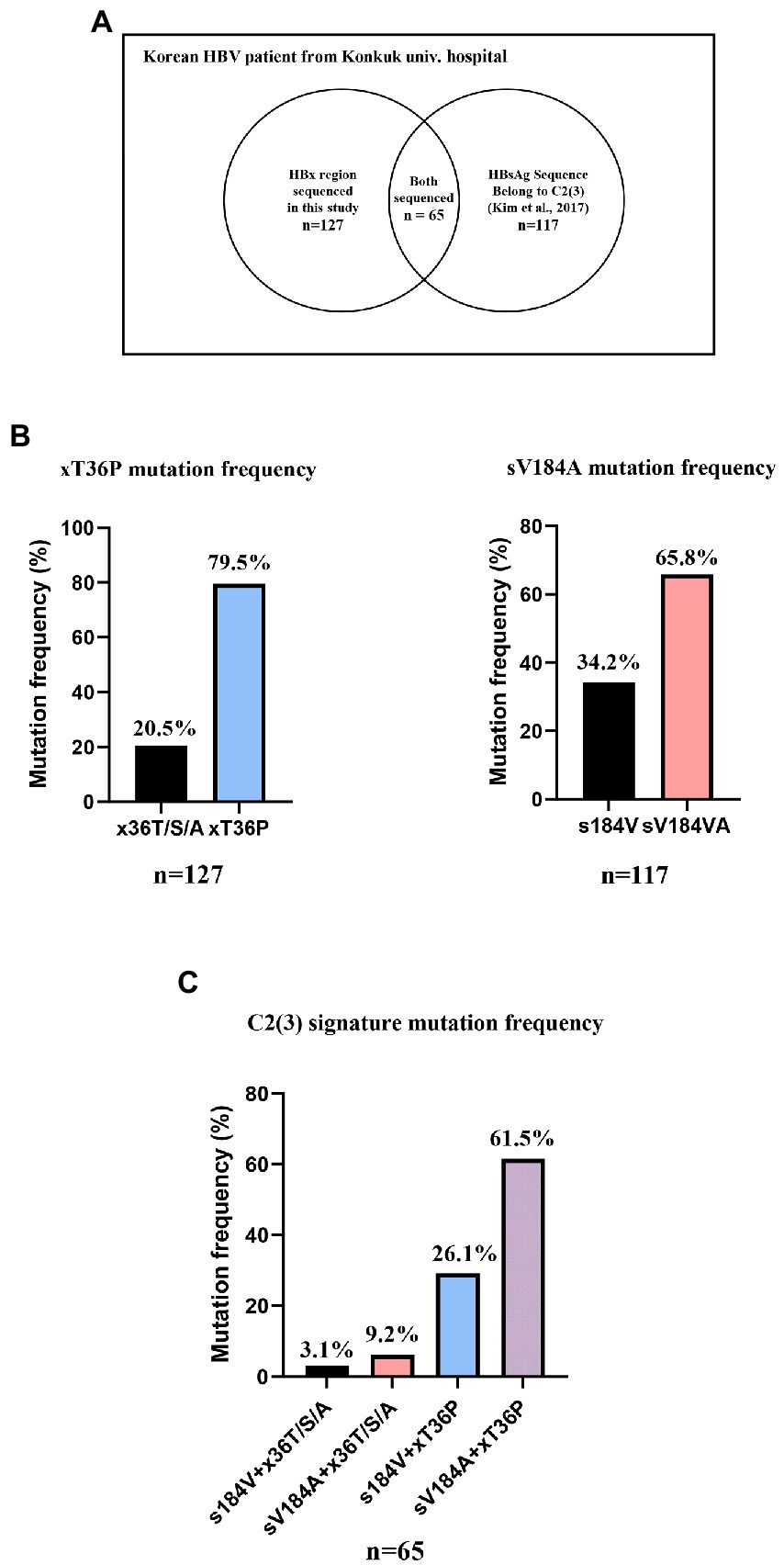
Figure 2. The frequency of two signature mutations specific for C2(3), sV184A and xT36P, from Korean chronic HBV infection patients from Konkuk university hospital. (A) A Venn diagram of 131 previously studied RT sequences and 127 HBx sequences generated in this study. (B) A total of 79.5% of Korean HBV C2(3) patients carried the xT36P mutation in HBx, and 65.8% of the patients carried sV184A in HBsAg. (C) A total of 65 DNA sequences of the HBsAg and HBx regions were accessible, and 61.5% showed both C2(3) signature sequences.
A 1,032 bp fragment of HBV DNA reverse transcriptase from 131 Korean patients was aligned to reverse transcriptase sequences extracted from the 683 subgenotype C2 sequence dataset using the MAFFT method, as described above. A phylogenetic tree was generated using the maximum likelihood method with the Tamura-Nei model (Tamura and Nei, 1993).
Statistical analysis was conducted using GraphPad Prism 8.0 software for Windows, GraphPad Software, San Diego, California United States, www.graphpad.com. Statistical significance of the number of amino acid differences was calculated by ANOVA (analysis of variance). For analysis of mutation sites, wild-type and mutated sequences were compared using the chi-square test, and the significance calculated is shown in related figures and tables.
A phylogenetic tree was created using a total of 1,315 HBV genotype C sequences, including 1,096 from a previous research dataset (McNaughton et al., 2020) and an additional 219 from “Korean” or “Japanese” patients downloaded for use in this study through the approximate maximum likelihood method (Supplementary Figure S1). The phylogenetic tree showed that 893 of the 1,315 sequences belonged to subgenotype C2 and were separated into three distinct clades, C2(1) (299 sequences), C2(2) (321 sequences), and C2(3) (273 sequences). We isolated 40 C2(1), 36 C2(2), and 34 C2(3) sequences in each clade and 133 sequences in other subgenotype C sequences from the tree. To clarify the branching between clades, a phylogenetic tree was created with 243 selected genotype C sequences, including 110 subgenotype C2 [40 C2(1), 36 C2(2), and 34 C2(3)] sequences, using the MrBayes method (Figure 3A). Our phylogenetic analysis showed separation of three phylogenetically distinctive subclades within subgenotype C2, consistent with previous findings (McNaughton et al., 2020). This separation and topology was also demonstrated in a tree produced using the maximum-likelihood method (Supplementary Figure S2). C2(1) and C2(3) form two independent clusters with C2(2) branching from C2(1), suggesting recent evolution of C2(2) from C2(1).
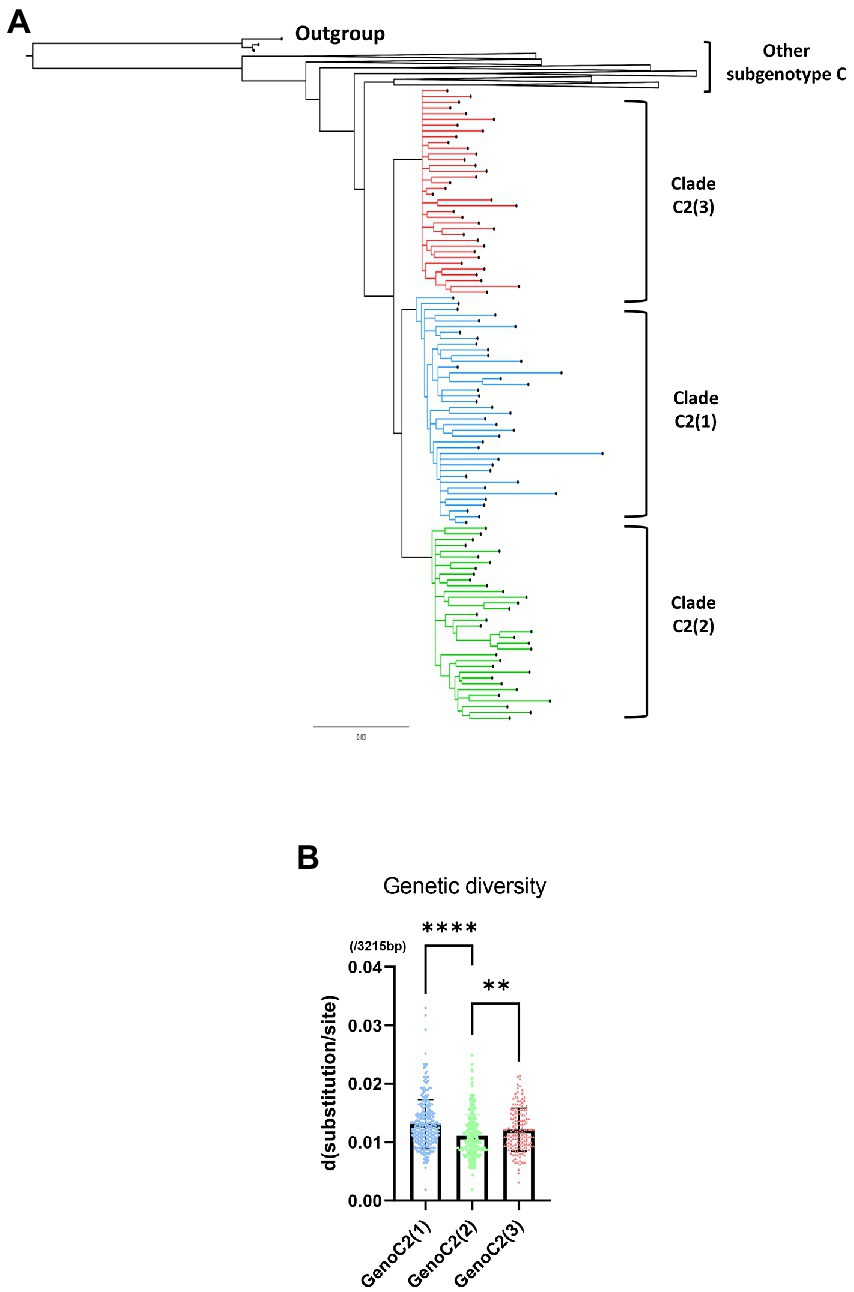
Figure 3. A phylogenetic tree of hepatitis B virus genotype C based on 243 genome sequences constructed using MrBayes. For phylogenetic analysis between three clades within subgenotype C2, 40 of 260 sequences from C2(1), 34 of 271 from C2(2) and 36 of 152 from C2(3) were selected. (A) Three clades of subgenotype C2 are shown in blue [C2(1)], green [C2(2)], and red [C2(3)]. (B) Comparison of HBV genetic diversity among clades C2(1), C2(2), and C2(3). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Regarding genetic diversity, the C2(2) HBV sequence exhibited the lowest average substitution per site (0.013 ± 0.004), which was statistically significant when compared to C2(1) (0.011 ± 0.004; p < 0.0001), and C2(3) (0.012 ± 0.004; p value < 0.01; Figure 3B). Geographical distribution analysis of 1,115 sequences available with country information among the genotype C sequence pool revealed subgenotype C2 strains to be predominant in three countries of East Asia: China (457/517 strains, 88.4%), Japan (111/114 strains, 97.4%) and South Korea (108/108 strains, 100%; Supplementary Table S5). Overall, geographical distribution at the clade level within subgenotype C2 indicated that subgenotype C2(3) is extremely dominant in South Korea (104/108, 96.3%) but that its prevalence is relatively low in China (28/457, 5.4%) and Japan (40/114, 35.1%; Figure 4). Together, our data indicate that subgenotype C2 infections are predominant in three nations of East Asia, China, Japan and South Korea, and that C2(3) infection is responsible for most chronic hepatitis B patients in South Korea.
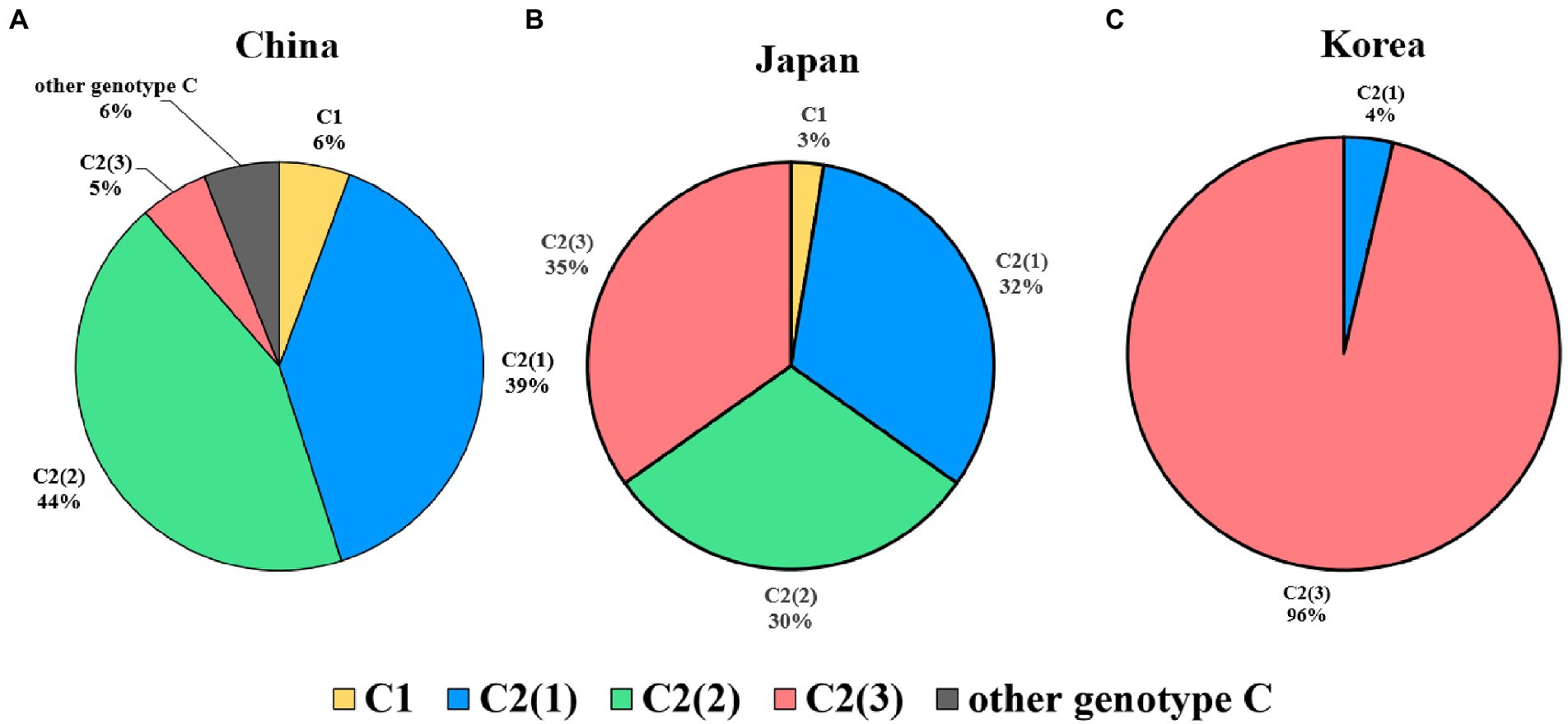
Figure 4. Global prevalence of three clades within hepatitis B virus subgenotype C2. (A) In China, C2(2) is the most prevalent genotype, followed by C2(1). (B) In Japan, the three subgenotypes are distributed at approximately 30%. (C) In Korea, the subgenotype C2(3) is overwhelmingly dominant and shows lower genotypic diversity than in the other two countries.
HBV strains belonging to subgenotype C2 have been reported to have higher frequencies of preC mutation (G1896A) and basal core promoter (BCP) double mutations (A1762T and G1764A), which are associated with abolition of HBeAg and its reduced expression, respectively (Park and Lee, 2016). Thus, mutation frequency was evaluated between clades C2(1), C2(2), and C2(3) using a total of 683 subgenotype C2 strains selected from the HBV sequence pool: C2(1) (n = 261), C2(2) (n = 270), and C2(3) (n = 152). Our data showed there was no significant difference between strains of the 3 clades with respect to frequency of the preC/C mutation (G1896A; Figure 5). However, for BCP double mutations A1762T and G1764A, strains belonging to C2(2) or C2(3) exhibited significantly higher frequency compared with C2(1), despite no significant difference between C2(2) and C2(3), consistent with the previous finding of higher frequency of BCP mutation in chronic HBV infection patients in South Korea (Lee et al., 2015). The frequency of the rtL269I polymorphism associated with the development of liver disease was significantly higher in clade C2(2) than in C2(1) and C2(3).
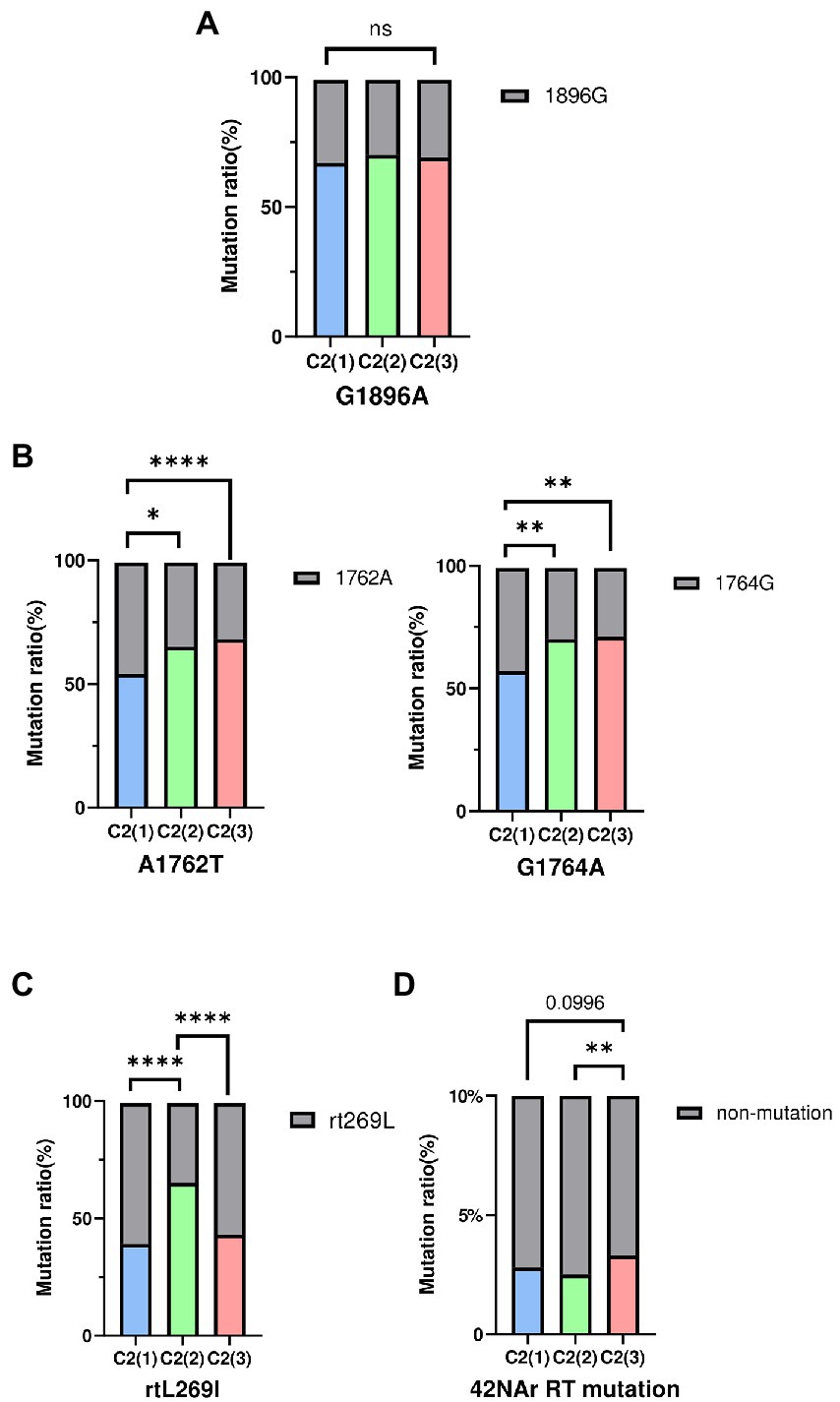
Figure 5. The frequency of HBV mutations between three clades. (A) Comparisons of preC/C mutation (G1896A) (B) Comparisons of BCP mutation (A1762T, G1764A). (C) Comparisons of reverse transcriptase polymorphism rtL269I. (D) Comparisons of total 42NAr mutation sites. Categorical variables were tested using the Chi-squared test. ns, nonsignificant, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
As genotype C HBV strains have been reported to have a higher frequency of reverse transcriptase (RT) mutations related to nucleos(t)ide analog (NAr) resistance (Kim et al., 2017), the amino acid mutation frequency of 42 potential NAr positions in HBV RT sequences (Liu et al., 2010) was evaluated between clades C2(1), C2(2) and C2(3) in a total of 683 subgenotype C2 strains. C2(3) showed a higher frequency of mutations at a total of 42 potential NAr positions than C2(1) or C2(2) (p < 0.05; Figure 5D). In particular, the frequency of YMDD-motif mutation at rt204 corresponding to the primary drug resistance mutation (category 1) was detected at a significantly higher rate in C2(3) than in C2(1) or C2(2) (p < 0.001; Table 1). The frequency of the L180M mutation, corresponding to a secondary drug resistance mutation, and the H126C/Y/Q mutation, a putative ADV NAr, were observed at a significantly higher frequency in C2(3) compared to C2(1) or C2(2) (p < 0.05, p < 0.001, respectively; Nakajima et al., 2021). The T128N/I mutation frequency among the putative NAr mutations was higher in C2(1) than in C2(2) or C2(3) (p < 0.01), and in the pretreatment mutation column, the mutation frequency of Y124H and D134E/N/C was higher in C2(1) than in other clades (p < 0.0.05, p < 0.001, respectively). To evaluate NAr mutation frequency in treatment-naïve patients of subgenotype C2, potential NAr mutation frequency was further analyzed using sequences of the 183 selected “treatment-naïve” patients, 72 C2(1), 47 C2(2), and 64 C2(3) sequences (Supplementary Table S6). Our data showed that of the putative NAr mutations, F221Y and N238D/S/H were significantly higher in C2(3) than in other clades (p < 0.01, p < 0.05, respectively). Among the pretreatment mutations, D134E/N/C was most common in C2(1) (p < 0.01), and C2(3) was statistically significant in N139K/H (p < 0.05). As shown in 683 subgenotype C2 strains, C2(3) also showed a significantly higher frequency of mutations at a total of 42 potential NAr positions compared to C2(1) or C2(2) in 183 selected “treatment-naïve” patients (p < 0.0001).
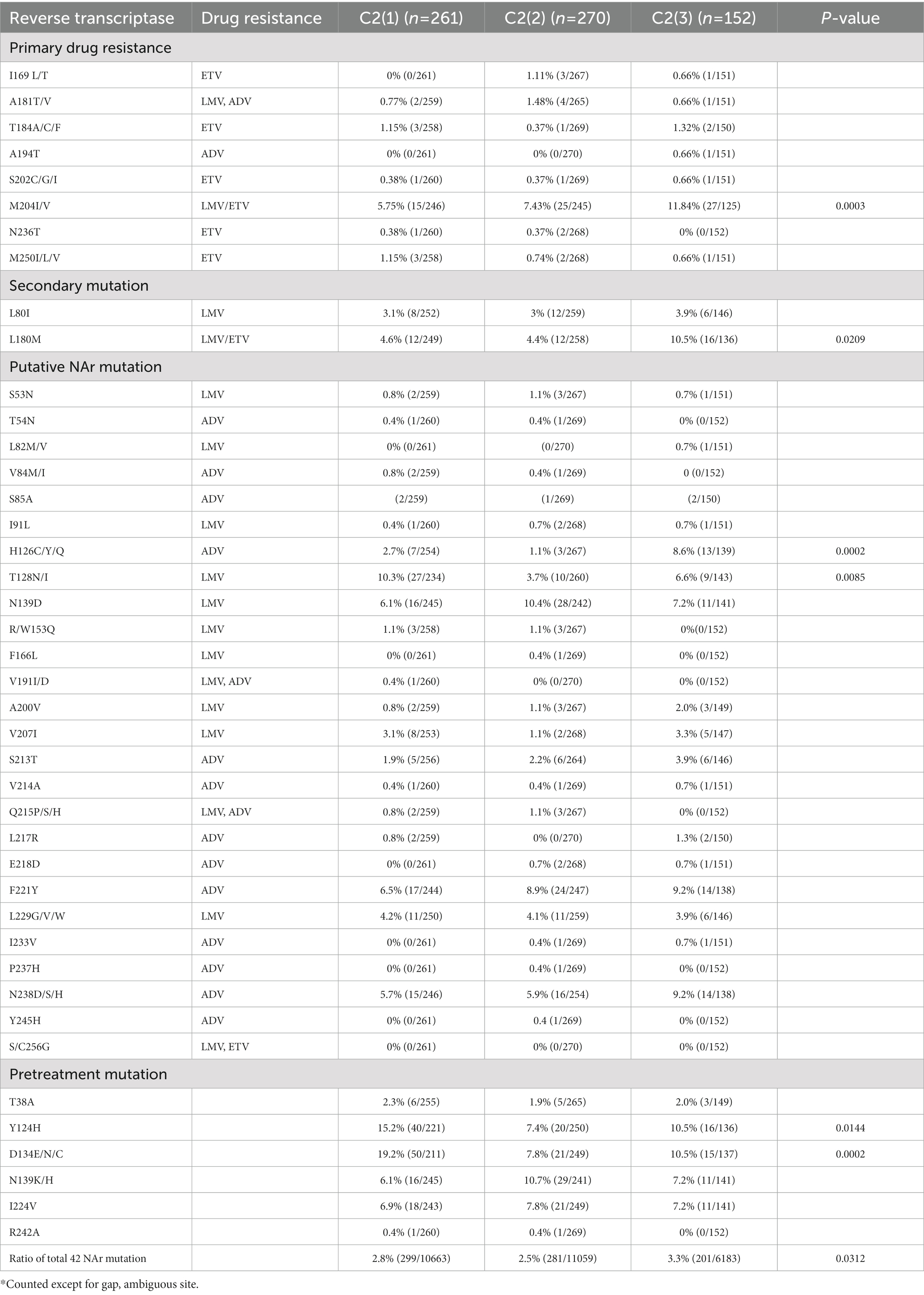
Table 1. Occurrence of NAr mutation in reverse transcriptase of hepatitis B virus genotype C2 by clade.
We identified signature mutations with significantly higher frequencies that are nonsynonymous and synonymous and specific to each clade. A total of 21 types of signature nonsynonymous (8) and synonymous mutations (13) specific for the respective clades were found (Supplementary Table S7). The signature mutations were present in the preS (4 types), S (1 types), X (3 types), PreC/C (2 types) and Pol (16 types) regions. Of these, 1 (1 NS mutation), 13 (3 NS mutations), and 7 (4 NS mutations) mutation types were specific to C2(1), C2(2), and C2(3), respectively (Table 2). L116V mutation in HBx was the only detected signature mutation of C2(1) (81.6% of strains). In C2(2), we observed 3 nonsynonymous mutations of 13 signature mutations: S314P in the Pol region (93.7%), F321L in the Pol spacer region (91.1%), and A90V in the preS1 region (89.3%). Of the 7 signature mutations in C2(3), 4 are signature nonsynonymous mutations: sV184A mutation in HBsAg (78.9%), T/S/A36P in HBx (82.9%), H81N mutation in the Pol terminal protein region (97.4%) and T272A in the Pol spacer region (76.3%).
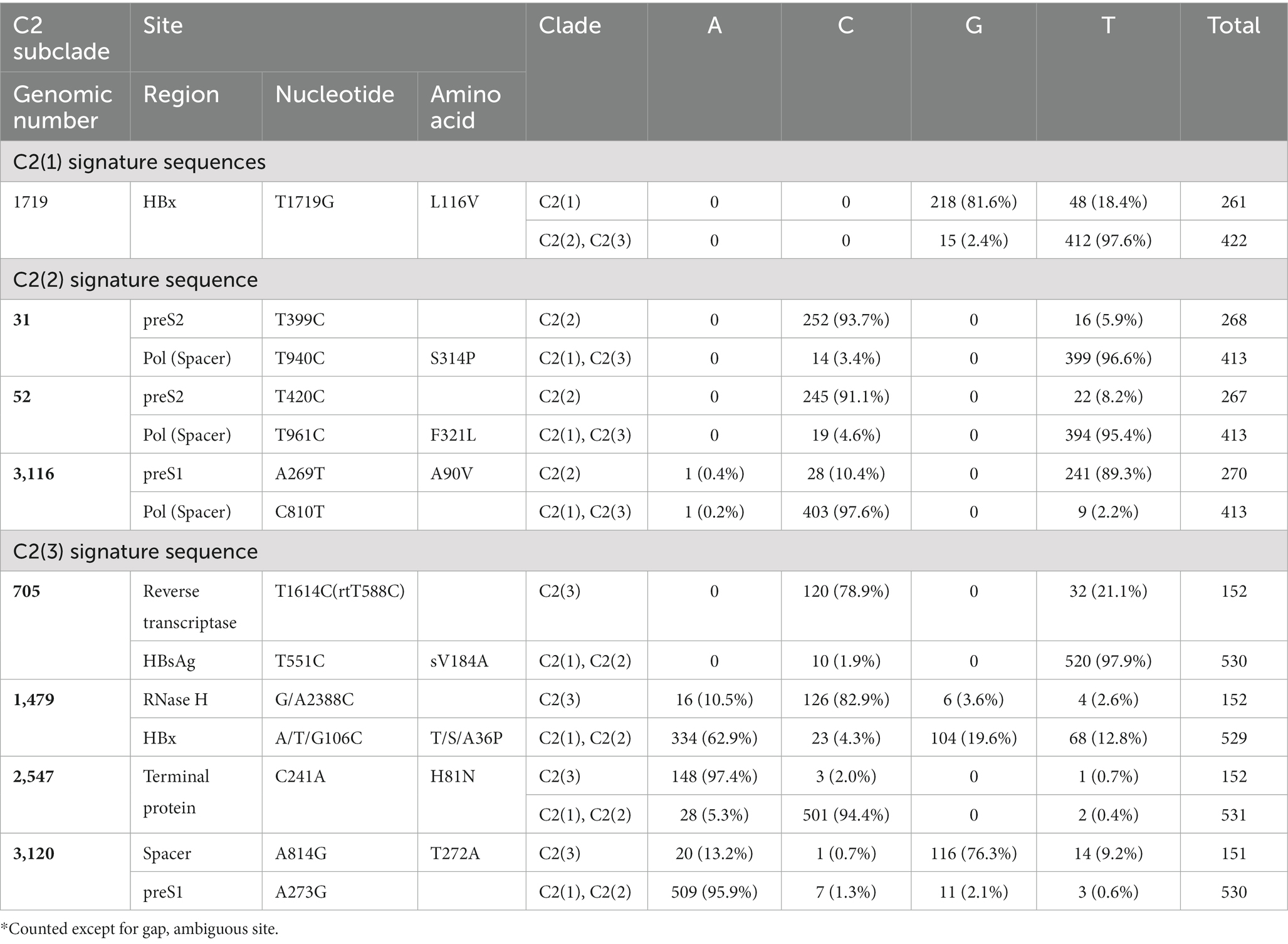
Table 2. Nonsynonymous mutation sites and their incidence rate in each clade of subgenotype C2.
To confirm the dominance of C2(3) in Korean patients with chronic HBV infection, we evaluated the clade distribution using 131 HBV RT sequences previously reported by our laboratory (Kim et al., 2017; Figure 6). Phylogenetic analysis using the maximum likelihood method showed that among the 131 patients, all belong to C2(3) (117/131 patients, 89.3%), except for 12 patients with C2(1) and 2 with C2(2), further supporting our finding of the dominance of C2(3) in South Korea. Of the 117 Korean patients infected with C2(3), 77 (65.8%) carried the sV184A mutation in the overlapping HBsAg region and 101 (79.5%) the xT36P mutation in HBx (Figure 2). Of a total of 65 patients for whom sequence information for both HBsAg and HBx regions was available, the proportion of those carrying both sV184A and xT36P was highest at 61.5% (40/65), followed by those with xT36P alone, at 26.1% (17/65), sV184A alone, at 9.2%, and s184V/+x36T/S/A, at 3.1%.
Figure 6. Differentiation at the clade level of 131 Korean chronic patients via phylogenetic analysis of the HBV reverse transcriptase region. Phylogenetic tree of reverse transcriptase from HBV subgenotype C2. Distribution of 131 Korean patients by alignment with 683 previously constructed subgenotype C2 reverse transcriptase sequences. Most Korean patients (89.3%) are infected with subgenotype C2(3). Korean patient sequences are shown in yellow branches.
HBV subgenotype C2 is responsible for most genotype C infections in three nations of East Asia, namely, China, Japan and South Korea, which are major HBV endemic areas (Cho et al., 2009; Wang et al., 2010; Demarchi et al., 2022). Subgenotype C2 is reported to have unique virological and clinical traits distinct from other genotypes and subgenotypes, including higher virulence, higher BCP or preC/C mutation frequency, and lower response to IFN-I therapy (Lin and Kao, 2011; Ito et al., 2018; Tang et al., 2018). Phylogenetic analysis based on entire HBV genome sequences to date indicates the existence of three distinct clades, C2(1), C2(2), and C2(3), within subgenotype C2 (McNaughton et al., 2020). However, studies regarding the global prevalence and molecular characteristics of these three distinct clades remain largely unknown. In this study, we for the first time determined the global prevalence and molecular characteristics of three distinct clades, C2(1), C2(2) and C2(3), within subgenotype C2.
First, we found differences between China, Japan and South Korea with regard to the clade distribution of subgenotype C2. C2(3) was found to be exclusively predominant in South Korea, distinct from China and Japan, showing coexistence of the three clades despite a discrepancy in their distribution (Figure 4). This epidemiologic finding may provide a likely explanation for the distinct findings of several studies using Korean cohorts, including a higher frequency of NAr or BCP mutation (Kim, 2014), the presence of rarely encountered HBV mutation types (Kim et al., 2017), and a higher prevalence of HBeAg-negative HCC patients (Jang et al., 2022).
Second, we identified 21 types of signature sequences specific for C2(1), C2(2) and C2(3), which may be used for differentiation of the 3 clades as genetic markers (Table 2). In particular, 8 types of nonsynonymous signature sequences might influence the distinct virological and clinical traits of each clade. Future studies should focus on nonsynonymous signature sequences to elucidate the underlying mechanism associated with the distinct virological and clinical traits of each clade.
Third, we found that compared to the other two clades, i.e., C2(1) and C2(2), C2(3) showed a higher frequency of NAr mutations, even in rt204 and rt180, which are related to primary and secondary drug resistance, respectively (Table 1). This suggests that C2(3) infection may lead to enhanced NA treatment failure compared to the other two clades. Indeed, our previous study using a cohort of treatment-naïve patients with chronic HBV infection revealed a higher prevalence of naturally occurring NAr mutations in Korea compared to other areas, including China (Kim et al., 2017). Our previous LNA-based RT–PCR assay also showed a higher prevalence of rt204I region YMDD variants, particularly in Korean HCC patients (Choe et al., 2019). Higher rates of relapse in Korean chronic HBV infection patients with HBeAg seroconversion after lamivudine treatment have also been found, supporting the above hypothesis (Song et al., 2000).
Fourth, despite no significant difference in the frequency of preC mutation, the BCP double mutation frequency was significantly higher in chronic HBV patients infected with C2(3) or C2(2) than in those infected with C2(1) (Figure 5). Given the relationships of BCP double mutation with liver disease progression and HBeAg-negative infection (Kim, 2014), the possibility that C2(3) or C2(2) vs. C2(1) may cause more advanced liver diseases, including HCC in HBeAg-negative chronic HBV patients, cannot be excluded (Zhang et al., 2010; Alexopoulou and Karayiannis, 2014). In fact, a recent study using a Korean cohort reported that HCC is more prevalent in HBeAg-negative patients without liver cirrhosis than in HBeAg-positive patients without liver cirrhosis, which is distinct from other studies showing that HBeAg is not an independent risk factor for HCC (Jang et al., 2022). This issue should be further assessed in future studies using Korean cohorts.
In conclusion, our data show that HBV subgenotype C2(3) is extremely prevalent in Korean patients with chronic HBV infection, which is distinct from two other East Asian countries, China and Japan, where diverse subgenotypes or clades within genotype C coexist. In addition, we found that HBV strain C2(3) shows a higher frequency of RT mutations related to NAr, including rtM204I and rtL180 M, than strains C2(1) and C2(2), suggesting an increased possibility of C2(3) infection in those with NA treatment failure. This epidemiologic trait might affect distinct virological and clinical traits in patients with chronic HBV infection in Korea, where C2(3) infection is exclusively predominant.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Seoul National University Hospital (1012-131-346). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
DK and B-JK designed the study. B-JK interpreted the research and wrote the first draft of the manuscript. DK performed the data analysis and revised the manuscript. Y-MC and JJ supported the data analysis and revised the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the National Research Foundation of Korea (NRF) and funded by the Ministry of Education (grant no. NRF-2022R1A2B5B01001421). This research was also supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), as funded by the Ministry of Health and Welfare, Republic of Korea (grant no. HI22C0476). Y-MC, DK, and JJ received a scholarship from the BK21-plus education program provided by the National Research Foundation of Korea.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1137084/full#supplementary-material
Alexopoulou, A., and Karayiannis, P. (2014). HBeAg negative variants and their role in the natural history of chronic hepatitis B virus infection. World J. Gastroenterol. 20, 7644–7652. doi: 10.3748/wjg.v20.i24.7644
Chan, H. L.-Y., Wong, M. L., Hui, A. Y., Hung, L. C.-T., Chan, F. K.-L., and Sung, J. J.-Y. (2003). Hepatitis B virus genotype C takes a more aggressive disease course than hepatitis B virus genotype B in hepatitis B e antigen-positive patients. J. Clin. Microbiol. 41, 1277–1279. doi: 10.1128/JCM.41.3.1277-1279.2003
Cho, J.-H., Yoon, K.-H., Lee, K.-E., Park, D.-S., Lee, Y.-J., Moon, H.-B., et al. (2009). Distribution of hepatitis B virus genotypes in Korea. Korean J. Hepatol. 15, 140–147. doi: 10.3350/kjhep.2009.15.2.140
Choe, W. H., Kim, K., Lee, S.-Y., Choi, Y.-M., Kwon, S. Y., Kim, J. H., et al. (2019). Tenofovir is a more suitable treatment than entecavir for chronic hepatitis B patients carrying naturally occurring rtM204I mutations. World J. Gastroenterol. 25, 4985–4998. doi: 10.3748/wjg.v25.i33.4985
da Fonseca, J. C. F. (2010). History of viral hepatitis. Rev. Soc. Bras. Med. Trop. 43, 322–330. doi: 10.1590/S0037-86822010000300022
Datta, S., Chatterjee, S., Veer, V., and Chakravarty, R. (2012). Molecular biology of the hepatitis B virus for clinicians. J. Clin. Exp. Hepatol. 2, 353–365. doi: 10.1016/j.jceh.2012.10.003
Demarchi, L. H. F., Bandeira, L. M., Taira, D. L., Zardin, M. C. S. U., Ibanhes, M. L., Esposito, A. O. P., et al. (2022). Hepatitis B virus infection among Japanese immigrants and descendants: the need to strengthen preventive and control measures. Viruses 14:1085. doi: 10.3390/v14051085
Gao, Z. Y., Li, T., Wang, J., Du, J. M., Li, Y. J., Li, J., et al. (2007). Mutations in preS genes of genotype C hepatitis B virus in patients with chronic hepatitis B and hepatocellular carcinoma. J. Gastroenterol. 42, 761–768. doi: 10.1007/s00535-007-2085-1
Guvenir, M., and Arikan, A. (2020). Hepatitis B virus: from diagnosis to treatment. Pol. J. Microbiol. 69, 391–399. doi: 10.33073/pjm-2020-044
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Ito, K., Yoneda, M., Sakamoto, K., and Mizokami, M. (2018). Virological and clinical characteristics of hepatitis B virus genotype a. J. Gastroenterol. 53, 18–26. doi: 10.1007/s00535-017-1367-5
Jang, H., Yoon, J. S., Park, S. Y., Lee, H. A., Jang, M. J., Kim, S. U., et al. (2022). Impact of HBeAg on hepatocellular carcinoma risk during Oral antiviral treatment in patients with chronic hepatitis B. Clin. Gastroenterol. Hepatol. 20, 1343–1353.e16. doi: 10.1016/j.cgh.2021.09.001
Jeong, H., Kim, D. H., Choi, Y. M., Choi, H., Kim, D., and Kim, B. J. (2021). rt269I type of hepatitis B virus (HBV) polymerase versus rt269L is more prone to mutations within HBV genome in chronic patients infected with genotype C2: evidence from analysis of full HBV genotype C2 genome. Microorganisms. 9:601. doi: 10.3390/microorganisms9030601
Kao, J.-H., Chen, P.-J., Lai, M.-Y., and Chen, D.-S. (2000). Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 118, 554–559. doi: 10.1016/S0016-5085(00)70261-7
Kim, B.-J. (2014). Hepatitis B virus mutations related to liver disease progression of Korean patients. World J. Gastroenterol. 20, 460–467. doi: 10.3748/wjg.v20.i2.460
Kim, H., Jee, Y. M., Song, B. C., Hyun, J. W., Mun, H. S., Kim, H. J., et al. (2007). Analysis of hepatitis B virus quasispecies distribution in a Korean chronic patient based on the full genome sequences. J. Med. Virol. 79, 212–219. doi: 10.1002/jmv.20789
Kim, J.-E., Lee, S.-Y., Kim, H., Kim, K.-J., Choe, W.-H., and Kim, B.-J. (2017). Naturally occurring mutations in the reverse transcriptase region of hepatitis B virus polymerase from treatment-naïve Korean patients infected with genotype C2. World J. Gastroenterol. 23, 4222–4232. doi: 10.3748/wjg.v23.i23.4222
Kurbanov, F., Tanaka, Y., and Mizokami, M. (2010). Geographical and genetic diversity of the human hepatitis B virus. Hepatol. Res. 40, 14–30. doi: 10.1111/j.1872-034X.2009.00601.x
Kyaw, Y. Y., Lwin, A. A., Aye, K. S., Thu, H. M., Htun, M. M., Soe, H. O., et al. (2020). Distribution of hepatitis B virus genotypes in the general population of Myanmar via nationwide study. BMC Infect. Dis. 20:552. doi: 10.1186/s12879-020-05269-z
Lee, J. H., Hong, S. P., Jang, E. S., Park, S. J., Hwang, S. G., Kang, S. K., et al. (2015). Analysis of HBV genotype, drug resistant mutations, and pre-core/basal core promoter mutations in Korean patients with acute hepatitis B. J. Med. Virol. 87, 993–998. doi: 10.1002/jmv.24148
Liang, T. J. (2009). Hepatitis B: the virus and disease. Hepatology 49, S13–S21. doi: 10.1002/hep.22881
Lin, C. L., and Kao, J. H. (2011). The clinical implications of hepatitis B virus genotype: recent advances. J. Gastroenterol. Hepatol. 26, 123–130. doi: 10.1111/j.1440-1746.2010.06541.x
Lin, S. Y. C., Magalis, B. R., Salemi, M., and Liu, H.-F. (2019). Origin and dissemination of hepatitis B virus genotype C in East Asia revealed by phylodynamic analysis and historical correlates. J. Viral Hepat. 26, 145–154. doi: 10.1111/jvh.13006
Liu, B.-M., Li, T., Xu, J., Li, X.-G., Dong, J.-P., Yan, P., et al. (2010). Characterization of potential antiviral resistance mutations in hepatitis B virus reverse transcriptase sequences in treatment-naive Chinese patients. Antiviral Res. 85, 512–519. doi: 10.1016/j.antiviral.2009.12.006
Liu, Z., Zhang, Y., Xu, M., Li, X., and Zhang, Z. (2021). Distribution of hepatitis B virus genotypes and subgenotypes: a meta-analysis. Medicine 100:e27941. doi: 10.1097/MD.0000000000027941
McNaughton, A. L., Revill, P. A., Littlejohn, M., Matthews, P. C., and Ansari, M. A. (2020). Analysis of genomic-length HBV sequences to determine genotype and subgenotype reference sequences. J. Gen. Virol. 101, 271–283. doi: 10.1099/jgv.0.001387
Nakajima, S., Watashi, K., Kato, T., Muramatsu, M., Wakita, T., Tamura, N., et al. (2021). Biochemical and structural properties of Entecavir-resistant hepatitis B virus polymerase with L180M/M204V mutations. J. Virol. 95:e0240120. doi: 10.1128/jvi.02401-20
Park, Y. M., and Lee, S. G. (2016). Clinical features of HBsAg seroclearance in hepatitis B virus carriers in South Korea: a retrospective longitudinal study. World J. Gastroenterol. 22, 9836–9843. doi: 10.3748/wjg.v22.i44.9836
Schweitzer, A., Horn, J., Mikolajczyk, R. T., Krause, G., and Ott, J. J. (2015). Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386, 1546–1555. doi: 10.1016/S0140-6736(15)61412-X
Shi, W., Zhu, C., Zheng, W., Zheng, W., Ling, C., Carr, M. J., et al. (2012). Subgenotyping of genotype C hepatitis B virus: correcting misclassifications and identifying a novel subgenotype. PLoS One 7:e47271. doi: 10.1371/journal.pone.0047271
Song, B. C., Suh, D. J., Lee, H. C., Chung, Y. H., and Lee, Y. S. (2000). Hepatitis B e antigen seroconversion after lamivudine therapy is not durable in patients with chronic hepatitis B in Korea. Hepatology 32, 803–806. doi: 10.1053/jhep.2000.16665
Sugauchi, F., Mizokami, M., Orito, E., Ohno, T., Kato, H., Suzuki, S., et al. (2001). A novel variant genotype C of hepatitis B virus identified in isolates from Australian aborigines. J. Gen. Virol. 82, 883–892. doi: 10.1099/0022-1317-82-4-883
Sunbul, M. (2014). Hepatitis B virus genotypes: global distribution and clinical importance. World J. Gastroenterol. 20, 5427–5434. doi: 10.3748/wjg.v20.i18.5427
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. doi: 10.1093/oxfordjournals.molbev.a040023
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tang, L. S., Covert, E., Wilson, E., and Kottilil, S. (2018). Chronic hepatitis B infection: a review. JAMA 319, 1802–1813. doi: 10.1001/jama.2018.3795
Tong, M. J., Blatt, L. M., Kao, J.-H., Cheng, J. T., and Corey, W. G. (2007). Basal core promoter T1762/A1764 and precore A1896 gene mutations in hepatitis B surface antigen-positive hepatocellular carcinoma: a comparison with chronic carriers. Liver Int. 27, 1356–1363. doi: 10.1111/j.1478-3231.2007.01585.x
Velkov, S., Ott, J. J., Protzer, U., and Michler, T. (2018). The global hepatitis B virus genotype distribution approximated from available genotyping data. Genes 9:495. doi: 10.3390/genes9100495
Wang, H. Y., Li, D., Liu, W., Jin, X., Du, B., Li, Y. P., et al. (2010). Hepatitis B virus subgenotype C2 is the most prevalent subgenotype in Northeast China. Clin. Microbiol. Infect. 16, 477–481. doi: 10.1111/j.1469-0691.2009.02834.x
Yim, S. Y., and Kim, J. H. (2019). The epidemiology of hepatitis B virus infection in Korea. Korean J. Intern. Med. 34, 945–953. doi: 10.3904/kjim.2019.007
Yin, Y., He, K., Wu, B., Xu, M., Du, L., Liu, W., et al. (2019). A systematic genotype and subgenotype re-ranking of hepatitis B virus under a novel classification standard. Heliyon 5:e02556. doi: 10.1016/j.heliyon.2019.e02556
Keywords: hepatitis B virus (HBV), subgenotype C2, C2(3), South Korea, sV184A
Citation: Kim DH, Choi Y-M, Jang J and Kim B-J (2023) Global prevalence and molecular characteristics of three clades within hepatitis B virus subgenotype C2: Predominance of the C2(3) clade in South Korea. Front. Microbiol. 14:1137084. doi: 10.3389/fmicb.2023.1137084
Edited by:
Anna Rosa Garbuglia, National Institute for Infectious Diseases Lazzaro Spallanzani (IRCCS), ItalyReviewed by:
Yongshui Fu, Guangzhou Blood Center, ChinaCopyright © 2023 Kim, Choi, Jang and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bum-Joon Kim, a2J1bWpvb25Ac251LmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.