 Tingting Ren1,2
Tingting Ren1,2 Wei Su
Wei Su Yingchun Mu
Yingchun Mu- 1School of Liquor and Food Engineering, Guizhou University, Guiyang, China
- 2Guizhou Provincial Key Laboratory of Fermentation Engineering and Biological Pharmacy, Guizhou University, Guiyang, China
The Chishui River basin is the main production area of the sauce-flavor Baijiu. Due to the particularity of sauce-flavor Baijiu technology, a large site of workshops needs to be built for brewing and storage. Therefore, used the natural karst caves of Guizhou province to manufacture the sauce-flavor Baijiu, which has enriched the connotation of sauce-flavor Baijiu and saved valuable land resources. In this study, the fermentation grains in the seven stages during the Xiasha round of the cave-brewed sauce-flavor Baijiu (CBSB) were detected using a combination of physicochemical analysis, Headspace solid-phase microextraction gas chromatography-mass detection, and Illumina HiSeq sequencing methods. The results showed Unspecified_Leuconostocaceae, Weissella, Unspecified_Bacillaceae, Saccharomycopsis, Thermomyces, and Unspecified_Phaffomycetaceae were the main bacterial and fungal genera in the stacking fermentation (SF). In the cellar fermentation (CF), the Lactobacillus, Unspecified_Lactobacillaceae, Thermoactinomyces, Saccharomycopsis, Unspecified_Phaffomycetaceae, and Wickerhamomyces were the main bacterial and fungal genera. A total of 72 volatiles were detected in the fermented grains. Linear discriminant analysis Effect Size (LEfSe) identified 23 significantly different volatile metabolites in the fermentation process, including 7 esters, 6 alcohols, 4 acids, 3 phenols, 1 hydrocarbon, and 2 other compounds. Redundancy analysis was used to explore the correlation between dominant microbial genera and physicochemical properties. Starch was the main physicochemical property affecting microbial succession in the SF. Acidity, moisture, and reducing sugar were the main driving factors of microbial succession in the CF. The Pearson correlation coefficient revealed the correlation between dominant microbial genera and significantly different volatile flavor substances. A total of 18 dominant microbial genera were associated with significantly different volatile metabolites, Lactobacillus, Weissella, Wickerhamomyces, and Aspergillus were shown to play crucial roles in metabolite synthesis. On this basis, a metabolic map of the dominant microbial genera was established. This study provides a theoretical basis for the production and quality control of sauce-flavor Baijiu brewed in natural karst caves and lays a foundation for studying the link between flavor formation and microorganisms.
1. Introduction
Distilled spirit has been used in China for over 2,000 years and occupies a very unmatched place in traditional Chinese culture (Liu and Sun, 2018). As one of the most complex and typical Baijiu in traditional Chinese Baijiu, sauce-flavor Baijiu is popular with many consumers (Wang Y. R. et al., 2021). The production process is based on a yearly production cycle, with two feeds, nine steams, eight fermentations, and seven distillations (Wang M. Y. et al., 2018). Due to its unique high-temperature fermentation process and particular environment, sauce-flavor Baijiu is distinguished from others by its complex flavors (Wang et al., 2017). The fermentation of sauce-flavor Baijiu is a complex process involving microbial interactions (Jin et al., 2017) that are influenced mainly by the region they are produced (Bokulich et al., 2014). Sauce-flavored Baijiu relies on unrepeatable ecological resources, and its main production areas are concentrated in Renhuai, Xishui, and Jinsha, of the Guizhou Province, China. Meanwhile, the Chishui River basin is the prime production area of the sauce-flavor Baijiu. The distinct manufacturing process of sauce-flavor Baijiu requires a large area for brewing and storage. The unique natural caves formed by the Guizhou karst terrain can be used for this purpose. Located on the banks of the Chishui River, the Guixian Cave is currently the only natural cave in the country for brewing sauce-flavor Baijiu. The cave's relatively stable temperature and humidity throughout the four seasons provide the unique conditions required for manufacturing cave-brewed sauce-flavor Baijiu (CBSB), thereby giving it more connotations.
The first grain-feeding stage of sauce-flavor Baijiu brewing is called the Xiasha round. Sorghum was gelatinized and decomposed to produce various flavors and precursor substances, which lays a foundation for the follow-up brewing process (Hao et al., 2021). The whole fermentation process is divided into two processes: stacking fermentation (SF) and cellar fermentation (CF). The physical and chemical properties (temperature, moisture, acidity, reducing sugar, starch) of the process are essential factors that affect the evolution of the microbial community (Wang H. et al., 2021). The microorganisms in fermented grains come from high-temperature Daqu and the production environment, including air, soils, water, and production tools (Wang et al., 2019; Zhang H. X. et al., 2021a). Bacteria can use protein and starch and contribute significantly to Baijiu's flavor formation (Zuo et al., 2020; Tu et al., 2022). Yeast mainly produces various flavor compounds and alcohol (Hu et al., 2020). Molds can secrete different enzymes to decompose raw materials and provide energy for the growth of other bacterial groups (Wang et al., 2014; Hu Y. L. et al., 2021).
Recent research on Chinese Baijiu has focused on the succession of microbial communities, the screening of functional microorganisms, and the correlation between the microbial communities and the physicochemical properties and flavor substances (Zou et al., 2018a; Jin et al., 2019; Song et al., 2019; Mu et al., 2022). These studies have revealed the core functional microorganisms of the fermentation process of Chinese Baijiu and their contribution to the flavor substances, laying a solid foundation for Chinese Baijiu research. However, these studies are all performed on the fermentation process in a conventional environment distillery, and rare research on brewing Baijiu in natural caves has been reported.
Physicochemical analysis, high-throughput sequencing, and headspace solid-phase micro-extraction gas chromatography-mass spectrometry (HS-SPME-GC-MS) were carried out to analyze the physicochemical properties, the microbial community structure, and the volatile flavor substances. In addition, redundancy analysis (RDA) and the Pearson coefficient analysis were performed to assess the correlation between microbiota and physicochemical properties with the volatile flavor substances. The results enabled metabolic mapping of the dominant genera during the Xiasha round of CBSB. This study can provide a theoretical basis for regulating the CBSB fermentation process.
2. Materials and methods
2.1. Sample collection
The fermented grain samples were manufactured between September 2020 and October 2020 during the Xiasha round at the Dongniangdongcang Winery, Guizhou Province, China. The fermentation process was carried out in two distinct phases: SF and CF. Samples of SF were collected on days 0, 1, 2, and 3 and marked as SF0, SF1, SF2, and SF3, respectively. For the CF stage, the samples were collected at 10-day intervals until the end of fermentation and marked as CF10, CF20, and CF30. The SF and CF sampling diagrams are shown in Figures 1A, B, respectively. The samples were collected from the upper, middle, and bottom layers of the fermentation piles and cellars at each sampling time point. One point was selected in the middle and four points at the edge of each layer, and at each point, 30 g of samples was collected. All samples collected from each point of all three layers were then evenly combined into one mixture to eliminate sampling error (Zhao et al., 2021). After mixing, they were immediately packed in a sterile sealed bag and stored in a low-temperature refrigerator at −80°C until analysis.

Figure 1. Spoil sampling points (A) sketch map of stacking fermentation sampling; (B) sketch map of cellar fermentation sampling.
2.2. Analysis of physicochemical indicators
To understand the fermentation processes, temperature, moisture, acidity, reducing sugar, and starch were detected. An alcohol thermometer was inserted into the fermented grains using a self-made tool, and the temperature was measured quickly after 5 min. The moisture of fermented grains was determined by a gravimetric method by drying samples to a constant weight at 105°C. The titratable acidity was determined by titration with a standard NaOH (0.1 mol/L) solution. A standard glucose solution (1.0 g/L) was used to titrate reducing sugar and starch in the fermented grains.
2.3. Analysis of volatile flavor substances
The fermented grain sample was ground and mixed, and 1.00 g of it was placed in a 20 ml headspace flask with 2 g of NaCl and 5 ml of purified water. Then, 10 μl of cyclohexanone (20 μg/ml) was added as the internal standard. The SPME fibers were extracted by 50/30 μm DVB/CAR/PDMS fiber (Sigma Aldrich, USA) at 40°C for 180 min, inserted into the injection port, and then separated by resolution at 230°C for 5 min.
Chromatographic conditions: A gas chromatography-mass spectrometer was used to analyze the fermented grain (Thermo Fisher Scientific, USA). The capillary column was DB-5MS (30 m × 0.25 mm × 0.25 μm, Agilent, USA) with helium (99.999%) as the carrier gas at a 1.0 ml/min flow rate in the non-split mode. Heating procedure: The column was maintained at 40°C for 5 min, then increased to 150°C at 5°C/min, later maintained at 150°C for 3 min, and finally increased to 240°C at 5°C/min and maintained for 5 min. Mass spectrometry conditions: electron bombardment ion source (EI), electron energy of 70 eV, a transmission line temperature of 280°C, and an ion source temperature of 230°C. The data were collected in the 50–450 amu at a rate of 1 scan/s. The constituents were tentatively identified by matching the mass spectrum with the NIST5 spectrum database and comparing their Kováts retention index (RI) with the RI reported in the literature, calculated using the C7–C40 n-alkanes, for verification.
2.4. Analysis of the microbial community
Total genomic DNA was extracted with the FastDNA® Spin Kit according to the manufacturer's protocol. DNA purity and concentration were checked using a NanoDrop 2000 ultra-micro spectrophotometer, followed by 1% agarose gel electrophoresis at 5 V/cm for 20 min to check DNA integrity. The V3-V4 region of the bacterial 16S rRNA gene was amplified with primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′), and the ITS1 region of the fungi was amplified with the primers ITS1F (5′- CTTGGTCATTTAGAGGAAGTAA-3′) and ITS1R (5′-GCTGCGTTCTTCATCGATGC-3′).
The total PCR amplification volume was 20 μl, including 0.8 μl of the forward primer (5 μm), 0.8 μl of the reverse primer (5 μm), 2 μl of 2.5 mm dNTPs, 2 μl of 10 × buffer, 0.2 μl of rTaq polymerase, 0.2 μl of BSA, 10 ng of template DNA, and 20 μl of ddH2O. The PCR conditions were as follows: initial denaturation at 95°C for 3 min, denaturation at 95°C for 30 s, annealing at 53°C and 55°C for 30 s for bacteria and fungi, respectively, followed by extension at 72°C for 45 s, 29 and 35 cycles for bacteria and fungi, respectively, and final extension at 72°C for 10 min and cooling to 10°C to end the reaction. All PCR products were tested for concentration and purity using NanoDrop2000, and 2% agarose gel electrophoresis was used for integrity testing.
The libraries were quantified and tested by Qubit. Then, the purified amplicons were pooled in equimolar amounts and paired-end sequenced (2 × 300) by using an Illumina MiSeq platform (Illumina, San Diego, USA) according to the standard protocols. Each sample was quality filtered, trimmed, denoised, and merged. Then, the chimeric sequences were identified and removed using the QIIME2 dada2 plugin to obtain the feature table of the amplicon sequence variant (ASV) (Callahan et al., 2016). Taxonomic annotation was carried out on the obtained ASVs based on the SILVA (bacteria) and UNITE (fungi) taxonomic databases.
2.5. Statistical analysis
The experiments were repeated three times; data analyses were performed using IBM SPSS statistics 26.0, and the obtained data were analyzed by ANOVA with a P-value of < 0.05 as the significance level. A principal component analysis was performed using SIMCA (version 14.1), and the graphs were drawn using Origin (version 9.8). Visualized correlation data painting was performed using the Cytoscape software (version 3.8.2).
3. Results and analysis
3.1. Changes in the physicochemical properties
As shown in Table 1, due to the enrichment and rapid growth of microorganisms, the temperature reached a maximum of 45.67 ± 8.02°C on day 3 of the SF process. We observed the time required by the sample to reach the highest temperature in the SF stage and found that it was consistent with that of the sauce-flavor Baijiu brewed in a conventional environment distillery (Hao et al., 2021). The acidity increased significantly (P < 0.05) on day 3 of SF, and then, the increase was more significant in the CF stage, which may be due to the accumulation of acid produced by the microorganisms during anaerobic fermentation (Cai et al., 2019). The starch content decreased significantly (P < 0.05) on day 1 of SF from 40.96 ± 0.04% to 39.24 ± 0.30%, with a decreasing trend in the CF stage. The reduced sugar content increased significantly (P < 0.05) on day 2 in the SF stage and significantly decreased in the CF stage (P < 0.05). The rapid growth and multiplication of microorganisms in the SF stage consumed starch to produce monosaccharides, which were further used to produce more flavors during the CF stage (Wu et al., 2013; Hu X. L. et al., 2021). The growth and reproduction of microorganisms produced a large amount of biological heat (Cai et al., 2019). Therefore, the moisture content tended to decrease during the SF process. The increased moisture content during CF was related to the interaction between microorganisms (Huang et al., 2020; Hu Y. L. et al., 2021).
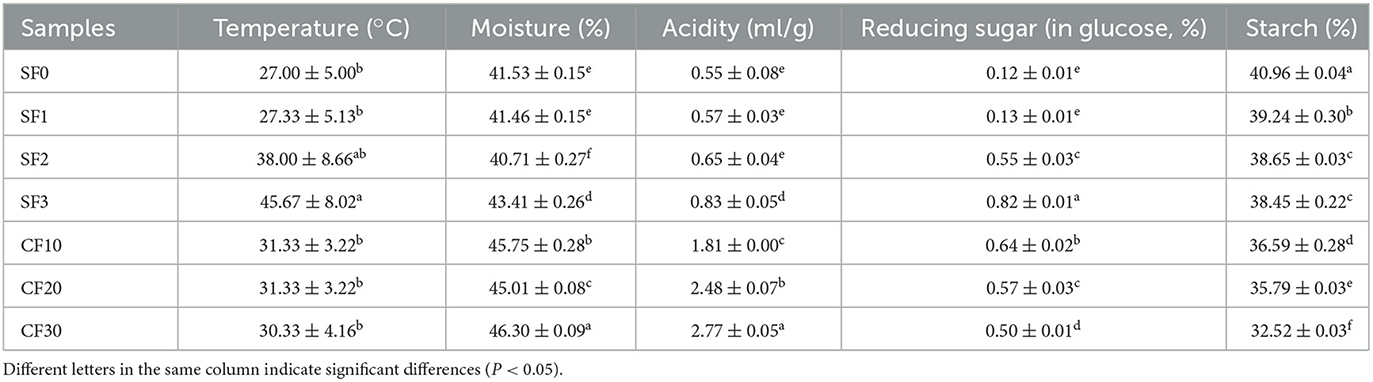
Table 1. Changes in the physicochemical properties during the fermentation process.
3.2. Differences in the microbial community structure during the fermentation process
The changes in the microbial community structure of the 21 samples from 7 time points were detected. The effective sequences of the bacteria and fungi ranged from 23,824 to 38,841 and 16,487 to 39,450, respectively. Although the number of fungal sequences in the samples was greater than that of bacteria, the total number of bacterial ASV was much greater than that of fungi (Supplementary Table S1). In addition, the sparsity curve reached a saturation plateau, indicating that the sequencing depth can represent the microbial diversity of the samples (Supplementary Figure S1). The samples were analyzed for α-diversity, and Supplementary Table S2 lists the species richness and diversity parameters, including the Chao1, Shannon, and Simpson indices. The diversity indices were consistent with the ASV results. Bacterial richness was greater than fungi in the SF, while the opposite was true in the CF stage. During the CF stage, bacterial diversity decreased but fungal diversity increased.
The results showed that, among 19 bacterial phyla and 185 bacterial genera, Firmicutes was the dominant bacterial phylum (90.18–99.73%). We defined the top 10 genera in relative abundance as the dominant microorganism genera to study community succession during fermentation (Figure 2). In all samples, the dominant bacterial genus contributed to 95.28% of the bacterial sequences, as shown in Figure 2A. The dominant microbial genera include Lactobacillus (28.46%), Unspecified_Leuconostocaceae (17.63%), Weissella (10.65%), Unspecified_Bacillaceae (9.60%), Unspecified_Lactobacillaceae (7.63%), Thermoactinomyces (6.64%), Pediococcus (6.43%), Bacillus (3.83%), Oceanobacillus (2.79%), and Leuconostoc (1.62%).
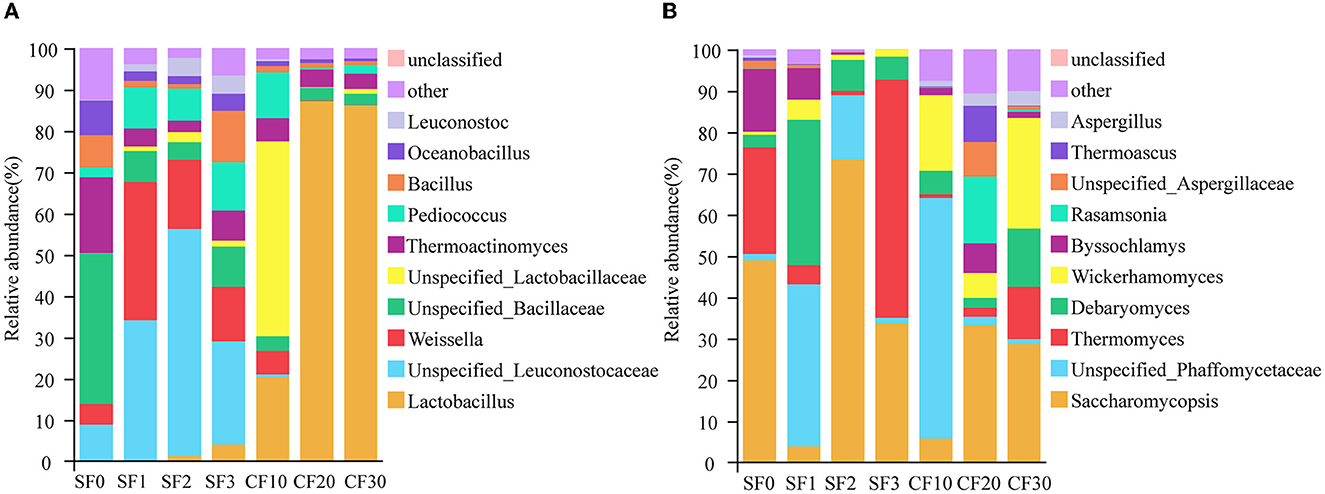
Figure 2. Relative abundance of bacterial (A) and fungal (B) genera in the Xiasha round of the cave-brew sauce-flavor Baijiu.
The bacterial community in the fermented grains changed significantly during the fermentation process (Figure 2A). Unspecified_Bacillaceae and Thermoactinomyces were the dominant genera during the SF1, which might be derived from high-temperature Daqu (Zuo et al., 2020). As SF proceeded, Unspecified_Leuconostocaceae and Weissella gradually replaced Unspecified_Bacillaceae and Thermoactinomyces as the dominant genera between days 1 and 3. The reason for the change in Weissella abundance may be due to the floor, tools, etc., in the operating environment (Wang X. S. et al., 2018). After the end of SF and into the cellars, which was anaerobic fermentation, the dominant bacterial genera were Lactobacillus, Unspecified_Lactobacillaceae, and Pediococcus at day 10 of CF. These acid-tolerant microorganisms gradually replaced the non-acid-tolerant ones as the acidity of the anaerobic fermentation system increased. During days 20–30 of CF, Lactobacillus became the dominant microbial genus. Lactobacillus can produce lactic acid and a variety of antimicrobial substances to inhibit the growth of pathogens and microorganism spoilage during brewing (Galati et al., 2014). Therefore, the abundance of some non-acid-tolerant and pathogenic bacteria, such as Enterococcus spp. and Klebsiella spp., was gradually decreased as Lactobacillus became the dominant bacteria genus during the fermentation process.
In the previous study of the conventional environment distillery, Wang et al. (2015) found that the main bacterial groups of sauce-flavor Baijiu SF and CF were Bacillaceae and Lactobacillales. In this study, we found that the dominant bacteria family was Leuconostocaceae, which was different from the conventional environment distillery of the SF. However, in the anaerobic fermentation, the dominant bacteria order also was Lactobacillales, which was the same as the conventional environment distillery in the CF. This indicates that, with the change in the environment, there is a change in the dominant microorganisms in the SF but has little impact on the CF.
A total of six fungal phyla and 102 fungal genera were identified, and Ascomycota was the dominant phylum (96.80–100.00%). Meanwhile, Saccharomycopsis (32.44%), Unspecified_Phaffomycetaceae (17.05%), Thermomyces (15.00%), Debaryomyces (10.52%), Wickerhamomyces (8.51%), Byssochlamys (4.86%), Rasamsonia (2.40%), Unspecified_Aspergillaceae (1.74%), Thermoascus (1.44%), and Aspergillus (1.22%) were the dominant fungal genera. During the SF, Saccharomycopsis, Thermomyces, Unspecified_Phaffomycetaceae, and Debaryomyces were predominant, accounting for 89.49% of the total abundance. The CF's main genera were Saccharomycopsis, Wickerhamomyces, and Unspecified_Phaffomycetaceae, accounting for 60.01% of the total abundance. The dominant fungi during SF and CF of sauce-flavor Baijiu in conventional environments were Pichia, Saccharomyces, and unidentified (Hao et al., 2021). The Xiasha round is the non-wine-producing round in the production cycle of CBSB. The role of microorganisms is mainly focused on the process of aroma production and precursor substance production (Wang M. Y. et al., 2018). For example, Saccharomycopsis, which is more abundant, although it is not a significant brewing yeast, produces a variety of enzymes to break down starch and is vital in terms of flavor determination and aroma complexity of the final product (Padilla et al., 2016; Boro et al., 2022). Wang H. et al. (2021) analyzed microbiome diversity and evolution during SF for seven rounds in conventional environment distilleries and found that the dominant fungal were Thermomyces, Byssochlamys, Thermoascus, unclassified_o_Eurotiales, and Aspergillus. Previous studies showed that the fungi in fermentation mainly come from the ground of the fermentation environment (Zhang H. X. et al., 2021a). Therefore, we suppose that the environment is the most crucial factor that affects the fungal community structure in the fermentation process of sauce-flavor Baijiu.
The community structure of microorganisms is closely related to the interactions between microorganisms (Wang Y. R. et al., 2021). Pearson correlation coefficients and p-values were calculated to evaluate the interactions at the microbial genus level whose relative abundance was >1%. We found 20 nodes and 33 edges (Supplementary Figure S2). The results of the correlation analysis between bacterial genera showed that Lactobacillus was significantly negatively correlated with Oceanobacillus and Unspecified_Leuconostocaceae (P < 0.01, r < −0.5). Lactobacillus is often found as the dominant bacteria in Baijiu fermentation, which has a regulatory function on the microbial flora in the Baijiu fermentation system and has a positive role in the production of Baijiu flavor substances (Cai et al., 2021; Wang H. et al., 2021). It can produce acetic acid by heteromorphic lactic acid fermentation during anaerobic metabolism (Sifeeldein et al., 2018). Bacillus was significantly positively correlated with Thermoactinomyces, Saccharopolyspora, Oceanobacillus, and Unspecified_Bacillaceae (P < 0.05, r > 0.5). Bacillus was identified as the dominant bacteria in the previous studies on the sauce-flavor Baijiu (Hao et al., 2021; Zhang H. X. et al., 2021b). In this study, it is not surprising that the average relative abundance of Bacillus has yet reached the maximum at the end of the SF. Bacillus can secrete various enzymes to degrade the fermentation substrate to meet its growth and produce Baijiu flavor substances (Cai et al., 2021; Xiao et al., 2021). The correlation results between fungi showed that Aspergillus was significantly positively correlated with Wickerhamomyces (P < 0.05, r > 0.5). In Baijiu fermentation, Aspergillus is a functional microorganism with a good saccharification effect (Wu et al., 2021) and is critical for flavor compound production (Jin et al., 2019; Liu et al., 2021). Saccharomycopsis was significantly negatively correlated with Unspecified_Phaffomycetaceae (P < 0.01, r < −0.5). Saccharomycopsis was the dominant fungal genus in high-temperature Daqu and plays an essential role in the formation of the unique flavor of Baijiu (Jiang et al., 2021; Zeng et al., 2022).
The results of the correlation between bacteria and fungi showed that Wickerhamomyces and Aspergillus were significantly positively correlated with Lactobacillus (P < 0.01, r > 0.5). Wickerhamomyces and Aspergillus can produce multiple enzymes to decompose raw materials and create conditions for the rapid growth of Lactobacillus. Thermomyces was significantly positively correlated with Saccharopolyspora and Bacillus (P < 0.05, r > 0.5). It can survive high temperatures and produce enzymatic decomposition materials (Liu and Miao, 2020). Saccharopolyspora and Bacillus can produce important biologically active substances to make the flavor more complex (Jiang et al., 2021). Aspergillus was significantly negatively correlated with Weissella and Unspecified_Leuconostocaceae (P < 0.01, r < −0.5), indicating that Aspergillus had an antagonistic effect on both. The flavor and quality of Baijiu can be affected by microbial interaction and the regulation of microbial structure (Zou et al., 2018b; Gao et al., 2022). In summary, these microorganisms coordinate and control each other during the fermentation process in the Xiasha round of CBSB. These mutual biological relationships constitute the primary biological control mechanism of the fermentation process of the CBSB, which is also the fundamental component of flavor formation.
3.3. Dynamic changes in flavor substances in fermented grains
In total, 72 volatile flavor substances were detected, including 23 esters, 13 alcohols, 11 acids, 8 phenols, 6 hydrocarbons, 4 aldehydes, and 7 other compounds (Supplementary Table S3). As shown in Figure 3A, the clusters on days 0, 1, 2, and 3 in SF were clearly different from those on day 10 of CF. This result is mainly due to changes in the oxygen level of the fermentation environment that eventually led to changes in the microbiota (Figure 2) and rapid changes in the metabolites in the fermented grains. Moreover, in the CF stage, the fermentation progressed very rapidly for the first 20 days and then changed slowly. This rapid metabolic change may be related to the favorable temperature and sufficient oxygen content available during CF for the first 20 days.
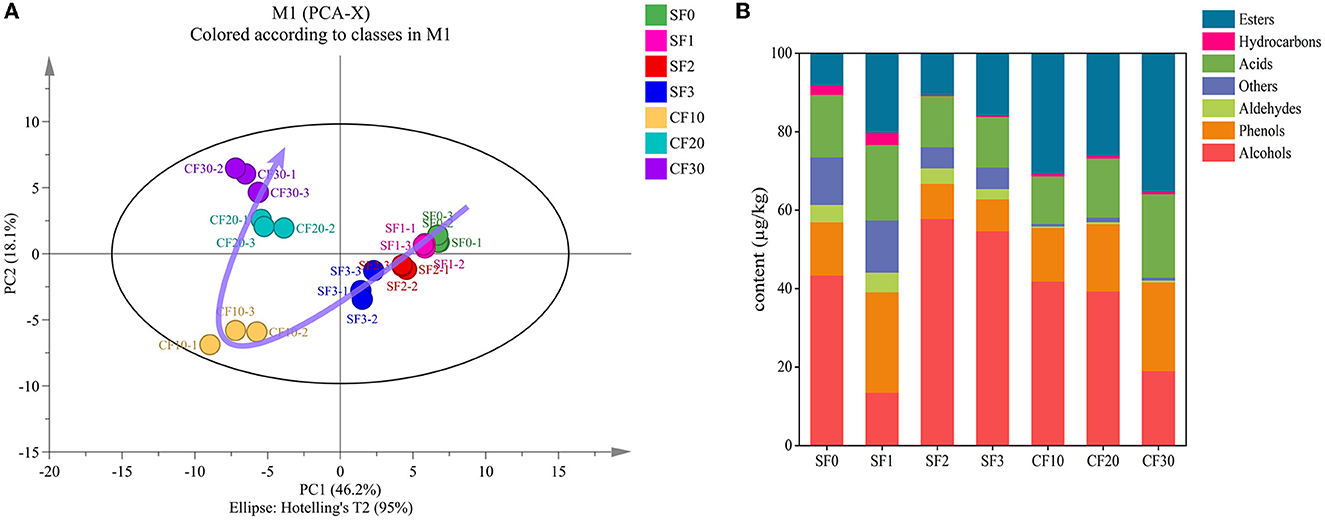
Figure 3. Principal component analysis of volatile compounds (A) and percent stacked column chart of volatile flavor substances (B).
The changes in the contents of volatile compounds during fermentation are shown in Figure 3B and Supplementary Table S4. Esters, acids, and phenols showed an increasing trend during the SF stage. Meanwhile, the alcohol content decreased significantly on day 1, which is closely related to the increase in other flavor substances. During SF, the alcohol content increased with the extension of fermentation time (Wei et al., 2020). While the high phenol content in the early period may be associated with the raw material sorghum (Rao et al., 2018; Li et al., 2021), the higher content of esters in the later period might be related to the accumulation of ester precursors and the production of metabolites by microorganisms (Xu et al., 2022). The significant increase in alcohol content in the CF10 might be caused by the rapid growth and reproduction of alcohol-producing yeast such as Saccharomyces (Cai et al., 2019). As oxygen depletion and the available substances inhibited the production of alcohol, the alcohol content decreases significantly. With the rapid growth of acid-producing microorganisms, the growth of ester-producing microorganisms is inhibited (Wu et al., 2017). Therefore, the content of esters decreased significantly in CF20. The contents of esters, phenols, and aldehydes were increased in CF30, which might be due to the transformation of alcohols into other substances by microorganisms and enzymes (Dai et al., 2020; Hong et al., 2020).
LEfSe analysis was performed to identify significantly different metabolites in each stage in the Xiasha round of the CBSB. A total of 23 compounds were significantly different in the Xiasha round of the CBSB (LDA = 4, P < 0.05) (Figure 4).
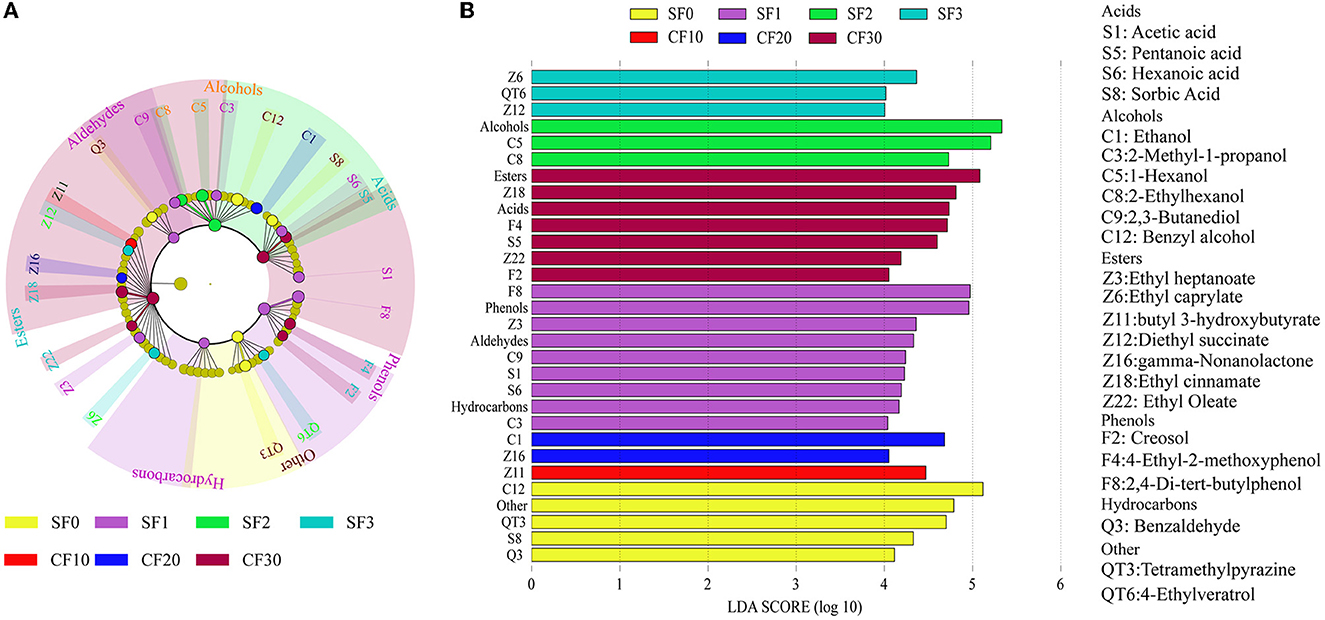
Figure 4. The LEfSe analysis of the flavor components in the Xiasha round of cave-brew sauce-flavor Baijiu. Cladogram (A) and LDA bar graph (B) results for flavor metabolite contents. Flavor compounds were evaluated by linear discriminant analysis coupled with effect size (LDA = 4, P < 0.05).
Ethyl heptanoate (Z3), ethyl caprylate (Z6), butyl 3-hydroxybutyrate (Z11), diethyl succinate (Z12), gamma-nonalactone (Z16), ethyl cinnamate (Z18), and ethyl oleate (Z22) were the differential eaters (Figure 4A). Among them, diethyl succinate, gamma-nonalactone, and ethyl oleate were detected throughout the fermentation process. Esters in Baijiu were usually synthesized by microbial metabolic pathways, with a small percentage coming from environmental and spontaneous chemical reactions (Xu et al., 2021). The levels of ethyl heptanoate, ethyl octanoate, and ethyl cinnamate gradually increased with increasing fermentation time during the Xiasha round, reaching a maximum on day 10 of CF and then decreased. Butyl 3-hydroxybutyrate was detected on day 0 and day 1 of SF and on days 20 and 30 of CF, which was not detected the rest of the time. Among them, ethyl heptanoate, ethyl octanoate, and ethyl cinnamate were not detected in the SF0, and their content increased with fermentation. This was sufficient to prove that these three esters were produced by microbial metabolism and esterification during the fermentation process (Dong et al., 2022). Moreover, because of the high content of ethanol produced during the fermentation of Chinese Baijiu, ethanol and acid undergo biochemical reactions to produce esters, which may also account for the great variety and concentration of ethyl esters. It can add fruit and floral aromas to Baijiu and make Baijiu taste soft, sweet, and astringent (Xu et al., 2022).
Fatty acids are crucial for the flavor of Chinese Baijiu, and microorganisms can synthesize low-molecular-weight fatty acids such as butyric, valeric, hexanoic acid, and octanoic acids, which are precursors of ester compounds (Wei et al., 2020). During the Xiasha round of CBSB, acetic acid (S1), pentanoic acid (S5), hexanoic acid (S6), and sorbic acid (S8) are the differential metabolites (Figure 4). While acetic acid and hexanoic acid were detected throughout the fermentation process, pentanoic acid was detected only on day 2 of SF and on day 10 of CF. In contrast, sorbic acid was detected only at the beginning and end of fermentation. Strong-flavored Baijiu is rich in hexanoic acid, which is a volatile flavoring substance that contributes much to the flavor (Fan et al., 2019). The acetic acid content was the highest throughout the fermentation stage. Although acetic acid produced a pungent odor, it could increase the flavor of Chinese Baijiu upon its combination with alcohol to produce esters, such as ethyl acetate (Cai et al., 2019).
Alcohols are auxiliary agents that coordinate Baijiu aroma and taste (Zhang M. Z. et al., 2021). In the differential alcohol metabolites during the Xiasha round of CBSB (Figure 4), the ethanol content showed an increasing trend during SF and CF, and it is an essential indicator of the fermentation performance of Baijiu (Wei et al., 2021). Furthermore, the higher alcohols 2-methyl-1-propanol (C3), benzyl alcohol (C12), 1-hexanol (C5), 2,3-butanediol (C9), and 2-ethylhexanol (C8) are present in many alcoholic beverages. Moderate amounts of higher alcohols increase the flavor and taste of Baijiu (Han et al., 2014).
Creosol (F2), 4-ethyl-2-methoxyphenol (F4), 2,4-di-tert-butylphenol (F8), tetramethylpyrazine (QT3), 4-ethylveratrol (QT6), and benzaldehyde (Q3) were also differential metabolites found during the Xiasha round of CBSB (Figure 4A). Creosol, 4-ethyl-2 methoxyphenol, and 2,4-di-tert-butylphenol showed an increasing trend during fermentation, while tetramethylpyrazine showed a decreasing trend. Benzaldehyde was detected throughout the fermentation stages, whereas 4-ethylveratrol was detected only in a few fermentation stages. Although these substances are a small part of the fermentation process, like phenolics, they are antioxidants and have beneficial effects on human health (Rao et al., 2018).
3.4. Correlation of microorganisms with physicochemical properties and volatile compounds
The effect of physicochemical characteristics on the dominant bacterial and fungal genera was revealed by the redundancy analysis (RDA), with the first two axes explaining 71.18 and 71.17% of the variations in community composition, respectively (Figures 5A, B). As shown in Figure 5C, among the bacterial communities, Unspecified_Leuconostocaceae, Weissella, Unspecified_Bacillaceae, Oceanobacillus, and Thermoactinomyces were significantly positively correlated with starch but negatively with acidity. Only Lactobacillus showed a highly significant negative correlation with starch (P < 0.001) but showed a highly significant positive correlation with acidity and moisture (P < 0.001). Bacillus and Unspecified_Bacillaceae were significantly positively and negatively correlated with temperature and reducing sugar, respectively. It shows that high moisture content and acidity can inhibit the growth of other microorganisms. Regarding fungi, Wickerhamomyces and Aspergillus showed a significant negative correlation with starch (P < 0.001) and a positive correlation with moisture and acidity (P < 0.001) (Figure 5C). Temperature showed a significant positive correlation and a significant negative correlation with Thermomyces and Byssochlamys, respectively. Only Saccharomycopsis has a negative correlation with moisture. Both Debaryomyces and Byssochlamys showed a significantly negative correlation with reducing sugar. Only Byssochlamys showed a significant positive correlation with starch. Lactobacillus and Aspergillus had a highly significant positive correlation with moisture and acidity, consistent with the research result of nong sauce–flavored Baijiu in a conventional environment (Zhuansun et al., 2022). They have a highly significant negative correlation with starch. These illustrate that they may all be related to the breakdown of starch. Aspergillus is an important filamentous fungus that produces a variety of enzymes to degrade starchy material into fermentable sugars during the fermentation of Chinese Baijiu (Chen et al., 2014). The RDA showed that microorganisms producing amylase have a significant influence on starch in the SF. At the same time, after entering the CF, the monosaccharide produced by starch decomposition provided the conditions for the growth of other microorganisms and influenced the acidity changes. Based on these results, starch was the main physicochemical property factor that affects microbial succession in the SF stage. At the same time, acidity, moisture, and reducing sugar were the main driving factors of microbial succession in the CF stage. This is different from the research results of the traditional brewing environment, in that reducing sugar is the key factor affecting the microbial succession during the SF process of sauce-flavor Baijiu, while acidity, alcoholicity, and temperature are the main factors affecting the microbial succession during the CF process (Hao et al., 2021). Previous research results of light flavor Baijiu showed that acidity, reducing sugar, and temperature were the decisive factors affecting its bacterial succession (Shen et al., 2021). During the fermentation of strong-flavor Baijiu, starch, moisture, and pH significantly affected microbial succession (Guan et al., 2020). These dominant microorganisms were regulated by the physicochemical properties by influencing the physicochemical properties.
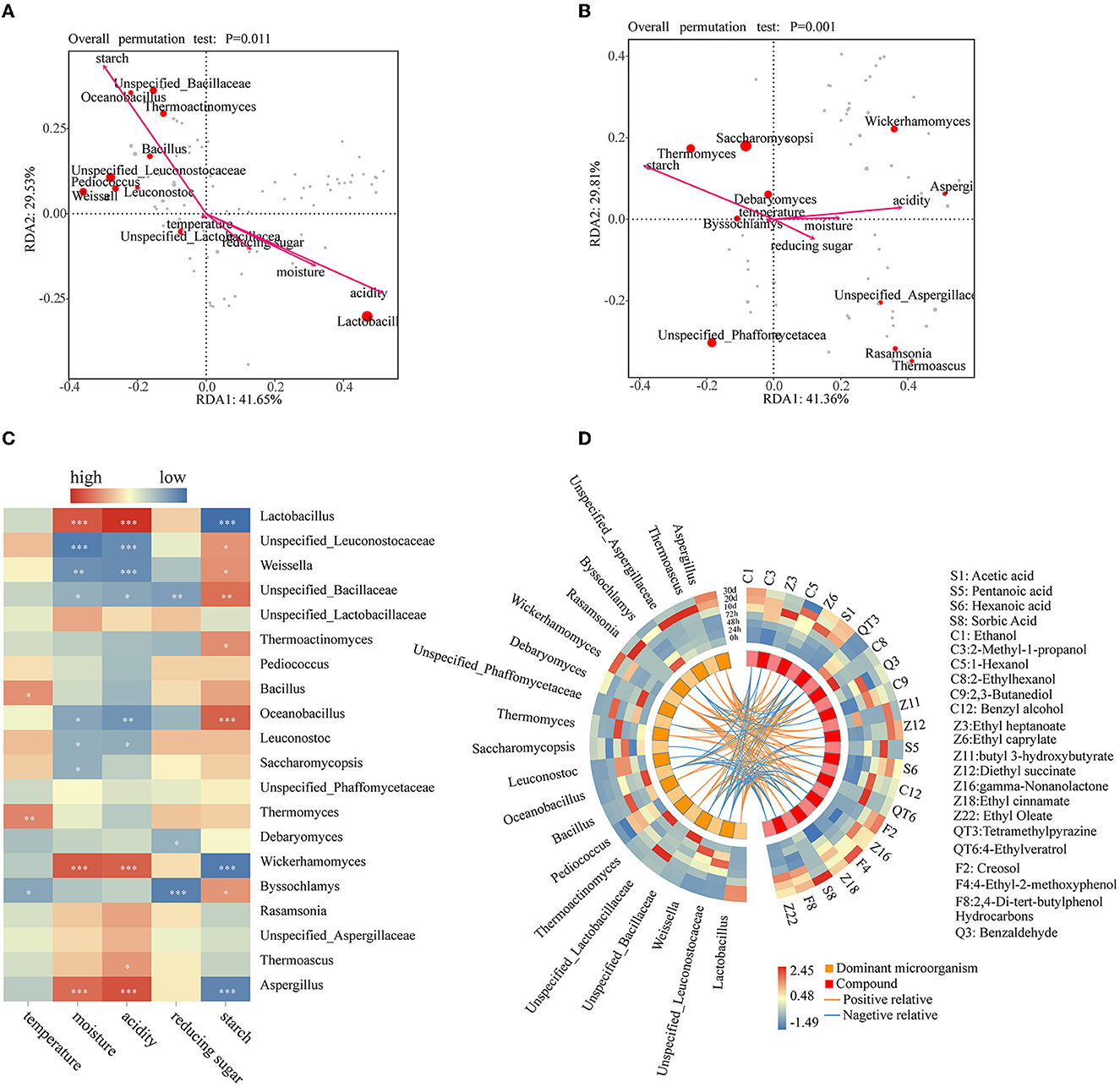
Figure 5. RDA analysis of the physicochemical properties and the bacterial (A) and fungal genera (B), the heat map of the correlation index between the top 10 microbial genera in relative abundance and physicochemical properties (C), and circos visualization figure between significantly different volatile compounds and dominant microbial genera in the Xiasha round of the cave brewing sauce-flavor Baijiu based on the Pearson correlation coefficient (D) [when the Pearson correlation coefficient is more significant than 0.5, it means that the absolute value of the Pearson correlation coefficient is positively correlated (orange line) or negatively correlated (blue line) with statistically significant differences (P < 0.05)].
Based on Pearson correlation analysis between the dominant microbial genera and the significantly different volatile metabolites visualized (Figure 5D), regarding the bacterial genera, Lactobacillus and Weissella were the genera with the maximum quantity of positive and negative correlations with 11 and 12 significantly different volatile metabolites, respectively. For fungi, Wickerhamomyces and Aspergillus were positively associated with 10 significantly different volatile metabolites, respectively, while Byssochlamys was negatively associated with four significantly different volatile metabolites.
Acetic acid (S1) and ethanol (C1) were the basis for the formation of various complex compounds, both of which showed significant negative correlations (P < 0.05, r < −0.5) with Weissella, Unspecified_Leuconostocaceae, Unspecified_Bacillaceae, and Oceanobacillus but highly significant positive correlations (P < 0.001, r > 0.6) with Wickerhamomyces, Lactobacillus, and Aspergillus. Wickerhamomyces and Lactobacillus were positively correlated with 10 and 11 significantly different metabolites, respectively, indicating that they were in the direction of anabolic products under anaerobic conditions with high acidity (Hu X. L. et al., 2021; Lin et al., 2022). Aspergillus was positively correlated with 10 significantly different metabolites. Aspergillus can secrete various enzymes to convert raw materials into small molecules and supply a material basis for the growth and metabolism of the microbial community (Ali et al., 2018; Zhao G. Z. et al., 2020). To the best of our knowledge, the contents of esters, phenols, alcohols, and acids are higher in CF. Previous studies indicated that Lactobacillus and Aspergillus vitally contribute to the synthesis of esters (He et al., 2022; Xu et al., 2023). Ethyl caprylate (Z6), diethyl succinate (Z12), gamma-nonalactone (Z16), and ethyl oleate (Z22) showed a positive correlation with Aspergillus (P < 0.01, r > 0.5). Diethyl succinate (Z12), gamma-nonalactone (Z16), ethyl cinnamate (Z18), and ethyl oleate (Z22) showed a positive correlation with Lactobacillus (P < 0.01, r > 0.5). These esters were significantly negatively correlated with Weissella. Unsurprisingly, this correlation is not consistent with the previous report that Weissella has the function of synthesizing esters (Hasan et al., 2006). Wickerhamomyces and Unspecified_Lactobacillaceae have a significant positive correlation with four esters separately. Compared with the SF stage, these microorganisms with many positive correlations with ester substances have higher relative abundance in the CF stage than in the SF stage. It shows that they have significant contributions to the synthesis of esters. Wickerhamomyces, Unspecified_Phaffomycetaceae, Unspecified_Lactobacillaceae, Thermoascus, Rasamsonia, Aspergillus, and Lactobacillus were significantly positively correlated with acids (acetic acid, pentanoic acid, hexanoic acid, and sorbic acid). This is consistent with the previous statement that Aspergillus and Lactobacillus have the ability to produce organic acids (Tang et al., 2019). Wickerhamomyces, Thermoascus, Rasamsonia, Lactobacillus, and Aspergillus have a significantly positive correlation with alcohols (ethanol, 1-hexanol, 2-ethylhexanol, and benzyl alcohol). Lactobacillus can produce ethanol by heterofermentation (Du et al., 2021). The microorganisms with a significant positive correlation with phenols (creosol, 4-ethyl-2-methoxyphenol, and 2,4-di-tert-butylphenol) were Wickerhamomyces, Unspecified_Lactobacillaceae, Thermoascus, Lactobacillus, and Aspergillus. The research results from a conventional environment showed that phenols are mainly related to Kroppenstedtia, Staphylococcus, and Thermoactinomyces (Zhuansun et al., 2022). The relative abundance of these microorganisms in the CF stage is higher than that in the SF stage. It shows that the CF stage is the main period for the accumulation of flavor substances, and the production of flavor substances is likely the result of the cooperation of microorganisms and biochemical reactions.
3.5. Metabolic mapping of dominant microorganism genera during the fermentation
To better understand the relationship between microorganisms and volatile flavors, metabolic mapping of dominant microorganisms during the Xiasha round of the CBSB was performed based on microbial annotated related enzymes and combined with the KEGG database and relevant literature (Figure 6A). Starch and cellulose as raw materials are metabolized by Saccharomycopsis, and Aspergillus and Bacillus secreted amylase and cellulase to form monosaccharides (Gopinath et al., 2017; Jin et al., 2019; Huang et al., 2020). Subsequently, monosaccharides are converted into the critical intermediate metabolite pyruvate, which is involved in the downstream of sugar metabolism. Pyruvate is converted into acetyl-CoA by the action of aldehyde dehydrogenase secreted by Saccharomycetales. Acetyl-CoA enters the TCA cycle with the participation of Lactobacillales, Bacillales, and Aspergillus to form alcohols, aldehydes, and ketones with amino acids. Among them, amino acids are derived from protein degradation by proteases secreted by Thermoascus, Thermomyces, Aspergillus, Bacillus, and Oceanobacillus (Ali et al., 2018; Jin et al., 2019; Park et al., 2019; Zhao G. Z. et al., 2020). Meanwhile, acetyl-CoA is converted into malonyl-CoA through the action of acetyl-CoA carboxylase to generate fatty acids and then forms an ester with ethanol with the participation of Weissella, Lactobacillales, Bacillus, Leuconostoc, and Aspergillus (Zhao C. et al., 2020). Lactobacillale is the main microorganism responsible for the production of lactic acid, which not only inhibits harmful microorganisms but also acts as a precursor for the formation of ethyl lactate (Jin et al., 2017; Zhang et al., 2018). In addition, Bacillus is associated with the synthesis of tetramethylpyrazine (Zhu and Xu, 2010a,b). The changes in the corresponding enzyme abundance are shown in Supplementary Table S5 and Figure 6B. Regarding the raw materials for Baijiu, the three main types of substances initially provided for microbial utilization are starch, cellulose, and protein. The average relative abundance of cellulase in SF was much higher than that in CF and increased significantly on day 3, which might be related to the significantly increased Bacillus during this period as it was the primary cellulase producer (Huang et al., 2020). The highest average relative abundance of amylase was observed on day 10 of CF, indicating that the content of starch computation was the largest on day 10, which was consistent with our aforementioned research result that significantly reduced the starch content on day 10 of the CF stage. The relative abundance of lactate dehydrogenase and acetyl CoA hydrolase in CF was higher than in the SF stage. We observed that the acid content increased significantly after entering the CF. Lactate, (S)-isopropyl lactate, and isobutyl lactate also have the same change trend, and Lactobacillus was the absolute dominant bacteria in the CF. As shown in Figure 6A, the average relative abundance of microorganisms producing carboxylesterase in CF was greater than that in the SF stage, which was consistent with the significant increase in ester content in the previous CF process.
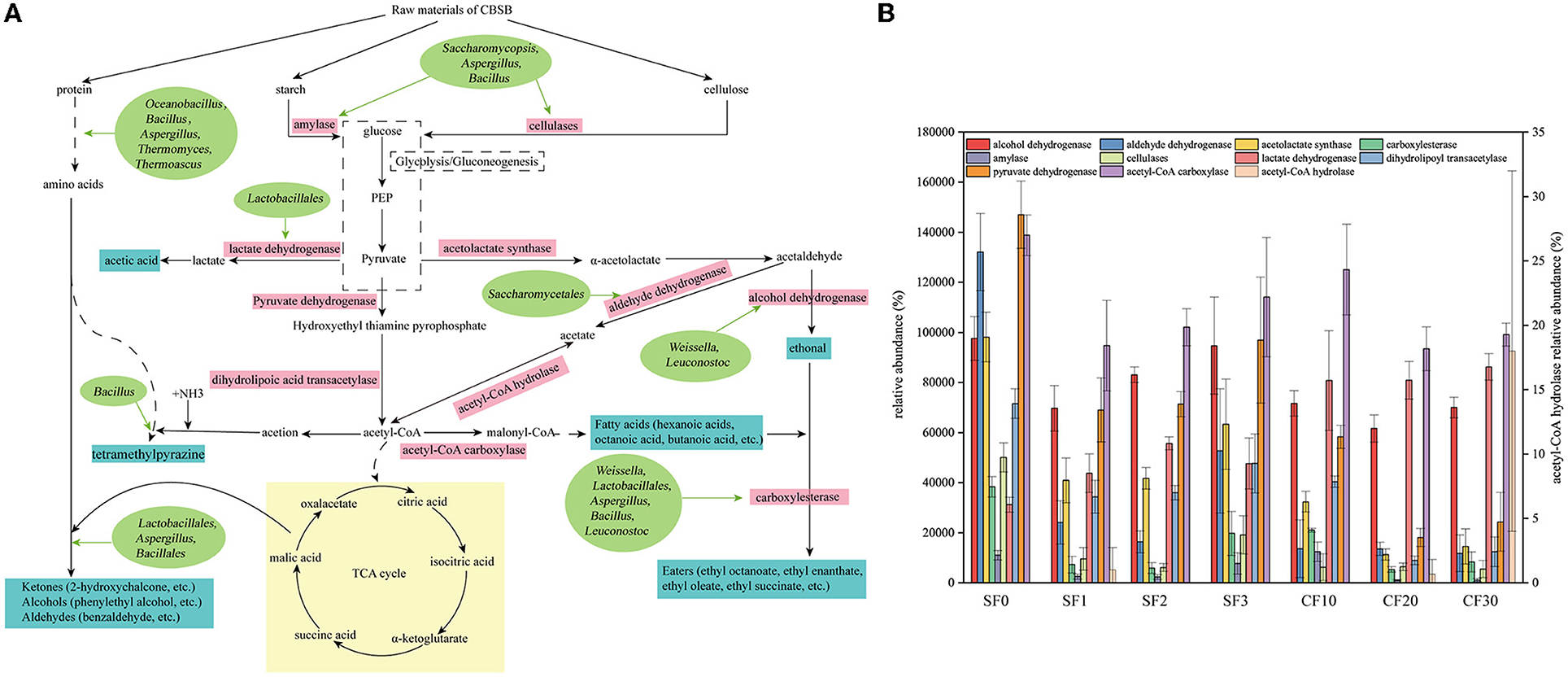
Figure 6. Metabolic profile of the dominant microorganisms (A) and relative abundance of enzymes (B) during the Xiasha round of the cave brew sauce-flavor Baijiu.
4. Conclusion
We investigated the dynamic changes of a microbial community, physicochemical properties, and volatile flavor compounds during the Xiasha round fermentation process of the CBSB. During the Xiasha round of the CBSB, the SF progress enriched the microflora necessary for flavor formation, and the flavor substance accumulation was mainly concentrated in the CF progress. This study clarified the microbial community structure in the fermentation process of Xiasha round in the CBSB. We revealed the correlation between microbial community, physicochemical properties, and flavor substances and established the correlation networks and metabolic maps to better understand the relationship between dominant microbial genera and critical flavor compounds in the fermentation process. We have demonstrated the potential role of dominant microorganism genera in flavor formation. In this study, most of the dominant microorganisms we detected also appeared in the traditional environmental fermentation process, which fully demonstrated the feasibility of brewing sauce-flavor Baijiu in the natural caves. Thus, these findings will provide theoretical bases for CBSB and thus lay a solid foundation for craft optimization and Baijiu quality improvement. Meanwhile, we are providing technical support for the further development of microbial resources for Baijiu brewing.
Data availability statement
The data presented in the study are deposited in the National Center for Biotechnology Information (NCBI) repository, accession number of bacteria and fungi are PRJNA928402 and PRJNA928447, respectively.
Author contributions
TTR and WS contributed the experimental design. TTR performed the statistical analysis and wrote the manuscript. WS and DWZ provided expertise and insight relating to Baijiu microbiology. TTR, WS, YCM, and QQ contributed to manuscript revision, read, and approved the submitted version. All authors contributed to the article and approved the submitted version.
Funding
The research was financially supported by the Regional Science Fund Project of the Natural Science Foundation under Grant No. 32160554, the Zunyi Innovative Talent Team Training Project (Zunyi Talents [2020] 9), and the Guizhou Fermentation Engineering and Baijiu Brewing Talent Base (Qianrenlingfa [2018] No. 3).
Acknowledgments
The authors thank Wekemo Tech (Group) Co., Ltd. (Shenzhen, China) for technical assistance in carrying out the analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1124817/full#supplementary-material
References
Ali, B., Yi, Z. L., Fang, Y., Chen, L. C., He, M. Z., Liu, D. Y., et al. (2018). Characterization of a fungal thermostable endoglucanase from Chinese Nong-flavor daqu by metatranscriptomic method. Int. J. Biol. Macromolec. 121, 183–190. doi: 10.1016/j.ijbiomac.2018.09.173
Bokulich, N. A., Thorngate, J. H., Richardson, P. M., and Mills, D. A. (2014). Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Nat. Acad. Sci. USA. 111, E139–E148. doi: 10.1073/pnas.1317377110
Boro, N., Borah, A., Sarma, R. L., and Narzary, D. (2022). Beer production potentiality of some non-Saccharomyces yeast obtained from a traditional beer starter emao. Braz. J. Microbiol. 53, 1515–1531. doi: 10.1007/s42770-022-00765-7
Cai, W. C., Wang, Y. R., Ni, H., Liu, Z. J., Liu, J. M., Zhong, J., et al. (2021). Diversity of microbiota, microbial functions, and flavor in different types of low-temperature Daqu. Food Res. Int. 150, 110734. doi: 10.1016/j.foodres.2021.110734
Cai, X. M., Shen, Y., Chen, M. Y., Zhong, M. Y., Zhou, Y. L., and Luo, A. M. (2019). Characterisation of volatile compounds in Maotai flavour liquor during fermentation and distillation. J. Inst. Brew. 125, 453–463. doi: 10.1002/jib.581
Callahan, B. J., Mcmurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chen, B., Wu, Q., and Xu, Y. (2014). Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 179, 80–84. doi: 10.1016/j.ijfoodmicro.2014.03.011
Dai, Y. J., Tian, Z. Q., Meng, W. N., and Li, Z. J. (2020). Microbial diversity and physicochemical characteristics of the maotai-flavored liquor fermentation process. J. Nanosci. Nanotechnol. 20, 4097–4109. doi: 10.1166/jnn.2020.17522
Dong, W. W., Zeng, Y. T., Cui, Y. X., Chen, P., Cai, K. Y., Guo, T. T., et al. (2022). Unraveling the composition and succession of microbial community and its relationship to flavor substances during Xin-flavor baijiu brewing. Int. J. Food Microbiol. 372, 109679. doi: 10.1016/j.ijfoodmicro.2022.109679
Du, H., Ji, M., Xing, M. Y., Wang, X. S., and Xu, Y. (2021). The effects of dynamic bacterial succession on the flavor metabolites during Baijiu fermentation. Food Res. Int. 140, 109860. doi: 10.1016/j.foodres.2020.109860
Fan, Q., Wang, X. L., Zhao, Y. F., Zheng, F. P., Li, H. H., Zhang, F. Y., et al. (2019). Characterization of key aroma compounds in Laobaigan Chinese Baijiu by GC × GC -TOF/MS and means of molecular sensory science. Flav. Fragr. J. 34, 514–525. doi: 10.1002/ffj.3533
Galati, A., Oguntoyinbo, F. A., Moschetti, G., Crescimanno, M., and Settanni, L. (2014). The cereal market and the role of fermentation in cereal-based food production in Africa. Food Rev. Int. 30, 317–337. doi: 10.1080/87559129.2014.929143
Gao, L., Zhou, J., and He, G. Q. (2022). Effect of microbial interaction on flavor quality in Chinese baijiu fermentation. Front. Nutr. 9, 960712. doi: 10.3389/fnut.2022.960712
Gopinath, S. C. B., Anbu, P., Arshad, M. K. M., Lakshmipriya, T., Voon, C. H., Hashim, U., et al. (2017). Biotechnological processes in microbial amylase production. Biomed. Res. Int. 2017, 1–9. doi: 10.1155/2017/1272193
Guan, T. W., Lin, Y. J., Chen, K. B., Ou, M. Y., and Zhang, J. X. (2020). Physicochemical factors affecting microbiota dynamics during traditional solid-state fermentation of chinese strong-flavor baijiu. Front. Microbiol. 11, 2090. doi: 10.3389/fmicb.2020.02090
Han, Q., Shi, J. L., Zhu, J., Lv, H. L., and Du, S. K. (2014). Enzymes extracted from apple peels have activity in reducing higher alcohols in Chinese liquors. J. Agric. Food Chem. 62, 9529–9538. doi: 10.1021/jf5018862
Hao, F., Tan, Y. W., Lv, X. B., Chen, L. Q., Yang, F., Wang, H. Y., et al. (2021). Microbial community succession and its environment driving factors during initial fermentation of maotai-flavor Baijiu. Front. Microbiol. 12, 669201. doi: 10.3389/fmicb.2021.669201
Hasan, F., Shah, A. A., and Hameed, A. (2006). Industrial applications of microbial lipases. Enzyme Microbial. Technol. 39, 235–251. doi: 10.1016/j.enzmictec.2005.10.016
He, M. W., Jin, Y., Zhou, R. Q., Zhao, D., Zheng, J., and Wu, C. D. (2022). Dynamic succession of microbial community in Nongxiangxing daqu and microbial roles involved in flavor formation. Food Res. Int. 159, 111559. doi: 10.1016/j.foodres.2022.111559
Hong, J. X., Tian, W. J., and Zhao, D. R. (2020). Research progress of trace components in sesame-aroma type of baijiu. Food Res. Int. 137, 109695. doi: 10.1016/j.foodres.2020.109695
Hu, X. L., Tian, R. J., Wang, K. L., Cao, Z. H., Yan, P. X., Li, F. Q., et al. (2021). The prokaryotic community, physicochemical properties and flavors dynamics and their correlations in fermented grains for Chinese strong-flavor Baijiu production. Food Res. Int. 148, 110626. doi: 10.1016/j.foodres.2021.110626
Hu, X. L., Wang, K. L., Chen, M. G., Fan, J. H., Han, S. N., Hou, J. G., et al. (2020). Profiling the composition and metabolic activities of microbial community in fermented grain for the Chinese strong-flavor Baijiu production by using the metatranscriptome, high-throughput 16S rRNA and ITS gene sequencings. Food Res. Int. 138, 109765. doi: 10.1016/j.foodres.2020.109765
Hu, Y. L., Wang, L. Y., Zhang, Z. J., Yang, Q., Chen, S. X., Zhang, L., et al. (2021). Microbial community changes during the mechanized production of light aroma Xiaoqu. Biotechnol. Biotechnol. Equip. 35, 487–495. doi: 10.1080/13102818.2021.1892525
Huang, X. N., Fan, Y., Lu, T., Kang, J. M., Pang, X. N., Han, B. Z., et al. (2020). Composition and metabolic functions of the microbiome in fermented grain during light-flavor Baijiu fermentation. Microorganisms 8, 1281. doi: 10.3390/microorganisms8091281
Jiang, Q. E., Wu, X. Y., Xu, Y. Q., Zhang, Y. L., Wang, Z. S., Shen, L. Y., et al. (2021). Microbial composition and dynamic succession during the Daqu production process of Northern Jiang-flavored liquor in China. 3 Biotech 11, 224. doi: 10.1007/s13205-021-02779-8
Jin, G. Y., Zhu, Y., and Xu, Y. (2017). Mystery behind Chinese liquor fermentation. Trends Food Sci. Technol. 63, 18–28. doi: 10.1016/j.tifs.2017.02.016
Jin, Y., Li, D. Y., Ai, M., Tang, Q. X., Huang, J., Ding, X. F., et al. (2019). Correlation between volatile profiles and microbial communities: A metabonomic approach to study Jiang-flavor liquor Daqu. Food Res. Int. 121, 422–432. doi: 10.1016/j.foodres.2019.03.021
Li, H. S., Han, X. L., Liu, H. R., Hao, J. Q., Jiang, W., and Li, S. Z. (2021). Silage fermentation on sweet sorghum whole plant for fen-flavor Baijiu. Foods 10, 1477. doi: 10.3390/foods10071477
Lin, B., Tang, J., Yang, Q., Su, Z. X., Zhu, L. P., Li, Q., et al. (2022). Microbial succession and its effect on key aroma components during light-aroma-type Xiaoqu Baijiu brewing process. World J. Microbiol. Biotechnol. 38, 166. doi: 10.1007/s11274-022-03353-x
Liu, C. J., Gong, X. W., Zhao, G., Soe Htet, M. N., Jia, Z. Y., Yan, Z. K., et al. (2021). Liquor flavour is associated with the physicochemical property and microbial diversity of fermented grains in waxy and non-waxy sorghum (Sorghum bicolor) during fermentation. Front. Microbiol. 12, 618458. doi: 10.3389/fmicb.2021.618458
Liu, H., and Sun, B. (2018). Effect of fermentation processing on the flavor of Baijiu. J. Agric. Food Chem. 66, 5425–5432. doi: 10.1021/acs.jafc.8b00692
Liu, P. L., and Miao, L. H. (2020). Multiple batches of fermentation promote the formation of functional microbiota in chinese miscellaneous-flavor baijiu fermentation. Front. Microbiol. 11, 75. doi: 10.3389/fmicb.2020.00075
Mu, Y., Huang, J., Zhou, R. Q., Mao, F. J., Pan, Q. L., Chen, S. Q., et al. (2022). Exploring the response patterns of strong-flavor baijiu brewing microecosystem to fortified Daqu under different pit ages. Food Res. Int. 155, 111062. doi: 10.1016/j.foodres.2022.111062
Padilla, B., Gil, J. V., and Manzanares, P. (2016). Past and future of non-saccharomyces yeasts: from spoilage microorganisms to biotechnological tools for improving wine aroma complexity. Front. Microbiol. 7, 411. doi: 10.3389/fmicb.2016.00411
Park, M. K., Seo, J., and Kim, Y. (2019). Comparative study on metabolic changes of Aspergillus oryzae isolated from fermented foods according to culture conditions. Int. J. Food Microbiol. 307, 108270. doi: 10.1016/j.ijfoodmicro.2019.108270
Rao, S. W. N., Santhakumar, A. B., Chinkwo, K. A., Wu, G. C., Johnson, S. K., and Blanchard, C. L. (2018). Characterization of phenolic compounds and antioxidant activity in sorghum grains. J. Cereal Sci. 84, 103–111. doi: 10.1016/j.jcs.2018.07.013
Shen, D. J., Shen, H. Y., Yang, Q., Chen, S. X., Dun, Y. H., Liang, Y. X., et al. (2021). Deciphering succession and assembly patterns of microbial communities in a two-stage solid-state fermentation system. Microbiol. Spectr. 9, e71821. doi: 10.1128/Spectrum.00718-21
Sifeeldein, A., Wang, S. R., Li, J. F., Dong, Z. H., Chen, L., Kaka, N. A., et al. (2018). Phylogenetic identificationI of lactic acid bacteria isolates and their effects on the fermentation quality of sweet sorghum (sorghum bicolor) silage. J. Appl. Microbiol. 126, 718–729. doi: 10.1111/jam.14123
Song, Z. W., Du, H., Zhang, M. H., Nie, Y., and Xu, Y. (2019). Schizosaccharomyces pombe can reduce acetic acid produced by baijiu spontaneous fermentation microbiota. Microorganisms 7, 606. doi: 10.3390/microorganisms7120606
Tang, Q. X., He, G. Q., Huang, J., Wu, C. D., Jin, Y., and Zhou, R. Q. (2019). Characterizing relationship of microbial diversity and metabolite in sichuan xiaoqu. Front. Microbiol. 10, 696. doi: 10.3389/fmicb.2019.00696
Tu, W. Y., Cao, X. N., Cheng, J., Li, L. J., Zhang, T., Wu, Q., et al. (2022). Chinese Baijiu: The perfect works of microorganisms. Front. Microbiol. 13, 919044. doi: 10.3389/fmicb.2022.919044
Wang, D. Q., Chen, L. Q., Yang, F., Wang, H. Y., and Wang, L. (2019). Yeasts and their importance to the flavour of traditional Chinese liquor: a review. J. Inst. Brewing 125, 214–221. doi: 10.1002/jib.552
Wang, H., Huang, Y. G., and Huang, Y. L. (2021). Microbiome diversity and evolution in stacking fermentation during different rounds of Jiang-flavoured Baijiu brewing. LWT 143, 111119. doi: 10.1016/j.lwt.2021.111119
Wang, L., Wang, Y. Y., Wang, D. Q., Xu, J., Yang, F., Liu, G., et al. (2015). Dynamic changes in the bacterial community in Moutai liquor fermentation process characterized by deep sequencing. J. Inst. Brew. 121, 603–608. doi: 10.1002/jib.259
Wang, M. Y., Yang, J. G., Zhao, Q. S., Zhang, K. Z., and Su, C. (2018). Research progress on flavor compounds and microorganisms of maotai flavor baijiu. J. Food Sci. 84, 6–18. doi: 10.1111/1750-3841.14409
Wang, X. D., Ban, S. D., Hu, B. D., Qiu, S. Y., and Zhou, H. X. (2017). Bacterial diversity of Moutai-flavour Daqu based on high-throughput sequencing method. J. Inst. Brew. 123, 138–143. doi: 10.1002/jib.391
Wang, X. S., Du, H., Zhang, Y., and Xu, Y. (2018). Environmental microbiota drives microbial succession and metabolic profiles during chinese liquor fermentation. Appl. Environ. Microbiol. 84, e2317–e2369. doi: 10.1128/AEM.02369-17
Wang, X. X., Fan, W. L., and Xu, Y. (2014). Comparison on aroma compounds in Chinese soy sauce and strong aroma type liquors by gas chromatography–olfactometry, chemical quantitative and odor activity values analysis. Eur. Food Res. Technol. 239, 813–825. doi: 10.1007/s00217-014-2275-z
Wang, Y. R., Cai, W. C., Wang, W. P., Shu, N., Zhang, Z. D., Hou, Q. C., et al. (2021). Analysis of microbial diversity and functional differences in different types of high-temperature Daqu. Food Sci. Nutr. 9, 1003–1016. doi: 10.1002/fsn3.2068
Wei, J. L., Du, H., Zhang, H. X., Nie, Y., and Xu, Y. (2021). Mannitol and erythritol reduce the ethanol yield during Chinese Baijiu production. Int. J. Food Microbiol. 337, 108933. doi: 10.1016/j.ijfoodmicro.2020.108933
Wei, Y., Zou, W., Shen, C. H., and Yang, J. G. (2020). Basic flavor types and component characteristics of Chinese traditional liquors: A review. J. Food Sci. 85, 4096–4107. doi: 10.1111/1750-3841.15536
Wu, C., Huang, J., and Zhou, R. (2017). Genomics of lactic acid bacteria: Current status and potential applications. Crit. Rev. Microbiol. 43, 393–404. doi: 10.1080/1040841X.2016.1179623
Wu, Q., Chen, L. Q., and Xu, Y. (2013). Yeast community associated with the solid state fermentation of traditional Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 166, 323–330. doi: 10.1016/j.ijfoodmicro.2013.07.003
Wu, Q., Zhu, Y., Fang, C., Wijffels, R. H., and Xu, Y. (2021). Can we control microbiota in spontaneous food fermentation? Chinese liquor as a case example. Trends Food Sci. Technol. 110, 321–331. doi: 10.1016/j.tifs.2021.02.011
Xiao, C., Yang, Y., Lu, Z. M., Chai, L. J., Zhang, X. J., Wang, S. T., et al. (2021). Daqu microbiota exhibits species-specific and periodic succession features in Chinese baijiu fermentation process. Food Microbiol. 98, 103766. doi: 10.1016/j.fm.2021.103766
Xu, Y. Q., Liu, X., Zhao, J. R., Huang, H. Q., Wu, M. Q., Li, X. T., et al. (2021). An efficient phthalate ester-degrading Bacillus subtilis: Degradation kinetics, metabolic pathway, and catalytic mechanism of the key enzyme. Environ. Pollut. 273, 116461. doi: 10.1016/j.envpol.2021.116461
Xu, Y. Q., Wu, M. Q., Niu, J. L., Lin, M. W., Zhu, H., Wang, K., et al. (2023). Characteristics and correlation of the microbial communities and flavor compounds during the first three rounds of fermentation in chinese sauce-flavor baijiu. Foods 12, 207. doi: 10.3390/foods12010207
Xu, Y. Q., Zhao, J. R., Liu, X., Zhang, C. S., Zhao, Z. G., Li, X. T., et al. (2022). Flavor mystery of Chinese traditional fermented baijiu: The great contribution of ester compounds. Food Chem. 369, 130920. doi: 10.1016/j.foodchem.2021.130920
Zeng, Y., Wang, Y., Chen, Q., Xia, X. J., Liu, Q., Chen, X. M., et al. (2022). Dynamics of microbial community structure and enzyme activities during the solid-state fermentation of Forgood Daqu: a starter of Chinese strong flavour Baijiu. Arch. Microbiol. 204, 577. doi: 10.1007/s00203-022-03198-w
Zhang, H. X., Wang, L., Tan, Y. W., Wang, H. Y., Yang, F., Chen, L. Q., et al. (2021a). Effect of Pichia on shaping the fermentation microbial community of sauce-flavor Baijiu. Int. J. Food Microbiol. 336, 108898. doi: 10.1016/j.ijfoodmicro.2020.108898
Zhang, H. X., Wang, L., Wang, H. Y., Yang, F., Chen, L. Q., Hao, F., et al. (2021b). Effects of initial temperature on microbial community succession rate and volatile flavors during Baijiu fermentation process. Food Res. Int. 141, 109887. doi: 10.1016/j.foodres.2020.109887
Zhang, J., Sun, J., Chen, X. Y., Nie, C. X., Zhao, J. B., Guan, W. Y., et al. (2018). Combination of clostridium butyricum and corn bran optimized intestinal microbial fermentation using a weaned pig model. Front. Microbiol. 9, 3091. doi: 10.3389/fmicb.2018.03091
Zhang, M. Z., Wu, X. F., Mu, D. D., Xu, B. Y., Xu, X. H., Chang, Q., et al. (2021). Profiling the influence of physicochemical parameters on the microbial community and flavor substances of zaopei. J. Sci. Food Agric. 101, 6300–6310. doi: 10.1002/jsfa.11299
Zhao, C., Su, W., Mu, Y., Mu, Y. C., and Jiang, L. (2021). Integrative Metagenomics–Metabolomics for Analyzing the Relationship Between Microorganisms and Non-volatile Profiles of Traditional Xiaoqu. Front. Microbiol. 11, 617030. doi: 10.3389/fmicb.2020.617030
Zhao, C., Su, W., Mu, Y. C., Jiang, L., and Mu, Y. (2020). Correlations between microbiota with physicochemical properties and volatile flavor components in black glutinous rice wine fermentation. Food Res. Int. 138, 109800. doi: 10.1016/j.foodres.2020.109800
Zhao, G. Z., Liu, C., Li, S., Wang, X. W., and Yao, Y. P. (2020). Exploring the flavor formation mechanism under osmotic conditions during soy sauce fermentation in Aspergillus oryzae by proteomic analysis. Food Funct. 11, 640–648. doi: 10.1039/C9FO02314C
Zhu, B. F., and Xu, Y. (2010a). A feeding strategy for tetramethylpyrazine production by Bacillus subtilis based on the stimulating effect of ammonium phosphate. Biopr. Biosyst. Eng. 33, 953–959. doi: 10.1007/s00449-010-0419-5
Zhu, B. F., and Xu, Y. (2010b). Production of tetramethylpyrazine by batch culture of Bacillus subtilis with optimal pH control strategy. J. Ind. Microbiol. Biotechnol. 37, 815–821. doi: 10.1007/s10295-010-0726-5
Zhuansun, W. W., Xu, J., Li, Z. Q., Liu, H. Z., Zhao, Y., Chen, L. L., et al. (2022). Dynamic changes in the microbial community, flavour components in jiupei of a novel Maotai-Luzhou–flavoured liquor under various daqu blending modes and their correlation analysis. LWT 172, 114167. doi: 10.1016/j.lwt.2022.114167
Zou, W., Ye, G. B., and Zhang, K. Z. (2018a). Diversity, function, and application of clostridium in chinese strong flavor baijiu ecosystem: a review. J. Food Sci. 83, 1193–1199. doi: 10.1111/1750-3841.14134
Zou, W., Zhao, C. Q., and Luo, H. B. (2018b). Diversity and function of microbial community in chinese strong-flavor baijiu ecosystem: a review. Front. Microbiol. 9, 671. doi: 10.3389/fmicb.2018.00671
Keywords: cave brewed sauce-flavor Baijiu, Xiasha round, microbial communities, physicochemical properties, volatile flavor substances
Citation: Ren T, Su W, Mu Y, Qi Q and Zhang D (2023) Study on the correlation between microbial communities with physicochemical properties and flavor substances in the Xiasha round of cave-brewed sauce-flavor Baijiu. Front. Microbiol. 14:1124817. doi: 10.3389/fmicb.2023.1124817
Received: 15 December 2022; Accepted: 30 January 2023;
Published: 01 March 2023.
Edited by:
M. Y. Sreenivasa, University of Mysore, IndiaReviewed by:
Jingrong Cheng, Guangdong Academy of Agricultural Sciences, ChinaXiangchen Meng, Northeast Agricultural University, China
Copyright © 2023 Ren, Su, Mu, Qi and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Su, c3V3ZWkxODg2QDE2My5jb20=