
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 26 January 2023
Sec. Microbe and Virus Interactions with Plants
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1119016
This article is part of the Research Topic Molecular Mechanisms of Evasion of Plant Immunity by Microbes View all 6 articles
 Kunqin Xiao1†
Kunqin Xiao1† Kaibin Qiao1†
Kaibin Qiao1† Wenjing Cui1
Wenjing Cui1 Xun Xu1
Xun Xu1 Hongyu Pan1
Hongyu Pan1 Fengting Wang1
Fengting Wang1 Shoudong Wang2
Shoudong Wang2 Feng Yang1
Feng Yang1 Yuanhu Xuan3
Yuanhu Xuan3 Anmo Li1
Anmo Li1 Xiao Han1
Xiao Han1 Zhuojian Song1
Zhuojian Song1 Jinliang Liu1*
Jinliang Liu1*Soybean sclerotinia stem rot (SSR) is a disease caused by Sclerotinia sclerotiorum that causes incalculable losses in soybean yield each year. Considering the lack of effective resistance resources and the elusive resistance mechanisms, we are urged to develop resistance genes and explore their molecular mechanisms. Here, we found that loss of GmSWEET15 enhanced the resistance to S. sclerotiorum, and we explored the molecular mechanisms by which gmsweet15 mutant exhibit enhanced resistance to S. sclerotiorum by comparing transcriptome. At the early stage of inoculation, the wild type (WT) showed moderate defense response, whereas gmsweet15 mutant exhibited more extensive and intense transcription reprogramming. The gmsweet15 mutant enriched more biological processes, including the secretory pathway and tetrapyrrole metabolism, and it showed stronger changes in defense response, protein ubiquitination, MAPK signaling pathway-plant, plant-pathogen interaction, phenylpropanoid biosynthesis, and photosynthesis. The more intense and abundant transcriptional reprogramming of gmsweet15 mutant may explain how it effectively delayed colonization by S. sclerotiorum. In addition, we identified common and specific differentially expressed genes between WT and gmsweet15 mutant after inoculation with S. sclerotiorum, and gene sets and genes related to gmsweet15_24 h were identified through Gene Set Enrichment Analysis. Moreover, we constructed the protein–protein interaction network and gene co-expression networks and identified several groups of regulatory networks of gmsweet15 mutant in response to S. sclerotiorum, which will be helpful for the discovery of candidate functional genes. Taken together, our results elucidate molecular mechanisms of delayed colonization by S. sclerotiorum after loss of GmSWEET15 in soybean, and we propose novel resources for improving resistance to SSR.
Soybean is an important oilseed and cash crop that is commonly cultivated for its rich protein and lipid content (Diers et al., 2018). In the past 10 years, the average annual soybean production in China has reached approximately 15 million tons. However, agricultural production of soybeans is frequently threatened by various diseases, including sclerotinia stem rot (SSR), which is a devastating plant fungal disease caused by Sclerotinia sclerotiorum that can damage soybean at the seedling, adult, and flowering stages. SSR mainly affects the upper parts of soybean plants and thereby affects quality and yield. In epidemic years, it can cause approximately 20%–30% reduction in soybean annual yield, with severely affected cultures experiencing 50% to complete loss (Wang et al., 2019d). The direct economic loss caused by SSR in the United States is approximately $200 million per year (Bolton et al., 2006). Sclerotinia sclerotiorum is a notorious necrotrophic phytopathogen that occurs in soybean cultures globally (Derbyshire et al., 2017), however, its epidemic mechanisms remain unclear, which limits the success of prevention and control measures (Hibbett et al., 2007). In recent years, SSR incidence increased, thus posing a considerable threat to soybean production. The mechanisms underlying soybean resistance to S. sclerotiorum remain unclear, and only few resistance resources have been explored so far.
To counteract pathogen infection, plants have evolved complex immune systems, including pathogen/microbe associated molecular patterns (P/MAMPs) triggered immunity (PTI; Wang et al., 2019d), effector protein triggered immunity (ETI; De Schutter and Van Damme, 2015; Thieffry et al., 2022), systemic acquired immunity (SAR), and complicated quantitative disease resistance (QDR). PTI acts as the first layer of defense, activating a series of immune responses through the sensing of PAMPs by plasma membrane-localized receptors, including the expression of defense genes, activation of MAPK, production of reactive oxygen species (ROS), callose deposition, and calcium influx (Wang et al., 2019d). In response to the evolution of pathogenic effectors, plants have evolved resistance (R) proteins that activate ETI through direct or indirect recognition of specific effector proteins, i.e., Avr proteins (Ngou et al., 2021; Yuan et al., 2021). In addition, plants also have a set of acquired immune mechanism termed SAR. These response are stimulated at the infection site, and distal tissues subsequently become resistant to potential pathogens through various mechanisms (Ding and Ding, 2020). QDR also occurs in plants when disease symptoms are mild but do not vanish easily. This type of resistance reduces selective pressure during pathogen evolution and is therefore generally considered more durable. QDR is typically provided by quantitative trait loci (QTL), controlled by one or multiple genes, or by multiple gene interactions (Corwin and Kliebenstein, 2017). More importantly, QDR seems to plays a more reliable role in the fight against broad host-range necrotrophic phytopathogens, such as S. sclerotiorum.
In recent years, a number of SSR resistance genes have been identified, including ectopic expression of polygalacturonase inhibitor protein (OsPGIP2) in rapeseed, which enhances resistance to S. sclerotiorum at the seedling and maturity stages (Wang Z. et al., 2018). Similarly, overexpression of AtPGIP1 in Arabidopsis reduces colonization ability of S. sclerotiorum (Wei et al., 2022). The kinases MPK3 and MPK4 of the MAPK signaling pathway and transcription factor WRKY33 play important roles in the resistance of rapeseed S. sclerotiorum (Wang et al., 2009, 2014, 2019c). BnMPK4-BnMKS1-BnWRKY33 exists in a nuclear localized complex that regulates resistance to S. sclerotiorum, which is most likely associated with the activation of salicylic acid (SA)- and jasmonic acid (JA)-mediated defense response and inhibition of H2O2 accumulation (Wang et al., 2014). Calcium signaling pathways are also important in plant immunity, and Ca2+ influx into the cytoplasm is an early responses of plants sensing pathogenic invasion. Cyclic nucleotide gated ion channels (CNGCs) in plants can generate Ca2+ signals and promote elevated Ca2+ levels in the cytoplasm in response to pathogen signals (Ma and Berkowitz, 2011). Several CNGCs have been reported to regulate the resistance to S. sclerotiorum, whereas several others negatively affect such resistance (Saand et al., 2015a,b). Similarly, other regulatory factors of calcium signaling positively regulate the resistance to S. sclerotiorum (Wang et al., 2015; Rahman et al., 2016; Wang et al., 2016). Many hormones are involved in signal transduction in plant defense responses to pathogens, such as JA, SA, ET, ABA, and growth hormones. Such hormones act as signaling molecules that transmit sensory signals which activate complex defense responses. In Arabidopsis, the JA/ET signaling pathway can positively regulate resistance to S. sclerotiorum, and ABA positively regulates JA biosynthesis. Arabidopsis mutants aba3-2, abi1-1, and abi2-1 are defective in ABA synthesis and exhibit complete loss of resistance to S. sclerotiorum (Guimaraes and Stotz, 2004; Perchepied et al., 2010). Several studies have identified QTLs associated with resistance to S. sclerotiorum (Wei et al., 2016; Wu et al., 2016; Wen et al., 2018). One study identified 39 candidate SSR resistance genes at three QTLs, and among these, the gene encoding ethylene response transcription factor 73 (ERF73, BnaC06g24360D) was considered a promising candidate for disease resistance (Wu et al., 2016). A different study identified 28 loci for a number of candidate SSR resistance genes, which interestingly were associated with different biological functions, and among these, two transferase-related genes were induced within 12 h after inoculation, which encoded acyltransferases (Glyma.04G198000) and a UDP-glucosyltransferase (Glyma.16G158100) involved in secondary metabolic biosynthesis (Wen et al., 2018). Despite successful identification of numerous SSR resistance genes, their resistance mechanisms remain to be further defined and, more importantly, SSR resistance genes are far from being comprehensively characterized.
Members of the Sugars Will Eventually be Exported Transporter (SWEET) family which possessing seven transmembrane helices can mediate the bidirectional transport of carbohydrates across membranes (Chen et al., 2012; Yuan and Wang, 2013; Eom et al., 2015). Previous studies have shown that SWEETs play crucial roles in plant growth, development, and responses to biological stress processes, such as phloem loading for long distance sugar transport, hormone transport, pollen development, fruit development, seed filling, plant-pathogen interactions, and plant-microbe interactions (Moriyama et al., 2006; Wellmer et al., 2006; Micali et al., 2008; Yuan et al., 2009; Chen et al., 2010; Ayre, 2011; Seo et al., 2011; Sosso et al., 2015; Kanno et al., 2016; Gao et al., 2018). The involvement of SWEETs in pathogen-host interactions is currently best understood with regard to rice OsSWEET11, which provides nutrients to the rice leaf blight pathogen, when Xanthomonas oryzae pv. oryzae (Xoo) infects rice, transcription activator-like effectors (TALEs) regulates their transcriptional expression by binding to a cis-acting element in the SWEETs promoters, thereby accelerating the transfer of sugars from rice to Xoo and promoting pathogen growth and colonization (Chu et al., 2006; Yang et al., 2006; Gao et al., 2018; Xue et al., 2022). Expression of AtSWEET2 localized in vesicles was significantly induced in Arabidopsis thaliana upon infestation with Pythium irregulare, and AtSWEET2 mutants showed reduced resistance to P. irregulare (Chen et al., 2015). IbSWEET10 of sweet potato is localized at the plasma membrane, and overexpression of IbSWEET10 in plants resulted in reduced sucrose content and increased resistance to Fusarium oxysporum (Li et al., 2017; Xue et al., 2022). However, no studies on the role of SWEETs with regard to the interactions between S. sclerotiorum and its host are available.
Fifty-two SWEETs have been identified in the soybean genome (Patil et al., 2015), however, many of their functions remain unexplored. Both soybean (Glycine max) GmSWEET15a and GmSWEET15b belong to branch III of the SWEETs. Previous studies have shown that these genes are highly expressed in the endosperm during the cotyledon stage. Knockout of GmSWEET15 in soybean results in delayed embryo development and endosperm persistence, leading to severe seed abortion. In addition, the embryonic sugar content is markedly reduced in these soybean knockout mutants, suggesting that the GmSWEET15 is essential for embryo development in soybean by mediating the export of sucrose from the endosperm to the embryo during seed development (Wang et al., 2019b). However, it is unclear whether GmSWEET15 also exerts other functions, particularly regarding those involved in resistance to SSR.
Here, we first illuminated that loss of GmSWEET15 enhanced resistance to S. sclerotiorum, and then we explored the molecular mechanisms of the gmsweet15 mutant enhancing the resistance to S. sclerotiorum by comparing transcriptome. Our comparative analyses revealed that the resistance phenotype of gmsweet15 mutant may be mainly related to the dynamic transcriptional reprogramming response during the early stage of inoculation with S. sclerotiorum. In addition, we identified a series of biological processes and functional genes that may play key roles in gmsweet15 mutant responses to S. sclerotiorum infection. Together, this study revealed the molecular mechanism enhanced soybean resistance to S. sclerotiorum in gmsweet15 mutant and proposes numerous candidate functional genes for elevating resistance to SSR.
Arabidopsis thaliana (Col-0) plants maintained at 16/8 h light/darkness, with 22°C during the day and 18°C at night, and soybean plants (Williams 82 and gmsweet15-1/Williams 82 mutant) maintained at 16/8 h light/darkness, with 30°C during the day and 25°C at night, were grown in a greenhouse. The gmsweet15-1 mutant was donated by Professor Huixia Shou, College of Life Sciences, Zhejiang University.
The S. sclerotiorum wild-type (WT) strain UF-1 was cultured on potato dextrose agar at 25°C for 36 h before inoculation. A 5-mm inoculation disk taken from the growing margin of S. sclerotiorum was then patched onto leaves which were wrapped in bags containing wet gauze to maintain humidity. Inoculated plants were incubated at a constant temperature of 22°C in a greenhouse with 75–90% relative humidity. Leaf samples were collected at various time points. The samples were directly frozen in liquid nitrogen, freeze-dried, and stored at-80°C until RNA sequencing (RNA-seq) and quantitative reverse-transcription PCR (qRT-PCR).
A first-strand cDNA synthesis kit (CWBIO, China) was used to reverse-transcribe 1 μg total RNA. The obtained cDNA was then diluted 10-fold and was used as a template for real-time PCR. qRT-PCR expression validation was performed, and specific primers were designed using the NCBI database (Supplementary Table S1). GmEF-1α and AtActin 8 (AT1G49240) were used as housekeeping genes of soybean and A. thaliana, respectively. Reaction mixtures (10 μL) comprised DEPC-treated water and primers (0.4 μL), template cDNA (1 μL), and 2× TransStart Top Green qPCR SuperMix (5 μL; CWBIO, China). The thermocycling program, carried out on a Pro Real-Time System, consisted of 2 min at 50°C and 2 min at 95°C for pre-denaturation, followed by 44 cycles of 15 s at 95°C denaturation and extension for 20 s at 60°C and a melting curve. Reaction products were evaluated using the Pro Study software. Relative expression values were calculated using the 2−ΔΔCT method, and the degrees of differential expression was expressed in terms of fold change (Ding et al., 2018).
Total RNA was extracted from all samples using TriZol reagent. RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, United States). The results were verified by agarose gel electrophoresis. Total RNA was used as input material for RNA sample preparation. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. PCR was performed using Phusion High-Fidelity DNA polymerase, universal PCR primers, and an index (X) Primer. PCR products were purified (AMPure XP system), and library quality was assessed using an Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina, CA, United States) according to the manufacturer’s instructions. After cluster generation, the libraries were sequenced on an Illumina NovaSeq platform. Raw reads in fastq format were first processed using in-house Perl scripts. In this step, clean reads were obtained by removing reads containing adapters, ploy-N’s, and low-quality reads. At the same time, the Q20, Q30, and GC content of the clean data was calculated. All the downstream analyses were performed using high-quality clean data.
Glycine max strain Williams 82 RefSeq Genome (GCA_000004515.5 Glycine_max_v4.0, https://www.ncbi.nlm.nih.gov/genome/5?genome_assembly_id=1605618) and gene model annotation files were downloaded directly from the genome website. The index of the reference genome was produced using Hisat2 v2.0.5 and clean paired-end reads were aligned to the reference genome using Hisat2 v2.0.5.
Feature Counts v1.5.0-p3 was used to count the reads numbers mapped to each gene. Then, the FPKM of each gene was calculated based on the length of the gene and the number of reads mapped to the respective gene. The count data were normalized and log-transformed using the function calcNormFactors (trimmed mean of M-values normalization) of the R package edgeR and the function voomWithQualityWeights of the package limma, respectively.
Differential expression analysis of the two conditions/groups (three biological replicates per condition) was performed using the R package DESeq2 (1.20.0). This program offers statistical pathways for determining differential expression in digital gene expression data using a model based on negative binomial distribution. The resulting p-values were adjusted using the Benjamini–Hochberg’s approach to control the false discovery rate. Differently expressed genes (DEGs) were identified using an adjusted p-value ≤ 0.05, and|log2[FoldChange]| ≥ 1 as the thresholds. Differential expression analysis in terms of the two conditions was performed using the R package edgeR (version 3.22.5).
Gene Ontology (GO) enrichment analysis of DEGs was performed using the R package clusterProfiler, during which gene length bias was corrected. GO terms with corrected p-values < 0.05 were considered significantly enriched with regard to DEGs. We used the R package clusterProfiler to test the statistical enrichment of DEGs in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and a local version of the Gene Set Enrichment Analysis (GSEA) analysis tool1 was used, GO and KEGG datasets were independently used for GSEA.
Protein–protein interaction (PPI) analysis of DEGs was executed using the on the STRING database, which contains known and predicted PPIs. Interactions with network data files were directly imported into Cytoscape software for visual editing. Each dot represents a protein, and the protein–protein interactions in the STRING protein interaction database are indicated by lines. When the number of interacting proteins of a given protein exceeded 20, this is indicated by larger circles, and different biological functions involved in the protein are represent by different colors.
Weighted correlation network analysis (WGCNA) is a systematic method to describe modes of gene associations among different samples. An adjacency matrix was constructed based on the normalized FPKM values. The R package for WGCNA was used for co-expression network analyses to evaluate modules in which genes showed high correlations. The adjacency matrix was further converted into a topological overlap matrix (TOM) using the WGCNA package. Transcripts with similar expression patterns were classified into one module (coefficient of dissimilarity < 0.25). The TOM and degree were subsequently calculated; the top 20 genes according to the degree were considered hub genes. Additionally, TOM > 0.3 indicated strong correlation and pronounced regulatory functions. The co-expression networks were visualized using Cytoscape software. Each dot represents a gene in the module and co-expression relationships are connected by lines. The network of the top hub genes is indicated by larger circles, and different biological functions involved in genes are represented by dots of different colors.
Previous studies have shown that GmSWEET15 is responsible for sucrose transport during seed development. However, the loss of GmSWEET15 also delays senescence of the whole plant and yellowing of leaves during the vegetative growth stage (Wang et al., 2019b). In the present study, gmsweet15 mutant plants and leaves were significantly smaller compared to WT plants after germination (Figures 1A, B), suggesting that GmSWEET15 may not exert only one isolated effect during seed development.
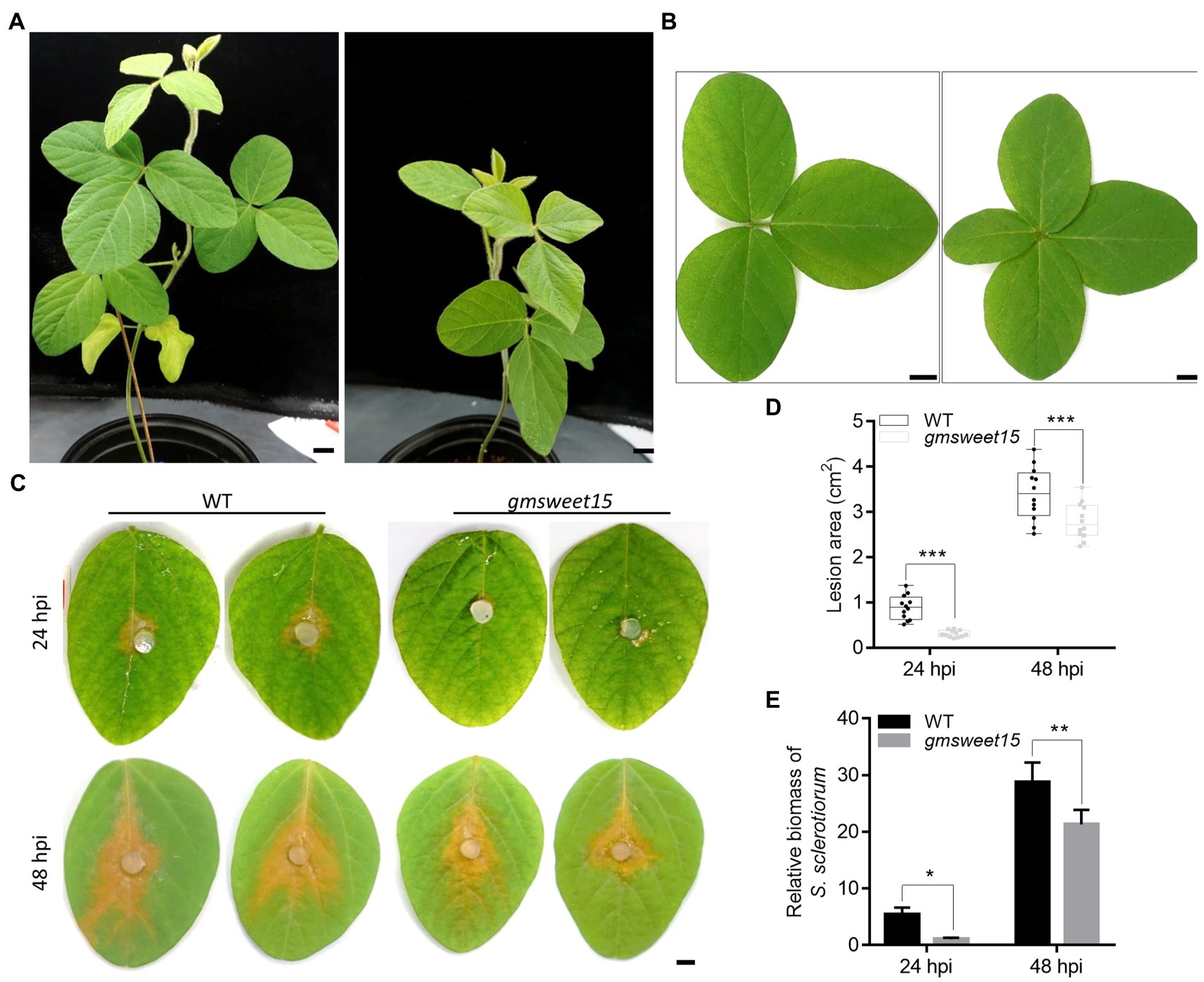
Figure 1. Loss of GmSWEET15 enhanced soybean resistance to Sclerotinia sclerotiorum. (A) Growth of WT and gmsweet15-1 mutant plants after 6 weeks in the greenhouse. Scale bar = 2 cm. (B) Leaf morphology of WT and gmsweet15-1 mutant second true leaves after 6 weeks. Scale bar = 1 cm. (C) Disease development on WT and gmsweet15-1 mutant leaves after inoculation of S. sclerotiorum for 24 and 48 h. hpi hours post inoculation. Scale bar = 0.5 cm. (D) Area of lesion on WT and gmsweet15-1 mutant leaves 24 and 48 hpi. (E) Relative biomass of S. sclerotiorum in WT and gmsweet15-1 mutant leaves 24 and 48 hpi. Genomic DNA quantitative PCR, for which the S. sclerotiorum β-tubulin was amplified and GmEF-1α was used as a reference gene for normalizing soybean biomass. All above experiments were performed three times with similar results. Four plants were in each replicate of A and B, and 12 leaves in C and D. Asterisks indicate a statistically significant difference (*p < 0.05, **p < 0.01, ***p < 0.001) according to a two-tailed comparisons t-test.
To explore whether GmSWEET15 affects the resistance of soybean to S. sclerotiorum, the lesion size of the WT and gmsweet15-1 mutant inoculated with S. sclerotiorum was evaluated. The results showed that the lesion size of gmsweet15-1 mutant was significantly smaller than that of the WT, at 24 h post inoculation (hpi) and at 48 hpi. Moreover, gmsweet15-1 mutant largely blocked early colonization by S. sclerotiorum (Figures 1C, D). The relative S. sclerotiorum biomass was based on ratios of S. sclerotiorum DNA to soybean DNA via genomic DNA quantitative PCR was estimated. Compared with the WT, gmsweet15-1 mutant exhibited a significantly decreased relative biomass of S. sclerotiorum (Figure 1E). These results suggested that GmSWEET15 is associated with soybean susceptibility to S. sclerotiorum.
To further explore the mechanism of gmsweet15 mutant resistance to S. sclerotiorum, comparative transcriptome sequencing was performed, including uninoculated WT and gmsweet15 mutant (WT_0h and gmsweet15_0h), WT and gmsweet15 mutant inoculated with S. sclerotiorum for 24 h (WT_24h and gmsweet15_24h), and WT and gmsweet15 mutant inoculated with S. sclerotiorum for 48 h (WT_48h and gmsweet15_48h). Clean data comprising 113.33 Gb form 18 samples were obtained in each group of three replicates (Supplementary Figure S1). After alignment of the reference genome of soybean (GCA_000004515.5), the alignment rate of per sample was 83.55–96.73% (Supplementary Figure S1). To evaluate the reliability of RNA-seq data, we randomly selected eight genes for qRT-PCR verification. The results showed that the transcriptome sequencing was reliable in terms of overall trends and expression levels (Figure 2A). We then carried out PCA (Figure 2B) and tested Pearson’s correlation coefficient (R; Figure 2C) between samples to evaluate differences between groups and between sample replicates within groups. The results showed that R2 between biological replicates exceeded 0.8, whereas the samples between different groups showed different degrees of dispersion. Combined hierarchical clustering of relative expression of all commonly detected sample genes (21,461 in total) in the RNA-seq analyses (Figure 2D; Supplementary Tables S2A, B) showed that the expression patterns were significantly different after inoculation, indicating that a large number of genes exhibited transcription changes, especially gmsweet15_24h, which was significantly different from gmsweet15_0h, whereas WT was mainly different from gmsweet15 mutant at 24 hpi, and there was little difference at uninoculated and 48 hpi, which suggested that the resistance phenotype of gmsweet15 mutant may be mainly related to the dynamic transcriptional reprogramming response at the early stage of inoculation with S. sclerotiorum.
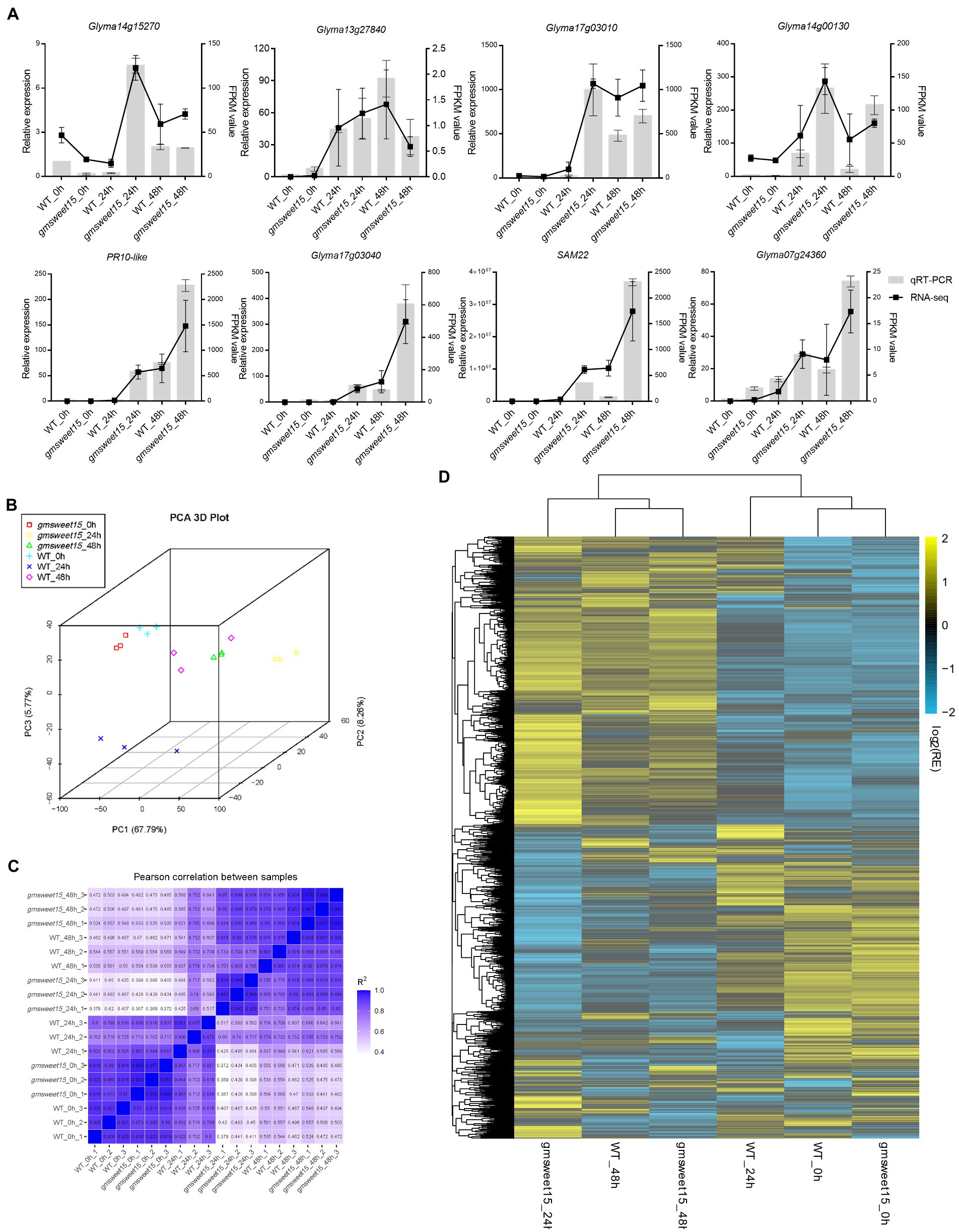
Figure 2. Assessment and full view of the RNA-seq. (A) qRT-PCR validation of eight randomly selected genes according to RNA-seq results. The qRT-PCR validation is shown as a bar chart, corresponding to the Y-axis on the left side, and EF-1α was used for each sample as an endogenous control. Relative expression values were determined against the average value of the WT_0h sample. The FPKM value was used as the expression level according to RNA-seq results, which is shown as a line on the right side. The standard deviation (n = 3) is represented by error bars. (B) Principal component analysis (PCA) of the transcriptome datasets. (C) Pearson correlation coefficient (R) of the transcriptome datasets. (D) Hierarchical clustering of relative expression (RE) of commonly detected all samples genes (in total, 21,461) in RNA-seq analysis. See Supplementary Tables S2A, B for gene expression data.
To explore which genes were affected by GmSWEET15 under inoculation with S. sclerotiorum, we analyzed the DEGs. A total of 24,993 DEGs were identified, and cluster heat maps were generated according to our screening criteria (Figures 3A, B; Supplementary Table S3). We found that 2,908 genes and 2,560 genes were upregulated and downregulated, respectively, in WT_24 h vs. WT_0 h (Figures 3B, C; Supplementary Tables S4A, B). While gmsweet15_24 h vs. gmsweet15_0 h showed stronger differential expression patterns, with 9,354 genes and 9,356 genes were upregulated and downregulated, respectively (Figures 3B, D; Supplementary Tables S4C, D). Comparing the gene expression levels at 48 and 0 hpi, 12,755 and 16,851 DEGs were identified in WT and gmsweet15 mutant, respectively (Figure 3B; Supplementary Table S5). Above all, when comparing the gene expression levels in WT and gmsweet15 mutant, only 1,405 and 1,501 DEGs were identified at uninoculated and 48 hpi, respectively, whereas 13,862 DEGs were observed at 24 hpi (Figure 3B; Supplementary Table S6). This suggests that gmsweet15 mutant exhibited stronger transcriptional changes in the early stage of inoculation than the WT, which was consistent with the analysis of the relative expression relationships of the samples.
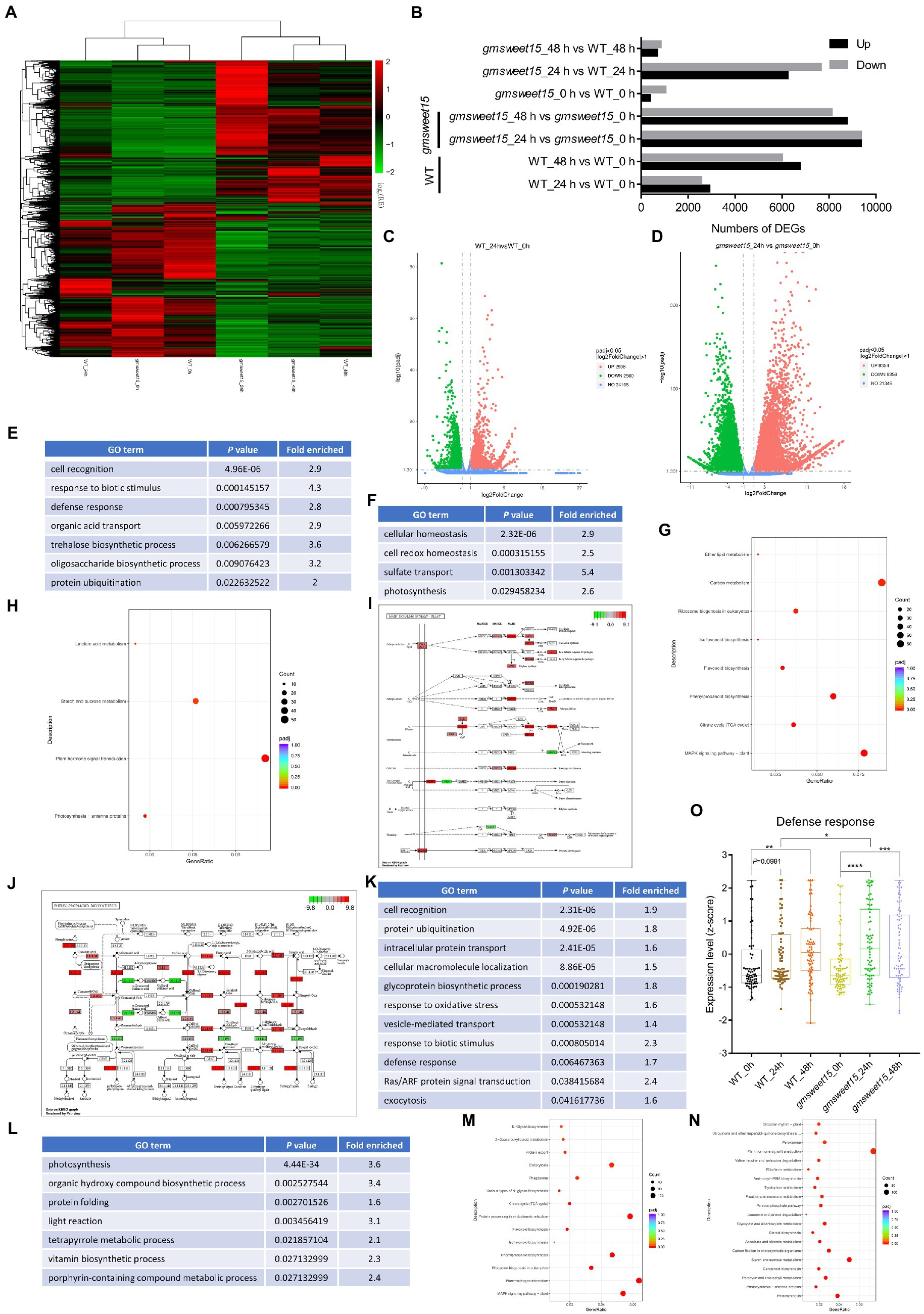
Figure 3. Pronounced transcription changes at the early stage of inoculation in gmsweet15 mutant. (A) Hierarchical clustering of the differentially expressed genes (DEGs) in RNA-seq. DEGs were identified using adjusted p-value ≤ 0.05 and |log2[FoldChange]| ≥ 1 as the threshold for the significance of the gene expression difference. See Supplementary Table S3 for gene expression data. (B) Statistical analysis of upregulated and downregulated DEGs in response to S. sclerotiorum infection and comparison between WT and gmsweet15 mutant. (C,D) Volcano plot of the distribution of DEGs in response to (Continued)FIGURE 3 (Continued)S. sclerotiorum 24 hpi in WT (C) and gmsweet15 mutant (D). The X-axis represents the change in gene expression in different samples, here represented by log2[FoldChange] value. The Y-axis indicates the statistical significance of changes in gene expression levels, here represented by-log10 (padj) value. The dots represent different genes. The blue dots represent genes with no significant difference, the red dots represent genes with significant differences in upregulation, and the green dots represent genes with significant differences in downregulation. (E,F) Significantly enriched gene ontology (GO) biological process terms in the upregulated (E) and downregulated (F) DEGs in WT_24 h vs. WT_0 h. (G,H) Significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in the upregulated (G) and downregulated (H) DEGs in WT_24 h vs. WT_0 h. (I,J) DEGs of significantly enriched MAPK signaling pathway-plant pathway (I) and phenylpropanoid biosynthesis pathway (J) in WT_24 h vs. WT_0 h. The ruler is the largest log2[FoldChange] value of all the differentially annotated genes into this pathway. Red indicates that the genes annotated to the pathway are upregulated, and green indicates that the genes annotated to the pathway are downregulated. (K,L) Significantly enriched GO biological process terms in the upregulated (K) and downregulated (L) DEGs in gmsweet15_24 h vs. gmsweet15_0 h. (M,N) Significantly enriched KEGG pathways in the upregulated (M) and downregulated (N) DEGs in gmsweet15_24 h vs. gmsweet15_0 h. (O) Box plots showing expression level (z-score) related to defense response in global gene. Box plots with boxes displaying the 25th–75th percentiles, the center line indicating the median. All individual data points are overlaid. Asterisks indicate a statistically significant difference (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) according to a two-tailed comparisons t-test. In E-N, the adjusted p-value ≤ 0.05 is used as the threshold of significant enrichment in GO and KEGG enrichment. For expression data and full GO or KEGG lists, see Supplementary Tables S4–S7.
GO and KEGG analyses were carried out to determine which biological processes and pathways were enriched in terms of the DEGs. Among the upregulated DEGs of WT_24 h vs. WT_0 h, cell recognition, response to biotic stimulus, and defense response, among others, were identified by GO enrichment analysis (Figure 3E; Supplementary Table S7A). Biological processes, such as cell redox homeostasis and photosynthesis were significantly downregulated (Figure 3F; Supplementary Table S7B). In addition, pathways such as MAPK signaling pathway-plant and phenylpropanoid biosynthesis were significantly upregulated, whereas photosynthesis-antenna proteins and plant hormone signal transduction were significantly downregulated (Figures 3G, H; Supplementary Tables S7C, D). Here, several genes of MAPK cascades were significantly upregulated (Figure 3I; Supplementary Table S7C), such as upstream receptor BAK1, intermediate cascade kinases MKK2 and MPK4, substrate MKS1, ACS6, and downstream transcription factors including WRKY29, WRKY33, and EIN3. Additionally, phenylpropanoid biosynthesis pathway had also undergone significant reprogramming (Figure 3J; Supplementary Table S7C), which included enzyme genes such as phenylalanine ammonia-lyase (PAL), trans-cinnamate 4-monooxygenase (C4H), cinnamoyl-CoA reductase (CHR), caffeoyl-CoA O-methyltransferase (CCoMT), lignin synthetases, cinnamyl alcohol dehydrogenase (CAD) and chalcone synthase (CHS). These results indicated that inoculation with S. sclerotiorum activated a certain number of defense-related signaling pathways, and the synthesis of secondary metabolites, but inhibited photosynthesis.
In contrast, gmsweet15 mutant showed more extensive and intense transcriptional reprogramming in the early stage of inoculation. Two major findings were produced. The first is that more specific biological processes were enriched in gmsweet15_24 h vs. gmsweet15_0 h, including intracellular protein transport, glycoprotein biosynthetic process, response to oxidative stress, Ras/ARF protein signal transduction, exocytosis, plant-pathogen interaction, protein processing in endoplasmic reticulum, and endocytosis were up-expressed (Figures 3K, M; Supplementary Tables S7E, G). Protein folding, tetrapyrrole metabolic process, steroid biosynthesis, and peroxisome processes were downregulated (Figures 3L, N; Supplementary Tables S7F, H). Secretory pathways such as endoplasmic reticulum processing, exocytosis, and protein transport were specifically upregulated, which implied that gmsweet15 mutant may have organized more abundant extracellular peptides such as PR1 as a weapon oppose primary infection of S. sclerotiorum, which was consistent with gmsweet15_24 h exhibiting higher PR1 expression than WT_24 h (Supplementary Table S3). The downregulation of some tetrapyrrole biosynthesis enzymes may lead to the accumulation of photosensitive intermediates in leaves and to cell death (Ishikawa et al., 2001; Chai et al., 2017). Such as coproporphyrinogen-III oxidase (CPX), was downregulated 3.4-fold (Supplementary Table S3), which may enhance immunity by reducing the biosynthesis of tetrapyrrole, as knock-out of GmCPX can significantly enhance the resistance of soybean to Phytophthora sojae (Ma et al., 2020). These results suggest that gmsweet15 mutant exhibited enrichment of more biological processes than WT in the early stage of inoculation with S. sclerotiorum, and the rearrangement of these pathways may play an important role in delaying infection with S. sclerotiorum in gmsweet15 mutant.
A significantly stronger upregulation of immunity and downregulation of photosynthesis occurred gmsweet15 mutant, although these processes were also enriched in WT after inoculation (Figure 3; Supplementary Table S7). To elucidate the biological processes or pathways that were differentially regulated under various conditions, all genes associated with a respective term in our transcriptome were selected, and standardized expression scores were calculated. The GO term defense response showed pronounced upregulation in gmsweet15 mutant after inoculation, especially at 24 hpi, while significant upregulation in WT occurred only at 48 hpi. Moreover, the expression was higher in the gmsweet15_24 h group than in the WT_24 h group (Figure 3O). This indicates that gmsweet15 mutant had a more powerful arsenal by upregulating the genes of defense response after sensing S. sclerotiorum, which may directly lead to gmsweet15 mutant gaining disease resistance.
To further explore the key genes and biological processes involved in gmsweet15 mutant resistance to S. sclerotiorum, we deployed the differential expression analysis of WT and gmsweet15 mutant before and after inoculation for the same time. Compared to the WT, 369 genes were upregulated and 1,036 genes were downregulated in the gmsweet15 mutant without inoculation (Figure 3B; Supplementary Tables S6A, B). In this case, GO terms such as polysaccharide catabolic process and response to chemical and drug transmembrane transport were upregulated, whereas protein ubiquitination and aminoglycan metabolic processes were downregulated (Supplementary Figure S2A; Supplementary Table S8A). In addition, terms such as MAPK signaling pathway-plant, plant-pathogen interaction, and plant hormone signal transduction were enriched (Supplementary Figures S2B, C; Supplementary Table S8B). Interestingly, the expression levels of genes such as MPK3, RBOHB, Pti1, SGT1 which have long been considered to contribute to disease resistance (Forzani et al., 2011; Hawamda et al., 2020; Yu G. et al., 2020; Zhang and Zhang, 2022), were lower in gmsweet15 mutant than in WT (Supplementary Figure S2D; Supplementary Table S6B). Considering that there were not many DEGs and no obvious functional enrichment in accordance with the basis of resistance, we propose that the difference in uninoculated is not the reason for the resistance of gmsweet15 mutant to S. sclerotiorum.
In contrast, a wealth of DEGs was identified in gmsweet15_24 h vs. WT_24 h, including 6,223 upregulated and 7,639 downregulated genes (Figure 3B; Supplementary Tables S6C, D). Functional analysis of these DEGs revealed that the enriched categories were almost the same as those enriched by gmsweet15_24 h vs. gmsweet15_0 h. They all were similarly upregulated in biological processes and pathways such as protein ubiquitination, protein transport, defense response, glycoprotein biosynthetic, plant-pathogen interaction, protein processing in endoplasmic reticulum, ribosome biogenesis, and MAPK signaling pathway-plant. Photosynthesis and tetrapyrrole metabolic processes were downregulated (Figures 4A–D; Supplementary Tables S8C–F). Notably, abundant genes in ribosome biosynthesis were significantly upregulated, including nucleolar protein 56 (NOP56), small nuclear ribonucleoprotein 13 (SNU13), and H/ACA ribonucleoprotein complex subunit 2 (NHP2) responsible for rRNA modification, U3 small nucleolar RNA-associated protein 14 (UTP14), nucleolar GTP-binding protein (Nog1), and 60S ribosomal export protein (NMD3) responsible for pre-ribosome cleavage and transport and almost all components of the 90s pre-ribosome precursor (Figures 4A, C; Supplementary Table S8E).
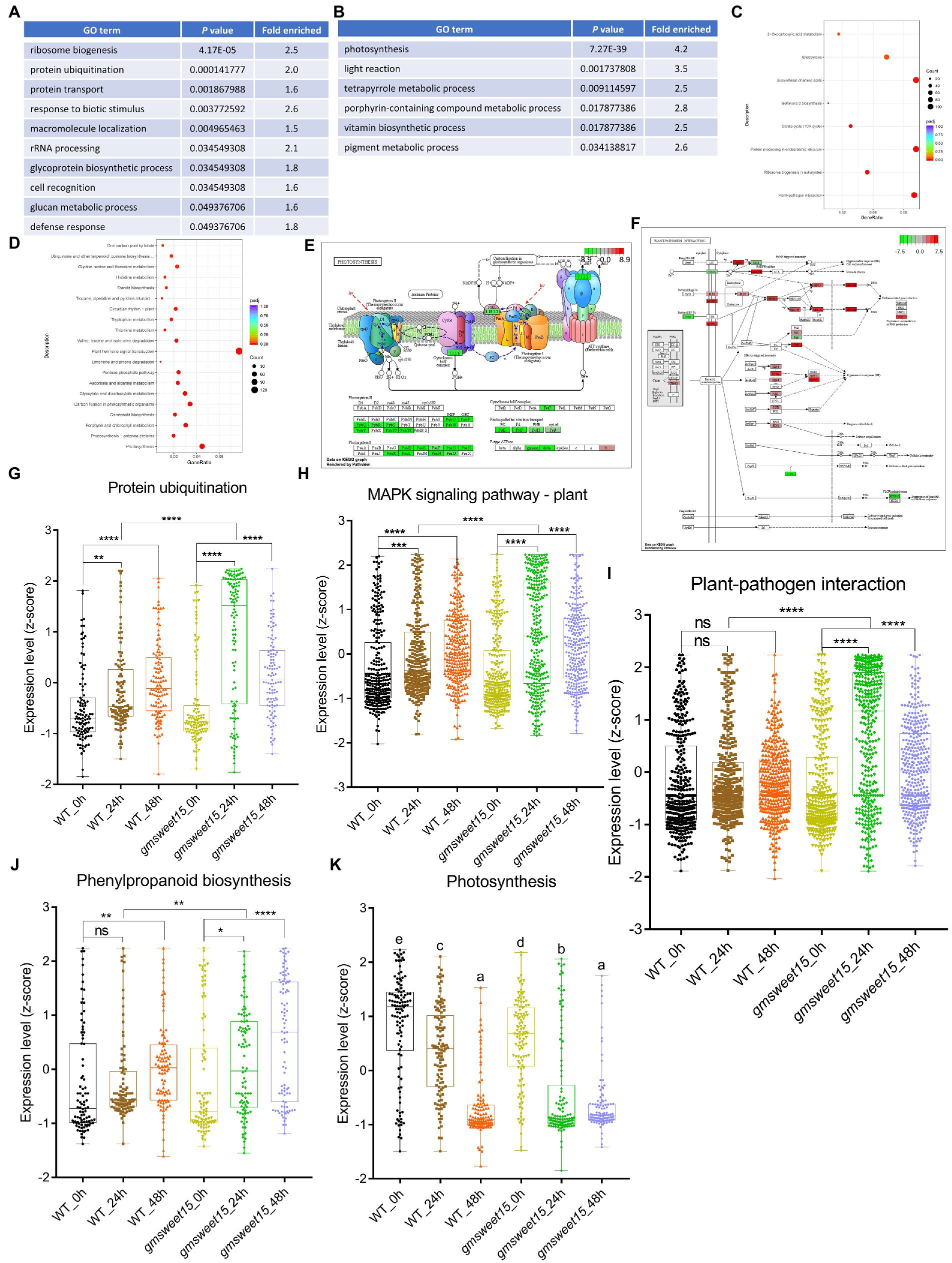
Figure 4. Characterization of the DEGs between gmsweet15 mutant and WT. (A,B) Significantly enriched GO biological process terms in the upregulated (A) and downregulated (B) DEGs in gmsweet15_24 h vs. WT_24 h. (C,D) Significantly enriched KEGG pathways in the upregulated (C) and downregulated (D) DEGs in gmsweet15_24 h vs. WT_24 h. (E, F) DEGs of significantly enriched photosynthesis pathway (E) and plant-pathogen interaction pathway (F) in gmsweet15_24 h vs. WT_24 h. (G–K) Box plots showing expression level (z-score) related to protein ubiquitination (G), MAPK signaling pathway-plant (H), plant-pathogen interaction (I), phenylpropanoid biosynthesis (J) and photosynthesis (K) in global gene. Asterisks indicate a statistically significant difference (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) according to a two-tailed comparisons t-test, and ns represent no statistical difference. For K, different letters indicate statistically significant differences (adjusted p < 0.05, two-tailed Student’s t-test, Benjamini–Hochberg method). For expression data and full GO or KEGG lists, see Supplementary Table S8.
Inhibition of photosynthesis is frequently observed in plants under various biotic stresses. Compared with the WT, gmsweet15 mutant showed more obvious photoinhibition, and all components of the photosynthetic apparatus were downregulated to different degrees, among which PsbO, PsbP and PsbQ of photosystem II oxygen-evolving enhancer protein were significantly downregulated (Figure 4E; Supplementary Table S8F). Furthermore, compared with the WT, gmsweet15 mutant also comprehensively enhanced the plant-pathogen interaction pathway, including membrane recognition receptors such as BAK1, chitin elicitor receptor kinase 1 (CERK1) and flagellin sensitive 2 (FLS2), various components of PTI, such as cyclic nucleotide gated channel (CNGCs), calcium-dependent protein kinase (CDPK), RBOH, calmodulin (CaM), MAPKs, WRKY33 and PR1, or some key nodes of ETI such as resistance to Pseudomonas maculicola 1 (RPM1), RPM1-interacting protein 4 (RIN4), AvrPphB susceptible 1 (PBS1), SGT1, heat shock protein 90 (HSP90), and enhanced disease susceptibility 1 (EDS1; Figure 4F; Supplementary Table S8E). The upregulated expression of these defense genes may largely explain why the gmsweet15 mutant was able to delay colonization by S. sclerotiorum.
Moreover, consistent with the GO enrichment analysis, the global gene of protein ubiquitination of gmsweet15 mutant was significantly upregulated, which was more pronounced than in the WT (Figure 4G). In addition, at the early stage of inoculation with S. sclerotiorum, the global genes of MAPK signaling pathway-plant, plant-pathogen interaction, and phenylpropanoid biosynthesis were markedly upregulated in gmsweet15 mutant (Figures 4H–J), while photosynthesis showed more significant downregulation (Figure 4K). These data are consistent with the previous analyses of DEGs (Figures 3, 4). In general, the more intense and abundant transcriptional changes in gmsweet15 mutant may explain why it was able to effectively delay colonization by S. sclerotiorum.
To explore the unique and common response mechanism of WT and gmsweet15 mutant after inoculation with S. sclerotiorum, we indicated the common and unique DEGs between WT and gmsweet15 mutant after inoculation in a Venn diagram. Under the condition of upregulation at 24 hpi, 1,794 common DEGs between WT and gmsweet15 mutant were identified, while 1,115 and 7,561 unique DEGs were identified in the WT and gmsweet15 mutant, respectively (Figure 5A; Supplementary Table S9A). In the context of downregulation at 24 hpi, 1,717 common DEGs were obtained, whereas 843 and 7,639 unique DEGs in WT and gmsweet15 mutant were identified, respectively (Figure 5B; Supplementary Table S9B). GO enrichment analysis of these upregulated DEGs showed that response to biotic stimulus and defense response terms were common biological processes, amino acid transport and organic acid transport were only upregulated in WT, while several secretion pathways and glycoprotein biosynthetic process were specifically upregulated in gmsweet15 mutant (Figure 5C; Supplementary Table S9C). With regard to the downregulated DEGs, cellular homeostasis and photosynthesis were common to both, and the WT specifically showed downregulated calcium ion binding terms, while the mutant not only showed downregulated another part of photosynthesis genes, but also inhibited the tetrapyrrole metabolic process (Figure 5D; Supplementary Table S9D).
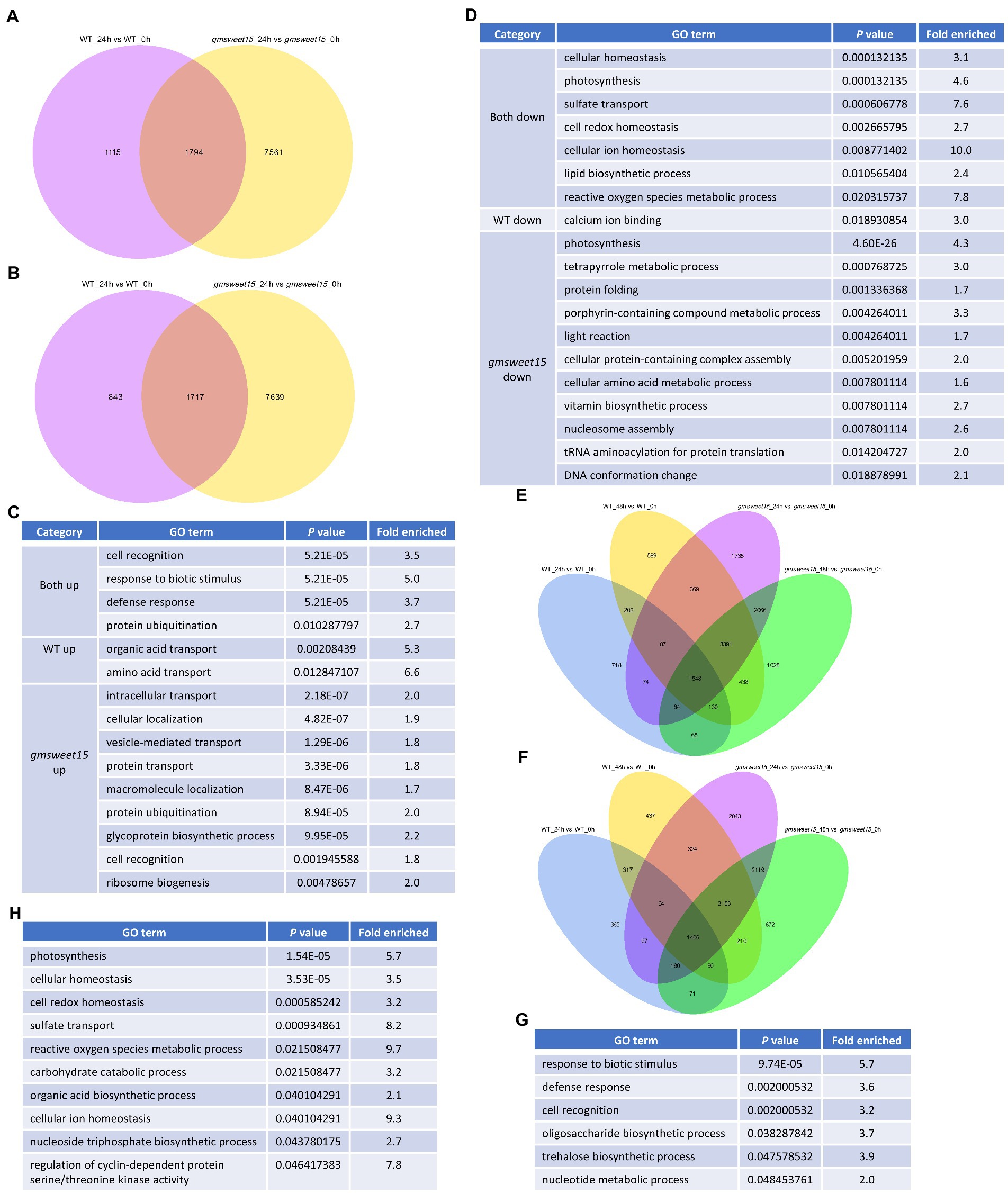
Figure 5. Characterization of common and specific DEGs between WT and gmsweet15 mutant. (A,B) Venn diagram showing the overlaps and specific upregulated (A) or downregulated (B) DEGs in WT and gmsweet15 mutant at 24 hpi. (C,D) Significantly enriched GO biological process terms in the upregulated (C) and downregulated (D) common and specific DEGs in WT and gmsweet15 mutant at 24 hpi. (E,F) Venn diagram showing the overlaps and specific upregulated (E) or downregulated (F) DEGs in WT and gmsweet15 mutant at 24 and 48 hpi. (G,H) Significantly enriched GO biological process terms in the upregulated (G) and downregulated (H) common and specific DEGs in WT and gmsweet15 mutant at 24 and 48 hpi. For expression data and full GO lists, see Supplementary Table S9.
Among the DEGs altered by S. sclerotiorum infection, 12,524 genes were induced, of which 1,548 genes were upregulated under all conditions (Figure 5E; Supplementary Table S9E). In contrast, 11,718 genes were inhibited and 1,406 genes were downregulated under all conditions (Figure 5F; Supplementary Table S9F). Among the 1,548 genes, defense response and trehalose biosynthetic process were enriched (Figure 5G; Supplementary Table S9G). Among the genes that were downregulated together, terms such as photosynthesis, cellular homeostasis, and reactive oxygen species metabolic process were enriched (Figure 5H; Supplementary Table S9H). Collectively, Venn diagrams identified common and specific DEGs between WT and gmsweet15 mutant after inoculation with S. sclerotiorum.
GSEA is frequently used to identify gene sets that are closely related to particular phenotypes. To explore which gene sets were related to early disease resistance of gmsweet15 mutant, we performed GSEA on two comparative combinations, i.e., gmsweet15_24 h vs. gmsweet15_0 h and gmsweet15_24 h vs. WT_24 h. In the former, 308 and 262 gene sets were significantly upregulated in gmsweet15_24h and gmsweet15_0 h, respectively (Supplementary Tables S10A, B). In the latter, 96 and 135 gene sets were significantly upregulated in gmsweet15_24 h and WT_24 h, respectively (Supplementary Tables S10C, D).
Selecting the top 20 gene sets with the highest enrichment degree for display, it can be seen that in addition to the gene sets of defense type described above, glucose metabolic process, glycosylation, calmodulin binding, phosphotransferases, ligand gated ion channel activity, and plasma membrane protein complex were upregulated in gmsweet15_24 h (Supplementary Figure S3A; Supplementary Table S10A). Correspondingly, the gene sets for interconverting aldoses and ketoses, heat shock protein binding, and DNA conformation change were enriched in gmsweet15_0 h (Supplementary Figure S3B; Supplementary Table S10B). In the comparative combination of gmsweet15_24 h and WT_24 h, gmsweet15_24 h was additionally enriched with vesicle mediated transport, organelle membrane and endoplasmic reticulum, although the enriched gene set of WT_24 h was similar that of gmsweet15_0 h (Supplementary Figures S3D, E; SupplementaryTables S10C, D).
Additionally, GSEA identified some genes that were strongly associated with the corresponding phenotypes in the two groups of comparative combinations. Genes such as terpene synthase 14 (TPS14), heat stress transcription factor (HSF5, HSF36), wound-induced protein (WIN), PR-10, and cysteine rich receptor-like protein kinase (CRK79, CRK63), heat shock protein 90-A1 (HSP90A1), cytochrome P450 82A2-like (CYP82A2), were positively correlated with the phenotype of gmsweet15_24 h, whereas the bZIP transcription factor (BZIP19), lipoxygenase-9 (LOX9), auxin transporter-like protein 12 (LAX12), CONSTANS-like 2a (COL2A), ammonium transporter AMT1.4, MYB transcription factor MYB164 and CBL-interacting protein kinase 23 were negatively correlated (Supplementary Figures S3C, F; Supplementary Tables S10E, F). Identification of these genes provides a promising breeding resource for to SSR resistance. Collectively, GSEA identified gene sets and genes related to gmsweet15_24 h.
First, we applied the interaction relationship in the STRING protein interaction database to construct a PPI network of the DEGs. Then, according to KEGG annotation and the number of interacting proteins, each protein was assigned a color and node size (Supplementary Figure S4). Proteins in the same pathway showed distinct aggregation, which suggested that genes not annotated by KEGG in a certain pathway may have similar functions. For example, GLYMA_04G101000 (referred to as PPP1) encodes a putative protein, although predicted to contain a domain of protein phosphatase 1 regulatory subunit 42, clustered in the plant hormone signal transduction pathway (Supplementary Figure S4). This suggested that it may also be involved in hormone signal transduction. Moreover, the PPI network showed that PPP1 interacted with GLYMA_04G147000 (EIN3-binding F-box protein 1, EBF1) and GLYMA_16G064100 (MDIS1-interacting receptor like kinase 2, MIK2), respectively, and MIK2 also interacted with EBF1 (Potuschak et al., 2003; Van der Does et al., 2017; Hou et al., 2021). These results imply that PPP1 may act as a phosphatase to fine-tune the ethylene response through dephosphorylation of EBF1 and phosphorylation of EBF1 by MIK2.
In addition, we have also identified some core proteins through the PPI network, and here, those with more than 20 interactions were designated core proteins. GLYMA_11G010500 (4-coumarate: CoA ligase isoenzyme 3, 4CL3) interacted with 27 proteins, most of which were involved in phenylpropanoid biosynthesis (Supplementary Figure S4), which emphasizes the important role of 4CL3 in phenylpropanoid biosynthesis. Collectively, we constructed a rich PPI network that was helpful for the discovery of candidate functional genes.
To deconvolute the gene regulatory network of the response to S. sclerotiorum in gmsweet15 mutant, we performed WGCNA on 3,2017 genes with expression levels. A total of 30 highly correlated modules were obtained (Supplementary Figures S5A–C). Distinct positive correlations between several modules and samples were observed (Supplementary Figure S5D). Notably, the blue module showed a specific high expression in gmsweet15_24 h, the brown module showed a trend of upregulation after inoculation, but gmsweet15 mutant exhibited a more substantial upregulation, while the turquoise module showed a downward trend after inoculation, contrary to the brown module, and the downregulation in gmsweet15 mutant was more pronounced (Figures 6A–C; Supplementary Tables S11A–C).
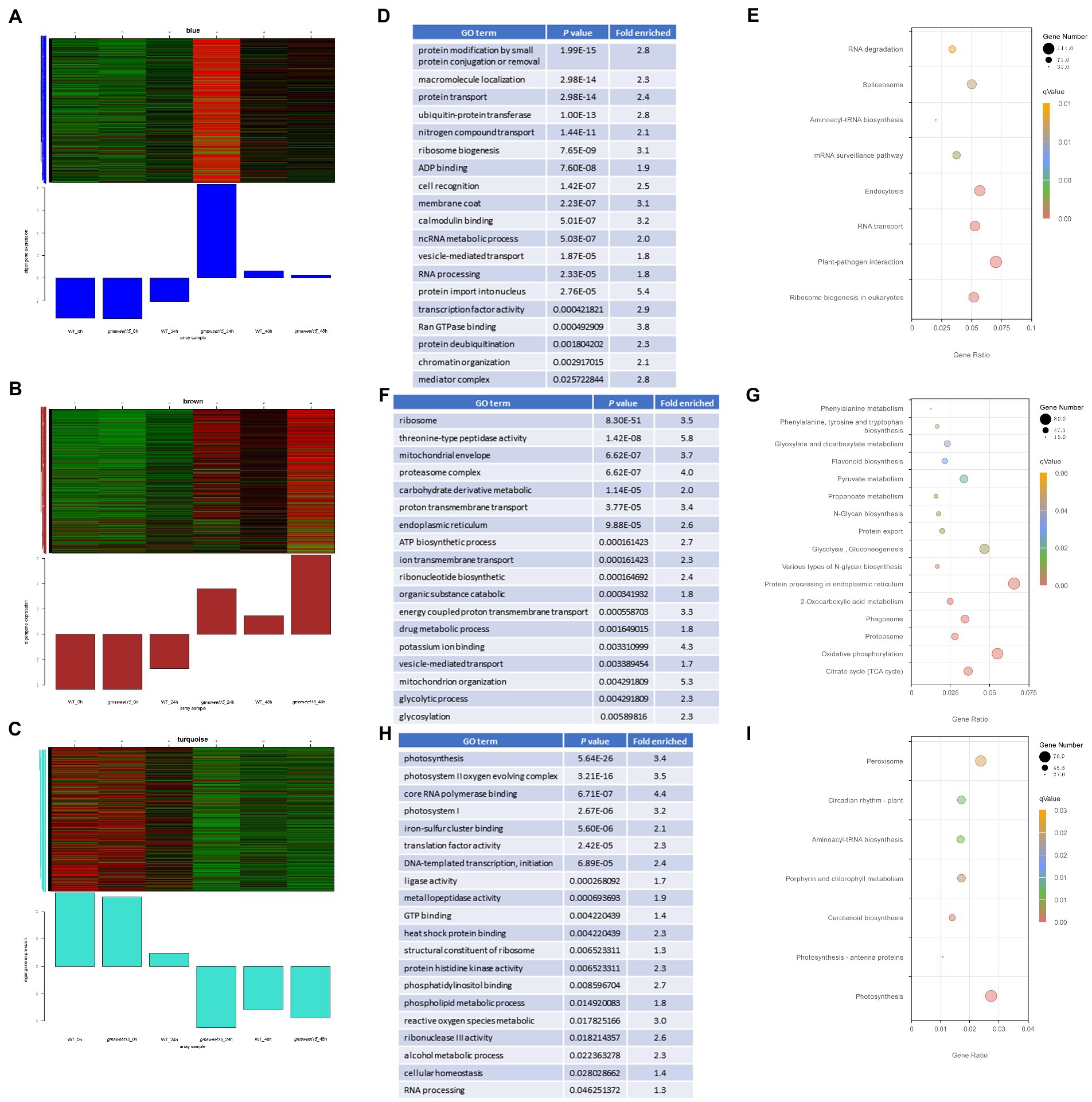
Figure 6. Characterization of gene modules associated with gmsweet15 mutant. (A–C) The co-expressed genes are shown in heatmaps and bar graphs in the blue module (A), brown module (B) and turquoise module (C). The red rectangles in the heatmap represent high expression; the green rectangles represent low expression. (D,E) Significantly enriched GO terms (D) or KEGG pathways (E) of the blue module. (F,G) Significantly enriched GO terms (F) or KEGG pathways (G) of the brown module. (H,I) Significantly enriched GO terms (H) or KEGG pathways (I) of the turquoise module. For expression data and full GO or KEGG lists, see Supplementary Table S11.
Functional enrichment analysis was performed for the three modules. The blue module was enriched with respect to terms including plant-pathogen interaction, ubiquitination modification, protein transport, ribosome biosynthesis, calcium signaling, and mediator complex (Figures 6D, E; Supplementary Table S11D). The enrichment of these terms in the blue module further confirmed that genes associated with this module are an important in the defense of gmsweet15 mutant during early S. sclerotiorum infection. In the brown module, terms such as proteasome, ATP biosynthetic process, glycosylation, endoplasmic reticulum, and flavonoid biosynthesis were enriched (Figures 6F, G; Supplementary Table S11E). Although the turquois module, terms such as photosynthesis, metallopeptidase activity, heat shock protein binding, phosphatidylinositol binding, ROS metabolism, circadian rhythm, and peroxisome were enriched (Figures 6H, I; Supplementary Table S11F).
Then, a co-expression network was constructed according to the correlation strength of the genes in each module, and we identified hub genes in each module where several biological processes clustered together (Figure 7). Notably, in the blue module, two stress-responsive genes, i.e., septum-promoting GTP-binding protein 1 (spg1; Guan and Nothnagel, 2004; Rizhsky et al., 2004; Sottosanto et al., 2007; Ascencio-Ibanez et al., 2008) and Pti1 kinase-like protein (PTI1B; Forzani et al., 2011), were considered hub genes (Figure 7A). In the brown module, MYB86 transcription factors, calnexin, 4CL3, and the three endoplasmic reticulum proteins GTP-binding protein SAR1A, translocon-associated protein subunit alpha (TRAP-alpha), and protein disulfide isomerase-like protein (PDIL-2) were identified as hub genes (Figure 7B). In addition, the NF-YA15 transcription factor was a hub gene in the turquois module (Figure 7C). We anticipated that transcription factors in hub genes should be highly co-expressed in a group with its target. Here, MYB86 was highly co-expressed with 268 genes, including 8 other transcription factors, 12 genes response to stress, 11 genes of phenylpropanoid biosynthesis, and 3 other hub genes, indicating that MYB86 may regulate the above biological processes and the expression of the respective genes. NF-YA15 may regulate the expression of 63 genes, including 16 photosynthesis genes, 2 redox homeostasis genes, and the transcription factor GLYMA_16G017400 (late elongated hypocotyl and circadian clock associated-1-like protein 1, LCL1) of the circadian rhythm. Collectively, we identified several groups of regulatory networks of gmsweet15 mutant in response to S. sclerotiorum infection using co-expression network analysis.
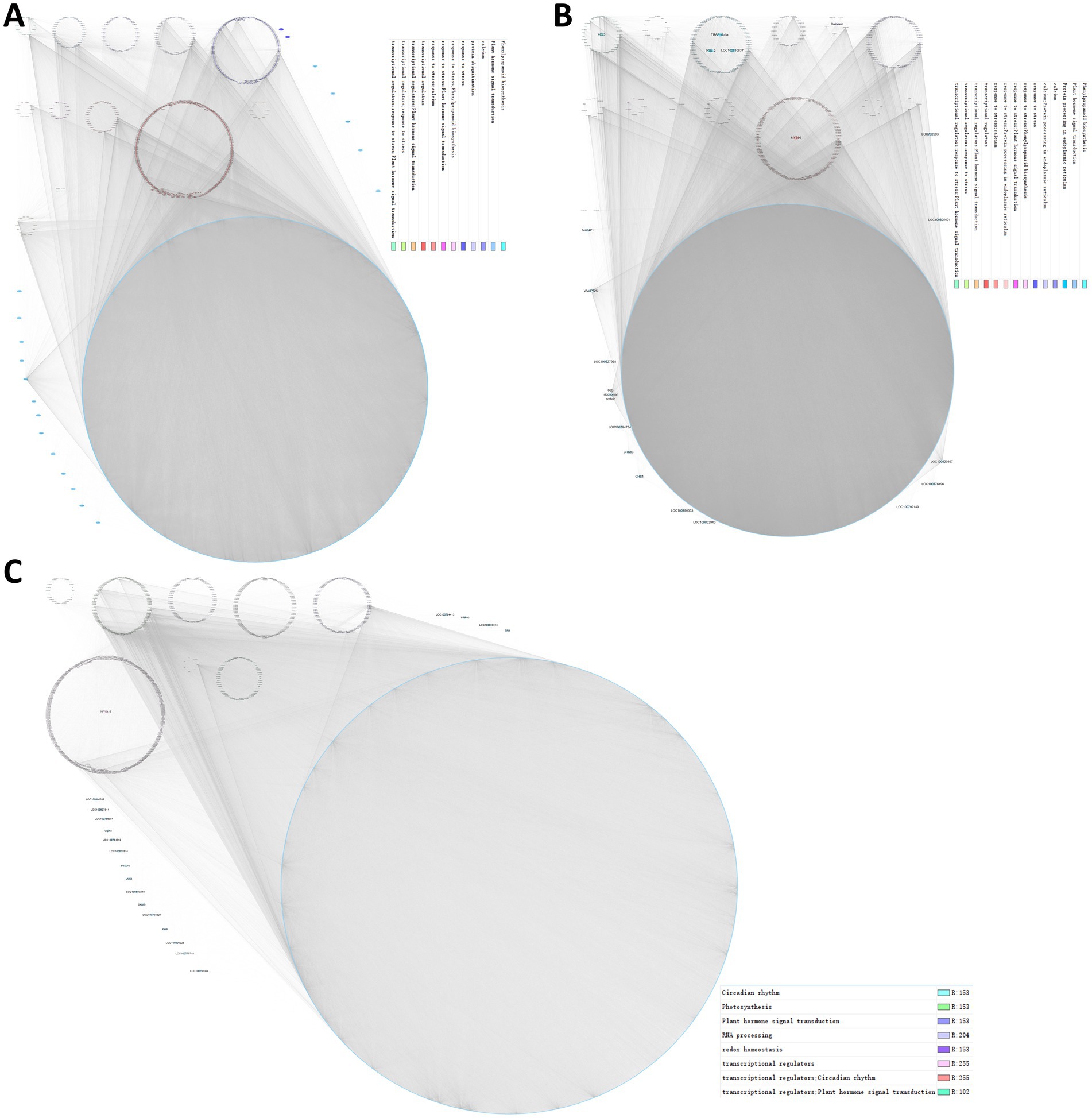
Figure 7. Regulatory networks of gmsweet15 mutant response to S. sclerotiorum infection. (A–C) Co-expression network of the blue module (A), brown module (B), and turquoise module (C). Each module was visualized using Cytoscape. Each dot represents a gene in the module, and the co-expression relationships are connected by lines. The network of top hub genes is indicated by larger circles and different biological functions involved in genes are represented by dots of different colors.
Starvation-induced resistance strategies using SWEETs as susceptibility genes have been applied in disease-resistant rice breeding (Li et al., 2012; Oliva and Quibod, 2017; Oliva et al., 2019). However, with regard to the interactions of soybean and S. sclerotiorum, the role of SWEET and potential molecular processes are unclear. In the current study, loss of GmSWEET15 enhanced soybean resistance to S. sclerotiorum, and a series of biological processes and functional genes that may play a key role in the response of gmsweet15 mutant response to S. sclerotiorum infection were identified by comparative transcriptome profiling. It was preliminarily determined that increased dynamic transcriptional reprogramming in the early stage of inoculation was an important reason for gmsweet15 mutant being able to decelerate colonization by S. sclerotiorum. Our research elucidated the molecular mechanism by which gmsweet15 mutant were able to increase their resistance to S. sclerotiorum, and we propose numerous candidate functional genes for elevating resistance to SSR.
SWEETs encode for sugar transporters, which are usually considered as susceptibility genes, while their recessive alleles provide resistance (Gao et al., 2018; Gupta, 2020; Xue et al., 2022). TALE effectors secreted by pathogenic bacteria induce the expression of SWEETs in the host and promote the release of more sugars into the apoplast, which are then consumed by pathogenic bacteria (Kay et al., 2009; Cox et al., 2017; Gupta et al., 2021). Unlike pathogenic bacteria, no similar TALE effectors have been identified in plant pathogenic fungi, although numerous studies reported that pathogenic fungi induce expression of SWEETs in the host (Miao et al., 2017; Li et al., 2018; Zhang et al., 2019). Interestingly, GmSWEET15 was not affected by inoculation with S. sclerotiorum, although its homologous gene AtSWEET15 was downregulated after inoculation (Supplementary Figure S6; Supplementary Table S2A). This observation differed from the results of previous studies, however, GmSWEET15 has also been identified as a susceptibility gene (Figures 1C, D), suggesting that gmsweet15 mutant may adopt different strategies to resist infection with S. sclerotiorum.
WT and gmsweet15 mutant upregulated defense response genes and inhibited photosynthesis at 24 hpi (Figures 3E, F, K, L, O, 4K). This is consistent with the results of many studies showing that plants inoculated with S. sclerotiorum activate the expression of defense response-related genes and downregulate the photosynthesis apparatus (Zhao et al., 2009; Ding et al., 2019; Ranjan et al., 2019; Sucher et al., 2020). Moreover, considering that enrichment analysis of DEGs depends only on the screening threshold, some important information may be lost. It is difficult to compare global expression patterns of certain biological processes under various conditions. Therefore, we combined the global defense response and photosynthesis genes to evaluate overall changes in trends. The results showed that gmsweet15 mutant exhibited more pronounced transcriptional changes (Figures 3O, 4K).
S. sclerotiorum can manipulate the redox homeostasis of host plants using pathogenic factors such as oxalic acid to promote infection (Williams et al., 2011; Girard et al., 2017; Ding et al., 2020). Here, the WT downregulated cell redox homeostasis at 24 hpi, whereas no respective enrichment occurred in gmsweet15 mutant. These results suggest that compared with WT, S. sclerotiorum easily destroyed the redox balance, making it in favor of colonization (Figure 3F). However, this process is likely associated with many obstacles that may have contributed to the failure of colonization in gmsweet15 mutant (Figure 3L).
The MAPK signaling pathway is the key signal module downstream of the receptor/sensor that perceives either endogenous or exogenous stimuli, such as P/MAMPs and effectors, and it is involved in plant defenses against pathogenic fungi, including S. sclerotiorum (Zhang and Zhang, 2022). Moreover, several genes of MAPK cascades have been proved in genetic evidence that they positively regulate the resistance of plant to S. sclerotiorum (Wang et al., 2009, 2014, 2019c). Here, the WT and gmsweet15 mutant upregulated this pathway at 24 hpi, which was more pronounced in gmsweet15 mutant (Figures 3G, I, M, 4H). Reprogramming of the phenylpropanoid pathway is considered an important resistance characteristic of soybean resistance to S. sclerotiorum (Piasecka et al., 2015; Ranjan et al., 2019), and abundant antibacterial metabolites are biosynthesized to target ergosterol biosynthesis in fungi. Accordingly, gmsweet15 mutant showed more substantial upregulation of this pathway (Figures 3G, J, M, 4J). These results imply that gmsweet15 mutant exhibited faster responses and accumulated more secondary defense metabolites upon perceiving invasion by S. sclerotiorum.
Compared with WT, gmsweet15 mutant showed more enriched biological processes at 24 hpi, such as upregulation of the secretory pathways and RAS/ARF protein signal transduction processes and inhibited tetrapyrrole metabolic process (Figures 3K, L). The mature processing and localization of PR1 seemed to be an important battlefield for pathogens in plants, and many effectors target this process (Yang et al., 2018; Sung et al., 2021; Li et al., 2022). In addition to being responsible for the mature processing and secretion of antibacterial peptides such as PR1, some endoplasmic reticulum and membrane transporters are associated with disease resistance capacity (Mao et al., 2017; Wang et al., 2021). Specific upregulation of secretion process may provide gmsweet15 mutant with an advantage in confrontation with possible effectors (Figure 3K). The adenosine diphosphate ribosylation factor guanine nucleotide exchange factor protein family is involved in regulating various signal transduction processes and plant immunity (Lozano-Duran et al., 2014; Zwiewka et al., 2019; Zhao et al., 2020). Upregulation of the proteins of this family may contribute to gmsweet15 mutant responses to S. sclerotiorum infection (Supplementary Figure S3E). Tetrapyrrole is closely related to chlorophyll degradation and leaf senescence, and it is an important retrograde signaling molecule that coordinates various stress responses by connecting chloroplasts with nucleus (Czarnecki et al., 2011; Barajas-Lopez Jde et al., 2013; Gao et al., 2016). Inhibition of tetrapyrrole metabolic process may lead to the accumulation of ROS in chloroplasts, but it does not exclude the effect of the retrograde signal from chloroplasts to nuclei, although the potential effects on interactions between soybean and S. sclerotiorum remain unclear. These biological processes of specific enrichment in gmsweet15 mutant suggest that loss of GmSWEET15 and superposition of the recognition of pathogens will specifically activate or inhibit these processes, which may play an important role in delaying early colonization by S. sclerotiorum. This also suggests that GmSWEET15 may inhibit the response of these processes to infection with S. sclerotiorum, although the mechanisms remain elusive.
Ribosome biosynthesis typically affects plant growth and development (Saez-Vasquez and Delseny, 2019). Although some abiotic stress responses have been reported, ribosome biogenesis can adapt to these stresses by increasing the transcript levels of respective key genes. However, the role of ribosome biosynthesis in plant immunity has rarely been reported (Yu H. et al., 2020; Tieu Ngoc et al., 2021). In this study, compared to WT_24 h, gmsweet15_24 h upregulated the abundance of genes related to ribosome biosynthesis (Figures 4A, C). Several studies have shown that ribosomal biosynthesis and rRNA processing are involved in the sugar/glucose response (Kojima et al., 2007; Ishida et al., 2016; Maekawa et al., 2017). This implies that GmSWEET15 loss may affect the communication between the nucleolar stress response pathway and the sugar response process during S. sclerotiorum infection, thus showing extraordinary upregulation of the ribosome biosynthesis pathway, compared with that in the WT. Ubiquitin regulates plant immunity in various ways, however, it has not been reported with regard to the interactions between plant and S. sclerotiorum (Dong et al., 2018; Wang J. et al., 2018; Ali et al., 2019; Feng et al., 2020; Liu et al., 2020; Gao et al., 2021). Here, the gmsweet15 mutant upregulated ubiquitination more strongly, indicating that these ubiquitinated enzymes may also contribute to the resistance of gmsweet15 mutant to S. sclerotiorum (Figure 4G). At present, it is inclined to think that plant immune response, especially MPK3/MPK6 activation, actively induces global downregulation of photosynthesis, thus coordinating the growth and defense trade-off (Bilgin et al., 2010; Ballare, 2014; Yang et al., 2014; de Torres Zabala et al., 2015). Moreover, photosynthetic inhibition is essential for the accumulation of ROS in chloroplasts and ETI activation (Su et al., 2018). Oxygen-evolving enhancer proteins have been implicated in stabilizing the photosystem, and their disruption leads to ROS accumulation in chloroplasts and enhances disease resistance (Lundin et al., 2007; Wang et al., 2019a). The rapid and drastic downregulation of these genes constituting the photosynthetic apparatus in gmsweet15 mutant at 24 hpi suggests that the gmsweet15 mutant may enhance its defense against S. sclerotiorum through higher intensity photoinhibition (Figure 4E). This intense transcriptional reprogramming may be an important strategy for gmsweet15 mutant to resist S. sclerotiorum, which also implies that GmSWEET15 loss may amplify the response of soybean to S. sclerotiorum in some ways.
According to WGCNA, the blue module was associated with high expression in gmsweet15_24 h and showed enriched calcium signaling and mediator complex processes (Supplementary Figure S5D; Figures 6A, D). Calcium influx is an important characteristic of plant immunity activation, and calcium signaling is involved in almost all aspects of plant immune signal transmission (Tian et al., 2020; Xu et al., 2022). The mediator complex also regulates the expression of defense genes through cooperative transcription complexes (Liao et al., 2016; Dolan et al., 2017; Mao et al., 2019; Agrawal et al., 2021; Chen et al., 2021; Kim et al., 2022). Considering the role of these two processes in plant immunity, it is obviously trustworthy for blue module to enrich them. Moreover, the genes involved in these two processes in the module are also candidate resistance to SSR genes. In addition, two hub genes, Spg1 and PTI1B, were identified in the blue module (Figure 7A). Considering the generally induced expression of Spg1 under various biotic and abiotic stresses (Guan and Nothnagel, 2004; Rizhsky et al., 2004; Sottosanto et al., 2007; Ascencio-Ibanez et al., 2008) and the role of PTI1B in plant immunity (Forzani et al., 2011), we propose that these hub genes are important candidates for resistance resources. Similarly, endoplasmic reticulum-related genes were enriched in the brown module (Figures 6F, G). Moreover, three endoplasmic reticulum proteins were identified in the 20 hub genes of this module (Figure 7B). This result further emphasizes the role of the endoplasmic reticulum in the response of gmsweet15 mutant to S. sclerotiorum infection. ROS metabolism and peroxisome were enriched in the turquois module (Figures 6H, I), suggesting that gmsweet15 may accumulate more ROS, which may be beneficial for blocking pathogen colonization. Collectively, we identified three modules related to the gmsweet15 mutant phenotype, the correspondingly enriched biological processes and hub genes using WGCNA, which provided a reliable reference for subsequent selection of resistance resources.
In the present study, through comparative transcriptome profiling, we preliminarily clarified the molecular mechanism of delaying the early colonization of S. sclerotiorum through more intense and extensive transcriptional reprogramming after the loss of GmSWEET15, and we propose numerous candidate functional genes for elevating SSR resistance.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
JL, KX, and KQ were responsible for experiment design and paper writing. SW, YX, and HP were responsible writing guidance. KQ, WC, XX, AL, XH, and ZS were responsible for experiment performance. KX, KQ, FW, and JL were responsible for experimental data processing and analysis. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (32172505), the Key Research and Development Program of Jilin Province (20210202131NC), and the Inter-Governmental International Cooperation Special Project of National Key R&D Program of China (2019YFE0114200).
We thank Baolei Jia for critically reading the manuscript. We would like to thank Editage (www.editage.cn) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1119016/full#supplementary-material
Agrawal, R., Jiri, F., and Thakur, J. K. (2021). The kinase module of the mediator complex: an important signalling processor for the development and survival of plants. J. Exp. Bot. 72, 224–240. doi: 10.1093/jxb/eraa439
Ali, M. R. M., Uemura, T., Ramadan, A., Adachi, K., Nemoto, K., Nozawa, A., et al. (2019). The ring-type E3 ubiquitin ligase JUL1 targets the VQ-motif protein JAV1 to coordinate Jasmonate signaling. Plant Physiol. 179, 1273–1284. doi: 10.1104/pp.18.00715
Ascencio-Ibanez, J. T., Sozzani, R., Lee, T. J., Chu, T. M., Wolfinger, R. D., Cella, R., et al. (2008). Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol. 148, 436–454. doi: 10.1104/pp.108.121038
Ayre, B. G. (2011). Membrane-transport systems for sucrose in relation to whole-plant carbon partitioning. Mol. Plant 4, 377–394. doi: 10.1093/mp/ssr014
Ballare, C. L. (2014). Light regulation of plant defense. Annu. Rev. Plant Biol. 65, 335–363. doi: 10.1146/annurev-arplant-050213-040145
Barajas-Lopez Jde, D., Kremnev, D., Shaikhali, J., Pinas-Fernandez, A., and Strand, A. (2013). PAPP5 is involved in the tetrapyrrole mediated plastid signalling during chloroplast development. PLoS One 8:e60305. doi: 10.1371/journal.pone.0060305
Bilgin, D. D., Zavala, J. A., Zhu, J., Clough, S. J., Ort, D. R., and Delucia, E. H. (2010). Biotic stress globally downregulates photosynthesis genes. Plant Cell Environ. 33, 1597–1613. doi: 10.1111/j.1365-3040.2010.02167.x
Bolton, M. D., Thomma, B. P., and Nelson, B. D. (2006). Sclerotinia sclerotiorum (lib.) de Bary: biology and molecular traits of a cosmopolitan pathogen. Mol. Plant Pathol. 7, 1–16. doi: 10.1111/j.1364-3703.2005.00316.x
Chai, Q., Shang, X., Wu, S., Zhu, G., Cheng, C., Cai, C., et al. (2017). 5-Aminolevulinic acid Dehydratase gene dosage affects programmed cell death and immunity. Plant Physiol. 175, 511–528. doi: 10.1104/pp.17.00816
Chen, L. Q., Hou, B. H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X. Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532. doi: 10.1038/nature09606
Chen, H. Y., Huh, J. H., Yu, Y. C., Ho, L. H., Chen, L. Q., Tholl, D., et al. (2015). The Arabidopsis vacuolar sugar transporter SWEET2 limits carbon sequestration from roots and restricts Pythium infection. Plant J. 83, 1046–1058. doi: 10.1111/tpj.12948
Chen, H., Li, M., Qi, G., Zhao, M., Liu, L., Zhang, J., et al. (2021). Two interacting transcriptional coactivators cooperatively control plant immune responses. Sci. Adv. 7:eabl7173. doi: 10.1126/sciadv.abl7173
Chen, L. Q., Qu, X. Q., Hou, B. H., Sosso, D., Osorio, S., Fernie, A. R., et al. (2012). Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335, 207–211. doi: 10.1126/science.1213351
Chu, Z., Fu, B., Yang, H., Xu, C., Li, Z., Sanchez, A., et al. (2006). Targeting xa13, a recessive gene for bacterial blight resistance in rice. Theor. Appl. Genet. 112, 455–461. doi: 10.1007/s00122-005-0145-6
Corwin, J. A., and Kliebenstein, D. J. (2017). Quantitative resistance: more than just perception of a pathogen. Plant Cell 29, 655–665. doi: 10.1105/tpc.16.00915
Cox, K. L., Meng, F., Wilkins, K. E., Li, F., Wang, P., Booher, N. J., et al. (2017). TAL effector driven induction of a SWEET gene confers susceptibility to bacterial blight of cotton. Nat. Commun. 8:15588. doi: 10.1038/ncomms15588
Czarnecki, O., Hedtke, B., Melzer, M., Rothbart, M., Richter, A., Schroter, Y., et al. (2011). An Arabidopsis GluTR binding protein mediates spatial separation of 5-aminolevulinic acid synthesis in chloroplasts. Plant Cell 23, 4476–4491. doi: 10.1105/tpc.111.086421
De Schutter, K., and Van Damme, E. J. (2015). Protein-carbohydrate interactions as part of plant defense and animal immunity. Molecules 20, 9029–9053. doi: 10.3390/molecules20059029
De Torres Zabala, M., Littlejohn, G., Jayaraman, S., Studholme, D., Bailey, T., Lawson, T., et al. (2015). Chloroplasts play a central role in plant defence and are targeted by pathogen effectors. Nat. Plants 1:15074. doi: 10.1038/nplants.2015.74
Derbyshire, M., Denton-Giles, M., Hegedus, D., Seifbarghy, S., Rollins, J., Van Kan, J., et al. (2017). The complete genome sequence of the phytopathogenic fungus Sclerotinia sclerotiorum reveals insights into the genome architecture of broad host range pathogens. Genome Biol. Evol. 9, 593–618. doi: 10.1093/gbe/evx030
Diers, B. W., Specht, J., Rainey, K. M., Cregan, P., Song, Q., Ramasubramanian, V., et al. (2018). Genetic architecture of soybean yield and agronomic traits. G3 8, 3367–3375. doi: 10.1534/g3.118.200332
Ding, P., and Ding, Y. (2020). Stories of salicylic acid: a plant defense hormone. Trends Plant Sci. 25, 549–565. doi: 10.1016/j.tplants.2020.01.004
Ding, Y., Jia, Y., Shi, Y., Zhang, X., Song, C., Gong, Z., et al. (2018). OST1-mediated BTF3L phosphorylation positively regulates CBFs during plant cold responses. EMBO J. 37:98288. doi: 10.15252/embj.201798228
Ding, Y., Mei, J., Chai, Y., Yang, W., Mao, Y., Yan, B., et al. (2020). Sclerotinia sclerotiorum utilizes host-derived copper for ROS detoxification and infection. PLoS Pathog. 16:e1008919. doi: 10.1371/journal.ppat.1008919
Ding, Y., Mei, J., Chai, Y., Yu, Y., Shao, C., Wu, Q., et al. (2019). Simultaneous Transcriptome analysis of host and pathogen highlights the interaction between Brassica oleracea and Sclerotinia sclerotiorum. Phytopathology 109, 542–550. doi: 10.1094/PHYTO-06-18-0204-R
Dolan, W. L., Dilkes, B. P., Stout, J. M., Bonawitz, N. D., and Chapple, C. (2017). Mediator complex subunits MED2, MED5, MED16, and MED23 genetically interact in the regulation of Phenylpropanoid biosynthesis. Plant Cell 29, 3269–3285. doi: 10.1105/tpc.17.00282
Dong, O. X., Ao, K., Xu, F., Johnson, K. C. M., Wu, Y., Li, L., et al. (2018). Individual components of paired typical NLR immune receptors are regulated by distinct E3 ligases. Nat. Plants 4, 699–710. doi: 10.1038/s41477-018-0216-8
Eom, J. S., Chen, L. Q., Sosso, D., Julius, B. T., Lin, I. W., Qu, X. Q., et al. (2015). SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 25, 53–62. doi: 10.1016/j.pbi.2015.04.005
Feng, H., Wang, S., Dong, D., Zhou, R., and Wang, H. (2020). Arabidopsis ubiquitin-conjugating enzymes UBC7, UBC13, and UBC14 are required in plant responses to multiple stress conditions. Plants 9:723. doi: 10.3390/plants9060723
Forzani, C., Carreri, A., La Fuente, D., Van Bentem, S., Lecourieux, D., Lecourieux, F., et al. (2011). The Arabidopsis protein kinase Pto-interacting 1-4 is a common target of the oxidative signal-inducible 1 and mitogen-activated protein kinases. FEBS J. 278, 1126–1136. doi: 10.1111/j.1742-4658.2011.08033.x
Gao, S., Gao, J., Zhu, X., Song, Y., Li, Z., Ren, G., et al. (2016). ABF2, ABF3, and ABF4 promote ABA-mediated chlorophyll degradation and leaf senescence by transcriptional activation of chlorophyll catabolic genes and senescence-associated genes in Arabidopsis. Mol. Plant 9, 1272–1285. doi: 10.1016/j.molp.2016.06.006
Gao, C., Sun, P., Wang, W., and Tang, D. (2021). Arabidopsis E3 ligase KEG associates with and ubiquitinates MKK4 and MKK5 to regulate plant immunity. J. Integr. Plant Biol. 63, 327–339. doi: 10.1111/jipb.13007
Gao, Y., Zhang, C., Han, X., Wang, Z. Y., Ma, L., Yuan, P., et al. (2018). Inhibition of OsSWEET11 function in mesophyll cells improves resistance of rice to sheath blight disease. Mol. Plant Pathol. 19, 2149–2161. doi: 10.1111/mpp.12689
Girard, I. J., Tong, C., Becker, M. G., Mao, X., Huang, J., De Kievit, T., et al. (2017). RNA sequencing of Brassica napus reveals cellular redox control of Sclerotinia infection. J. Exp. Bot. 68, 5079–5091. doi: 10.1093/jxb/erx338
Guan, Y., and Nothnagel, E. A. (2004). Binding of arabinogalactan proteins by Yariv phenylglycoside triggers wound-like responses in Arabidopsis cell cultures. Plant Physiol. 135, 1346–1366. doi: 10.1104/pp.104.039370
Guimaraes, R. L., and Stotz, H. U. (2004). Oxalate production by Sclerotinia sclerotiorum deregulates guard cells during infection. Plant Physiol. 136, 3703–3711. doi: 10.1104/pp.104.049650
Gupta, P. K. (2020). SWEET genes for disease resistance in plants. Trends Genet. 36, 901–904. doi: 10.1016/j.tig.2020.08.007
Gupta, P. K., Balyan, H. S., and Gautam, T. (2021). SWEET genes and TAL effectors for disease resistance in plants: present status and future prospects. Mol. Plant Pathol. 22, 1014–1026. doi: 10.1111/mpp.13075
Hawamda, A. I. M., Zahoor, A., Abbas, A., Ali, M. A., and Bohlmann, H. (2020). The Arabidopsis RboHB encoded by At1g09090 is important for resistance against nematodes. Int. J. Mol. Sci. 21:5556. doi: 10.3390/ijms21155556
Hibbett, D. S., Binder, M., Bischoff, J. F., Blackwell, M., Cannon, P. F., Eriksson, O. E., et al. (2007). A higher-level phylogenetic classification of the fungi. Mycol. Res. 111, 509–547. doi: 10.1016/j.mycres.2007.03.004
Hou, S., Liu, D., Huang, S., Luo, D., Liu, Z., Xiang, Q., et al. (2021). The Arabidopsis MIK2 receptor elicits immunity by sensing a conserved signature from phytocytokines and microbes. Nat. Commun. 12:5494. doi: 10.1038/s41467-021-25580-w
Ishida, T., Maekawa, S., and Yanagisawa, S. (2016). The pre-rRNA processing complex in Arabidopsis includes two WD40-domain-containing proteins encoded by glucose-inducible genes and plant-specific proteins. Mol. Plant 9, 312–315. doi: 10.1016/j.molp.2015.11.003
Ishikawa, A., Okamoto, H., Iwasaki, Y., and Asahi, T. (2001). A deficiency of coproporphyrinogen III oxidase causes lesion formation in Arabidopsis. Plant J. 27, 89–99. doi: 10.1046/j.1365-313x.2001.01058.x
Kanno, Y., Oikawa, T., Chiba, Y., Ishimaru, Y., Shimizu, T., Sano, N., et al. (2016). AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat. Commun. 7:13245. doi: 10.1038/ncomms13245
Kay, S., Hahn, S., Marois, E., Wieduwild, R., and Bonas, U. (2009). Detailed analysis of the DNA recognition motifs of the Xanthomonas type III effectors AvrBs3 and AvrBs3Δrep16. Plant J. 59, 859–871. doi: 10.1111/j.1365-313X.2009.03922.x
Kim, J. H., Castroverde, C. D. M., Huang, S., Li, C., Hilleary, R., Seroka, A., et al. (2022). Increasing the resilience of plant immunity to a warming climate. Nature 607, 339–344. doi: 10.1038/s41586-022-04902-y
Kojima, H., Suzuki, T., Kato, T., Enomoto, K., Sato, S., Kato, T., et al. (2007). Sugar-inducible expression of the nucleolin-1 gene of Arabidopsis thaliana and its role in ribosome synthesis, growth and development. Plant J. 49, 1053–1063. doi: 10.1111/j.1365-313X.2006.03016.x
Li, H., Li, X., Xuan, Y., Jiang, J., Wei, Y., and Piao, Z. (2018). Genome wide identification and expression profiling of SWEET genes family reveals its role during Plasmodiophora brassicae-induced formation of Clubroot in Brassica rapa. Front. Plant Sci. 9:207. doi: 10.3389/fpls.2018.00207
Li, T., Liu, B., Spalding, M. H., Weeks, D. P., and Yang, B. (2012). High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 30, 390–392. doi: 10.1038/nbt.2199
Li, J., Ma, X., Wang, C., Liu, S., Yu, G., Gao, M., et al. (2022). Acetylation of a fungal effector that translocates host PR1 facilitates virulence. Elife 11:e82628. doi: 10.7554/eLife.82628
Li, Y., Wang, Y., Zhang, H., Zhang, Q., Zhai, H., Liu, Q., et al. (2017). The plasma membrane-localized sucrose transporter IbSWEET10 contributes to the resistance of sweet potato to Fusarium oxysporum. Front. Plant Sci. 8:197. doi: 10.3389/fpls.2017.00197
Liao, C. J., Lai, Z., Lee, S., Yun, D. J., and Mengiste, T. (2016). Arabidopsis HOOKLESS1 regulates responses to pathogens and Abscisic acid through interaction with MED18 and acetylation of WRKY33 and ABI5 chromatin. Plant Cell 28, 1662–1681. doi: 10.1105/tpc.16.00105
Liu, Y., Wang, K., Cheng, Q., Kong, D., Zhang, X., Wang, Z., et al. (2020). Cysteine protease RD21A regulated by E3 ligase SINAT4 is required for drought-induced resistance to pseudomonas syringae in Arabidopsis. J. Exp. Bot. 71, 5562–5576. doi: 10.1093/jxb/eraa255
Lozano-Duran, R., Bourdais, G., He, S. Y., and Robatzek, S. (2014). The bacterial effector HopM1 suppresses PAMP-triggered oxidative burst and stomatal immunity. New Phytol. 202, 259–269. doi: 10.1111/nph.12651
Lundin, B., Hansson, M., Schoefs, B., Vener, A. V., and Spetea, C. (2007). The Arabidopsis PsbO2 protein regulates dephosphorylation and turnover of the photosystem II reaction Centre D1 protein. Plant J. 49, 528–539. doi: 10.1111/j.1365-313X.2006.02976.x
Ma, W., and Berkowitz, G. A. (2011). Ca2+ conduction by plant cyclic nucleotide gated channels and associated signaling components in pathogen defense signal transduction cascades. New Phytol. 190, 566–572. doi: 10.1111/j.1469-8137.2010.03577.x
Ma, J., Yang, S., Wang, D., Tang, K., Feng, X. X., and Feng, X. Z. (2020). Genetic mapping of a light-dependent lesion mimic mutant reveals the function of Coproporphyrinogen III oxidase homolog in soybean. Front. Plant Sci. 11:893. doi: 10.3389/fpls.2020.00893
Maekawa, S., Ishida, T., and Yanagisawa, S. (2017). Reduced expression of APUM24, encoding a novel rRNA processing factor, induces sugar-dependent Nucleolar stress and altered sugar responses in Arabidopsis thaliana. Plant Cell 30, 209–227. doi: 10.1105/tpc.17.00778
Mao, H., Aryal, B., Langenecker, T., Hagmann, J., Geisler, M., and Grebe, M. (2017). Arabidopsis BTB/POZ protein-dependent PENETRATION3 trafficking and disease susceptibility. Nat. Plants 3, 854–858. doi: 10.1038/s41477-017-0039-z
Mao, X., Kim, J. I., Wheeler, M. T., Heintzelman, A. K., Weake, V. M., and Chapple, C. (2019). Mutation of mediator subunit CDK8 counteracts the stunted growth and salicylic acid hyperaccumulation phenotypes of an Arabidopsis MED5 mutant. New Phytol. 223, 233–245. doi: 10.1111/nph.15741
Miao, H., Sun, P., Liu, Q., Miao, Y., Liu, J., Zhang, K., et al. (2017). Genome-wide analyses of SWEET family proteins reveal involvement in fruit development and abiotic/biotic stress responses in banana. Sci. Rep. 7:3536. doi: 10.1038/s41598-017-03872-w
Micali, C., Gollner, K., Humphry, M., Consonni, C., and Panstruga, R. (2008). The powdery mildew disease of Arabidopsis: a paradigm for the interaction between plants and biotrophic fungi. Arabidop. Book 6:e0115. doi: 10.1199/tab.0115
Moriyama, E. N., Strope, P. K., Opiyo, S. O., Chen, Z., and Jones, A. M. (2006). Mining the Arabidopsis thaliana genome for highly-divergent seven transmembrane receptors. Genome Biol. 7:R96. doi: 10.1186/gb-2006-7-10-r96
Ngou, B. P. M., Ahn, H. K., Ding, P., and Jones, J. D. G. (2021). Mutual potentiation of plant immunity by cell-surface and intracellular receptors. Nature 592, 110–115. doi: 10.1038/s41586-021-03315-7
Oliva, R., Ji, C., Atienza-Grande, G., Huguet-Tapia, J. C., Perez-Quintero, A., Li, T., et al. (2019). Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat. Biotechnol. 37, 1344–1350. doi: 10.1038/s41587-019-0267-z
Oliva, R., and Quibod, I. L. (2017). Immunity and starvation: new opportunities to elevate disease resistance in crops. Curr. Opin. Plant Biol. 38, 84–91. doi: 10.1016/j.pbi.2017.04.020
Patil, G., Valliyodan, B., Deshmukh, R., Prince, S., Nicander, B., Zhao, M., et al. (2015). Soybean (Glycine max) SWEET gene family: insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genomics 16:520. doi: 10.1186/s12864-015-1730-y
Perchepied, L., Balagué, C., Riou, C., Claudel-Renard, C., Rivière, N., Grezes-Besset, B., et al. (2010). Nitric oxide participates in the complex interplay of defense-related signaling pathways controlling disease resistance to Sclerotinia sclerotiorum in Arabidopsis thaliana. Mol. Plant Microbe Interact. 23, 846–860. doi: 10.1094/MPMI-23-7-0846
Piasecka, A., Jedrzejczak-Rey, N., and Bednarek, P. (2015). Secondary metabolites in plant innate immunity: conserved function of divergent chemicals. New Phytol. 206, 948–964. doi: 10.1111/nph.13325
Potuschak, T., Lechner, E., Parmentier, Y., Yanagisawa, S., Grava, S., Koncz, C., et al. (2003). EIN3-dependent regulation of plant ethylene hormone signaling by two Arabidopsis F box proteins: EBF1 and EBF2. Cells 115, 679–689. doi: 10.1016/S0092-8674(03)00968-1
Rahman, H., Xu, Y. P., Zhang, X. R., and Cai, X. Z. (2016). Brassica napus genome possesses extraordinary high number of CAMTA genes and CAMTA3 contributes to PAMP triggered immunity and resistance to Sclerotinia sclerotiorum. Front. Plant Sci. 7:581. doi: 10.3389/fpls.2016.00581
Ranjan, A., Westrick, N. M., Jain, S., Piotrowski, J. S., Ranjan, M., Kessens, R., et al. (2019). Resistance against Sclerotinia sclerotiorum in soybean involves a reprogramming of the phenylpropanoid pathway and up-regulation of antifungal activity targeting ergosterol biosynthesis. Plant Biotechnol. J. 17, 1567–1581. doi: 10.1111/pbi.13082
Rizhsky, L., Davletova, S., Liang, H., and Mittler, R. (2004). The zinc finger protein Zat12 is required for cytosolic ascorbate peroxidase 1 expression during oxidative stress in Arabidopsis. J. Biol. Chem. 279, 11736–11743. doi: 10.1074/jbc.M313350200
Saand, M. A., Xu, Y. P., Li, W., Wang, J. P., and Cai, X. Z. (2015a). Cyclic nucleotide gated channel gene family in tomato: genome-wide identification and functional analyses in disease resistance. Front. Plant Sci. 6:303. doi: 10.3389/fpls.2015.00303
Saand, M. A., Xu, Y. P., Munyampundu, J. P., Li, W., Zhang, X. R., and Cai, X. Z. (2015b). Phylogeny and evolution of plant cyclic nucleotide-gated ion channel (CNGC) gene family and functional analyses of tomato CNGCs. DNA Res. 22, 471–483. doi: 10.1093/dnares/dsv029
Saez-Vasquez, J., and Delseny, M. (2019). Ribosome biogenesis in plants: from functional 45S ribosomal DNA organization to ribosome assembly factors. Plant Cell 31, 1945–1967. doi: 10.1105/tpc.18.00874
Seo, P. J., Park, J. M., Kang, S. K., Kim, S. G., and Park, C. M. (2011). An Arabidopsis senescence-associated protein SAG29 regulates cell viability under high salinity. Planta 233, 189–200. doi: 10.1007/s00425-010-1293-8
Sosso, D., Luo, D., Li, Q. B., Sasse, J., Yang, J., Gendrot, G., et al. (2015). Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 47, 1489–1493. doi: 10.1038/ng.3422
Sottosanto, J. B., Saranga, Y., and Blumwald, E. (2007). Impact of AtNHX1, a vacuolar Na+/H+ antiporter, upon gene expression during short-and long-term salt stress in Arabidopsis thaliana. BMC Plant Biol. 7:18. doi: 10.1186/1471-2229-7-18
Su, J., Yang, L., Zhu, Q., Wu, H., He, Y., Liu, Y., et al. (2018). Active photosynthetic inhibition mediated by MPK3/MPK6 is critical to effector-triggered immunity. PLoS Biol. 16:e2004122. doi: 10.1371/journal.pbio.2004122
Sucher, J., Mbengue, M., Dresen, A., Barascud, M., Didelon, M., Barbacci, A., et al. (2020). Phylotranscriptomics of the Pentapetalae reveals frequent regulatory variation in plant local responses to the fungal pathogen Sclerotinia sclerotiorum. Plant Cell 32, 1820–1844. doi: 10.1105/tpc.19.00806
Sung, Y. C., Outram, M. A., Breen, S., Wang, C., Dagvadorj, B., Winterberg, B., et al. (2021). PR1-mediated defence via C-terminal peptide release is targeted by a fungal pathogen effector. New Phytol. 229, 3467–3480. doi: 10.1111/nph.17128
Thieffry, A., Lopez-Marquez, D., Bornholdt, J., Malekroudi, M. G., Bressendorff, S., Barghetti, A., et al. (2022). PAMP-triggered genetic reprogramming involves widespread alternative transcription initiation and an immediate transcription factor wave. Plant Cell 34, 2615–2637. doi: 10.1093/plcell/koac108
Tian, W., Wang, C., Gao, Q., Li, L., and Luan, S. (2020). Calcium spikes, waves and oscillations in plant development and biotic interactions. Nat. Plants 6, 750–759. doi: 10.1038/s41477-020-0667-6
Tieu Ngoc, L. N., Jung Park, S., Thi Huong, T., Lee, K. H., and Kang, H. (2021). N4-methylcytidine ribosomal RNA methylation in chloroplasts is crucial for chloroplast function, development, and abscisic acid response in Arabidopsis. J. Integr. Plant Biol. 63, 570–582. doi: 10.1111/jipb.13009
Van Der Does, D., Boutrot, F., Engelsdorf, T., Rhodes, J., Mckenna, J. F., Vernhettes, S., et al. (2017). The Arabidopsis leucine-rich repeat receptor kinase MIK2/LRR-KISS connects cell wall integrity sensing, root growth and response to abiotic and biotic stresses. PLoS Genet. 13:e1006832. doi: 10.1371/journal.pgen.1006832
Wang, Z., Bao, L. L., Zhao, F. Y., Tang, M. Q., Chen, T., Li, Y., et al. (2019c). BnaMPK3 is a key regulator of defense responses to the devastating plant pathogen Sclerotinia sclerotiorum in oilseed rape. Front. Plant Sci. 10:91. doi: 10.3389/fpls.2019.00091
Wang, Z., Fang, H., Chen, Y., Chen, K., Li, G., Gu, S., et al. (2014). Overexpression of BnWRKY33 in oilseed rape enhances resistance to Sclerotinia sclerotiorum. Mol. Plant Pathol. 15, 677–689. doi: 10.1111/mpp.12123
Wang, J., Grubb, L. E., Wang, J., Liang, X., Li, L., Gao, C., et al. (2018). A regulatory module controlling homeostasis of a plant immune kinase. Mol. Cell 69:e496, 493–504.e6. doi: 10.1016/j.molcel.2017.12.026
Wang, H., Guo, B., Yang, B., Li, H., Xu, Y., Zhu, J., et al. (2021). An atypical Phytophthora sojae RxLR effector manipulates host vesicle trafficking to promote infection. PLoS Pathog. 17:e1010104. doi: 10.1371/journal.ppat.1010104
Wang, S., Li, Q. P., Wang, J., Yan, Y., Zhang, G. L., Yan, Y., et al. (2019a). YR36/WKS1-mediated phosphorylation of PsbO, an extrinsic member of photosystem II, inhibits photosynthesis and confers stripe rust resistance in wheat. Mol. Plant 12, 1639–1650. doi: 10.1016/j.molp.2019.10.005
Wang, Z., Ma, L. Y., Cao, J., Li, Y. L., Ding, L. N., Zhu, K. M., et al. (2019d). Recent advances in mechanisms of plant defense to Sclerotinia sclerotiorum. Front. Plant Sci. 10:1314. doi: 10.3389/fpls.2019.01314
Wang, Z., Mao, H., Dong, C., Ji, R., Cai, L., Fu, H., et al. (2009). Overexpression of Brassica napus MPK4 enhances resistance to Sclerotinia sclerotiorum in oilseed rape. Mol. Plant Microbe Interact. 22, 235–244. doi: 10.1094/MPMI-22-3-0235
Wang, J. P., Munyampundu, J. P., Xu, Y. P., and Cai, X. Z. (2015). Phylogeny of plant calcium and Calmodulin-dependent protein kinases (CCaMKs) and functional analyses of tomato CCaMK in disease resistance. Front. Plant Sci. 6:1075. doi: 10.3389/fpls.2015.01075
Wang, Z., Wan, L., Xin, Q., Chen, Y., Zhang, X., Dong, F., et al. (2018). Overexpression of OsPGIP2 confers Sclerotinia sclerotiorum resistance in Brassica napus through increased activation of defense mechanisms. J. Exp. Bot. 69, 3141–3155. doi: 10.1093/jxb/ery138
Wang, J. P., Xu, Y. P., Munyampundu, J. P., Liu, T. Y., and Cai, X. Z. (2016). Calcium-dependent protein kinase (CDPK) and CDPK-related kinase (CRK) gene families in tomato: genome-wide identification and functional analyses in disease resistance. Mol. Genet. Genomics 291, 661–676. doi: 10.1007/s00438-015-1137-0
Wang, S., Yokosho, K., Guo, R., Whelan, J., Ruan, Y. L., Ma, J. F., et al. (2019b). The soybean sugar transporter GmSWEET15 mediates sucrose export from endosperm to early embryo. Plant Physiol. 180, 2133–2141. doi: 10.1104/pp.19.00641
Wei, L., Jian, H., Lu, K., Filardo, F., Yin, N., Liu, L., et al. (2016). Genome-wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol. J. 14, 1368–1380. doi: 10.1111/pbi.12501
Wei, W., Xu, L., Peng, H., Zhu, W., Tanaka, K., Cheng, J., et al. (2022). A fungal extracellular effector inactivates plant polygalacturonase-inhibiting protein. Nat. Commun. 13:2213. doi: 10.1038/s41467-022-29788-2
Wellmer, F., Alves-Ferreira, M., Dubois, A., Riechmann, J. L., and Meyerowitz, E. M. (2006). Genome-wide analysis of gene expression during early Arabidopsis flower development. PLoS Genet. 2:e117. doi: 10.1371/journal.pgen.0020117
Wen, Z., Tan, R., Zhang, S., Collins, P. J., Yuan, J., Du, W., et al. (2018). Integrating GWAS and gene expression data for functional characterization of resistance to white mould in soyabean. Plant Biotechnol. J. 16, 1825–1835. doi: 10.1111/pbi.12918
Williams, B., Kabbage, M., Kim, H. J., Britt, R., and Dickman, M. B. (2011). Tipping the balance: Sclerotinia sclerotiorum secreted oxalic acid suppresses host defenses by manipulating the host redox environment. PLoS Pathog. 7:e1002107. doi: 10.1371/journal.ppat.1002107
Wu, J., Zhao, Q., Liu, S., Shahid, M., Lan, L., Cai, G., et al. (2016). Genome-wide association study identifies new loci for resistance to Sclerotinia stem rot in Brassica napus. Front. Plant Sci. 7:1418. doi: 10.3389/fpls.2016.01418
Xu, G., Moeder, W., Yoshioka, K., and Shan, L. (2022). A tale of many families: calcium channels in plant immunity. Plant Cell 34, 1551–1567. doi: 10.1093/plcell/koac033
Xue, X., Wang, J., Shukla, D., Cheung, L. S., and Chen, L. Q. (2022). When SWEETs turn tweens: updates and perspectives. Annu. Rev. Plant Biol. 73, 379–403. doi: 10.1146/annurev-arplant-070621-093907
Yang, B., Sugio, A., and White, F. F. (2006). Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc. Natl. Acad. Sci. USA 103, 10503–10508. doi: 10.1073/pnas.0604088103
Yang, G., Tang, L., Gong, Y., Xie, J., Fu, Y., Jiang, D., et al. (2018). A cerato-platanin protein SsCP1 targets plant PR1 and contributes to virulence of Sclerotinia sclerotiorum. New Phytol. 217, 739–755. doi: 10.1111/nph.14842
Yang, C., Zhang, Z. S., Gao, H. Y., Liu, M. J., and Fan, X. L. (2014). Mechanisms by which the infection of Sclerotinia sclerotiorum (lib.) de Bary affects the photosynthetic performance in tobacco leaves. BMC Plant Biol. 14:240. doi: 10.1186/s12870-014-0240-4
Yu, H., Kong, X., Huang, H., Wu, W., Park, J., Yun, D. J., et al. (2020). STCH4/REIL2 confers cold stress tolerance in Arabidopsis by promoting rRNA processing and CBF protein translation. Cell Rep. 30, 229–242 e225. doi: 10.1016/j.celrep.2019.12.012
Yu, G., Xian, L., Xue, H., Yu, W., Rufian, J. S., Sang, Y., et al. (2020). A bacterial effector protein prevents MAPK-mediated phosphorylation of SGT1 to suppress plant immunity. PLoS Pathog. 16:e1008933. doi: 10.1371/journal.ppat.1008933
Yuan, M., Chu, Z., Li, X., Xu, C., and Wang, S. (2009). Pathogen-induced expressional loss of function is the key factor in race-specific bacterial resistance conferred by a recessive R gene xa13 in rice. Plant Cell Physiol. 50, 947–955. doi: 10.1093/pcp/pcp046
Yuan, M., Jiang, Z., Bi, G., Nomura, K., Liu, M., Wang, Y., et al. (2021). Pattern-recognition receptors are required for NLR-mediated plant immunity. Nature 592, 105–109. doi: 10.1038/s41586-021-03316-6
Yuan, M., and Wang, S. (2013). Rice MtN3/saliva/SWEET family genes and their homologs in cellular organisms. Mol. Plant 6, 665–674. doi: 10.1093/mp/sst035
Zhang, W., Wang, S., Yu, F., Tang, J., Shan, X., Bao, K., et al. (2019). Genome-wide characterization and expression profiling of SWEET genes in cabbage (Brassica oleracea var. capitata L.) reveal their roles in chilling and clubroot disease responses. BMC Genomics 20:93. doi: 10.1186/s12864-019-5454-2
Zhang, M., and Zhang, S. (2022). Mitogen-activated protein kinase cascades in plant signaling. J. Integr. Plant Biol. 64, 301–341. doi: 10.1111/jipb.13215
Zhao, J., Buchwaldt, L., Rimmer, S. R., Sharpe, A., Mcgregor, L., Bekkaoui, D., et al. (2009). Patterns of differential gene expression in Brassica napus cultivars infected with Sclerotinia sclerotiorum. Mol. Plant Pathol. 10, 635–649. doi: 10.1111/j.1364-3703.2009.00558.x
Zhao, Z., Yang, X., Lu, S., Fan, J., Opiyo, S., Yang, P., et al. (2020). Deciphering the novel role of AtMIN7 in cuticle formation and defense against the bacterial pathogen infection. Int. J. Mol. Sci. 21:5547. doi: 10.3390/ijms21155547
Keywords: GmSWEET15, Sclerotinia sclerotiorum, transcriptome, gene co-expression network, susceptibility
Citation: Xiao K, Qiao K, Cui W, Xu X, Pan H, Wang F, Wang S, Yang F, Xuan Y, Li A, Han X, Song Z and Liu J (2023) Comparative transcriptome profiling reveals the importance of GmSWEET15 in soybean susceptibility to Sclerotinia sclerotiorum. Front. Microbiol. 14:1119016. doi: 10.3389/fmicb.2023.1119016
Edited by:
Xiuling Yang, Institute of Plant Protection (CAAS), ChinaReviewed by:
Fuhao Cui, China Agricultural University, ChinaCopyright © 2023 Xiao, Qiao, Cui, Xu, Pan, Wang, Wang, Yang, Xuan, Li, Han, Song and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinliang Liu,  amxsaXVAamx1LmVkdS5jbg==
amxsaXVAamx1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.