 Xue Bai
Xue Bai Yvjing Li
Yvjing Li Pengyu Zhao
Pengyu Zhao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 22 February 2023
Sec. Microbiological Chemistry and Geomicrobiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1116535
Introduction: A growing amount of heavy metal contamination in soil disturbs the ecosystem’s equilibrium, in which microbial populations play a key role in the nutrient cycle of soils. However, given the different sensitivity of microbial communities to different spatial and temporal scales, microbial community structure and function also have varied response mechanisms to different heavy metal contaminated habitats.
Methods: In this study, samples were taken prior to Cr stress (CK) and 6 h and 6 days after Cr stress (Cr_6h, Cr_6d) in laboratory experiments. High-throughput sequencing revealed trends in the structure and diversity of the bacterial communities, and real-time fluorescence quantitative polymerase chain reaction (qPCR) was used to analyze trends in nitrogen cycle functional genes (AOA-amoA, AOB-amoA, narG, nirK, and nifH).
Results: The findings showed that (1) the composition structure of the soil bacterial community changed considerably in Cr–stressed soils; α-diversity showed significant phase transition characteristic from stress to stability (p < 0.05). (2) With an overall rising tendency, the abundance of the nitrogen cycle functional genes (AOA-amoA and AOB-amoA) decreased considerably before increasing, and α-diversity dramatically declined (p < 0.05). (3) The redundancy analysis (RDA) and permutational multivariate analysis of variance (PERMANOVA) tests results showed that the soil physicochemical parameters were significantly correlated with the nitrogen cycle functional genes (r: 0.4195, p < 0.01). Mantel analysis showed that available nitrogen (N), available potassium (K), and available phosphorus (P) were significantly correlated with nifH (p = 0.006, 0.008, 0.004), and pH was highly significantly correlated with nifH (p = 0.026). The PLS-ME (partial least squares path model) model further demonstrated a significant direct effect of the soil physicochemical parameters on the nitrogen cycling functional genes.
Discussion: As a result, the composition and diversity of the bacterial community and the nitrogen cycle functional genes in Cr–stressed agricultural soils changed considerably. However, the influence of the soil physicochemical parameters on the functional genes involved in the nitrogen cycle was greater than that of the bacterial community. and Cr stress affects the N cycling process in soil mainly by affecting nitrification. This research has significant practical ramifications for understanding the mechanisms of microbial community homeostasis maintenance, nitrogen cycle response mechanisms, and soil remediation in heavy metal–contaminated agricultural soils.
Chromium (Cr) has remained one of the most prevalent heavy metal pollutants in groundwater and soil since the industrialization of society in the middle and late 20th century as a result of mining, metallurgy, and tanning (Chen et al., 2014). According to studies, China has a total exceedance rate of 16.1% for heavy metal contamination in agricultural soils, and the amount of arable land that has been contaminated by Cr has surpassed 20 million hm2 (square hectometer) (Duan et al., 2016). As the main heavy metal pollutant in China’s soil, Cr can enter the organism through the cell plasma membrane to change the genetic material or disrupt the expression of metal proteins due to its high mobility, toxicity, persistence, and solubility (Mathivanan et al., 2021), posing a major threat to soil microbes, plant growth, and food safety. As a result, heavy metal contamination has emerged as a challenging aspect of environmental management and has sparked global concern.
Although microbial communities in soils only make up a small portion of organic matter, they play an important role in energy flow, nutrient cycling, material transformation, and pollutant degradation and are highly sensitive to heavy metals (Xie et al., 2016). Therefore, the accumulation of heavy metals in soils impacts the structure and function of soil ecosystems, and their structural and functional alterations are reliable indicators of soil quality and ecological functions (Wu et al., 2017). Heavy metal pollution, such as that caused by lead (Pb), cadmium (Cd), copper (Cu), or complex heavy metals, greatly affects the abundance, composition, and diversity of microbial communities in soil, and the degree of the impact is directly correlated with the type of heavy metals, heavy metal concentration microbial communities can improve plant tolerance by reducing the effectiveness of heavy metals in soil (Zhang et al., 2016; Huang et al., 2021; Mao et al., 2022). Additionally, soil physicochemical properties have a close relationship with soil microbial communities, which act as key nutrient regulators. The abundance and diversity of the microbial community are strongly linked to soil organic carbon (SOC), pH, available nitrogen (N) and available phosphorus (P), indicating that heavy metals and soil physicochemical properties have a synergistic effect on the structure and function of the microbial community (Deng et al., 2015; Kasemodel et al., 2019; Hu et al., 2021). As a result, research on the methods by which soil microbial communities respond to heavy metal stress is a hot topic right now, but there is presently a dearth of data on the microbial communities in soils that are stressed by Cr.
One of the components necessary for microbial growth is nitrogen (N), which is also critical for soil nitrogen supply, crop growth, and food security (Robertson and Vitousek, 2009). Soil nitrogen turnover is a crucial aspect of agroecosystems. In addition, the nitrogen cycle is a network of redox processes of nitrogen compounds that are catalyzed jointly by plants and bacteria, fungus, and archaea (Devrim et al., 2017). Soil microorganisms are usually involved in the main processes of the nitrogen cycle such as nitrogen fixation, ammonification, nitrification, and denitrification. Moreover, soil microorganisms constitute the primary driving force in the nitrogen cycle process, which can directly influence the process of nitrogen transformation and supply (Van der Heijden et al., 2008). Among soil microorganisms, the nitrogen cycle mediated by nitrogen-fixing bacteria and nitrifying bacteria plays an important role in ecosystem function (Kuypers et al., 2018). Through the expression of functional genes such as encoding key enzymes, nitrogen cycle functional microorganisms in soil can regulate the soil nitrogen cycle, and the abundance of their genes is frequently used to estimate the number of functional microorganisms and population fluctuations related to the soil nitrogen cycle (Li X. M. et al., 2020). The investigation of the ecological processes of the nitrogen cycle and associated functional microorganisms or functional gene clusters is one of the hottest areas of current microbial ecology research.
Numerous studies have examined the ecological processes of nitrogen cycling in various habitats by examining the expression of genes such as nifH (nitrogen-fixing enzyme), amoA (ammonia monooxygenase), and narG (nitrate reductase) during nitrogen fixation, nitrification, and denitrification (Ouyang et al., 2018; Li P. P. et al., 2019, 2020). For example, high levels of heavy metal contamination may lead to greater functional diversity and abundance of resistance genes to strengthen microbial community resistance (Jie et al., 2016); narG gene abundance in biochar-remediated heavy metal-contaminated soils was most sensitive to changes in soil samples’ physicochemical properties (Li M. Y. et al., 2019); the abundance of bacterial functional genes associated with the carbon and nitrogen cycles increased in Cd-contaminated soils with soil amendments applied by GeoChip; and in eutrophic soils with urea addition under Cd stress conditions, Cd altered microorganisms’ C/N metabolism from catabolic to anabolic dominance. However, there is little research on the functional microbial communities associated with nitrogen cycling in Cr-contaminated soils (Xu et al., 2021). The nitrogen transformation process of microbial communities induced by heavy metal pollution is controlled by a variety of soil physicochemical parameters and complicated interactions between organisms (Giannopoulos et al., 2020; Rijk and Ekblad, 2020), and the sensitivity of nitrogen cycling-related microbial communities to different geographical and temporal scales is diverse, so their response processes are difficult to predict.
Because ammonia-oxidizing and denitrifying microorganisms serve as model organisms, both functional gene abundances are frequently employed to predict the potential rates of total soil nitrification and denitrification (Wang et al., 2022); additionally, the ammonia-oxidizing process consists only of a small number of phylogenetically aggregated ammonia-oxidizing archaea (AOA) and ammonia-oxidizing bacteria (AOB), which are better suited for investigating their relationship with the ecosystem (Khanal and Lee, 2020). Therefore, in this study, we collected rhizosphere soil samples of cereals under Cr stress and used high-throughput sequencing analysis and the real-time fluorescence quantitative PCR (q-PCR) technique to analyze the microbial community and the abundance of functional nitrogen cycle genes (nifH, AOA-amoA, AOB-amoA, narG, nirK [nitrate reductase]) in soil. In order to analyze the impacts of Cr stress on the nitrogen cycle in agricultural soils holistically, it is helpful to study the mechanisms by which functional microbial communities in the rhizosphere soils of cereals respond to time series of Cr stress. This research provides a theoretical framework for anticipating how microorganisms will respond, adapt, and feedback to heavy metal contamination in the environment, which is crucial for attaining sustainable agriculture and soil management.
The seeds used for planting were of the high-quality, high-yielding cereal ‘Jingu 21,’ the most recent variety grown by the Shanxi Academy of Agricultural Science and the Industrial Crop Institute; the soil used for planting was grass charcoal substrate soil (for seedlings). And according to previous research (Sharma et al., 2020), the reagent used in the experiments was 1 mmol∙L−1 of Cr6+ (K2Cr2O7) to ensure that high concentrations of Cr were toxic to cereal crops.
In this study’s greenhouse pot experiment, the test soil was cleaned of stones and other contaminants before being left aside and air-drying for 3 days in the laboratory. Selected high-quality seeds were cleaned with tap water, shielded from light, and immersed in water for approximately 48 h to promote germination. For the planting process, 18 pots (15 cm × 20 cm) were used, and the soil moisture was regulated to 60% of the water-holding capacity required for crop growth. Each pot included 1,300 g of soil and 50–60 seeds that were uniformly dispersed, and the top layer was covered with soil to protect the seeds from light and promote germination and was wrapped with plastic wrap to prevent contamination by airborne bacteria. The pots were then placed in a growth chamber at a constant temperature of 23°C with 16–8 h of light and dark per day. After 15–20 days of incubation, each pot received 300 ml of 1 mmol·L−1 Cr6+ (The Cr6+ solution was added to the trays and applied every other day at a fixed time. And distilled water was sprayed on the soil surface daily to ensure soil moisture, no fertilizer was applied to the soil during the experiment).
Three groups made up the experiment: the group used before treatment with Cr solution are the control (CK, equal amount of distilled water was applied to the control group), Cr stress treatment for 6 h (Cr_6h), and Cr stress treatment for 6 days (Cr_6d) (Leng et al., 2020). The setting of the sampling time was done because the microbial community in the soil will respond to the heavy metal stress to a certain extent when Cr stress is applied for approximately 6 h. Due to its inherent resistance and resilience, the microbial community eventually displayed a stable state after around 6 days of the Cr stress treatment. No follow-up analysis was done since heavy metal pollution is a long-term problem. Each experimental group had six biological replicates, and the soil in each pot was randomly selected as the sampling target for rhizosphere soil, for a total of 18 samples. The sampling process was carried out by gently grating the soil with a small spade to expose all the roots of the plant as much as possible and to ensure the integrity of the root soil. The cereal seedlings were removed as a whole and the bulk soils of the root system were shaken off, and about 1 g of soil was collected from each plant. The collected soil was stored in sealed sterile bags and marked properly. The sterile bags were placed in a biological sample sampling box for low temperature storage (in advance in ice packs or dry ice). All samples were then stored in a −80°C refrigerator until use; some were used for DNA extraction and some for determination of soil physical and chemical indicators.
The physical and chemical properties of soil were measured with a JXBS-3001-SCPT-SC (Weihai, Shandong Province, China), which is a high-precision environmental sensor that determines available nitrogen (N), available phosphorus (P), available potassium (K), electrical conductivity (EC) and pH in soil.
A Beijing Zhongke Weihe chlorophyll meter (TYS-3 N, Haidian District, Beijing, China), a pressure-head sensor that emits red and infrared light with peak wavelengths of 650 mm and 940 mm, respectively, was used to measure the chlorophyll content and nitrogen content of the leaves of living seedlings. The samples were measured for stem length, root length, and fresh weight; dried and heated at 105°C for 30 min, then dried at 60°C until a constant weight to calculate their dry weight, after which they were photographed to record seedling growth.
A TIANamp Bacteria DNA kit (Tiangen Biochemical Technology Co., Beijing, China) was used to extract and purify genomic DNA from the samples, which totaled 1 g of soil. Agarose gel electrophoresis was used to assess the extracted DNA, and a enzyme-labeled instrument (Epoch, Xinghui Biotechnology Co., Ltd., Tianjin, China) was used to gauge the extracts’ purity and quantity.
Genomic DNA was amplified by polymerase chain reaction (PCR). The following components made up the amplification system (50 μl): Taq DNA polymerase (5 U∙μL-1) 1.0 μl, 10 × PCR buffer 10.0 μl, dNTP solution 8.0 μl, template DNA 10.0 μl and primers (50 μmol∙L-1). PCR reaction conditions: pre-denaturation at 98°C for 1 min, followed by 98°C for 10 s, 50°C for 30 s, and 72°C for 30 s, for a total of 30 cycles; with a final extension at 72°C for 5 min (Zeng and An, 2021).
The PCR products were then delivered to Shanghai Meiji Biotechnology Engineering Co., Ltd. for Illumina MiSeq PE300 platform high-throughput sequencing analysis. The bacterial community in soil was sequenced using the sequence amplification region 338F_806R (338F: ACTCCTACGGGAGGCAGCAG; 806R: GGACTACHVGGGTWTCTAAT) (Caporaso et al., 2011), and the raw data from the sequencing platform were homogenized. The Flash (1.2.111) and Fastp (0.19.62) software programs were used to perform double-end sequence splicing, filter the reads’ tails for bases with quality values below 20, and eliminate reads that contained N bases to produce high-quality sequences. The overlap between PE readings was used to guide the splicing of the sequence. Uparse (7.0.10901) software was used to cluster non-repeated sequences (excluding single sequences) into OTUs based on 97% similarity, and chimeras were eliminated during the process to generate representative sequences of OTUs (operational classification units). Taxonomic analysis of representative sequences of OTUs with a 97% similarity level was carried out using the RDP classifier (2.112) Bayesian technique. Based on OTU abundance data, a annotation analysis was carried out to determine the makeup of the bacterial community in soil samples. Qiime (1.9.13) software was used to create a table of the taxonomic abundance and the calculation of the β-diversity distance. Mothur (1.30.24) software was used to conduct the α-diversity study.
The target segments of five nitrogen cycle functional genes, i.e., nitrogen-fixing enzyme (nifH), bacterial ammonia monooxygenase (AOB-amoA), archaeal ammonia monooxygenase (AOA-amoA), nitrate reductase (narG), and nitrite reductase (nirK), were quantified by qPCR. In Table 1, the specifics of the primer sequences for the nitrogen cycle functional genes are displayed.

Table 1. The primer sets for the amplification of functional genes involved in the nitrogen cycle.
An ABI Model 7,300 Fluorescent Quantitative PCR instrument (Applied Biosystems, United States) and ChamQ SYBR Color qPCR Master Mix (2X) reagent (Nanjing Novozymes Biotechnology Co., Ltd.) were used to perform real-time fluorescence quantitative PCR (q-PCR). The final reaction mixture volume for the quantification technique was 20 μl and contained 10 μl 2X Taq Plus Master Mix, 1 μl template DNA, 0.8 μ primer F (5 μM), 8 μl primer R (5 μM), and 7.4 μl ddH2O. Each qPCR reaction was run in triplicate for each set of primers, and the non-template control served as a negative control. Furthermore, q-PCR was carried out using a three-step thermal cycling method that involved 35 cycles of 95°C pre-denaturation for 5 min, 95°C denaturation for 30 s, 58°C annealing for 30 s, and 72°C extension for 1 min (Li et al., 2022). Standard curves were created by diluting pMD18-T, a plasmid for five functional genes, in a 10-fold gradient. Additionally, the R2 value of each standard curve exceeded 0.95, demonstrating a linear relationship throughout the concentration range used in this work, with amplification effectiveness ranging from 89.28 to 103.81% (mean 96.31%).
R (4.1.2), SPSS (19.0), and Excel 2019 were used for data analysis and graphing. The variations in the soil physicochemical parameters, as well as the compositional structure and diversity of the soil bacterial communities and the nitrogen cycle functional genes, in the Cr stress time series were evaluated using ANOVA. The ‘ggtern’ package in R was used to create ternary phase diagrams to illustrate the distribution variations in the phylum and genus levels of the bacterial communities in different samples (Hamilton and Ferry, 2018). Mothur (V1.30.2) was used to calculate the α-diversity indices (Shannon index and Simpson index) to reflect the variation in the α-diversity of the soil bacterial communities and the functional genes involved in the nitrogen cycle. QIIME software was used to calculate the weighted (Bray–Curtis) and unweighted (UniFrac) diversity indices in order to compare and contrast the makeup of the soil bacterial communities and functional genes involved in the nitrogen cycle in the Cr stress time series. Non-metric multidimensional scale analysis (NMDS) was used to rank and analyze the temporal distribution patterns of the soil bacterial communities and nitrogen cycle functional genes in the Cr stress time series; the results were verified by stress values, and the graphs were typically thought to have some explanatory significance when the stress<0.2. Redundancy analysis (RDA) was used to examine the relationship between the bacterial community, nitrogen cycle functional genes, and environmental factors (Oksanen et al., 2020). The ‘ggcor’ package in R was used to map the relationships between the dominant bacterial phylum, functional genes involved in the nitrogen cycle, and soil physicochemical parameters. Mantel analysis and the PLS-EM model were used to further investigate the strength of the correlations between the soil physicochemical parameters, bacterial communities, and functional genes involved in the nitrogen cycle (Li Y. et al., 2019).
The results of ANOVA (Figure 1) of the physicochemical parameters of the soil revealed that there was no significant difference (p > 0.05) between the CK and Cr_6h groups, but that there was a difference between the Cr_6h and Cr_6d groups (p < 0.05). At the Cr_6d stage, the soil physicochemical parameters pH and EC value, N, P, K content increased substantially by 135.79, 131.02, 135.34, and 3.55%, respectively (p < 0.05).
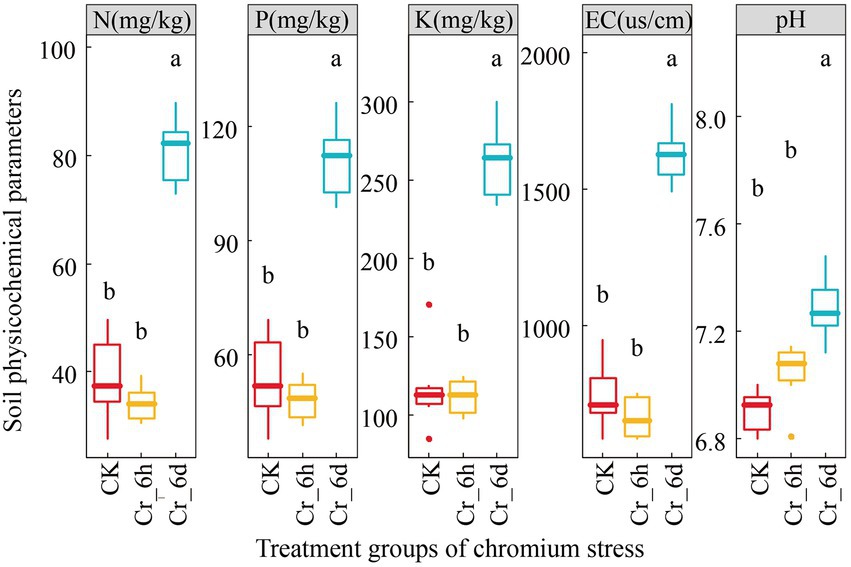
Figure 1. Variation trend of the soil physicochemical parameters in chromium-contaminated soils. CK, control treatment; Cr_6h, Cr stress for 6 h; Cr_6d, Cr stress for 6 days. Different letters indicate significant differences among treatments at the p < 0.05 level. The upper, middle, and lower lines of the box chart represent the upper quartile, median, and lower quartile, respectively, and the vertical line is the error bar.
In order to assess the impact of Cr stress on grain growth, measurements of the plant stem length, root length, dry weight, fresh weight, chlorophyll content, and nitrogen content were conducted (Supplementary Figure S1). ANOVA (Supplementary Table S1) revealed that stem length and chlorophyll of cereals significantly decreased by 22.40 and 19.07% (p < 0.05) at Cr_6h stage; stem length significantly increased by 8.67% (p < 0.05) at Cr_6d stage; and root length significantly decreased by 26.31% (p < 0.05) at Cr_6d stage compared to the CK.
A total of 791,427 high-quality sequences were discovered for the bacterial community in soil according to the high-throughput sequencing. The sparsity curve trend tended to be flat, indicating that the sequencing volume matched the analytical requirements, and 3,509 taxonomic units of bacteria were compared based on a 97% threshold for sequence similarity.
The ternary phase diagrams were plotted to represent the distribution of population types at the level of bacterial phylum (Supplementary Figure S2A) and genus (Supplementary Figure S2B) communities in soil across the time period of Cr stress. The contributions of the three groups of samples to the relative abundance of each microorganism led to the varied points in the plots or the locations of the various bacterial communities. All of the bacterial phylum and genus level communities were dispersed between 20 and 40% of the data, showing that the distribution of the bacterial phylum and genus level communities was more consistent in the Cr stress time series.
ANOVA was used to examine trends of the top 10 bacteria phylum and genus communities in soil (Supplementary Table S2), as shown in Figure 2. The top five bacterial phyla were Actinobacterota (31.11%), Firmicutes (19.00%), Proteobacteria (18.82%), Chloroflexi (11.87%), and Bacteroidota (4.57%). In the time series of Cr stress, the phylum Actinomycetes (p < 0.01) and Chloroflexi (p < 0.05) both displayed highly significant increases followed by decreases, while Firmicutes (p < 0.05) and Myxococcota (p < 0.01) displayed significant decreases; the phyla Proteobacteria (p < 0.01) and Gemmatimonadota (p < 0.01) both displayed highly significant decreases followed by increases.
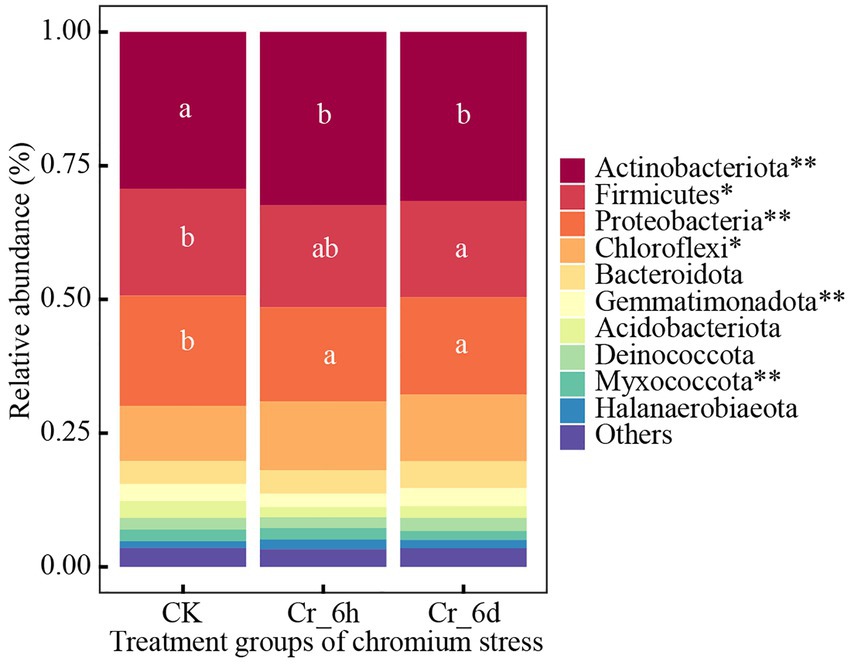
Figure 2. Phylum level bacterial community structures and differences among treatments in chromium-contaminated soils. Significance (P), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; CK, control treatment; Cr_6h, Cr stress for 6 h; Cr_6d, Cr stress for 6 days. Different lowercase letters indicate significant differences between components (p < 0.05). ‘Others’ represents the sum of the relative abundance of the remaining bacteria.
According to Supplementary Figure S3 and Supplementary Table S3, the top five bacterial genera were Planifilum (6.34%), Longispora (4.59%), Actinomadura (2.66%), Haloactinopolyspora (2.51%), and Truepera (2.22%). In the Cr stress time series, the genus Truepera (p < 0.001) displayed a highly significant decline, whereas Haloactinopolyspora (p < 0.05) displayed a significant increase followed by a decrease.
The expression levels of the nitrogen cycle functional genes AOA-amoA, AOB-amoA, narG, nirK, and nifH were measured by q-PCR to clarify the impact of Cr stress on these genes (Figure 3). In the Cr stress time series, the gene abundances of AOA-amoA and AOB-amoA dramatically decreased by 5.78 and 3.75% (p < 0.05) at the Cr_6h stage and increased considerably by 7.89 and 8.30% (p < 0.05) at the Cr_6d stage. In contrast, the gene abundances of nifH, narG, and nirK increased at the Cr_6h stage and by 4.97, 1.84, 1.27%, respectively, at the Cr_6d stage. As a result, there was an overall increase in the expression of the nitrogen cycle–related functional genes in soil under Cr stress.
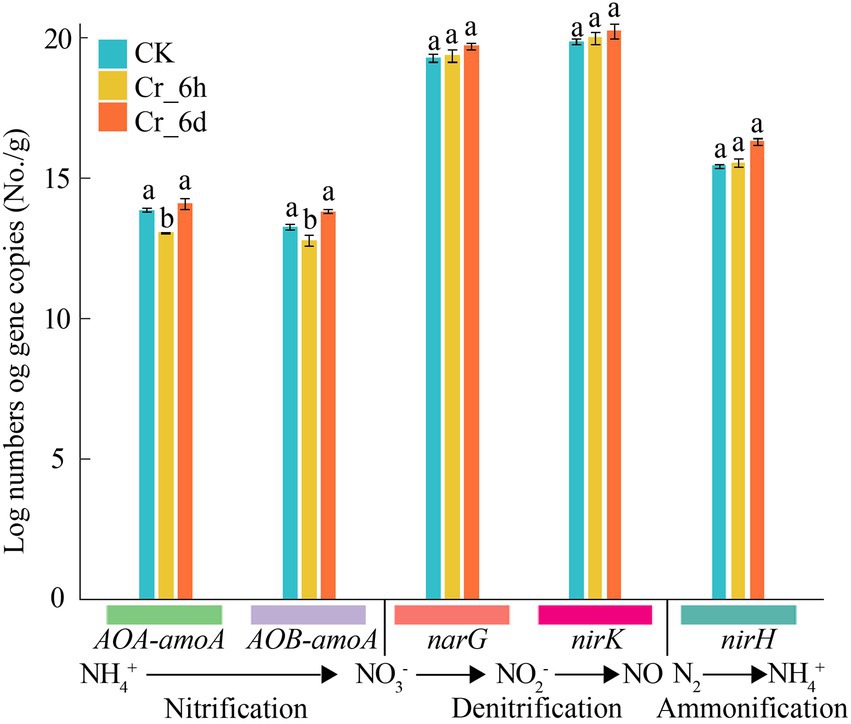
Figure 3. Abundance of functional genes involved in nitrogen cycling in chromium-contaminated soils. Error bars represent standard errors (n = 3); different lowercase letters indicate significant differences between components (p < 0.05).
ANOVA was used to analyze the patterns of the bacterial community and the diversity of nitrogen cycle functional genes in agricultural soils before and after Cr stress (Shannon index, Simpson index, Figure 4). The Shannon index of the bacterial community showed a significant decrease followed by an increase (6.09, 5.93, 6.05, p < 0.05), while the Simpson index revealed a significant increase followed by a decrease (0.0068, 0.0078, 0.0068, p < 0.05). In contrast, only the Simpson index of the functional genes involved in the nitrogen cycle increased significantly at Cr_6d stage (0.794, 0.793, 0.795, p < 0.05). According to the results, the bacterial community displayed a phase trend of increasing and then decreasing, while the variety of nitrogen cycle functional gene diversity overall significantly decreased. As a result, the α-diversity of soil bacterial communities and nitrogen cycle functional genes in agricultural fields changed significantly before and after Cr stress.
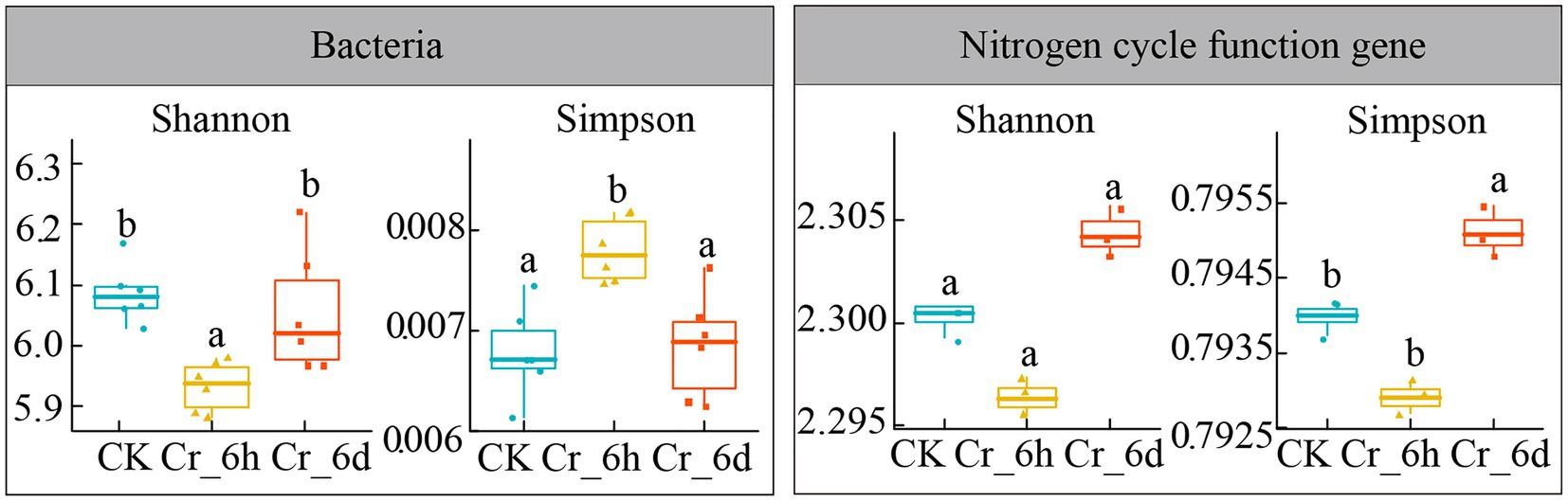
Figure 4. Analysis of the bacterial community α-diversity indexes in chromium-contaminated soils. CK, control treatment; Cr_6h, Cr stress for 6 h; Cr_6d, Cr stress for 6 days. Different letters indicate significant differences among treatments at the p < 0.05 level. Dithered points indicate the remaining data points, preventing points of the same value from being hidden. The upper, middle, and lower lines of the box chart represent the upper quartile, median, and lower quartile, respectively, and the vertical line is the error bar.
Based on the abundance of the bacterial phylum level community and nitrogen cycle functional gene expression in soil, non-metric multidimensional scale analysis (NMDS) and analysis of similarities (ANOSIM) were used to examine the diversity of both in the Cr stress time series (i.e., temporal distribution pattern, Figure 5). To further illustrate unique inter-group variability, grouped box plots with PC1 values as the y-axis (right side of Figure 5) and PC2 values as the y-axis (top side of Figure 5) were also presented. The results showed that the soil bacterial community structure (Figure 5A, Stress = 0.071, p = 0.001) and the nitrogen cycle functional genes (Figure 5B, Stress = 0.007, p = 0.001) were clustered within the same stage of Cr stress, but they were more distinct and spread at various stages. In Figure 5A, the box line plot on the right side demonstrates a significant difference in bacterial communities between the CK&Cr_6h and CK&Cr_6d groups (p-values 0.013 and 0.0018, respectively), and the box line plot on the top side demonstrates a significant difference in bacterial communities between the CK&Cr_6h groups (p = 0.034). In Figure 5B, only the box line plot (top side) revealed a significant difference in the functional genes between the Cr_6h and Cr_6d groups (p = 0.035). All of the aforementioned results show that the temporal distribution patterns of the bacterial communities and functional genes involved in the nitrogen cycle in soil changed noticeably in the Cr stress time series.
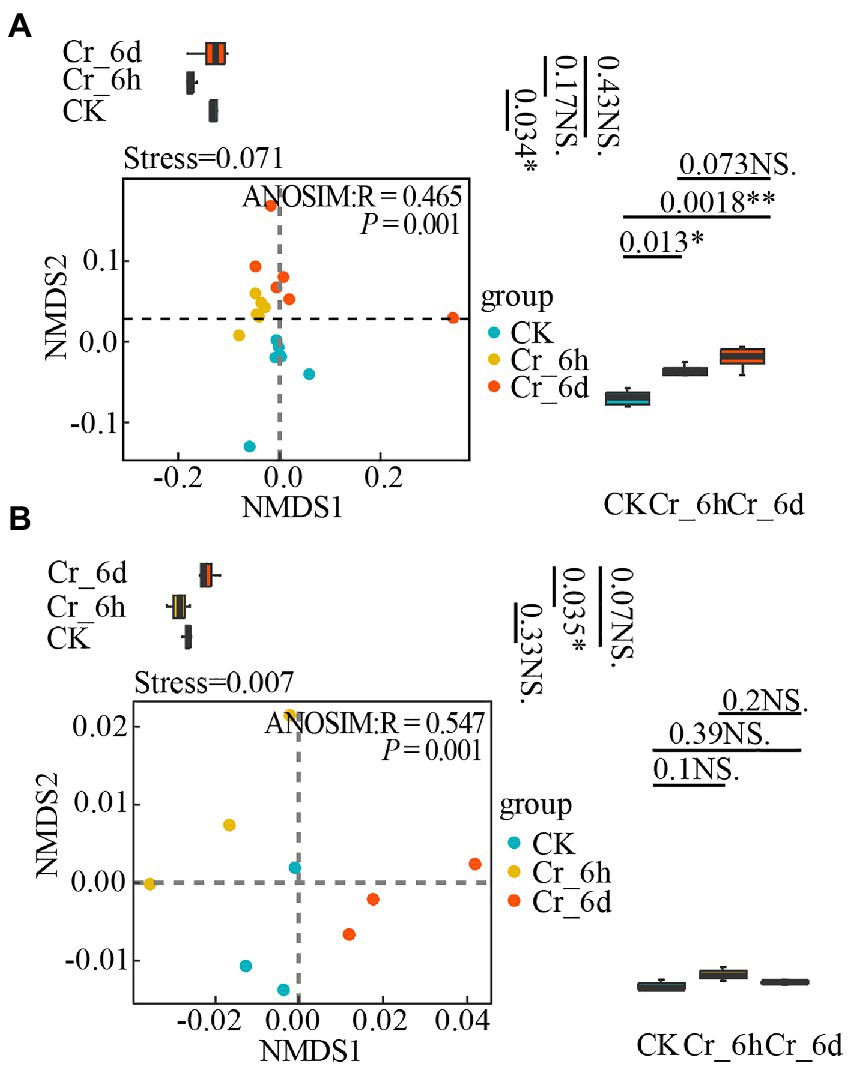
Figure 5. Non-metric multidimensional scale analysis of bacterial communities (A) and nitrogen cycle functional genes (B) in chromium-contaminated soils. Significance (P), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, NS, no significant difference; CK, control treatment; Cr_6h, Cr stress for 6 h; Cr_6d, Cr stress for 6 days. The R value indicates whether there is a difference between different groups, and R > 0 indicates that the difference between groups is greater than the difference within groups. Points of different colors or shapes represent samples of different treatments. The closer the two sample points, the more similar the species composition of the two samples. The upper, middle, and lower lines of the box chart represent the upper quartile, median, and lower quartile, respectively, and the vertical line is the error bar.
According to the results of the RDA (PERMANOVA, p < 0.01) of the soil bacterial phylum level communities with physicochemical parameters, the first two RDA axes combined accounted for 70.52% of the total variation (Figure 6A). The pH of the soil had a greater impact on Chloroflexi and others, while Gemmatimonadota, Bacteroidota, and others were more impacted by the soil N, P, K, and EC concentrations. The ‘vegan’ package was also used to perform the Mantel test (Pearson correlation index) to examine the relationship between various environmental factors and changes in the bacterial community structure. The results of the test between the bacterial community and environmental factors (Supplementary Figure S3; Supplementary Table S4) were Mantel statistic r: 0.1967, p > 0.05, indicating that there was no statistically significant association between the environmental factors and the overall structural alterations of the bacterial community.
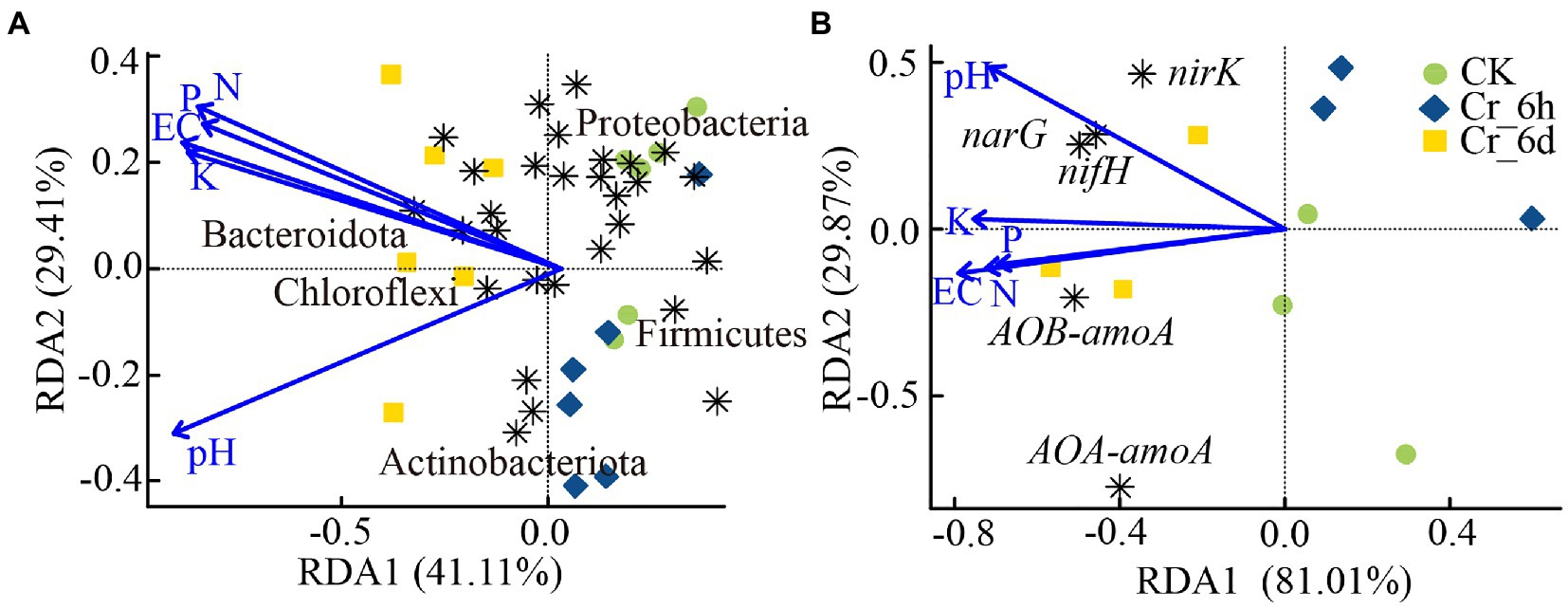
Figure 6. Relationship between soil physicochemical parameters and bacterial communities (A) and nitrogen cycle functional genes (B) revealed by redundancy analysis. The horizontal and vertical coordinates in the Figure indicate the degree to which the soil environmental factors explain the bacterial community structure (A) or phenotype (B). The arrow in the Figure indicates the environmental factor, and the length of the line indicates the degree of correlation between the environmental factor and the bacterial community structure (A) or phenotype (B). The longer the line, the greater the correlation, and vice versa. The angle between the line of the arrow and the axis indicates the correlation; the smaller the angle, the higher the correlation. An acute angle indicates a positive correlation, and an obtuse angle indicates a negative correlation.
Redundancy analysis of the nitrogen cycle functional genes and soil physiochemical properties revealed (Figure 6B) that the first two RDA axes combined explained 90.88% of the overall variation. Among these properties, pH had a greater impact on nifH, narG, and nirK, whereas EC, N, and P had a greater impact on AOB-amoA. Further analysis of the relationship between various environmental factors and the abundance of the nitrogen cycle-functional genes was carried out using the Mantel test (Pearson correlation index). The results of the test between functional genes involved in the nitrogen cycle and environmental factors (Supplementary Figure S4; Supplementary Table S5) were Mantel statistic r: 0.4195, p < 0.01, indicating that the abundance of the nitrogen cycle functional genes was statistically significantly correlated with environmental factors. Additionally, the abundance of functional genes was strongly linked to N (r = 0.36, p = 0.024), K (r = 0.38, p = 0.025), and EC (r = 0.42, p = 0.01), with EC having a significant influence.
Because all the bacterial phyla in soil were analyzed using RDA, the ggcor package was used to further map the correlation analysis. The Bray–Curtis distance was calculated for the dominant bacterial species and functional genes, and the Euclidean distance matrix of the soil physicochemical parameters was developed to clarify the Mantel association among these three, allowing an extensive investigation of how environmental factors influence functional genes and dominant bacterial communities.
The correlation analysis of the soil dominant bacterial community with the nitrogen cycle functional genes and soil physicochemical parameters (Figure 7; Supplementary Table S6) revealed that EC, N, and K in the soil were highly significantly correlated with nifH (p-values were 0.008, 0.006, and 0.01, respectively); pH was significantly correlated with nifH (p = 0.026, p < 0.05); while P and K were significantly correlated with AOB-amoA (p-values were 0.02, 0.015, respectively, p < 0.05). Firmicutes were substantially negatively associated (p < 0.05) with Chloroflexi, narG, and nifH, whereas nifH was significantly negatively correlated (p < 0.05) with AOA-amoA and nirK and extremely significantly positively correlated (p < 0.01) with AOA-amoA and narG; narG was significantly positively correlated with nirK. According to the aforementioned findings, there was a significant correlation between the soil physicochemical parameters, dominant bacterial phylum, and genes that function in the nitrogen cycle. However, the soil physicochemical parameters were significantly connected only with the nitrogen cycle function genes.
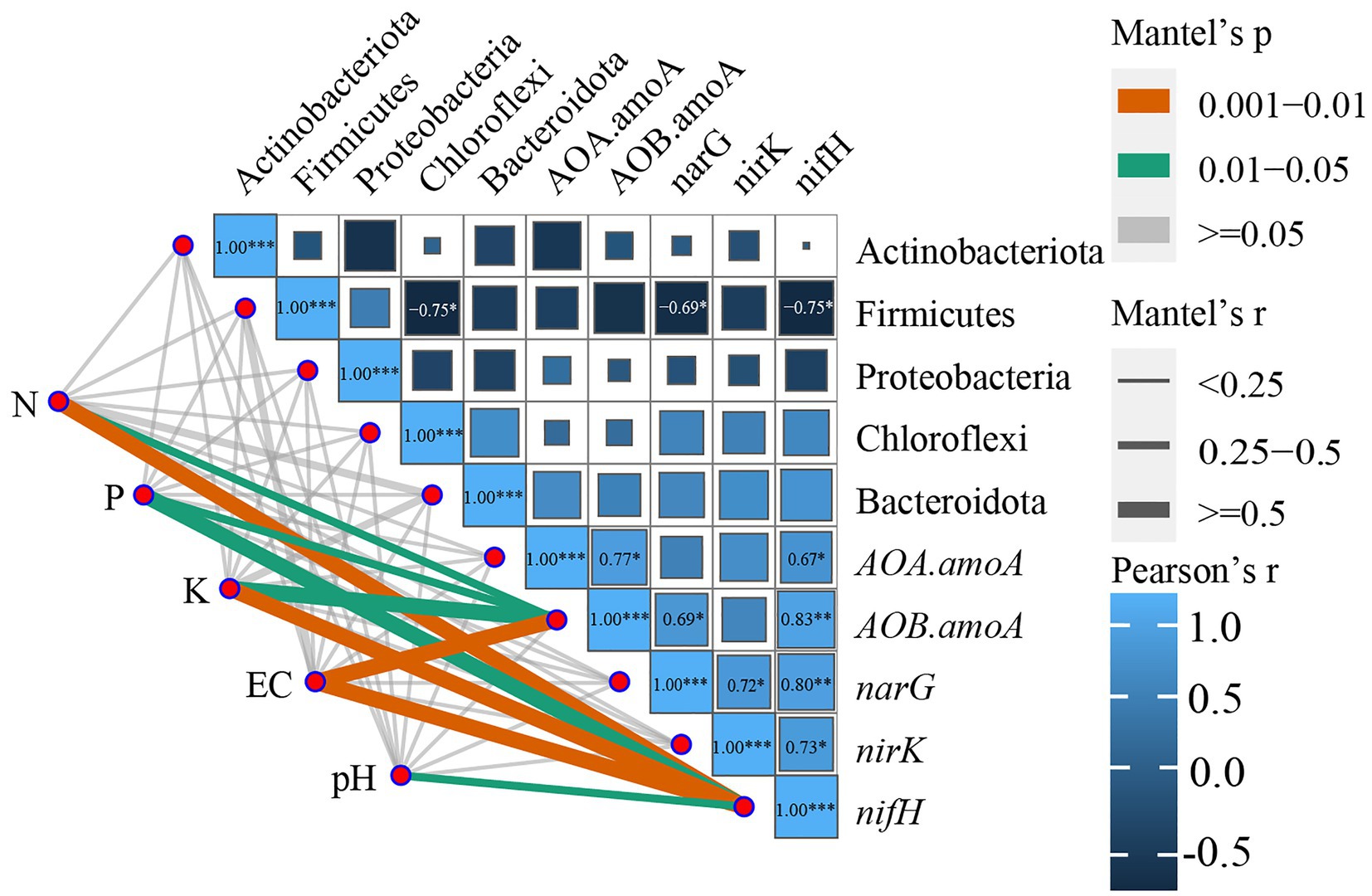
Figure 7. Correlation analysis of functional genes of the nitrogen cycle with environmental factors and the dominant bacterial community. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Only significant correlations are presented as colored lines, and the color of the line corresponds to the strength of the significance of the correlation; the thickness of the line represents the magnitude of the Mantel test r-value.
With the use of the ‘plspm’ package in R (4.1.2), a reflective measurement model of the effects of the soil physicochemical parameters on the dominant bacterial population and functional genes involved in the nitrogen cycle was created based on the partial least squares path model (PLS-PM, Figure 8). The model had a good fit, as indicated by the goodness of fit (GOF), which was 0.6034. The dominant bacterial community and the functional genes involved in the nitrogen cycle were directly influenced by the soil physicochemical parameters (positive path and path coefficient: 0.784 and 0.6369, respectively), and the nitrogen cycle functional genes were significantly impacted (p = 0.012). The PLS-EM results further demonstrated that physicochemical parameters in soil had a stronger influence on functional genes than bacterial communities.
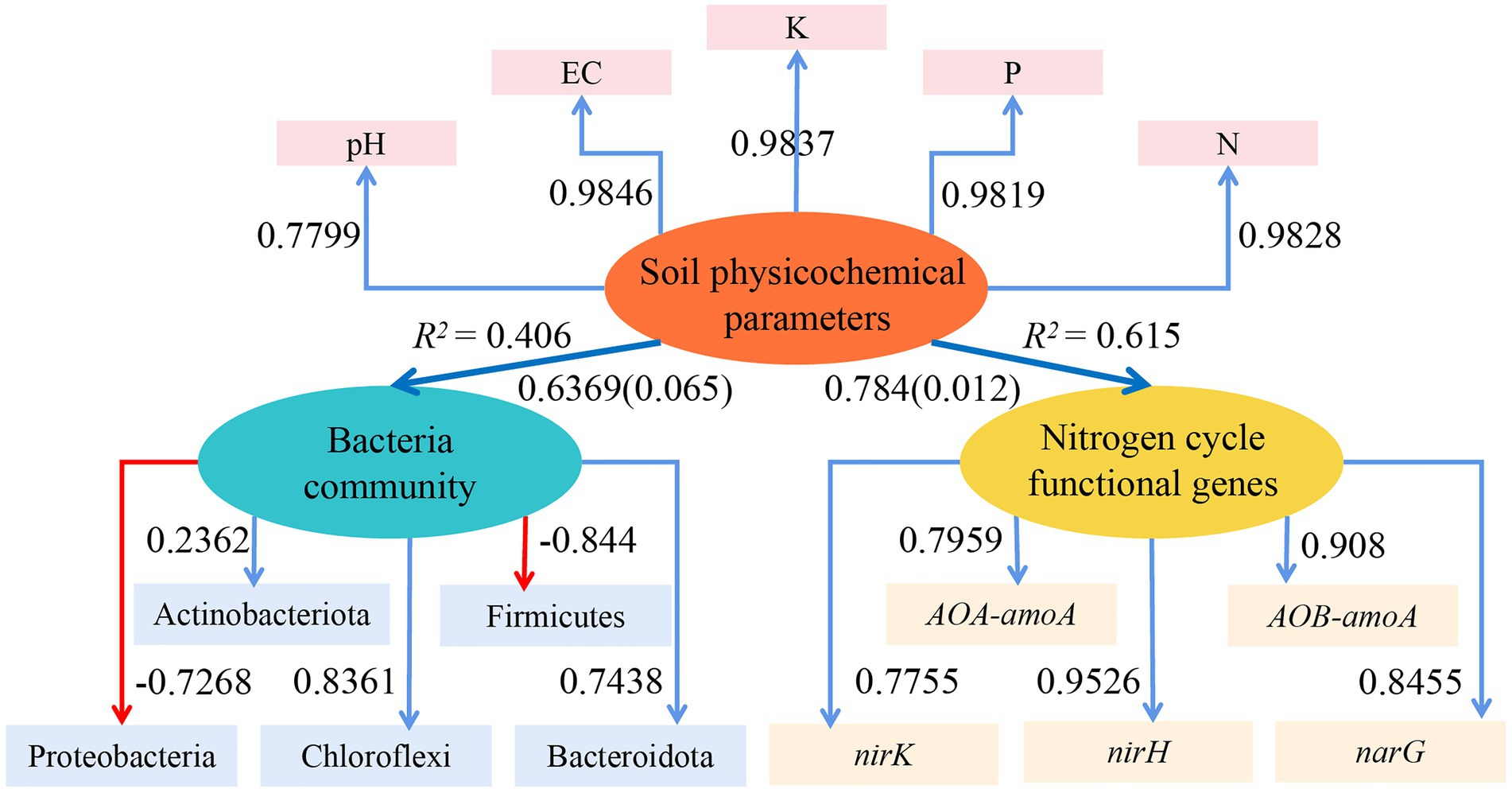
Figure 8. PLS-EM model to analyze the effect of the soil physicochemical parameters on the dominant bacterial community and functional genes of the nitrogen cycle. Blue arrows in the Figure indicate positive correlations, red arrows indicate negative correlations, numbers on the arrows indicate standardized path coefficients, and values in parentheses are significant p-values; ovals indicate potential variables, and squares are measured variables; R2 next to the variables is the explained variation.
According to the experimental findings, there were significant differences between the soil bacterial communities at the phylum and genus levels in the Cr stress time series in terms of their composition, structure, and diversity.
Similar to the findings of Zhang et al. (2021), Actinobacteriota, Firmicutes, and Proteobacteria were the three taxa with the highest abundance at the phylum level, and Planifilum, Longispora, and Actinomadura were the three with the highest abundance at the genus level. At the Cr_6d stage, the abundances of Actinobacteriota, Firmicutes, and Chloroflexi in the soil significantly decreased as the Cr stress time increased, whereas the abundances of Proteobacteria and Gemmatiminadota significantly increased. This may be due to the ability of soil microorganisms to adjust to various levels of heavy metal contamination by altering the structure of the microbial community (Li et al., 2017). Previous studies have demonstrated that the bacterial communities transferred across contamination levels in Cr-affected soils are predominantly Proteobacteria, Actinobacteriota, and Chloroflexi (Liu et al., 2019). Proteobacteria, the dominant phylum in soils contaminated with Cd and many other heavy metals, have considerable resistance to heavy metals (Jiang et al., 2019), and some of them have good Cr(VI) biotransformation efficiency (Garavaglia et al., 2010). Thus, Proteobacteria are frequently utilized as markers of Cr contamination, and their abundance increased significantly in the Cr_6d phase of Cr stress.
In addition, Bacillus in the Firmicutes and Streptomyces in the Actinobacteriota can reduce the toxic effect of Cr by reducing Cr(VI) (Liu et al., 2019) and have strong Cr tolerance, making them the dominant genera in Cr-stressed soils, and their abundance is more stable over time when Cr stress is present. At the stage of Cr_6d stress, however, the abundance of Actinobacteriota and Firmicutes decreased considerably, most likely because the concentration of heavy metals was higher than the community could tolerate, which inhibited growth and reduced the abundance of the community. Both Chloroflexi and Myxococcota have been demonstrated to be dominant in a variety of heavy metal-contaminated soils due to their tolerance and resistance to heavy metal stress (Berg et al., 2012; Hao et al., 2021); Chloroflexi, as low-nutrient bacteria, have an advantage in nutrient poor soils (Islam et al., 2019). Additionally, Actinobacteriota have been demonstrated to play a role in soil nutrient cycling (Narendrula-Kotha and Nkongolo, 2017), while Acidobacteriota are a dominant phylum in acidic environments or locations that are contaminated with heavy metals, using a variety of nitrogenous compounds as nitrogen sources for their growth (Eichorst et al., 2018). Therefore, as the main bacterial phyla, Acidobacteriota and Actinobacteriota may control the N cycle in Cr-stressed soils.
According to the α-diversity results, the Shannon and Simpson indices significantly increased and then decreased, and initially decreased and then increased, respectively, which also revealed that the diversity of bacterial communities in soil exhibited a phase change from stress to stability over the Cr stress time series. On the one hand, this might be related to the fact that the total amount of heavy metals in soils contaminated with Cd, zinc (Zn), Pb, and Cr decreases the biomass and diversity of bacterial communities and even inhibits bacterial enzymatic activities (Chen et al., 2020; Tang et al., 2022). The stage-specific changes in bacterial communities can be characterized by the extensive substrate utilization properties and high metabolic activity of some bacterial communities, which are better adapted to the toxic effects in heavy metal-contaminated soils. On the other hand, changes in α-diversity may be closely related to soil nutrients such as N, P, and SOC (Soil Organic Carbon), as soil nutrients may be the main source of energy in promoting the growth and metabolic processes of bacterial communities (Li et al., 2021). Additionally, soil microbes are crucial for material and nutrient cycling in plant and soil ecosystems, but the mechanisms driving the composition and diversity of microbial communities vary depending on the kind of vegetation. According to research (Xu et al., 2021), vegetation traits and soil physicochemical features may synergistically regulate microbial communities in soils. Furthermore, because heavy metals have more complex natural origins and soil biogeochemical characteristics and result in complex interactions between microbial communities, multiple factors combine to determine whether an ecosystem stimulated by metals would produce a positive or negative response (Abdu et al., 2017). In summary, bacterial communities in soils adapt to Cr contamination through phase changes in composition, structure, and diversity; rather than solely being influenced by the toxic effects of heavy metals, these phase shift characteristics of bacterial community diversity in soil may be influenced by a number of factors, including heavy metal contamination, soil nutrients, and vegetation conditions.
According to the experimental data, there were significant differences in the composition structure and diversity of functional genes involved in the nitrogen cycle in soil under conditions of Cr stress.
The AOA-amoA and AOB-amoA abundances exhibited a significant decrease and then an increase (p < 0.05). In particular, the abundance of the AOB-amoA gene increased by 8.30% in the Cr_6d stage compared with the Cr_6h stage; the abundances of nifH, narG, and nirK showed an increasing tendency; and the gene abundance was ranked as denitrification > nitrogen fixation > nitrification. The results of α-diversity analysis revealed that from Cr_6h to Cr_6d, the diversity of the nitrogen cycle functional genes significantly decreased. Similar to the bacterial community diversity, the functional gene abundance displayed a stress to stability phase transition. This is most likely because high Cr pollution temporarily inhibits the nitrogen cycle function in soil, which is caused by the soil microbial community’s immediate damage response to external stimuli and the gradual recovery of the nitrogen cycle function with increasing Cr stress time (Zhou et al., 2016). It has been demonstrated that Cd stress promotes N cycling activities in soil and that the degree of stress, microbial response mechanisms, and other mechanisms change with time (Zhao et al., 2021), which is consistent with the present study’s findings. The initial and rate-limiting phase of soil nitrification is the ammonia oxidation process, which is mediated by AOA and AOB genes with significant changes in abundance. As specialized chemoautotrophic bacteria, AOA and AOB can oxidize ammonia to nitrite and generate huge amounts of ATP, which promotes the survival of microbial communities in soil (Prosser et al., 2020). Similar to other research, the results showed that AOA was more abundant than AOB, probably because AOA is the dominating microbe in soil ammonia oxidation (Leininger et al., 2006) and is better adapted to the challenging environment of low oxygen and oligotrophic growth (David et al., 2014). Although AOA and AOB populations exhibit ecological niche differentiation and functional complementarity (Prosser and Nicol, 2012), AOB populations are more vulnerable to Cr stress than AOA, and therefore their abundance changes more significantly.
In the Cr stress time series, the abundance of the nitrogen cycle functional genes generally increased. This may be explained by the fact that soil microbial communities in Cr-stressed environments protect themselves from metal toxicity using a variety of resistance mechanisms, such as intercellular isolation and enzymatic detoxification, while dominant phyla such as Actinomycetes and Acidobacteria may enhance the transcriptional processes of genes related to amino acid biosynthesis and uptake pathways for protein detoxification (Viti et al., 2014). Thus, as the succession process advances, the soil microbial community gradually adjusts to external pressures at the physiological, genotypic, or community level (Spang et al., 2012; Azarbad et al., 2015) and inevitably requires more energy and irreversible overall metabolic transformation in order to maintain higher energy expenditures in organism growth and metabolic activities (Singh et al., 2014); thus, the nitrogen use and transformation process may be expedited. However, during the Cr_6h phase, Cr stress inhibited bacterial communities and nitrogen cycle functional gene abundance in soil, probably because most denitrifying bacteria, diazotrophs, and soil bacteria are mostly heterotrophic and primarily take up ammonia to biosynthesize compounds required for metal detoxification. Based on the energy saving strategy of the soil microbial community, in order to save energy costs, the soil microbial community may inhibit processes that require high energy, such as cell division (Chen et al., 2016), so its abundance tends to decrease. In conclusion, Cr stress may accelerate the process of N utilization and transformation in soil, and soil microbial communities may adopt energy production and conservation strategies to survive in Cr-stressed environments.
The findings revealed that at the Cr_6d stage, all soil physicochemical parameters increased significantly (p < 0.05); pH had a greater impact on Chloroflexi, while the N, P, K, and EC contents primarily affected Gemmatimonadota and Bacteroidota, and the functional genes involved in the nitrogen cycle nifH and AOB-amoA were significantly correlated with EC, N, and K, with EC having the strongest effect. Similar to the findings of Xu et al. (2019), pH had an impact on the bacterial community and was significantly correlated with nifH functional genes. First, it is possible that pH directly imposes a certain degree of physiological restriction on soil microorganisms; its value outside of a certain range will change how microorganisms interact, which will have an impact on the abundance of microbial taxa that cannot grow individually (Lauber et al., 2009). Second, previous research has demonstrated that soil pH affects the transport and bioavailability of heavy metals (Liang et al., 2017), and in addition to influencing the composition of the microbial community, it also regulates the diversity and abundance of functional genes involved in the nitrogen cycle (Lauber et al., 2009; Rousk et al., 2010). Additionally, Kandeler et al. (2006) demonstrated that pH had little to no impact on the functional genes involved in denitrification, which is in line with the findings of the present study. A wide-ranging variable in soil, pH is strongly related to properties such as the moisture content and nutrient availability. By modifying soil conditions, pH may cause variations in microbial composition; consequently, other soil physicochemical parameters other than soil pH may also have a limiting impact on the spread of microbial communities. For example, it has been demonstrated that the microbial community and available potassium in soil are significantly and negatively correlated (Pan et al., 2020), which may result from the enrichment of microorganisms that prefer potassium, such as Paenibacillus that dominate the soil, thus reducing microbial diversity.
The nitrification process of soil bacteria is favored by an elevated N content in the soil (Ouyang et al., 2017); hence, AOB is strongly associated with N when the N concentration increases. One study demonstrated that AOB are susceptible to environmental factors, and high pH and EC directly affect the variety of AOB in soil (Wang and Gu, 2014). In contrast, AOA require less energy and ammonium than AOB, and they have mechanisms for adapting to pH and severe settings, making AOA better suited for surviving in soil environments that are stressed by heavy metals. In Cr-contaminated soils, the soil may constitute a boundary habitat affected by heavy metals at the initial stage of stress, when nutrients are scarce and the concentration of effective carbon and nitrogen is low, thus the colonization of nitrogen-fixing bacteria enhances the input of nitrogen into the soil (Schrama et al., 2012). Furthermore, the nifH genes in this study had a high initial abundance and gradually increased, indicating that the input of heterotrophic nitrogen may be crucial in the time series.
1. In Cr-contaminated soils, the bacterial community’s composition and structure at the phylum and genus levels changed significantly; the diversity of the community displayed a phase change characteristic from stress to stability. Resistant bacterial phyla such as Proteobacteria and Chloroflexi were enriched to maintain community homeostasis and adapted to Cr stress by changes in compositional structure and diversity. Their abundance and diversity alterations may be jointly influenced by heavy metals, soil physiochemical properties, and vegetation factors.
2. In Cr-contaminated soils, the quantity of the nitrogen cycle functional genes was ranked as denitrification > nitrogen fixation > nitrification, and the overall abundance typically increased while the diversity noticeably decreased. Cr stress had a considerable impact on the abundance of the AOA-amoA and AOB-amoA genes in the nitrification process, and the abundance of both genes was characterized by a transition from stress to stability. Based on the energy production and conservation strategies of microbial communities, Cr stress may have sped up the transformation and use of nitrogen in soil while reducing the abundance of the microbial communities.
3. The correlation analysis with soil physicochemical parameters, including RDA, ggcor correlation analysis, and PLS-PM, revealed that environmental factors had a greater impact on functional genes involved in nitrogen cycling than bacterial communities. Additionally, the high abundance of nifH and strong connection with the soil physicochemical parameters suggested that the colonization of nitrogen-fixing bacteria during the Cr stress time series may have facilitated nitrogen fixation in the environment to increase soil nutrients and facilitate the survival of the microbial community in the soil.
The original contributions presented in the study are publicly available. This data can be found here: National Center for Biotechnology Information (NCBI), accession no. PRJNA930506.
XB, PZ, XZ, and XJ designed this study. XB and YL participated in the operation process of the experiment and carried out data processing. XB performed the statistical analysis and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the Science and Technology Innovation Programs of Higher Education Institutions in Shanxi (2020L0535), and the Construction of Innovation Discipline Cluster Servicing Valley Ecological Governance Industry (Shanxi “1331 Project”). This research was also supported by the Department of Biological Sciences and Technology.
We thank teachers Pengyu Zhao, Xiuqing Jing, and Xiaodong Zhao for their guidance during the experimental design and experimental manipulation. We thank Yujing Li for her help in writing the paper. We also thank the LetPub platform reviewers for their valuable comments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1116535/full#supplementary-material
2. ^http://sourceforge.net/projects/rdp-classifier/
Abdu, N., Abdullahi, A. A., and Abdulkadir, A. (2017). Heavy metals and soil microbes. Environ. Chem. Lett. 15, 65–84. doi: 10.1007/s10311-016-0587-x
Azarbad, H., Niklińska, M., Nikiel, K., van Straalen, N. M., and Röling, W. F. M. (2015). Functional and compositional responses in soil microbial communities along two metal pollution gradients: does the level of historical pollution affect resistance against secondary stress? Biol. Fertil. Soils 51, 879–890. doi: 10.1007/s00374-015-1033-0
Barta, J., Melichova, T., Vanek, D., Picek, T., and Santruckova, H. (2010). Effect of pH and dissolved organic matter on the abundance of nirK and nirS denitrifiers in spruce forest soil. Biogeochemistry 101, 123–132. doi: 10.1007/s10533-010-9430-9
Berg, J., Brandt, K. K., al-Soud, W. A., Holm, P. E., Hansen, L. H., Sørensen, S. J., et al. (2012). Selection for cu-tolerant bacterial communities with altered composition, but unaltered richness, via long-term cu exposure. Appl. Environ. Microbiol. 78, 7438–7446. doi: 10.1128/AEM.01071-12
Bru, D., Sarr, A., and Philippot, L. (2007). Relative abundances of proteobacterial membrane-bound and periplasmic nitrate reductases in selected environments. Appl. Environ. Microbiol. 73, 5971–5974. doi: 10.1128/aem.00643-07
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U. S. A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chen, Y. M., Chao, Y. Q., Li, Y. Y., Lin, Q. Q., Bai, J., Tang, L., et al. (2016). Survival strategies of the plant-associated bacterium Enterobacter sp. strain EG16 under cadmium stress. Appl. Environ. Microbiol. 82, 1734–1744. doi: 10.1128/AEM.03689-15
Chen, J. H., He, F., Zhang, X. H., Sun, X., Zheng, J. F., and Zheng, J. W. (2014). Heavy metal pollution decreases microbial abundance, diversity and activity within particle-size fractions of a paddy soil. FEMS Microbiol. Ecol. 87, 164–181. doi: 10.1111/1574-6941.12212
Chen, X. M., Zhao, Y., Zhao, X. Y., Wu, J. Q., Zhu, L. J., Zhang, X., et al. (2020). Selective pressures of heavy metals on microbial community determine microbial functional roles during composting: sensitive, resistant and actor. J. Hazard. Mater. 398:122858. doi: 10.1016/j.jhazmat.2020.122858
David, J. L. -B., Cindy, E. P., and Susan, J. G. (2014). Microbial functional genes involved in nitrogen fixation, nitrification and denitrification in forest ecosystems. Soil Biol. Biochem. 75, 11–25. doi: 10.1016/j.soilbio.2014.03.021
Deng, L. J., Zeng, G. M., Fan, C. Z., Lu, L. H., Chen, X. F., Chen, M., et al. (2015). Response of rhizosphere microbial community structure and diversity to heavy metal co-pollution in arable soil. Appl. Microbiol. Biotechnol. 99, 8259–8269. doi: 10.1007/s00253-015-6662-6
Devrim, C., Dev, T. B., Weiming, S., and Herbert, J. K. (2017). How plant root exudates shape the nitrogen cycle. Trends Plant Sci. 22, 661–673. doi: 10.1016/j.tplants.2017.05.004
Duan, Q., Lee, J., Liu, Y., Chen, H., and Hu, H. Y. (2016). Distribution of heavy metal pollution in surface soil samples in China: a graphical review. Bull. Environ. Contam. Toxicol. 97, 303–309. doi: 10.1007/s00128-016-1857-9
Eichorst, S. A., Trojan, D., Roux, S., Herbold, C., Rattei, T., and Woebken, D. (2018). Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 20, 1041–1063. doi: 10.1111/1462-2920.14043
Garavaglia, L., Cerdeira, S. B., and Vullo, D. L. (2010). Chromium (VI) biotransformation by β- and γ-Proteobacteria from natural polluted environments: a combined biological and chemical treatment for industrial wastes. J. Hazard. Mater. 175, 104–110. doi: 10.1016/j.jhazmat.2009.09.134
Giannopoulos, G., Hartop, K. R., Brown, B. L., Song, B., Elsgaard, L., and Franklin, R. B. (2020). Trace metal availability affects greenhouse gas emissions and microbial functional group abundance in freshwater wetland sediments. Front. Microbiol. 11:560861. doi: 10.3389/fmicb.2020.560861
Hamilton, N. E., and Ferry, M. (2018). Ggtern: ternary diagrams using ggplot2. J. Stat. Softw. 87, 1–17. doi: 10.18637/jss.v087.c03
Hao, X., Zhu, J. J., Rensing, C., Liu, Y., Gao, S. H., Chen, W. L., et al. (2021). Recent advances in exploring the heavy metal(loid) resistant microbiome. Comput. Struct. Biotechnol. J. 19, 94–109. doi: 10.1016/J.CSBJ.2020.12.006
Hu, X. W., Wang, J. L., Lv, Y., Liu, X. Y., Zhong, J., Cui, X. L., et al. (2021). Effects of heavy metals/metalloids and soil properties on microbial communities in farmland in the vicinity of a metals smelter. Front. Microbiol. 12:707786. doi: 10.3389/FMICB.2021.707786
Huang, C. C., Liang, C. M., Yang, T. I., Chen, J. L., and Wang, W. K. (2021). Shift of bacterial communities in heavy metal-contaminated agricultural land during a remediation process. PLoS One 16:e0255137. doi: 10.1371/journal.pone.0255137
Islam, Z. F., Cordero, P. R. F., Feng, J., Chen, Y. -J., Bay, S. K., Jirapanjawat, T., et al. (2019). Two Chloroflexi classes independently evolved the ability to persist on atmospheric hydrogen and carbon monoxide. ISME J. 13, 1801–1813. doi: 10.1038/s41396-019-0393-0
Jiang, B., Adebayo, A., Jia, J. L., Xing, Y., Deng, S. Q., Guo, L., et al. (2019). Impacts of heavy metals and soil properties at a Nigerian e-waste site on soil microbial community. J. Hazard. Mater. 362, 187–195. doi: 10.1016/j.jhazmat.2018.08.060
Jie, S. Q., Li, M. M., Gan, M., Zhu, J. Y., Yin, H. Q., and Liu, X. D. (2016). Microbial functional genes enriched in the Xiangjiang River sediments with heavy metal contamination. BMC Microbiol. 16:179. doi: 10.1186/s12866-016-0800-x
Kandeler, E., Deiglmayr, K., Tscherko, D., Bru, D., and Philippot, L. (2006). Abundance of narG, nirS, nirK, and nosZ genes of denitrifying bacteria during primary successions of a glacier foreland. Appl. Environ. Microbiol. 72, 5957–5962. doi: 10.1128/AEM.00439-06
Kasemodel, M. C., Sakamoto, I. K., Varesche, M. B. A., and Rodrigues, V. G. S. (2019). Potentially toxic metal contamination and microbial community analysis in an abandoned Pb and Zn mining waste deposit. Sci. Total Environ. 675, 367–379. doi: 10.1016/j.scitotenv.2019.04.223
Khanal, A., and Lee, J. H. (2020). Functional diversity and abundance of nitrogen cycle-related genes in paddy soil. Appl. Biol. Chem. 63:17. doi: 10.1186/s13765-020-00500-6
Kuypers, M. M. M., Marchant, H. K., and Kartal, B. (2018). The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16, 263–276. doi: 10.1038/nrmicro.2018.9
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/aem.00335-09
Leininger, S., Urich, T., Schloter, M., Schwark, L., Qi, J., Nicol, G. W., et al. (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442, 806–809. doi: 10.1038/nature04983
Leng, Y., Li, Y., Wen, Y., Zhao, H., Wang, Q., and Li, S. W. (2020). Transcriptome analysis provides molecular evidences for growth and adaptation of plant roots in cadimium-contaminated environments. Ecotoxicol. Environ. Saf. 204:111098. doi: 10.1016/j.ecoenv.2020.111098
Li, Y., Bao, W. K., Bongers, F., Chen, B., Chen, G. K., Guo, K., et al. (2019). Drivers of tree carbon storage in subtropical forests. Sci. Total Environ. 654, 684–693. doi: 10.1016/j.scitotenv.2018.11.024
Li, P. P., Han, Y. L., He, J. Z., Zhang, S. Q., and Zhang, L. M. (2019). Soil aggregate size and long-term fertilization effects on the function and community of ammonia oxidizers. Geoderma 338, 107–117. doi: 10.1016/j.geoderma.2018.11.033
Li, Y., Kang, E., Song, B., Wang, J. S., Zhang, X. D., Wang, J. Z., et al. (2021). Soil salinity and nutrients availability drive patterns in bacterial community and diversity along succession gradient in the Yellow River Delta. Estuar. Coast. Shelf Sci. 262:107621. doi: 10.1016/j.ecss.2021.107621
Li, S., Liang, H., Wang, Y., Zhang, Z., Zhang, L., Zhou, G., et al. (2022). Responses of functional genes involved in nitrogen cycling to green manuring in different paddy soils in South China. Plant Soil 478, 519–532. doi: 10.1007/s11104-022-05491-5
Li, X. Q., Meng, D. L., Li, J., Yin, H. Q., Liu, H. W., Liu, X. D., et al. (2017). Response of soil microbial communities and microbial interactions to long-term heavy metal contamination. Environ. Pollut. 231, 908–917. doi: 10.1016/j.envpol.2017.08.057
Li, X. M., Qiao, J. T., Li, S., Häggblom, M. M., Li, F. B., and Hu, M. (2020). Bacterial communities and functional genes stimulated during anaerobic Arsenite oxidation and nitrate reduction in a Paddy soil. Environ. Sci. Technol. 54, 2172–2181. doi: 10.1021/acs.est.9b04308
Li, M. Y., Ren, L. H., Zhang, J. C., Luo, L., Qin, P. F., Zhou, Y. Y., et al. (2019). Population characteristics and influential factors of nitrogen cycling functional genes in heavy metal contaminated soil remediated by biochar and compost. Sci. Total Environ. 651, 2166–2174. doi: 10.1016/j.scitotenv.2018.10.152
Li, P. P., Zhang, S. Q., Li, F., Zhang, Y. T., and Han, Y. L. (2020). Long term combined fertilization and soil aggregate size on the denitrification and community of denitrifiers. Appl. Soil Ecol. 156:103718. doi: 10.1016/j.apsoil.2020.103718
Liang, J., Yang, Z. X., Tang, L., Zeng, G. M., Yu, M., Li, X. D., et al. (2017). Changes in heavy metal mobility and availability from contaminated wetland soil remediated with combined biochar-compost. Chemosphere 181, 281–288. doi: 10.1016/j.chemosphere.2017.04.081
Liu, B., Su, G. R., Yang, Y. R., Yao, Y., Huang, Y. G., Hu, L., et al. (2019). Vertical distribution of microbial communities in chromium-contaminated soil and isolation of Cr(VI)-reducing strains. Ecotoxicol. Environ. Saf. 180, 242–251. doi: 10.1016/j.ecoenv.2019.05.023
Long, X., Chen, C. R., Xu, Z. H., Oren, R., and He, J.-Z. (2012). Abundance and community structure of ammonia-oxidizing bacteria and archaea in a temperate forest ecosystem under ten-years elevated CO2. Soil Biol. Biochem. 46, 163–171. doi: 10.1016/j.soilbio.2011.12.013
Mao, Y. B., Tan, H. F., Wang, M. M., Jiang, T. H., Wei, H. W., Xu, W. P., et al. (2022). Research Progress of soil microorganisms in response to heavy metals in Rice. J. Agric. Food Chem. 70, 8513–8522. doi: 10.1021/acs.jafc.2c01437
Mathivanan, K., Chandirika, J. U., Vinothkanna, A., Yin, H., Liu, X., and Meng, D. (2021). Bacterial adaptive strategies to cope with metal toxicity in the contaminated environment-a review. Ecotoxicol. Environ. Saf. 226:112863. doi: 10.1016/j.ecoenv.2021.112863
Narendrula-Kotha, R., and Nkongolo, K. K. (2017). Bacterial and fungal community structure and diversity in a mining region under long-term metal exposure revealed by metagenomics sequencing. Ecol. Genet. Genom. 2, 13–24. doi: 10.1016/j.egg.2016.11.001
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2020). Vegan: Community Ecology Package. R Package Version 2.5–7. Available at: https://CRAN.R-project.org/package=vegan (Accessed November 20, 2022)
Ouyang, Y., Norton, J. M., and Stark, J. M. (2017). Ammonium availability and temperature control contributions of ammonia oxidizing bacteria and archaea to nitrification in an agricultural soil. Soil Biol. Biochem. 113, 161–172. doi: 10.1016/j.soilbio.2017.06.010
Ouyang, Y., Reeve, J. R., and Norton, J. M. (2018). Soil enzyme activities and abundance of microbial functional genes involved in nitrogen transformations in an organic farming system. Biol. Fertil. Soils 54, 437–450. doi: 10.1007/s00374-018-1272-y
Pan, X. M., Zhang, S. R., Zhong, Q. M., Gong, G. S., Wang, G. Y., Guo, X., et al. (2020). Effects of soil chemical properties and fractions of Pb, cd, and Zn on bacterial and fungal communities. Sci. Total Environ. 715:136904. doi: 10.1016/j.scitotenv.2020.136904
Prosser, J. I., Hink, L., Gubry-Rangin, C., and Nicol, G. W. (2020). Nitrous oxide production by ammonia oxidizers: physiological diversity, niche differentiation and potential mitigation strategies. Glob. Chang. Biol. 26, 103–118. doi: 10.1111/gcb.14877
Prosser, J. I., and Nicol, G. W. (2012). Archaeal and bacterial ammonia-oxidisers in soil: the quest for niche specialisation and differentiation. Trends Microbiol. 20, 523–531. doi: 10.1016/j.tim.2012.08.001
Rijk, I. J. C., and Ekblad, A. (2020). Carbon and nitrogen cycling in a lead polluted grassland evaluated using stable isotopes (δ 13 C and δ 15 N) and microbial, plant and soil parameters. Plant Soil 449, 249–266. doi: 10.1007/s11104-020-04467-7
Robertson, G. P., and Vitousek, P. M. (2009). Nitrogen in agriculture: balancing the cost of an essential resource. Annu. Rev. Environ. Resour. 34, 97–125. doi: 10.1146/annurev.environ.032108.105046
Rösch, C., and Bothe, H. (2005). Improved assessment of denitrifying, N2-fixing, and total-community bacteria by terminal restriction fragment length polymorphism analysis using multiple restriction enzymes. Appl. Environ. Microbiol. 71, 2026–2035. doi: 10.1128/aem.71.4.2026-2035.2005
Rousk, J., Bååth, E., Brookes, P. C. B., Lauber, C. L., Lozupone, C., Caporaso, J. G., et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4, 1340–1351. doi: 10.1038/ismej.2010.58
Schrama, M., Berg, M. P., and Olff, H. (2012). Ecosystem assembly rules: the interplay of green and brown webs during salt marsh succession. Ecology 93, 2353–2364. doi: 10.1890/11-1102.1
Sharma, A., Kapoor, D., Wang, J., Shahzad, B., Kumar, V., Bali, A. S., et al. (2020). Chromium bioaccumulation and its impacts on plants: An overview. Plan. Theory 9:100. doi: 10.3390/plants9010100
Singh, B. K., Quince, C., Macdonald, C. A., Khachane, A., Thomas, N., Al-Soud, W. A., et al. (2014). Loss of microbial diversity in soils is coincident with reductions in some specialized functions. Environ. Microbiol. 16, 2408–2420. doi: 10.1111/1462-2920.12353
Spang, A., Poehlein, A., Offre, P., Zumbrägel, S., Haider, S., Rychlik, N., et al. (2012). The genome of the ammonia-oxidizing Candidatus Nitrososphaera gargensis: insights into metabolic versatility and environmental adaptations. Environ. Microbiol. 14, 3122–3145. doi: 10.1111/j.1462-2920.2012.02893.x
Szukics, U., Hackl, E., Zechmeister-Boltenstern, S., and Sessitsch, A. (2011). Rapid and dissimilar response of ammonia oxidizing archaea and bacteria to nitrogen and water amendment in two temperate forest soils. Microbiol. Res. 167, 103–109. doi: 10.1016/j.micres.2011.04.002
Tang, B., Hao, P. X., Feng, M. X., Ge, H. G., and Yue, S. Y. (2022). Effects of heavy metals on microorganisms and enzymes in soils of lead–zinc tailing ponds. Environ. Res. 207:112174. doi: 10.1016/j.envres.2021.112174
Van der Heijden, M. G. A., Bardgett, R. D., and Van Straalen, N. M. (2008). The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 11, 296–310. doi: 10.1111/j.1461-0248.2007.01139.x
Viti, C., Marchi, E., Decorosi, F., and Giovannetti, L. (2014). Molecular mechanisms of Cr(VI) resistance in bacteria and fungi. FEMS Microbiol. Rev. 38, 633–659. doi: 10.1111/1574-6976.12051
Wang, S. Z., Chen, W. W., Gao, Q. Q., and Zhou, C. F. (2022). Dynamic changes of Rhizosphere soil microbiome and functional genes involved in carbon and nitrogen cycling in Chinese fir monoculture. Forests 13:1906. doi: 10.3390/f13111906
Wang, Y. F., and Gu, J. D. (2014). Effects of allylthiourea, salinity, and pH on ammonia/ammonium-oxidizing prokaryotes in mangrove sediment incubated in laboratory microcosms. Appl. Microbiol. Biotechnol. 98, 3257–3274. doi: 10.1007/s00253-013-5399-3
Wu, W. C., Dong, C. X., Wu, J. H., Liu, X. W., Wu, Y. X., Chen, X. B., et al. (2017). Ecological effects of soil properties and metal concentrations on the composition and diversity of microbial communities associated with land use patterns in an electronic waste recycling region. Sci. Total Environ. 601–602, 57–65. doi: 10.1016/j.scitotenv.2017.05.165
Xie, Y., Fan, J. B., Zhu, W. X., Amombo, E., Lou, Y. H., Chen, L., et al. (2016). Effect of heavy metals pollution on soil microbial diversity and bermudagrass genetic variation. Front. Plant Sci. 7:755. doi: 10.3389/fpls.2016.00755
Xu, M., Hao, X., Xiong, Z., Liao, H., Wang, L., Zhang, T., et al. (2021). Soil amendments change bacterial functional genes more than taxonomic structure in a cadmium-contaminated soil. Soil Biol. Biochem. 154:108126. doi: 10.1016/J.SOILBIO.2020.108126
Xu, Y., Wang, H. M., Xiang, X., Wang, R. C., and Tian, W. (2019). Vertical variation of nitrogen fixers and ammonia oxidizers along a sediment profile in the Dajiuhu Peatland, Central China. J. Earth Sci. 30, 397–406. doi: 10.1007/s12583-018-0982-2
Xu, M. P., Wang, J. Y., Zhu, Y. F., Han, X. H., Ren, C. J., and Yang, G. H. (2021). Plant biomass and soil nutrients mainly explain the variation of soil microbial communities during secondary succession on the loess plateau. Microb. Ecol. 83, 114–126. doi: 10.1007/s00248-021-01740-9
Zeng, Q., and An, S. (2021). Identifying the biogeographic patterns of rare and abundant bacterial communities using different primer sets on the loess plateau. Microorganisms 9:139. doi: 10.3390/microorganisms9010139
Zhang, X. P., Gai, X., Zhong, Z. K., Bian, F. Y., Yang, C. B., Li, Y. F., et al. (2021). Understanding variations in soil properties and microbial communities in bamboo plantation soils along a chromium pollution gradient. Ecotoxicol. Environ. Saf. 222:112507. doi: 10.1016/J.ECOENV.2021.112507
Zhang, C., Nie, S., Liang, J., Zeng, G. M., Wu, H. P., Hua, S. S., et al. (2016). Effects of heavy metals and soil physicochemical properties on wetland soil microbial biomass and bacterial community structure. Sci. Total Environ. 557-558, 785–790. doi: 10.1016/j.scitotenv.2016.01.170
Zhao, H. C., Lin, J. H., Wang, X. H., Shi, J. C., Dahlgren, R. A., and Xu, J. M. (2021). Dynamics of soil microbial N-cycling strategies in response to cadmium stress. Environ. Sci. Technol. 55, 14305–14315. doi: 10.1021/ACS.EST.1C04409
Keywords: heavy metal stress, microbial community, high-throughput sequencing, qPCR, PLS-EM
Citation: Bai X, Li Y, Jing X, Zhao X and Zhao P (2023) Response mechanisms of bacterial communities and nitrogen cycle functional genes in millet rhizosphere soil to chromium stress. Front. Microbiol. 14:1116535. doi: 10.3389/fmicb.2023.1116535
Edited by:
Chris Yeager, Los Alamos National Laboratory (DOE), United StatesReviewed by:
Xiaomin Li, South China Normal University, ChinaCopyright © 2023 Bai, Li, Jing, Zhao and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengyu Zhao, ✉ Mzk0MzgyNTU3QHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.