 Hui Cao
Hui Cao Suying Li
Suying Li Yichao Wu
Yichao Wu Qiaoyun Huang
Qiaoyun Huang Peng Cai
Peng Cai Chun-Hui Gao
Chun-Hui Gao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 March 2023
Sec. Terrestrial Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1115300
Soil microbial diversity is important for maintaining ecosystem functions. However, the linkage between microbial diversity, especially rare and abundant bacterial diversity, and carbon decomposition remains largely unknown. In this study, we assessed the establishment and maintenance of rare and abundant bacterial α-diversities at the taxonomic and phylogenetic levels and their linkages with soil carbon decomposition separately in four Chinese woodlands. Compared to abundant bacteria, rare bacteria showed higher community diversity, tighter phylogenetic clustering, wider environmental breadth, stronger phylogenetic signals, and higher functional redundancy. The assembly of the abundant bacterial subcommunity was governed by stochastic (59.2%) and deterministic (41.8%) processes, whereas the assembly of the rare bacterial subcommunity was mainly dominated by deterministic processes (85.8%). Furthermore, total phosphorus, soil pH, and ammonium nitrogen balanced stochastic and deterministic processes in both rare and abundant bacterial subcommunities. Our results reveal that rare bacteria displayed stronger environmental adaptability and environmental constraint. Importantly, the α-diversities of rare taxa, rather than abundant taxa, were significantly related to carbon decomposition. This study provides a holistic understanding of biogeographic patterns of abundant and rare bacteria and their α-diversities in relation to carbon decomposition, thus helping us better predict and regulate carbon dynamics under the background of global climate change.
Woodlands, one of the stable terrestrial carbon sinks, play a significant role in the global carbon cycle and consequently affect global climate change (Dixon et al., 1994; Bonan, 2008). Dynamic changes in woodland carbon pools are induced by carbon assimilation and decomposition (Dan et al., 2020). The carbon decomposition of woodland releases enormous amounts of carbon dioxide (CO2), increasing global warming (Rustad et al., 2000). Soil microorganisms (e.g., arbuscular mycorrhizal fungi and Acidobacteria) are the main engines of carbon decomposition and their microbial diversity is vital for maintaining ecosystem functions (Trivedi et al., 2016; Liang et al., 2017; Zhang et al., 2018; Tláskal and Baldrian, 2021). Therefore, understanding the relationship between microbial diversity and carbon decomposition would be helpful in the mitigation of global climate change.
Microorganisms usually show skewed abundance distributions, consisting of some species with higher abundance (abundant taxa) and most species with lower abundance (rare taxa; Nemergut et al., 2011). The abundant taxa are traditionally considered to dominate microbial communities and perform major ecological functions (Pedrós-Alió, 2012). Recent studies have shown a significant positive correlation between rare species diversity and multifunctionality, which may be attributed to a limited microbiota performing a specific ecological function (Chen et al., 2019; Zhang et al., 2022). For instance, Desulfosporosinus spp. is the most critical sulfate-reducing agent in peatlands, but its relative abundance is only 0.0006% (Pester et al., 2010). In acid mine drainage, rare bacteria (e.g., Acidithiobacillus sp. FKB1 and FKB2) are closely related to the processes of nitrogen fixation and sulfur oxidation (Hua et al., 2015). However, the relative contribution of rare and abundant bacterial diversity to the functions possessed by such a broad range of microorganisms as carbon decomposition is unclear.
Microbial diversity maintenance would be effectively evaluated by the microbial environmental adaptability and ecological assembly process. Environmental adaptability, which was clarified by the environmental breadth and phylogenetic signal, reflects the resistance of species to environmental change (Goberna and Verdú, 2016). The abundant and rare taxa show different abilities of environmental adaptability. For instance, abundant rather than rare microbial taxa show stronger environmental adaptability in wetland soils of the Qinghai-Tibet Plateau (Wan et al., 2021a). Ecological assembly processes, including stochastic and deterministic processes, shape the composition and coexistence pattern of microbial community and couple microbial community function with ecosystem function (Feng et al., 2018). Both stochastic and deterministic processes influence bacterial community assemblies (Gao et al., 2020; Jiao and Lu, 2020b). Specifically, stochastic processes determine the assembly of abundant bacterial subcommunity, whereas deterministic processes dominate the assembly of rare ones in Chinese farmland soils (Jiao and Lu, 2020b). Yet, few works have revealed the ecological assembly process and environmental adaptability of rare and abundant bacteria in woodland ecosystems.
We chose four woodlands across eastern China and collected soils covering four soil types, showing distinct differences in physicochemical properties (Supplementary Table S1). This study set out to i) evaluate the linkage of rare and abundant taxa with carbon decomposition in woodlands and ii) reveal the environmental adaptability and assembly process of rare and abundant bacterial subcommunities. Since microbial diversity is closely connected to ecosystem function (Tilman et al., 2014; Morris et al., 2020), the contributions of rare and abundant bacterial diversity to carbon decomposition should both exist. Given the highly competitive and metabolic capacity of abundant bacteria (Jousset et al., 2017), we hypothesized that abundant bacteria would have greater environmental fitness than those of rare bacteria and may contribute more to carbon decomposition in soils. To test our hypotheses, we performed 16S rRNA gene sequencing and determined the enzymatic activities related to carbon decomposition.
We chose four woodlands covering four soil types (i.e., black, brown, cinnamonic, and red soils), and these four woodlands were mainly covered with poplar, poplar, poplar and pine trees, respectively. The four sites were separately located in Qiqihar in Heilongjiang Province (124°78′E, 48°19′N), Laiyang in Shandong Province (120°43′E, 36°58′N), Fengqiu in Henan Province (114°33′E, 35°2′N), and Qiyang in Hunan Province (111°53′E, 26°46′N; Supplementary Figure S1). Four sampling sites (50 × 50 m) were established in each woodland. Soil cores were taken from the soil surface (0–20 cm) at each sampling site using the five-point sampling method. The samples were immediately covered with ice packs and sent back to the laboratory within 24 h. Impurities such as plant and animal residues, stones and roots were manually removed. Then, the samples were passed through a sieve with a 2 mm pore size. A portion of the samples was stored for physicochemical properties and enzyme activities determination and the remainder were kept at -80°C for DNA extraction.
Standard analytical methods were followed to measure thirteen soil physicochemical indicators (Bao, 2000), including soil moisture content (Moi), water holding capacity (WHC), pH, soil organic matter (SOM), total carbon (TC), total nitrogen (TN), nitrate-nitrogen (NO3−-N), ammonium nitrogen (NH4+-N), total phosphorus (TP), total potassium (TK), available phosphorus (AP) and available potassium (AK) and electrical conductivity (EC). Details of the assays were given in the supplementary information (Supplementary Method).
Enzyme activities related to carbon cycling, including β-1,4-glucosidase (βG), β-D-cellobiohydrolase (CB), β-xylosidase (XYL) and α-glucosidase (αG), were measured using 4-methylumbelliferone (MUB)-linked substrate by a microplate reader (Spark; Tecan, Switzerland; Marx et al., 2001; Bell et al., 2013). The carbon cycling index (CCI) was determined by employing Z-score transformation (Luo et al., 2018; Wan et al., 2021c) based on the following equation. The equation is EVi = (Ei – Eave)/SDi, where EV is the standardized variable for factor i (i.e., TC, βG, CB, XYL, and αG), Ei is the factor i, and Eave and SDi are the mean value and standard deviation of factor i over all samples, respectively. The CCI is the mean of the EV values for the five selected factors.
The DNeasy® PowerSoil® Kit (Qiagen, Germantown, MD) was used to extract total microbial genome DNA from 0.25 g soil. The extracted DNA was quantified using a NanoDrop NC2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States).
The forward primer 338F (5′-ACT CCT ACG GGA GGC AGC A-3′) and the reverse primer 806R (5′-GGA CTA CHV GGG TWT CTA AT-3′) were employed to amplify the V3-V4 region of bacterial 16S rRNA genes (Mori et al., 2014; Wan et al., 2022). The following PCR procedure was used: 98°C for 5 min, followed by 25 cycles of denaturation at 98°C for 30 s, annealing at 53°C for 30 s, and extension at 72°C for 45 s, with a final extension of 5 min at 72°C. PersonalBio (Shanghai, China) performed Illumina MiSeq sequencing. The raw sequences were imported into QIIME2 and the “DADA2” plugin was used for denoising and quality control, resulting in amplicon sequence variants (ASVs; Bolyen et al., 2019; Prodan et al., 2020). The Greengene 13_8 99% database (DeSantis et al., 2006; McDonald et al., 2012) was used as a reference to classify and annotate the ASVs using the QIIME 2 feature classifier plugin (Bokulich et al., 2018). The sequencing datasets were deposited in the SRA database1 under the accession numbers SRR22803208–SRR22803223.
First, we deleted ASVs (excluding archaea and fungal ASVs) with few than 20 reads (Jiao and Lu, 2020a). Then, ASVs with a relative abundance >0.1% of total sequences were assigned to “abundant taxa,” and ASVs with a relative abundance <0.01% of total sequences were assigned to “rare taxa,” according to previous definition criteria (Wan et al., 2021b,c).
To evaluate the phylogenetic clustering, the standardized effect sizes of the mean nearest taxa distance (SES.MNTD) were determined based on the null model using the “ses.mntd” function of the “picante” R package (Kembel et al., 2010). The β-mean nearest taxon distance (βMNTD) was obtained via the “comdistnt” function of the “picante” package to quantify the abundance-weighted phylogenetic distance between the two closest ASVs in the two communities (Kembel et al., 2010). Ordinary least squares between phylogenetic similarity and geographic distance was used to characterize the distance attenuation relationship. Spearman’s correlation was implemented with the “vegan” package (Dixon, 2003) in R software (version 4.2.2).
Environmental breadths were assessed by the response thresholds of rare and abundant bacteria to the environmental gradient, which was implemented using the “TITAN2” package (Baker and King, 2010). The ecological preferences of species were obtained by calculating their correlations with environmental variables. Subsequently, we determined the phylogenetic signals by the Fritz-Purvis D test (Goberna and Verdú, 2016). Based on 16S rRNA gene sequence similarity, functional predictions were carried out using “Tax4Fun2” package, and functional redundancy indices (FRI) were calculated for different functions (Wemheuer et al., 2020).
The β-nearest taxon index (βNTI) and Bray–Curtis-based Raup-Crick (RCbray) were used to evaluate rare and abundant bacterial subcommunity assemblies based on the null model (Chase and Myers, 2011). When |βNTI| > 2, the selection effect dominates the difference in community composition. Among them, if βNTI < −2, it is considered that homogeneous selection leads to a more similar community phylogeny, and if βNTI > +2, it is considered variable selection. When |βNTI| < 2, the community composition differences are due to dispersal limitation, homogenizing dispersal, or undominant processes. If RCbray < −0.95, it indicates that homogenizing dispersal plays a decisive role in community assembly, and if RCbray >0.95, it is mainly dispersal limitation; −0.95 < RCbray <0.95 is used to estimate the undominant assembly process, including weak selection role and dispersal, diversification and drift process (Stegen et al., 2013). The correlation between physicochemical factors and βNTI was determined by the Mantel test.
A total of 831,328 reads were divided into 6,360 ASVs (ASVs with <20 reads were deleted). Rare (3,356 ASVs) and abundant taxa (132 ASVs) contributed 52.8% and 2.1% of the total community richness, with total relative abundances of 1.57% and 25.1%, respectively. Occupancy was significantly and positively related to abundance for rare bacteria (R2 = 0.18, p < 0.001) but not for abundant bacteria (R2 = 0.013, p > 0.05; Figure 1A). In more than half of all the samples, 52.3% of the abundant taxa and 3% of the rare taxa were detected. Abundant taxa were assigned to 8 phyla and rare taxa to 26 phyla. The abundant taxa mainly included Actinobacteria (51%), Proteobacteria (21.1%) and Chloroflexi (18.8%), while Actinobacteria (28.7%), Proteobacteria (28.1%), Acidobacteria (16.3%) and Chloroflexi (12.3%) were dominant in the rare taxa (Figure 1B). These findings revealed significant differences in the rare and abundant bacterial distribution patterns. Rare taxa had noticeably lower mean values of SES.MNTD than those of abundant taxa (p < 0.001, Wilcoxon rank-sum test), with all values less than 0 (Figure 1C), indicating that rare taxa have a tighter phylogenetic distribution than that of abundant one. According to the Mantel test for correlations between environmental variables and bacterial subcommunities, soil moisture, pH and TP were significantly correlated with community diversity and composition (Table 1).
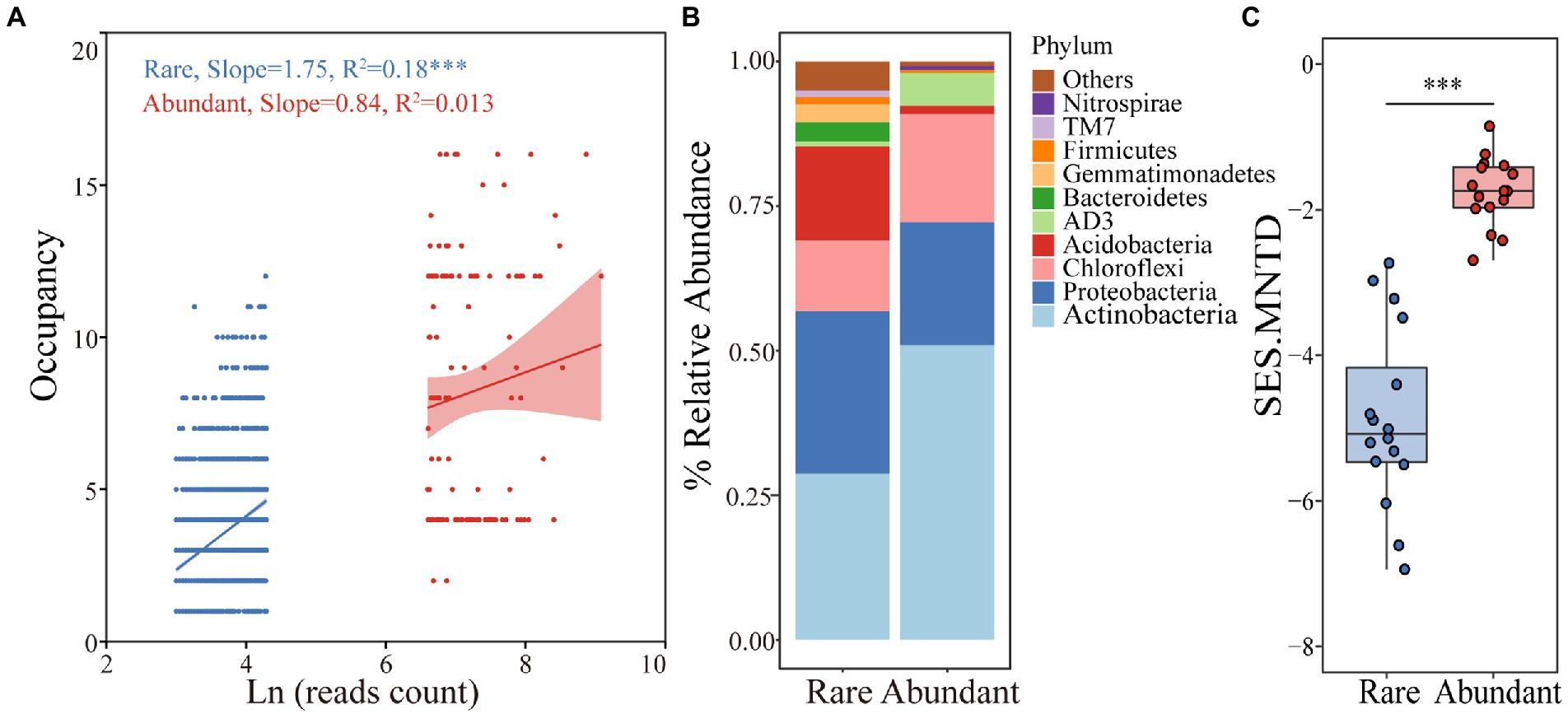
Figure 1. Distribution patterns of rare and abundant taxa in woodland soils. (A) Relationships between abundance and occupancy of rare and abundant ASVs. (B) Distributions of rare and abundant bacteria at the phylum level. (C) Comparison of the differences in SES.MNTD values between rare and abundant bacteria.
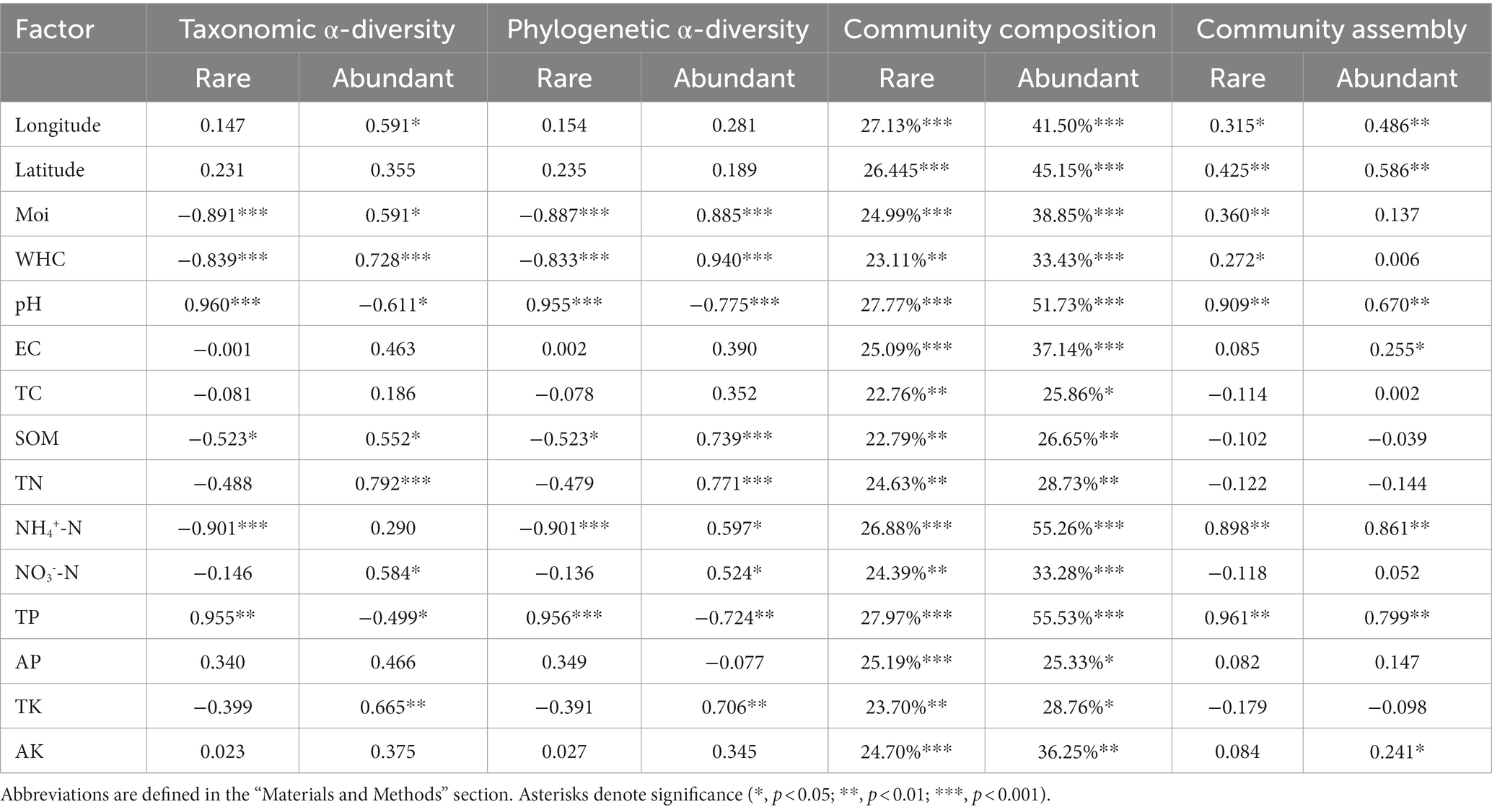
Table 1. Pearson’s correlations between environmental factors and α-diversities, PERMANOVA showing the pure effect of each environmental factor on community composition, and Mantel’s test of environmental variables against the βNTI of rare and abundant bacterial subcommunities.
Both rare and abundant taxa exhibited distance decay relationships, as their taxonomic similarities were significantly negatively correlated with geographic distances. However, the attenuation coefficients of the two were different. The abundant taxa’s attenuation coefficient (0.32) was larger than the rare taxa’s (0.18), suggesting that abundant taxa had a higher species turnover rate. Similarly, the phylogenetic similarities of the rare and abundant bacteria significantly decreased with increasing geographical distances. The abundant taxa (0.14) had a larger attenuation coefficient than that of rare taxa (0.08; Figure 2A). The taxonomic and phylogenetic distances of rare bacterial subcommunities were remarkably higher than those of abundant ones (p < 0.01 for both; Figure 2B). Furthermore, significant correlations were observed between taxonomic and phylogenetic distances of both abundant and rare bacterial subcommunities. The correlation was stronger for the abundant bacterial subcommunity (Supplementary Figure S2), implying that abundant and rare bacterial subcommunity phylogenies were differentially sensitive to environmental changes.
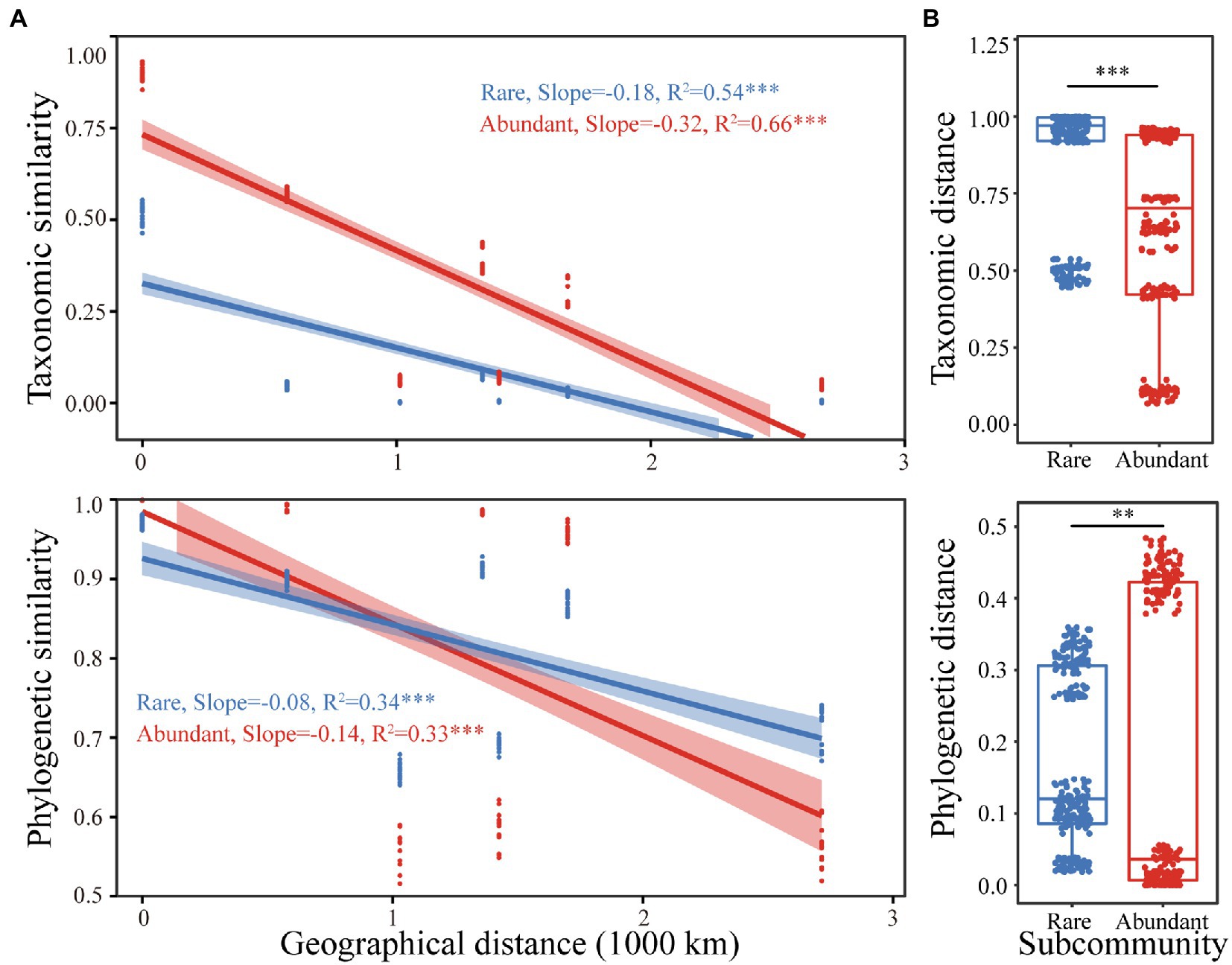
Figure 2. (A) The relationships between geographical distance and the taxonomic and phylogenetic similarity of rare and abundant bacteria. (B) Boxplots show differences in taxonomic and phylogenetic distances between rare and abundant taxa. Asterisks indicate significance (**, p < 0.01, ***, p < 0.001).
The relative abundances of the top 20 ASVs of abundant taxa were more significantly correlated with the activities of carbon-cycling-related enzymes, whereas the relative abundances of the top 20 ASVs of rare taxa were more significantly correlated with TC and SOM (Supplementary Figure S3). Both the Shannon–Wiener index and PD index were higher for rare bacteria than for abundant bacteria (Supplementary Figure S4). The taxonomic α-diversity of rare taxa was significantly positively related to the carbon cycle index (R2 = 0.28, p < 0.05), while the taxonomic α-diversity of abundant taxa was insignificantly negatively related to the carbon cycle index (R2 = 0.02, p > 0.05; Figure 3). Similarly, there was a significantly positive correlation between rare taxa’s phylogenetic α-diversity and carbon cycle index (R2 = 0.28, p < 0.05), whereas there was an insignificant negative correlation between abundant taxa’s phylogenetic α-diversity and carbon cycle index (R2 = 0.04, p > 0.05; Figure 3). The above results indicated that the α-diversities of rare and abundant taxa were related differently to the carbon decomposition function.
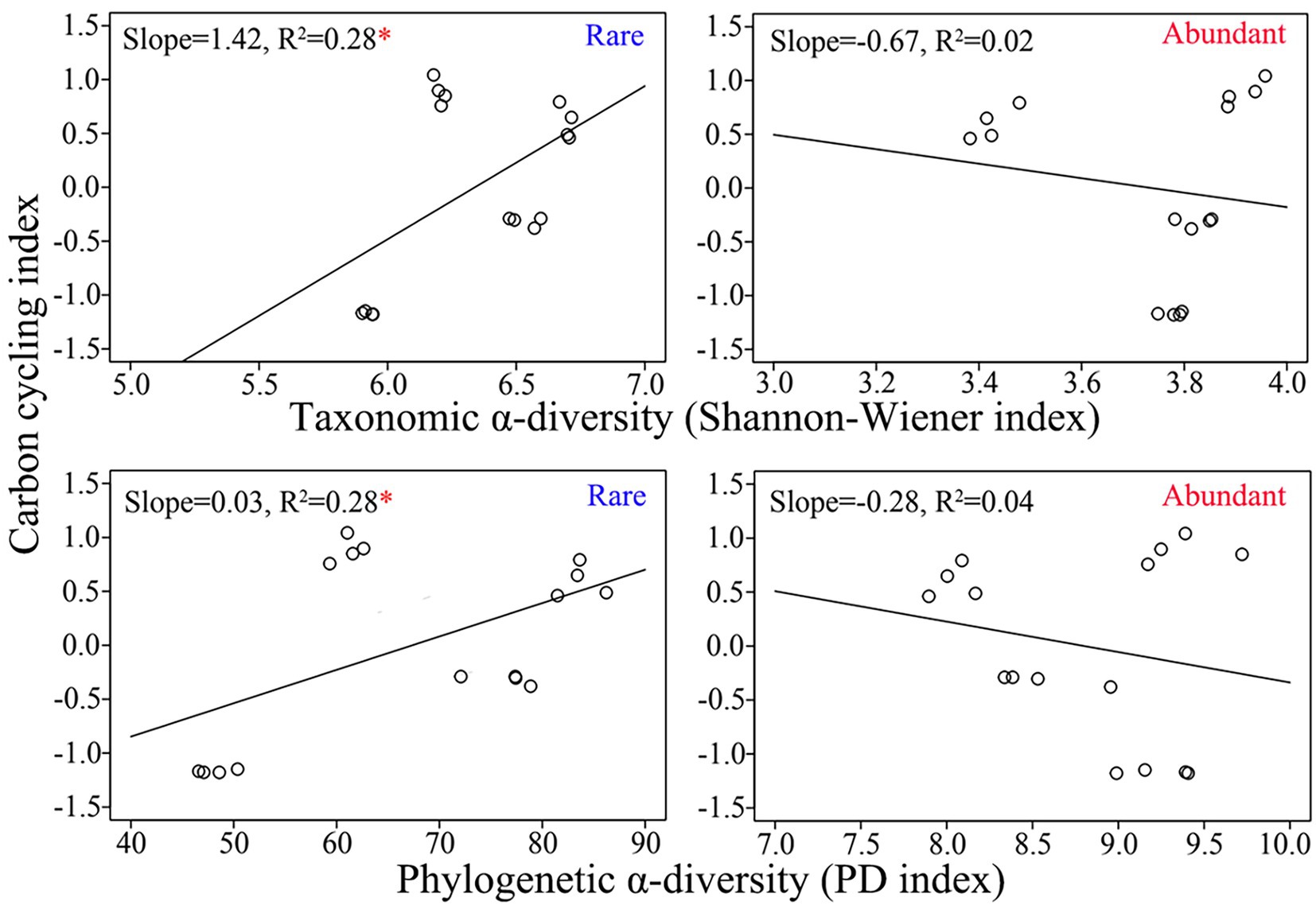
Figure 3. Correlations between the carbon cycle index and α-diversity of rare and abundant taxa. The asterisk indicates significance (*, p < 0.05).
Environmental threshold analysis was employed to investigate the responses of rare and abundant taxa to each geospatial and physicochemical variable tested. Rare taxa showed broader environmental thresholds for all factors tested compared to abundant taxa (Figure 4A). According to the Fritz-Purvis D test, rare taxa had stronger phylogenetic signals than those of abundant taxa for all environmental variables examined (Figure 4B).
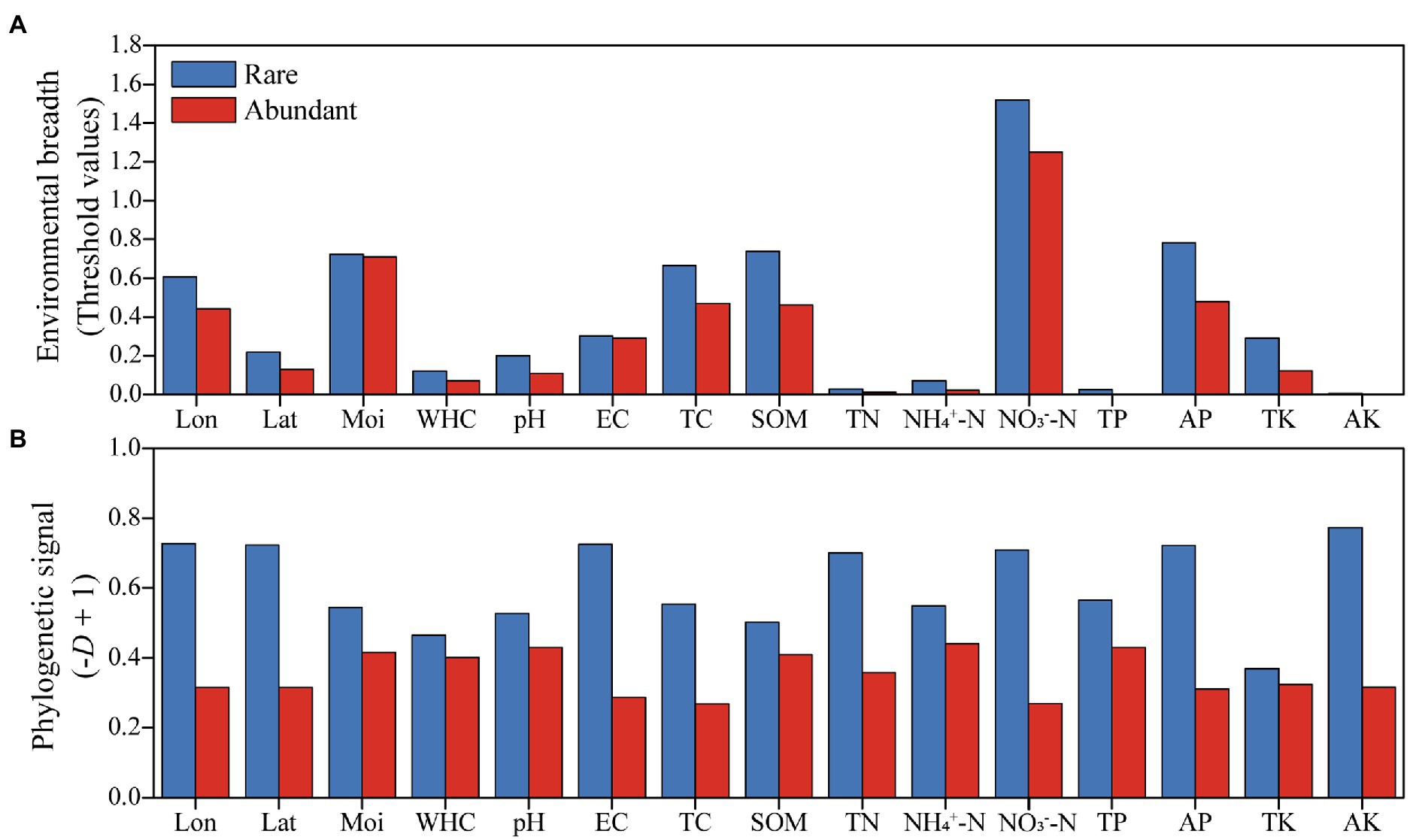
Figure 4. Environmental adaptability of abundant and rare bacteria in woodland soils. (A) Environmental breadths were assessed by the response thresholds of rare and abundant bacteria to environmental factors. (B) Phylogenetic signals reflect the conservation of environmental preference traits in rare and abundant taxa. Abbreviations for environmental variables were described in the “materials and methods” section.
In both rare and abundant bacterial subcommunities, the phylogenetic Mantel correlogram displayed a significant correlation between ASV niche differences and phylogenetic distance along with phylogenetic distance (p < 0.05; Figure 5A). The median βNTI values were 4.371 for rare taxa and-0.198 for abundant taxa (Figure 5B). Null model analysis results showed that ecological processes contributed differently to rare and abundant bacterial subcommunity assemblies (Figure 5C). Variable selection (61.7%) had the largest contribution to the subcommunity assembly of rare bacteria, but variable selection (40%) and dispersal limitation (25.8%) contributed most to the subcommunity assembly of abundant bacteria. Homogeneous selection, homogenizing dispersal and undominated processes had little influences on rare and abundant bacterial subcommunity assemblies. Therefore, deterministic (85.8%) and differentiating (71.7%) processes governed the subcommunity assembly of rare bacteria, while the processes of differentiating (65.8%) and stochastic (59.2%) dominated abundant bacterial subcommunity assembly. The value of the sorting to dispersal limitation for rare taxa was approximately 8.58, which was greater than the value of 1.58 for abundant taxa, indicating that rare taxa were subject to stricter environmental constraints than abundant ones. According to Mantel tests, pH, NH4+-N and TP were more closely linked with βNTI than other physicochemical variables (Table 1).
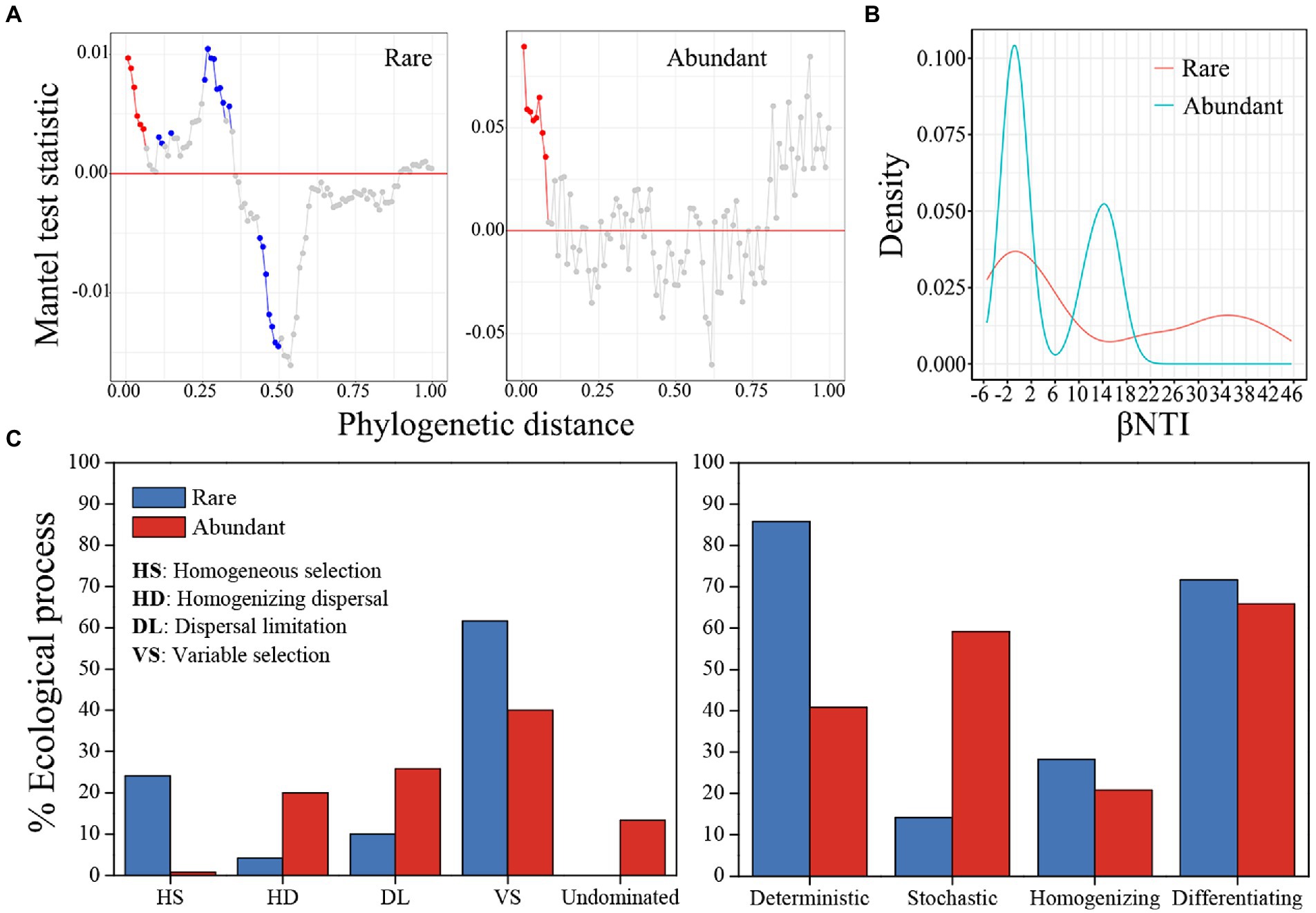
Figure 5. The rare and abundant bacterial subcommunity assembly. (A) Phylogenetic Mantel correlogram evaluating phylogenetic signals in rare and abundant bacterial subcommunities. Each point denotes the Mantel correlation coefficient between phylogenetic distances and ASV niche differences. Colors indicate significance (red, p < 0.01, blue, p < 0.05 and gray, p > 0.05). (B) Kernal density estimates for βNTI distributions for rare and abundant bacteria. (C) Null model-based ecological assembly processes of rare and abundant bacterial subcommunities.
At KEGG pathway level 3, there were a total of 304 functions based on the function prediction results. The rare taxa showed higher functional redundancy (177 functions) than the abundant taxa (127 functions; Figure 6A). Some carbon catabolic functions were significantly higher for rare rather than abundant taxa, including pentose and glucuronate interconversions, inositol phosphate metabolism, galactose metabolism, carbohydrate digestion and absorption, and ascorbate and aldarate metabolism. However, glyoxylate and dicarboxylate metabolism and C5-Branched dibasic acid metabolism were higher in abundant bacteria than in rare bacteria (Figure 6B). More significant correlations were found between carbon-cycling-related enzyme activity and carbon-cycling-related functions of abundant bacterial subcommunities than rare ones (Supplementary Figure S5). However, significant correlations were observed almost only between total carbon, soil organic matter and carbon-cycling-related functions of rare bacterial subcommunities.
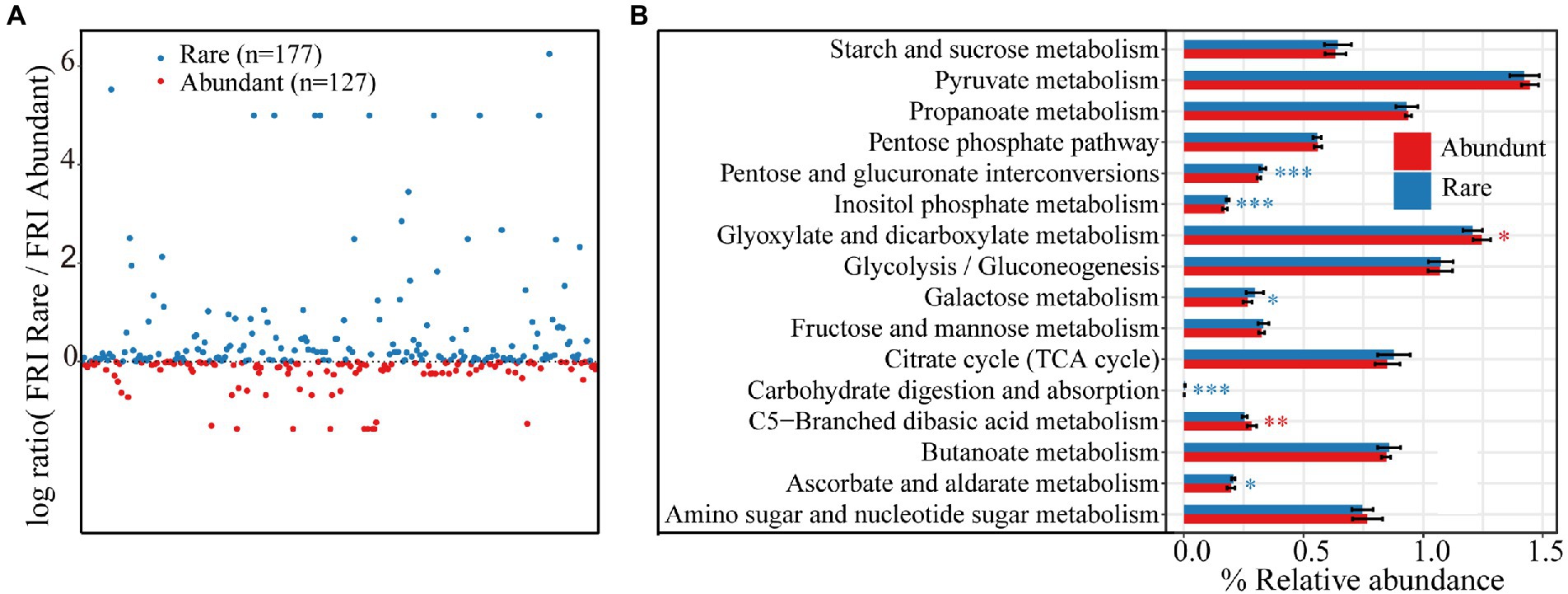
Figure 6. (A) Functional redundancy of rare and abundant taxa at KEGG pathway level 3. (B) Differences in functional redundancy among 16 carbon decomposition functions between rare and abundant bacteria. Asterisks indicate significance (*p < 0.05; **p < 0.01; ***p < 0.001).
The rare and abundant bacterial subcommunities had various species compositions in the woodland ecosystem. Rare bacterial subcommunity had much higher taxonomic and phylogenetic α-diversity than those of abundant bacterial subcommunity, supporting the view that rare bacteria are pivotal to α-diversity (Lynch and Neufeld, 2015). “Environmental filtration” affects bacterial diversity and thus the ecological functions of the bacterial community (O’Malley, 2008). Here, soil moisture, pH and TP are the most critical factors influencing the rare and abundant bacterial subcommunity diversity and structure in woodland soils. pH is closely correlated with microbial community composition in woodland soils (Borchers et al., 2022). Additionally, an earlier study reported that TN and pH affected the abundant bacterial subcommunity composition, whereas organic carbon shapes the rare bacterial subcommunity composition in oil-contaminated soils (Jiao et al., 2017). The discrepancies may be due to differences in geographical and environmental factors.
Soil carbon decomposition depends on microbial activity and diversity (Monteux et al., 2020; Xu et al., 2021). Carbon-cycling-related functions of abundant and rare taxa showed different potential effects on carbon-decomposing-related enzymes and soil organic matter. This phenomenon might be due to the large population of abundant bacteria and the high redundancies of some carbon-cycling-related functions of rare bacteria. Microbial diversity can respond to changes in environmental factors and serve as a predictor of ecosystem multifunctionality. Recent research has shown that rare taxa are the main driving factors for ecosystem multifunctionality (Chen et al., 2019; Zhang et al., 2022). Furthermore, rare bacteria are proven to be more central to the nitrogen and phosphorus cycles than abundant bacteria (Wei et al., 2019; Liang et al., 2020). Our research showed a stronger linkage of diversity-carbon decomposition for rare rather than abundant bacteria in woodland soils. The different linkages of rare and abundant bacterial diversity with carbon decomposition might be attributed to distinct mechanisms for their establishment and maintenance. Future studies will validate these findings on large spatial and long temporal scales for better generalization.
Recently, researchers have focused on the environmental adaptability of rare and abundant taxa in farmland, grassland, wetland and other ecosystems (Ji et al., 2020; Jiao and Lu, 2020a; Wan et al., 2021a,c). Nevertheless, there are few studies on the environmental adaptability of rare and abundant bacteria in woodland soils. Contrary to our hypothesis, rare bacteria had a wider environmental breadth than that of abundant bacteria for all physical and chemical factors, indicating that rare taxa can effectively use a broader range of resources in woodlands. This could be explained by the fact that rare bacteria have a higher richness than that of abundant bacteria, allowing a large number of specialists to adapt to environments and thus obtain a broader environmental breadth (Pedrós-Alió, 2012). This finding differs from the case in which abundant species adapt to a wider environmental gradient than rare species in eastern China farmland (Jiao and Lu, 2020a). The discrepancy may be caused by different kinds of microorganisms and relative abundances of rare subcommunities. Similar to several previous studies, abundant bacteria were more omnipresent in woodland soils than rare bacteria and could be detected in almost all samples (Ji et al., 2020; Jiao and Lu, 2020b). Most rare bacteria were found in only a few soil samples, consistent with previous results (Jousset et al., 2017), which may be due to their low competitiveness and metabolic capacity, limiting their habitat breadth (Barberán et al., 2014).
Rare bacteria exhibited stronger phylogenetic signals than those of abundant bacteria, consistent with a finding in coastal wetlands (Gao et al., 2020), indicating that closely related species have more similar ecological preferences in rare taxa. The functional characteristics of microorganisms based on ecological preferences are greatly influenced by their evolutionary history, in contrast to the limited influence of environmental heterogeneity (Thomas et al., 2016; Saladin et al., 2019). For instance, the growth and metabolism of soil microorganisms are affected more by evolutionary history than by environmental variables such as temperature and precipitation in four forest habitats (Morrissey et al., 2019). Interestingly, a stronger positive correlation was observed between taxonomic distance and phylogenetic distance for abundant bacteria than for rare bacteria (Supplementary Figure S2). This may be owing to the fact that the phylogeny of rare microbial subcommunities is less susceptible to persistent environmental changes (Ji et al., 2020). The potential potentiality of a community to maintain phylogeny can reflect its ability to conserve ecological niches (Miller et al., 2013). Hence the decoupling between taxonomic and phylogenetic distances suggests that rare bacteria maintain ecological niches better than abundant bacteria. These findings probably explain why rare bacteria had a wider environmental breadth and a unique biogeographic pattern. In short, the above two findings indicated that rare bacteria were more environmentally adaptable than abundant bacteria in woodland soils at the level of taxonomy and phylogeny, which may be a crucial reason for the stronger linkage of diversity-carbon decomposition for rare rather than abundant bacteria.
Deterministic processes (mainly variable selection) dominated rare bacterial subcommunity assembly, while stochastic processes (e.g., dispersal limitation) contributed more to abundant bacterial subcommunity assembly in woodland soils. This is consistent with the finding that abundant taxa are more environmentally constrained in mountain forests in northern Xinjiang, China (Wang et al., 2021). Nevertheless, rare and abundant bacterial subcommunity assemblies in eastern Chinese farmland are dominated by homogeneous selection belonging to deterministic processes (Jiao and Lu, 2020b). Rare bacterial subcommunity assembly in Tibetan Plateau grassland was mainly controlled by stochastic processes, whereas abundant bacterial subcommunity assembly was governed by both stochastic and deterministic processes (Ji et al., 2020). These inconsistent results might be due to geographic heterogeneity and different environmental adaptations of microorganisms (Shi et al., 2018). In addition, the lower SES.MNTD of rare taxa in our study indicates tighter phylogenetic clustering, consistent with prior research (Jiao and Lu, 2020a; Wan et al., 2021c). An earlier study have documented that environmental filtering mediates phylogenetic clustering in microbial communities (Horner-Devine and Bohannan, 2006). This seems to be confirmed by our findings that rare taxa subject to more variable selection controls are more phylogenetically clustered.
Soil pH, NH4+-N and TP played vital roles in rare and abundant bacterial subcommunity assemblies. In-depth studies more recently have reported that pH balances deterministic and stochastic processes in rare and abundant bacterial subcommunities of Chinese farmland (Jiao and Lu, 2020b) and dryland montane forest soils (Wang et al., 2021). Additionally, pH strongly affected community assemblies of bacteria in grassland soils (Wang et al., 2017) and functional bacteria (e.g., aerobic methanotrophs) in forest soils (Li et al., 2021). The decisive role of pH in shaping bacterial community assembly may be attributable to its strong influence on cell growth and metabolism. A small change in 1.5 units of in-situ pH reduces by 50% bacterial activity (Fernández-Calviño and Bååth, 2010). Ammonium nitrogen, another critical factor in bacterial community assembly, was supported by another study (Wan et al., 2021a). Soil NH4+-N may mediate microbial community assembly by affecting soil microbial activity or altering soil bulk density (Pan et al., 2018). In addition, it is worth noting that TP was momentous. Phosphorus increases total soil carbon by promoting plant growth, thereby increasing microbial biomass and changing community structure. On the other hand, phosphorus can also affect microbial growth by changing the pH and osmotic pressure of the soil (Huang et al., 2016). Considering the coupling process of element cycles such as carbon, nitrogen, and phosphorus (Mooshammer et al., 2017), more elements may be involved in mediating rare and abundant bacterial subcommunity assemblies. In the future, we will consider more environmental samples and physicochemical factors to verify our conjecture at larger spatial scales.
Our study explored the diversity, environmental adaptations and community assembly mechanisms of rare and abundant bacteria in woodland soils and their correlations with carbon decomposition. Rare taxa had more community diversity, higher phylogenetic signal, wider environmental breadth, tighter phylogenetic cluster and more environmental constraints than those of abundant ones. Rare bacterial diversity was strongly related to carbon decomposition at taxonomic and phylogenetic levels, and its functional redundancy contributed more to carbon cycle-related functions. These findings are helpful in better understanding rare and abundant bacterial diversity maintenance and predicting carbon dynamics under global climate change.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
HC: conceptualization, methodology, formal analysis, visualization, and writing – original draft. SL: investigation, methodology, and data curation. HH and YS: writing – review and editing. YW: supervision, and writing – review and editing. QH: resources and supervision. PC and C-HG: resources, supervision, funding acquisition, and writing – review and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (42177281 and 32100090), Royal Society-Newton Advanced Fellowship (NAF\R1\191017), and Fundamental Research Funds for the Central Universities (2662021JC012 and 2662022ZHQD001).
We also thank Wenjie Wan from the Chinese Academy of Sciences Wuhan Botanical Garden for helpful comments and suggestions on the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1115300/full#supplementary-material
Baker, M. E., and King, R. S. (2010). A new method for detecting and interpreting biodiversity and ecological community thresholds. Methods Ecol. Evol. 1, 25–37. doi: 10.1111/j.2041-210X.2009.00007.x
Bao, S. D. (2000). Soil and agricultural chemistry analysis. Available at: http://www.researchgate.net/publication/301822463_Soil_and_agricultural_chemistry_analysis (Accessed March 14, 2022).
Barberán, A., Ramirez, K. S., Leff, J. W., Bradford, M. A., Wall, D. H., and Fierer, N. (2014). Why are some microbes more ubiquitous than others? Predicting the habitat breadth of soil bacteria. Ecol. Lett. 17, 794–802. doi: 10.1111/ele.12282
Bell, C. W., Fricks, B. E., Rocca, J. D, Steinweg, J. M., McMahon, S. K., and Wallenstein, M. D. (2013). High-throughput fluorometric measurement of potential soil extracellular enzyme activities. JoVE J. Vis. Exp. e50961. doi: 10.3791/50961
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bonan, G. B. (2008). Forests and climate change: forcings, feedbacks, and the climate benefits of forests. Science 320, 1444–1449. doi: 10.1126/science.1155121
Borchers, N., Hannam, J., and Pawlett, M. (2022). Microbial community composition of translocated ancient woodland soil: a case study. Restor. Ecol. 30:e13618. doi: 10.1111/rec.13618
Chase, J. M., and Myers, J. A. (2011). Disentangling the importance of ecological niches from stochastic processes across scales. Philos. Trans. R. Soc. B Biol. Sci. 366, 2351–2363. doi: 10.1098/rstb.2011.0063
Chen, Q. L., Ding, J., Zhu, D., Hu, H. W., and Zhu, Y. G. (2019). Rare microbial taxa as the major drivers of ecosystem multifunctionality in long-term fertilized soils. Soil Biol. Biochem. 141:107686. doi: 10.1016/j.soilbio.2019.107686
Dan, N., Sadler, N., Bhattacharjee, A., Graham, E. B., and Jansson, J. K. (2020). Soil microbiomes under climate change and implications for carbon cycling. Annu. Rev. Environ. Resour. 45, 29–59. doi: 10.1146/annurev-environ-012320-082720
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Dixon, R. K., Solomon, A. M., Brown, S., Houghton, R. A., Trexier, M. C., and Wisniewski, J. (1994). Carbon pools and flux of global Forest ecosystems. Science 263, 185–190. doi: 10.1126/science.263.5144.185
Feng, Y., Chen, R., Stegen, J. C., Guo, Z., Zhang, J., Li, Z., et al. (2018). Two key features influencing community assembly processes at regional scale: initial state and degree of change in environmental conditions. Mol. Ecol. 27, 5238–5251. doi: 10.1111/mec.14914
Fernández-Calviño, D., and Bååth, E. (2010). Growth response of the bacterial community to pH in soils differing in pH. FEMS Microbiol. Ecol. 73, 149–156. doi: 10.1111/j.1574-6941.2010.00873.x
Gao, G.-F., Peng, D., Tripathi, B. M., Zhang, Y., and Chu, H. (2020). Distinct community assembly processes of abundant and rare soil bacteria in coastal wetlands along an inundation gradient. Microbial systems 5, e01150–e01120. doi: 10.1128/mSystems.01150-20
Goberna, M., and Verdú, M. (2016). Predicting microbial traits with phylogenies. ISME J. 10, 959–967. doi: 10.1038/ismej.2015.171
Horner-Devine, M. C., and Bohannan, B. J. M. (2006). Phylogenetic clustering and overdispersion in bacterial communities. Ecology 87, S100–S108. doi: 10.1890/0012-9658(2006)87[100:PCAOIB]2.0.CO;2
Hua, Z.-S., Han, Y.-J., Chen, L.-X., Liu, J., Hu, M., Li, S.-J., et al. (2015). Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 9, 1280–1294. doi: 10.1038/ismej.2014.212
Huang, J., Hu, B., Qi, K., Chen, W., Pang, X., Bao, W., et al. (2016). Effects of phosphorus addition on soil microbial biomass and community composition in a subalpine spruce plantation. Eur. J. Soil Biol. 72, 35–41. doi: 10.1016/j.ejsobi.2015.12.007
Ji, M., Kong, W., Stegen, J., Yue, L., Wang, F., Dong, X., et al. (2020). Distinct assembly mechanisms underlie similar biogeographical patterns of rare and abundant bacteria in Tibetan plateau grassland soils. Environ. Microbiol. 22, 2261–2272. doi: 10.1111/1462-2920.14993
Jiao, S., Chen, W., and Wei, G. (2017). Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol. Ecol. 26, 5305–5317. doi: 10.1111/mec.14218
Jiao, S., and Lu, Y. (2020a). Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields. Glob. Change Biol. 26, 4506–4520. doi: 10.1111/gcb.15130
Jiao, S., and Lu, Y. (2020b). Soil pH and temperature regulate assembly processes of abundant and rare bacterial communities in agricultural ecosystems. Environ. Microbiol. 22, 1052–1065. doi: 10.1111/1462-2920.14815
Jousset, A., Bienhold, C., Chatzinotas, A., Gallien, L., Gobet, A., Kurm, V., et al. (2017). Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 11, 853–862. doi: 10.1038/ismej.2016.174
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Li, C., Tu, B., Kou, Y., Wang, Y., Li, X., Wang, J., et al. (2021). The assembly of methanotrophic communities regulated by soil pH in a mountain ecosystem. Catena 196:104883. doi: 10.1016/j.catena.2020.104883
Liang, C., Schimel, J. P., and Jastrow, J. D. (2017). The importance of anabolism in microbial control over soil carbon storage. Nat. Microbiol. 2, 17105–17106. doi: 10.1038/nmicrobiol.2017.105
Liang, Y., Xiao, X., Nuccio, E. E., Yuan, M., Zhang, N., Xue, K., et al. (2020). Differentiation strategies of soil rare and abundant microbial taxa in response to changing climatic regimes. Environ. Microbiol. 22, 1327–1340. doi: 10.1111/1462-2920.14945
Luo, G., Rensing, C., Chen, H., Liu, M., Wang, M., Guo, S., et al. (2018). Deciphering the associations between soil microbial diversity and ecosystem multifunctionality driven by long-term fertilization management. Funct. Ecol. 32, 1103–1116. doi: 10.1111/1365-2435.13039
Lynch, M. D. J., and Neufeld, J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13, 217–229. doi: 10.1038/nrmicro3400
Marx, M.-C., Wood, M., and Jarvis, S. C. (2001). A microplate fluorimetric assay for the study of enzyme diversity in soils. Soil Biol. Biochem. 33, 1633–1640. doi: 10.1016/S0038-0717(01)00079-7
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Miller, E. T., Zanne, A. E., and Ricklefs, R. E. (2013). Niche conservatism constrains Australian honeyeater assemblages in stressful environments. Ecol. Lett. 16, 1186–1194. doi: 10.1111/ele.12156
Monteux, S., Keuper, F., Fontaine, S., Gavazov, K., Hallin, S., Juhanson, J., et al. (2020). Carbon and nitrogen cycling in Yedoma permafrost controlled by microbial functional limitations. Nat. Geosci. 13, 794–798. doi: 10.1038/s41561-020-00662-4
Mooshammer, M., Hofhansl, F., Frank, A. H., Wanek, W., Hämmerle, I., Leitner, S., et al. (2017). Decoupling of microbial carbon, nitrogen, and phosphorus cycling in response to extreme temperature events. Sci. Adv. doi: 10.1126/sciadv.1602781
Mori, H., Maruyama, F., Kato, H., Toyoda, A., Dozono, A., Ohtsubo, Y., et al. (2014). Design and experimental application of a novel non-degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Res. 21, 217–227. doi: 10.1093/dnares/dst052
Morris, A., Meyer, K., and Bohannan, B. (2020). Linking microbial communities to ecosystem functions: what we can learn from genotype–phenotype mapping in organisms. Philos. Trans. R. Soc. B Biol. Sci. 375:20190244. doi: 10.1098/rstb.2019.0244
Morrissey, E. M., Mau, R. L., Hayer, M., Liu, X.-J. A., Schwartz, E., Dijkstra, P., et al. (2019). Evolutionary history constrains microbial traits across environmental variation. Nat. Ecol. Evol. 3, 1064–1069. doi: 10.1038/s41559-019-0918-y
Nemergut, D. R., Costello, E. K., Hamady, M., Lozupone, C., Jiang, L., Schmidt, S. K., et al. (2011). Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 13, 135–144. doi: 10.1111/j.1462-2920.2010.02315.x
O’Malley, M. A. (2008). ‘Everything is everywhere: but the environment selects’: ubiquitous distribution and ecological determinism in microbial biogeography. Stud. Hist. Philos. Sci. Part C stud. Hist. Philos. Biol. Biomed. Sci. 39, 314–325. doi: 10.1016/j.shpsc.2008.06.005
Pan, H., Liu, H., Liu, Y., Zhang, Q., Luo, Y., Liu, X., et al. (2018). Understanding the relationships between grazing intensity and the distribution of nitrifying communities in grassland soils. Sci. Total Environ. 634, 1157–1164. doi: 10.1016/j.scitotenv.2018.04.117
Pedrós-Alió, C. (2012). The rare bacterial biosphere. Annu. Rev. Mar. Sci. 4, 449–466. doi: 10.1146/annurev-marine-120710-100948
Pester, M., Bittner, N., Deevong, P., Wagner, M., and Loy, A. (2010). A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 4, 1591–1602. doi: 10.1038/ismej.2010.75
Prodan, A., Tremaroli, V., Brolin, H., Zwinderman, A. H., Nieuwdorp, M., and Levin, E. (2020). Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS One 15:e0227434. doi: 10.1371/journal.pone.0227434
Rustad, L. E., Huntington, T. G., and Boone, R. D. (2000). Controls on soil respiration: implications for climate change. Biogeochemistry 48, 1–6. doi: 10.1023/A:1006255431298
Saladin, B., Thuiller, W., Graham, C. H., Lavergne, S., Maiorano, L., Salamin, N., et al. (2019). Environment and evolutionary history shape phylogenetic turnover in European tetrapods. Nat. Commun. 10:249. doi: 10.1038/s41467-018-08232-4
Shi, Y., Li, Y., Xiang, X., Sun, R., Yang, T., He, D., et al. (2018). Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China plain. Microbiome 6:27. doi: 10.1186/s40168-018-0409-4
Stegen, J. C., Lin, X., Fredrickson, J. K., Chen, X., Kennedy, D. W., Murray, C. J., et al. (2013). Quantifying community assembly processes and identifying features that impose them. ISME J. 7, 2069–2079. doi: 10.1038/ismej.2013.93
Thomas, M. K., Kremer, C. T., and Litchman, E. (2016). Environment and evolutionary history determine the global biogeography of phytoplankton temperature traits. Glob. Ecol. Biogeogr. 25, 75–86. doi: 10.1111/geb.12387
Tilman, D., Isbell, F., and Cowles, J. M. (2014). Biodiversity and ecosystem functioning. Annu. Rev. Ecol. Evol. Syst. 45, 471–493. doi: 10.1146/annurev-ecolsys-120213-091917
Tláskal, V., and Baldrian, P. (2021). Deadwood-inhabiting bacteria show adaptations to changing carbon and nitrogen availability during decomposition. Front. Microbiol. 12:685303. doi: 10.3389/fmicb.2021.685303
Trivedi, P., Delgado-Baquerizo, M., Trivedi, C., Hu, H., Anderson, I. C., Jeffries, T. C., et al. (2016). Microbial regulation of the soil carbon cycle: evidence from gene–enzyme relationships. ISME J. 10, 2593–2604. doi: 10.1038/ismej.2016.65
Wan, W., Gadd, G. M., Yang, Y., Yuan, W., Gu, J., Ye, L., et al. (2021a). Environmental adaptation is stronger for abundant rather than rare microorganisms in wetland soils from the Qinghai-Tibet plateau. Mol. Ecol. 30, 2390–2403. doi: 10.1111/mec.15882
Wan, W., Gadd, G. M., He, D., Liu, W., Xiong, X., Ye, L., et al. (2022). Abundance and diversity of eukaryotic rather than bacterial community relate closely to the trophic level of urban lakes. Environ. Microbiol. doi: 10.1111/1462-2920.16317
Wan, W., He, D., Li, X., Xing, Y., Liu, S., Ye, L., et al. (2021b). Linking rare and abundant phoD-harboring bacteria with ecosystem multifunctionality in subtropical forests: From community diversity to environmental adaptation. Sci. Total Environ. 796:148943. doi: 10.1016/j.scitotenv.2021.148943
Wan, W., Liu, S., Li, X., Xing, Y., Chen, W., and Huang, Q. (2021c). Bridging rare and abundant bacteria with ecosystem multifunctionality in salinized agricultural soils: from community diversity to environmental. ADA Forecast 6, e01221–e01220. doi: 10.1128/mSystems.01221-20
Wang, Y., Li, C., Kou, Y., Wang, J., Tu, B., Li, H., et al. (2017). Soil pH is a major driver of soil diazotrophic community assembly in Qinghai-Tibet alpine meadows. Soil Biol. Biochem. 115, 547–555. doi: 10.1016/j.soilbio.2017.09.024
Wang, J., Li, M., and Li, J. (2021). Soil pH and moisture govern the assembly processes of abundant and rare bacterial communities in a dryland montane Forest. Environ. Microbiol. Rep. 13, 862–870. doi: 10.1111/1758-2229.13002
Wei, X., Hu, Y., Razavi, B. S., Zhou, J., Shen, J., Nannipieri, P., et al. (2019). Rare taxa of alkaline phosphomonoesterase-harboring microorganisms mediate soil phosphorus mineralization. Soil Biol. Biochem. 131, 62–70. doi: 10.1016/j.soilbio.2018.12.025
Wemheuer, F., Taylor, J. A., Daniel, R., Johnston, E., Meinicke, P., Thomas, T., et al. (2020). Tax4Fun2: prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 15:11. doi: 10.1186/s40793-020-00358-7
Xu, M., Li, X., Kuyper, T. W., Xu, M., Li, X., and Zhang, J. (2021). High microbial diversity stabilizes the responses of soil organic carbon decomposition to warming in the subsoil on the Tibetan plateau. Glob. Change Biol. 27, 2061–2075. doi: 10.1111/gcb.15553
Zhang, L., Feng, G., and Declerck, S. (2018). Signal beyond nutrient, fructose, exuded by an arbuscular mycorrhizal fungus triggers phytate mineralization by a phosphate solubilizing bacterium. ISME J. 12, 2339–2351. doi: 10.1038/s41396-018-0171-4
Keywords: rare versus abundant bacteria, stochasticity versus determinism, carbon decomposition, environmental adaptability, environmental breadth, phylogenetic signal
Citation: Cao H, Li S, He H, Sun Y, Wu Y, Huang Q, Cai P and Gao C-H (2023) Stronger linkage of diversity-carbon decomposition for rare rather than abundant bacteria in woodland soils. Front. Microbiol. 14:1115300. doi: 10.3389/fmicb.2023.1115300
Edited by:
Jian Yang, China University of Geosciences Wuhan, ChinaReviewed by:
Yu Shi, Chinese Academy of Sciences, ChinaCopyright © 2023 Cao, Li, He, Sun, Wu, Huang, Cai and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-Hui Gao, Z2FvY2hAbWFpbC5oemF1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.