 Wenyuan Zhou1,2Yajie Li1Xuechao Xu1
Wenyuan Zhou1,2Yajie Li1Xuechao Xu1 Shengqi Rao1Hua Wen1Yeiling Han1Aiping Deng1Zhenwen Zhang3
Shengqi Rao1Hua Wen1Yeiling Han1Aiping Deng1Zhenwen Zhang3 Zhenquan Yang1*
Zhenquan Yang1* Guoqiang Zhu2*
Guoqiang Zhu2*- 1College of Food Science and Engineering, Yangzhou University, Yangzhou, Jiangsu, China
- 2College of Veterinary Medicine, Yangzhou University, Yangzhou, Jiangsu, China
- 3Medical College, Yangzhou University, Yangzhou, Jiangsu, China
Prophages as a part of Staphylococcus aureus genome contribute to the genetic diversity as well as survival strategies of their host. Some S. aureus prophages also have an imminent risk of host cell lysis and become a lytic phage. Nonetheless, interactions among S. aureus prophages, lytic phages, and their hosts, as well as the genetic diversity of S. aureus prophages, remain unclear. We identified 579 intact and 1,389 incomplete prophages in the genomes of 493 S. aureus isolates obtained from the NCBI database. The structural diversity and gene content of intact and incomplete prophages were investigated and compared with 188 lytic phages. Mosaic structure comparison, ortholog group clustering, phylogenetic analysis, and recombination network analysis were performed to estimate genetic relatedness among S. aureus intact prophages, incomplete prophages, and lytic phages. The intact and incomplete prophages harbored 148 and 522 distinct mosaic structures, respectively. The major difference between lytic phages and prophages was the lack of functional modules and genes. Compared to the lytic phages, both the S. aureus intact and incomplete prophages harbored multiple antimicrobial resistance (AMR) and virulence factor (VF) genes. Several functional modules of lytic phages 3_AJ_2017 and 23MRA shared more than 99% nucleotide sequence identity with S. aureus intact (ST20130943_p1 and UTSW_ MRSA_55_ip3) and incomplete prophages (SA3_LAU_ip3 and MRSA_FKTN_ip4); other modules showed little nucleotide sequence similarity. Ortholog and phylogenetic analyses revealed a common gene pool shared between the prophages and lytic Siphoviridae phages. Moreover, most shared sequences existed within intact (43428/137294, 31.6%) and incomplete prophages (41248/137294, 30.0%). Therefore, the maintenance or loss of functional modules in intact and incomplete prophages is key to balance the costs and benefits of large prophages harboring various AMR and VF genes in the bacterial host. The shared identical functional modules between S. aureus lytic phages and prophages are likely to result in the exchange, acquisition, and loss of functional modules, and therefore contribute to their genetic diversity. Moreover, constant recombination events within prophages globally were responsible for the coevolution of lytic phages and their bacterial hosts.
Introduction
The Staphylococcus aureus pathogen causes nosocomial and community-acquired infections in humans and animals owing to several immune resistance and evasion factors, toxins, and invasive mechanisms (Zhou et al., 2018; Guerillot et al., 2019). The threat of S. aureus can be attributed to a highly variable genome associated with phenotypic diversity and varying epidemiologic factors in different strains (Moon et al., 2015), with prophage genetic material accounting for 10–20% of the host genome (Johnson et al., 2022). Moreover, S. aureus can rapidly acquire antimicrobial resistance (AMR) and virulence factor (VF) genes via prophage transduction (Kondo et al., 2021). Studies on S. aureus prophage diversity at the nucleotide, structural, and genomic levels are necessary to understand the genomic plasticity and potential threats of S. aureus isolates.
Prophages originate from temperate bacteriophages (phages) that undergo a lysogenic cycle and integrate into the host chromosome, acting as a genetic reservoir and promoting diversity of their bacterial host (Ramisetty and Sudhakari, 2019; Johnson et al., 2022). A previous study indicated that distinct distribution of AMR and VF genes has been observed in intact and incomplete prophages in the genomes of seven pathogens, including 424 S. aureus isolates, derived from a public database (Kondo et al., 2021). Intact prophages can be characterized as genetic elements that can transmit VF genes and other genetic traits to S. aureus isolates (Moon et al., 2016). Despite the deletion of most phage genes, incomplete prophages or prophage-like elements occur stably in the genome of Lactococcus lactis IL1403 (Aucouturier et al., 2018). Nonetheless, the genetic relationship among lytic phages, intact prophages and incomplete prophages that is vital for elucidating the evolutionary relationships between S. aureus prophages and their bacterial host remains unclear.
Previously, the association of S. aureus prophages with increased bacterial pathogenicity and fitness has been examined (Kondo et al., 2021). The presence of different prophage-encoded VF and AMR genes among methicillin-resistant S. aureus (MRSA) strains enables them to produce a broad range of diseases (Dini et al., 2019). Furthermore, as genetic reservoirs, prophages promote diversity in hosts via enhanced recombination (Nadeem and Wahl, 2017). Besides, prophages mediate active horizontal gene transfer (HGT) via transduction, resulting in a large common gene pool shared by lytic phage genomes (Ramisetty and Sudhakari, 2019; Dion et al., 2020). Therefore, S. aureus prophages can remarkably influence the diversity and evolution of lytic phages and their hosts.
In this study, we identified 1968 prophages in 493 S. aureus isolate genomes and compared their sequences to those of 188 previously published lytic phages to (i) better elucidate structural diversity and gene content of S. aureus prophages, (ii) explore the genetic relationship between S. aureus intact and incomplete prophages, and (iii) understand the coevolutionary strategy of lytic phages and their hosts involving interaction with prophages.
Materials and methods
Collection of Staphylococcus aureus prophage metadata
The genomic sequences of 500 S. aureus isolates including 83 complete genome sequences and 417 incomplete genomes generated by high-throughput sequencing technologies were downloaded from the NCBI Genome database (April 2022). PATRIC was used to access the genome quality, as previously described (Johnson et al., 2022). In the assessment, 493 sequences derived from 58 countries globally (North America, South America, Asia, Europe, Africa, and Oceania) were selected (Supplementary Table S1). These strains were isolated from various samples (humans, n = 346; bovine, n = 39; porcine, n = 20; poultry, n = 12; sheep, n = 5; canine, n = 5; other origins, n = 27; unknown origin, n = 36). The cohort comprised genomes of 188 methicillin-sensitive S. aureus and 302 MRSA isolates. Phage sequences were predicted using the PHASTER software (Arndt et al., 2016) and confirmed by CheckV (Nayfach et al., 2021). The prophage regions were classified as intact (Supplementary Table S2) or incomplete (Supplementary Table S3) using the PHASTER criteria.
Prediction and annotation of Staphylococcus aureus isolates and prophages
The genomes of 493 S. aureus isolates, 1968 predicted prophages (Supplementary Tables S2, S3), and 188 lytic phages (previously published) (Zhou et al., 2022) were analyzed using FGENESB (Liu et al., 2016), Glimmer v3.02 (Delcher et al., 2007), and GeneMarkS (Borodovsky and Lomsadze, 2014) to predict open reading frames (ORFs). ORFs were annotated using the ‘NCBI non-redundant (nr) protein database’ (Tatusova et al., 2016), ‘Clusters of Orthologous Groups’ (Galperin et al., 2019), ‘InterProScan’ (Jones et al., 2014), and ‘eggNOG’ functions (Cantalapiedra et al., 2021). AMR and VF genes were predicted using the Comprehensive Antibiotic Resistance Database (mcmaster.ca) (CARD, version 4.0.0) (Alcock et al., 2020) and virulence factor database (VFDB) (Liu et al., 2019), respectively. The latest data involving all AMR and VF genes were downloaded from CARD1 and VFDB2 websites. BLAST was used for local analysis via blastn with a threshold of >80% identity and coverage. CRISPR arrays in S. aureus isolate, prophage, and lytic phage genomes, were detected and classified using CRISPRCasFinder (version 1.6.4) with default settings (Russel et al., 2020). Genes encoding tRNAs in prophages were screened using tRNAScan-SE (Chan and Lowe, 2019).
Phylogenetic analysis
Phylogenetic trees were constructed based on single-nucleotide polymorphisms (SNPs) to assess evolutionary relationships of S. aureus intact and incomplete prophages, respectively. Another phylogenetic analysis was performed to estimate the genetic relationship among the representative 148 intact prophages (Supplementary Table S4), 522 incomplete prophages (Supplementary Table S5), and 188 previously published lytic phages. Phylogenetic trees of S. aureus prophage and lytic phage genomes were generated using the Erwinia phage phiEa2809 as an outgroup. SNPs were determined for all genomes using the kSNP3 (version 3.1) software package (Gardner et al., 2015). The k-mer size estimated using the Kchooser software was set to 15 (Gardner et al., 2015). The SNP-based phylogenetic trees were constructed based on the Maximum Likelihood methods by RAxML (v 8.2.12) with 1,000 bootstraps and the GTRCAT nucleotide substitution model (Stamatakis, 2014). The phylogenetic trees were rooted using the outgroup and annotated using iTOL (Letunic and Bork, 2021). The mosaic structure of the S. aureus prophages was aligned using progressive MAUVE (Zhou et al., 2022).
Orthogroup clustering
The proteomes of 188 lytic phages, 579 intact prophages, and 1,389 incomplete prophages were used for orthogroup clustering (Emms and Kelly, 2019). In total, 93,038 protein sequences (lytic phages, 20,293; intact prophages, 38,809; incomplete prophages, 33,936) were clustered using OrthoFinder v2 with the default parameters. The resulting orthogroups were annotated using the ‘NCBI nr protein database’, ‘Clusters of Orthologous Groups’, ‘InterProScan’, ‘eggNOG’ functions, CARD, and VFDB.
Recombination network
The recombination events between the 188 lytic phages and 1968 prophages were estimated as previously described (Belcaid et al., 2010). Each sequence was queried against each other in the database using local BLAST. Homologous results with a nucleotide sequences identity of >95% were considered as the shared segments (Owen et al., 2017). The shared segments among intact prophages, incomplete prophages and lytic phages from different geographic were used to establish a worldwide recombination network as previously described (Göller et al., 2021).
Statistical analyses
Pearson’s chi-squared test (two-tailed) was performed to analyze the differences in the distribution of genes encoding for phage morphogenesis, DNA metabolism, host cell lysis, DNA packaging, lysogeny, AMR genes and VF among intact, incomplete, and lytic phages using SPSS software (version 26).
Results
Staphylococcus aureus intact and incomplete prophages exhibit distinct mosaic structures
In total, 493 S. aureus sequences deposited in GenBank were analyzed in this study (Supplementary Table S1). Except for eight isolates without any prophages, S. aureus genomes had 4.0 ± 0.2 prophages. A total of 1968 prophages, comprising 579 intact (Supplementary Table S2) and 1,389 incomplete prophage sequences (Supplementary Table S3), were identified. Except for 20 prophages harboring 4.5 ± 2.2 tRNA genes, 1948 prophages had no tRNA genes. The genome size of intact prophages was 48.8 ± 1.5 kb with 67 ± 2 ORFs (Supplementary Table S2). The G + C content of the intact prophages was 33.5 ± 0.2%. The genome size of incomplete prophages was 18.5 ± 0.6 kb with 24 ± 1 ORFs (Supplementary Table S3). The G + C content of incomplete prophages was 31.0 ± 0.2%.
We determined two phylogenies for 579 S. aureus intact and 1,389 incomplete prophages, respectively. Except for 8 singletons, the phylogenetic tree based on the 49,189 SNPs from genome sequences of 579 S. aureus intact prophages and the Erwinia phage phiEa2809 revealed 2 major genetic lineages (I–II; Supplementary Figure S1). Lineage I consisted of 35 intact prophages and lineage II concluded 536 intact prophages. However, statistical analyses based on the Pearson’s chi-square test showed no significant differences in the distribution of intact prophages from distinct geographic origin, host and isolation source between the two lineages (p > 0.05). Another phylogenetic tree based on the 292,450 SNPs from genome sequences of 1,389 S. aureus incomplete prophages and the Erwinia phage phiEa2809 revealed 51 singletons and 2 main lineages (I–II, Supplementary Figure S2). Lineage I consisted of 46 incomplete prophages and lineage II conclude 1,292 incomplete prophages. Consistently, there was no significant differences in the distribution of incomplete prophages from distinct geographic origin, host and isolation source between the two lineages (p > 0.05). The mosaic structure and gene content were compared across phylogenetic groups. Colinary and MARVE analyses revealed 148 mosaic structures among intact S. aureus prophages (Supplementary Table S4). Five mosaic structures (types CP1–CP5) were found in 5.9% (34/579), 4.7% (27/579), 4.7% (27/579), 4.1% (24/579), and 3.6% (21/579) of the intact prophages, respectively (Supplementary Table S4 and Supplementary Figures S3–S7). Among incomplete S. aureus prophages, 522 mosaic structures were identified (Supplementary Table S5). Five mosaic structures (IP1–IP5) were found in 3.4% (47/1389), 3.2% (45/1389), 3.2% (44/1389), 2.7% (38/1389), and 2.5% (35/1389) of incomplete prophages, respectively (Supplementary Table S5 and Supplementary Figures S8–S12).
All five intact prophages (CP1–CP5) typically consisted of genes associated with four functional modules: phage morphogenesis, host cell lysis, DNA packaging, and DNA metabolism (Figure 1). CP1 was consist of three functional regions: host cell lysis and phage morphogenesis, DNA packaging, and DNA metabolism. Despite CP2 and CP3 comprised only one module, this module included genes associated all four functions. CP4 comprised four functional regions including 2 DNA metabolism modules, 1 DNA packaging module, and 1 phage morphogenesis and host cell lysis module. CP5 comprised two functional regions: DNA metabolism, and phage morphogenesis and host cell lysis. However, incomplete prophages IP1–IP5 comprised only genes associated with phage morphogenesis, DNA metabolism, lysogeny, and virulence, but genes associated with host cell lysis and DNA packaging (Figure 2).
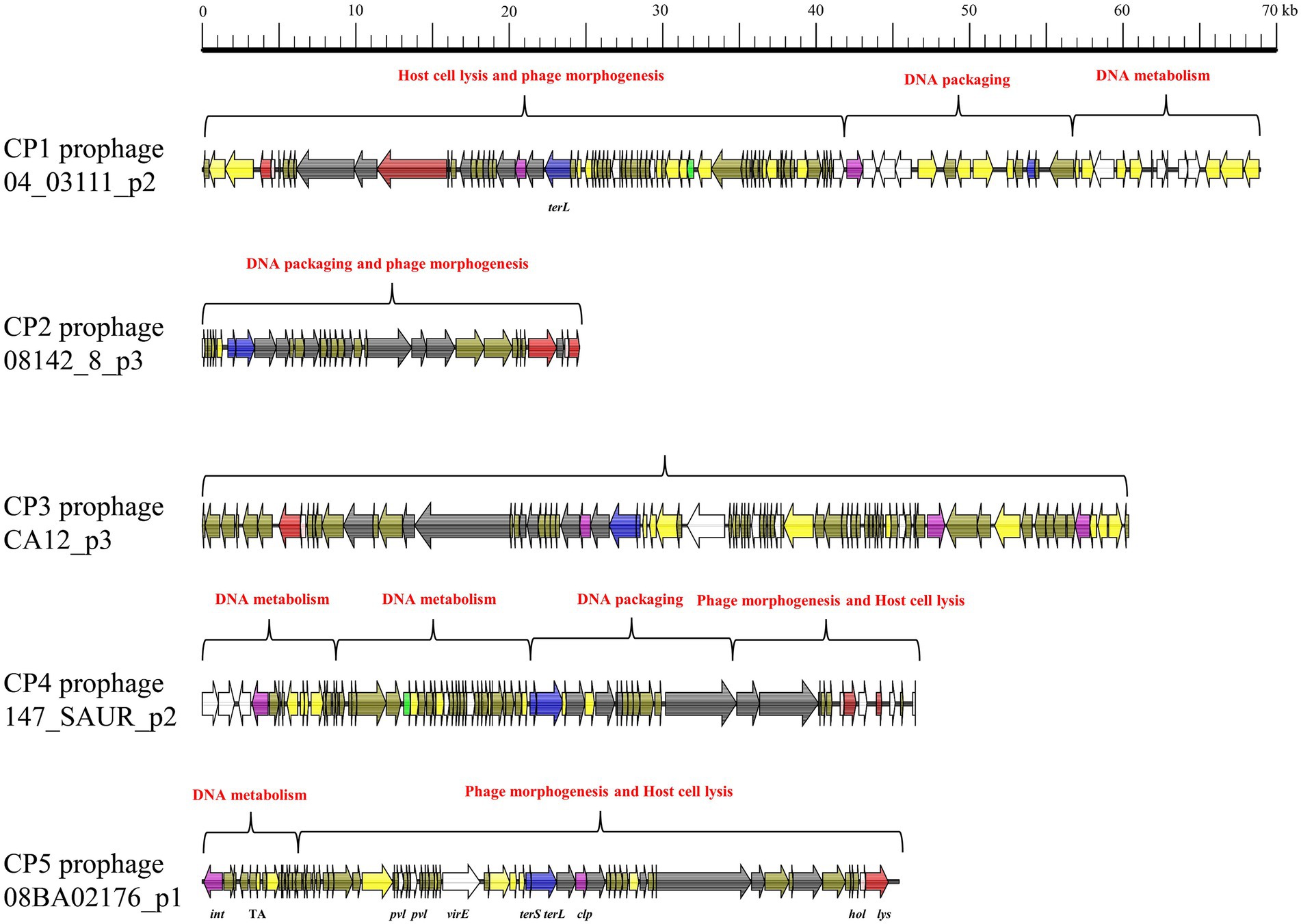
Figure 1. Mosaic pattern of the primary structure of intact prophages CP1–5. Functional modules are annotated with different colors. ORFs are represented by arrows indicating the transcription direction, and the colors of the arrows represent different genes. Gene color: virulence determinants, white; holin-encoding gene, pink; lysin-encoding gene, red; genes associated with lysogeny, purple; bla, green; DNA packaging genes, blue; DNA metabolism genes, yellow; and hypothetical protein genes, brown.
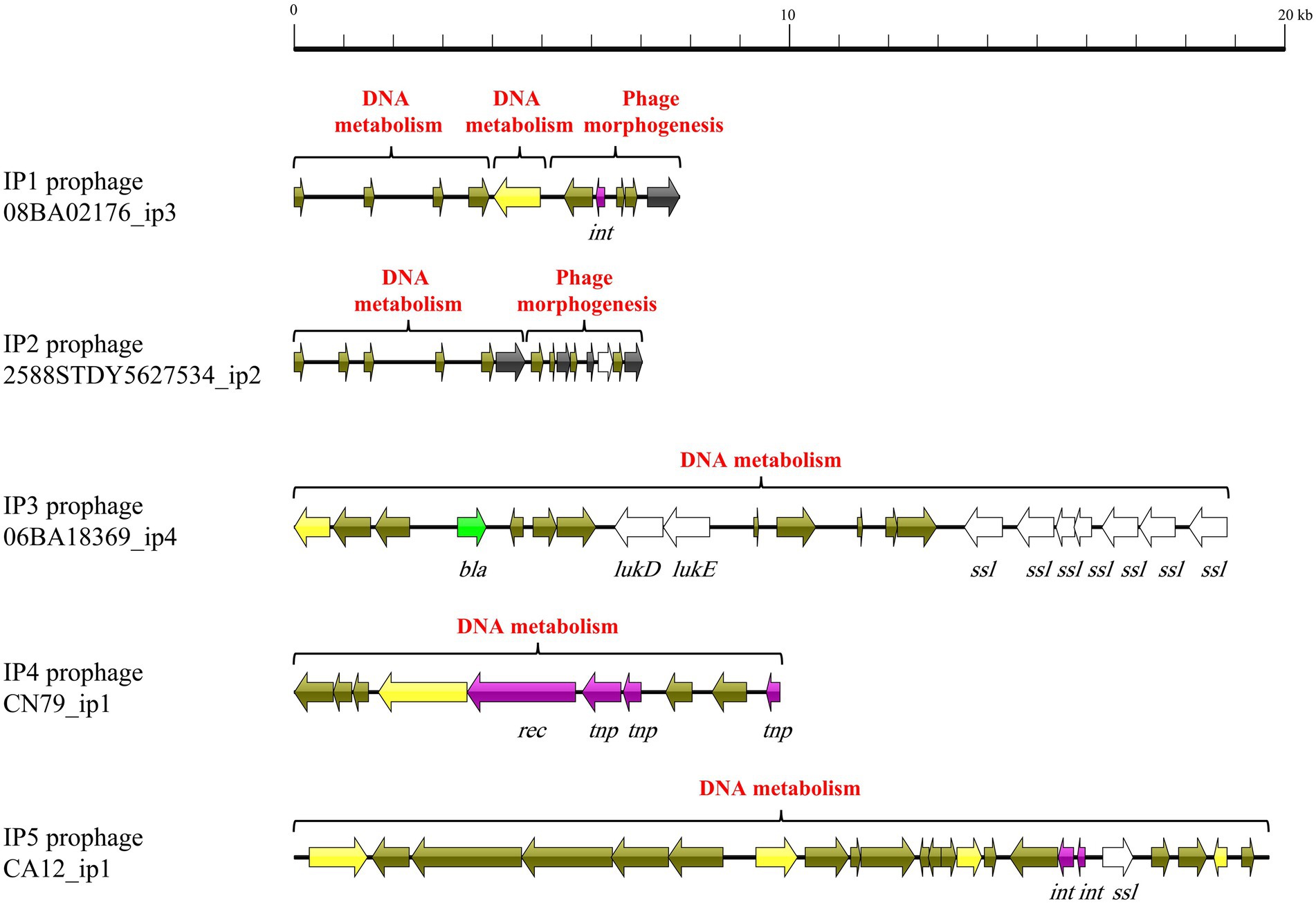
Figure 2. Mosaic structure of the main structure of incomplete prophages IP1–5.
Functional gene loss contributes to a major difference between intact and incomplete prophages
We compared the typical mosaic structures (Supplementary Tables S4, S5) of 148 intact and 522 incomplete prophages of S. aureus with those of 16 S. aureus lytic phages (Zhou et al., 2022). Genes encoding head and tail morphogenetic and portal proteins were found in all lytic phages (16/16; 100%; p ≤ 0.001; Figure 3). Such genes were found in 92/148 (62.2%), 125/148 (84.5%), and 121/148 (81.8%) intact prophages and 74/522 (14.2%), 72/522 (13.8%), and 62/522 (11.9%) incomplete prophages, respectively. Moreover, the prevalence of genes encoding the head-tail connector protein was much higher in lytic phages (37.5%, 6/16) than that in intact (18.9%, 28/148) and incomplete prophages (10.7%, 56/522; p ≤ 0.001). Genes encoding baseplate proteins were found in 50.0% (8/16) of lytic phages and 0.7% (1/148, p ≤ 0.001) of intact prophages. However, genes encoding tail tube proteins were more prevalent in intact prophages (37.2%, 55/148) than those in lytic phages (12.5%, 2/16) and incomplete prophages (8.2%, 43/522; p ≤ 0.001). As for host cell lysis module genes, lys and hol were more prevalent in lytic phages (lysin, 16/16, 100%; holin, 15/16, 93.8%) than those in intact prophages (lysin, 84/148, 56.8%; holin, 35/148, 23.6%) and incomplete prophages (lysin, 55/522, 10.5%; holin, 23/522, 4.4%; p ≤ 0.001). Genes encoding DNA metabolism and synthesis proteins, RNA polymerase, and type III restriction enzymes were only found in 5/16 (31.3%), 2/16 (12.5%), and 4/16 (25.0%) lytic phages, respectively. Genes encoding DNA helicase were considerably more prevalent in lytic phages (10/16, 62.5%) when compared with those in intact prophages (15/148, 10.1%) and incomplete prophages (34/522, 6.5%; p ≤ 0.001). Genes encoding DNA primase were only found in 11 genomes, including 31.3% (5/16), 1.4% (2/148), and 0.8% (4/522) of lytic phages, intact prophages, and incomplete prophages, respectively (p ≤ 0.001). The DNA packaging gene was more prevalent in lytic phages (16/16, 100%) than that in intact (15/148, 10.1%) and incomplete prophages (34/522, 22.0%; p ≤ 0.001). The lysogeny genes (rec, tnp and int) encoding for recombinase, transposase, and integrase were observed at the following rates: 16.8% (115/686), 17.3% (119/686), and 42.6% (292/686). The genes encoding Clp protease were found in 65 phages (lytic phages, 18.8%, 3/16; intact prophages, 30.4%, 45/148; and incomplete prophages, 3.3%, 17/522) (p ≤ 0.001).
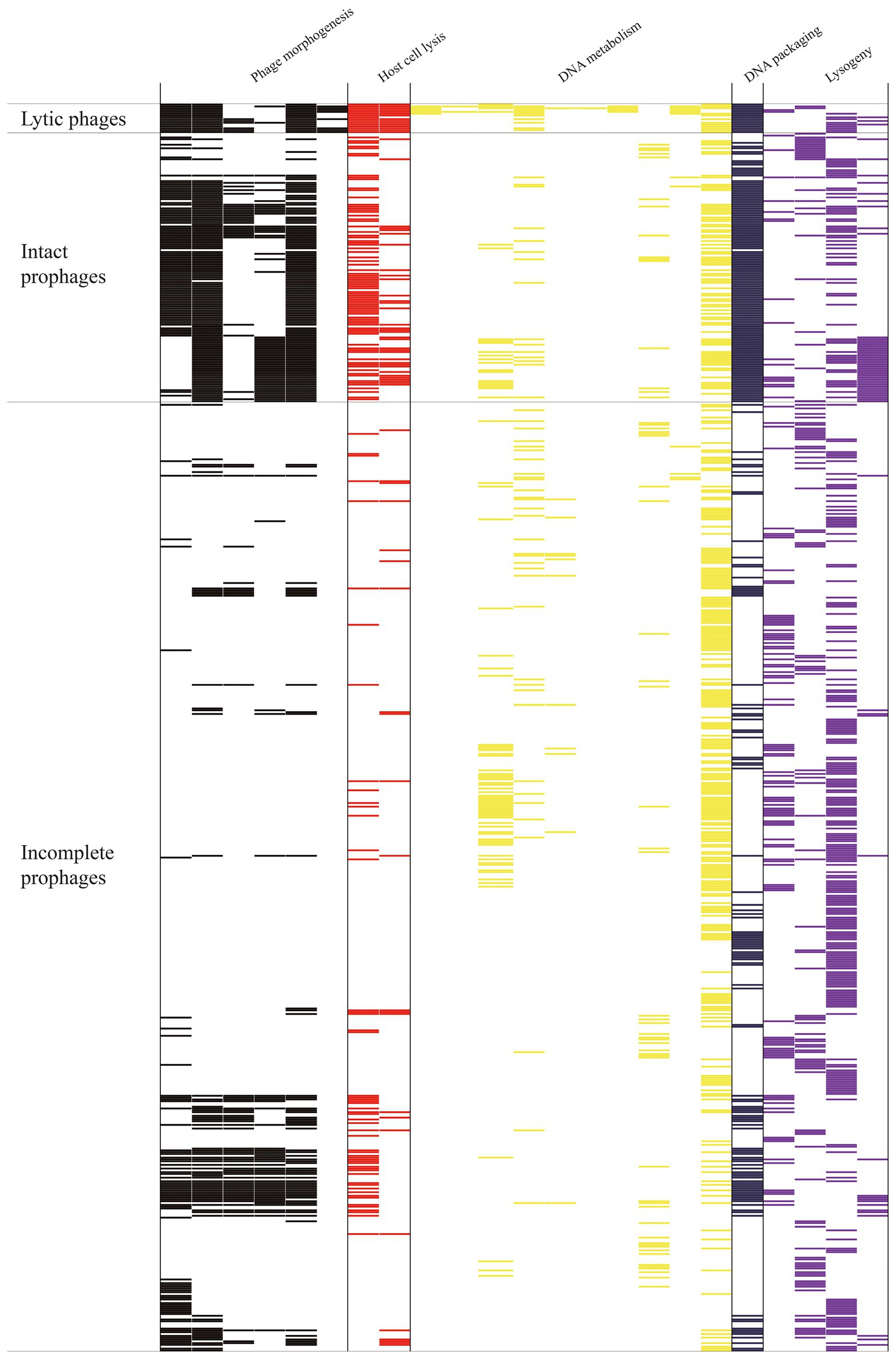
Figure 3. Heat map showing gene distribution associated with phage morphogenesis, host cell lysis, DNA metabolism, DNA packaging, and lysogeny of S. aureus 148 intact prophages, 522 incomplete prophages, and 16 lytic phages. Gene presence (colored blocks) or absence (white) is shown.
Staphylococcus aureus intact and incomplete prophages harbored multiple AMR and VF genes
AMR gene distribution was compared in 579 intact prophages, 1,389 incomplete prophages and their host genomes, respectively. Conferring resistance against 19 types of antimicrobials, 85 AMR genes were identified in these S. aureus isolates (Supplementary Table S6). Moreover, a total of 28 (32.9%, 28/85) and 26 (30.6%, 26/85) AMR genes were found in the genomes of intact and incomplete prophages, respectively. All of the AMR genes observed in the prophages were also found in their host genome. Notably, both intact and incomplete prophages harbored 20 genes associated with resistance against beta-lactamases, aminoglycosides, fluoroquinolones, trimethoprim, tetracycline, and multidrug use. Besides, the distribution of AMR genes among intact prophages (n = 148), incomplete prophages (n = 522), and lytic phages (n = 16) were also compared in this study (Supplementary Table S7). Two AMR genes were found in intact prophages (ermC, 2/148, 1.4%; mepA, 2/148, 1.4%), but absent in incomplete prophages and lytic phages (p ≤ 0.05). Moreover, 12 AMR genes were more frequently observed in tnp-carrying phages (blaZ, 4/119, 3.4%; aac6_Ie_aph2_Ia, 21/119, 17.6%; ant(6)-Ia, 12/119, 10.1%; ant(9)-Ia, 3/119, 2.5%; aph(2″)-Ia, 1/119, 0.8%; dfrC, 6/119, 5.0%; cat-TC, 1/119, 0.8%; tet(K), 2/119, 1.7%; mphC, 1/119, 0.8%; ermA, 3/119, 2.5%; ermC, 2/119, 1.7%; msrA, 1/119, 0.8%) than those in phages without tnp (blaZ, 3/567, 0.5%; aac6_Ie_aph2_Ia, 4/567, 0.7%; ant(6)-Ia, 3/567, 0.5%; ant(9)-Ia, 3/567, 0.5%; aph(2″)-Ia, 0/567, 0%; dfrC, 6/567, 1.1%; cat-TC, 0/567, 0%; tet(K), 1/567, 0.2%; mphC, 0/567, 0%; ermA, 3/567, 0.5%; ermC, 0/567, 0%; msrA, 0/567, 0%; p ≤ 0.05).
A total of 493 S. aureus isolates were screened for VF genes (Supplementary Table S6), and 146 VF genes associated with adherence (13.7%, 20/146), immune modulation (17.1%, 25/146), exoenzymes (12.3%, 18/146), effector delivery system (8.9%, 13/146), nutritional/metabolic factors (11.6%, 17/146), exotoxins (30.1%, 44/146), biofilms (4.8%, 7/146), stress survival (0.7%, 1/146), and antimicrobial activity/competitive advantage (0.7%, 1/146) were observed. Notably, 59.6% (87/146) and 60.3% (88/146) of VF genes were found in the genomes of intact and incomplete prophages, respectively. Both intact and incomplete prophages harbored 20 VF genes associated with adherence, immune modulation, exoenzymes, nutritional/metabolic factors, exotoxins, and biofilms. As shown in Supplementary Table S7, some VF genes were more prevalent in intact prophages (ebp, 6/148, 4.1%; fnbA, 5/148, 3.4%; sdrD, 8/148, 5.4%; sea, 6/148, 4.1%; seh, 2/148, 1.4%; set8, 5/148, 3.4%; set10, 2/148, 1.4%) than those in incomplete prophages (ebp, 2/522, 0.4%; fnbA, 3/522, 0.6%; sdrD, 8/522, 1.5%; sea, 4/522, 0.8%; seh, 0/522, 0%; set8, 3/522, 0.6%; set10, 0/522, 0%), but absent in lytic phages (p ≤ 0.05). However, gene encoding for staphylococcal enterotoxin A precursor (sea) was more prevalent in incomplete prophages (23/522, 4.4%) than that in intact prophages (1/148, 0.7%) and lytic phages (0/16, 0%; p ≤ 0.05).
Besides, 13 VF genes were more frequently detected in rec-harboring phages (ebp, 4/115, 3.5%; map, 1/115, 0.9%; sbi, 1/115, 0.9%; adsA, 2/115, 1.7%; isdA, 1/115, 0.9%; isdF, 1/115, 0.9%; isdG, 1/115, 0.9%; sfaD, 1/115, 0.9%; srtB, 1/115, 0.9%; hlgA, 1/115, 0.9%; sea, 4/115, 3.5%; selp, 2/115, 1.7%; set9, 1/115, 0.9%) than those in phages without gene encoding for recombinase (ebp, 4/571, 0.7%; map, 0/571, 0%; sbi, 0/571, 0%; adsA, 1/571, 0.2%; isdA, 0/571, 0%; isdF, 0/571, 0%; isdG, 0/571, 0%; sfaD, 0/571, 0%; srtB, 0/571, 0%; hlgA, 0/571, 0%; sea, 6/571, 1.1%; selp, 1/571, 0.2%; set9, 0/571, 0%; p ≤ 0.05). Moreover, 24 VF genes were more frequently detected in in tnp-containing phages (map, 1/119, 0.8%; fnbA, 8/119, 6.7%; fnbB, 4/119, 3.4%; atl, 1/119, 0.8%; sasA, 1/119, 0.8%; sdrC, 4/119, 3.4%; sdrD, 8/119, 6.7%; adsA, 2/119, 1.7%; splD, 10/119, 8.4%; splE, 9/119, 7.6%; splF, 11/119, 9.2%; sspA, 1/119, 0.8%; sspB, 1/119, 0.8%; esaG, 1/119, 0.8%; hla, 5/119, 4.2%; seg, 11/119, 9.2%; sei, 10/119, 8.4%; seln, 1/119, 0.8%; set4, 1/119, 0.8%; set5, 1/119, 0.8%; set7, 5/119, 4.2%; set8, 5/119, 4.2%; set9, 1/119, 0.8%; set10, 2/119, 1.7%) than those in the phages without tnp (map, 0/567, 0%; fnbA, 0/567, 0%; fnbB, 2/567, 0.4%; atl, 0/567, 0%; sasA, 0/567, 0%; sdrC, 5/567, 0.9%; sdrD, 8/567, 1.4%; adsA, 1/567, 0.2%; splD, 17/567, 3.0%; splE, 13/567, 2.3%; splF, 13/567, 2.3%; sspA, 0/567, 0%; sspB, 0/567, 0%; esaG, 0/567, 0%; hla, 4/567, 0.7%; seg, 14/567, 2.5%; sei, 14/567, 2.5%; seln, 0/567, 0%; set4, 0/567, 0%; set5, 0/567, 0%; set7, 3/567, 0.5%; set8, 3/567, 0.5%; set9, 0/567, 0%; set10, 0/567, 0%; p ≤ 0.05). Furthermore, 10 VF genes were more prevalent in int- harboring phages (eno, 8/292, 2.7%; geh, 12/292, 4.1%; hlb, 54/292, 18.5%; lukS-PV, 7/292, 2.4%; lukF-PV, 7/292, 2.4%; lukG, 53/292, 18.2%; lukH, 53/292, 18.2%; sec, 9/292, 3.1%; sell, 9/292, 3.1%; tsst-1, 8/292, 2.7%) than those in phages without gene encoding for integrase (eno, 3/394, 0.8%; geh, 1/394, 0.3%; hlb, 29/394, 7.4%; lukS-PV, 1/394, 0.3%; lukF-PV, 1/394, 0.3%; lukG, 5/394, 1.3%; lukH, 5/394, 1.3%; sec, 1/394, 0.3%; sell, 1/394, 0.3%; tsst-1, 1/394, 0.3%; p ≤ 0.05).
Staphylococcus aureus lytic phages and prophages shared identical functional modules
The mosaic structures of intact prophage ST20130943_p1, lytic phage 3_AJ_2017, and incomplete prophage SA3_LAU_ip3 were analyzed (Figure 4). The prophage ST20130943_p1 genome comprised 47,507 bp and three functional modules (host cell lysis module, phage morphogenesis and DNA packaging module, and DNA metabolism module). The host cell lysis module comprised eight ORFs with four virulence genes (hlb, scn, chp, and sak) and three host cell lysis genes (lys and hol). The phage morphogenesis and DNA packaging module comprised eight genes encoding phage morphogenesis proteins and two DNA packaging genes. The DNA metabolism module comprised four virulence genes (dut, hlb, lukG, and lukH) and one lysogeny gene (int). The lytic phage-3_AJ_2017 genome comprised 43,922 bp and three functional modules (host cell lysis module, phage morphogenesis and DNA packaging module, and DNA metabolism module). The DNA metabolism module comprised one lysogeny (int), one AMR gene (bla), and two virulence (dut and hlb) genes. The prophage SA3_LAU_ip3 genome comprised only 28,267 bp and two functional modules (phage morphogenesis and DNA packaging module, and host cell lysis module). Notably, region A of the intact prophage ST20130943_p1 comprised 27,929 bp and 35 ORFs, including two functional modules (host cell lysis module, and phage morphogenesis and DNA packaging module). This region showed more than 99.0% nucleotide sequence identity with region B in the lytic phage 3_AJ_2017 and region C in the incomplete prophage SA3_LAU_ip3.
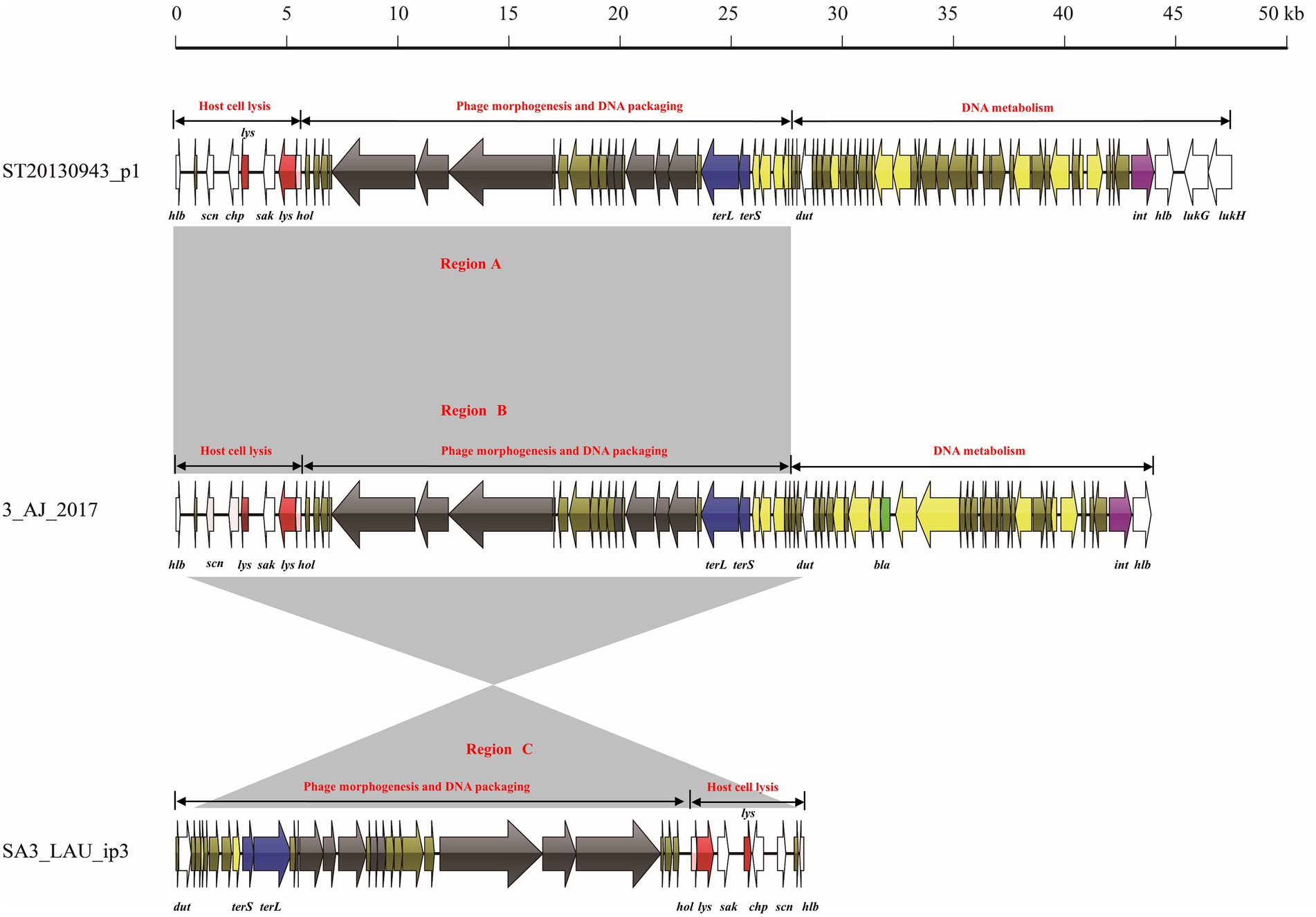
Figure 4. Comparative structural analyses of lytic phage-23MRA with intact prophage UTSW_MRSA_55_p3 and incomplete prophage MRSA_FKTN_ip4.
The mosaic structures of intact prophage UTSW_MRSA_55_p3, lytic phage 23MRA, and incomplete prophage MRSA_FKTN_ip4 were also compared (Figure 5). The incomplete prophage-UTSW_MRSA_55_p3 comprised 70,093 bp and four functional modules including three DNA metabolism modules, and one host cell lysis and phage morphogenesis module. The first DNA metabolism module comprised one virulence gene (map). The host cell lysis and phage morphogenesis module comprised one lysogeny gene (clp), two host cell lysis genes (lys and hol), seven phage morphogenesis genes, four virulence genes (hlb, scn, chp, and sak) and one DNA packaging gene (terL). The second DNA metabolism module comprised three virulence genes (dut, pvl and hlb), and one lysogeny gene (int). The third DNA metabolism module comprised two virulence genes (lukG, and lukH), and one DNA packaging gene (terS). The lytic phage 23MRA genome comprised two modules (DNA metabolism module, and host cell lysis and phage morphogenesis module). The DNA metabolism module comprised two virulence genes (dut, and pvl), and one lysogeny gene (int). The host cell lysis and phage morphogenesis module comprised two lysin genes (lys), one holin gene (hol), one lysogeny gene (clp), four virulence genes (hlb, scn, chp, and sak) and one DNA packaging gene (terL). The incomplete prophage-MRSA_FKTN_ip4 genome was 27,240 bp in size and comprised one DNA packaging gene (terL), one lysogeny gene (clp), two host cell lysis genes (hol and lys), and four virulence genes (sak, chp, scn, and hlb). Notably, region A in the UTSW_MRSA_55_p3 genome comprised one functional module (host cell lysis and phage morphogenesis module), which showed more than 99.0% nucleotide sequence identity with region D of the lytic phage 23MRA and region E of the incomplete prophage MRSA_FKTN_ip4. Moreover, region B in the UTSW_MRSA_55_p3 genome comprised another functional module (DNA metabolism module) and showed more than 99.0% nucleotide sequence identity with region C of the lytic phage 23MRA.
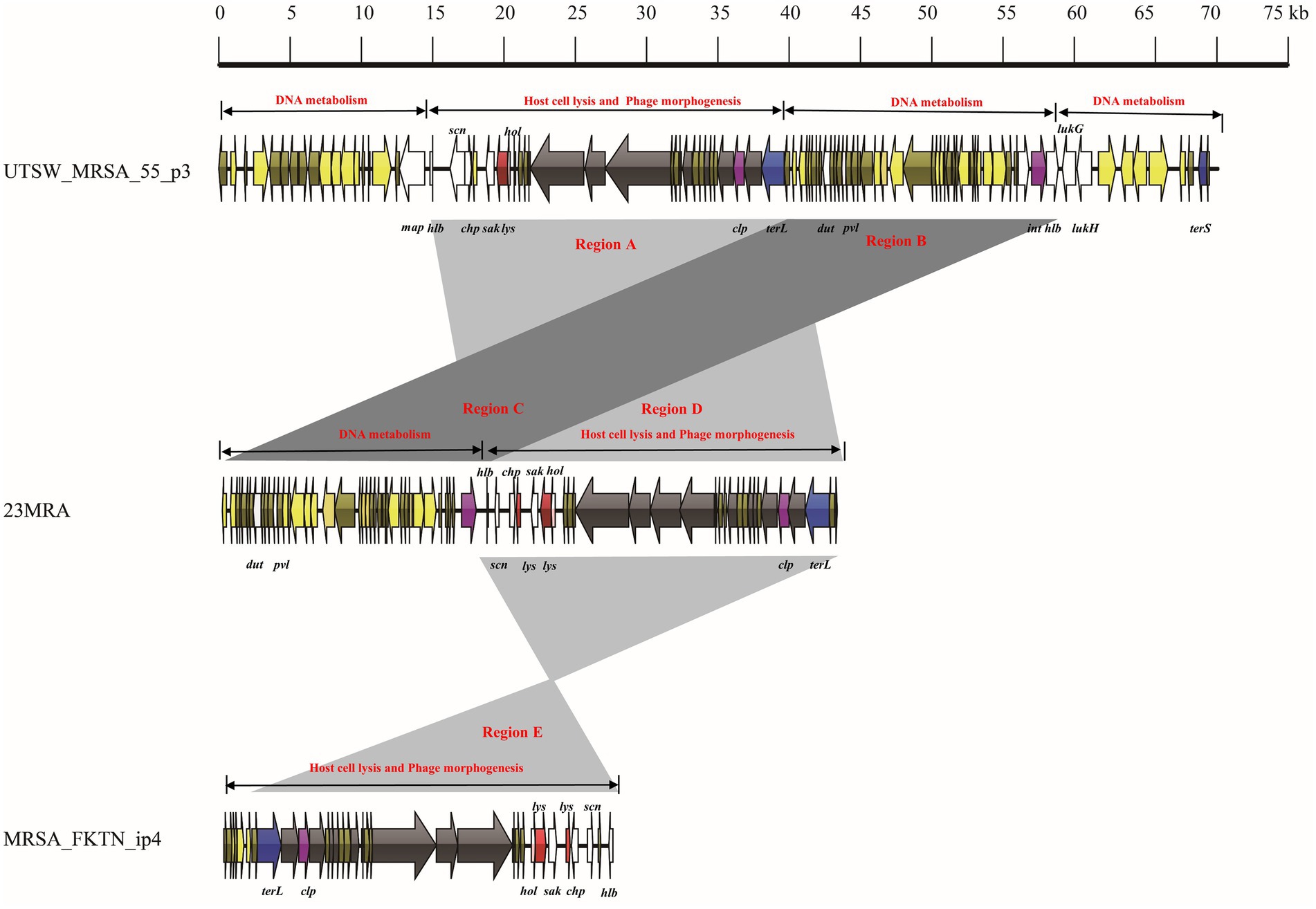
Figure 5. Comparative structural analyses of lytic phage-3_AJ_2017 with intact prophage ST20130943_p1 and incomplete prophage SA3_LAU_ip3.
High prevalence of recombination events among Staphylococcus aureus prophages promotes coevolution of the Staphylococcus aureus lytic phage and its host
Ortholog clusters were analyzed among 188 lytic phages, 579 intact prophages, and 1,389 incomplete prophages (Figure 6). Orthofinder revealed 26,800 orthogroups in these sequences. Notably, 2,466 orthogroups were found in S. aureus lytic phages including 1,219 orthogroups, which were also found in prophage genomes. Moreover, 834 orthogroups were found among all lytic phages and prophages, including genes encoding phage morphogenesis proteins (head proteins, 9; capsid proteins, 5; portal proteins, 5; head-tail joint proteins, 5; tail proteins, 21), host cell lysis (lysins, 2; holins, 8), DNA metabolism (DNA polymerases, 3; DNA-binding proteins, 15; DNA helicases, 2; type III restriction enzyme, 1), DNA packaging proteins, lysogeny proteins (recombinase, 1; transposases, 2; integrases, 12; Clp proteases, 2), and virulence factors (Staphylokinase, 2; beta-hemolysin, 2; Panton-Valentine leukocidin chain S precursor, 2; chemotaxis-inhibiting protein CHIPS, 1; complement inhibitor SCIN, 1; staphylococcal enterotoxin A precursor, 1; staphylococcal enterotoxin type P, 1; glycerol ester hydrolase, 1).
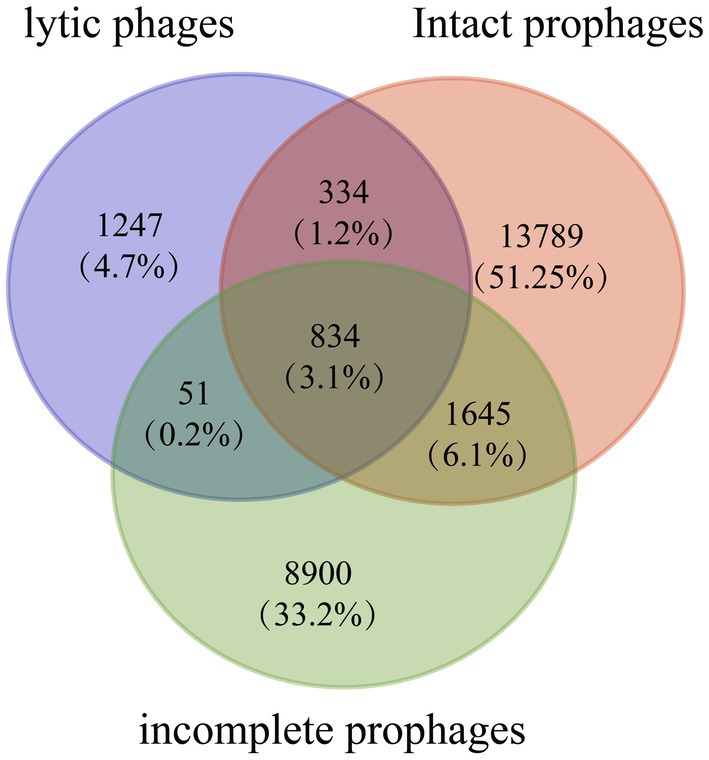
Figure 6. Venn diagrams showing the number of orthologous groups among 579 intact prophage sequences, 1,389 incomplete prophages, and 188 lytic phages.
Phylogenetic analysis was based on 439,973 SNPs from the genome sequences of 188 lytic phages, 148 representative intact prophages (Supplementary Table S4), 522 representative incomplete prophages (Supplementary Table S5), and the Erwinia phage phiEa2809. Except for five singletons, this tree revealed two major genetic lineages (I–II; Figure 7). The most ancestral sequences (singletons) comprised five lytic phages, including four Siphoviridae and one Herelleviridae phage. Lineage I comprised exclusively 79 lytic phages, including 19 and 60 phages belonging to Podoviridae and Herelleviridae, respectively. However, lineage II comprised the genomes of 148 intact prophages, 522 incomplete prophages, 101 Siphoviridae lytic phages, and three unclassified lytic phage genomes.
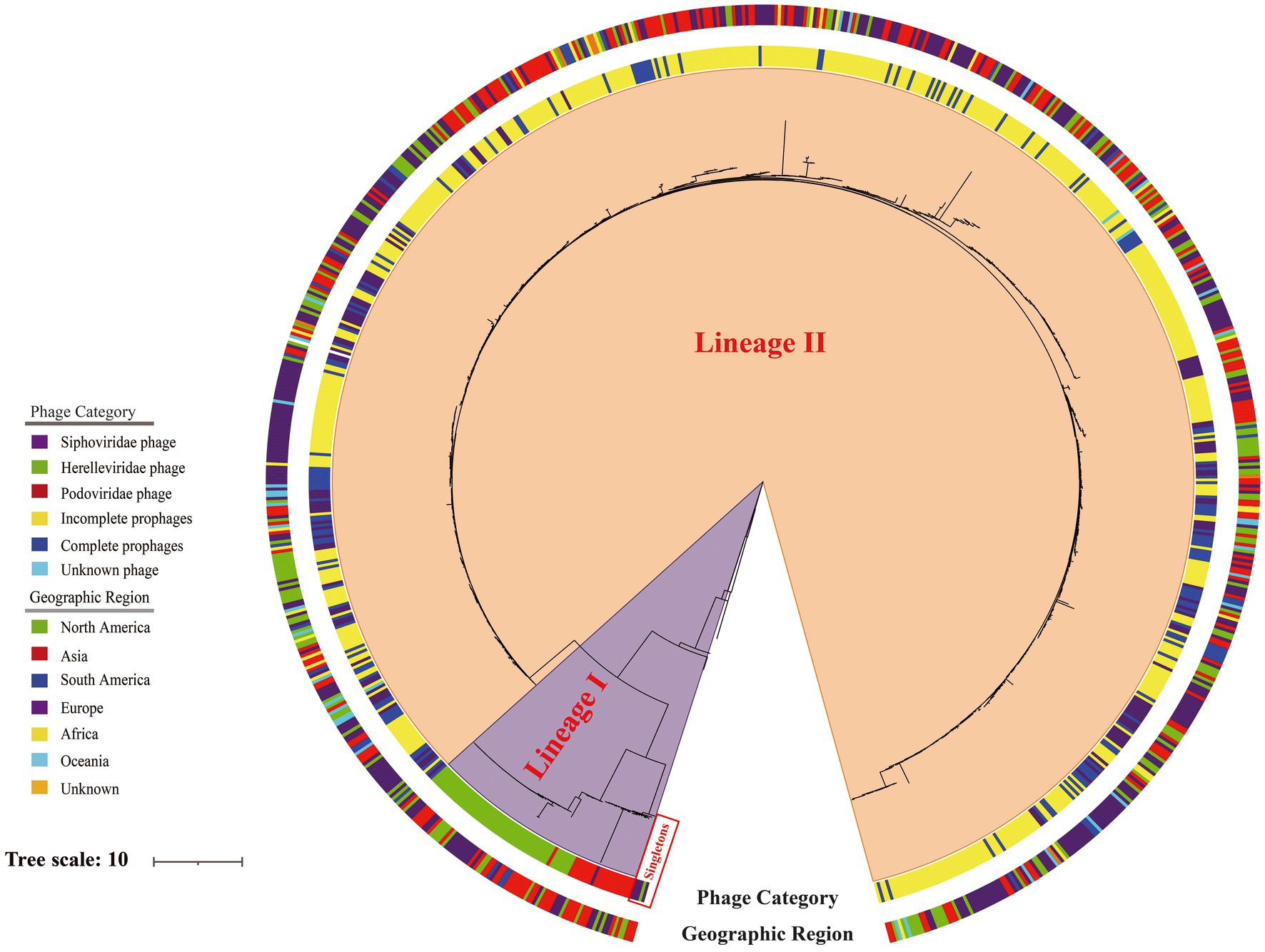
Figure 7. Phylogeny of 188 lytic phages, 148 representative intact prophages and 522 representative incomplete prophages analyzed using Erwinia phage phiEa2809 as the outgroup based on 439,973 single-nucleotide polymorphisms. The inner ring is colored according to type of the sequence and the outer ring according to the geographic region.
The recombination events were analyzed among intact prophages, incomplete prophages, and lytic phages (Figure 8). Phages and prophages from different geographic regions were represented as nodes, and the edges between the two nodes were colored based on the number of shared sequences. The BLAST algorithm revealed 137,294 shared segments with a query length of 5,003–86,192 bp among the 2,156 sequences. Most shared sequences existed within intact (43428/137294, 31.6%) and incomplete prophages (41248/137294, 30.0%). The predominant interplays occurred in 235 intact prophages (11347/137294, 8.3%) and 532 incomplete prophages (10099/137294, 7.4%) from Europe. Notably, 11.6% (15879/137294) of the shared sequences existed between lytic phages and prophages (intact prophages, 11571/137294, 8.4%; incomplete prophages, 4308/137294, 3.1%). Furthermore, predominant interplay occurred between the lytic phages from North America and intact prophages from Europe (1545/137294, 1.1%), and the lytic phages from Asia and intact prophages from Europe (1178/137294, 0.9%).
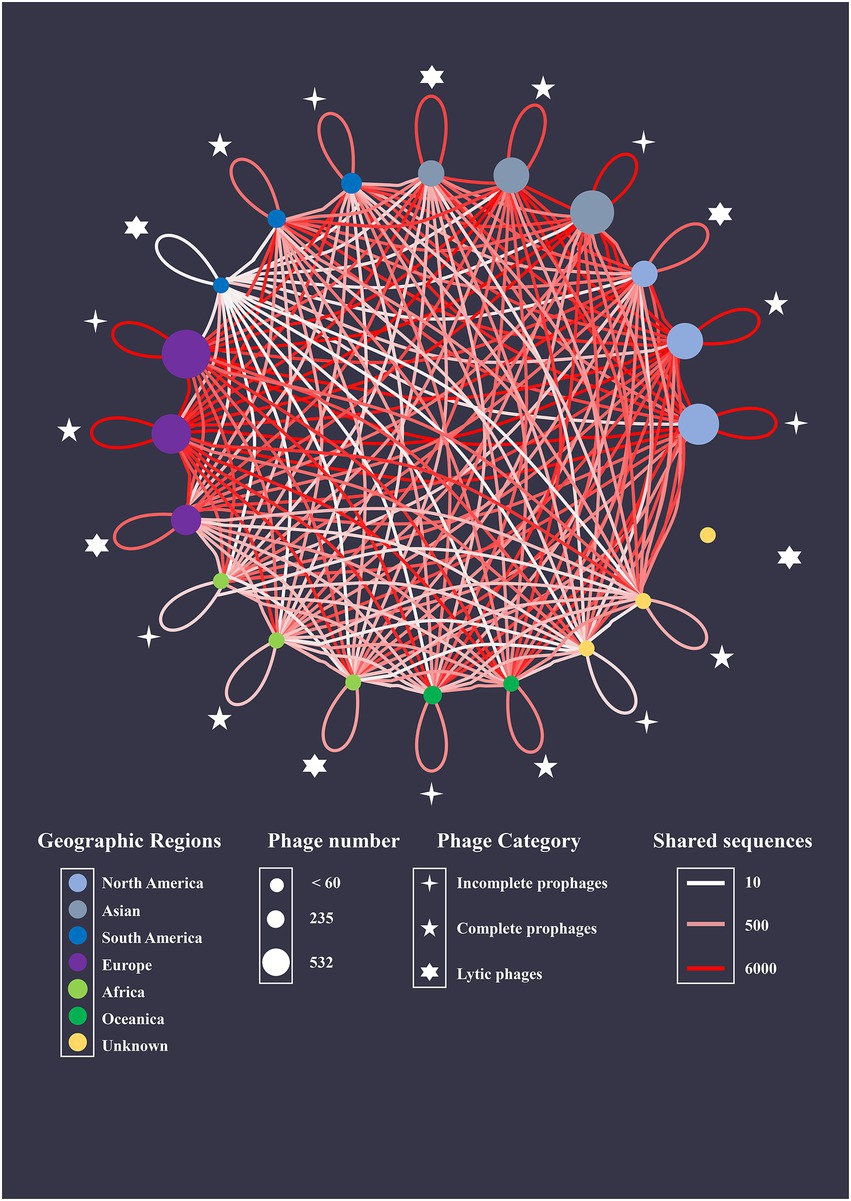
Figure 8. Genetic relationship network with shared sequences as coupling links. S. aureus intact prophage, incomplete prophages, and lytic phages isolated from different geographic regions are represented as nodes. The area of each node directly correlates with the average number of shared sequences connected to a species. The number of shared sequences between nodes is represented by colored edges.
Besides, we assessed the distribution of genes associated with CRISPR-Cas in 493 S. aureus isolates, 1968 prophages, and 188 lytic phages. Notably, only 7 S. aureus isolates (08BA02167, APS210, APS211, JS395, M1169, M1216, and ST1358) harbored type III-A CRISPR-Cas proteins and operons. However, none of the CRISPR-Cas proteins were predicted in the prophages and lytic phages.
Discussion
Prophages play potent roles in the pathogenicity, fitness and diversity of their S. aureus hosts via HGT. However, the genetic diversity of prophages at the nucleotide, structural, and genomic levels remains unknown. The both phylogenies for S. aureus intact and incomplete prophages revealed that prophages from distinct geographic origin and host were clustered together, indicating the genetic diversity of the S. aureus prophages. Besides, our previous study analyzed the genetic diversity of 188 S. aureus lytic phages and observed 16 mosaic structures (Zhou et al., 2022), while in this study, we identified as many as 670 distinct mosaic structures (148 intact and 522 incomplete prophages) in the genomes of prophages, indicating the extraordinary structural diversity of S. aureus prophages. The transduction of temperate bacteriophage phiSaBov is accompanied by mobilization of the prophages vSaα, vSaβ, and vSaγ (Moon et al., 2015, 2016). Therefore, distinct mosaic structures of intact and incomplete prophages of S. aureus promote genomic plasticity in hosts and genetic diversity in S. aureus lytic phages. Intact and incomplete prophages exhibited distinct mosaic structures, which was consistent with those of a previous study indicating that mosaicism of phage genomes is perhaps one of the few common features (Dion et al., 2020). Our previous study revealed that the mosaic structure that harbored the most VF genes among the 16 main structures of S. aureus lytic phages was the clade IIIa (hlg, pvl, dut, and virE), clade IIIb (dut, pvl, scn, and sak) and clade IIIc (hlb, sak, and dut) phages (Zhou et al., 2022). However, higher prevalence of the VF genes was observed in the genomes of CP1 (dut, hlb, lukG, lukH, and hld), CP3 (virE, dut, lukH, lukG, hlb, scn and sak), CP4 (lukH, lukG, hlb, scn, sak, and dut), and IP3 (splA, splB, splC, lukD, lukE, seg, seln, yent1, yent2, sei, selm and selo). Therefore, the S. aureus prophages probably contributes to the transmission of VF genes in their hosts.
The major difference of the major mosaic structures between lytic phages, and the intact and incomplete prophages was the functional modules as well as genes. The 16 major mosaic structures of lytic phages normally harbored four functional modules and other genes associated with the VF and lysogeny (Zhou et al., 2022). However, the intact and incomplete prophages were lack of functional genes associated with typical phage modules including phage morphogenesis, host cell lysis, DNA metabolism, and DNA packaging. These results were inconsistent with those of previous studies, suggesting that large-scale deletions occurred in the accessory genes of prophages such as integrase and cargo genes, rather than conserved genes involved in lytic gene repression, replication, expression of capsid proteins, and packaging (Ramisetty and Sudhakari, 2019). S. aureus isolates harboring large prophages and related genes impose an extra metabolic burden but may promote bacterial adaptation and habitat colonization (Fillol-Salom et al., 2019; Ramisetty and Sudhakari, 2019). Therefore, we hypothesized that the maintenance or loss of functional genes is key to balancing the costs and benefits of large prophages, thus expediting bacterial genome evolution. Besides, phage-induced lysis involves the destruction of the inner membrane, peptidoglycan layer, and outer membrane of the host cell through the phage encoded holin and lysin (Young, 2014). Double-stranded DNA bacteriophages package their genome at high pressure inside a procapsid through the DNA packaging proteins (Cuervo et al., 2019). These results indicated the loss of functional modules in prophages is the possible mechanism to ground the phages and, thus protect the S. aureus isolates from the lysis cycle of phages, which is consistent with a previous study (Bernheim and Sorek, 2020).
AMR in S. aureus is a serious growing problem associated with enormous human and medical costs, and resistance transmission is promoted by mobile genetic elements (Allen et al., 2010). Furthermore, S. aureus can survive robust activation of the host’s innate immune system as a result of the expression of a wide array of VFs, disabling the acquired immune response (Goldmann and Medina, 2018). Compared to the only 1 AMR gene (bla) and 5 VF genes observed in the genomes of 188 lytic phages (Zhou et al., 2022), this study revealed various AMR and VF genes identified in the genomes of S. aureus prophages. These results indicates that prophages act as a genetic reservoir of AMR and VF for S. aureus and that transduction is the major means of HGT for AMR and VF, consistent with the results of a recent study (Kondo et al., 2021). Over the past 75 years, beta-lactams have been the most important antibiotics used in treating S. aureus infections (Vestergaard et al., 2019). A previous study surveyed global antibiotic consumption from 2000 to 2010 and suggested that antimicrobial drugs, such as aminoglycosides, fluoroquinolones, trimethoprim, and tetracycline, were the most abundantly consumed antibiotics globally (Van Boeckel et al., 2014). S. aureus strains that secrete exotoxins and exoenzymes can disrupt endothelial and epithelial barriers via cell lysis and junction protein cleavage (Tam and Torres, 2019). Adhesins allow S. aureus to attach to host cells and the extracellular matrix during the early colonization phase (Negron et al., 2022). Nutritional/metabolic factors are critical for the fitness of S. aureus and its adaptation to nutritionally diverse environments (Balasubramanian et al., 2017). The formation of S. aureus biofilms in vivo renders it highly resistant to chemotherapeutics, leading to chronic diseases (Paharik and Horswill, 2016). Hence, intact and incomplete prophages of S. aureus contribute to adaptation to the host environment and the severity of S. aureus infections.
In addition, our analysis revealed that the prevalence of several AMR and VF genes in the intact prophages were significantly higher than those in incomplete prophages and lytic phages. These results suggested the easily spreading of AMR and virulence via prophage transduction, which was consistent with previous studies (Moon et al., 2015, 2016). Besides, the numbers of some AMR and VF genes in lysogeny genes (rec, tnp, and int) harboring phages were significantly higher than those in phages without lysogeny genes, which is inconsistent with a previous study revealing that prophages containing VF-encoding genes are not likely to possess recombination-related genes near to VF genes (Kondo et al., 2021). These results indicated that the AMR genes and VF in the prophages may be acquired by various mechanism as well as phage transduction.
Phage 3-AJ-2017, isolated in Colombia, belongs to the Siphoviridae family. The host cell lysis module, and phage morphogenesis and DNA packaging module of phage 3-AJ-2017 shared high nucleotide sequence identity with an intact prophage in S. aureus strain ST20130943 isolated from humans in Brazil and an incomplete prophage in S. aureus strain SA3_LAU isolated from humans in Lebanon. However, the DNA metabolism module of phage-3-AJ-2017 shared little nucleotide sequence identity with ST20130943_p1. Consistently, the host cell lysis and phage morphogenesis module of phage 23MRA were similar to those of an intact prophage in S. aureus strain UTSW_MRSA_55 and an incomplete prophage in S. aureus strain MRSA_FKTN isolated from humans in the United States. The DNA metabolism module of phage 23MRA shared high nucleotide sequence identity with UTSW_MRSA_55_p3. However, the genome of UTSW_MRSA_55_p3 harbored other genes associated with DNA metabolism. These results revealed the identical functional modules between the lytic phages and prophages, which is likely to result in the easily homologous recombination events between the lytic phages and prophages (Dion et al., 2020). A previous study indicated that extensive mosaicism with genes organized into functional modules that are frequently exchanged between phages because they coexist in a common host (Deghorain and Van Melderen, 2012). Therefore, exchange, acquisition, and loss of functional modules resulting from homologous recombination may augment the genetic diversity of the lytic phages, prophages and their hosts. However, a limitation of this study can be attributed to the numbers of S. aureus lytic phage genomes analyzed, therefore a more accurate determination of the genetic relationship between the prophage and lytic phage is required.
Consistently, ortholog and phylogenetic analyses indicated that prophages and lytic phages shared a common core gene pool and therefore could easily exchange, acquire, and lose genetic material in the host genomes. In the phylogenetic tree, the prophages were exclusively clustered with the S. aureus lytic Siphoviridae phages, indicating that the Siphoviridae lytic phage and prophages shared a common ancestor. These results suggest that a broad recombination events probably occurred between prophages and S. aureus Siphoviridae phages, consistent with the previous study (Deghorain and Van Melderen, 2012). The phylogenetic tree obtained via whole-genome phylogenetic analysis revealed that the S. aureus prophages and lytic Siphoviridae phages from different regions belonged to the same lineage, indicating that the recombination events between S. aureus prophages and lytic Siphoviridae phages occurred globally. However, future research should elucidate the taxonomic classification of these prophages and their evolutionary relationship with Siphoviridae phages based on their genetic features.
The worldwide genome interaction network revealed the majority of shared sequences in S. aureus prophages, representing the complex recombination events may easily occur in these prophages (Belcaid et al., 2010). Recombination events, called HGT, are major mediators of phage evolution and render the understanding of evolutionary relationships difficult (Dion et al., 2020). Therefore, frequent recombination events within S. aureus prophages resulting from homologous recombination promoted the genetic diversity of S. aureus isolates as well as prophages. Consistent with these results, region A in the genome of 09_01244_p4 comprised 43,514 bp and shared 99.0% nucleotide sequence identity with region B in the S. aureus prophage BSAR202_p1 (Supplementary Figure S13). Moreover, region B in the MAL9_ip4 genome comprised 29,477 bp and shared 98.0% nucleotide sequence identity with region D in the prophage MRGR3_ip9 (Supplementary Figure S14). Incomplete prophages that were considered genetic junks confer advantageous phenotypes to bacterial hosts with respect to virulence, stress resistance, or mutation rate (Ramisetty and Sudhakari, 2019). The present study illustrates that incomplete prophages can also be reservoirs of genetic material for lytic phages. These results identified that the constant recombination between S. aureus prophages in host chromosomes and lytic phages tightly links their evolution and indirectly accelerates bacterial evolution. However, future studies should elucidate the exact mechanism underlying recombination events among S. aureus intact prophages, incomplete prophages and lytic phages.
The CRISPR-Cas system is an important constraint for HGT, and ensuring the maintenance or loss of prophages is key to the environmental adaption of this pathogen (Wheatley and MacLean, 2021). The non-specific DNase activity of the staphylococcal type III-A CRISPR-Cas system contributes to mutation rates in bacteria and AMR in S. aureus and S. epidermidis (Mo et al., 2021). Therefore, our study suggested that type III-A CRISPR-Cas proteins may mediate the genetic diversity of S. aureus prophages as well as their hosts. Considering the low detection rate of CRISPR-Cas genes in S. aureus genomes, recombination within S. aureus prophages via HGT was responsible for the genetic diversity and coevolution in S. aureus prophages, lytic phages, and their hosts.
Conclusion
In summary, our analysis suggests that intact and incomplete S. aureus prophages exhibited distinct mosaic structures, and the major difference between lytic phages, and intact and incomplete prophages was the loss of functional modules and genes. Compared to the lytic phages, both the S. aureus intact and incomplete prophages harbored multiple AMR and VF genes and contributed to the environmental adaption of its hosts and infection severity. Hence, the maintenance or loss of functional modules in intact and incomplete prophages is key to balance the costs and benefits of large prophages harboring various AMR and VF genes in the bacterial host. Besides, exchange, acquisition, and loss of functional modules between lytic phages and prophages resulting from homologous recombination contribute to the genetic diversity of S. aureus prophages. Constant recombination within prophages globally were responsible for the genetic diversity of S. aureus lytic phages and their host. Through these evolutionary strategies, the S. aureus prophage accelerates the evolution itself and the coevolution of lytic phages and their bacterial hosts.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
WZ and ZY generated the research and critically reviewed the final version of manuscript. WZ and GZ designed and conceptualized the research. WZ and YL performed the research. HW, YH, and AD analyzed the data. XX, SR, and ZZ supervised and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (grant numbers: 32102100 and 32001661), the China Postdoctoral Science Foundation (grant number: 2022M712695), Natural Science Foundation of Jiangsu Province, China (grant number: BK20190890), Jiangsu Key Research & Development Program (grant number: BE2019436-5), Yangzhou University food quality and safety talents project (grant number: YZUJX2020-A3), and Yangzhou city talents project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1088125/full#supplementary-material
Footnotes
References
Alcock, B. P., Raphenya, A. R., Lau, T., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Allen, H. K., Donato, J., Wang, H. H., Cloud-Hansen, K. A., Davies, J., and Handelsman, J. (2010). Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 8, 251–259. doi: 10.1038/nrmicro2312
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aucouturier, A., Chain, F., Langella, P., and Bidnenko, E. (2018). Characterization of a prophage-free derivative strain of Lactococcus lactis ssp. Lactis IL1403 reveals the importance of prophages for phenotypic plasticity of the host. Front. Microbiol. 9:2032. doi: 10.3389/fmicb.2018.02032
Balasubramanian, D., Harper, L., Shopsin, B., and Torres, V. J. (2017). Staphylococcus aureus pathogenesis in diverse host environments. Pathog. Dis. 75:ftx0005. doi: 10.1093/femspd/ftx0005
Belcaid, M., Bergeron, A., and Poisson, G. (2010). Mosaic graphs and comparative genomics in phage communities. J. Comput. Biol. 17, 1315–1326. doi: 10.1089/cmb.2010.0108
Bernheim, A., and Sorek, R. (2020). The pan-immune system of bacteria: Antiviral defence as a community resource. Nat. Rev. Microbiol. 18, 113–119. doi: 10.1038/s41579-019-0278-2
Borodovsky, M., and Lomsadze, A. (2014). Gene identification in prokaryotic genomes, phages, metagenomes, and EST sequences with GeneMarkS suite. Curr. Protoc. Microbiol. 32:Unit 1E.7. doi: 10.1002/9780471729259.mc01e07s32
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). EggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
Chan, P. P., and Lowe, T. M. (2019). TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Cuervo, A., Fàbrega-Ferrer, M., Machón, C., Conesa, J. J., Fernández, F. J., Pérez-Luque, R., et al. (2019). Structures of T7 bacteriophage portal and tail suggest a viral DNA retention and ejection mechanism. Nat. Commun. 10:3746. doi: 10.1038/s41467-019-11705-9
Deghorain, M., and Van Melderen, L. (2012). The staphylococci phages family: An overview. Viruses 4, 3316–3335. doi: 10.3390/v4123316
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
Dini, M., Shokoohizadeh, L., Jalilian, F. A., Moradi, A., and Arabestani, M. R. (2019). Genotyping and characterization of prophage patterns in clinical isolates of Staphylococcus aureus. BMC. Res. Notes 12:669. doi: 10.1186/s13104-019-4711-4
Dion, M. B., Oechslin, F., and Moineau, S. (2020). Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138. doi: 10.1038/s41579-019-0311-5
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Fillol-Salom, A., Alsaadi, A., Sousa, J., Zhong, L., Foster, K. R., Rocha, E., et al. (2019). Bacteriophages benefit from generalized transduction. PLoS Pathog. 15:e1007888. doi: 10.1371/journal.ppat.1007888
Galperin, M. Y., Kristensen, D. M., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2019). Microbial genome analysis: The COG approach. Brief. Bioinform. 20, 1063–1070. doi: 10.1093/bib/bbx117
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). KSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Goldmann, O., and Medina, E. (2018). Staphylococcus aureus strategies to evade the host acquired immune response. Int. J. Med. Microbiol. 308, 625–630. doi: 10.1016/j.ijmm.2017.09.013
Göller, P. C., Elsener, T., Lorgé, D., Radulovic, N., Bernardi, V., Naumann, A., et al. (2021). Multi-species host range of staphylococcal phages isolated from wastewater. Nat. Commun. 12:6965. doi: 10.1038/s41467-021-27037-6
Guerillot, R., Kostoulias, X., Donovan, L., Li, L., Carter, G. P., Hachani, A., et al. (2019). Unstable chromosome rearrangements in Staphylococcus aureus cause phenotype switching associated with persistent infections. Proc. Natl. Acad. Sci. U. S. A. 116, 20135–20140. doi: 10.1073/pnas.1904861116
Johnson, G., Banerjee, S., and Putonti, C. (2022). Diversity of Pseudomonas aeruginosa temperate phages. mSphere 7:e101521. doi: 10.1128/msphere.01015-21
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kondo, K., Kawano, M., and Sugai, M. (2021). Distribution of antimicrobial resistance and virulence genes within the prophage-associated regions in nosocomial pathogens. mSphere 6:e45221. doi: 10.1128/mSphere.00452-21
Letunic, I., and Bork, P. (2021). Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Liu, Z., Liu, S., Yao, J., Bao, L., Zhang, J., Li, Y., et al. (2016). The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 7:11757. doi: 10.1038/ncomms11757
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Mo, C. Y., Mathai, J., Rostol, J. T., Varble, A., Banh, D. V., and Marraffini, L. A. (2021). Type III-A CRISPR immunity promotes mutagenesis of staphylococci. Nature 592, 611–615. doi: 10.1038/s41586-021-03440-3
Moon, B. Y., Park, J. Y., Hwang, S. Y., Robinson, D. A., Thomas, J. C., Fitzgerald, J. R., et al. (2015). Phage-mediated horizontal transfer of a Staphylococcus aureus virulence-associated genomic island. Sci. Rep. 5:9784. doi: 10.1038/srep09784
Moon, B. Y., Park, J. Y., Robinson, D. A., Thomas, J. C., Park, Y. H., Thornton, J. A., et al. (2016). Mobilization of genomic islands of Staphylococcus aureus by temperate bacteriophage. PLoS One 11:e151409. doi: 10.1371/journal.pone.0151409
Nadeem, A., and Wahl, L. M. (2017). Prophage as a genetic reservoir: Promoting diversity and driving innovation in the host community. Evolution 71, 2080–2089. doi: 10.1111/evo.13287
Nayfach, S., Camargo, A. P., Schulz, F., Eloe-Fadrosh, E., Roux, S., and Kyrpides, N. C. (2021). CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39, 578–585. doi: 10.1038/s41587-020-00774-7
Negron, O., Hur, W. S., Prasad, J., Paul, D. S., Rowe, S. E., Degen, J. L., et al. (2022). Fibrin(ogen) engagement of S. aureus promotes the host antimicrobial response and suppression of microbe dissemination following peritoneal infection. PLoS Pathog. 18:e1010227. doi: 10.1371/journal.ppat.1010227
Owen, S. V., Wenner, N., Canals, R., Makumi, A., Hammarlof, D. L., Gordon, M. A., et al. (2017). Characterization of the prophage repertoire of African Salmonella Typhimurium ST313 reveals high levels of spontaneous induction of novel phage BTP1. Front. Microbiol. 8:235. doi: 10.3389/fmicb.2017.00235
Paharik, A. E., and Horswill, A. R. (2016). The staphylococcal biofilm: Adhesins, regulation, and host response. Microbiol. Spectr. 4, 1–27. doi: 10.1128/microbiolspec.VMBF-0022-2015
Ramisetty, B., and Sudhakari, P. A. (2019). Bacterial 'grounded' prophages: Hotspots for genetic renovation and innovation. Front. Genet. 10:65. doi: 10.3389/fgene.2019.00065
Russel, J., Pinilla-Redondo, R., Mayo-Munoz, D., Shah, S. A., and Sorensen, S. J. (2020). CRISPRCasTyper: Automated identification, annotation, and classification of CRISPR-Cas loci. CRISPR J. 3, 462–469. doi: 10.1089/crispr.2020.0059
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tam, K., and Torres, V. J. (2019). Staphylococcus aureus secreted toxins and extracellular enzymes. Microbiol. Spectr. 7, 1–34. doi: 10.1128/microbiolspec.GPP3-0039-2018
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Van Boeckel, T. P., Gandra, S., Ashok, A., Caudron, Q., Grenfell, B. T., Levin, S. A., et al. (2014). Global antibiotic consumption 2000 to 2010: An analysis of national pharmaceutical sales data. Lancet Infect. Dis. 14, 742–750. doi: 10.1016/S1473-3099(14)70780-7
Vestergaard, M., Frees, D., and Ingmer, H. (2019). Antibiotic resistance and the MRSA problem. Microbiol. Spectr. 7, 1–23. doi: 10.1128/microbiolspec.GPP3-0057-2018
Wheatley, R. M., and MacLean, R. C. (2021). CRISPR-Cas systems restrict horizontal gene transfer in Pseudomonas aeruginosa. ISME J. 15, 1420–1433. doi: 10.1038/s41396-020-00860-3
Young, R. (2014). Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 52, 243–258. doi: 10.1007/s12275-014-4087-z
Zhou, W., Li, X., Osmundson, T., Shi, L., Ren, J., and Yan, H. (2018). WGS analysis of ST9-MRSA-XII isolates from live pigs in China provides insights into transmission among porcine, human and bovine hosts. J. Antimicrob. Chemother. 73, 2652–2661. doi: 10.1093/jac/dky245
Keywords: prophages, Staphylococcus, bacteriophage, genomes, evolution
Citation: Zhou W, Li Y, Xu X, Rao S, Wen H, Han Y, Deng A, Zhang Z, Yang Z and Zhu G (2023) Whole-genome analysis showed the promotion of genetic diversity and coevolution in Staphylococcus aureus lytic bacteriophages and their hosts mediated by prophages via worldwide recombination events. Front. Microbiol. 14:1088125. doi: 10.3389/fmicb.2023.1088125
Edited by:
Antony T. Vincent, Laval University, CanadaReviewed by:
Haiyan Zeng, Guangdong University of Technology, ChinaJens Andre Hammerl, Bundesinstitut für Risikobewertung, Germany
Copyright © 2023 Zhou, Li, Xu, Rao, Wen, Han, Deng, Zhang, Yang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenquan Yang, eWFuZ3pxQHl6dS5lZHUuY24=; Guoqiang Zhu, eXpncXpodUB5enUuZWR1LmNu