 Agnieszka Klonowska1
Agnieszka Klonowska1 Julie Ardley2*
Julie Ardley2* Lionel Moulin1
Lionel Moulin1 Jaco Zandberg2
Jaco Zandberg2 Delphine Patrel1
Delphine Patrel1 Margaret Gollagher3
Margaret Gollagher3 Dora Marinova3T. B. K. Reddy4
Dora Marinova3T. B. K. Reddy4 Neha Varghese4Marcel Huntemann4
Neha Varghese4Marcel Huntemann4 Tanja Woyke4
Tanja Woyke4 Rekha Seshadri4
Rekha Seshadri4 Natalia Ivanova4
Natalia Ivanova4 Nikos Kyrpides4
Nikos Kyrpides4 Wayne Reeve2*
Wayne Reeve2*- 1Université de Montpellier, IRD, CIRAD, INRAE, Institut AgroPHIM Plant Health Institute, Montpellier, France
- 2Centre for Crop and Food Innovation, Food Futures Institute, College of Science, Health, Engineering and Education, Murdoch University, Murdoch, WA, Australia
- 3Curtin University Sustainability Policy Institute, Curtin University, Bentley, WA, Australia
- 4Department of Energy Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, CA, United States
Integrated virus genomes (prophages) are commonly found in sequenced bacterial genomes but have rarely been described in detail for rhizobial genomes. Cupriavidus taiwanensis STM 6018 is a rhizobial Betaproteobacteria strain that was isolated in 2006 from a root nodule of a Mimosa pudica host in French Guiana, South America. Here we describe features of the genome of STM 6018, focusing on the characterization of two different types of prophages that have been identified in its genome. The draft genome of STM 6018 is 6,553,639 bp, and consists of 80 scaffolds, containing 5,864 protein-coding genes and 61 RNA genes. STM 6018 contains all the nodulation and nitrogen fixation gene clusters common to symbiotic Cupriavidus species; sharing >99.97% bp identity homology to the nod/nif/noeM gene clusters from C. taiwanensis LMG19424T and “Cupriavidus neocalidonicus” STM 6070. The STM 6018 genome contains the genomes of two prophages: one complete Mu-like capsular phage and one filamentous phage, which integrates into a putative dif site. This is the first characterization of a filamentous phage found within the genome of a rhizobial strain. Further examination of sequenced rhizobial genomes identified filamentous prophage sequences in several Beta-rhizobial strains but not in any Alphaproteobacterial rhizobia.
Introduction
Root nodule bacteria, collectively known as rhizobia, are the dinitrogen-fixing microsymbionts of legumes. Their taxonomic diversity encompasses both the Alpha- and Beta- subclasses of Proteobacteria, and the names Alpha- and Beta-rhizobia are used for convenience to distinguish each group (Moulin et al., 2001; Gyaneshwar et al., 2011). To date, Beta-rhizobia include three genera: Paraburkholderia, Cupriavidus (formerly Ralstonia; Vandamme and Coenye, 2004) and Trinickia (Estrada-de los Santos et al., 2018; de Lajudie et al., 2019). Beta-rhizobia have a particular affinity for the genus Mimosa and related South American mimosoid legumes and nodulate at least 68 Mimosa species, including in particular the invasive species M. pudica, M. pigra, and M. bimucronata in Asia, Australia, and Central and South America (Barrett and Parker, 2005; Parker et al., 2007; Bontemps et al., 2010; dos Reis Jr et al., 2010; Gehlot et al., 2013; Estrada-de los Santos et al., 2018). Based on a comparison of house-keeping and nodulation gene phylogenies, Paraburkholderia species have been postulated to be ancestral symbionts of South American Mimosa and Piptadenia group species (Bontemps et al., 2010; Bournaud et al., 2013), while symbiotic Cupriavidus appear to have recently acquired nodulation genes from a Paraburkholderia ancestor (Chen et al., 2003; da Silva et al., 2012; Mishra et al., 2012). Cupriavidus symbionts have been isolated from invasive M. pudica and M. diplotricha in Central America, China, Christmas Island, French Guiana, India, New Caledonia and Taiwan (Chen et al., 2003; Andam et al., 2007; Klonowska et al., 2012; Mishra et al., 2012; Gehlot et al., 2013; De Meyer et al., 2018; Liu et al., 2020), as well as from native Mimosa spp. in southern United States and Uruguay (Andam et al., 2007; Platero et al., 2016) and Parapiptadenia rigida in Uruguay (Taulé et al., 2012).
Whether Paraburkholderia or Cupriavidus strains are the dominant nodule occupants of these mimosoid hosts appears to depend primarily on edaphic factors, and to some extent on host preference. In general, hosts growing in acidic soils are nodulated by Paraburkholderia strains, whereas Cupriavidus predominates in soils that are neutral-alkaline and/or have high heavy metal content (Klonowska et al., 2012; Mishra et al., 2012; Platero et al., 2016; Liu et al., 2020). Competition studies have revealed the dominance of Paraburkholderia phymatum and Paraburkholderia tuberum within M. pudica nodules compared to Cupriavidus or Rhizobium strains, however, the Taiwanese accession M. pudica var. unijuga displayed a higher affinity for C. taiwanensis strains, suggesting local co-adaptation (Melkonian et al., 2014).
Recent analyses of non-symbiotic Paraburkholderia and Cupriavidus strains have identified diverse integrated bacteriophages (prophages) that comprise a considerable portion of some of their genomes (Pratama et al., 2018; Van Houdt et al., 2018). Bacteriophages are ubiquitous in bacterial populations, and although they can kill or impose metabolic burdens on their hosts, phages that are integrated into the chromosome can supply benefits to the host bacterium as agents of horizontal gene transfer (HGT), sources of genetic variation, in bacterial competition, and by supplying phage-encoded virulence genes; they may also affect bacterial motility, biofilm production and relationships with hosts (Figueroa-Bossi et al., 2001; Wagner and Waldor, 2002; Allison, 2007; Harrison and Brockhurst, 2017; Secor et al., 2020). Although the recent expansion of both phage-derived and bacterial sequence databases has uncovered a high abundance of prophages within Proteobacterial genomes in particular (López-Leal et al., 2022), their role in rhizobial lifestyles is currently not well understood (Ford et al., 2021).
Cupriavidus taiwanensis strain STM 6018 was isolated from a M. pudica trap host using a soil sample from near the town of Remire, French Guiana, South America (Mishra et al., 2012) and was selected for sequencing at the US Department of Energy Joint Genome Institute (JGI) as part of the Genomic Encyclopedia of Bacteria and Archaea-Root Nodule Bacteria (GEBA-RNB) project (Reeve et al., 2015). Preliminary analysis of the genome identified that it had the unusual property of containing two prophages, one being a capsular phage and the other a filamentous phage. Here we describe the symbiotic and genomic features of STM 6018, and specifically focus on characterizing the two prophages and examining features in the STM 6018 genome that are putatively important for phage interactions with this host. We furthermore survey the genomes of other rhizobial strains for the presence of filamentous prophages and compare the STM 6018 filamentous prophage with those found in two other Beta-rhizobia strains.
Results and discussion
STM 6018 morphology and growth, genome classification, and species assignment
C. taiwanensis STM 6018 is a motile, Gram-negative rod (Supplementary Figures S1A,B) in the order Burkholderiales of the class Betaproteobacteria. It is fast growing, forming colonies within 3–4 days when grown on half strength Lupin Agar (½LA; Howieson et al., 1988), tryptone-yeast extract agar (TY; Beringer, 1974) or a modified yeast-mannitol agar (YMAm; Howieson and Dilworth, 2016) at 28°C. Colonies on ½LA are white-opaque, slightly domed and moderately mucoid with smooth margins (Supplementary Figure S1C).
The STM 6018 genome project is deposited in the Genomes OnLine Database (GOLD; Mukherjee et al., 2020) and a draft genome sequence is available in the Integrated Microbial Genomes System (Chen et al., 2021). Minimum Information about the Genome Sequence (MIGS) is provided in Supplementary Table S1. Phylogenetic analysis based on an intragenic fragment of the 16S rRNA gene shows that the C. taiwanensis strains STM 6018 and LMG 19424T are most closely related to Cupriavidus nantongensis X1T, Cupriavidus alkaliphilus ASC-732T and the rhizobial strain “Cupriavidus neocaledonicus” STM 6070 (Supplementary Figure S2). The STM 6018 16S rRNA gene shares 99.6, 99.37, 99.14, 99.02 and 98.5% sequence identity (over 1,426 bp) to the 16S rRNA genes of the type strains C. taiwanensis LMG19424T (Amadou et al., 2008), C. nantongensis X1T (Sun et al., 2016), “C. neocaledonicus” STM 6070 (Klonowska et al., 2020), C. alkaliphilus ASC-732T (Estrada-de los Santos et al., 2012) and C. necator ATCC43291T (N-1; Poehlein et al., 2011), respectively.
The species assignment for STM 6018 was further assessed by calculating the average nucleotide identity (ANI) values of this genome to other Cupriavidus genomes (Table 1). Analysis of the ANIb and ANIm values (>98%, over 92% conserved DNA) and ANIg values (>99%) showed that STM 6018 belongs to the same species as the M. pudica-nodulating C. taiwanensis LMG19424T, consistent with the 16S rRNA gene analysis. The next closest rhizobial species is “Cupriavidus neocaledonicus” STM 6070, also a microsymbiont of M. pudica (Klonowska et al., 2020), with ANIb and ANIm values (>93% over >82% conserved DNA) and ANIg values (>94%). In contrast, the sequenced rhizobial strains Cupriavidus sp. AMP6, isolated from native Mimosa asperata in Texas (Andam et al., 2007) and C. necator UYPR2.512, which nodulates Parapiptadenia rigida in Uruguay (Taulé et al., 2012), have ANIb, ANIm or ANIg values <90%.
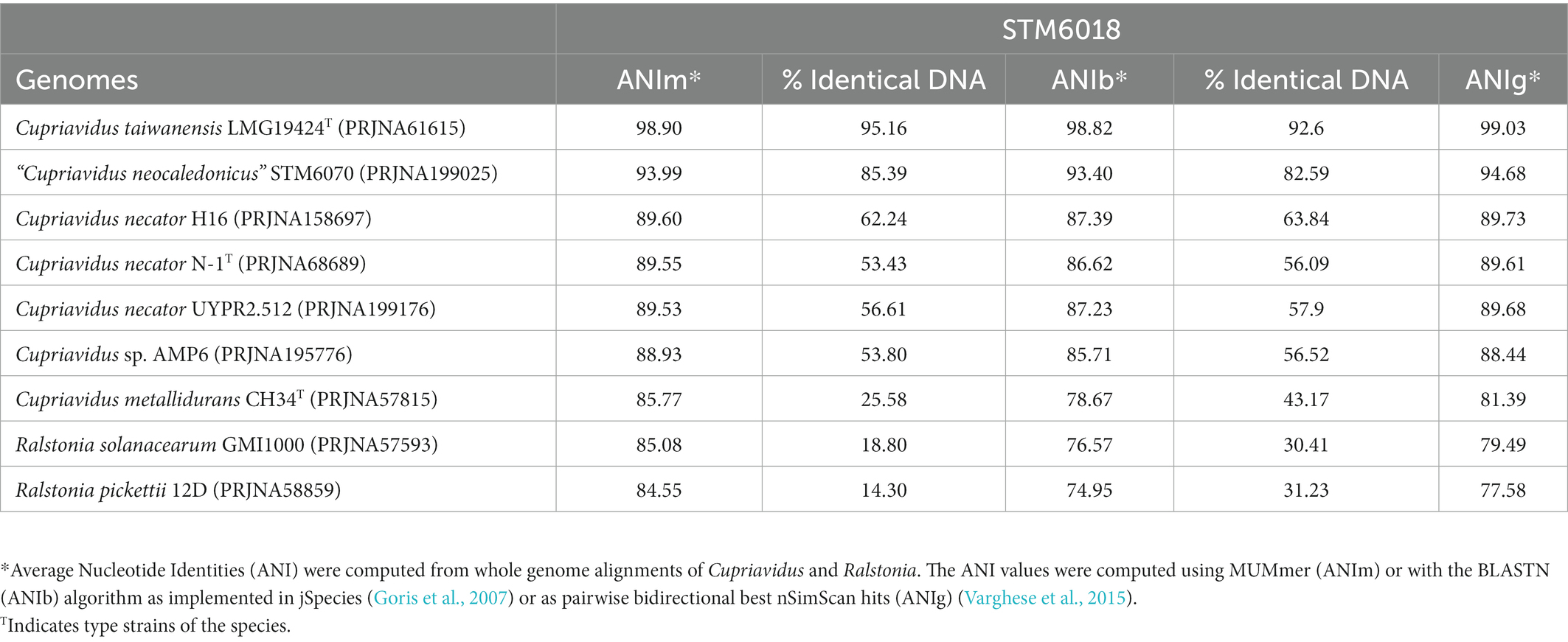
Table 1. Average nucleotide identities* (ANI) of STM6018 genome compared to close relatives.
General properties and features of the STM 6018 genome
The STM 6018 draft genome is given as 6,553,639 nucleotides with 66.90% GC content (Supplementary Table S2) and comprised of 80 scaffolds of 80 contigs (449x sequence coverage) with a total of 5,925 annotated genes, of which 5,864 are protein encoding and 61 RNA only encoding genes. Most of the protein encoding genes were predicted to have functions (80.69%), while the remaining genes were annotated as hypothetical. The distribution of genes into functional COG categories is presented in Supplementary Table S3.
Pangenome analysis: Comparisons of STM 6018 with other sequenced symbiotic Cupriavidus genomes
A pangenome analysis comparing the STM 6018 genome to the genomes of the closely related C. taiwanensis LMG19424T and “C. neocaledonicus” STM 6070 revealed that the pan- and variable genomes consisted of 5,205 and 438 genes, respectively, while 244 genes were unique to STM 6018 (Supplementary Figure S3). A previous progressiveMauve alignment of the draft genome of STM 6018 to the finished genome of LMG19424T showed a high degree of synteny between the STM 6018 scaffolds and the LMG19424T chromosome 1, chromosome 2 and symbiotic plasmid (Klonowska et al., 2020), suggesting that the STM 6018 replicons also consist of chromosome 1, chromosome 2 and a symbiotic plasmid.
The 244 genes unique to STM 6018 included two intact prophage regions, identified as a Mu-like phage and, surprisingly, a filamentous phage. Among the 5,205 genes that make up the C. taiwanensis/ “C. neocaledonicus” pangenome, the symbiotic, bacterial secretion system, and pilus system genes were highly conserved. Because these systems are important components of rhizobial interactions with legume hosts (Amadou et al., 2008; Deakin and Broughton, 2009; Zatakia et al., 2014), and of phage interactions with bacterial hosts, either for phage adsorption during infection (Bertozzi Silva et al., 2016; Hay and Lithgow, 2019), for secretion of filamentous phage virions from the host (Davis et al., 2000; Bille et al., 2005), or for secretion of phage-encoded toxins (Davis et al., 2000; Nakamura et al., 2021), we targeted them for further analysis. Below, we describe the STM 6018 symbiotic genotype and phenotype, secretion system and pilus system genes, and finally the two prophage genomes, concentrating on the filamentous phage, which has not previously been described in any rhizobial strain.
Symbiotic genotype and phenotype of STM 6018
The STM 6018, LMG19424T and STM 6070 nod/nif/noeM gene clusters, along with characteristic mobile elements that are present in these clusters, were highly conserved in all three strains (Figure 1). Additional analysis of the nodulation (nod and noe) and nitrogen fixation (nif, fix and fdx) genes showed that these were well conserved in mimosoid-nodulating Cupriavidus strains (C. taiwanensis STM 6018 and LMG19424T, “C. neocaledonicus” STM 6070, Cupriavidus sp. AMP6, and C. necator UYPR2.512), with a high degree of both synteny and % identity (Figure 1). STM 6018, LMG19424T and STM 6070 shared nearly 100% sequence identity (>99.97% bp identity over 100% coverage of the sequence of these genes), while AMP6 and UYPR2.512 shared lower % identities (>92% bp identity over >90% coverage and > 73% bp identity over >50% coverage, respectively) and lacked most of the transposase genes that are a feature of this gene neighborhood in the C. taiwanensis and “C. neocaledonicus” strains. This supports the hypothesis that symbiotic Cupriavidus populations have arisen via horizontal gene transfer (Parker, 2015) and suggests that the M. pudica isolates STM 6018, LMG19424T and STM 6070 share well conserved symbiotic plasmids. All strains contained the nodulation genes nodDBCIKHASUQ and noeM, consistent with C. taiwanensis Nod factors being pentameric chito-oligomers with C18:1 or C16:0 fatty acyl chains, N-methylated and C-6 carbamoylated on the non-reducing end and sulfated on the reducing terminal residue, with additional production of atypical Nod factors with an open-chain oxidized terminal residue (Daubech et al., 2019). The recently described noeM, which is involved in the biosynthesis of these atypical Nod factors, is predominantly found in M. pudica microsymbionts and is important for symbiotic competitiveness of C. taiwanensis on M. pudica (Daubech et al., 2019). The putative NoeM proteins in STM 6018, STM 6070, AMP6 and UYPR2.512 were located and found to have high identity with the characterized NoeM of LMG19424T (>70% identity in amino acid sequence, aligning >76% of the query protein). The rhizobial Cupriavidus nitrogen fixation genes were arranged in operons and included nifA, nifENfdxBnifQ, nifX, nifVWfixABCX, nifBfdxNnifZfixU, and nifHDK (Figure 1), as has been described for LMG19424T (Amadou et al., 2008). Although these genes were all syntenic, the translated NifV of AMP6 and UYPR2.512 had low identity (77.89 and 48.13%, respectively) compared with the 100% identity shared by STM 6018, LMG19424T and STM 6070 NifV (Figure 1). All five strains are capable of fixing nitrogen with M. pudica (Saad et al., 2012a; Klonowska et al., 2020), in accordance with the conservation of the nod, nif, fix, fdx and noeM gene regions.
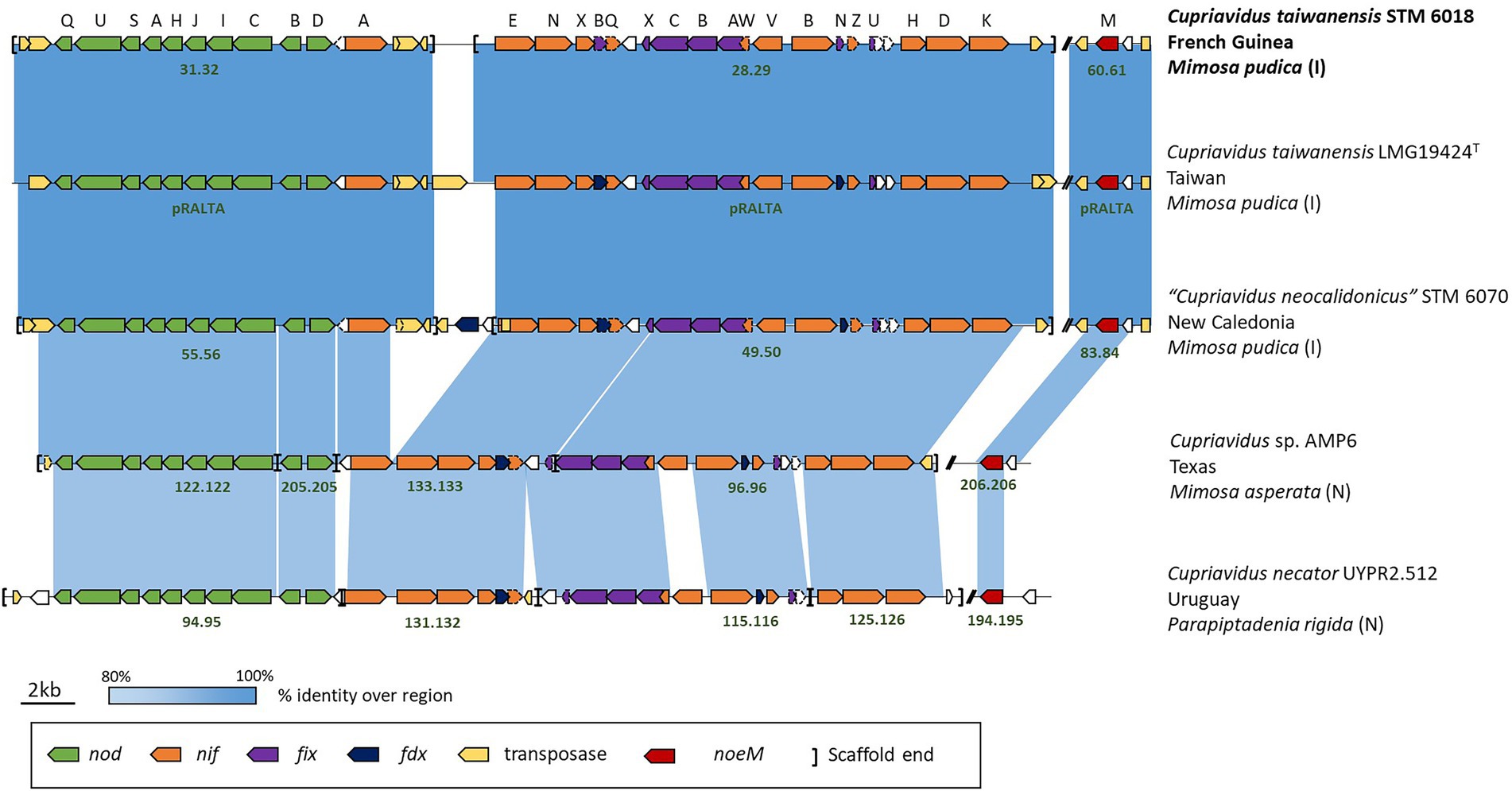
Figure 1. Schematic of the gene neighborhoods of symbiotic Cupriavidus species, showing nodulation (nod), nitrogen fixation (nif, fdx and fix) and transposase gene cluster alignments. Information on the microsymbiont’s host species, geographical location, and whether the host is introduced (I) or native (N) to the location is included. The scaffold number or replicon name is in dark green text and two bars (//) represent gaps in the sequence between the genes of interest.
C. taiwanensis STM 6018 forms nodules (Nod+) and effectively fixes N2 (Fix+) on a diverse range of Mimosa species. It proficiently fixes nitrogen with Mimosa pudica and Mimosa pigra, nodulates but does not fix nitrogen with Mimosa caesalpiniaefolia and Mimosa acustipulata and does not nodulate Mimosa scabrella (Table 2). Furthermore, STM 6018 has also been shown to out-compete a gfp marked derivative of Paraburkholderia phymatum STM815T for the nodulation of M. pudica var. unijuga from Taiwan (80% nodule occupation), in contrast to the nodulation of M. pudica var. tetrandra (30% nodule occupation) or hispida (5% nodule occupation; Melkonian et al., 2014).
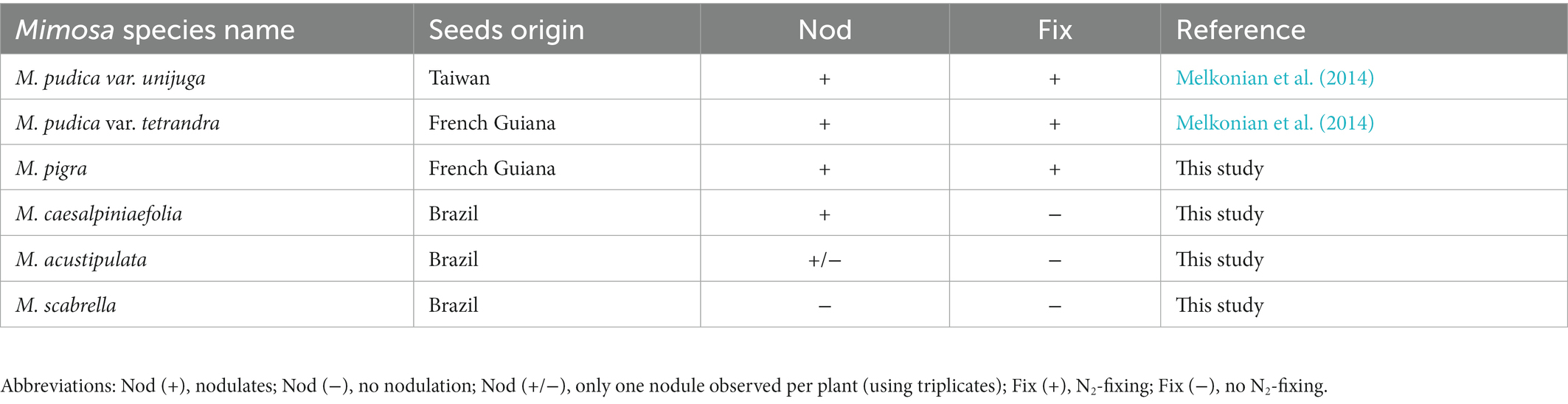
Table 2. Symbiotaxonomy of STM6018 using different species of Mimosa.
STM 6018 secretion and pilus systems
Genomes of the C. taiwanensis strains STM 6018 and LMG19424T contained loci encoding components of type I, II, III, IV and VI secretion systems and Type IV pilus systems (Supplementary Table S4), as well as loci for the Sec, Tat and SRP protein secretion systems.
Type I secretion system (T1SS)
The T1SS is a heterotrimer composed of an outer membrane protein (TolC), a periplasmic membrane fusion protein (HlyD) and an inner membrane ATP-binding cassette transporter (HlyB; Kanonenberg et al., 2018). We identified five genes encoding the TolC outer membrane protein in the STM 6018 genome; four of these were associated with gene clusters that also contained hlyB and hlyD homologs (Supplementary Table S4). One cluster (A3AADRAFT_03604-A3AADRAFT_05738, syntenic with RALTA_B1439-RALTA_B1442) contained a gene encoding a putative exported metalloprotease with cadherin domains, while another cluster (A3AADRAFT_03425–03433, with no homolog in LMG19424T) contained sequences encoding a putative exported adhesin.
Type II secretion system and type IV pilus systems
The T2SS is encoded by 12 core genes designated gsp (for General Secretory Pathway) and consists of a cytoplasmic hexameric ATPase (GspE); an inner membrane platform (GspC, GspF, GspL, GspM); a pseudopilus (GspG, GspH, GspI, GspJ, and GspK); a prepilin peptidase (GspO), required for proteolytic processing of the prepilin molecules, and an outer membrane pore (GspD), termed the secretin (Korotkov et al., 2012). In STM 6018, genes encoding the required T2SS components are clustered in a region (A3AADRAFT_04757–04770) that is syntenic with one on LMG19424T chromosome 1 (RALTA_A2985-2,998). gspO was not present in this cluster, however, a gene encoding the prepilin peptidase homolog PilD was found in another region of the genome (A3AADRAFT_02062, RALTA_A2712) along with genes encoding the type IV pilus assembly proteins PilB and PilC (Supplementary Table S4). This appears to be similar to the arrangement in Pseudomonas aeruginosa, which requires PilD to process both type II and type IV pilin precursors (Nunn and Lory, 1992).
The TFP systems are evolutionarily related to the T2SS, having homologous components and similar architectures; specific types of TFP mediate adhesion, protein secretion, DNA uptake, and twitching motility, and are important for the formation of biofilms and in host colonization (Mattick, 2002; Denise et al., 2019). Bacterial pili also serve as the primary receptors of filamentous phage infection (Hay and Lithgow, 2019) – different phages capable of infecting the same organism adsorb specifically to different types of pilus during infection (Holland et al., 2006). STM 6018 and LMG19424T contained six gene clusters encoding components of TFP systems (Supplementary Table S4). Two separate clusters (A3AADRAFT_01126–01139, RALTA_A0688-0702; A3AADRAFT_01206–01218, RALTA_B0189-0201) encoded components of a tight adherence (Tad) adhesive pilus system. The remaining clusters encoded separate components of a TFP system: the previously mentioned PilBCD, the alignment proteins PilMNOP and secretin PilQ (A3AADRAFT_04540–04544, RALTA_A2899-2,895), three clusters of the pilin proteins PilVWXYE and FimT, and the pilus retraction ATPases PilT and PilU (A3AADRAFT_02194–02195, RALTA_A2578-2,579). A monocistronic gene (A3AADRAFT_01884, RALTA_A0505) encoded the PilA major pilin. Pilus biogenesis pil loci are present on the symbiotic plasmid pRALTA of C. taiwanensis LMG 19424T (Amadou et al., 2008) and on a syntenic region of STM 6018 (Supplementary Table S4). Genes encoding proteins involved in root attachment (pilVWXYE, pilQPONM and a monocistronic gene coding for a PilX-related protein) were shown to be up-regulated in LMG 19424T cultures exposed to M. pudica root exudates (Klonowska et al., 2018), suggesting that they may play a role in host colonization.
Type III secretion system
The T3SS is a complex nanomachine that injects effector proteins directly into the cytosol of host cells. T3SSs are important virulence determinants for bacterial pathogens and are also used by some rhizobial strains in symbiotic interactions with legume hosts (Marie et al., 2001). STM 6018 possesses a T3SS (A3AADRAFT_03420–03406) that is syntenic with that found on LMG19424T chromosome 2 (RALTA_B1250–1,264; Amadou et al., 2008; Supplementary Table S4). We did not identify gene(s) in either STM 6018 or LMG19424T encoding homologs of the SctA needle filament, or of the translocon that forms a pore in the host membrane through which effectors can enter, however, two hypothetical proteins within the T3SS gene cluster may be candidates for these roles (Supplementary Table S4). It has previously been shown that the T3SS of C. taiwanensis is not induced by M. pudica root exudates (Klonowska et al., 2018); moreover, regulation of C. taiwanensis T3SS genes was mediated by glutamate rather than legume flavonoids, and T3SS inactivation had no effect on M. pudica nodulation, but did allow nodulation and N2 fixation with Leucaena leucocephala (Saad et al., 2012b).
Type IV secretion system
T4SSs are functionally diverse machineries for transporting DNA, proteins, or other macromolecules to bacterial or eukaryotic cell targets. There are two main subfamilies: (i) conjugation systems that mediate DNA transfer between bacterial cells, and (ii) translocators that deliver effector macromolecules into prokaryotic or eukaryotic cells (Costa et al., 2021). The trb and tra loci related to conjugative plasmid transfer are present on the symbiotic plasmid pRALTA of C. taiwanensis LMG 19424T (Amadou et al., 2008) and on a syntenic region of STM 6018 (Supplementary Table S4).
Type VI secretion system
The type VI secretion system (T6SS) is widely distributed among Gram-negative bacteria. It delivers effector toxins directly into a target cell and is usually deployed in competition against rival bacteria. The effector proteins are carried on a Vgr spike protein, which is fired into the target cell (Cianfanelli et al., 2016). We identified two separate T6SS systems in the genome of STM 6018, which were also present in LMG19424T. The first was on STM 6018 scaffold 0.1 in a neighborhood that was syntenic with that found on LMG19424T chromosome 2 (Supplementary Table S4). In addition to the required T6SS machinery genes, this cluster contained a gene (locus tag A3AADRAFT_00194) encoding a novel type VI secretion protein, peptidoglycan L-alanyl-D-glutamate endopeptidase, which had 63% protein identity at the C-terminal end to the peptidoglycan-degrading enzyme TagX, required for type VI secretion in Acinetobacter baumannii (Weber et al., 2016). The second T6SS cluster, on STM 6018 scaffold 3.4 (LMG19424T chromosome 1; Supplementary Table S4) was annotated as Sci-like proteins, from the T6SS cluster first identified in the Salmonella enterica subspecies I centisome 7 genomic island (Folkesson et al., 2002). The Vgr proteins all included RHS (rearrangement hot-spot) domains, often associated with type VI Vgr proteins as part of a protein secretion module with variable C-terminal toxic domains (Jackson et al., 2009). In addition to the vgr genes associated with these two clusters, a separate vgr gene (locus tag A3AADRAFT_01651, RALTA_A0273) was located upstream of genes encoding a putative effector protein – cognate immunity protein pair. STM 6018 also contained a vgr gene (A3AADRAFT_01451, scaffold 2.3) that was not found in LMG19424T.
Characterization of the STM 6018 Mu-like prophage genome
The intact Mu-like prophage was identified on scaffold 19.20 of STM 6018, inserted between a gene encoding an acyl-CoA dehydrogenase (locus tag A3AADRAFT_05251) and a gene encoding salicylate hydroxylase (locus tag A3AADRAFT_05309). The gene neighborhoods on each side of the Mu-like prophage are syntenic with those found on LMG19424T chromosome 2. Bacteriophage Mu is a member of the Myoviridae family of tailed bacteriophages and has a number of distinctive properties, including the ability to integrate into nearly random chromosomal locations (Morgan et al., 2002). The STM 6018 Mu-like prophage had an estimated size of 36,733 bp with a similar genome architecture to Mu and contained most of the core genes (Morgan et al., 2002) but had low core protein identity (25–40%) and lacked homologs to several core genes, including those encoding the G-segment invertase Gin, the adenine modification enzyme Mom, and an invertible segment encoding tail fibers (Figure 2). NCBI BLASTN analysis of the complete STM 6018 Mu-like prophage region identified similar prophages in the genomes of several other C. taiwanensis strains. LMG 19424T lacks this Mu-like prophage but instead has previously been characterized as containing a prophage on Chromosome 1 that is similar to the Phi CTX phage of P. aeruginosa (Amadou et al., 2008).
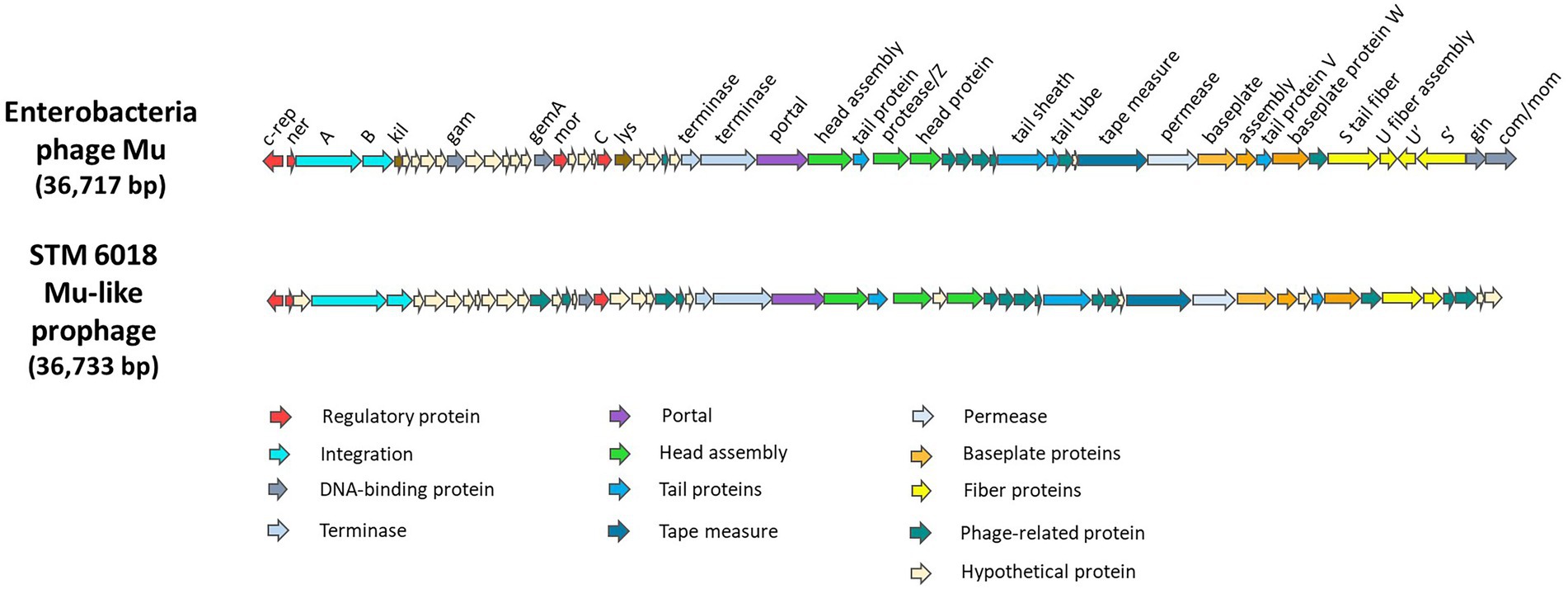
Figure 2. Genes and genome organization of Enterobacteria phage Mu (Morgan et al., 2002) and the Mu-like phage found in STM 6018.
Characterization of the STM 6018 filamentous prophage genome
Although a recent study has highlighted the diversity of filamentous phages and their presence in a broad range of bacterial and archaeal hosts (Roux et al., 2019), and filamentous phages have been reported to infect Sinorhizobium (Ensifer) meliloti (Cubo et al., 2020), to the best of our knowledge this is the first characterization of a filamentous prophage from the genome of a rhizobial strain. Filamentous phages currently belong to the Inoviridae family. Well-studied examples include the Escherichia coli Ff phages (M13, fd and F1; Marvin and Hoffmann-Berling, 1963), the Vibrio cholerae CTXϕ phage (Waldor and Mekalanos, 1996), and the Pf phages of P. aeruginosa (Knezevic et al., 2015). Filamentous phages replicate by a rolling circle mechanism (Baas, 1985) and possess several unique morphological and genetic features. Within the host cell, they can exist as either episomes or as integrated prophages, and can replicate without causing cell death, as viral particles are secreted across the cell envelope rather than released via lysis (Marvin, 1998). Whereas most bacteriophages have double-stranded DNA genomes packaged within a protein capsid, the virions of filamentous phages are long filaments, with circular, single-stranded DNA genomes of ~5–15 kb surrounded by several thousand major coat protein subunits arranged in a helical array (Mai-Prochnow et al., 2015; Hay and Lithgow, 2019).
The genomes of filamentous phages are modularly organized, mosaic structures. The encoded proteins have low identity across different phages, however, the order of the core genes and their sizes and membrane topology tends to be conserved, allowing putative identification of the genes, based on those of the well-studied Ff phages of E. coli (Mai-Prochnow et al., 2015). Figure 3 shows a general diagram of a filamentous phage, the phage genome, genes, and encoded proteins. The core genes include modules for (1) replication (R), (2) structural components (S), and (3) assembly and secretion [AS; reviewed in Mai-Prochnow et al. (2015) and Hay and Lithgow (2019)]. R genes encode the pII protein that is required for DNA replication, the pV protein that binds to phage ssDNA in the cytoplasm and the pX protein that binds to dsDNA and prevents hydrolysis. pX is translated from an internal start site within the pII gene. S proteins include the major coat protein pVIII and the minor coat proteins pIII, pVI, pVII and pIX. pIII additionally plays an essential role in host infection, as it specifically mediates adsorption of the phage, firstly to the host cell pilus primary receptor and then to the TolA component of the inner membrane TolQRA complex, which forms part of the Tol-Pal complex involved in cell division and membrane integrity. S proteins are all integrated into the host inner membrane. The AS module includes pI, pIV and pXI. These proteins form a trans-envelope complex that is essential for assembly and secretion of the phage, where pI/pXI form the inner membrane component and pIV is an outer membrane secretin that belongs to the same family as type II and type III outer membrane channels and the type IV pilus assembly system. pXI is translated from an internal start site within the pI gene. pXI and pIV are not always conserved in filamentous phages, and in cases where the gene encoding pIV is absent, a host-encoded secretin is used instead. pI contains ATP-binding Walker A and Walker B motifs and is presumed to be an ATPase that powers the assembly and transport of the phage out of the bacterial cell (Loh et al., 2017). Regulatory genes may also be present in the phage genome, as well as accessory genes. A notable example is the CTXϕ phage, where accessory genes encode the CtxAB toxin, a primary virulence factor that converts V. cholerae strains carrying the phage to deadly pathogens (Waldor and Mekalanos, 1996). Only the pI protein gene is universally conserved across all members of the Inoviridae (Roux et al., 2019).
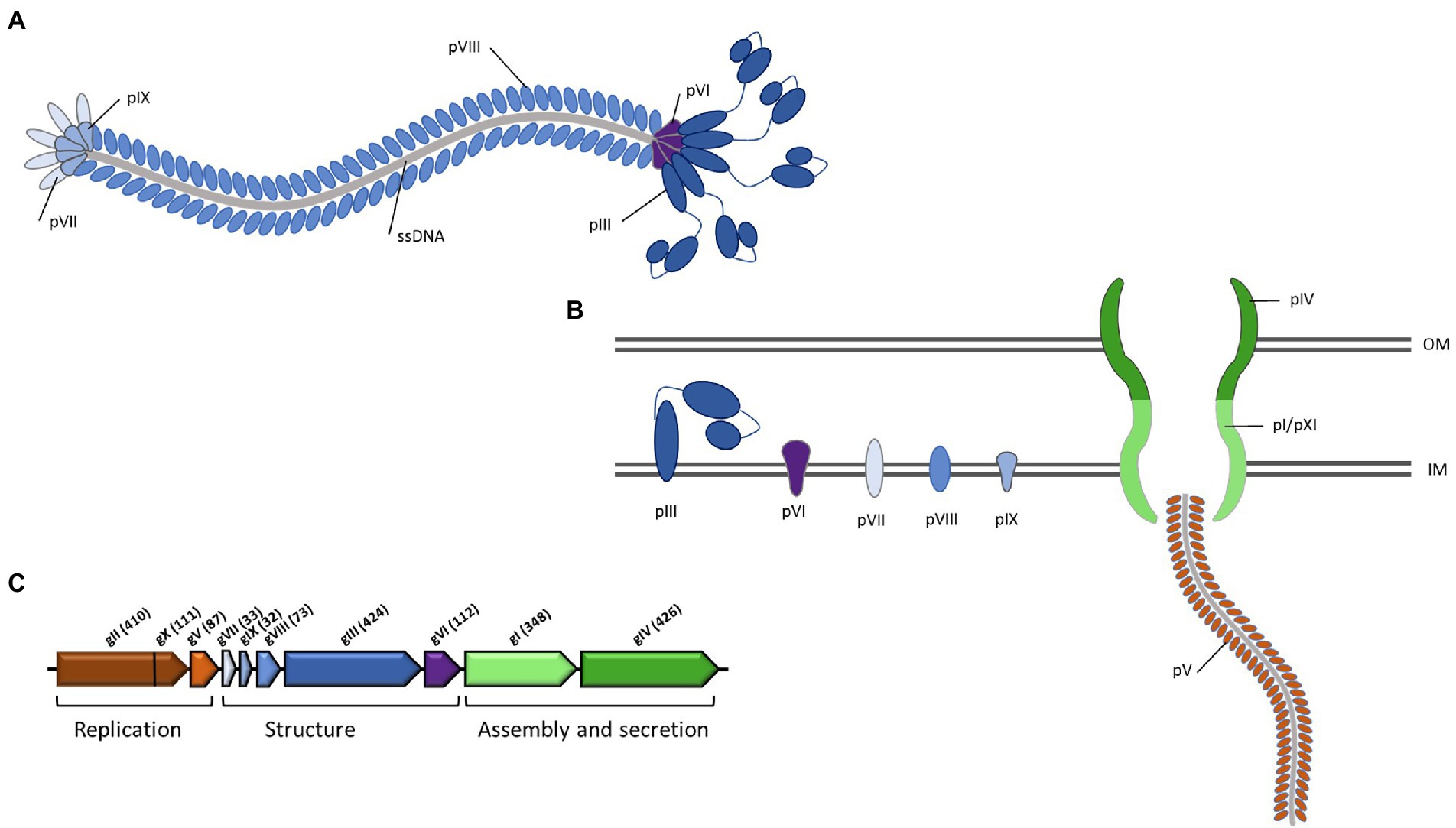
Figure 3. General schema of genes and genome organization of filamentous phages. The genes and the proteins they encode are color-coded according to function, where brown indicates replication; blue indicates structural; green indicates assembly and secretion. (A) the mature virion; (B) the virion within the bacterial host cell; associated phage structural, assembly and secretion proteins are embedded within the bacterial membrane; (C) genome organization of the model Ff filamentous phage, with the standard gene notations of gI to gX. Figure adapted from Mai-Prochnow et al. (2015).
The filamentous prophage identified in the genome of STM 6018 was located on scaffold 0.1, in a region that was syntenic with that of C. taiwanensis LMG 19424T chromosome II. The prophage genome has an estimated size of 7,540 bp in length, has a GC content of 61.1% and includes 11 predicted protein-encoding genes along with flanking regions, based on the annotations in IMG and NCBI (Supplementary Table S4). Based on these annotations and the order, size and membrane topology of the core genes, we identified the putative replication, structural, assembly and secretion, regulatory and accessory genes in the STM 6018 prophage, and compared its genome with those of other well-characterized filamentous phages (Mai-Prochnow et al., 2015; Figure 4; Supplementary Table S4).
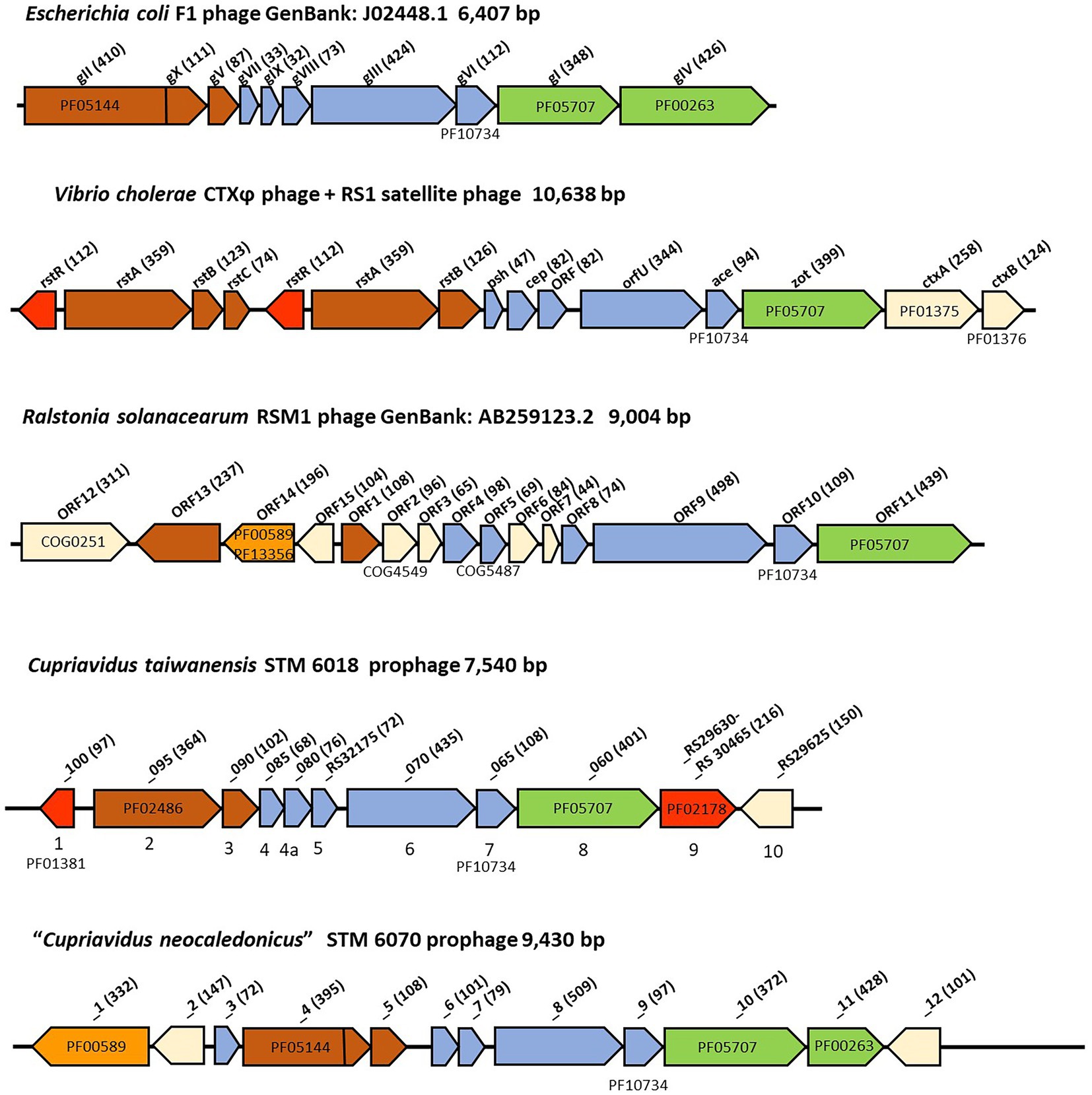
Figure 4. Comparison of filamentous phage genomes. The genes are colored according to function, where red indicates regulatory genes, brown indicates replication genes, orange indicates genes encoding integrases, blue indicates structural genes, and green indicates assembly and secretion genes. White indicates genes that are unique for each phage. The amino acid sequence length of the encoded proteins is in brackets. Where relevant, COG and pfam numbers of the encoded proteins are shown.
Genes encoding putative pII, pV, pVII, pIX, pVIII, pIII, pVI and pI proteins were all present, however, as is typical with filamentous phages, they had low sequence identity (< 30%) with the core proteins of characterized filamentous phages (Mai-Prochnow et al., 2015; Supplementary Table S4). There are some notable differences between the STM 6018 filamentous phage and the E. coli F1 phage (Figure 4). The STM 6018 filamentous phage lacks a gene encoding the pIV secretin and therefore presumably relies on a host secretin to transport virions out of the cell, as has been found for V. cholerae and Neisseria meningitidis filamentous phages (Davis et al., 2000; Bille et al., 2005). The putative pII protein was identified as DNA relaxase NicK (Rep_trans endonuclease pfam02486) rather than the phage replication protein CRI (pfam05144) present in Ff phages; moreover, the STM 6018 phage pII lacked a domain that was functionally equivalent to the pX protein. Rolling circle replication initiator proteins are known to be diverse (Wawrzyniak et al., 2017), as demonstrated by the various pII proteins found in characterized filamentous phages (Figure 4).
The pI proteins in some pathogenic V. cholerae and Campylobacter concisus strains are additional virulence determinants, due to cleavage and secretion of a toxic C-terminal domain that binds to tight junctions (zonula occludens) between intestinal epithelial cells and increases permeability (Fasano et al., 1991; Mahendran et al., 2016), causing pI proteins to be generally annotated as zonula occludens toxins (Zot). Because rhizobial strains are generally classified as Risk Group 1 (organisms that do not cause disease in healthy adult humans) and because they are often used as inoculants for legume crops and pastures, we examined the sequence of the STM6018 phage pI protein for the presence of this toxic domain. However, the C-terminus of V. cholerae Zot that contains the biologically active fragment (FCIGRL; Di Pierro et al., 2001) is missing in the pI of the STM 6018 filamentous prophage. and a phylogenetic tree of pI proteins from phages of characterized bacterial animal and plant pathogens, symbionts, and environmental strains places the STM 6018 pI protein well away from the V. cholerae Zot (Figure 5). Similarly, the CTXϕ protein Ace (accessory cholera enterotoxin) that is functionally equivalent to the pVI protein (Waldor and Mekalanos, 1996) is an additional V. cholerae virulence determinant that activates calcium-dependent chloride-bicarbonate secretion (Trucksis et al., 1993, 2000) but this role in virulence has not been reported for any other filamentous phage pVI protein. The STM 6018 prophage also contains accessory genes encoding a transcriptional regulator (A3AADRAFT_00018), a putative regulator that contains a DNA-binding AT-hook motif (A3AADRAFT_00010), and a hypothetical protein (A3AADRAFT_0009; Figure 4).
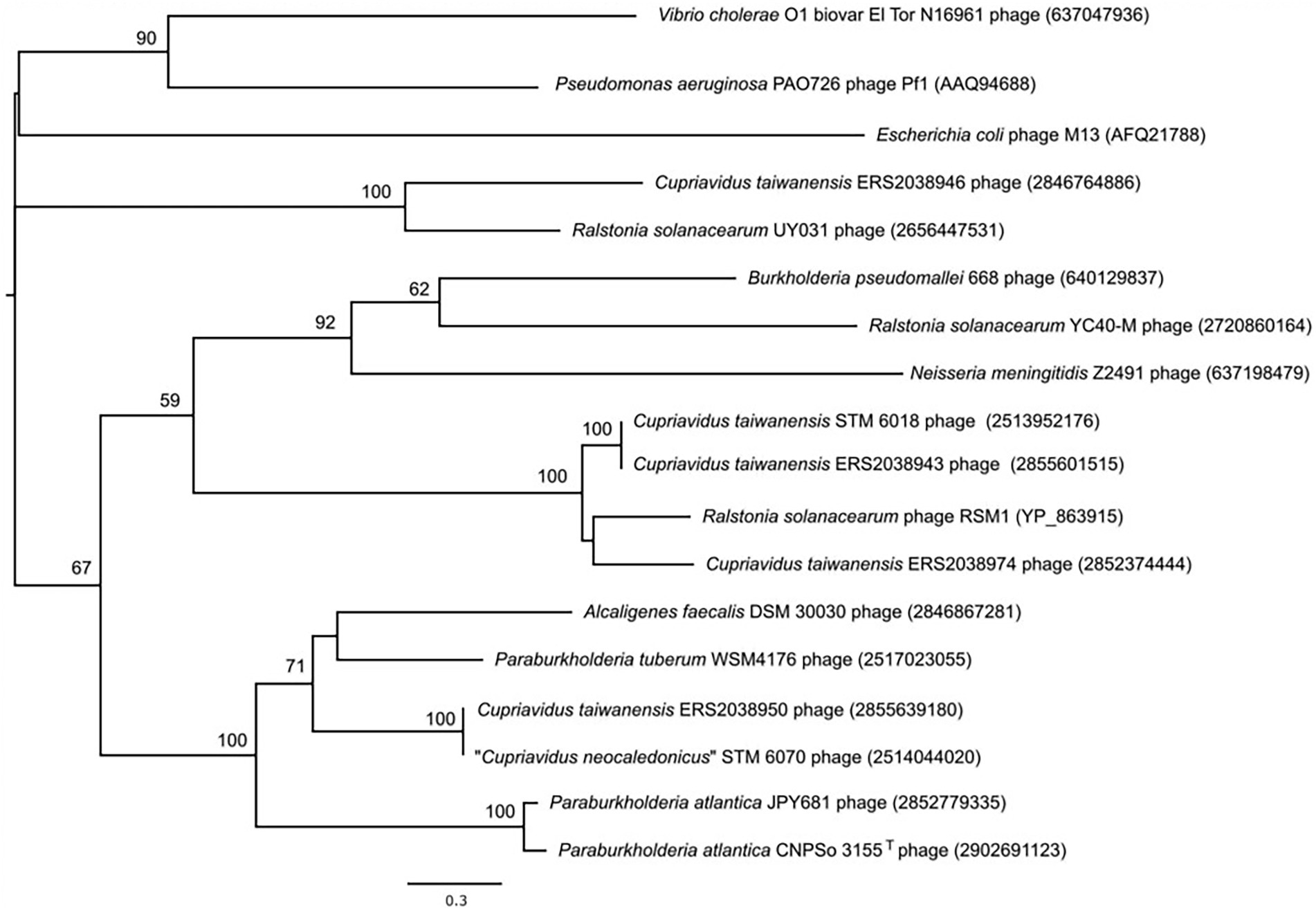
Figure 5. Phylogenetic tree based on the amino acid sequences of pI (zonula occludens toxin) proteins of filamentous phages from the genomes of Cupriavidus taiwanensis STM 6018 and diverse bacterial animal and plant pathogens, symbionts, and environmental strains. NCBI and IMG accession numbers are given in parenthesis. A total of 547 amino acid positions were used to reconstruct the tree. The evolutionary history was inferred by using the Maximum Likelihood method and a discrete Gamma distribution was used to model evolutionary rate differences among sites. Human pathogens, or potentially pathogenic species in the tree include Burkholderia pseudomallei, Escherichia coli, Neisseria meningitidis, Pseudomonas aeruginosa and Vibrio cholerae; Ralstonia solanacearum is a plant pathogen; the Cupriavidus and Paraburkholderia spp. are rhizobial symbionts; and Alcaligenes faecalis is an environmental species.
The AT-hook (pfam02178) is a small DNA-binding motif that contains a core GRP or Q/RGR sequence. It binds to the minor groove of AT-rich DNA regions and is found in many eukaryotic nuclear proteins (Aravind and Landsman, 1998) and in bacterial nucleoid-associated proteins such as H-NS (histone-like nucleoid structuring protein) from some Gram-negative bacteria and Lsr2 from mycobacteria (Gordon et al., 2011). However, the STM 6018 prophage AT-hook motif protein is unrelated to H-NS or Lsr2 proteins and moreover is structurally different, as the canonical RGR motif is found in the N-terminal region of the protein instead of the N-terminal oligomerization domain and AT-hook C-terminal DNA binding domain of H-NS- and Lsr2-like proteins (Gordon et al., 2011; Pfeifer et al., 2016). BLASTp analysis indicated that this protein is conserved in several filamentous prophages, as we identified genes encoding similar proteins containing an AT-hook motif located downstream of the gene encoding the pI protein in the genomes of various Cupriavidus and Ralstonia strains.
Although the STM 6018 filamentous prophage does not contain homologs of H-NS-like proteins, C. taiwanensis does contain several genes annotated in IMG as encoding H-NS or H-NS-like proteins (pfam00816; Supplementary Table S4). H-NS and Lsr2 specifically target and downregulate gene expression from AT-rich regions of DNA (Gordon et al., 2011; Singh et al., 2016). This includes genes acquired by horizontal gene transfer, which are relatively AT-rich compared with the rest of the bacterial genome and need to be tightly regulated to prevent fitness costs from their unregulated expression. H-NS and Lsr2 therefore play significant roles as xenogenic silencers of this foreign DNA, facilitating integration of horizontally acquired genes into the host regulatory network and contributing to bacterial evolution (Navarre, 2016). Xenogenic silencing also appears to be required for the survival of bacterial cells that contain prophages. In P. aeruginosa, loss of the xenogenic silencing proteins MvaT and MvaU, which are functionally equivalent to H-NS, results in an increase in gene expression and subsequent increase in the production of the filamentous phage Pf4, leading to cell death or inhibition of cell growth (Castang and Dove, 2012). Recent papers have also identified genes encoding H-NS- and Lsr2-like proteins in phage genomes, with either predicted roles in repressing host defense mechanisms against phage infection (Skennerton et al., 2011) or a demonstrated role in silencing gene expression of the prophage and consequent maintenance of the lysogenic state within the host genome (Pfeifer et al., 2016). We postulate that the AT-hook motif-containing protein encoded by A3AADRAFT_00010 may play a similar role in regulating expression of either host or prophage genes.
Because filamentous prophages have not previously been described in rhizobia, we evaluated their occurrence in sequenced genomes of genera that are known to contain rhizobial strains, using the conserved pI (Zot) protein (COG4128, pfam05707) as a marker for the presence of Inoviridae prophages (Roux et al., 2019) and then further establishing whether the gene encoding the Zot domain was within an intact prophage. We identified genes that encoded a Zot domain in the genomes of some alphaproteobacterial strains, however, these proteins were small (79–142 amino acids) and the genes were not associated with a prophage. In contrast, we identified intact filamentous prophages in rhizobial and non-rhizobial strains of betaproteobacterial Cupriavidus, Paraburkholderia and Trinickia. A total of 13 of these genomes were rhizobial strains (Supplementary Table S5). The phylogenetic tree of these pI proteins (Figure 5) demonstrates that highly diverse prophages can infect a given bacterial species, as shown here by the C. taiwanensis prophages. Rhizobial strains that contained intact filamentous prophages included the well-characterized “C. neocaledonicus” STM 6070 (Klonowska et al., 2020) and Paraburkholderia tuberum WSM4176 (Howieson et al., 2013; Reeve et al., 2015). We therefore selected the prophages of these strains for further analysis, with an emphasis on the prophages within the two Cupriavidus genomes.
Comparison of the filamentous prophages in STM 6070 and WSM4176 with the STM 6018 prophage
The STM 6070 filamentous prophage was located on scaffold 19.20 in a region that was syntenic with LMG 19424T Chromosome 1 and STM 6018 scaffold 9.10. There was very low sequence identity between the prophage found in STM 6018 and that of STM 6070; moreover, the STM 6070 prophage genome contained more genes, had a different gene order for some of the core phage proteins and had a different pII protein to that of the STM 6018 filamentous prophage (Figure 4; Supplementary Table S4). The genome included genes encoding a XerD-like site-specific tyrosine recombinase of the phage integrase family, a pIV secretin and two hypothetical proteins (Figure 4; Supplementary Table S4). The pII phage replication protein had the same pfam (pfam05144) as that of the pII CRI protein of Ff phages and similarly included a pX domain within the pII gene.
The P. tuberum WSM4176 prophage was located on scaffold 3.7 and similarly had low sequence identity with the STM 6018 and STM 6070 prophages. In addition to the core genes, the genome included genes encoding a Lambda repressor-like DNA-binding domain, a pIV secretin and three hypothetical proteins (Supplementary Table S4). The pII phage replication protein was a DNA relaxase NicK (pfam02486).
The STM 6018 and STM 6070 phages integrate into different sites within the Cupriavidus genome
To further characterize the STM 6018 and STM 6070 prophages, we examined where they integrated in the Cupriavidus genome. Integration of filamentous phages into host chromosomes occurs by site specific recombination and employs diverse strategies, according to the type of phage. Characterized phages may use dif, tRNA or inverted repeat target sequences and either host- or phage-encoded recombination or transposase systems (Hay and Lithgow, 2019). In C. taiwanensis, the STM 6018 prophage located at position 19,342–26,881 on scaffold 0.1 is integrated into a putative dif (deletion induced filamentation) site between genes encoding L-amino acid N-acyltransferase YncA (A3AADRAFT_00019; = RALTA_B1219 in LMG19424T) and a putative 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase (DAHPS; A3AADRAFT_0008; = RALTA_B1221 in LMG19424T; Figure 6). This region lacks synteny with STM 6070, as the genes encoding YncA and putative DAHPS homologs are found on separate scaffolds in the STM 6070 genome. The dif site is normally the site where resolution of concatenated chromosomes occurs via XerC and XerD site-specific recombination, but is also used by various filamentous phages for integration/excision (Carnoy and Roten, 2009). In STM 6018, the left-hand and right-hand flanking regions of the filamentous prophage contain DNA sequences that are homologous to the C. taiwanensis dif sequence (Figure 6). The STM 6018 filamentous phage lacks genes encoding Xer tyrosine recombinases and presumably relies on the host XerCD to integrate into the chromosome, which is the mechanism employed by V. cholerae CTXϕ (McLeod and Waldor, 2004). The STM 6018 genome contains several genes encoding XerC and XerD homologs (Supplementary Table S4). In contrast, the STM 6070 filamentous phage integrates into a tRNA-Met site within STM 6070, presumably via its phage-encoded XerD-like site-specific tyrosine recombinase. Similarly, characterized Pseudomonas phages integrate into tRNA-Gly, −Met and -Sec sites (Fiedoruk et al., 2020), while the Ralstonia phage RSM1 reversibly integrates into a tRNA-Ser site (Hay and Lithgow, 2019).
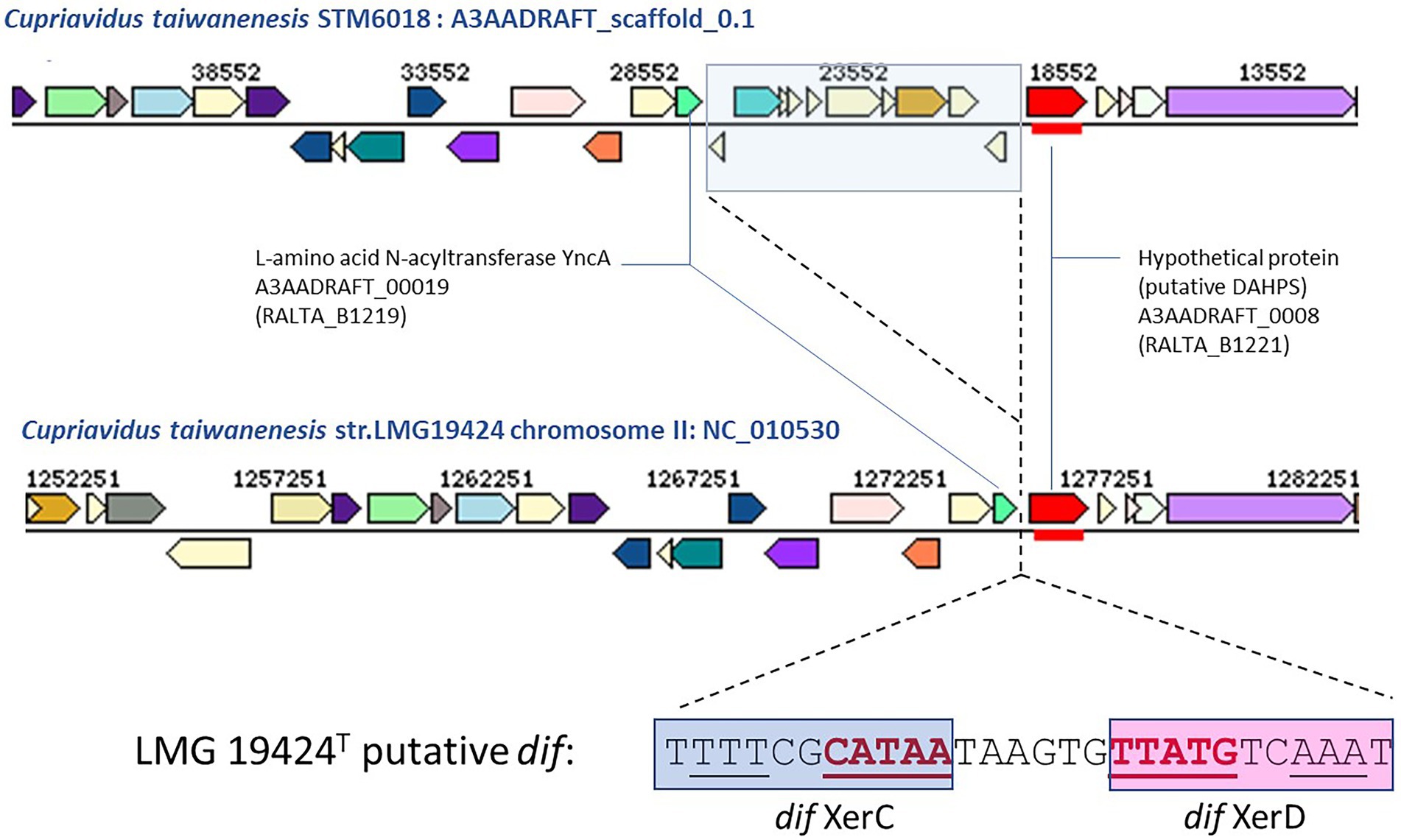
Figure 6. Comparison of the gene neighborhoods of Cupriavidus taiwanensis strains STM 6018 and LMG 19424T showing the site of integration of the filamentous phage found in STM 6018 and the DNA sequence of the putative dif site and Xer binding sites. Xer binding sites were identified using the highly conserved XerD binding sites of closely related Ralstonia spp. reported by Carnoy and Roten (2009).
Effects of phage infection on bacterial hosts and symbiotic implications
Filamentous phages can have marked effects on the virulence of their pathogenic bacterial hosts. A well-known example is V. cholerae, where strains that acquire the CTXϕ phage become toxigenic due to phage-encoded cholera toxin (Waldor and Mekalanos, 1996). Filamentous phages have also been shown to affect biofilm formation, exopolysaccharide (EPS) biosynthesis, motility, bacterial cell aggregation and expression of virulence genes, which can either enhance or reduce bacterial colonization, infectivity, or pathogenicity of a eukaryote host [reviewed in Mai-Prochnow et al. (2015)]. For example, Ralstonia solanacearum cells infected with the ϕRSS1 phage had enhanced virulence on tobacco and tomato plants, whereas cells infected with φRSM-type phages showed loss of virulence (Askora and Yamada, 2015). Similarly, other types of prophage can have significant effects on bacterial interactions, and may provide ecological and evolutionary benefits (Harrison and Brockhurst, 2017). Indeed, it has been suggested that the relationship between filamentous phages and their bacterial hosts, rather than being parasitic, can be one of facultative mutualism, with fitness benefits to the host in certain environments (Shapiro et al., 2016). Little is currently known of the effects of phage infection on rhizobial hosts. However, as biofilms, EPS, motility and expression of symbiotic genes are important for rhizobial lifestyles (Rinaudi and Giordano, 2010), we suggest that if phage infection causes changes in these attributes it will affect rhizobial ecology and relationships with legume hosts. More widely, it has been proposed that the tripartite relationship between phages, bacteria, and plants be explored further due to the putative effects of filamentous phages in improving the fitness of bacterial inocula to promote ecorestoration (Sharma et al., 2019). As the legume-rhizobia symbiosis is of particular importance in the restoration of degraded ecosystems (Chen et al., 2008; Teng et al., 2015; Jach et al., 2022), further research should be performed to explore the role of filamentous prophages in rhizobial colonization and competition for nodulation of the host plant, ecological fitness, and bacterial population structure. STM 6018 and other rhizobial strains that contain filamentous prophages will be useful resources in this enterprise.
Materials and methods
Growth conditions, genomic DNA isolation and nodulation, and phenotypic assay
C. taiwanensis STM 6018 was isolated from a soil sample from French Guiana, South America, as previously described, using the trap host M. pudica (Mishra et al., 2012). Bacterial isolates were cultured on ½LA (Howieson et al., 1988), TY (Beringer, 1974) or YMA (Howieson and Dilworth, 2016) at 28°C. For long-term maintenance, bacterial strains were grown in YM broth and preserved in 20% glycerol at −80°C.
For bacterial genomic DNA isolation C. taiwanensis STM 6018 was streaked onto TY solid medium and grown at 28°C for 3 days to obtain well grown separated colonies, and then a single colony was selected and used to inoculate 5 ml TY broth medium. The culture was grown for 48 h on a gyratory shaker (200 rpm) at 28°C. Subsequently, 1 ml was used to inoculate 60 ml TY broth medium that was incubated on a gyratory shaker (200 rpm) at 28°C until an OD600nm of 0.6 was reached. DNA was isolated from 60 ml of cells using a CTAB bacterial genomic DNA isolation method (Howieson and Dilworth, 2016). Final concentration of the DNA was set to 0.5 mg ml−1.
For nodulation assay of Mimosa spp. plants seedlings were prepared as previously described by Mishra et al. (2012) and were inoculated 7 days after germination with 1 ml of bacterial cell suspensions from cultures in exponential growth phase, as described in Melkonian et al. (2014).
Genome sequencing and assembly
The genome of C. taiwanensis STM 6018 was sequenced at the JGI using the Illumina technology (Bennett, 2004). An Illumina standard shotgun library was constructed and sequenced using the Illumina HiSeq 2000 platform which generated 14,977,300 reads totaling 2,245 Mb. Genome sequencing project information is shown in Supplementary Table S6.
All raw Illumina sequence data was passed through DUK, a filtering program developed at JGI, which removes known Illumina sequencing and library preparation artifacts (Mingkun, L., Copeland, A. and Han, J.).1 The following steps were then performed for assembly: (1) filtered Illumina reads were assembled using Velvet (Zerbino, 2010; version 1.1.04), (2) 1–3 Kb simulated paired end reads were created from Velvet contigs using wgsim,2 (3) Illumina reads were assembled with simulated read pairs using Allpaths–LG (Gnerre et al., 2011; version r39750). Parameters for assembly steps were: (1) Velvet (−-v --s 51 --e 71 --i 2 --t 1 --f “-shortPaired -fastq $FASTQ” --o “-ins_length 250 -min_contig_lgth 500” 10) (2) wgsim (−e 0–1 76–2 76 –r 0 –R 0 –X 0 3) Allpaths–LG (PrepareAllpathsInputs:PHRED64 = 1 PLOIDY = 1 FRAGCOVERAGE = 125 JUMPCOVERAGE = 25 LONGJUMPCOV = 50, RunAllpath-sLG: THREADS = 8 RUN = stdshredpairs TARGETS = standard VAPIWARNONLY = True OVERWRITE = True).
Genome annotation
Genes were identified using Prodigal (Hyatt et al., 2010) as part of the JGI annotation pipeline (Huntemann et al., 2015). The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database,3 TIGRFams database,4 UniProt,5 Pfam (now hosted by InterPro),6 PRIAM (Claudel-Renard et al., 2003), KEGG,7 COG,8 and InterPro9 databases. The tRNAScanSE tool (Lowe and Eddy, 1997) was used to find tRNA genes, whereas ribosomal RNA genes were found by searches against models of the ribosomal RNA genes built from SILVA (Pruesse et al., 2007). Other non–coding RNAs such as the RNA components of the protein secretion complex and the RNase P were identified by searching the genome for the corresponding Rfam profiles using INFERNAL.10 Additional gene prediction analysis and manual functional annotation was performed within the Integrated Microbial Genomes (IMG-ER) platform11 (Markowitz et al., 2009) and the genome was released through the Integrated Microbial Genomes System (Chen et al., 2021).
Genome analyses
The species assignment for STM 6018 was assessed by calculating the Average Nucleotide Identity (ANI) values of this genome to taxonomically proximal Cupriavidus and Ralstonia genomes. ANIb or ANIm values were computed with jSpecies (Richter and Rosselló-Móra, 2009) using either alignments produced by BLASTN or MUMmer, respectively. ANIg values were also computed as pairwise bidirectional best nSimScan hits of genes having 70% or more identity and at least 70% coverage of the shorter gene (Varghese et al., 2015). Species affiliation cut-off used was set at >95% ANI over 69% of the conserved DNA for ANIb or ANIm values (Goris et al., 2007) or 96.5% for ANIg values (Varghese et al., 2015).
The comparison of gene orthologs of STM 6018 with those of the symbiotic Cupriavidus strains LMG 19424T and STM 6070 was performed using the “Gene Phyloprofile” tool in the Microscope MaGe platform (Vallenet et al., 2019). The orthologous counterparts in the genomes were detected by applying parameters of a minimum of 30% for protein sequence identity over a minimum of 80% of the protein length (>30% protein MinLrap 0.8). Additional searches for homologous proteins were performed using the BLAST program in IMG with parameters of >30% protein sequence identity over a minimum of 80% of the protein length.
To identify the presence of prophages within other rhizobial genomes, we used the pI protein (COG4128, pfam05707) as a marker for the presence of Inoviridae prophages and searched for this marker within the genomes of both Alpha- and Betaproteobacterial rhizobial genera within the IMG database, using the Function Search tool in IMG. We then examined the gene neighborhoods of genes encoding a pfam05707 domain to determine whether the gene was within an intact filamentous prophage.
Phylogenetic analyses
We assessed the phylogenetic position of STM 6018 using a 1,301 bp internal region of the 16S rRNA gene. Ralstonia solanacearum LMG2299T was used as an outgroup. Sequences were aligned using MUSCLE as implemented in MEGA, version X (Kumar et al., 2018). Phylogenetic analyses were performed in MEGA X (Kumar et al., 2018) using the Neighbor-Joining method (Saitou and Nei, 1987). The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., 2004) and are in the units of the number of base substitutions per site. Bootstrap analysis (Felsenstein, 1985) with 1,000 replicates was performed to assess the support of the clusters.
We analyzed the phylogeny of the pI protein from the STM 6018 filamentous prophage using the amino acid sequences of characterized phage pI proteins and translated amino acid sequences of the pI gene identified in filamentous prophages present in Betaproteobacterial rhizobial genomes. Sequences were aligned using MUSCLE as implemented in MEGA, version X (Kumar et al., 2018). The evolutionary history was inferred using the Maximum Likelihood method based on the Le_Gascuel_2008 model (Le and Gascuel, 2008). The tree with the highest log likelihood (−13502.17) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites [5 categories (+G, parameter = 5.0459)]. A total of 547 amino acid positions were used in the final dataset. Evolutionary analyses were conducted in MEGA X (Kumar et al., 2018).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
AK isolated and performed the initial study of the STM 6018 strain. WR coordinated the project and supplied the DNA to the JGI for sequencing. All authors were involved in sequencing, annotation and analysis of the genome and/or editing the final paper. JA, LM, AK, JZ, and WR drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work (proposal: 10.46936/10.25585/60007492) conducted by the US Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, is supported by the Office of Science of the US Department of Energy operated under Contract No. DE-AC02-05CH11231.
Acknowledgments
We gratefully acknowledge the funding received from the French National Agency of Research (Project BETASYM ANR-09-JCJ-0046), Curtin University Sustainability Policy Institute, and the funding received from Murdoch University Small Research Grants Scheme. We thank Gordon Thompson (Murdoch University) for the preparation of SEM and TEM photos.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1082107/full#supplementary-material
Abbreviations
½LA, 1/2 strength Lupin Agar; ANI, Average Nucleotide Identity; DoE, Department of Energy; EPS, Exopolysaccharide; GEBA-RNB, Genomic Encyclopedia for Bacteria and Archaea-Root Nodule Bacteria; GOLD, Genomes OnLine Database; MIGS, Minimum Information about the Genome Sequence; TY, Tryptone-Yeast extract agar; YMAm, Yeast-Mannitol Agar (modified).
Footnotes
1. ^https://www.osti.gov/biblio/1016000
2. ^https://github.com/lh3/wgsim
3. ^https://www.ncbi.nlm.nih.gov/
4. ^https://www.ncbi.nlm.nih.gov/genome/annotation_prok/tigrfams/
6. ^https://www.ebi.ac.uk/interpro/search/text/398365647/#table
7. ^https://www.genome.jp/kegg/
8. ^https://www.ncbi.nlm.nih.gov/research/cog-project/
9. ^https://www.ebi.ac.uk/interpro/
References
Allison, H. E. (2007). Stx-phages: drivers and mediators of the evolution of STEC and STEC-like pathogens. Future Microbiol. 2, 165–174. doi: 10.2217/17460913.2.2.165
Amadou, C., Pascal, G., Mangenot, S., Glew, M., Bontemps, C., Capela, D., et al. (2008). Genome sequence of the beta-rhizobium Cupriavidus taiwanensis and comparative genomics of rhizobia. Genome Res. 18, 1472–1483. doi: 10.1101/gr.076448.108
Andam, C. P., Mondo, S. J., and Parker, M. A. (2007). Monophyly of nodA and nifH genes across Texan and Costa Rican populations of Cupriavidus nodule symbionts. Appl. Environ. Microbiol. 73, 4686–4690. doi: 10.1128/AEM.00160-07
Aravind, L., and Landsman, D. (1998). AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 26, 4413–4421. doi: 10.1093/nar/26.19.4413
Askora, A., and Yamada, T. (2015). Two different evolutionary lines of filamentous phages in Ralstonia solanacearum: their effects on bacterial virulence. Front. Genet. 6:217. doi: 10.3389/fgene.2015.00217
Baas, P. D. (1985). DNA replication of single-stranded Escherichia coli DNA phages. Biochim. Biophys. Acta 825, 111–139. doi: 10.1016/0167-4781(85)90096-X
Barrett, C. F., and Parker, M. A. (2005). Prevalence of Burkholderia sp. nodule symbionts on four mimosoid legumes from Barro Colorado Island, Panama. Systematic and Applied Microbiology 28, 57–65. doi: 10.1016/j.syapm.2004.09.002
Beringer, J. E. (1974). R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84, 188–198.
Bertozzi Silva, J., Storms, Z., and Sauvageau, D. (2016). Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 363:fnw002. doi: 10.1093/femsle/fnw002
Bille, E., Zahar, J.-R., Perrin, A., Morelle, S., Kriz, P., Jolley, K. A., et al. (2005). A chromosomally integrated bacteriophage in invasive meningococci. J. Exp. Med. 201, 1905–1913. doi: 10.1084/jem.20050112
Bontemps, C., Elliott, G. N., Simon, M. F., Dos Reis Júnior, F. B., Gross, E., Lawton, R. C., et al. (2010). Burkholderia species are ancient symbionts of legumes. Mol. Ecol. 19, 44–52. doi: 10.1111/j.1365-294X.2009.04458.x
Bournaud, C., De Faria, S. M., Dos Santos, J. M. F., Tisseyre, P., Silva, M., Chaintreuil, C., et al. (2013). Burkholderia species are the most common and preferred nodulating symbionts of the Piptadenia group (tribe Mimoseae). PLoS One 8:e63478. doi: 10.1371/journal.pone.0063478
Carnoy, C., and Roten, C.-A. (2009). The dif/Xer recombination systems in Proteobacteria. PLoS One 4:e6531. doi: 10.1371/journal.pone.0006531
Castang, S., and Dove, S. L. (2012). Basis for the essentiality of H-NS family members in Pseudomonas aeruginosa. J. Bacteriol. 194, 5101–5109. doi: 10.1128/JB.00932-12
Chen, I.-M. A., Chu, K., Palaniappan, K., Ratner, A., Huang, J., Huntemann, M., et al. (2021). The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 49, D751–D763. doi: 10.1093/nar/gkaa939
Chen, W. M., Moulin, L., Bontemps, C., Vandamme, P., Béna, G., and Boivin-Masson, C. (2003). Legume symbiotic nitrogen fixation by beta-proteobacteria is widespread in nature. J. Bacteriol. 185, 7266–7272. doi: 10.1128/JB.185.24.7266-7272.2003
Chen, W.-M., Wu, C.-H., James, E. K., and Chang, J.-S. (2008). Metal biosorption capability of Cupriavidus taiwanensis and its effects on heavy metal removal by nodulated Mimosa pudica. J. Hazard. Mater. 151, 364–371. doi: 10.1016/j.jhazmat.2007.05.082
Cianfanelli, F. R., Monlezun, L., and Coulthurst, S. J. (2016). Aim, load, fire: the type VI secretion system, a bacterial nanoweapon. Trends Microbiol. 24, 51–62. doi: 10.1016/j.tim.2015.10.005
Claudel-Renard, C., Chevalet, C., Faraut, T., and Kahn, D. (2003). Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 31, 6633–6639. doi: 10.1093/nar/gkg847
Costa, T. R. D., Harb, L., Khara, P., Zeng, L., Hu, B., and Christie, P. J. (2021). Type IV secretion systems: advances in structure, function, and activation. Mol. Microbiol. 115, 436–452. doi: 10.1111/mmi.14670
Cubo, M. T., Alías-Villegas, C., Balsanelli, E., Mesa, D., De Souza, E., and Espuny, M. R. (2020). Diversity of Sinorhizobium (Ensifer) meliloti bacteriophages in the rhizosphere of Medicago marina: myoviruses, filamentous and N4-like podovirus. Front. Microbiol. 11:22. doi: 10.3389/fmicb.2020.00022
Da Silva, K., Florentino, L. A., Da Silva, K. B., De Brandt, E., Vandamme, P., and Moreira, F. M. D. (2012). Cupriavidus necator isolates are able to fix nitrogen in symbiosis with different legume species. Syst. Appl. Microbiol. 35, 175–182. doi: 10.1016/j.syapm.2011.10.005
Daubech, B., Poinsot, V., Klonowska, A., Capela, D., Chaintreuil, C., Moulin, L., et al. (2019). noeM, a new nodulation gene involved in the biosynthesis of nod factors with an open-chain oxidized terminal residue and in the symbiosis with Mimosa pudica. Mol. Plant-Microbe Interact. 32, 1635–1648. doi: 10.1094/MPMI-06-19-0168-R
Davis, B. M., Lawson, E. H., Sandkvist, M., Ali, A., Sozhamannan, S., and Waldor, M. K. (2000). Convergence of the secretory pathways for cholera toxin and the filamentous phage, CTXϕ. Science 288, 333–335. doi: 10.1126/science.288.5464.333
De Lajudie, P. M., Andrews, M., Ardley, J., Eardly, B., Jumas-Bilak, E., Kuzmanović, N., et al. (2019). Minimal standards for the description of new genera and species of rhizobia and agrobacteria. Int. J. Syst. Evol. Microbiol. 69, 1852–1863. doi: 10.1099/ijsem.0.003426
de Meyer, S. E., Ruthrof, K. X., Edwards, T., Hopkins, A. J. M., Hardy, G., O’Hara, G., et al. (2018). Diversity of endemic rhizobia on Christmas Island: implications for agriculture following phosphate mining. Syst. Appl. Microbiol. 41, 641–649. doi: 10.1016/j.syapm.2018.07.004
Deakin, W. J., and Broughton, W. J. (2009). Symbiotic use of pathogenic strategies: rhizobial protein secretion systems. Nat. Rev. Microbiol. 7, 312–320. doi: 10.1038/nrmicro2091
Denise, R., Abby, S. S., and Rocha, E. P. C. (2019). Diversification of the type IV filament superfamily into machines for adhesion, protein secretion, DNA uptake, and motility. PLoS Biol. 17:e3000390. doi: 10.1371/journal.pbio.3000390
Di Pierro, M., Lu, R., Uzzau, S., Wang, W., Margaretten, K., Pazzani, C., et al. (2001). Zonula occludens toxin structure-function analysis. Identification of the fragment biologically active on tight junctions and of the zonulin receptor binding domain. J. Biol. Chem. 276, 19160–19165. doi: 10.1074/jbc.M009674200
Dos Reis Jr, F. B., Simon, M. F., Gross, E., Boddey, R. M., Elliott, G. N., Neto, N. E., et al. (2010). Nodulation and nitrogen fixation by mimosa spp. in the Cerrado and Caatinga biomes of Brazil. New Phytol. 186, 934–946. doi: 10.1111/j.1469-8137.2010.03267.x
Estrada-de los Santos, P., Martínez-Aguilar, L., López-Lara, I. M., and Caballero-Mellado, J. (2012). Cupriavidus alkaliphilus sp. nov., a new species associated with agricultural plants that grow in alkaline soils. Syst. Appl. Microbiol. 35, 310–314. doi: 10.1016/j.syapm.2012.05.005
Estrada-de los Santos, P., Palmer, M., Chávez-Ramírez, B., Beukes, C., Steenkamp, E., Briscoe, L., et al. (2018). Whole genome analyses suggests that Burkholderia sensu lato contains two additional novel genera (Mycetohabitans gen. Nov., and Trinickia gen. Nov.): implications for the evolution of diazotrophy and nodulation in the Burkholderiaceae. Genes 9:389. doi: 10.3390/genes9080389
Fasano, A., Baudry, B., Pumplin, D. W., Wasserman, S. S., Tall, B. D., Ketley, J. M., et al. (1991). Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. U. S. A. 88, 5242–5246. doi: 10.1073/pnas.88.12.5242
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.2307/2408678
Fiedoruk, K., Zakrzewska, M., Daniluk, T., Piktel, E., Chmielewska, S., and Bucki, R. (2020). Two lineages of Pseudomonas aeruginosa filamentous phages: structural uniformity over integration preferences. Genome Biol. Evol. 12, 1765–1781. doi: 10.1093/gbe/evaa146
Figueroa-Bossi, N., Uzzau, S., Maloriol, D., and Bossi, L. (2001). Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in salmonella. Mol. Microbiol. 39, 260–272. doi: 10.1046/j.1365-2958.2001.02234.x
Folkesson, A., Löfdahl, S., and Normark, S. (2002). The salmonella enterica subspecies I specific centisome 7 genomic island encodes novel protein families present in bacteria living in close contact with eukaryotic cells. Res. Microbiol. 153, 537–545. doi: 10.1016/S0923-2508(02)01348-7
Ford, S., Moeskjær, S., Young, P., Santamaría, R. I., and Harrison, E. (2021). Introducing a novel, broad host range temperate phage family infecting rhizobium leguminosarum and beyond. Front. Microbiol. 12:5271. doi: 10.3389/fmicb.2021.765271
Gehlot, H. S., Tak, N., Kaushik, M., Mitra, S., Chen, W. M., Poweleit, N., et al. (2013). An invasive mimosa in India does not adopt the symbionts of its native relatives. Ann. Bot. 112, 179–196. doi: 10.1093/aob/mct112
Gnerre, S., Maccallum, I., Przybylski, D., Ribeiro, F. J., Burton, J. N., Walker, B. J., et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U. S. A. 108, 1513–1518. doi: 10.1073/pnas.1017351108
Gordon, B. R. G., Li, Y., Cote, A., Weirauch, M. T., Ding, P., Hughes, T. R., et al. (2011). Structural basis for recognition of AT-rich DNA by unrelated xenogeneic silencing proteins. Proc. Natl. Acad. Sci. U. S. A. 108, 10690–10695. doi: 10.1073/pnas.1102544108
Goris, J., Konstantinidis, K. T., Klappenbach, J., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Gyaneshwar, P., Hirsch, A. M., Moulin, L., Chen, W. M., Elliott, G. N., Bontemps, C., et al. (2011). Legume-nodulating betaproteobacteria: diversity, host range, and future prospects. Mol. Plant-Microbe Interact. 24, 1276–1288. doi: 10.1094/MPMI-06-11-0172
Harrison, E., and Brockhurst, M. A. (2017). Ecological and evolutionary benefits of temperate phage: what does or doesn’t kill you makes you stronger. BioEssays 39:1700112. doi: 10.1002/bies.201700112
Hay, I. D., and Lithgow, T. (2019). Filamentous phages: masters of a microbial sharing economy. EMBO Rep. 20:e47427. doi: 10.15252/embr.201847427
Holland, S. J., Sanz, C., and Perham, R. N. (2006). Identification and specificity of pilus adsorption proteins of filamentous bacteriophages infecting Pseudomonas aeruginosa. Virology 345, 540–548. doi: 10.1016/j.virol.2005.10.020
Howieson, J. G., De Meyer, S. E., Vivas-Marfisi, A., Ratnayake, S., Ardley, J. K., and Yates, R. J. (2013). Novel Burkholderia bacteria isolated from Lebeckia ambigua – a perennial suffrutescent legume of the fynbos. Soil Biol. Biochem. 60, 55–64. doi: 10.1016/j.soilbio.2013.01.009
Howieson, J. G., and Dilworth, M. J. (2016). Working with rhizobia. Australia: Australian Centre for International Agricultural Research (ACIAR).
Howieson, J. G., Ewing, M. A., and D’antuono, M. F. (1988). Selection for acid tolerance in rhizobium meliloti. Plant Soil 105, 179–188. doi: 10.1007/BF02376781
Huntemann, M., Ivanova, N. N., Mavromatis, K., Tripp, H. J., Paez-Espino, D., Palaniappan, K., et al. (2015). The standard operating procedure of the DOE-JGI microbial genome annotation pipeline (MGAP v.4). Stand. Genomic Sci. 10:86. doi: 10.1186/s40793-015-0077-y
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jach, M. E., Sajnaga, E., and Ziaja, M. (2022). Utilization of legume-nodule bacterial symbiosis in phytoremediation of heavy metal-contaminated soils. Biology (Basel) 11:676. doi: 10.3390/biology11050676
Jackson, A. P., Thomas, G. H., Parkhill, J., and Thomson, N. R. (2009). Evolutionary diversification of an ancient gene family (rhs) through C-terminal displacement. BMC Genomics 10:584. doi: 10.1186/1471-2164-10-584
Kanonenberg, K., Spitz, O., Erenburg, I. N., Beer, T., and Schmitt, L. (2018). Type I secretion system—it takes three and a substrate. FEMS Microbiol. Lett. 365, fny094. doi: 10.1093/femsle/fny094
Klonowska, A., Chaintreuil, C., Tisseyre, P., Miché, L., Melkonian, R., Ducousso, M., et al. (2012). Biodiversity of Mimosa pudica rhizobial symbionts (Cupriavidus taiwanensis, rhizobium mesoamericanum) in New Caledonia and their adaptation to heavy metal-rich soils. FEMS Microbiol. Ecol. 81, 618–635. doi: 10.1111/j.1574-6941.2012.01393.x
Klonowska, A., Melkonian, R., Miché, L., Tisseyre, P., and Moulin, L. (2018). Transcriptomic profiling of Burkholderia phymatum STM815, Cupriavidus taiwanensis LMG19424 and rhizobium mesoamericanum STM3625 in response to Mimosa pudica root exudates illuminates the molecular basis of their nodulation competitiveness and symbiotic evolutionary history. BMC Genomics 19:105. doi: 10.1186/s12864-018-4487-2
Klonowska, A., Moulin, L., Ardley, J. K., Braun, F., Gollagher, M. M., Zandberg, J. D., et al. (2020). Novel heavy metal resistance gene clusters are present in the genome of Cupriavidus neocaledonicus STM 6070, a new species of Mimosa pudica microsymbiont isolated from heavy-metal-rich mining site soil. BMC Genomics 21, 1–18. doi: 10.1186/s12864-020-6623-z
Knezevic, P., Voet, M., and Lavigne, R. (2015). Prevalence of Pf1-like (pro)phage genetic elements among Pseudomonas aeruginosa isolates. Virology 483, 64–71. doi: 10.1016/j.virol.2015.04.008
Korotkov, K. V., Sandkvist, M., and Hol, W. G. J. (2012). The type II secretion system: biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 10, 336–351. doi: 10.1038/nrmicro2762
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Liu, X., You, S., Liu, H., Yuan, B., Wang, H., James, E. K., et al. (2020). Diversity and geographic distribution of microsymbionts associated with invasive mimosa species in southern China. Front. Microbiol. 11:3389. doi: 10.3389/fmicb.2020.563389
Loh, B., Haase, M., Mueller, L., Kuhn, A., and Leptihn, S. (2017). The transmembrane morphogenesis protein gp1 of filamentous phages contains Walker a and Walker B motifs essential for phage assembly. Viruses 9:73. doi: 10.3390/v9040073
López-Leal, G., Camelo-Valera, L. C., Hurtado-Ramírez, J. M., Verleyen, J., Castillo-Ramírez, S., and Reyes-Muñoz, A. (2022). Mining of thousands of prokaryotic genomes reveals high abundance of prophages with a strictly narrow host range. mSystems 7:e00326–00322.
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Mahendran, V., Liu, F., Riordan, S. M., Grimm, M. C., Tanaka, M. M., and Zhang, L. (2016). Examination of the effects of campylobacter concisus zonula occludens toxin on intestinal epithelial cells and macrophages. Gut Pathogens 8:18. doi: 10.1186/s13099-016-0101-9
Mai-Prochnow, A., Hui, J. G. K., Kjelleberg, S., Rakonjac, J., Mcdougald, D., and Rice, S. A. (2015). Big things in small packages: the genetics of filamentous phage and effects on fitness of their host. FEMS Microbiol. Rev. 39, 465–487. doi: 10.1093/femsre/fuu007
Marie, C., Broughton, W. J., and Deakin, W. J. (2001). Rhizobium type III secretion systems: legume charmers or alarmers? Curr. Opin. Plant Biol. 4, 336–342. doi: 10.1016/S1369-5266(00)00182-5
Markowitz, V. M., Mavromatis, K., Ivanova, N. N., Chen, I. M., Chu, K., and Kyrpides, N. C. (2009). IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 25, 2271–2278. doi: 10.1093/bioinformatics/btp393
Marvin, D. A. (1998). Filamentous phage structure, infection and assembly. Curr. Opin. Struct. Biol. 8, 150–158. doi: 10.1016/S0959-440X(98)80032-8
Marvin, D. A., and Hoffmann-Berling, H. (1963). Physical and chemical properties of two new small bacteriophages. Nature 197, 517–518. doi: 10.1038/197517b0
Mattick, J. S. (2002). Type IV pili and twitching motility. Annu. Rev. Microbiol. 56, 289–314. doi: 10.1146/annurev.micro.56.012302.160938
Mcleod, S. M., and Waldor, M. K. (2004). Characterization of XerC- and XerD-dependent CTX phage integration in vibrio cholerae. Mol. Microbiol. 54, 935–947. doi: 10.1111/j.1365-2958.2004.04309.x
Melkonian, R., Moulin, L., Béna, G., Tisseyre, P., Chaintreuil, C., Heulin, K., et al. (2014). The geographical patterns of symbiont diversity in the invasive legume Mimosa pudica can be explained by the competitiveness of its symbionts and by the host genotype. Environ. Microbiol. 16, 2099–2111. doi: 10.1111/1462-2920.12286
Mishra, R. P., Tisseyre, P., Melkonian, R., Chaintreuil, C., Miché, L., Klonowska, A., et al. (2012). Genetic diversity of Mimosa pudica rhizobial symbionts in soils of French Guiana: investigating the origin and diversity of Burkholderia phymatum and other beta-rhizobia. FEMS Microbiol. Ecol. 79, 487–503. doi: 10.1111/j.1574-6941.2011.01235.x
Morgan, G. J., Hatfull, G. F., Casjens, S., and Hendrix, R. W. (2002). Bacteriophage mu genome sequence: analysis and comparison with mu-like prophages in Haemophilus, Neisseria and Deinococcus. J. Mol. Biol. 317, 337–359. doi: 10.1006/jmbi.2002.5437
Moulin, L., Munive, A., Dreyfus, B., and Boivin-Masson, C. (2001). Nodulation of legumes by members of the beta-subclass of Proteobacteria. Nature 411, 948–950. doi: 10.1038/35082070
Mukherjee, S., Stamatis, D., Bertsch, J., Ovchinnikova, G., Sundaramurthi, J. C., Lee, J., et al. (2020). Genomes OnLine database (GOLD) v.8: overview and updates. Nucleic Acids Res. 49, D723–D733. doi: 10.1093/nar/gkaa983
Nakamura, K., Ogura, Y., Gotoh, Y., and Hayashi, T. (2021). Prophages integrating into prophages: a mechanism to accumulate type III secretion effector genes and duplicate Shiga toxin-encoding prophages in Escherichia coli. PLoS Pathog. 17:e1009073. doi: 10.1371/journal.ppat.1009073
Navarre, W. W. (2016). “The impact of gene silencing on horizontal gene transfer and bacterial evolution” in Advances in microbial physiology. ed. R. K. Poole (Cambridge: Academic Press), 157–186.
Nunn, D. N., and Lory, S. (1992). Components of the protein-excretion apparatus of Pseudomonas aeruginosa are processed by the type IV prepilin peptidase. Proc. Natl. Acad. Sci. U. S. A. 89, 47–51. doi: 10.1073/pnas.89.1.47
Parker, M. A. (2015). A single sym plasmid type predominates across diverse chromosomal lineages of Cupriavidus nodule symbionts. Syst. Appl. Microbiol. 38, 417–423. doi: 10.1016/j.syapm.2015.06.003
Parker, M. A., Wurtz, A. K., and Paynter, Q. (2007). Nodule symbiosis of invasive Mimosa pigra in Australia and in ancestral habitats: a comparative analysis. Biol. Invasions 9, 127–138. doi: 10.1007/s10530-006-0009-2
Pfeifer, E., Hünnefeld, M., Popa, O., Polen, T., Kohlheyer, D., Baumgart, M., et al. (2016). Silencing of cryptic prophages in Corynebacterium glutamicum. Nucleic Acids Res. 44, 10117–10131. doi: 10.1093/nar/gkw692
Platero, R., James, E. K., Rios, C., Iriarte, A., Sandes, L., Zabaleta, M., et al. (2016). Novel Cupriavidus strains isolated from root nodules of native Uruguayan mimosa species. Appl. Environ. Microbiol. 82, 3150–3164. doi: 10.1128/AEM.04142-15
Poehlein, A., Kusian, B., Friedrich, B., Daniel, R., and Bowien, B. (2011). Complete genome sequence of the type strain Cupriavidus necator N-1. J. Bacteriol. 193:5017. doi: 10.1128/JB.05660-11
Pratama, A. A., Chaib De Mares, M., and Van Elsas, J. D. (2018). Evolutionary history of bacteriophages in the genus Paraburkholderia. Front. Microbiol. 9:835. doi: 10.3389/fmicb.2018.00835
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. D. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Reeve, W., Ardley, J., Tian, R., Eshragi, L., Yoon, J. W., Ngamwisetkun, P., et al. (2015). A genomic encyclopedia of the root nodule bacteria: assessing genetic diversity through a systematic biogeographic survey. Stand. Genomic Sci. 10:14. doi: 10.1186/1944-3277-10-14
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rinaudi, L. V., and Giordano, W. (2010). An integrated view of biofilm formation in rhizobia. FEMS Microbiol. Lett. 304, 1–11. doi: 10.1111/j.1574-6968.2009.01840.x
Roux, S., Krupovic, M., Daly, R. A., Borges, A. L., Nayfach, S., Schulz, F., et al. (2019). Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 4, 1895–1906. doi: 10.1038/s41564-019-0510-x
Saad, M. M., Crevecoeur, M., Masson-Boivin, C., and Perret, X. (2012). The type 3 protein secretion system of Cupriavidus taiwanensis strain LMG19424 compromises symbiosis with Leucaena leucocephala. Appl. Environ. Microbiol. 78, 7476–7479. doi: 10.1128/AEM.01691-12
Secor, P. R., Burgener, E. B., Kinnersley, M., Jennings, L. K., Roman-Cruz, V., Popescu, M., et al. (2020). Pf bacteriophage and their impact on pseudomonas virulence, mammalian immunity, and chronic infections. Front. Immunol. 11:244. doi: 10.3389/fimmu.2020.00244
Shapiro, J. W., Williams, E. S., and Turner, P. E. (2016). Evolution of parasitism and mutualism between filamentous phage M13 and Escherichia coli. PeerJ 4:e2060. doi: 10.7717/peerj.2060
Sharma, R. S., Karmakar, S., Kumar, P., and Mishra, V. (2019). Application of filamentous phages in environment: a tectonic shift in the science and practice of ecorestoration. Ecol. Evol. 9, 2263–2304. doi: 10.1002/ece3.4743
Singh, K., Milstein, J. N., and Navarre, W. W. (2016). Xenogeneic silencing and its impact on bacterial genomes. Annu. Rev. Microbiol. 70, 199–213. doi: 10.1146/annurev-micro-102215-095301
Skennerton, C. T., Angly, F. E., Breitbart, M., Bragg, L., He, S., Mcmahon, K. D., et al. (2011). Phage encoded H-NS: a potential Achilles heel in the bacterial defence system. PLoS One 6:e20095. doi: 10.1371/journal.pone.0020095
Sun, L.-N., Wang, D.-S., Yang, E.-D., Fang, L.-C., Chen, Y.-F., Tang, X.-Y., et al. (2016). Cupriavidus nantongensis sp. nov., a novel chlorpyrifos-degrading bacterium isolated from sludge. Int. J. Syst. Evol. Microbiol. 66, 2335–2341. doi: 10.1099/ijsem.0.001034
Tamura, K., Nei, M., and Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U. S. A. 101, 11030–11035. doi: 10.1073/pnas.0404206101
Taulé, C., Zabaleta, M., Mareque, C., Platero, R., Sanjurjo, L., Sicardi, M., et al. (2012). New Betaproteobacterial rhizobium strains able to efficiently nodulate Parapiptadenia rigida (Benth.) Brenan. Appl. Environ. Microbiol. 78, 1692–1700. doi: 10.1128/AEM.06215-11
Teng, Y., Wang, X., Li, L., Li, Z., and Luo, Y. (2015). Rhizobia and their bio-partners as novel drivers for functional remediation in contaminated soils. Front. Plant Sci. 6:32. doi: 10.3389/fpls.2015.00032
Trucksis, M., Conn, T. L., Wasserman, S. S., and Sears, C. L. (2000). Vibrio cholerae ACE stimulates Ca2+−dependent cl−/HCO3 −secretion in T84 cells in vitro. Am. J. Phys. Cell Phys. 279, C567–C577. doi: 10.1152/ajpcell.2000.279.3.C567
Trucksis, M., Galen, J. E., Michalski, J., Fasano, A., and Kaper, J. B. (1993). Accessory cholera enterotoxin (ace), the third toxin of a vibrio cholerae virulence cassette. Proc. Natl. Acad. Sci. U. S. A. 90, 5267–5271. doi: 10.1073/pnas.90.11.5267
Vallenet, D., Calteau, A., Dubois, M., Amours, P., Bazin, A., Beuvin, M., et al. (2019). MicroScope: an integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucleic Acids Res. 48, D579–D589. doi: 10.1093/nar/gkz926
Van Houdt, R., Provoost, A., Van Assche, A., Leys, N., Lievens, B., Mijnendonckx, K., et al. (2018). Cupriavidus metallidurans strains with different mobilomes and from distinct environments have comparable phenomes. Genes 9:507. doi: 10.3390/genes9100507
Vandamme, P., and Coenye, T. (2004). Taxonomy of the genus Cupriavidus: a tale of lost and found. Int. J. Syst. Evol. Microbiol. 54, 2285–2289. doi: 10.1099/ijs.0.63247-0
Varghese, N. J., Mukherjee, S., Ivanova, N., Konstantinidis, K. T., Mavrommatis, K., Kyrpides, N. C., et al. (2015). Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771. doi: 10.1093/nar/gkv657
Wagner, P. L., and Waldor, M. K. (2002). Bacteriophage control of bacterial virulence. Infect. Immun. 70, 3985–3993. doi: 10.1128/IAI.70.8.3985-3993.2002
Waldor, M. K., and Mekalanos, J. J. (1996). Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914. doi: 10.1126/science.272.5270.1910
Wawrzyniak, P., Płucienniczak, G., and Bartosik, D. (2017). The different faces of rolling-circle replication and its multifunctional initiator proteins. Front. Microbiol. 8:2353. doi: 10.3389/fmicb.2017.02353
Weber, B. S., Hennon, S. W., Wright, M. S., Scott, N. E., Berardinis, V. D., Foster, L. J., et al. (2016). Genetic dissection of the type VI secretion system in Acinetobacter and identification of a novel peptidoglycan hydrolase, TagX, required for its biogenesis. MBio 7, e01253–e01216. doi: 10.1128/mBio.01253-16
Zatakia, H. M., Nelson, C. E., Syed, U. J., Scharf, B. E., and Parales, R. E. (2014). ExpR coordinates the expression of symbiotically important, bundle-forming Flp pili with quorum sensing in Sinorhizobium meliloti. Appl. Environ. Microbiol. 80, 2429–2439. doi: 10.1128/AEM.04088-13
Keywords: Betaproteobacteria, root-nodule bacteria, bacteriophage, filamentous phage, symbiosis
Citation: Klonowska A, Ardley J, Moulin L, Zandberg J, Patrel D, Gollagher M, Marinova D, Reddy TBK, Varghese N, Huntemann M, Woyke T, Seshadri R, Ivanova N, Kyrpides N and Reeve W (2023) Discovery of a novel filamentous prophage in the genome of the Mimosa pudica microsymbiont Cupriavidus taiwanensis STM 6018. Front. Microbiol. 14:1082107. doi: 10.3389/fmicb.2023.1082107
Edited by:
Alejandra Prieto-Davó, National Autonomous University of Mexico, MexicoReviewed by:
Diogo Antonio Tschoeke, Federal University of Rio de Janeiro, BrazilRosario Gil, University of Valencia, Spain
Copyright © 2023 Klonowska, Ardley, Moulin, Zandberg, Patrel, Gollagher, Marinova, Reddy, Varghese, Huntemann, Woyke, Seshadri, Ivanova, Kyrpides and Reeve. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie Ardley, ✉ Si5BcmRsZXlAbXVyZG9jaC5lZHUuYXU=; Wayne Reeve, ✉ Vy5SZWV2ZUBtdXJkb2NoLmVkdS5hdQ==