
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
TECHNOLOGY AND CODE article
Front. Microbiol. , 30 March 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1050130
 Xuemei Liu1†
Xuemei Liu1† Ziqi Cheng2†
Ziqi Cheng2† Guohao Xu3
Guohao Xu3 Jiemin Xie3
Jiemin Xie3 Xudong Liu1
Xudong Liu1 Bozhen Ren3
Bozhen Ren3 Dongmei Ai4
Dongmei Ai4 Yangxin Chen5,6*
Yangxin Chen5,6* Li Charlie Xia3*
Li Charlie Xia3*Phylogenetic tools are fundamental to the studies of evolutionary relationships. In this paper, we present Ksak, a novel high-throughput tool for alignment-free phylogenetic analysis. Ksak computes the pairwise distance matrix between molecular sequences, using seven widely accepted k-mer based distance measures. Based on the distance matrix, Ksak constructs the phylogenetic tree with standard algorithms. When benchmarked with a golden standard 16S rRNA dataset, Ksak was found to be the most accurate tool among all five tools compared and was 19% more accurate than ClustalW2, a high-accuracy multiple sequence aligner. Above all, Ksak was tens to hundreds of times faster than ClustalW2, which helps eliminate the computation limit currently encountered in large-scale multiple sequence alignment. Ksak is freely available at https://github.com/labxscut/ksak.
Phylogenetic analysis is the cornerstone of evolutionary biology and taxonomy. Phylogeny based on molecular sequence similarity has become the de facto standard. All subsequences of size k derived from a molecular sequence are called its k-mers. Numerous studies demonstrated that k-mers of molecular sequences, such as genomic DNA and proteins are conserved within closely related organisms, and diverge with speciation (Fan et al., 2015; Zhang et al., 2019). Thus k-mer statistics are efficient and effective phylogenetic distance measures (Bussi et al., 2021).
We developed Ksak – a tool that not only efficiently computes seven widely accepted k-mer statistics: Chebyshev (Ch), Manhattan (Ma), Euclidian (Eu), Hao (Qi et al., 2004), d2, d2S, and d2star (Song et al., 2013), but also performs alignment-free phylogenetic analysis. By applying Ksak to the golden standard 16S rRNA dataset, we extensively benchmarked its accuracy and efficiency by comparing to Muscle (Edgar, 2004), ClustalW2 (Patel et al., 2012), Mafft (Katoh and Standley, 2014), Cafe (Lu et al., 2017) and Afann (Tang et al., 2019) – six popular multiple sequence aligners and phylogenetic analysis tools. We made the software of Ksak open source with this paper. Ksak runs on MS Windows operating systems with a graphical user interface (see Supplementary Figure 1).
Ksak constructs a phylogenetic tree from the input of N molecular sequences in four steps: (1) it counts the k-mer frequency in each input source sequence; (2) it merges the obtained k-mer frequencies into a 4k-by-N frequency matrix; (3) it applies the user-specified distance measures, and parameters k and M (if needed) to calculate the pairwise distance, obtaining an N-by-N distance matrix, where M is the order of background Markov model (see Supplementary Methods); (4) it applies the Unweighted Pair Group Method with Arithmetic Mean (UPGMA; Sokal, 1958) or the Neighbour Joining (NJ; Saitou and Nei, 1987) algorithms to the distance matrix and constructs the phylogenetic tree (Figure 1A).
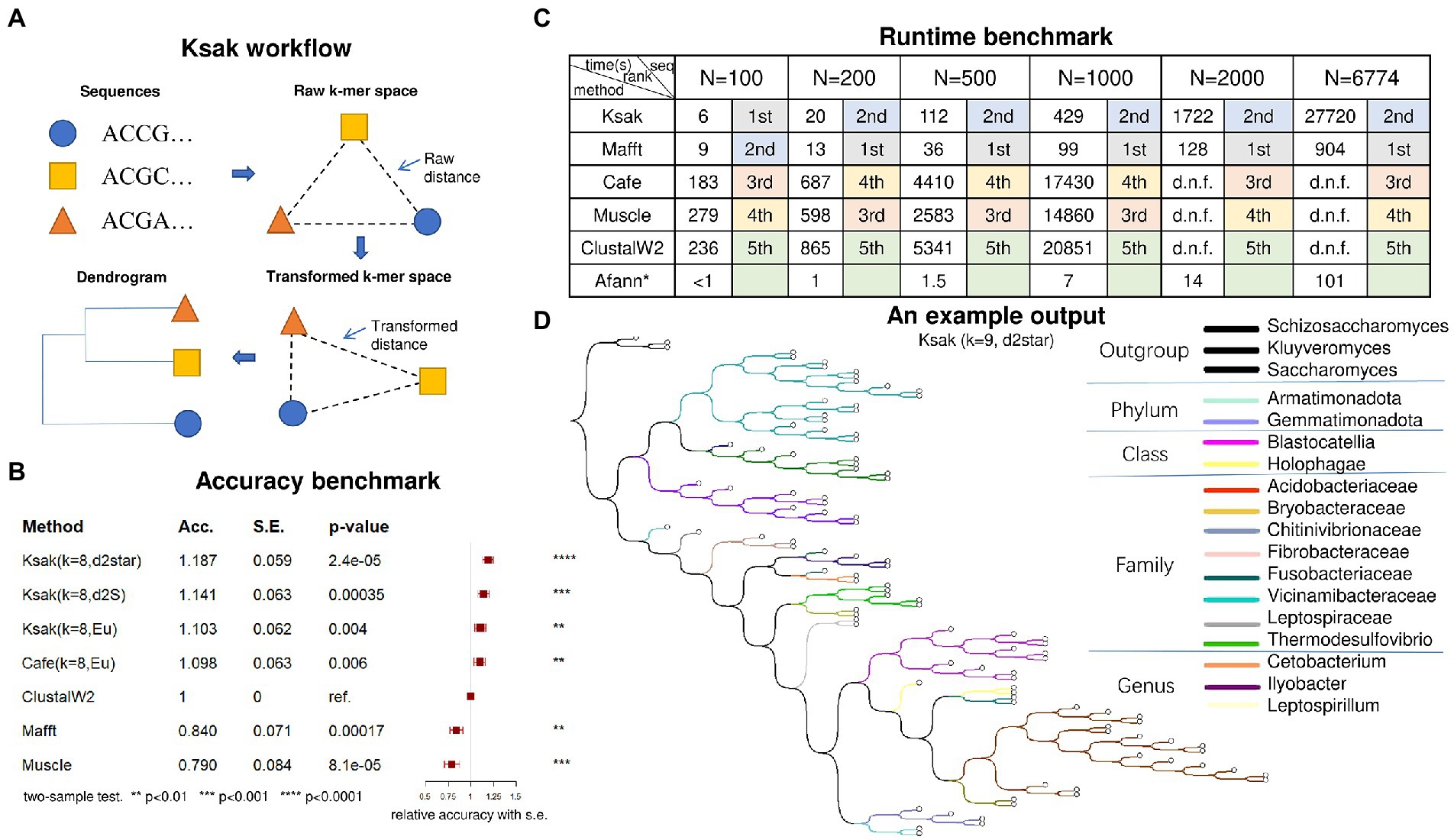
Figure 1. (A) The computation framework of Ksak; (B) Accuracy benchmark results; (C) Efficiency benchmark results; (D) An example phylogeny tree output of Ksak.
Efficient counting of k-mers is the basis for k-mer based statistical tools. In recent years, a variety of applications with many methods to count k-mers were developed (Crusoe et al., 2015; Bize et al., 2021; Cattaneo et al., 2022). Given the fact that the most useful k for alignment-free phylogenetics is relatively small, typically <10, we implemented a neat yet ultra-efficient algorithm to count k-mer frequency (see Supplementary Figure 2 for an illustrated example), which mathematically transforms a k-mer to its index based on the powers of 4. Accordingly, this index can randomly address and efficiently operate on an integer array of k-mer counts in computer memory. This neat algorithm allows Ksak to process thousands of sequences in minutes with only a personal computer.
We downloaded a 16S rRNA sequence dataset containing an expert-curated phylogenetic tree from All-species Living Tree Project (LTP) as the golden standard for benchmarking (Yilmaz et al., 2014; Beccati et al., 2017). However, it is important to note that performing an alignment-free phylogenetic analysis with Ksak is not restricted to any specific genes or genomes. The LTP has >6,700 sequences which spans the kingdoms of archaea and bacteria. We applied a series of subsampled LTP data for computation efficiency benchmarks. We also randomly selected 100 sequences from the LTP tree to form a subsampled, balanced ground truth tree, and also selected 3 yeast sequences (Kluyve-romyces, Schizosaccharomyces, Saccharomyces) outgroup to the truth tree (see Supplementary Tables 1, 2 for the full list of sequences).
We applied Ksak to calculate the transformed k-mer distance matrix of the sequences included in the truth tree (Figure 1D). Based on that we inferred their phylogenetic tree and compared it to the truth tree by relative accuracy, with the performance of ClustalW2 specified as the reference (Figure 1B). The relative accuracy is defined as the ratio of target’s and reference method’s symmetric difference – an error measuring the inner branches difference between the inferred and the truth tree. The result showed that Ksak was the most accurate tool of all tools compared and was 19% more accurate than ClustalW2 – a widely used multiple sequence aligner, when Ksak was configured with k = 8 and using the d2star measure. The Ape package in R was used to compute the symmetric differences (Robinson and Foulds, 1981).
To evaluate the computational efficiency of Ksak, we compared its run time cost to the other five alignment and non-alignment tools: Mafft, Muscle, Cafe, Afann, and ClustalW2. While Afann is ultra-fast in counting k-mers, it was not ranked because it does not produce an output tree. Among the other five tools did output phylogenetic trees, we found Ksak was significantly faster than Muscle, Cafe and ClustalW2, for all input sequence sets size N ranging from 100 to 6,774 (Figure 1C). It was ranked in the top tier (1st or 2nd) with Mafft while Ksak is 35% more accurate than Mafft (Figure 1B). At N = 6,774, the run time of Ksak was capped at 27, 720 s while ClustalW2, the most accurate multiple sequence aligner, did not accomplish the job given 8 h. At N = 1,000, the largest input size that all tools can accomplish in time, Ksak has a 40-times run time cost reduction over Cafe. In the benchmark, all the tools were given the same input sequences to generate phylogenetic tree with the measure Eu (k = 8). All the run time cost computation was done on a personal computer, with Intel Core i7-4790K CPU @ 4.00GHz, 32G mem, Windows 11 and Ubuntu 20.
In this paper, we presented Ksak, a novel high-throughput tool for alignment-free phylogenetic analysis. We found that the measure d2star (k = 9) is generally the most accurate for alignment-free phylogenetics (see Supplementary Figures 3, 4). For user application of Ksak, we provided an evolutionary relationship analysis of 27 coronavirus sequences with guides and explanations in the Supplementary Figure 1. To further prove the computing power and speed of Ksak, we also provided a full-scale phylogenetic analysis of 50 bacteria whole genome sequences, which was finished within 32.85 s using d2star (k = 9), see Supplementary Figure 5. Given its neat and easy-to-use quality, we hope Ksak to be a handy tool for the research community when analyzing large-scale microbiome data.
Publicly available datasets were analyzed in this study. This data can be found at: http://www.arb-silva.de.
XML, YC, and LCX conceived the project and designed the study. ZC and XML implemented the software. XML, GX, JX, XDL, BR, and DA performed the analysis. All authors contributed to the article and approved the submitted version.
YC was supported by National Natural Science Foundation of China (81970200, 82271609). LCX was supported by the Guangdong Basic and Applied Basic Research Foundation (2022A1515-011426), National Natural Science Foundation of China (61873027).
We would like to thank Yunhui Xiong, at the School of Mathematics, South China University of Technology, Guangzhou, Guangdong, China who provided us with technical support.
ZC was employed by Guangzhou Boguan Telecommunication Technology Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer NJ declared a shared affiliation with the author YC to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1050130/full#supplementary-material
Beccati, A., Gerken, J., Quast, C., Yilmaz, P., and Glockner, F. O. (2017). SILVA tree viewer: interactive web browsing of the SILVA phylogenetic guide trees. Bmc Bioinformatics 18:433. doi: 10.1186/s12859-017-1841-3
Bize, A., Midoux, C., Mariadassou, M., Schbath, S., Forterre, P., and Da Cunha, V. (2021). Exploring short k-mer profiles in cells and mobile elements from archaea highlights the major influence of both the ecological niche and evolutionary history. BMC Genomics 22:ARTN 186. doi: 10.1186/s12864-021-07471-y
Bussi, Y., Kapon, R., and Reich, Z. (2021). Large-scale k-mer-based analysis of the informational properties of genomes, comparative genomics and taxonomy. PLoS One 16:e0258693. doi: 10.1371/journal.pone.0258693
Cattaneo, G., Petrillo, U. F., Giancarlo, R., Palini, F., and Romualdi, C. (2022). The power of word-frequency-based alignment-free functions: a comprehensive large-scale experimental analysis. Bioinformatics 38, 925–932. doi: 10.1093/bioinformatics/btab747
Crusoe, M. R., Alameldin, H. F., Awad, S., Boucher, E., Caldwell, A., Cartwright, R., et al. (2015). The khmer software package: enabling efficient nucleotide sequence analysis. F1000Res 4:900. doi: 10.12688/f1000research.6924.1
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. Bmc Bioinformatics 5, 113–119. doi: 10.1186/1471-2105-5-113
Fan, H., Ives, A. R., Surget-Groba, Y., and Cannon, C. H. (2015). An assembly and alignment-free method of phylogeny reconstruction from next-generation sequencing data. BMC Genomics 16:522. doi: 10.1186/s12864-015-1647-5
Katoh, K., and Standley, D. M. (2014). MAFFT: iterative refinement and additional methods. Methods Mol. Biol. 1079, 131–146. doi: 10.1007/978-1-62703-646-7_8
Lu, Y. Y., Tang, K. J., Ren, J., Fuhrman, J. A., Waterman, M. S., and Sun, F. Z. (2017). CAFE: aCcelerated alignment-FrEe sequence analysis. Nucleic Acids Res. 45, W554–W559. doi: 10.1093/nar/gkx351
Patel, S., Panchal, H., and Anjaria, K. (2012). “Phylogenetic analysis of some leguminous trees using CLUSTALW2 bioinformatics tool,” in 2012 IEEE International Conference on Bioinformatics and Biomedicine Workshops (Bibmw).
Qi, J., Wang, B., and Hao, B. I. (2004). Whole proteome prokaryote phylogeny without sequence alignment: a K-string composition approach. J. Mol. Evol. 58, 1–11. doi: 10.1007/s00239-003-2493-7
Robinson, D. F., and Foulds, L. R. (1981). Comparison of phylogenetic trees. Math. Biosci. 53, 131–147. doi: 10.1016/0025-5564(81)90043-2
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sokal, R. R. (1958). A statistical method for evaluating systematic relationships. Univ. Kansas, Sci. Bull. 38, 1409–1438.
Song, K., Ren, J., Zhai, Z. Y., Liu, X. M., Deng, M. H., and Sun, F. Z. (2013). Alignment-free sequence comparison based on next-generation sequencing reads. J. Comput. Biol. 20, 64–79. doi: 10.1089/cmb.2012.0228
Tang, K., Ren, J., and Sun, F. (2019). Afann: bias adjustment for alignment-free sequence comparison based on sequencing data using neural network regression. Genome Biol. 20, 266–217. doi: 10.1186/s13059-019-1872-3
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and "all-species living tree project (LTP)" taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Keywords: k-mer, phylogentic tree, alignment free, open source, microbiome
Citation: Liu X, Cheng Z, Xu G, Xie J, Liu X, Ren B, Ai D, Chen Y and Xia LC (2023) Ksak: A high-throughput tool for alignment-free phylogenetics. Front. Microbiol. 14:1050130. doi: 10.3389/fmicb.2023.1050130
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Na Jiao, Sun Yat-sen University, ChinaCopyright © 2023 Liu, Cheng, Xu, Xie, Liu, Ren, Ai, Chen and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Charlie Xia, bGN4aWFAc2N1dC5lZHUuY24=; Yangxin Chen, Y2hlbnl4MzlAbWFpbC5zeXN1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.