 Wenfeng Guo1,2
Wenfeng Guo1,2 Wei Wang
Wei Wang Xiaoqiong Li
Xiaoqiong Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 16 February 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1048910
Fungus-farming mutualisms are models for studying co-evolutionary among species. Compared to well-documented fungus-farming in social insects, the molecular aspects of fungus-farming mutualisms in nonsocial insects have been poorly explored. Euops chinensis is a solitary leaf-rolling weevil feeding on Japanese knotweed (Fallopia japonica). This pest has evolved a special proto-farming bipartite mutualism with the fungus Penicillium herquei, which provide nutrition and defensive protection for the E. chinensis larvae. Here, the genome of P. herquei was sequenced, and the structure and specific gene categories in the P. herquei genome were then comprehensively compared with the other two well-studied Penicillium species (P. decumbens and P. chrysogenum). The assembled P. herquei genome had a 40.25 Mb genome size with 46.7% GC content. A diverse set of genes associating with carbohydrate-active enzymes, cellulose and hemicellulose degradation, transporter, and terpenoid biosynthesis were detected in the P. herquei genome. Comparative genomics demonstrate that the three Penicillium species show similar metabolic and enzymatic potential, however, P. herquei has more genes associated with plant biomass degradation and defense but less genes associating with virulence pathogenicity. Our results provide molecular evidence for plant substrate breakdown and protective roles of P. herquei in E. chinensis mutualistic system. Large metabolic potential shared by Penicillium species at the genus level may explain why some Penicillium species are recruited by the Euops weevils as crop fungi.
A number of insects have established various associations with microorganisms, and among these, insect-fungus mutualisms are one of the better-studied in nature (Biedermann and Vega, 2020). Many eusocial and subsocial insects such as ants (Mueller et al., 2001), termites (Aanen, 2006), and ambrosia beetles (Kirkendall et al., 2015) exhibit sophisticated forms of mutualistic relationships with fungi (fungus-farming mutualisms) that allow these insects to cultivate their fungal crops on appropriate substrates, and in turn these insects obligatorily rely on consumption of their fungal crops for key nutrients and/or certain services such as protection from enemies (Biedermann and Vega, 2020). Mutualisms between fungi and fungus-farming insects are model systems for studying co-evolutionary interactions between species (Nygaard et al., 2016; Solomon et al., 2019; Biedermann and Vega, 2020; Pereira and Kjellberg, 2021). Compared to the well-documented fungus-farming mutualisms in some social insects, fungus farming by non-social organisms is uncommon, but includes some examples such as a lizard beetle Doubledaya bucculenta (Toki et al., 2012), weevils in the genus Euops (Coleoptera: Attelabidae) (Sawada and Morimoto, 1986; Kobayashi et al., 2008; Li et al., 2012), a marine snail (Silliman and Newell, 2003), and several species of damselfish (Hata and Kato, 2006). The biological and molecular aspects of fungus-farming mutualisms in solitary, non-social insects have been poorly explored.
Bioinformatic analysis of the genomic sequence of a fungal symbiote can be used to assist the discovery of molecular mechanisms responsible for symbiotic association, nutrition, pathogenicity, and/or defensive protection (Fan et al., 2015; Fu et al., 2020). Moreover, comparative genomics is a useful tool to better understand the molecular basis of fungus-farming mutualism in insects (Nygaard et al., 2011; Poulsen et al., 2014). Many molecular and genomic studies have provided new insights to our understanding of the establishment (Mueller et al., 2018), persistence (Schmidt et al., 2022), recognition and specificity adaptations (da Costa et al., 2019; Skelton et al., 2019; Goes et al., 2020) of fungus-growing insects and their mutualistic fungal symbiotes in recent years. The available evidence has shown that mutualistic symbiotes are crucial for the survival of the host insects (Schmidt et al., 2022), the generation of phenotypic diversity (Gohli et al., 2017; Solomon et al., 2019), and in the origin of various niches (Six, 2020). Fungus-farming mutualisms can result in specialized organs with unique development (Mayers et al., 2022), novel metabolic capabilities (Huang et al., 2019; Schmidt et al., 2022), better defense against natural enemies or other stresses (Pathak et al., 2019), and contraction or expansion of novel genes (Nygaard et al., 2016). Elucidating how fungus-farming mutualisms influence genes and genomes is essential to better understand fungal evolution from a molecular sequence perspective.
Weevils of the genus Euops (Attelabidae) present interesting model systems for studying fungus-farming mutualism in nonsocial insects (Sakurai, 1985; Kobayashi et al., 2008; Grebennikov and Leschen, 2010; Li et al., 2012). All species of this genus have developed mutualistic relationships with fungi (Sawada and Morimoto, 1986). Euops chinensis Voss (Coleoptera: Attelabidae) is a solitary leaf-rolling weevil on Japanese knotweed Fallopia japonica (Houtt.) Ronse Decraene (Wang et al., 2010). This pest has evolved a special proto-farming bipartite mutualism with the fungus Penicillium herquei that the female adults carry the fungus in a specialized fungus transport organ called mycetangium (Francke-Grosmann, 1967; Grebennikov and Leschen, 2010), and inoculate the fungal spores to the leaf-rolls before laying eggs (Li et al., 2012). Interesting, the other two Euops leaf-rolling weevils, Euops lespedezae (Kobayashi et al., 2008) and Euops splendida (Sakurai, 1985), have also been reported to form fungus-farming mutualisms with Penicillium spp. However, Euops - Penicillium system remains to be poorly studied compared to other fungus-farming insects like ants and termites.
Although E. chinensis is obligatory dependency on P. herquei for nutrition and protection like other fungus-farming insects, its fungus proto-farming behavior pparently differs from that of social insects in that the female weevils do not continuously tend the cultivated material as the leaf-rolls are cut from the plants and gradually decay (Li et al., 2012; Wang et al., 2015), and therefore, P. herquei has a saprophytic life style when growing on the leaf-rolls. This fungal cultivar has been demonstrated to be vertically transmitted by the weevil (Li et al., 2016), and obligatorily benefit the development of E. chinensis by altering the chemical composition of leaf-rolls (Li et al., 2012) and protecting leaf-rolls against plant pathogens (Wang et al., 2015), it also acts as a food resource for the larvae (Wang et al., 2010; Li et al., 2012). Thus, the growth, development, or survival of E. chinensis are strongly influenced by P. herquei (Li et al., 2012).
In this study, we provide the first detailed description of the genome of P. herquei. The structure, metabolic capabilities, secondary metabolite gene clusters, and important pathogenic characteristics of the P. herquei genome were then comprehensively compared with the other two well-studied Penicillium species, P. chrysogenum and P. decumbens. The results would provide new insights into the molecular basis of fungus-farming mutualism in Euops - Penicillium system.
Leaf-rolls constructed by E. chinensis were collected from Jiangxi Province (N27°46′16.33, E114°23′38.30), China, in early May in 2021. Penicillium herquei on the leaf-rolls was isolated according to the methods of Li et al. (2012), and the fungus was cultured on potato dextrose agar (PDA) plates. The strain Penicillium herquei XQL_2021 was purified by single-spore isolation, and was preserved at −80°C prior to use. For genome DNA extraction and sequencing, the fungal strain was cultured on PDA plates for 2 weeks at 25°C, the spores were then harvested by flooding with sterile distilled water, a 1-ml aliquot of a spore suspension was added to 20 ml potato dextrose broth (PDB) medium in 50-ml conical flasks and was cultured at 25°C on a rotary shaker at 200 rpm for 3 days. Fungal hyphae were collected in sterile tubes by filtering the culture liquid thoroughly with sterilized water. The hyphae collected were then washed thoroughly with sterilized water, immediately frozen with liquid nitrogen, and stored at −80°C until used.
About 2.0 g aliquots of hyphae (fresh weight) was collected, and genomic DNA was extracted based on the cetyltrimethylammonium bromide (CTAB) methods (Porebski et al., 1997). The quantity and quality of the extracted genomic DNA were checked using a Nanodrop (Thermo Scientific, United States).
For short reads, sequencing was done on a NovaSeq 6000 platform, and a large fragment library was prepared using a NEBNext® Ultra™ II DNA Library Prep Kit for Illumina (NEB, United States). The library was quantified using an Agilent 2100 bioanalyzer instrument (Agilent DNA 1000 reagents; Agilent, Santa Clara, CA, United States) and real-time quantitative PCR (RT-qPCR). The qualified libraries were amplified within the flow cell on an Illumina cBOT instrument for cluster generation (NovaSeq 6000 PE cluster kit; Illumina). The clustered flow cell was loaded onto a NovaSeq 6000 sequencer (NovaSeq 6000 SBS kit; Illumina) for paired-end sequencing with recommended read lengths of 150 bp. Raw reads were filtered using the SOAPnuke (v2.1.4) tool1 to remove reads with adaptors or unknown nucleotides, and low-quality reads with ≥50% low-quality bases. After data filtering, clean data were used for subsequent analyses.
For long reads, sequencing was done on a Nanopore PromethION platform, and the libraries were prepared with an Oxford Nanopore ligation kit (SQK-LSK109) according to a standard protocol. The purified library was loaded onto a primed R9.4 Spot-On Flow cell (FLO-MIN106), and sequencing was performed with a PromethION sequencer (Oxford Nanopore Technologies, Oxford, United Kingdom) running for 48 h at Wuhan Benagen Technology Company Limited (Wuhan, China). Resulting FAST5 files were base-called using the Oxford Nanopore GUPPY software (v0.3.0), and reads with a quality ≤7 were discarded.
Genomic assembly was performed using NECAT2. Two rounds of error correction were performed using Racon (v1.4.3) (Vaser et al., 2017) and Pilon (v1.23) based on the nanopore and the Illumina Novaseq sequencing data (Walker et al., 2014), respectively. The heterozygous sequences were removed using the Purge_haplotigs pipeline (v1.0.4) (Roach et al., 2018). Homology-based gene prediction was performed using SNAP (Johnson A.D. et al., 2008), AUGUSTUS v 3.2.1 (Stanke et al., 2006), and GeneMark-ES v4.21 (Ter-Hovhannisyan et al., 2008). BUSCO (v4.1.2) based on the fungi_odb10 reference database was employed to evaluate the quality of the prediction (Waterhouse et al., 2018). The tRNA regions and secondary structures were detected using tRNAscan-SE v1.23. The rRNAs were analyzed using RNAmmer software (Lagesen et al., 2007), and the small RNAs were predicted using Infernal v1.1.2 to search against the Rfam 9.1 database (Gardner et al., 2009). To evaluate the transposable elements within the P. herquei genome, the transposable elements were searched with the Repbase database (Bao et al., 2015) using RepeatMasker v4.0.93.
For functional annotation, BLASTP searched against a series of protein databases, including UniProt/Swiss-Prot (Bairoch et al., 2005), Non-Redundant Protein Sequence Database (NR) in NCBI4, Gene Ontology (GO) (Gene Ontology Consortium, 2004), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000), and Cluster of Orthologous Groups (COG) (Tatusov et al., 2003) with a cut-off of <1e–05, and the best hit was used to infer the gene’s biological function.
The genome reference sequences of the other 14 Ascomycetes species (see Supplementary Tables S1, S2 in the supplemental materials) were downloaded from the NCBI database and were used to construct gene families. Sequence alignment was done with MUSCLE (Edgar, 2004), and positions containing gaps of ≥80% in multiple sequence alignment were trimmed using TrimaAI v1.4. rev22 (Capella-Gutiérrez et al., 2009). The comparison and annotation of orthologous gene clusters were carried out using OrthoFinder 2.2.7 (Emms and Kelly, 2019). Phylogenetic trees were constructed using the maximum-likelihood approach implemented in RAxML v8.0 (Stamatakis, 2014), the sequence of Coccidioides immitis (Stiles) (GenBank accession number GCA_000146045.2) was used as an outgroup. Gene family expansion and contraction were identified using CAFE v4.2 (De Bie et al., 2006). A time-calibrated phylogeny was inferred under the Bayesian framework employing fossil information (Drummond and Rambaut, 2007). The chronograms shown were calculated using the median clade credibility tree and 95% confidence intervals. The PAML mcmctree v4.5 program (Yang and Rannala, 2006) was used to compute split times using the approximate likelihood calculation algorithm. The model of sequence evolution was determined using Modeltest 3.75. Tracer v1.5.06 was applied to examine convergence, and two independent runs were performed for confirmation.
For a better understanding of genomic characters of P. herquei within the context of the genus Penicillium, the genome of P. herquei was compared with the genomes of the other two well-studied Penicillium species, P. chrysogenum (GenBank accession no.: GCA_000149335.2) and P. decumbens (GenBank accession no.: GCA_002072245.1). Gene families were generated by MCL software (v12-068) using an inflation value of 2.0 (Enright et al., 2002). BLASTp was used to compare all protein sequences from the 3 selected species, the results were filtered using threshold limits of e values ≤1e–5, alignment identity ≥30%, and an alignment coverage ≥50%. GO annotation was performed by the use of Blast2GO, which assigned homologous sequences aligned by BLAST with Uniprot and the NR database to GO terms. The number of shared and specific gene families among the three Penicillium species were than analyzed.
Genes related to cellulose and hemicellulose degradation: Carbohydrate-active enzymes (CAZymes) in the three Penicillium species were annotated using BLAST (Johnson M. et al., 2008). The dbCAN annotation program HMMER 3 (Finn et al., 2011) was used to search against the CAZy (carbohydrate-active enzyme) database (Lombard et al., 2014). The results were combined when e values ≤1e–5. The class II peroxidases and dye-decolorizing peroxidases were further confirmed by BLAST searches against PeroxiBase (Fawal et al., 2012).
Secondary metabolism genes: Candidate transporter genes in the three Penicillium species were identified based on searches of the Transporter Classification Database (TCDB) (Saier et al., 2014) with e values ≤1e–5 and identity values ≥40%. The secondary metabolism biosynthesis genes and gene clusters in the genomes of the three Penicillium species were predicted with AntiSMASH 6.0 (Blin et al., 2021). The Comprehensive Antibiotic Research Database (CARD) (McArthur et al., 2013) was used to compare coding genes (of the three Penicillium species) involved in antimicrobial resistance.
Virulence associated genes: Candidate pathogen-host interactions (PHI) genes within the genome of the three Penicillium species were identified using BLASTp to search against PHI-base v4.37, and protein alignments were performed to identify putative virulence-associated genes in the three Penicillium species with identity ≥40% and query coverage ≥70%.
After quality control, we obtained 10.829 Gb of NovaSeq data (269 coverage) and 16.343 Gb of Nanopore data (406 coverage). Combined sequences from the two platforms were assembled into 65 scaffolds with an N50 value of 414,225 bp to obtain a total genome size of 40.25 Mb (46.72% GC content) (Table 1). We predicted 14,532 genes with an average length of 2,275 bp, and 96.12% of the protein-coding genes had significant sequence similarity to previously documented fungal sequences (Table 1). BUSCO was used to calculate the completeness of assembly and annotation. Among 1875 single-copy orthologs, 75.6% of contigs were complete and 16.4% of contigs were complete duplicated BUSCOs, while only 0.7% were fragmented and 7.4% were missing.
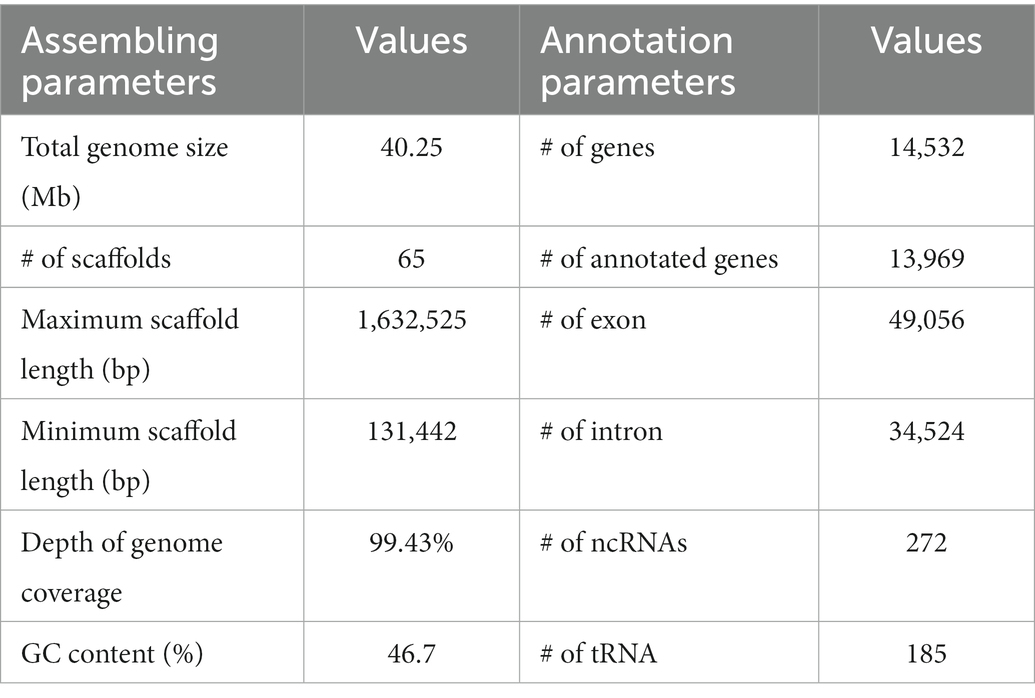
Table 1. Genome features of Penicillium herquei.
Of the predicted genes, 8,751 (60.22%) showed similarity to known proteins in the NR database, and 70.87% of these genes show significant matches to known proteins of 9 Penicillium species (Supplementary Table S3). We detected 49,056 exons with an average length of 421.53 bp. The average length of the introns was 100.23 nucleotides. For noncoding RNAs (ncRNAs), 185 tRNA, 41 RNA, and 40 snRNA genes were identified in the P. herquei genome (Supplementary Table S4). We identified 458,687 bp of repetitive elements in the P. herquei genome (1.14% of the genomic sequence). Tandem repeat sequences accounted for 0.04% and transposable elements for 1.69% of the assembled genome, and unknown and other repetitive elements accounted for 0.02% of the genome. Long terminal repeats (LTRs) were the most abundant transposable elements and accounted for about 0.43% of the genomic sequence (Supplementary Table S4).
COG annotation results showed that 1,227 genes were classified into 24 gene types, accounting for 8.44% of the total genes in P. herquei, as shown in Figure 1. The functional annotation results in the GO database showed that 8,665 genes (59.63% of all the genes) could be classified into three types, with 7,114 being genes related to cellular components, 7,585 being genes related to molecular functions, and 7,086 being genes related to the biological processes. The GO terms with the highest numbers of genes that were classified as related to biological processes were genes related to transmembrane transport (405 genes). The GO terms with the highest numbers of genes that were classified as related to cellular components were the nucleus (2055 genes) or were integral components of membranes (641 genes). The GO terms with the highest numbers of genes classified as related to molecular function were for ATP binding (944 genes, 16.17%) and metal ion binding (873 genes, 14.95%) (Supplementary Figure S1). The results of KEGG pathway analysis showed that 6,638 (45.67%) genes were classified as related to 338 known metabolic pathways. The metabolic pathway with the largest number of genes was amino acid metabolism (771 genes), followed by carbohydrate metabolism (743 genes) and signal transduction (593 genes). Cluster analysis showed that the relevant 338 metabolic pathways could be categorized into the five groupings of metabolism, genetic information processing, organismal systems, cellular processes, and environmental information processing (Supplementary Figure S2).
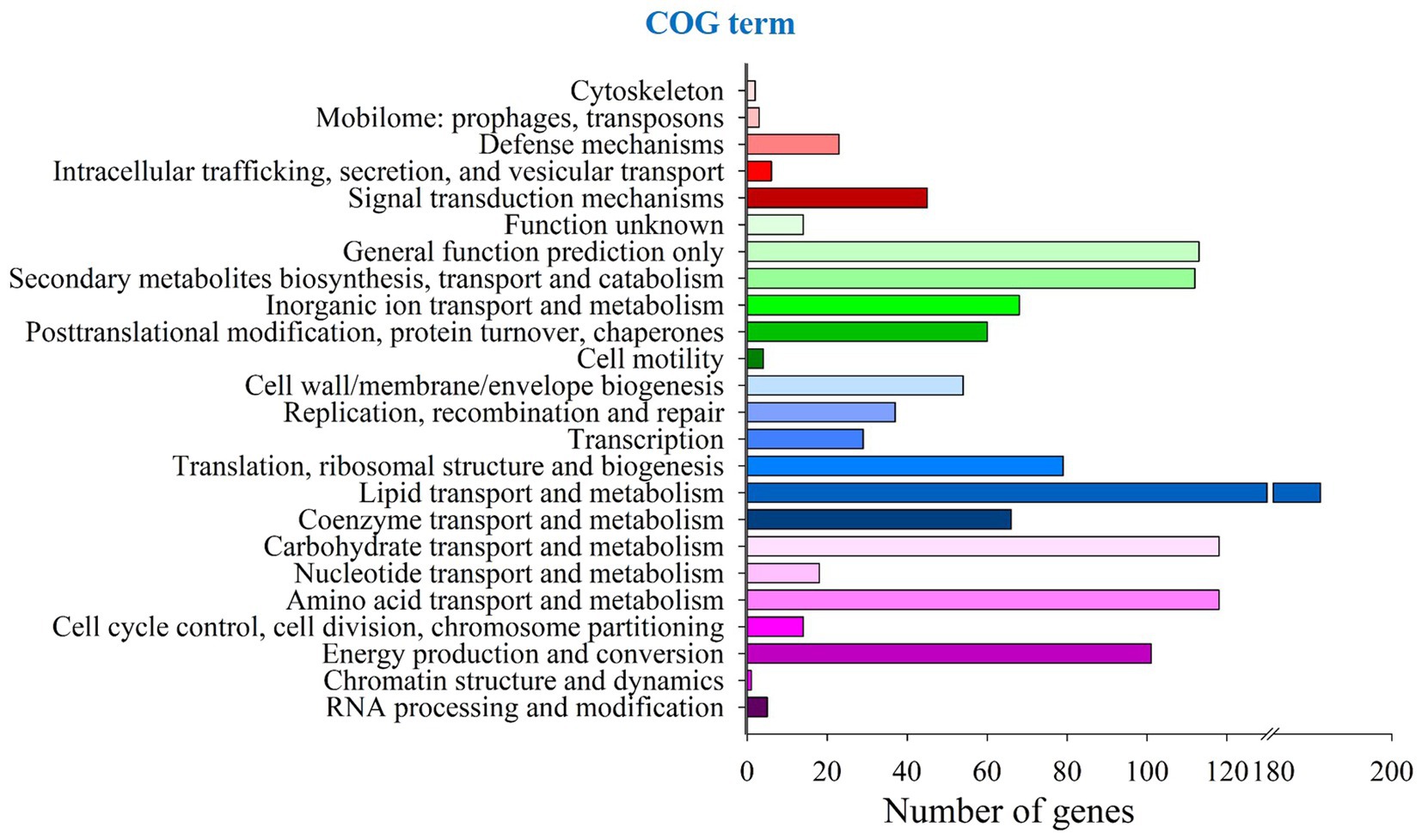
Figure 1. COG functional annotation of coding sequences in the whole genome of Penicillium herquei.
OrthoFinder identified 2,852 gene clusters, among which were included 48,701 orthologous genes shared among all the 15 fungal species considered. From these shared gene clusters, 23,220 single-copy orthologous genes were chosen to analyze the evolutionary relationship of P. herquei with the other 14 fungal reference genomes. It is noteworthy that the P. herquei genome possesses more multiple-copy orthologs but fewer sigle-copy orthologs than the genomes of other 14 fungal species examined (Supplementary Figure S3). Phylogenetic analysis revealed that P. herquei clustered with other Penicillium species and was closest to the plant-pathogenic fungus P. decumbens (Figure 2). Moreover, 49 genes and 419 gene families were significantly contracted but 4,878 genes and 1900 gene families were significantly expanded in the P. herquei genome. Compared with other fungal species, P. herquei had the highest ratio of expanded gene families to contracted ones (Figure 2).
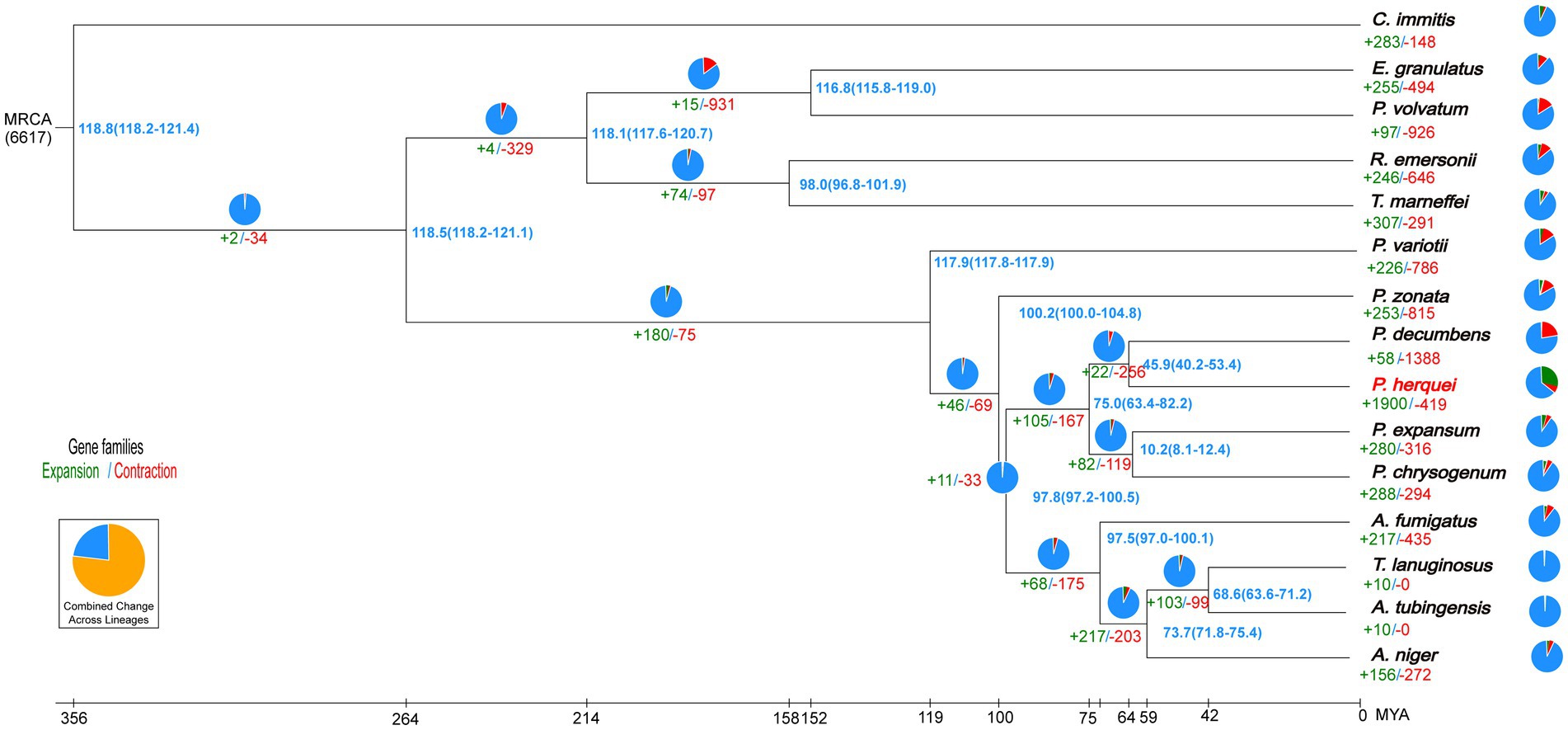
Figure 2. Phylogenetic tree of Penicillium herquei and sequences of the 14 fungal species available from GenBank. The blue numbers on the nodes are divergence times in units of million years ago. Numbers for significantly expanded (+, green) and contracted (−, red) gene families are shown below branches or taxon names with percentages indicated by pie charts.
Genome size (40.25 Mb) was larger but GC content (46.7%) was lower for P. herquei compared to the other two Penicillium species examined (P. chrysogenum: 32.52 Mb with 48.9% GG content; P. decumbens: 23.94 Mb with 50.2% GC content). The three Penicillium species shared 5,309 gene families (Figure 3A). In addition to the core gene families that are all present in the three species, P. herquei shared more gene families with P. chrysogenum than with P. decumbens. There were 239 gene families that were uniquely shared P. herquei and P. decumbens, while 1,675 gene families were uniquely shared by P. herquei and P. chrysogenum. The shared and specific gene families in the three Penicillium species were classified by GO analysis according to their related biological processes (Figure 3B). The majority of the 5,309 core families (Circle I) were classified into the functional categories of cellular and metabolic processes, but approximately half of the species-specific gene families (Circles V to VII) could not be assigned into a designated GO category (Figure 3B). The results of COG annotation showed that P. herquei had more genes involved in matter transportation and metabolism, including those that clustered under the groups of “lipid transport and metabolism,” “carbohydrate transport and metabolism,” “amino acid transport and metabolism,” and “secondary metabolite biosynthesis, transport, and catabolism” (Figure 3C).
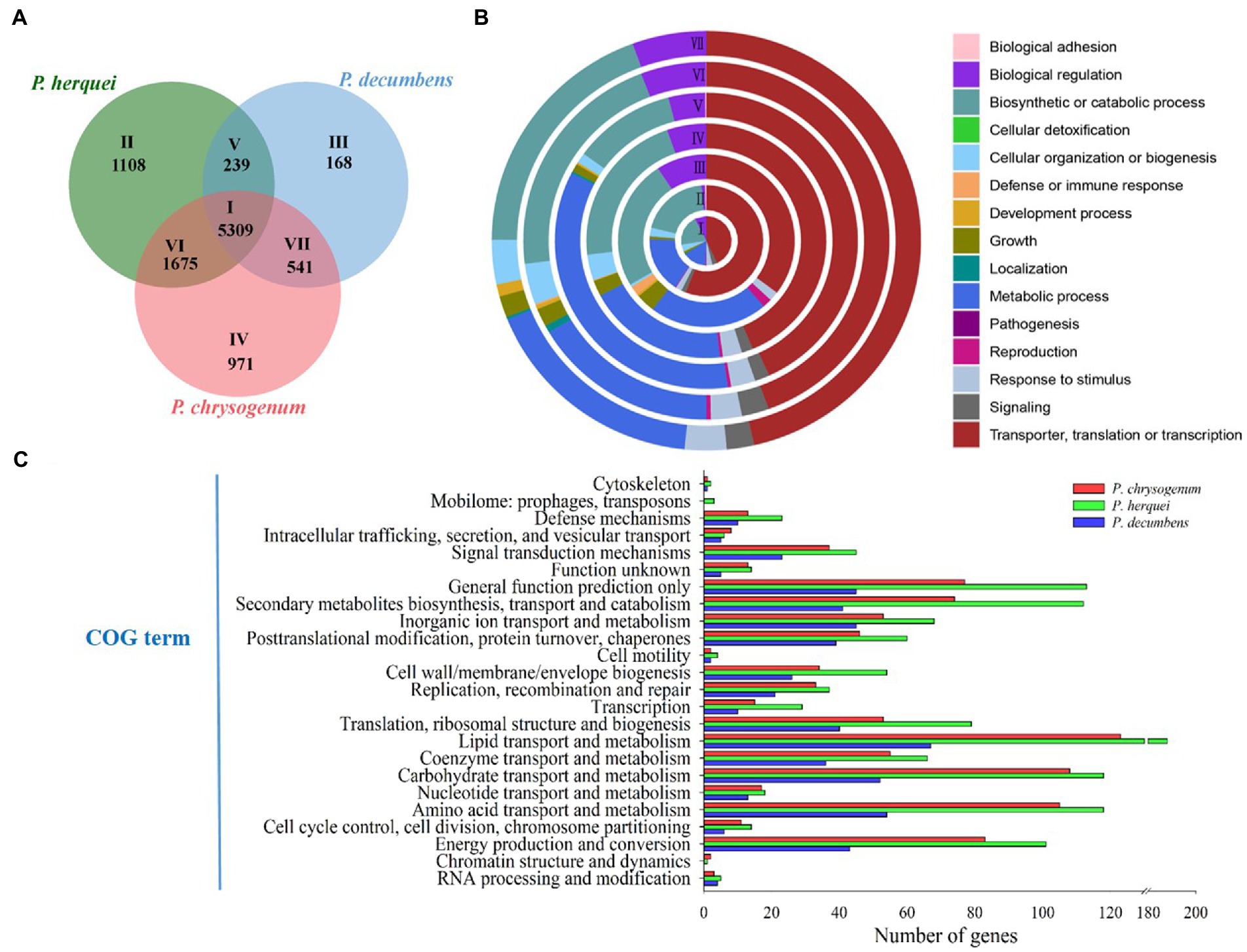
Figure 3. (A) Venn diagram showing the number of shared and specific gene families among three Penicillium species, P. herquei (Pher), P. chrysogenum (Pchr), and P. decumbens (Pdec). (B) Gene Ontology (GO) categories of gene families according to biological process. Circles I to VII correspond to areas I to VII in A. (C) Comparative analysis of genes by functional Cluster of Orthologous Groups (COG) categories, from the genomes of P. herquei, P. chrysogenum, and P. decumbens.
Genes related to cellulose and hemicellulose degradation: We identified 608 CAZymes in the P. herquei genome, consisting of 285 glycoside hydrolases (GHs), 103 glycosyl transferases (GTs), 95 enzymes with auxiliary activities (AAs), 92 carbohydrate esterases (CEs), 28 carbohydrate-binding modules (CBMs), and 5 polysaccharide lyases (PLs) (Figure 4A). Penicillium herquei had more CAZymes genes (especially for genes encoding GHs, CEs, and AAs) than the other two Penicillium species (P. chrysogenum, 477 and P. decumbens, 249) (Figure 4A). Moreover, many genes encoding for GHs and CEs related to cellulose and hemicellulose degradation were also found in the P. herquei genome. In particular, there were more GH6 and GH7 genes related to cellulose degradation as well as GH35, GH67, CE16 and CE1 genes related to hemicellulose degradation in the P. herquei genome than in the other two Penicillium species (Figures 4B,C).
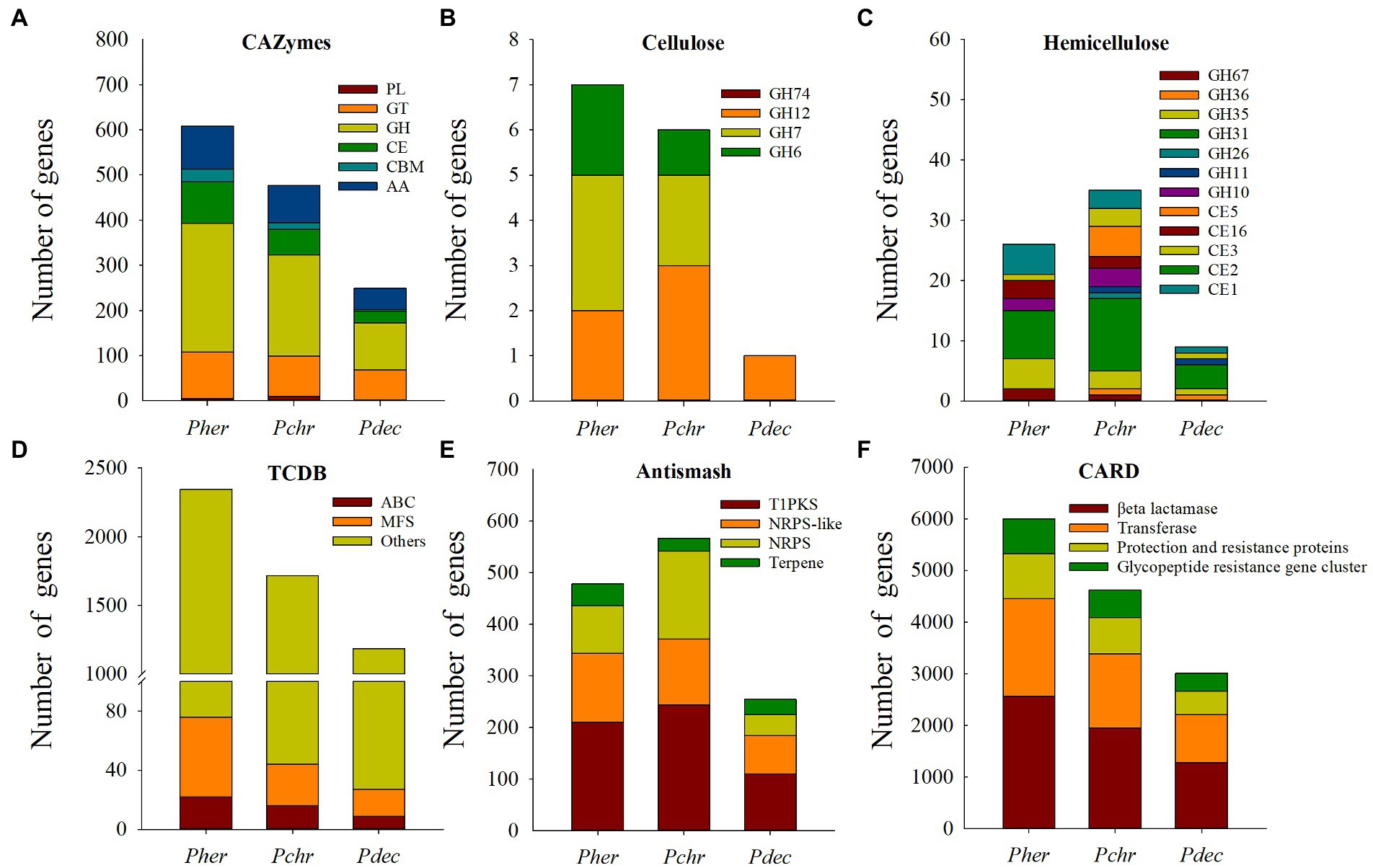
Figure 4. Comparison of genes encoding carbohydrate active enzymes (CAZymes) (A–C) and secondary metabolites (D–F) in Penicillum herquei (Pher), Penicillium chrysogenum (Pchr) and Penicillium decumbens (Pdec). (A) CAZymes identified in the genome of Pher, Pchr and Pdec. PL, polysaccharide lyase; GT, glycosyltransferase; GH, glycoside hydrolase; CE, carbohydrate esterase; CBM, carbohydrate-binding module; AA, auxiliary activity. CAZymes involved in cellulose degradation (B) and hemicellulose degradation (C). (D) Secondary metabolite backbone genes. PKS, polyketide synthase; NRPS, nonribosomal peptide synthetase. (E) Genes encoding transporters. MFS: major facilitator superfamily of transporters, ATP-binding cassette (ABC) transporters. (F) Antimicrobial resistance genes identified in CARD database.
Secondary metabolism genes: We identified 2,346 transport proteins in the P. herquei genome, more than in the other two Penicillium species. Among these transporters, 54 genes encoded major facilitator superfamily (MFS) proteins and 22 encoded ATP-binding cassette (ABC) proteins (Figure 4D). AntiSMASH analysis revealed that the number of secondary metabolite genes predicted for the P. herquei (520) genome was comparable with the number of these genes in the P. chrysogenum genome (566), but was higher than in the P. decumbens genome (254). These secondary metabolite genes included genes encoding type-I polyketide synthase (T1PKS), nonribosomal peptide synthetases (NRPS), and nonribosomal peptide synthetase-like (NRPS-like) enzyme (Figure 4E). In particular, the P. herquei genome contained 42 terpene synthase genes, which was more than the number of such enzymes in the other two Penicillium species (Figure 4E). CARD identified four antimicrobial resistance genes in the genomes of the three Penicillium species, including βeta lactamase, transferase, protection and resistance proteins, and a glycopeptide-resistance gene cluster. The numbers of these genes in P. herquei were all higher than in the other two Penicillium species (Figure 4F).
Virulence associated genes: We predicted a total of 99,031 PHI genes in the P. herquei genome; the highest proportion of the PHI genes was related to “reduced virulence” (38.00%), followed by “unaffected pathogenicity” (34.65%), “loss of pathogenicity” (9.95%), and “mixed outcome” (10.90%) (Figure 5A). Although P. herquei had more PHI genes than P. chrysogenum (77,638) and P. decumbens (49,326), PHI genes related to reduced virulence, unaffected pathogenicity, or loss of pathogenicity were all more abundant in the P. herquei genome than in the other two Penicillium species (Figure 5B).
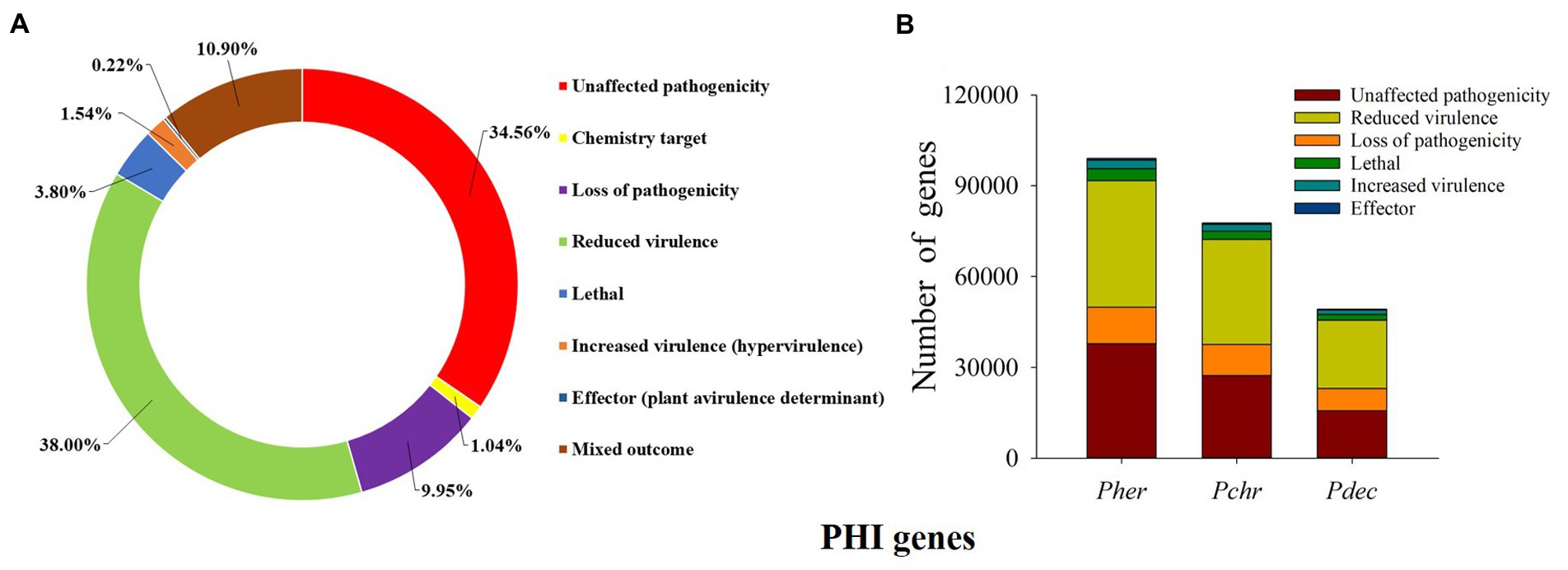
Figure 5. Putative pathogen-host interactions (PHI) genes. (A) The distribution of phenotypic categories of PHI gene orthologs in the Penicillium herquei genome. (B) Comparison of PHI genes in the three Penicillium species, P. herquei (Pher), P. chrysogenum (Pchr), and P. decumbens (Pdec).
The assembly quality highly depends on the genome size. We found the genome size of P. herquei (40.25 Mb) was larger than that of the two Penicillium species examined, P. chrysogenum (23.94 Mb) and P. decumbens (32.52 Mb). Similarly, Raffaelea ambrosiae, a symbiote of Platypus ambrosia beetles, had a larger genome size (40.78 Mb) (Vanderpool et al., 2018) than that of its close non-ambrosia relative, Ophiostoma piceae (32.84 Mb) (Haridas et al., 2013). However, Ambrosiella cleistominuta, the fungal symbiote of the ambrosia beetle Anisandrus maiche, had a similar genome size (27.08 Mb) (Wilken et al., 2020) with its close non-ambrosia relative Ceratocystis fimbriata (31.61 Mb) (Santos et al., 2020). As many of fungi are highly heterozygous and polyploid, the assemblies of their genomes are often highly fragmented, and the different assemblers may generate different assembly sizes. Kooij and Pellicer (2020) considered that published fragmented genome assemblies have overestimated the genome sizes of polyploid fungal symbiotes.
The genome and gene evolution of fungi enable them to exist in diverse environments, patterns of gene family expansion or contraction can reflect particular selection pressure that a species has been subjected to over evolutionary time scales (Nygaard et al., 2011, 2016). Both gene family expansion and reduction were found in the P. herquei genome, consistent with the genomes of other fungal cultivars of termites (Poulsen et al., 2014) and leaf-cutting ants (Aylward et al., 2013). For example, the genome of the Termitomyces sp., cultivated by the termite Macrotermes natalensis, showed 10 gene family expansions but 4 gene family contractions (Poulsen et al., 2014). The fungus Leucoagaricus gongylophorus, cultivated by leaf-cutting ants has an expansion of lignocellulases (Aylward et al., 2013). The expansion or contraction of gene families has likely been driven by mutualisms between fungus-farming insects and their obligate cultivars (Poulsen et al., 2014; Nygaard et al., 2016). Evolutionary modifications in the attine ant genomes include unprecedented rates of genome-wide structural rearrangement, early loss of arginine biosynthesis and positive selection on chitinase pathways, correspondingly, reciprocal genomic evolution of their fungal cultivars includes loss of a key ligninase domain, changes in chitin synthesis and a reduction in carbohydrate-degrading enzymes (Nygaard et al., 2016).
Penicillium herquei belongs to Penicillium section Sclerotiora, although fungal species in this section are commonly found in soil, plants, and insects (Houbraken and Samson, 2011), phylogeny of this section is still poorly investigated. Moreover, the genus Penicillium is affiliated to the family Aspergillaceae, and it contains two subgenera, Aspergilloides and Penicillium (Houbraken and Samson, 2011). We have also noticed that P. herquei belongs phylogenetically to Aspergilloides, whereas the other two Penicillium species we compared belong phylogenetically to subgenus Penicillium (Houbraken and Samson, 2011). Thus, future study of genomes of other closer relatives in subgenus Aspergilloides would be helpful for learning more about P. herquei.
The plant cell wall is an important barrier against insect attack, however, most of insects lack endogenous enzymes for plant cell wall digestion (Tokuda, 2019). To overcome the barrier of the plant cell wall, many insects have established close associations with fungi (Calderón-Cortés et al., 2012), because of which can secrete a diverse array of enzymes, such as cellulose, hemicellulose, and pectin etc., that are capable of degrading cell wall polymers (Kubicek et al., 2014). Among these, CAZymes are responsible for the metabolism of glycoconjugates, oligosaccharides, and polysaccharides, and they play crucial roles in the synthesis and degradation of carbohydrates (Hage and Rosso, 2021) and in host-pathogen interactions (Kubicek et al., 2014). In our analysis, P. herquei had more CAZymes-related genes than did the two Penicillium species examined and it also has a large number of genes associated with cellulose and hemicellulose degradation, which play important roles in effectively degrading plant cell walls into nutrients such as glucose, mannose, galactose, acetic acid, and xylose (Knowles et al., 1987; Yang et al., 2007). These genes help degrade cell walls of the leaf-rolls that the larvae of E. chinensis consume, providing additional nutrients for the growth of the larvae. We have also demonstrated that P. herquei can alter leaf chemical components by lowering the cellulose content of the leaf-rolls (Li et al., 2012). GHs are common enzymes that can degrade cellulose, hemicellulose, and starch, and they are involved in the hydrolysis of the glycosidic bond between or within carbohydrate molecules (Wyman et al., 2005; Sammond et al., 2012). In our study, genes encoding GH7, GH6, GH35, and GH67 class enzymes outnumbered those encoding other GH enzymes, indicating that this fungus has a stronger ability to degrade cellulose and hemicellulose than the other two Penicillium species studied.
The higher number of CAZymes-related genes and genes associating with cellulose and hemicellulose degradation in the P. herquei genome may suggest it natural history. Besides of being a symbiote of E. chinensis, P. herquei occurs widely in soil, litter, fruits, and as plant endophytic fungus in nature (Tansakul et al., 2014; Zhou et al., 2019). Thus, this fungus has possibly evolved these genes associating with plant decomposition to thrive in natural environment, but keeps these genomic features after cultivated by E. chinensis as a fungal crop. Further work on comparison of both gene expression and compound production between P. herquei isolates from E. chinensis mycantagium and free-living counterparts is thus necessary to further elaborate how this fungus have adapted to its lifestyles and ecological niches in E. chinensis.
The three Penicillium species examined in this study show similar metabolic and enzymatic potential, alternatively, Penicillium spp. have also been reported to form fungus-farming mutualism with the other two Euops leaf-rolling weevils, Euops lespedezae (Kobayashi et al., 2008) and Euops splendida (Sakurai, 1985). Penicillium are one of the most chemically inventive genera, and are well known for their ability to produce a wide range of secondary metabolites and small molecules that function as antibiotics, toxins and pigments (Nielsen et al., 2017). The most well-known member of the genu is Penicillium chrysogenum because of its role in the production of penicillin and as a contaminant of indoor environments, food, and feedstuffs (van den Berg et al., 2008). Penicillium decumbens has been widely used in biorefinery due to its high production of cellulase and hemicellulose (Liu et al., 2013). The large enzymatic and secondary metabolic potential of Penicillium fungi (Nielsen et al., 2017) is consistent with their wide existence in natural ecosystems (Yadav et al., 2018), and also makes them an ideal candidate cultivar that can be recruited by fungus-farming insects.
The mutualistic Euops-Penicillium system shares a similar feature with those famous fungus-farming insects that they all depend fungi to produce the majority of the plant biomass-degrading enzymes. Genomic analyses of Leucoagaricus gongylophorus, a basidiomycetous fungus that serve as a food source for fungus-farming ants, have confirmed the presence of genes predicted to encode biomass-degrading enzymes for the digestion of cellulose, xylan and other plant polymers (Schiøtt et al., 2008), extracellular cellulases of L. gongylophorus include GH6 and GH7, GH15, and CE5 (Aylward et al., 2013). The fungus-farming termites primarily provision their fungal cultivars with decaying plant material, Termitomyces fungi and garden bacteria are responsible for lignin, cellulose, and hemicellulose degradation (Hyodo et al., 2003). The ambrosia fungi appear to preferentially degrade hemicellulose and other simple sugars (De Fine Licht and Biedermann, 2012). Using microbial symbiotes to access plant polysaccharides appears to be also shared by some nonsocial fungus-farming insects, such as a Eurasian woodwasp Sirex noctilio (F.) (Fu et al., 2020).
Transporter molecules are integral membrane proteins that facilitate movement of macromolecules, ions, or small molecules across a biological membrane (Perlin et al., 2014). MFS and ABC transporters are the two biggest families of fungal transporters that mediate transport of intermediates and toxic molecules in the secondary metabolism pathway (Perlin et al., 2014). In our study, although only a few ABC and MFS proteins from the three fungal species have been functionally characterized, we found that P. herquei had more ABC and MFS proteins than did the other two Penicillium species. Conversely, Fu et al. (2020) found that Amylostereum areolatum (Fr.) Boidin, a fungal symbiote of S. noctilio, had few ABC proteins in comparison with free-living fungi, possibly because A. areolatum is mainly transferred by its insect hosts. In contrast, although P. herquei is also carried in a special mycetangium by the female E. chinensis weevils before being used to inoculate leaf-rolls, it is also free-living after being inoculated on the leaf-rolls (Wang et al., 2015). Moreover, the leaf-roll is cut from the plant by the female weevil and drops to the soil, where it gradually decays in the moist environment (Li et al., 2012). Thus, P. herquei on the leaf-roll has a relatively long saprophytic phase in its life cycle, and thus it has evolved a strong ability to transport and discharge the intermediate and toxic substrates of leaf-rolls.
Maintaining monocultural fungus cultivar without other fungi present requires highly effective defense functions in fungus-farming systems (Schmidt et al., 2022). The production of toxic secondary metabolites by fungal symbiotes can provide protection to their hosts (Clay, 2014). Penicillium herquei can produce a vast array of biologically active secondary metabolites, including antibiotic phenalenones, norherqueinone and herqueinone, alkaloids, and some triene derivatives (Tansakul et al., 2014), that have shown significant antifungal and antiinfluenza activities (Zhou et al., 2019). We have demonstrated that P. herquei can suppress the growth of two pathogenic Rhizopus spp. frequently isolated from the leaf-rolls (Li et al., 2012). Wang et al. (2015) also found that P. herquei produces the antibiotic (+)-scleroderolide, which can protect the leaf-roll against potential infection. In line with this, our study found the P. herquei genome includes genes producing PKS, NRPS, T1PKS compounds, as well as terpenoid biosynthetic genes, which likely function in secondary metabolism and may serve to help the host to defend against toxins and parasites. PKS and NRPS are known to be symbiote-produced compounds that function in the defense of some insects and marine invertebrates (Piel, 2002). Penicillium herquei possesses a larger number of backbone genes associated with secondary metabolites, which may have antagonistic effects on pathogens and parasites that occur in both the leaf-rolls and the host insect. Notably, we found that P. herquei has significantly more genes for terpenoid biosynthesis than do the other two Penicillium species we studied. Terpenoids are the largest, most diverse group of secondary metabolites, exceeding phenolics, flavonoids, and alkaloids (Boncan et al., 2020). Terpenoids mediate various antagonistic and beneficial interactions among organisms (Gershenzon and Dudareva, 2007), and are used by many species of plants, animals and microorganisms to defend themselves against predators, pathogens, or competitors (Boncan et al., 2020). The possession of more terpenoid biosynthetic genes by P. herquei may contribute to higher terpene accumulation in P. herquei, leading to improved protection of leaf-rolls and E. chinensis larvae.
Pathogen-insect communications are essential for stimulating insect immune responses and inducing expression of virulence factors by fungal pathogens (Noman et al., 2021). Many studies have demonstrated that genes mediating pathogen-plant communications in signaling pathways are critical for fungal pathogenicity (Shang et al., 2016). The reduction in PHI genes in the P. herquei genome suggests that selection to maintain virulence genes has been relaxed, likely due to its mutualistic association with E. chinensis, and the fungal spores are carried in a special mycetangium on the female weevils. The female weevils protect the fungus before using it to inoculate the larval leaf-rolls. Moreover, P. herquei acts as an important nutritional resource for the E. chinensis larvae (Wang et al., 2010; Li et al., 2012). In this study, we found only a few virulence-associated genes (associated with increased virulence and lethal genes) in the P. herquei genome, and the inactivation or reduction of the expression of these genes can reduce or eliminate pathogenicity of this fungus to both the adults and the larvae of E. chinensis. Similarly, the absence of expression or low expression of lethal genes is also observed in the genome of the symbiote of the Eurasian wood wasp S. noctilio (Fu et al., 2020).
The genome of P. herquei contains a diverse set of genes associating with carbohydrate-active enzymes, cellulose and hemicellulose degradation, transporter, and terpenoid biosynthesis. Comparative genomics demonstrate that the three Penicillium species show similar metabolic and enzymatic potential, however, P. herquei has more genes associated with plant biomass degradation and defense but less genes associating with virulence pathogenicity. Taken together, our results provide molecular evidence for plant substrate degradation and protective roles of P. herquei in its host E. chinensis. The large enzymatic and secondary metabolic potential shared by Penicillium genera is likely an important property that some Penicillium fungi are recruited by the Euops fungus-farming weevils as fungal crops.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/ GenBank: JANAWU000000000 BioProject: PRJNA854755 BioSample: SAMN29446570.
XL and WG designed the experiments, analyzed the data, and wrote the paper. WW and WG sampled the leaf rolls. JT carried out fungal isolation. JT and TL contributed to the assembly of P. herquei and bioinformatics analysis. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (31800423 and 331460115) and the Natural Science Foundation of Guangxi Province (2018GXNSFBA281172).
We are grateful to Wuhan Benagen Technology Company Limited for assistance with genome sequence and bio-information analysis. We thank Hu Chen, Xiao Zhang, Pengsen Cai, and Xiaojing Li for their help on data analysis and on project management. We also appreciate Roy Van Driesche for the language edits. The authors especially thank three reviewers for their valuable comments on earlier versions of this paper.
TL was employed by Wuhan Benagen Technology Company Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1048910/full#supplementary-material
1. ^https://github.com/BGI-flexlab/SOAPnuke
2. ^https://github.com/xiaochuanle/NECAT
3. ^http://www.repeatmasker.org
4. ^https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/
5. ^http://darwin.uvigo.es/software/modeltest.html
Aanen, D. K. (2006). As you reap, so shall you sow: coupling of harvesting and inoculating stabilizes the mutualism between termites and fungi. Biol. Lett. 2, 209–212. doi: 10.1146/annurev-ento-011019-024910
Aylward, F. O., Burnum-Johnson, K. E., Tringe, S. G., Teiling, C., Tremmel, D. M., Moeller, J. A., et al. (2013). Leucoagaricus gongylophorus produces diverse enzymes for the degradation of recalcitrant plant polymers in leaf-cutter ant fungus gardens. Appl. Environ. Microbiol. 79, 3770–3778. doi: 10.1128/AEM.03833-12
Bairoch, A., Apweiler, R., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., et al. (2005). The universal protein resource (UniProt). Nucleic Acids Res. 33, D154–D159. doi: 10.1093/nar/gki070
Bao, W. D., Kojima, K. K., and Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6:11. doi: 10.1186/s13100-015-0041-9
Biedermann, P. H., and Vega, F. E. (2020). Ecology and evolution of insect-fungus mutualisms. Annu. Rev. Entomol. 65, 431–455. doi: 10.1146/annurev-ento-011019-024910
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., Van Wezel, G. P., Medema, M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi: 10.1093/nar/gkab335
Boncan, D. A. T., Tsang, S. S. K., Li, C., Lee, I. H. T., Lam, H. M., Chan, T. F., et al. (2020). Terpenes and terpenoids in plants: interactions with environment and insects. Int. J. Mol. Sci. 21:7382. doi: 10.3390/ijms21197382
Calderón-Cortés, N., Quesada, M., Watanabe, H., Cano-Camacho, H., and Oyama, K. (2012). Endogenous plant cell wall digestion: a key mechanism in insect evolution. Annu. Rev. Ecol. Evol. Syst. 43, 45–71. doi: 10.1146/annurev-ecolsys-110411-160312
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Clay, K. (2014). Defensive symbiosis: a microbial perspective. Funct. Ecol. 28, 293–298. doi: 10.1111/1365-2435.12258
da Costa, R. R., Vreeburg, S. M., Shik, J. Z., Aanen, D. K., and Poulsen, M. (2019). Can interaction specificity in the fungus-farming termite symbiosis be explained by nutritional requirements of the fungal crop? Fungal Ecol. 38, 54–61. doi: 10.1016/j.funeco.2018.08.009
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
De Fine Licht, H. H., and Biedermann, P. H. (2012). Patterns of functional enzyme activity in fungus farming ambrosia beetles. Front. Zool. 9:13. doi: 10.1186/1742-9994-9-13
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Enright, A. J., Van Dongen, S., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Fan, H., Noda, H., Xie, H., Suetsugu, Y., Zhu, Q., and Zhang, C. (2015). Genomic analysis of an ascomycete fungus from the rice planthopper reveals how it adapts to an endosymbiotic lifestyle. Genome Biol. Evol. 7, 2623–2634. doi: 10.1093/gbe/evv169
Fawal, N., Li, Q., Savelli, B., Brette, M., Passaia, G., Fabre, M., et al. (2012). PeroxiBase: a database for large-scale evolutionary analysis of peroxidases. Nucleic Acids Res. 41, D441–D444. doi: 10.1093/nar/gks1083
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Francke-Grosmann, H. (1967). “Ectosymbiosis in wood-inhabiting insects” in Symbiosis. ed. S. M. Henry (New York and London: Academic Press), 141–203.
Fu, N., Wang, M., Wang, L., Luo, Y., and Ren, L. (2020). Genome sequencing and analysis of the fungal symbiont of Sirex noctilio, Amylostereum areolatum: revealing the biology of fungus-insect mutualism. mSphere 5, e00301–e00320. doi: 10.1128/mSphere.00301-20
Gardner, P. P., Daub, J., Tate, J. G., Nawrocki, E. P., Kolbe, D. L., Lindgreen, S., et al. (2009). Rfam: updates to the RNA families database. Nucleic Acids Res. 37, D136–D140. doi: 10.1093/nar/gkn766
Gene Ontology Consortium (2004). The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, 258D–2261D. doi: 10.1093/nar/gkh036
Gershenzon, J., and Dudareva, N. (2007). The function of terpene natural products in the natural world. Nat. Chem. Biol. 3, 408–414. doi: 10.1038/nchembio.2007.5
Goes, A. C., Barcoto, M. O., Kooij, P. W., Bueno, O. C., and Rodrigues, A. (2020). How do leaf-cutting ants recognize antagonistic microbes in their fungal crops? Front Eco Evol 8:95. doi: 10.3389/fevo.2020.00095
Gohli, J., Kirkendall, L. R., Smith, S. M., Cognato, A. I., Hulcr, J., and Jordal, B. H. (2017). Biological factors contributing to bark and ambrosia beetle species diversification. Evolution 71, 1258–1272. doi: 10.1111/evo.13219
Grebennikov, V. V., and Leschen, R. A. B. (2010). External exoskeletal cavities in Coleoptera and their possible mycangial functions. Entomol Sci 13, 81–98. doi: 10.1111/j.1479-8298.2009.00351.x
Hage, H., and Rosso, M. N. (2021). Evolution of fungal carbohydrate-active enzyme portfolios and adaptation to plant cell-wall polymers. J Fungi 7:185. doi: 10.3390/jof7030185
Haridas, S., Wang, Y., Lim, L., Massoumi Alamouti, S., Jackman, S., Docking, R., et al. (2013). The genome and transcriptome of the pine saprophyte Ophiostoma piceae, and a comparison with the bark beetle-associated pine pathogen Grosmannia clavigera. BMC Genomics 14, 1–15. doi: 10.1186/1471-2164-14-373
Hata, H., and Kato, M. (2006). A novel obligate cultivation mutualism between damselfish and Polysiphonia algae. Biol. Lett. 2, 593–596. doi: 10.1098/rsbl.2006.0528
Houbraken, J., and Samson, R. (2011). Phylogeny of Penicillium and the segregation of Trichocomaceae into three families. Stud. Mycol. 70, 1–51. doi: 10.3114/sim.2011.70.01
Huang, Y. T., Skelton, J., and Hulcr, J. (2019). Multiple evolutionary origins lead to diversity in the metabolic profiles of ambrosia fungi. Fungal Ecol. 38, 80–88. doi: 10.1016/j.funeco.2018.03.006
Hyodo, F., Tayasu, I., Inoue, T., Azuma, J. I., Kudo, T., and Abe, T. (2003). Differential role of symbiotic fungi in lignin degradation and food provision for fungus-growing termites (Macrotermitinae: Isoptera). Funct. Ecol. 17, 186–193. doi: 10.1046/j.1365-2435.2003.00718.x
Johnson, A. D., Handsaker, R. E., Pulit, S. L., Nizzari, M. M., O’Donnell, C. J., and De Bakker, P. I. (2008). SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24, 2938–2939. doi: 10.1093/bioinformatics/btn564
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9. doi: 10.1093/nar/gkn201
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kirkendall, L. R., Biedermann, P. H., and Jordal, B. H. (2015). “Evolution and diversity of bark and ambrosia beetles” in Bark Beetles. eds. F. E. Vega and R. Hofstetter (New York: Academic Elsevier), 85–156.
Knowles, J., Lehtovaara, P., and Teeri, T. (1987). Cellulase families and their genes. Trends Biotechnol. 5, 255–261. doi: 10.1016/0167-7799(87)90102-8
Kobayashi, C., Fukasawa, Y., Hirose, D., and Kato, M. (2008). Contribution of symbiotic mycangial fungi to larval nutrition of a leaf-rolling weevil. Evol. Ecol. 22, 711–722. doi: 10.1007/s10682-007-9196-2
Kooij, P. W., and Pellicer, J. (2020). Genome size versus genome assemblies: are the genomes truly expanded in polyploid fungal symbionts? Genome Biol. Evol. 12, 2384–2390. doi: 10.1093/gbe/evaa217
Kubicek, C. P., Starr, T. L., and Glass, N. L. (2014). Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 52, 427–451. doi: 10.1146/annurev-phyto-102313-045831
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Li, X. Q., Guo, W. F., and Ding, J. Q. (2012). Mycangial fungus benefits the development of a leaf-rolling weevil, Euops chinesis. J. Insect Physiol. 58, 867–873. doi: 10.1016/j.jinsphys.2012.03.011
Li, X. Q., Guo, W. F., Wen, Y. G., Solanki, M. K., and Ding, J. Q. (2016). Transmission of symbiotic fungus with a nonsocial leaf-rolling weevil. J. Asia Pac. Entomol. 19, 619–624. doi: 10.1016/j.aspen.2016.06.006
Liu, G., Zhang, L., Wei, X., Zou, G., Qin, Y., Ma, L., et al. (2013). Genomic and secretomic analyses reveal unique features of the lignocellulolytic enzyme system of Penicillium decumbens. PLoS One 8:e55185. doi: 10.1371/journal.pone.0055185
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Mayers, C. G., Harrington, T. C., and Biedermann, P. H. W. (2022). “Mycangia define the diverse ambrosia beetle-fungus symbioses” in The Convergent Evolution of Agriculture in Humans and Insects. eds. T. R. Schultz, R. Gawne, and P. N. Peregrine (Cambridge: The MIT Press), 1–38.
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob Agents Ch 57, 3348–3357. doi: 10.1128/AAC.00419-13
Mueller, U. G., Kardish, M. R., Ishak, H. D., Wright, A. M., Solomon, S. E., Bruschi, S. M., et al. (2018). Phylogenetic patterns of ant-fungus associations indicate that farming strategies, not only a superior fungal cultivar, explain the ecological success of leafcutter ants. Mol. Ecol. 27, 2414–2434. doi: 10.1111/mec.14588
Mueller, U. G., Schultz, T. R., Currie, C. R., Adams, R. M., and Malloch, D. (2001). The origin of the attine ant-fungus mutualism. Q. Rev. Biol. 76, 169–197. doi: 10.1086/393867
Nielsen, J. C., Grijseels, S., Prigent, S., Ji, B., Dainat, J., Nielsen, K. F., et al. (2017). Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat. Microbiol. 2:17044. doi: 10.1038/nmicrobiol.2017.44
Noman, A., Aqeel, M., Islam, W., Khalid, N., Akhtar, N., Qasim, M., et al. (2021). Insects-plants-pathogens: toxicity, dependence and defense dynamics. Toxicon 197, 87–98. doi: 10.1016/j.toxicon.2021.04.004
Nygaard, S., Hu, H., Li, C., Schiøtt, M., Chen, Z., Yang, Z., et al. (2016). Reciprocal genomic evolution in the ant-fungus agricultural symbiosis. Nat. Commun. 7:12233. doi: 10.1038/ncomms12233
Nygaard, S., Zhang, G., Schiøtt, M., Li, C., Wurm, Y., Hu, H., et al. (2011). The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res. 21, 1339–1348. doi: 10.1101/gr.121392.111
Pathak, A., Kett, S., and Marvasi, M. (2019). Resisting antimicrobial resistance: lessons from fungus farming ants. Trends Ecol. Evol. 34, 974–976. doi: 10.1016/j.tree.2019.08.007
Pereira, R. A. S., and Kjellberg, F. (2021). “Mutualism as a source of evolutionary innovation: insights from insect-plant interactions” in Plant-Animal Interactions. eds. K. Del-Claro and H. Torezan-Silingardi (Cham: Springer), 307–332.
Perlin, M. H., Andrews, J., and San Toh, S. (2014). Essential letters in the fungal alphabet: ABC and MFS transporters and their roles in survival and pathogenicity. Adv. Genet. 85, 201–253. doi: 10.1016/B978-0-12-800271-1.00004-4
Piel, J. (2002). A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc. Natl. Acad. Sci. U. S. A. 99, 14002–14007. doi: 10.1073/pnas.222481399
Porebski, S., Bailey, L. G., and Baum, B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Peporter 15, 8–15. doi: 10.1007/BF02772108
Poulsen, M., Hu, H., Li, C., Chen, Z., Xu, L., Otani, S., et al. (2014). Complementary symbiont contributions to plant decomposition in a fungus-farming termite. Proc. Natl. Acad. Sci. U. S. A. 111, 14500–14505. doi: 10.1073/pnas.131971811
Roach, M. J., Schmidt, S. A., and Borneman, A. R. (2018). Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19:460. doi: 10.1186/s12859-018-2485-7
Saier, J., Milton, H., Reddy, V. S., Tamang, D. G., and Västermark, Å. (2014). The transporter classification database. Nucleic Acids Res. 42, D251–D258. doi: 10.1093/nar/gkt1097
Sakurai, K. (1985). An attelabid weevil (Euops splendida) cultivates fungi. J. Ethol. 3, 151–156. doi: 10.1007/BF02350306
Sammond, D. W., Payne, C. M., Brunecky, R., Himmel, M. E., Crowley, M. F., and Beckham, G. T. (2012). Cellulase linkers are optimized based on domain type and function: insights from sequence analysis, biophysical measurements, and molecular simulation. PLoS One 7:e48615. doi: 10.1371/journal.pone.0048615
Santos, S. A., Vidigal, P. M., Thrimawithana, A., Betancourth, B. M., Guimarães, L. M., Templeton, M. D., et al. (2020). Comparative genomic and transcriptomic analyses reveal different pathogenicity-related genes among three eucalyptus fungal pathogens. Fungal Genet. Biol. 137:103332. doi: 10.1016/j.fgb.2019.103332
Sawada, Y., and Morimoto, K. (1986). The mycetangia and the mode of the fungus transmission in the weevil genus Euops (Coleoptera: Attelabidae). Sci Bull Fac Agric Kyushu Univ 40, 197–205.
Schiøtt, M., De Fine Licht, H. H., Lange, L., and Boomsma, J. J. (2008). Towards a molecular understanding of symbiont function: identification of a fungal gene for the degradation of xylan in the fungus gardens of leaf-cutting ants. BMC Microbiol. 8, 1–10. doi: 10.1186/1471-2180-8-40
Schmidt, S., Kildgaard, S., Guo, H., Beemelmanns, C., and Poulsen, M. (2022). The chemical ecology of the fungus-farming termite symbiosis. Nat. Prod. Rep. 39, 231–248. doi: 10.1039/D1NP00022E
Shang, Y., Xiao, G., Zheng, P., Cen, K., Zhan, S., and Wang, C. (2016). Divergent and convergent evolution of fungal pathogenicity. Genome Biol. Evol. 8, 1374–1387. doi: 10.1093/gbe/evw082
Silliman, B. R., and Newell, S. Y. (2003). Fungal farming in a snail. Proc. Natl. Acad. Sci. U. S. A. 100, 15643–15648. doi: 10.1073/pnas.2535227100
Six, D. L. (2020). Niche construction theory can link bark beetle-fungus symbiosis type and colonization behavior to large scale causal chain-effects. Curr Opin Insect Sci 39, 27–34. doi: 10.1016/j.cois.2019.12.005
Skelton, J., Johnson, A. J., Jusino, M. A., Bateman, C. C., Li, Y., and Hulcr, J. (2019). A selective fungal transport organ (mycangium) maintains coarse phylogenetic congruence between fungus-farming ambrosia beetles and their symbionts. Proc Royal Soc B 286:20182127. doi: 10.1098/rspb.2018.2127
Solomon, S. E., Rabeling, C., Sosa-Calvo, J., Lopes, C. T., Rodrigues, A., Vasconcelos, H. L., et al. (2019). The molecular phylogenetics of Trachymyrmex Forel ants and their fungal cultivars provide insights into the origin and coevolutionary history of ‘higher-attine’ant agriculture. Syst. Entomol. 44, 939–956. doi: 10.1111/syen.12370
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Tansakul, C., Rukachaisirikul, V., Maha, A., Kongprapan, T., Phongpaichit, S., Hutadilok-Towatana, N., et al. (2014). A new phenalenone derivative from the soil fungus Penicillium herquei PSU-RSPG93. Nat. Prod. Res. 28, 1718–1724. doi: 10.1080/14786419.2014.941363
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. doi: 10.1101/gr.081612.108
Toki, W., Tanahashi, M., Togashi, K., and Fukatsu, T. (2012). Fungal farming in a non-social beetle. PLoS One 7:e41893. doi: 10.1371/journal.pone.0041893
Tokuda, G. (2019). Plant cell wall degradation in insects: recent progress on endogenous enzymes revealed by multi-omics technologies. Adv Insect Physiol 57, 97–136. doi: 10.1016/bs.aiip.2019.08.001
van den Berg, M. A., Albang, R., Albermann, K., Badger, J. H., Daran, J. M., Driessen, M., et al. (2008). Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat. Biotechnol. 26, 1161–1168. doi: 10.1038/nbt.1498
Vanderpool, D., Bracewell, R. R., and McCutcheon, J. P. (2018). Know your farmer: ancient origins and multiple independent domestications of ambrosia beetle fungal cultivars. Mol. Ecol. 27, 2077–2094. doi: 10.1111/mec.14394
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, L., Feng, Y., Tian, J., Xiang, M., Sun, J., Ding, J., et al. (2015). Farming of a defensive fungal mutualist by an attelabid weevil. ISME J. 9, 1793–1801. doi: 10.1038/ismej.2014.263
Wang, Y. Z., Wu, K., and Ding, J. Q. (2010). Host specificity of Euops chinesis, a potential biological control agent of Fallopia japonica, an invasive plant in Europe and North America. BioControl 55, 551–559. doi: 10.1007/s10526-010-9279-9
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2018). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Wilken, P. M., Aylward, J., Chand, R., Grewe, F., Lane, F. A., Sinha, S., et al. (2020). IMA genome-F13: Draft genome sequences of Ambrosiella cleistominuta,Cercospora brassicicola, C.citrullina, Physcia stellaris, and Teratosphaeria pseudoeucalypti. IMA Fungus 11, 19–17. doi: 10.1186/s43008-020-00039-7
Wyman, C. E., Decker, S. R., Himmel, M. E., Brady, J. W., Skopec, C. E., and Viikari, L. (2005). “Hydrolysis of cellulose and hemicellulose” in Polysaccharides: Structural Diversity and Functional Versatility. ed. S. Dumitriu (New York: Marcel Dekker, Inc), 1023–1062.
Yadav, A. N., Verma, P., Kumar, V., Sangwan, P., Mishra, S., Panjiar, N., et al. (2018). “Biodiversity of the genus Penicillium in different habitats” in New and Future Developments in Microbial Biotechnology and Bioengineering. eds. V. Gupta and S. Rodriguez-Couto (Amsterdam: Elsevier), 3–18.
Yang, Z. H., and Rannala, B. (2006). Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 23, 212–226. doi: 10.1093/molbev/msj024
Yang, H., Yan, R., Chen, H., Lee, D. H., and Zheng, C. (2007). Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 86, 1781–1788. doi: 10.1016/j.fuel.2006.12.013
Keywords: Euops, nonsocial insect, fungus farming, mutualism, molecular, genome, Penicillium
Citation: Guo W, Wang W, Tang J, Li T and Li X (2023) Genome analysis and genomic comparison of a fungal cultivar of the nonsocial weevil Euops chinensis reveals its plant decomposition and protective roles in fungus-farming mutualism. Front. Microbiol. 14:1048910. doi: 10.3389/fmicb.2023.1048910
Edited by:
Peter H. W. Biedermann, University of Freiburg, GermanyReviewed by:
Xia Zhichao, Hangzhou Normal University, ChinaCopyright © 2023 Guo, Wang, Tang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqiong Li, ✉ bGl4aWFvcWlvbmcxMDBAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.