
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 November 2022
Sec. Microbial Symbioses
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.996938
This article is part of the Research Topic Spiroplasma, Mycoplasma, Phytoplasma, and Other Genome-Reduced and Wall-Less Mollicutes: Their Genetics, Genomics, Mechanics, Interactions and Symbiosis with Insects, Other Animals and Plants View all 12 articles
 Peng Liu1
Peng Liu1 Yuxin Li1Youyuan Ye1Jiaxin Chen1Rong Li1Qinyi Zhang1Yuan Li1Wen Wang2
Yuxin Li1Youyuan Ye1Jiaxin Chen1Rong Li1Qinyi Zhang1Yuan Li1Wen Wang2 Qingguo Meng2Jingyu Ou1Zhujun Yang1Wei Sun3*Wei Gu2*
Qingguo Meng2Jingyu Ou1Zhujun Yang1Wei Sun3*Wei Gu2*Spiroplasma mirum, small motile wall-less bacteria, was originally isolated from a rabbit tick and had the ability to infect newborn mice and caused cataracts. In this study, the whole genome and antigen proteins of S. mirum were comparative analyzed and investigated. Glycolysis, pentose phosphate pathway, arginine metabolism, nucleotide biosynthesis, and citrate fermentation were found in S. mirum, while trichloroacetic acid, fatty acids metabolism, phospholipid biosynthesis, terpenoid biosynthesis, lactose-specific PTS, and cofactors synthesis were completely absent. The Sec systems of S. mirum consist of SecA, SecE, SecDF, SecG, SecY, and YidC. Signal peptidase II was identified in S. mirum, but no signal peptidase I. The relative gene order in S. mirum is largely conserved. Genome analysis of available species in Mollicutes revealed that they shared only 84 proteins. S. mirum genome has 381 pseudogenes, accounting for 31.6% of total protein-coding genes. This is the evidence that spiroplasma genome is under an ongoing genome reduction. Immunoproteomics, a new scientific technique combining proteomics and immunological analytical methods, provided the direction of our research on S. mirum. We identified 49 proteins and 11 proteins (9 proteins in common) in S. mirum by anti-S. mirum serum and negative serum, respectively. Forty proteins in S. mirum were identified in relation to the virulence. All these proteins may play key roles in the pathogeny and can be used in the future for diagnoses and prevention.
Spiroplasmas, belonging to the class Mollicutes that includes Mycoplasma, Phytoplasma, and so on, featured by small genome sizes and a lack of peptidoglycan layer (Liu et al., 2018; Sasajima and Miyata, 2021). Spiroplasmas are geographically widespread as bacterial pathogens or symbionts in insects and plants (Liu P. et al., 2017) as well as important pathogens in commercially exploited aquatic crustaceans (Wang et al., 2004, 2005; Nunan et al., 2005). Spiroplasmas perform helical structure with well-defined cytoskeleton-like structure (Kürner et al., 2005; Trachtenberg, 2006) also show snake-like motility (Shaevitz et al., 2005) and unique infective characters including male-killing endosymbionts (Watts et al., 2009), which possibly involved in certain neurodegenerative diseases such as transmissible spongiform encephalopathies (Bastian et al., 2007). In recent years, this microorganism was studied as an attractive minimal model system (Trachtenberg, 2004), and its ultrastructural and macro-molecular characters have been well-described (Trachtenberg, 2004; Shaevitz et al., 2005; Trachtenberg et al., 2008).
Genome research was necessary and useful technique to investigate many species, it helps us to answer previously intractable biological questions (Sussman, 2015). Many bacterial whole genomes have been sequenced in the class of Mollicutes (including Mycoplasma, Phytoplasma, and Spiroplasma), the whole genome sequence of a few spiroplasma bacteria has yet to be published (Liu P. et al., 2017; Chen et al., 2019). Genomic research can provide us with more information on the characteristics of the morphology, structure, physiological metabolism, and genetic evolution of the bacteria (Chen et al., 2019). Spiroplasma eriocheiris, a pathogen of Chinese mitten crab Eriocheir sinensis and close to Spiroplasma mirum (Wang et al., 2011), performs chemotaxis without the conventional two-component system which was commonly found in bacterial chemotaxis. The cells are polarized by a tip structure and a dumbbell-shaped core in the tip connected by a fat ribbon, they are forming the internal structure of S. eriocheiris. The spiroplasma genomes reported so far do not have orthologs of other bacterial motility systems but have one tubulin homolog-FtsZ and five to seven MreB protein. MreB is related to actin, which is responsible for many eukaryotic motility systems (Takahashi et al., 2020). Sixteen proteins were identified as the components of the internal structure by mass spectrometry, including Fibril protein and four types of MreB proteins (Liu P. et al., 2017). MreB5, one of the five MreB paralogs, was proven the contributing to cell elongation and is essential for the transition from rod-to-helical shape in Spiroplasma (Harne et al., 2020). Cytoskeleton-like proteins (Fibril, Mrebs, and EF-Tu) were also phosphorylated suggesting that phosphorylation may play a crucial role in the formation of the cytoskeleton-like structure (Liu et al., 2020).
With the completion of a large number of biological genome sequences, while it is found that genome research alone does not explain the fundamental problem, because the ultimate function of various physiological functions in organisms is protein. Immunoproteomics as an emerging field of proteome analysis plays a key role in screening immunogenic proteins. Such a method has been used to successfully identify antigenic epitopes in several pathogens, such as Mycoplasma bovis, Brucella abortus, Neisseria meningitidis, and S. eriocheiris (Liu Y. et al., 2017).
Therefore, the study of proteomics has become the main research topic and direction in the post-genome era (Vandemoortele et al., 2016), and the most important issue for pathogenic microorganisms is to find the main immune proteins or antigen proteins (Aslam et al., 2017). That is to find the protein that plays a major role in the pathogenic process. The main method to study microbial immunoproteomics is to extract the whole protein or membrane protein of microorganism, electrophoresis, then hybridization with antiserum, so as to screen out antigen protein and then identify it by mass spectrometry or sequencing (Vandemoortele et al., 2016). Several mycoplasma immunoproteomics studies have been carried out using this method, such as Mycoplasma genitalium, Mycoplasma pneumoniae, Mycoplasma hyopneumoniae, Mycoplasma synoviae, and Mycoplasma mycoides (Khan et al., 2018).
Spiroplasma mirum originally isolated from a rabbit tick (Haemaphysalis leporis-palustis ATCC 29335 = CIP 103925) (Megraud et al., 1983). S. mirum and S. eriocheiris are close to each other in the phylogenetic tree and both can cause the cataract of neonatal rat (Hou et al., 2017). In this study, we mainly conducted comparative genomics studies on S. mirum and S. eriocheiris to find out the characteristics of the physiological metabolism, and genetic evolution of the spiroplasma, and to determine their different pathogenicity. We screened the antigenic proteins and provide a comprehensive view of the immunogenic proteins of S. mirum and also certainly provide valuable information for the identification of virulent proteins or diagnosis of pathogenic mechanisms.
Spiroplasma mirum, Suckling Mouse Cataract Agent (SMCA) (Megraud et al., 1983), was originally isolated from rabbit ticks (Haemaphysalis leporis-palustis). We purchased SMCA [CIP 103925] from ATCC and cultured and cloned it with the same method (Itoh et al., 1989).
The complete genome sequences of S. mirum were firstly determined by high-throughput sequencing via pyrosequencing. A total of 170,589 sequences with an average length of 255 bp were obtained for the S. mirum genome, resulting in 37-fold genome coverage.
Assembly was performed using Newbler software of the 454-suite package, producing 19 contigs ranging from 0.5 to 490 kb for S. mirum.
Multiplex PCR was performed to determine the relationship of contigs, and closure of the gaps was performed by sequencing the PCR products. Phred, Phrap, and Consed software package1 was used for the final assembly and edition. Regions with poor sequencing quality and homopolymers were resequenced. The final consensus accuracy was 99.9999% for the S. mirum genome.
Putative protein-coding sequences (CDSs) were identified by Glimmer3 (Delcher et al., 1999) and ZCURVE 1.0 (Guo et al., 2003). Functional annotation of CDSs was performed through BLASTP searches against GenBank’s non-redundant (nr) protein database, followed by manual inspection. Transfer RNA genes were predicted by tRNAScan-SE (v1.23) (Lowe and Eddy, 1997). The metabolic pathways were constructed using KEGG database (Juncker et al., 2003; Kanehisa et al., 2004). Lipoprotein was determined by LipoP 1.0 (Juncker et al., 2003) and searching PROSITE motif PS51257. Protein domain prediction and COG assignment were performed by RPS-BLAST using NCBI CDD library.
An all-to-all BLASTP was applied to determine the paralogs. Two sequences in a pair are paralogs if the remaining HSPs cover at least 80% of the shorter protein’s length and if the identity is greater or equal to 50%.
Orthologous proteins between S. eriocheiris and S. mirum were identified by all-vs-all reciprocal-BLASTP search, with criteria of similarity >20% and coverage >80%. Orthologs of known Mollicutes genomes were obtained from the MBGD database (Uchiyama, 2003). Unique genes were verified by TBLASTN search using protein sequences of each strain against the other strain’s genome sequence. Comparison of the gene order between S. eriocheiris and S. mirum was analyzed by Artemis Comparison Tool (ACT) (Carver et al., 2005). Protein synteny plot of S. eriocheiris and S. mirum was constructed by blast-score-ratio (Rasko et al., 2005).
The spiroplasma cells were activated in R2 medium to exponential phase and collected by centrifugation. After washing and resuspension, the sample was sonicated on ice and centrifuged to remove the cellular debris. Rabbit antiserum against S. mirum was prepared by following the methods described by Liu Y. et al. (2017). The serum was conjugated to Protein A Beads at 4°C for 4 h with gentle shaking. Conjugated antibody was added to the solution protein of spiroplasma and incubated at 4°C for 4 h. After a wash, the proteins were eluted by Glycine from the beads.
Dried protein pellets were reconstituted in 100 mM ammonium bicarbonate and protein concentrations were determined by Bio-Rad protein assay. Proteins were digested with trypsin and inactivated by heating at 99°C for 5 min. The solution was cleared by centrifugation and vacuum dried to remove bicarbonate salts.
Peptides were continuously separated by strong cation column (SCX) followed by C18 columns (Dionex) before being subjected to MS/MS analysis in an LTQ-Orbitrap mass spectrometer (Thermo Electron, Bremen, Germany). The mass spectrum data were initially searched against the database constructed using the predicted proteins of S. mirum with the aid of the Sequest search engine.
We analyzed the genome of S. mirum, it revealed that S. mirum shares a total of 84 common proteins with S. eriocheiris, 14 Mycoplasmas, 4 Phytoplasmas, 3 Ureaplasmas, 1 Acholeplasma, and part of the genome sequence analysis of Spiroplasma citri. Concatenated protein sequences of 84 orthologous proteins of mycoplasma species were first aligned using MUSCLE (Edgar, 2004), then the conserved alignment blocks were extracted by the Gblocks program (Castresana, 2000). The phylogenetic tree was built using the maximum likelihood method implemented in PHYML (Guindon and Gascuel, 2003) using the following parameters: 100 replications for bootstrap analysis, “JTT” for substitution model, “estimated” for the proportion of invariable sites, “estimated” for gamma distribution parameters, “4” for the number of substitution categories, “yes” to optimize tree topology, and “BIONJ” for starting tree(s). Graphical representation and edition of the phylogenetic tree were performed with MEGA4.
The complete genome sequence and annotation of S. mirum were deposited in GenBank under accession numbers CP002082.
The genomes of S. mirum contain a single, circular chromosome with 1,132,591 bp in length. The base numbering start point was chosen at the first base of the dnaA gene, adjacent to the replication origin (oriC; Figure 1). The basic characteristics of the genomes of S. mirum and S. eriocheiris are shown in Table 1. S. mirum genome length is 1, 132, 591 bp, and G + C content is 29.41%. S. mirum and S. eriocheiris are close to each other in the phylogenetic tree (Figure 2), both species could cause cataracts in rats (Hou et al., 2017). The S. eriocheiris genome size is 1, 364,757 bp, with 29.79% G + C content (Liu P. et al., 2017). The specific genes of S. mirum genomes are listed in Supplementary Table 1. Generally, there is a complete base excision repair system in the Spiroplasma genomes. S. eriocheiris genome contains several genes involved in homologous recombination, including recA, ruvA/B, recD, recO, recR, and recU, while in S. mirum only the recA and recD genes remain complete (Supplementary Table 2), giving us a hint that S. mirum lost the capacity for recombinational repair. We identified 22 transcription factors and one sigma factor in the S. eriocheiris genome, and 13 transcription factors and one sigma factor in the S. mirum genome (Supplementary Table 3).

Figure 1. Chromosome Atlas of Spiroplasma mirum. Moving inside, each concentric circle represents a different genomic feature. The outermost circle shows predicted protein-coding sequences on both strands, colored by functional categories according to COG classification. The second circle displays specific genes of S. mirum genome compared with the other. The third circle illustrates tRNA genes on plus strand (blue) and minus strand (red), and ribosomal RNA genes (green). The fourth circle presents IS elements. The fifth circle shows mean centered G + C content (red-above mean, blue-below mean). The sixth circle (innermost) represents GC skew (G - C)/(G + C) calculated using a 1 kb window.
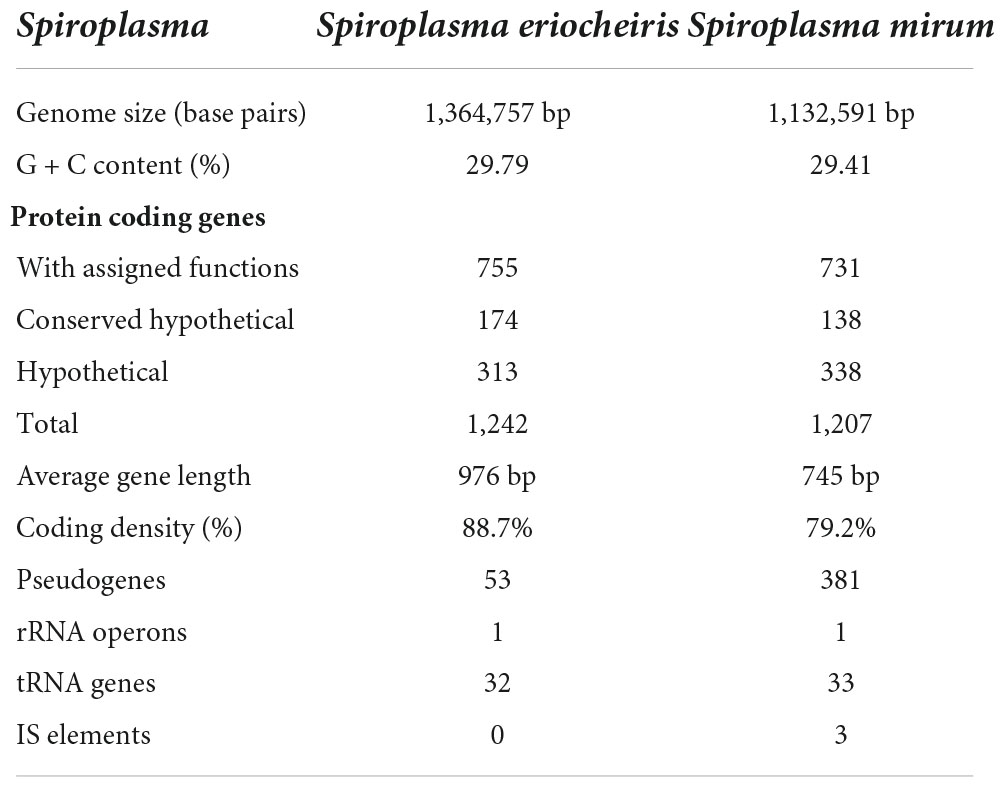
Table 1. General features of Spiroplasma eriocheiris and Spiroplasma mirum genome.
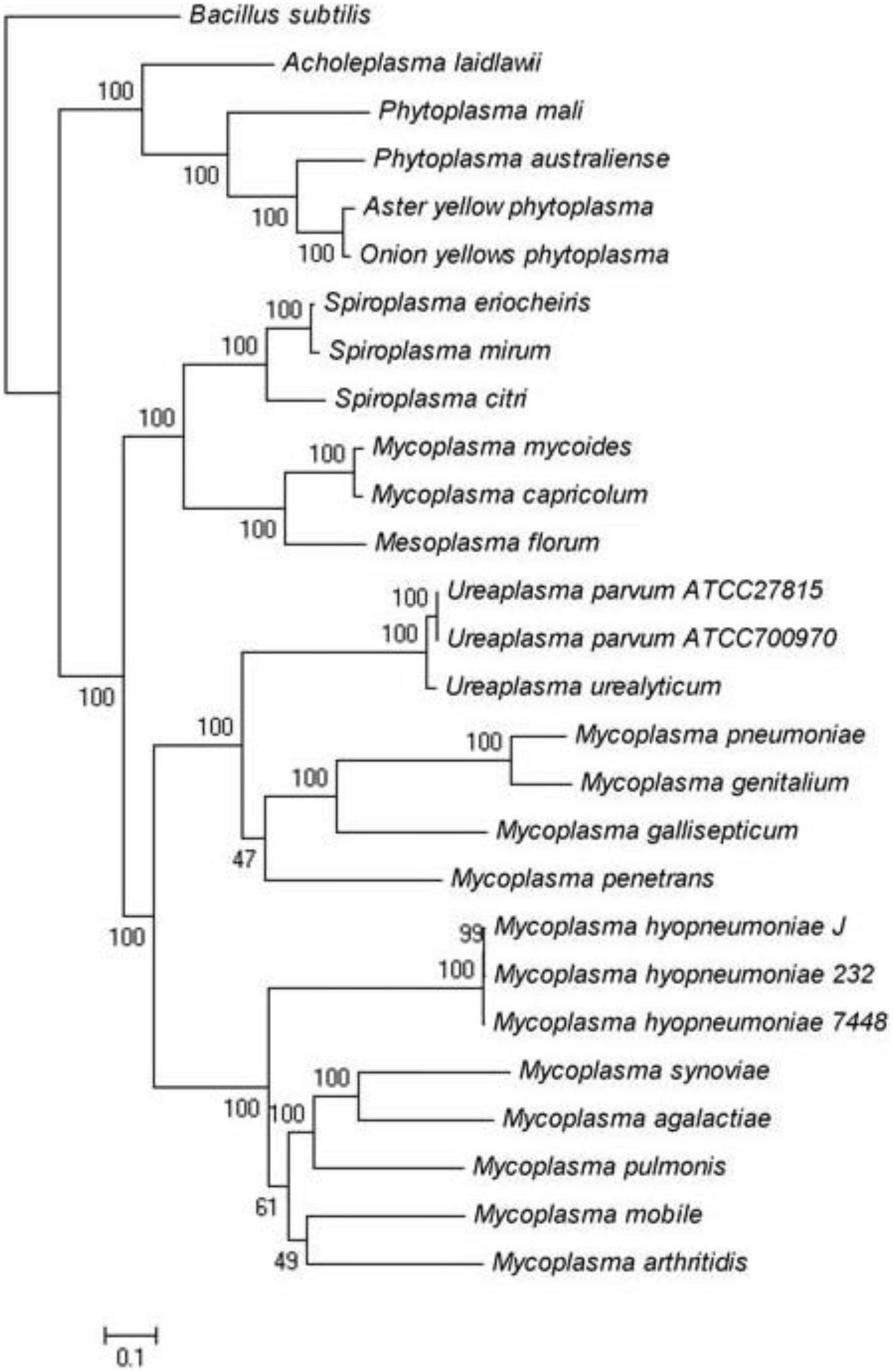
Figure 2. Maximum-likelihood tree of Spiroplasma and Mycoplasma species for which complete genomes are available.
The S. mirum contains 1, 207 CDSs, 1 ribosomal RNA operon, and 33 tRNA genes. The average length of CDSs is 745 bp, and CDSs and the totality of rRNA and tRNA genes accounted for 79.2 and 0.6% of the genome, respectively. The GC contents on the GC and the third codon were 30.58 and 20.93%, respectively. A total of 731 CDSs (60.6%) have clear biological functions, 138 CDSs (11.4%) are similar to conservative proteins with unknown function, 338 CDSs (28%) are hypothetical proteins, COG classification was possible for 735 CDSs (60.9%). We assume that the S. eriocheiris contains 1, 242 CDSs, with an average length of 976 bp, and the GC contents on the GC and the third codon were 30.66 and 20.65%, respectively. A total of 755 CDSs (60.8%) have been identified, which have clear biological functions, another 174 CDSs (14%) are similar to conservative proteins with unknown function, 313 CDSs (25.2%) are hypothetical proteins, and 769 CDSs (61.9%) can be classified by COG (Figure 3). The genome contains 1 ribosomal RNA operon and 32 tRNA genes. CDSs and stable RNA genes accounted for 88.7 and 0.5% of the genome, respectively. There are no insertion sequences (IS element) or phages in the S. eriocheiris genome, while there are three IS elements in the S. mirum genome, one of which may be a phage. The phage with a length of 16,008 bp includes 21 genes (From SMM_0576 to SMM_0597). Particularly, SMM_578, SMM_579, SMM_580, SMM_581, and SMM_583 were identified as putative adhesin p58, P12, P54, P123, and P18 of S. citri, respectively. Thus, the phage may be acquired from S. citri. The phage may have facilitated extensive genome rearrangements in S. mirum and contributed to horizontal gene transfers that led to species-specific adaptation to different eukaryotic hosts and acquisition of pathogenicity. In addition, the common ancestor of the Spiroplasma, Entomoplasma, and Mycoplasma clade may have had a relatively large genome and flexible metabolic capacity; the extremely reduced genomes of Mycoplasma and Spiroplasma species are likely to be the result of independent gene losses (Lo et al., 2013). It is thought that genome reduction is aided by genetic isolation-bacteria that live in monocultures in special host organs, or inside host cells, have less access to other bacterial species from which they can obtain genes (Waterworth et al., 2020).
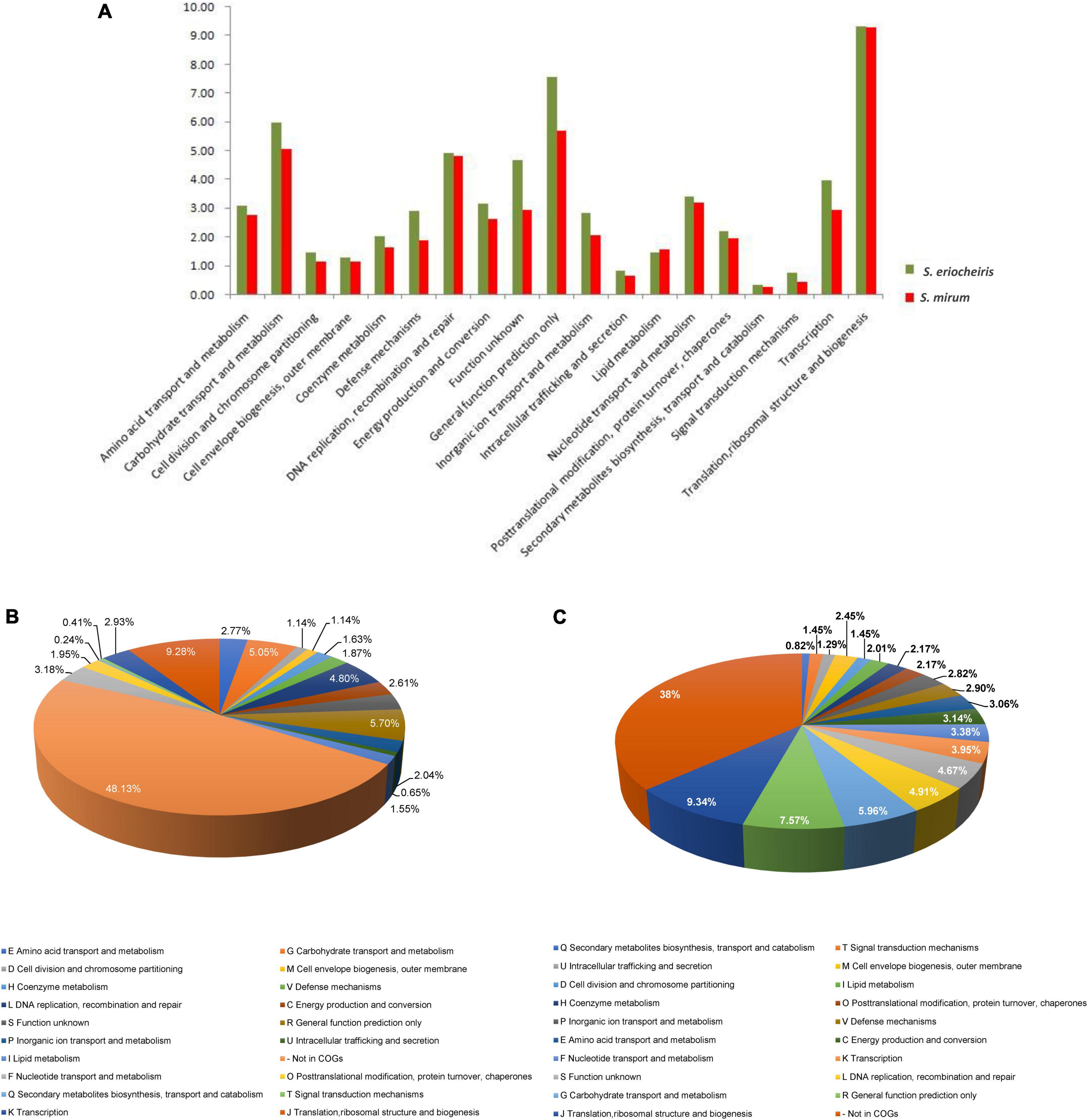
Figure 3. (A) Comparison of COG distribution of Spiroplasma eriocheiris and Spiroplasma mirum. (B,C) COG classification of S. mirum and S. eriocheiris, respectively.
The genome of S. mirum is 232,166 bp smaller than S. eriocheiris. Genome reduction is a common phenomenon in numerous pathogens. The pattern of genome reduction observed in S. atrichopogonis has been found in several other arthropod-associated bacteria, such as S. poulsonii (Lo et al., 2015). Generally, the stable establishment of a bacterium in a host cell results in genome reduction (Campbell et al., 2015). The loss of functions required for living independently but not within a host gives rise to diminished genomes in various symbionts. Although the phenomenon of genome reduction can be explained by existing evolutionary models, the initiation of the process is not thoroughly understood. The key step preceding genome reduction in the symbiont was likely the horizontal acquisition of the putative lagriamide lga biosynthetic gene cluster. The parasitic bacterium does need to produce nutrients that the host provides, leading to the loss of genes that it would need to live independently and to a consequent reduction in genome size (McCutcheon and Moran, 2011).
Basic feature of S. mirum metabolism elucidated by genomic analysis permitted the metabolic reconstruction of spiroplasma (Figure 4). Spiroplasmas are parasitic in specific hosts and their specific tissues, indicating that they have special nutritional requirements and parasitic history. Genomic sequences will provide evidence to reveal the complex nutritional requirement of spiroplasma. Both S. eriocheiris and S. mirum can utilize glucose in the Embden–Meyerhof–Parnas (EMP) pathway, The oxidative branch of the pentose phosphate pathway was completely absent, as well as the TCA cycle, and finally, pyruvate can be converted into lactic acid, formic acid, and acetic acid. S. eriocheiris can also produce acetaldehyde. However, the arginine dihydrolase pathway was found in both strains. Compared with other flexor-membranous microorganisms, S. eriocheiris has fructose 1,6-diphosphate fructose in the glycolysis pathway, which can catalyze 1,6-diphosphate to form fructose 6-phosphate in gluconeogenesis. However, due to the absence of citric acid cycle, the gluconeogenesis pathway is still incomplete (Figure 4). Both genomes show a complete absence of pentose phosphate pathway oxidation pathways and incomplete non-oxidation pathways, this is similar to other mycoplasmas (Pollack et al., 2002; Westberg et al., 2004).
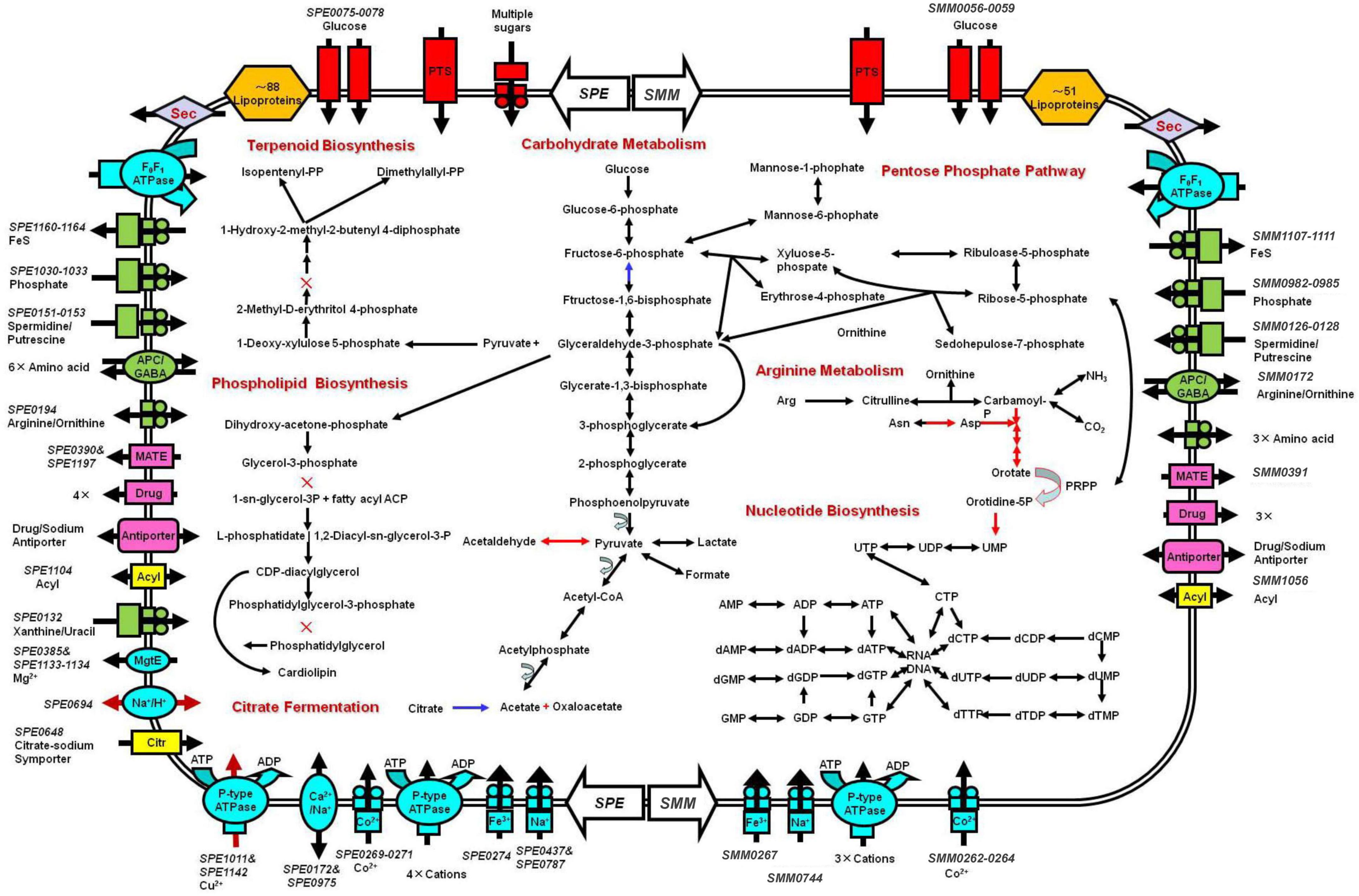
Figure 4. Schematic representation of metabolic pathways of Spiroplasma eriocheiris (left side) and Spiroplasma mirum (right side). Shown are transporters and the main elements of metabolic pathways, deduced from the set of genes with predicted functions. Black arrows: pathways found in both S. eriocheiris and S. mirum genomes. Blue arrows: S. eriocheiris special in Mollicutes. Red arrows: pathways found in the S. eriocheiris genome but not in the S. mirum genome. × : absent enzyme in pathways. Transporters are colored according to their substrates: blue, cations; green, anions and amino acids; rad, carbohydrates; pink, multidrug, and metabolic product efflux. Arrows indicate the direction of substrate transport. In some cases, we cannot identify all the components of these transporters.
The tricarboxylic acid cycle is completely absent in the genome, so do mycoplasma. Arginine hydrolysis pathway exists in both spiroplasma genomes, resulting in ornithine and ATP, CO2, and ammonia. Both purine and pyrimidine cannot be synthesized in S. mirum, but S. eriocheiris can synthesize pyrimidine through 1-diphosphate-5-phosphate ribose (PRPP). The pyrimidine is synthesized. Most amino acids cannot be synthesized in spiroplasma, oxaloacetic acid (produced by citric acid fermentation) can produce aspartic acid, pyruvate can produce serine, asparagine, and glycine can be converted from aspartic acid and serine, respectively. Consistent with their parasitic life characteristics, spiroplasma loses most of their synthetic abilities, such as fatty acids and various cofactors (except CoA). GDSL family lipases can catalyze the decomposition of lipids to obtain fatty acids in S. eriocheiris and S. mirum, and then fatty acids can be oxidized, but the enzymes that catalyze fatty acids β-oxidation are missing in the genome. terpenoids and phospholipids cannot be synthesized in spiroplasma because of the lack of some enzymes, which must be obtained from the host or medium to maintain their survival (Figure 4).
We used the IgG and antibodies of different antigens in serum can specifically bind to other antigens and Protein A, we use polyclonal antibodies to screen antigens in spiroplasma. In the experiment, S. mirum were tested, and negative serum control group was set up to remove non-specific immune binding. We identified 49 and 11 proteins of positive and negative serum, including 9 common proteins. Therefore, 40 antigenic proteins of S. mirum were screened, and the results are shown in Table 2. While 45 antigenic proteins of S. eriocheiris were identified (Liu Y. et al., 2017). There are six antigenic proteins of S. mirum are homologous proteins that can be used as antigen proteins of S. eriocheiris, including extension factor EF-G, pyruvate dehydrogenase (β subunit, E2 subunit, E3 subunit), thioredoxin reductase and DegV family proteins.
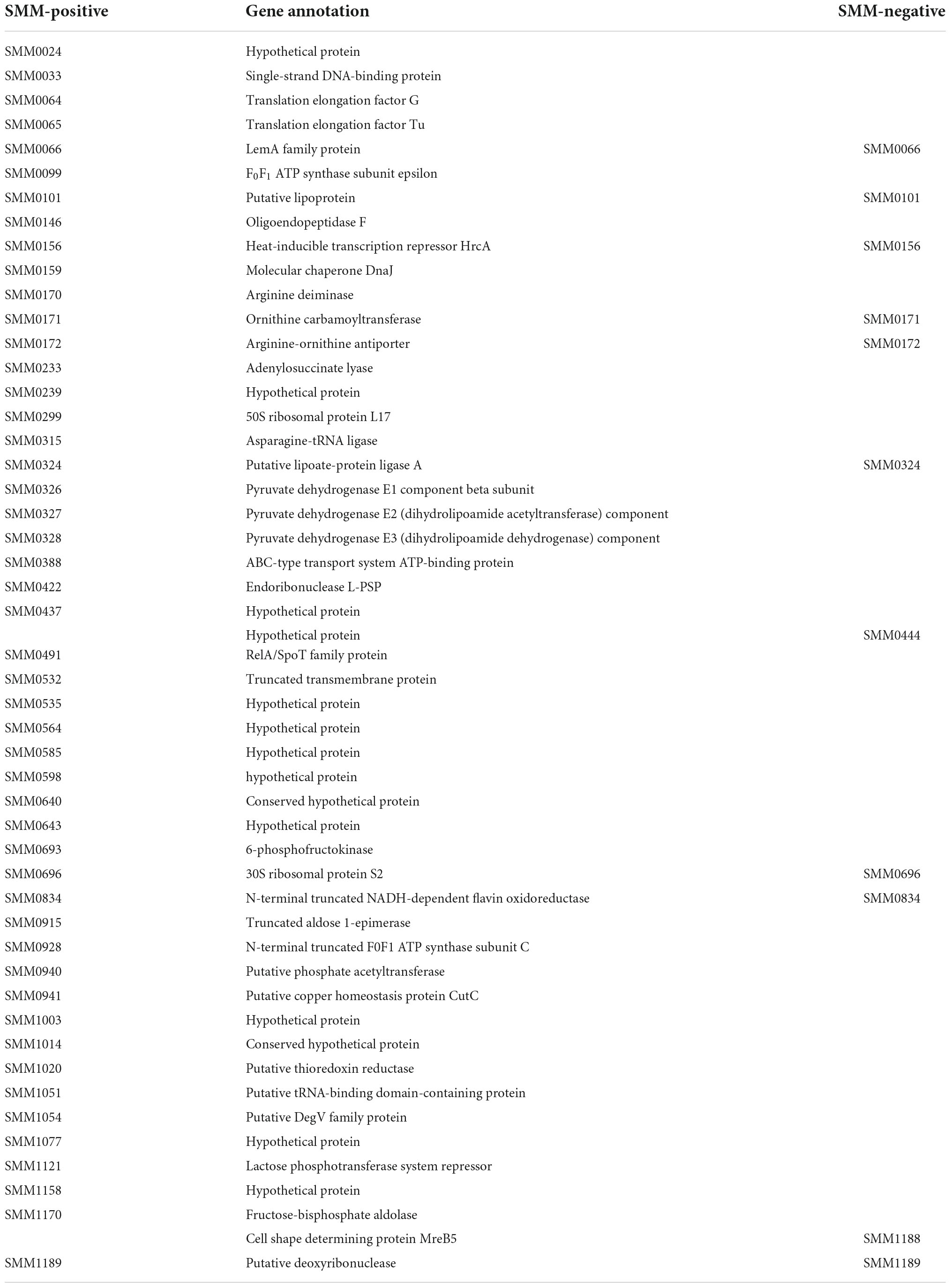
Table 2. Immunogenic proteins of Spiroplasma mirum identified by anti-S. mirum serum and negative serum.
The oligoendopeptidase F (SMM0146), belonging to thimet oligopeptidase family member, was proved as a surface-exposed proteases of M. hyopneumoniae (Jarocki et al., 2019). EF-Tu (SMM_0065) and EF-G (SMM_0064) were identified as antigenic proteins. EF-Tu has evolved the ability to perform diverse functions on the extracellular surface of a wide variety of pathogenic bacteria. While moonlighting functions vary among microbial species, there is a common theme for roles in adherence and in immune regulation (Harvey et al., 2019). EF-G, which is the third most conserved trGTPase among all domains of life. The elongation phase of translation is an important regulatory node in health and disease (Xu et al., 2021). F0F1 ATP synthase (SMM_0099) has transmembrane proton transport function (Feniouk and Junge, 2005) and can be considered candidate antigens to minimize cross reaction in the diagnosis of brucellosis and useful sources for Brucella vaccine development (Ko et al., 2012). Only one lipoprotein (SMM_0101) was screened, lipoprotein is a surface-exposed molecule that is immunodominant and is used as the major antigen for serological diagnosis of Mycoplasma penetrans infection (Sasaki et al., 2002). RelA/SpoT family protein (SMM_0491) is considered as enzymes, which control bacterial physiology through synthesis and degradation of the nucleotide alarmone, and acts as toxins of toxin–antitoxin modules (Kurata et al., 2021). Furthermore, putative copper homeostasis protein CutC (SMM_0941) was found in the antigenic protein. Numerous exciting studies have revealed that copper plays an indispensable role in the microbial pathogen-host axis for entities ranging from pathogenic bacteria to deadly fungal species (Li et al., 2019). Fructose 1,6-bisphosphate aldolase is a ubiquitous cytosolic enzyme that catalyzes the fourth step of glycolysis. Apart from their conserved role in carbohydrate metabolism, aldolases have been reported to perform numerous non-enzymatic functions. Fructose-1,6-bisphosphate aldolase of M. bovis is a plasminogen-binding adhesion (Gao et al., 2018).
We analyzed the genome of S. mirum, 2 spiroplasmas, 14 mycoplasmas, 4 phytoplasmas, 3 ureaplasmas, and 1 acholeplasma, which revealed that S. mirum share a total of 84 common proteins with them (Supplementary Table 4). Bacillus subtilis as an external group. M. mycoides, Mycoplasma capricolum, and Mesoplasma florum have a closer evolutionary relationship to spiroplasma than other Mollicutes, this is consistent with the analysis results of the phylogenetic tree constructed with 16S rRNA (Figure 2) (Stülke et al., 2009). We found 684 spiroplasma-specific proteins, including 187 S. eriocheiris-specific proteins, 182 S. mirum-specific proteins, and 315 common proteins exist in two spiroplasma (Supplementary Table 5). No restriction-modification (RM) system was revealed in Spiroplasma; thus, the barrier of horizontal gene transfer does not exist. Although the functions of most common proteins are unknown, several adhesion-like lipoprotein may be associated with spiroplasma-specific virulence or host-specific invasion.
From the comparative analysis of the S. mirum and S. eriocheiris genomes, it can be found that S. eriocheiris genome is 232 kb bigger than S. mirum, but those two genomes have a highly conserved genome structure and gene sequence (Figure 5). 273 CDSs of S. eriocheiris genomes did not find homologous sequences in S. mirum. These CDSs include two lactose PTS systems and some gene clusters. One of the gene clusters functions as citric acid fermentation, consisting of genes such as CitC, CitDEF, CitG, CitS, and CitX, the sodium citrate transporter (CitS) is responsible for the upregulation of citric acid in anaerobic conditions (van der Rest et al., 1992). Citric acid lyase (CitDEF) catalyzes the decomposition of citric acid into acetic acid and oxaloacetic acid. However, oxaloacetic acid cannot further become pyruvate, malic acid, or isocitrate, so citric acid cannot be used as a carbon source and energy source. It is worth to notice that this gene cluster has not been found in the other Mollicutes, which means that S. eriocheiris may have some special characteristics. Glucuronic acid can be synthesized in S. eriocheiris, but it cannot be synthesized in S. mirum because of the lack of D-mannose redox enzyme. Another gene cluster catalyzes the first four steps of uracil nucleoside synthesis, from carbamoyl phosphate to lactate 5-phosphate, which is also present in the M. penetrans (hf-2) genome (Sasaki et al., 2002). A total of 129 CDSs of S. mirum did not find homologous sequences in S. eriocheiris genome, and the function of these specific proteins is still unknown.
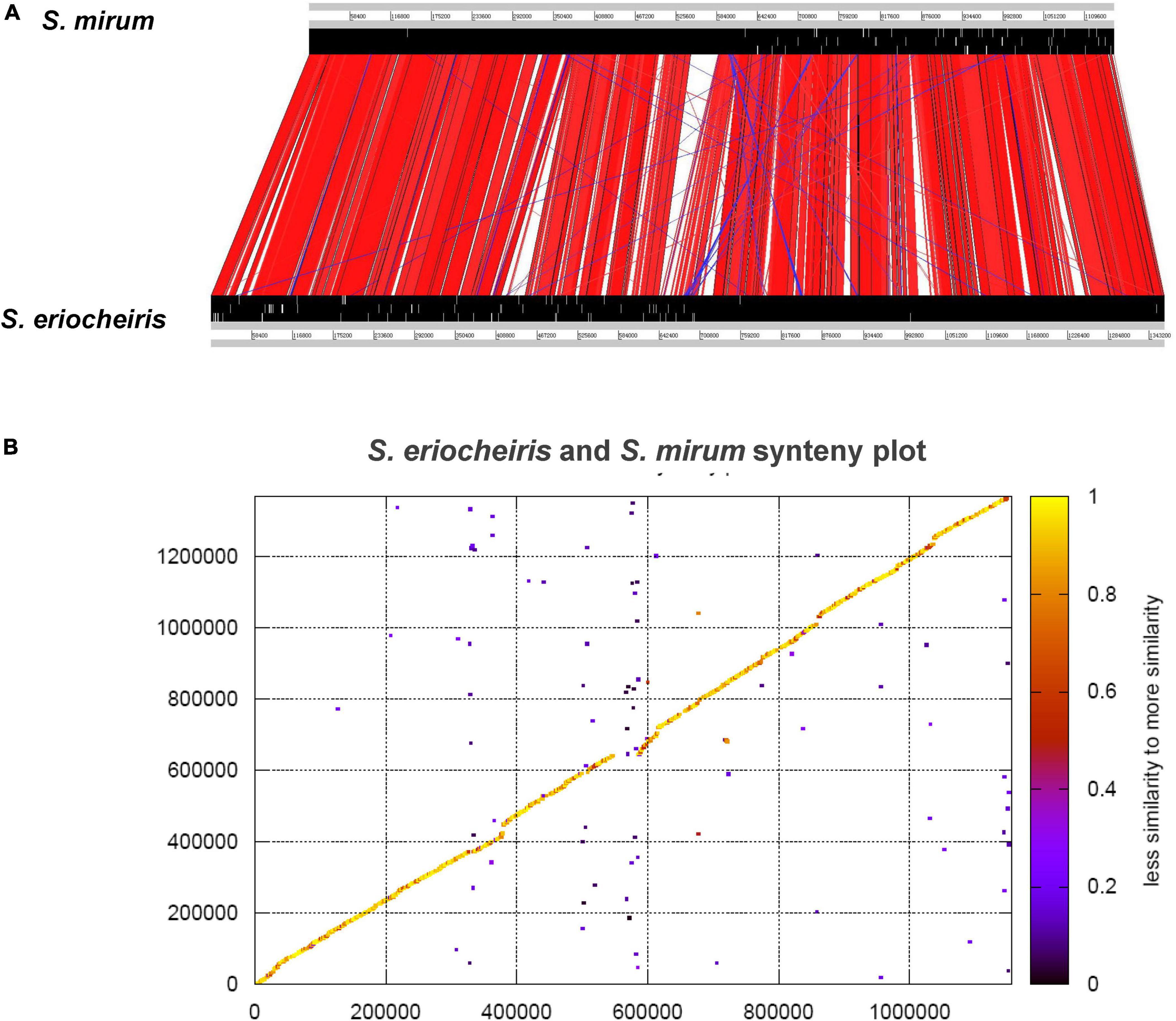
Figure 5. (A) Comparison of the gene order between Spiroplasma eriocheiris and Spiroplasma mirum. (B) Protein synteny plot of S. eriocheiris and S. mirum.
Phosphotransferase system (PTS system) in bacteria consists of two parts: Histidine-containing protein (HPr) that can phosphorylate all carbohydrates and enzyme I. Enzyme II can only catalyze specific substrates, which are composed of 3 functional subunits AII, IIB, IIC, and IID only exist in mannose. Therefore, enzyme II can be divided into four categories: glucose, lactose, mannitol, and mannose. There are 9 categories of PTS systems in the genome of S. eriocheiris, including 3 lactose PTS system and 3 fructose PTS system. While in the S. mirum genome, most of the genes of the PTS system have been lacking, and only 9 of the 23 genes may have functions except for one glucose and one phage fructose PTS system (Supplementary Table 6). Both fructose and glucose PTS systems are encoded by single genes in both genomes.
Because spiroplasma lacks many metabolic and substance synthesis pathways, most of the nutrients they need are obtained from host or artificial medium. The need for a lot of exotic nutrients means that spiroplasma must have a lot of transfer systems. But from the genome, we find that the number of genes in the ATP-binding cassette transport system (ABC transport system) of these two spiroplasma and their proportion to the total number of genes is much less than those of other bacteria, and many are broken genes or pseudogenes. ABC transporters in the genomes of S. eriocheiris and S. mirum are shown in Supplementary Table 7. Five proteins in S. eriocheiris (SPE_0075, SPE_1062, SPE_1069, SPE_1074, and SPE_1075) and one protein in S. mirum (SMM_0056) are similar to the solute-binding protein of an ABC transporter in S. citri. It has been reported that the ability of S. citri to be transmitted by Circulifer haematoceps is clearly lost by disruption of a gene encoding a putative solute-binding protein of an ABC transporter and restored by the addition of this gene (Boutareaud et al., 2004). The ATP copper ion transporter in S. eriocheiris may be related to the specific infection of mitten crab, because the concentration of copper ion in hemocytes of Chinese mitten crab is particularly high. Moreover S. eriocheiris there are more sodium ion-related transporters than S. mirum, which is consistent with S. eriocheiris physiological characteristics such as high salinity tolerance, because Chinese mitten crab lives in seawater (high salinity) during spawning period.
The secretory system plays a very important role in the excretion of toxins and pathogenicity in bacteria. The spiroplasma genome only has Sec secretory pathway, which consists of SecA, SecE, SecDF, SecG, SecY, and YidC (Supplementary Table 8). These proteins also constitute the simplest Sec secretion pathway in spiroplasma, although the Sec secretion pathway of spiroplasma is incomplete when compared to the secretion pathway in Escherichia coli, these proteins are consistent with the minimal required set of secretion and translocation proteins. The genome analysis indicates that the composition of Sec secretion pathway of spiroplasma is very similar to that of mycoplasma, but the important feature is the lack of SecB proteins in spiroplasma. It is speculated that there are other molecular chaperones to perform SecB functions, like Bacillus subtilis. The other special thing is that most of the Mollicutes have double-functional SecDF proteins. It is located on the membrane and pumps secretory proteins out of the membrane by hydrolysis ATP with SecA and SecY support. Because the spiroplasma have very special motility, the contribution of Sec system in spiroplasma motility is not as great as mycoplasma.
Signal peptidase is divided into signal peptidase I and signal peptidase II, which are responsible for disconnecting signal peptides at the N terminal of protein, but we only found signal peptidase II in the spiroplasma genome, the same as other Mollicutes members. Signal peptidase is considered to play an important role in the pathogenesis of Mollicutes, because it plays a crucial role in the formation of adhesion proteins in M. hyopneumoniae and M. pneumoniae; 37 and 15 extracellular proteins were found in S. eriocheiris and S. mirum, respectively. These proteins may be secreted by the Sec system, but the function of most proteins is unknown. Besides the hypothetical proteins and transporters, endonuclease I and GDSL family lipases have been found in S. eriocheiris genome and these two proteins may play a role in host invading. Verotoxin, a known functional extramembrane protein, which is an important toxin in bacteria, was found in S. mirum.
There are the S. mirum contains 243 membrane proteins, 51 lipoproteins, and 15 extra-membrane proteins, while 282 membrane proteins, 88 lipoproteins, and 37 extra-membrane proteins in the genome of S. eriocheiris. Lipoproteins are usually located on the surface of the cell membrane of Gram-positive bacteria, but because spiroplasma evolved from clostridium, and has no cell wall. Lipoproteins located in the outer membrane of spiroplasma are considered to be involved in the colonization of the hosts, ensuring vertical transmission (Paredes et al., 2015). While only one lipoprotein (SMM0101) was identified by antigen proteomics. This result may imply that not all of the lipoprotein is immunogenic. Spiralin is the most studied protein in spiroplasma, and it is also a unique protein in spiroplasma. There are two Spiralin protein (SPE0615 and SPE1172) in the genome of S. eriocheiris, while only 87 bp fragments at the C terminal of one Spiralin protein exist in the S. mirum. The similarity of Spiralin protein between S. mirum, S. citri, Spiroplasma kunkelii, Spiroplasma melliferum and Spiroplasma phoenicerum is low, which may be caused by the difference of host and parasitic environment, and the Spiralin protein has polymorphism.
The Pre-lipoprotein acetyltransferase is a membrane lipoprotein, which can catalyze the first step of lipoprotein synthesis, transfer the acetyl group to the N terminal of the protein, and connect with cysteine to form lipoprotein. Prolipoprotein diacylglyceryl transferase (Lgt) and lipoprotein signal peptidase II (Lsp II) are unique enzymes (Hutchings et al., 2009) and associated with lipoprotein metabolism in prokaryotes. Alterations in the expression of pre-lipoprotein acetyltransferase and signal peptidase can cause changes in virulence, transport, and signaling systems. There are two Lgt and one Lsp II encoding genes in both genomes of S. eriocheiris and S. mirum (Supplementary Table 9).
According to the genome analysis, there are two kinds of adhesion proteins (SARPs) (SPE0025 and SPE0529) in S. eriocheiris (Supplementary Table 9). Adhesion proteins play a very important role in the interaction between spiroplasma and insect cells (Yu et al., 2000). The transposons in S. mirum contain five adhesion-like proteins (SMM_0578-0581 and SMM_0583; Supplementary Table 9), these proteins are homologous to P58, P123, P54, and P18 in S. citri, and these proteins are specific in spiroplasma (Bai and Hogenhout, 2002). The ability of S. citri to infect leafhoppers depends on a protein, identified as a substrate-binding protein for ABC transporters (Boutareaud et al., 2004). There are 5 Substrate-binding proteins (SPE0075, SPE 1062, SPE 1069, SPE 1074, and SPE 1075) and 1 ABC transporters homologous (SMM0056) present in S. eriocheiris and S. mirum. Those proteins may promote the invasion process.
Endo-beta-N-acetylglucosaminidase (SPE_0847 and SMM_ 0801) was shown to be a pathogenicity determinant of mammal pathogens and depressed immune response and interfered with host defense (Valisena et al., 1991). SPE_0813 and SMM_0767 encoded a truncated toxin protein, which is lethal to mice and toxic to Vero cells (Amimoto et al., 2007). SPE_0973 encodes a truncated toxin A, which is S. eriocheiris specific and is homologous with the N-terminal of toxin A of Clostridium difficile. SMM_0541 encodes an extracellular verocytotoxin 1, which is S. mirum specific and shows homology with the shiga toxin 1A subunit (Supplementary Table 9). The presence of domains similar to proteins of the incomplete sec secretion system in pathogenic bacteria suggests that S. mirum possesses a related translocation system. The Virulence substances may be delivered by the sec secretion system.
FeoB, a high-affinity ferrous iron uptake membrane protein, is present in many bacteria except for Mollicute genomes and contributes considerably to bacterial virulence (Cartron et al., 2006). We identified a FeoB-encoding gene in spiroplasma genomes (SPE_0274 and SMM_0267), which might enable spiroplasmas to compete for iron from the host and contribute to their pathogenicity.
Two copper-transporting P-type ATPases (SPE_1011, SPE_1142) of S. eriocheiris might be associated with its pathogenicity and host specificity. It is known that S. eriocheiris is transmitted in blood of crabs (Wang et al., 2004), which consists of a large amount of copper cation. NADH peroxidase (SMM_0037) and putative thiol peroxidase (SMM_1137 and SMM_0497) were found in the genome of S. mirum, these enzymes may protect S. mirum from oxidative. The presence of domains similar to proteins of the sec secretion system in pathogenic bacteria suggests that spiroplasma possesses a related translocation system.
Spiroplasma mirum have 383 pseudogenes, which covered 11.7% genome length, 31.6% gene numbers (Supplementary Table 10). This indicates that the genome of S. mirum is degenerating. In contrast, only 53 pseudogenes were revealed in the S. eriocheiris genome, and 23 pseudogenes can be compensated for by other collateral homologous genes. We found 282 genes in the genome of S. eriocheiris constitute 90 collateral homologous gene families, while 188 genes in S. mirum can form 56 collateral homologous gene families (Supplementary Table 11). The complete collateral homologous genes of 96 pseudogenes in S. mirum were found. This means that other pseudogenes are not necessary for S. mirum. The main function of pseudogenes is related to carbohydrate transport and metabolism in S. mirum. The encoding density of S. mirum genome is 79.2%, which is much lower than that of other bacteria. A large amount of non-coding DNA may be caused by genetic degradation. The GC content of the non-coding regions in S. eriocheiris and S. mirum was 22.9 and 24.9%, respectively, which was lower than the coding region. Pseudogenes may be caused by previously important genes that are not strictly evolved and gradually accumulated mutations (Lawrence et al., 2001). Usually, the high deletion rate of chromosomes will lead to the formation of pseudogenes, and the intracellular bacterium will lead to an increase in deletion rate and mutation rate due to the constant environment. However, the limited function of mismatch repair system and the deletion of homologous recombinant gene may be the main reasons for the increase in pseudogenes. The pathogen has acquired genes from other bacteria despite going through genome reduction, suggesting that isolation has not yet played a major role in this case of genome reduction, with horizontal gene gains still offering a potential route for adaptation (Waterworth et al., 2020).
Because there is no restriction modification system in the spiroplasma genome, the hindrance of horizontal transfer gene does not exist. 51 S. eriocheiris proteins and 15 S. mirum proteins are homologous to non-mollicutes bacteria (Supplementary Table 12). These genes may obtain from other bacteria genes after the evolution of spiroplasma. These horizontal transfer genes can be divided into 6 gene clusters in S. eriocheiris genome, including 3 PTS systems, 1 citric acid fermentation and 1 glucuronic acid fermentation. The acquisition of some horizontal transfer genes represents some special properties of spiroplasma, such as fructose 1,6-bisphosphate enzyme mentioned in metabolism. In addition, we also found some virulence factors in the horizontal transfer system. Endo-Beta-N-acetylglucosidase has been found in both spiroplasma genome, which is an important pathogenic factor of animal pathogen and can reduce host immune function (Valisena et al., 1991). Both S. eriocheiris and S. mirum contain this protein consistent with their characteristics as animal pathogen.
One disconnected toxin protein was found in S. eriocheiris (SPE_0319) and S. mirum (SMM_0306) genomes and which is homologous to the N terminal of the cytotoxic TpeL protein of Clostridium perfringens. TpeL protein, a novel virulence factor, plays an important role in mouse lethal processes (Amimoto et al., 2007). Because the N terminal sequence is conserved in many clostridium toxins, we speculate that TpeL protein may be an important virulence factor of spiroplasma. A disconnected toxin A protein is also present in the genome of S. eriocheiris, which is homologous to the N terminal of Clostridium difficile toxin A protein. As discussed in Sec system, the presence of Verotoxin I (SMM_0541) in the genome of S. mirum, which is homologous to the virulence factors of Shigella dysenteriae and Shigatoxigenic E. coli, named shigella toxin IA subunit. Ferritin FeoB is a very important virulence factor in many bacteria. SPE0274 and SMM0267 can encode complete FeoB, which may be helpful for spiroplasma to despoil ions in host and produce virulence. These horizontal transfer genes do not exist in other Mollicutes members but are important virulence factors in some Clostridium and bacilli. This reveals that the pathogenic characteristics of spiroplasma are similar to intestinal pathogens such as Clostridium or bacillus.
The S. eriocheiris and S. mirum genomics research reveals the commonness and difference in spiroplasma in aquatic environment and terrestrial environment. In an evolutionary sense, we can continue to study the evolutionary origin and classification of cell wall-less microbes in the Mollicutes, as well as their differences and evolutionary relationships with other bacteria. Further genomic analysis of two spiroplasma may identify factors associated with their special living environment (parasitic history) and pathogenicity due to aquatic and terrestrial habitat differences.
Antigen proteomics is a new research field of pathogen in recent years. At present, there is still less study of spiroplasma antigen proteomics, and the study of mycoplasma antigen proteomics has just begun. The current research on mycoplasma has M. genitalium, M. pneumoniae, M. hyopneumoniae, and M. synoviae. S. mirum belongs to serological group V, while S. eriocheiris belongs to a new Spiroplasma group XLIII (Wang et al., 2011). The two species have very different hosts and environments. So, it is not surprising that these two strains have very different cell surface proteins and antigen-related genes. We used immunoprecipitation to screen the antigenic proteins of S. mirum. Negative serum was used as a control to determine the main antigen protein. We’ve screened out 40 antigen proteins of S. mirum were screened out, six of them are homologous proteins that can be used as antigen proteins of S. eriocheiris, including extension factor EF-G, pyruvate dehydrogenase (β subunit, E2 subunit, and E3 subunit), thioredoxin reductase, and DegV family proteins. Elongation factor thermo unstable (EF-Tu) and pyruvate dehydrogenase E1-β subunit are also M. hyopneumoniae antigen proteins (Pinto et al., 2007). The EF-Tu is also the antigen protein of M. synoviae (Bercic et al., 2008). Mmm SC PG1 of M. mycoides shared with extraordinary EF-Tu, extension factors EF-G, pyruvate dehydrogenase, fatty acids protein ligase A and fructose diphosphate aldolase as antigen proteins (Jores et al., 2009), M. mycoides subsp. mycoides strain B237 shared pyruvate dehydrogenase as antigen protein (Naseem et al., 2010). In addition, translation elongation factor G is likely to be a drug target for the malarial parasite. Therefore, the EF-G could be evaluated as potential proteins of diagnostic markers and target proteins of research on pathogenicity (Liu Y. et al., 2017).
Previously, we have screened out 45 antigen proteins of S. eriocheiris (Liu Y. et al., 2017), many of which are homologous to proteins found in mycoplasma antigen proteomics, such as pyruvate dehydrogenase E1-β subunit is also M. pneumoniae antigen protein (Pinto et al., 2007); enolases, ATP synthetases β subunits, pyruvate kinases, and DanK can act as antigen proteins of M. synoviae (Bercic et al., 2008). Mycoplasma, which has the closest evolutionary relationship with spiroplasma, has been studied in immunoproteomics. Jores et al. screened the immunogenic protein of M. mycoides subsp. mycoides small clonal strain MmmSC PG1T and used experiments. The serum prepared in the laboratory screened out 24 immunogenic proteins. 11 of these 24 antigenic proteins were homologous to the antigenic proteins of S. eriocheiris sinensis. They are elongation factor-G, leucyl peptidase, Pyruvate dehydrogenase (α subunit, β subunit, E2 subunit, and E3 subunit), acetate kinase, glyceraldehyde 3-phosphate dehydrogenase, triose phosphate isomerase, phosphoglycerate mutase, and ATP synthase β subunit and others (Jores et al., 2009). Naseem et al. screened 22 immunogenic proteins in the small clonal strain B237 of M. mycoides subsp. mycoides, including DNA-guided RNA polymerase proteins such as β subunit and acyl carrier protein phosphodiesterase, are also antigen proteins of S. eriocheiris (Naseem et al., 2010). The other antigenic proteins specific to the microbes of the class lamina are mostly lipoproteins or membrane proteins with variable morphology and structure. A putative copper homeostasis protein CutC (SMM0941) was identified in the antigenic protein. The bacterial copper efflux system plays a predominant role in regulating pathogen fitness during infection. Analyses of copper homeostasis in bacteria and fungi extensively demonstrate that copper is utilized by the host immune system as an anti-microbial agent. The expression of copper efflux and detoxification from microbial pathogens is induced to counteract the host’s copper bombardment, which in turn disrupts these machineries, resulting in the attenuation of microbial survival in host tissue (Li et al., 2019).
Now we have entered the post-genome era because there are no technical obstacles to genome sequencing, so proteomics (including immunoproteomics and antigenic histology) has also entered a period of rapid development. Regarding pathogenic microorganisms, people are most concerned about which proteins of microorganisms play a major role in the pathogenic process, so it is of great significance for us to carry out this antigen-screening experiment. However, we only preliminarily screened the antigen proteins of spiroplasma, and the specific functions of each protein need to be further studied.
Spiroplasma was attractive for research due to their very small, helical structure and unusual mode of motility by means of a contractile cytoskeleton-like structure, functioning as a linear motor (Trachtenberg, 2006; Trachtenberg et al., 2008). Some structural, dynamic, and proteomic approaches have elucidated the basic organizing principles of their minimal, yet functional, cytoskeleton-like architecture (Liu P. et al., 2017). The full genome information of spiroplasma should provide further significant opportunities to understand the pathogenesis and mechanism of this unique cytoskeleton-like architecture. The comparative genomic analysis of S. eriocheiris and S. mirum reveals some remarkable differences in genes related to their special characteristics associated with their living environments. The complete genome sequence of S. mirum from rabbit tick reveals that Glycolysis, pentose phosphate pathway, arginine metabolism, nucleotide biosynthesis, and citrate fermentation were found in S. mirum. Trichloroacetic acid, fatty acids metabolism, phospholipid biosynthesis, terpenoid biosynthesis, and cofactors synthesis were completely absent. But lactose-specific PTS was not identified in S. mirum. A total of 273 S. eriocheiris CDSs had no homologous in S. mirum genome, including lactose-specific PTS and citrate fermentation gene cluster. S. mirum genome had 381 pseudogenes, accounting for 31.6% of total protein-coding genes. This is the evidence that spiroplasma genome is under an ongoing genome reduction. Interestingly, S. mirum was infective for suckling mice and produced cataracts. But we could not find genes associated with this character up to now. However, further analyses and experiments based on the genomic data should be helpful to probe the mechanisms of S. mirum infections in vertebrate hosts.
We identified the antigen proteins and antigen membrane proteins of S. mirum. 40 proteins in S. mirum are identified in relation to virulence. The results reported in this study will elucidate the immune relationship between the host and the pathogen S. mirum, as well as benefit analytical techniques for identifying bioindicators and more precise diagnosis of the pathology. Currently, there is no explicit understanding of the exact role of these potential immunogenic proteins; future experiments should reveal the immunogenicity of these immunogenic proteins, and the interaction between these proteins and host proteins can be identified and proved.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
PL, YXL, YY, JC, WS, WG (Antigen proteome analysis parts), RL, QZ, YAL, WW, QM, JO, and ZY (Genome part) contributed to the conception and design. PL, YXL (Antigen proteome analysis parts), and YY (Genome part) contributed to the collection and assembly of data. PL, YXL, and YY wrote the manuscript. PL, WS, and WG approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Hunan Province, China (2019JJ50493); Scientific Research Foundation of Hunan Provincial Education Department, China (19B489); Research Foundation of University of South China (190XQD015); and Hunan Provincial College Students’ Innovation and Entrepreneurship Training Program (S202210555220 and 202210555076).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.996938/full#supplementary-material
Amimoto, K., Noro, T., Oishi, E., and Shimizu, M. (2007). A novel toxin homologous to large clostridial cytotoxins found in culture supernatant of Clostridium perfringens type C. Microbiology 153(Pt 4), 1198–1206. doi: 10.1099/mic.0.2006/002287-0
Aslam, B., Basit, M., Nisar, M. A., Khurshid, M., and Rasool, M. H. (2017). Proteomics: Technologies and their applications. J. Chromatogr. Sci. 55, 182–196.
Bai, X., and Hogenhout, S. A. (2002). A genome sequence survey of the mollicute corn stunt spiroplasma Spiroplasma kunkelii. FEMS Microbiol. Lett. 210, 7–17. doi: 10.1111/j.1574-6968.2002.tb11153.x
Bastian, F. O., Sanders, D. E., Forbes, W. A., Hagius, S. D., Walker, J. V., Henk, W. G., et al. (2007). Spiroplasma spp. from transmissible spongiform encephalopathy brains or ticks induce spongiform encephalopathy in ruminants. J. Med. Microbiol. 56(Pt 9), 1235–1242.
Bercic, R. L., Slavec, B., Lavric, M., Narat, M., Bidovec, A., Dovc, P., et al. (2008). Identification of major immunogenic proteins of Mycoplasma synoviae isolates. Vet. Microbiol. 127, 147–154. doi: 10.1016/j.vetmic.2007.07.020
Boutareaud, A., Danet, J. L., Garnier, M., and Saillard, C. (2004). Disruption of a gene predicted to encode a solute binding protein of an ABC transporter reduces transmission of Spiroplasma citri by the leafhopper Circulifer haematoceps. Appl. Environ. Microbiol. 70, 3960–3967. doi: 10.1128/AEM.70.7.3960-3967.2004
Campbell, M. A., Van Leuven, J. T., Meister, R. C., Carey, K. M., Simon, C., and McCutcheon, J. P. (2015). Genome expansion via lineage splitting and genome reduction in the cicada endosymbiont Hodgkinia. Proc. Natl. Acad. Sci. U.S.A. 112, 10192–10199. doi: 10.1073/pnas.1421386112
Cartron, M., Maddocks, S., Gillingham, P., Craven, C., and Andrews, S. (2006). Feo - transport of ferrous iron into bacteria. Biometals 19, 143–157.
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M. A., Barrell, B. G., and Parkhill, J. (2005). ACT: The artemis comparison tool. Bioinformatics 21, 3422–3423.
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552.
Chen, S., Hao, H., Yan, X., Liu, Y., and Chu, Y. (2019). Genome-wide analysis of Mycoplasma dispar provides insights into putative virulence factors and phylogenetic relationships. G3 9, 317–325. doi: 10.1534/g3.118.200941
Delcher, A. L., Harmon, D., Kasif, S., White, O., and Salzberg, S. L. (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641.
Edgar, R. (2004). MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Feniouk, B. A., and Junge, W. (2005). Regulation of the F0F1-ATP synthase: The conformation of subunit epsilon might be determined by directionality of subunit gamma rotation. FEBS Lett. 579, 5114–5118. doi: 10.1016/j.febslet.2005.08.030
Gao, X., Bao, S., Xing, X., Fu, X., Zhang, Y., Xue, H., et al. (2018). Fructose-1,6-bisphosphate aldolase of Mycoplasma bovis is a plasminogen-binding adhesin. Microb. Pathog. 124, 230–237. doi: 10.1016/j.micpath.2018.08.032
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704.
Guo, F. B., Ou, H. Y., and Zhang, C. T. (2003). ZCURVE: A new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res. 31, 1780–1789. doi: 10.1093/nar/gkg254
Harne, S., Duret, S., Pande, V., Bapat, M., Béven, L., and Gayathri, P. (2020). MreB5 is a determinant of rod-to-helical transition in the cell-wall-less bacterium spiroplasma. Curr. Biol. 30, 4753–4762.e7. doi: 10.1016/j.cub.2020.08.093
Harvey, K. L., Jarocki, V. M. I, Charles, G., and Djordjevic, S. P. (2019). The diverse functional roles of elongation factor Tu (EF-Tu) in microbial pathogenesis. Front. Microbiol. 10:2351. doi: 10.3389/fmicb.2019.02351
Hou, L., Gu, W., Zhu, H., Yao, W., Wang, W., and Meng, Q. (2017). Spiroplasma eriocheiris induces mouse 3T6-Swiss albino cell apoptosis that associated with the infection mechanism. Mol. Immunol. 91, 75–85. doi: 10.1016/j.molimm.2017.08.002
Hutchings, M. I., Palmer, T., Harrington, D. J., and Sutcliffe, I. C. (2009). Lipoprotein biogenesis in Gram-positive bacteria: Knowing when to hold ‘em, knowing when to fold ‘em. Trends Microbiol. 17, 13–21. doi: 10.1016/j.tim.2008.10.001
Itoh, K. I, Pan, J., and Koshimizu, K. (1989). A proposed life cycle model of Spiroplasma mirum based on scanning electron microscopical observations of growth in liquid culture. Microbiol. Immunol. 33, 821–832. doi: 10.1111/j.1348-0421.1989.tb00968.x
Jarocki, V. M., Raymond, B. B. A., Tacchi, J. L., Padula, M. P., and Djordjevic, S. P. (2019). Mycoplasma hyopneumoniae surface-associated proteases cleave bradykinin, substance P, neurokinin A and neuropeptide Y. Sci. Rep. 9:14585. doi: 10.1038/s41598-019-51116-w
Jores, J., Meens, J., Buettner, F. F., Linz, B., Naessens, J., and Gerlach, G. F. (2009). Analysis of the immunoproteome of Mycoplasma mycoides subsp. mycoides small colony type reveals immunogenic homologues to other known virulence traits in related Mycoplasma species. Vet. Immunol. Immunopathol. 131, 238–245. doi: 10.1016/j.vetimm.2009.04.016
Juncker, A. S., Willenbrock, H., Von Heijne, G., Brunak, S., Nielsen, H., and Krogh, A. (2003). Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12, 1652–1662.
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280.
Khan, F. A., Zhao, G., Guo, Y., Faisal, M., Chao, J., Chen, X., et al. (2018). Proteomics identification and characterization of MbovP730 as a potential DIVA antigen of Mycoplasma bovis. Oncotarget 9, 28322–28336. doi: 10.18632/oncotarget.22265
Ko, K. Y., Kim, J. W., Her, M., Kang, S. I., Jung, S. C., Cho, D. H., et al. (2012). Immunogenic proteins of Brucella abortus to minimize cross reactions in brucellosis diagnosis. Vet. Microbiol. 156, 374–380. doi: 10.1016/j.vetmic.2011.11.011
Kurata, T., Brodiazhenko, T., Alves Oliveira, S. R., Roghanian, M., Sakaguchi, Y., Turnbull, K. J., et al. (2021). RelA-SpoT Homolog toxins pyrophosphorylate the CCA end of tRNA to inhibit protein synthesis. Mol. Cell 81, 3160–3170.e9. doi: 10.1016/j.molcel.2021.06.005
Kürner, J., Frangakis, A. S., and Baumeister, W. (2005). Cryo-electron tomography reveals the cytoskeletal structure of Spiroplasma melliferum. Science 307, 436–438. doi: 10.1126/science.1104031
Lawrence, J. G., Hendrix, R. W., and Casjens, S. (2001). Where are the pseudogenes in bacterial genomes? Trends Microbiol. 9, 535–540.
Li, C., Li, Y., and Ding, C. (2019). The role of copper homeostasis at the host-pathogen axis: From bacteria to fungi. Int. J. Mol. Sci. 20:175.
Liu, P., Du, J., Zhang, J., Wang, J., Gu, W., Wang, W., et al. (2018). The structural and proteomic analysis of Spiroplasma eriocheiris in response to colchicine. Sci. Rep. 8:8577. doi: 10.1038/s41598-018-26614-y
Liu, P., Hou, L., Liu, M., Xu, X., Gao, Q., Deng, J., et al. (2020). Phosphoproteomic analysis of Spiroplasma eriocheiris and crosstalk with acetylome reveals the role of post-translational modifications in metabolism. Curr. Proteomics 17, 392–403.
Liu, P., Zheng, H., Meng, Q., Terahara, N., Gu, W., Wang, S., et al. (2017). Chemotaxis without conventional two-component system, based on cell polarity and aerobic conditions in helicity-switching swimming of Spiroplasma eriocheiris. Front. Microbiol. 8:58. doi: 10.3389/fmicb.2017.00058
Liu, Y., Xu, Y., Li, S., Xu, X., Gao, Q., Yuan, M., et al. (2017). Identification of proteome, antigen protein and antigen membrane protein from Spiroplasma eriocheiris. Lett. Appl. Microbiol. 65, 395–402. doi: 10.1111/lam.12784
Lo, W. S., Chen, L. L., Chung, W. C., Gasparich, G. E., and Kuo, C. H. (2013). Comparative genome analysis of Spiroplasma melliferum IPMB4A, a honeybee-associated bacterium. BMC Genomics 14:22. doi: 10.1186/1471-2164-14-22
Lo, W. S., Gasparich, G. E., and Kuo, C. H. (2015). Found and lost: The fates of horizontally acquired genes in arthropod-symbiotic spiroplasma. Genome Biol. Evol. 7, 2458–2472. doi: 10.1093/gbe/evv160
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964.
McCutcheon, J. P., and Moran, N. A. (2011). Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26.
Megraud, F., Gamon, L. B., and McGarrity, G. J. (1983). Characterization of Spiroplasma mirum (suckling mouse cataract agent) in a rabbit lens cell culture. Infect. Immun. 42, 1168–1175. doi: 10.1128/iai.42.3.1168-1175.1983
Naseem, S., Meens, J., Jores, J., Heller, M., Dübel, S., Hust, M., et al. (2010). Phage display-based identification and potential diagnostic application of novel antigens from Mycoplasma mycoides subsp. mycoides small colony type. Vet. Microbiol. 142, 285–292. doi: 10.1016/j.vetmic.2009.09.071
Nunan, L. M., Lightner, D. V., Oduori, M. A., and Gasparich, G. E. (2005). Spiroplasma penaei sp. nov., associated with mortalities in Penaeus vannamei, Pacific white shrimp. Int. J. Syst. Evol. Microbiol. 55(Pt 6), 2317–2322. doi: 10.1099/ijs.0.63555-0
Paredes, J. C., Herren, J. K., Schüpfer, F., Marin, R., Claverol, S., Kuo, C. H., et al. (2015). Genome sequence of the Drosophila melanogaster male-killing Spiroplasma strain MSRO endosymbiont. mBio 6:e02437-14. doi: 10.1128/mBio.02437-14
Pinto, P. M., Chemale, G., de Castro, L. A., Costa, A. P., Kich, J. D., Vainstein, M. H., et al. (2007). Proteomic survey of the pathogenic Mycoplasma hyopneumoniae strain 7448 and identification of novel post-translationally modified and antigenic proteins. Vet. Microbiol. 121, 83–93. doi: 10.1016/j.vetmic.2006.11.018
Pollack, J. D., Myers, M. A., Dandekar, T., and Herrmann, R. (2002). Suspected utility of enzymes with multiple activities in the small genome Mycoplasma species: The replacement of the missing household nucleoside diphosphate kinase gene and activity by glycolytic kinases. Omics 6, 247–258. doi: 10.1089/15362310260256909
Rasko, D. A., Myers, G. S., and Ravel, J. (2005). Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics 6:2. doi: 10.1186/1471-2105-6-2
Sasajima, Y., and Miyata, M. (2021). Prospects for the mechanism of spiroplasma swimming. Front. Microbiol. 12:706426. doi: 10.3389/fmicb.2021.706426
Sasaki, Y., Ishikawa, J., Yamashita, A., Oshima, K., Kenri, T., Furuya, K., et al. (2002). The complete genomic sequence of Mycoplasma penetrans, an intracellular bacterial pathogen in humans. Nucleic Acids Res. 30, 5293–5300. doi: 10.1093/nar/gkf667
Shaevitz, J., Lee, J., and Fletcher, D. (2005). Spiroplasma swim by a processive change in body helicity. Cell 122, 941–945. doi: 10.1016/j.cell.2005.07.004
Stülke, J., Eilers, H., and Schmidl, S. R. (2009). “Mycoplasma and spiroplasma,” in Encyclopedia of microbiology, 3rd Edn, ed. M. Schaechter (Oxford: Elsevier), 208–219.
Sussman, H. E. (2015). 20 years of genome research. Preface. Genome Res. 25:xv. doi: 10.1101/gr.199026.115
Takahashi, D., Fujiwara, I., and Miyata, M. (2020). Phylogenetic origin and sequence features of MreB from the wall-less swimming bacteria Spiroplasma. Biochem. Biophys. Res. Commun. 533, 638–644. doi: 10.1016/j.bbrc.2020.09.060
Trachtenberg, S. (2006). The cytoskeleton of spiroplasma: A complex linear motor. J. Mol. Microbiol. Biotechnol. 11, 265–283. doi: 10.1159/000094060
Trachtenberg, S., Dorward, L. M., Speransky, V. V., Jaffe, H., Andrews, S. B., and Leapman, R. D. (2008). Structure of the cytoskeleton of Spiroplasma melliferum BC3 and its interactions with the cell membrane. J. Mol. Biol. 378, 778–789. doi: 10.1016/j.jmb.2008.02.020
Uchiyama, I. (2003). MBGD: Microbial genome database for comparative analysis. Nucleic Acids Res. 31, 58–62.
Valisena, S., Varaldo, P., and Satta, G. (1991). Staphylococcal endo-beta-N-acetylglucosaminidase inhibits response of human lymphocytes to mitogens and interferes with production of antibodies in mice. J. Clin. Invest. 87, 1969–1976. doi: 10.1172/JCI115224
van der Rest, M. E., Siewe, R. M., Abee, T., Schwarz, E., Oesterhelt, D., and Konings, W. N. (1992). Nucleotide sequence and functional properties of a sodium-dependent citrate transport system from Klebsiella pneumoniae. J. Biol. Chem. 267, 8971–8976.
Vandemoortele, G., Gevaert, K., and Eyckerman, S. (2016). Proteomics in the genome engineering era. Proteomics 16, 177–187.
Wang, W., Gu, W., Ding, Z., Ren, Y., Chen, J., and Hou, Y. (2005). A novel spiroplasma pathogen causing systemic infection in the crayfish Procambarus clarkii (Crustacea: Decapod), in China. FEMS Microbiol. Lett. 249, 131–137. doi: 10.1016/j.femsle.2005.06.005
Wang, W., Gu, W., Gasparich, G. E., Bi, K., Ou, J., Meng, Q., et al. (2011). Spiroplasma eriocheiris sp. nov., associated with mortality in the Chinese mitten crab, Eriocheir sinensis. Int. J. Syst. Evol. Microbiol. 61, 703–708. doi: 10.1099/ijs.0.020529-0
Wang, W., Wen, B., Gasparich, G. E., Zhu, N., Rong, L., Chen, J., et al. (2004). A spiroplasma associated with tremor disease in the Chinese mitten crab (Eriocheir sinensis). Microbiology 150(Pt 9), 3035–3040. doi: 10.1099/mic.0.26664-0
Waterworth, S. C., Flórez, L. V., Rees, E. R., Hertweck, C., Kaltenpoth, M., and Kwan, J. C. (2020). Horizontal gene transfer to a defensive symbiont with a reduced genome in a multipartite beetle microbiome. mBio 11:e02430-19. doi: 10.1128/mBio.02430-19
Watts, T., Haselkorn, T. S., Moran, N. A., and Markow, T. A. (2009). Variable incidence of Spiroplasma infections in natural populations of Drosophila species. PLoS One 4:e5703. doi: 10.1371/journal.pone.0005703
Westberg, J., Persson, A., Holmberg, A., Goesmann, A., Lundeberg, J., Johansson, K. E., et al. (2004). The genome sequence of Mycoplasma mycoides subsp. mycoides SC type strain PG1T, the causative agent of contagious bovine pleuropneumonia (CBPP). Genome Res. 14, 221–227. doi: 10.1101/gr.1673304
Xu, B., Liu, L., and Song, G. (2021). Functions and regulation of translation elongation factors. Front. Mol. Biosci. 8:816398. doi: 10.3389/fmolb.2021.816398
Keywords: genome, antigen proteome, Spiroplasma mirum, spiroplasma, pathogenesis
Citation: Liu P, Li Y, Ye Y, Chen J, Li R, Zhang Q, Li Y, Wang W, Meng Q, Ou J, Yang Z, Sun W and Gu W (2022) The genome and antigen proteome analysis of Spiroplasma mirum. Front. Microbiol. 13:996938. doi: 10.3389/fmicb.2022.996938
Received: 18 July 2022; Accepted: 10 October 2022;
Published: 02 November 2022.
Edited by:
Shigeyuki Kakizawa, National Institute of Advanced Industrial Science and Technology (AIST), JapanReviewed by:
Maxuel Andrade, National Center for Research in Energy and Materials, BrazilCopyright © 2022 Liu, Li, Ye, Chen, Li, Zhang, Li, Wang, Meng, Ou, Yang, Sun and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Sun, c3dqc2NkY0BhbGl5dW4uY29t; Wei Gu, c2t5d2VpNDI2QHNpbmEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.