 Yuannuo Wu
Yuannuo Wu Zihan Li
Zihan Li Jingru Zhao1,3
Jingru Zhao1,3 Xingjia Xiang
Xingjia Xiang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 September 2022
Sec. Microorganisms in Vertebrate Digestive Systems
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.991998
The intestinal microbiota play vital roles for health of wild birds in many ways. Migratory birds with unique life history might increase the risk of pathogenic transmission across the regions. However, few studies have clarified the fungal community structure and inferred the potential pathogens in guts of migratory birds. The high-throughput sequencing method was applied to analyze the fungal community structure and detect the potential fungal pathogens in guts of hooded cranes among different wintering stages. Significant differences were found in gut fungal community composition of hooded cranes among three wintering stages, with the lowest fungal diversity in the late wintering stage. In the late stage, hooded cranes harbored higher relative abundance of plant saprotroph, contributing to food digestion for hosts. Hooded cranes were associated with the lowest diversity and relative abundance of animal pathogens in the late wintering stage. There was an increasing trend of deterministic process for gut fungal community assembly, suggesting that hosts interaction with their fungal communities changed by enhanced gut selection/filtering along wintering periods. Hooded crane was associated with the strongest gut selection/filtering to obtain defined gut fungal community with retaining probiotics (i.e., plant saprotroph) and exclusion of certain pathogens in the late wintering stage. Overall, these results demonstrated that hooded cranes might regulate their gut microbiota to enhance digestion and decrease gut pathogens in preparation for long-term migration.
The guts of vertebrates contain multiple and complex microbial communities (Walter et al., 2011). Intestinal microbiota have numerous functions for their hosts (Li et al., 2016), such as maintaining nutrition (Duca et al., 2021), regulating metabolism and immunity (Sonnenburg and Bäckhed, 2016; Behary et al., 2021). In addition, the gut microbiota are highly dynamic, and plenty factors can significantly affect the intestinal microbial community (Chi et al., 2021), including diet (David et al., 2014), season (Maurice et al., 2015), sex (Org et al., 2016), life style (Vujkovic-Cvijin et al., 2020) and environment (Rothschild et al., 2018).
Avian gut microbiota differ from the majority of vertebrates, as they have extremely complex and unique diet, physiological characteristics and developmental strategy (Song et al., 2020). Correlative network analyses have confirmed the existence of a diverse and complex microbial network in the gut of birds, and the members within microbial network harbored certain interaction (Trosvik and de Muinck, 2015; Paul et al., 2021). Previous study has shown that bird host species are the strongest factor determining their gut microbial community composition (Waite and Taylor, 2014). The diet also has a primary effect on avian gut microbial community (Pan and Yu, 2014). Migratory birds inhabit different environments during their annual cycling, and their diets vary greatly throughout the year, with fluctuation in their gut microbial composition (Grond et al., 2019). The diet can also influence microbial diversity of migratory birds by mediating community assembly process (Youngblut et al., 2019).
As migratory birds would travel long distances and use different habitats before reaching the wintering site, they might contact various pathogens and might be regarded as important vectors of infectious pathogens (Ryu et al., 2014). These pathogens in migratory birds could be transmitted among wintering birds when they forage together among wintering periods (Bodawatta et al., 2022). Studies have shown that migratory birds would pose certain risks to animals and even human health by the spread of disease (Sturm-Ramirez et al., 2005; Zhao et al., 2018). Therefore, understanding the potential pathogens in guts of migratory birds might be helpful to clarify the potential pathogenic transmission cross species.
The Shengjin Lake is an internationally important wetland, offering perfect habitats for migratory birds during wintering periods (Xiang et al., 2019). Hooded crane is one of large long-distance wading wild birds, belong to Threatened Species in the IUCN (International Union for Conservation of Nature and Natural Resources) Red List and the first-class national protected wild animal in China (Zheng et al., 2015). Current studies have focused on the bacterial community structure and their roles in health or disease of hooded cranes (Xiang et al., 2019; Fu et al., 2020). However, the ecological roles or potential pathogens of gut fungal community in hooded cranes have not been well estimated.
The study of gut microbiota was helpful to better understand avian wintering ecology and their environmental adaptation. In this study, the high-throughput sequencing method was used to analyze the gut fungal community structure and infer the potential pathogens in guts of hooded cranes among different wintering stages. The aims of this study were (1) to clarify the effect of wintering periods on gut fungal community structure, (2) to evaluate the potential functions of fungal community for hooded cranes, and (3) to identify potential pathogens in guts of hooded cranes in different wintering stages.
The study site was located in the Shengjin lake, an internationally important wetland. The Shengjin lake served as an indispensable wintering area for migratory birds (Fox et al., 2011). According to the natural climatic characteristics of the lake, the wintering period was divided into three stages, named the early, middle and late stages (Wei et al., 2020). Fecal samples were collected from three wintering stages: the early stage from November 1st to November 2nd, 2018; the middle stage from December 28th to December 29th, 2018; the late stage from February 19th to February 20th, 2019. Before sampling, the telescope was used to search the flocks of hooded crane. Fresh fecal samples were collected immediately after foraging. DNA extractions of fecal samples were performed by using the Qiagen DNA Stool Mini Kit.
Primers BIRDF1/BIRDR1 were used to amplify the COL gene to identify bird species (Hebert et al., 2004). PCR reaction was carried out in 50 μl reaction mixtures, with the parameters as follows: 95°C for 5 min, a total of 35 cycles, 95°C for 30 s, 55°C for 45 s, and 72°C for 90 s, with a final extension period at 72°C for 10 min. The PCR products was sequenced and blasted (>99% identity) in National Center for Biotechnology Information (NCBI) to identify bird species. Only hooded crane samples were retained for high-throughput sequencing.
PCR reaction was conducted using primer ITS1/ITS2 for sequencing to get the fungal data. Raw data were analyzed by QIIME software (QIIME 1.9; Caporaso et al., 2010). High-quality sequences (average mass fraction more than 30 and length > 250 bp) were clustered into Operational Taxonomic Units (OTU) by the UCLUST (Edgar, 2010). OTUs of chimera and monomer were deleted. The most abundant sequence within each OTU was selected as the representative sequence identified by the ribosomal database project Classifier, and aligned by PyNAST (Caporaso et al., 2010). Randomly selecting subsets of 43,000 sequences per sample were used to compare fungal community compositions and diversity for all samples.
The differences of fungal community compositions were shown by non-metric multidimensional scaling (NMDS). Biomarkers of intestinal fungi in each stage were identified by linear discriminant analysis (LDA) effect size (Segata et al., 2011). The fungal functional guilds (i.e., functional group) were assigned using the FUNGuild pipeline (Nguyen et al., 2016). The abundance-based β-null model was used to distinguish the relative importance of deterministic and stochastic processes by the value of null deviation (NDV, Luan et al., 2020). Co-occurrence network analysis was used to clarify the interaction of intestinal fungi in guts of hooded cranes among three wintering stages. The detail information of Materials and Methods can be found in Supporting Information.
A total of 3,754,368 quality-filtered fungal sequences and a total of 3,469 fungal OTUs were detected in all samples, ranging from 43,786 to 72,614 sequences and 73 to 688 OTUs per sample, respectively. There were 574 OTUs (16.5%) sharing in guts of hooded cranes across three wintering stages (Supplementary Figure S1). The unique gut fungal OTUs were 607 (17.5%), 1,093 (31.5%) and 191 (5.5%) for the early, middle and late stages, respectively. The result of One-way ANOVA showed that gut fungal alpha-diversity (i.e., OTU richness and Chao1 index) was the lowest in the late stage compared to early and middle stages (Figure 1).
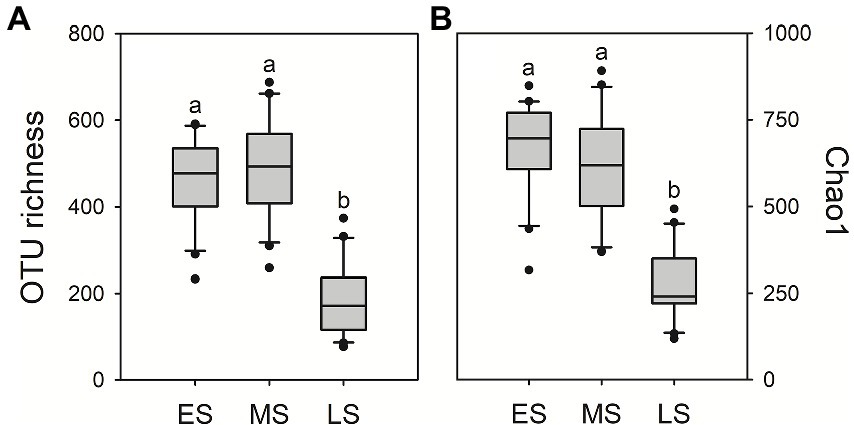
Figure 1. Intestinal fungal alpha-diversity in the three stages of hooded crane. Gut fungal OTU richness (A) and Chao1 index (B). The bottom and top of the box denote the first and third quartiles; the band inside the box denotes median. Letters represent significant differences from the One-way ANOVA test (P < 0.05). ES: early stage; MS: middle stage; LS: late stage. OTU: operational taxonomic units. ANOVA: Analysis of Variance.
The dominant gut fungal phyla were Ascomycota (80.13%), Basidiomycota (11.71%), Zygomycota (5.72%) and Rozellomycota (1.10%). The late stage had the highest relative abundance of Ascomycota. The middle stage had higher relative abundance of Basidiomycota and Rozellomycota relative to early and late stages. The relative abundance of Zygomycota was the highest in the early stage (Supplementary Figure S2). There were significant differences in the gut fungal community composition of hooded cranes among three wintering stages (p < 0.001 in all cases; Figure 2). The abundance-based β-null model was used to distinguish the relative importance of deterministic and stochastic processes (Supplementary Figure S3). The result showed an increasing trend of null deviation value (NDV) with the greater influence of deterministic process along wintering periods.
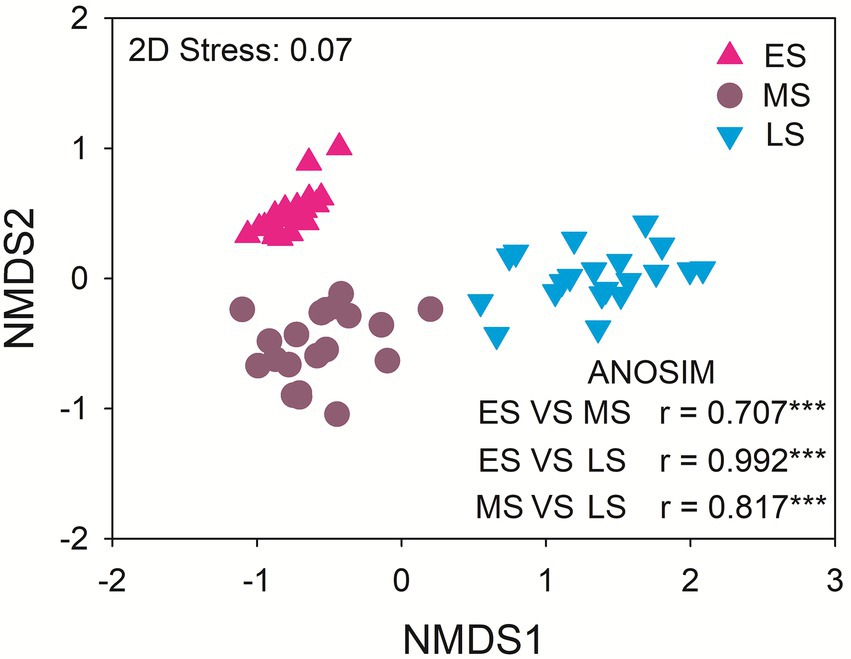
Figure 2. The intestinal fungal community composition among the three stages. ES: early stage; MS: middle stage; LS: late stage. ANOSIM: analysis of similarity. ***P < 0.001.
The result of LEfSe showed that fungi in one phylum (i.e., Zygomycota), 2 classes (i.e., Dothideomycetes and Sordariomycetes), and 2 orders (i.e., Pleosporales and Hypocreales) were enriched in the early stage (Figure 3; Supplementary Figure S4). Fungi from two phyla (i.e., Basidiomycota and Rozellomycota), three classes (i.e., Agaricomycetes, Microbotryomycetes and Tremellomycetes) and five orders (i.e., Onygenales, Agaricales, Polyporales, Microbotryales and Cystofilobasidiales) were more abundant in the middle stage (Figure 3; Supplementary Figure S4). One phylum (i.e., Ascomycota), 2 classes (i.e., Leotimycetes and Chytridiomycetes), and 1 order (i.e., Theleboales) were enriched in the late stage (Figure 3; Supplementary Figure S4).
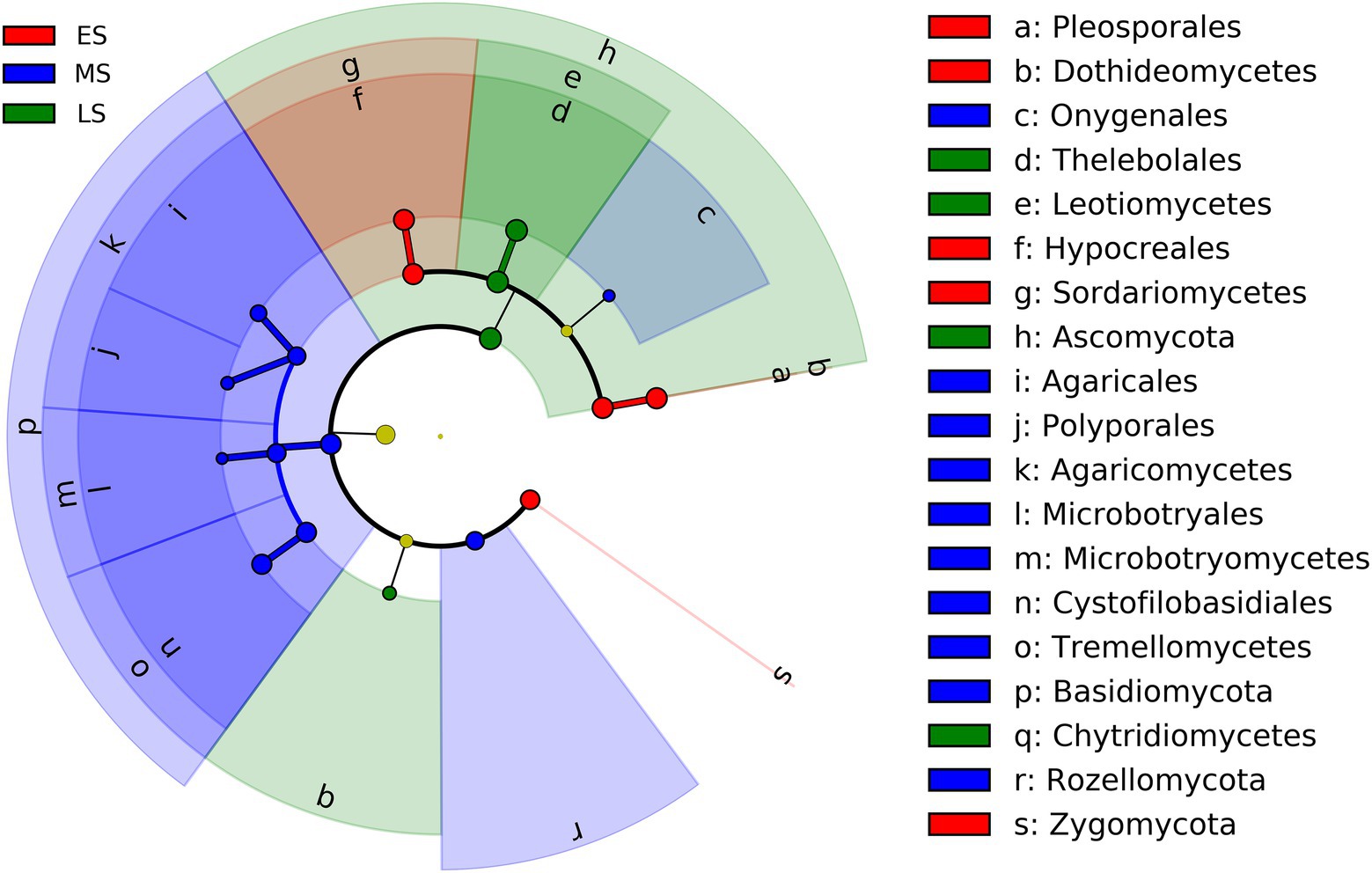
Figure 3. LEfSe analysis showing intestinal fungal biomarkers associated with each stage (the effect size >2 and the alpha value was <0.05). ES, early stage; MS, middle stage; LS, late stage.
There were 12 (e.g., Phoma, Gibberella, Mortierella, etc), 9 (i.e., Acremonium, Cystofilobasidium, Rhodotorula, etc) and 7 (i.e., Schizothecium, Didymella, Botrytis, etc) indicator genera in the early, middle and late stages, respectively (Table 1), and there were 14 (e.g., Phoma calidophila, Gibberella fujikuroi, Mortierella camargensis, etc), 13 (i.e., Cystofilobasidium infirmominiatum, Acremonium dichromosporum, Acremonium nepalense, etc) and 10 (i.e., Thelebolales sp., Schizothecium carpinicola, Didymella exigua, etc) indicator species in the early, middle and late stages, respectively (Supplementary Table S1). Simper analysis indicated that Phoma calidophila (21%) and Gibberella fujikuroi (16%) contributed largely to the differences of the fungal community composition between the early and middle stages. The Thelebolales sp. (37%) and Phoma calidophila (18%) primarily triggered the difference of fungal community composition between the early and middle stages. The Thelebolales sp. (41%) and Cystofilobasidium infirmominiatum (9.1%) were the main taxa explained the difference of fungal community composition between middle and late stages (Supplementary Table S2).
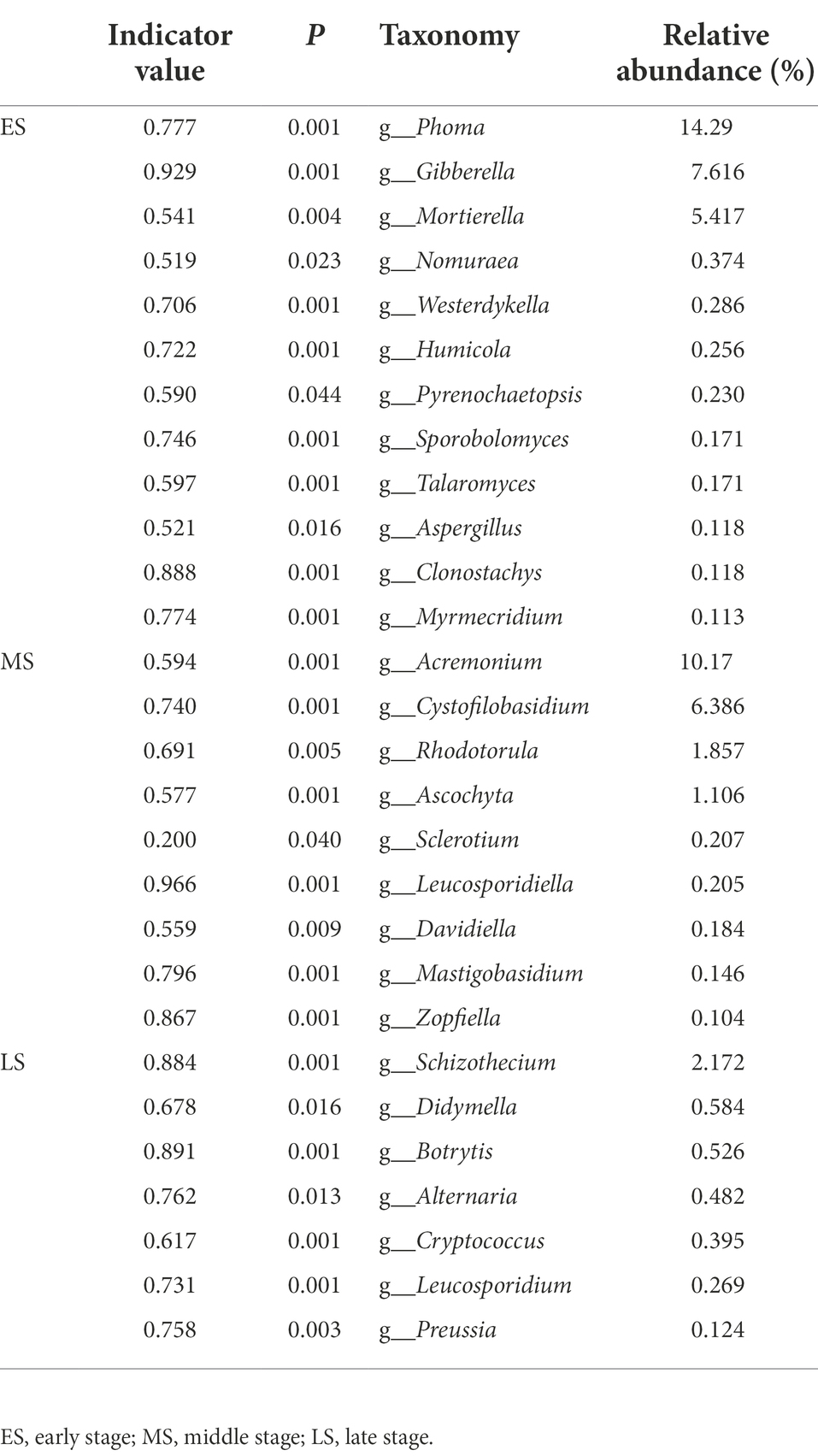
Table 1. Indicator analysis was conducted to show indicator genera with relative abundance >0.1% of each stage.
The FUNGuild analysis was used to infer the plant saprotroph, animal pathogens and endosymbionts in guts of hooded cranes. A total of 12 plant saprotrophic OTUs, 28 animal pathogenic OTUs and 10 animal endosymbiotic OTUs were detected across all samples, ranging from 1 to 10, 2 to 21 and 0 to 8 OTUs per sample, respectively (Supplementary Figure S5). There was litter difference in gut saprotrophic and endosymbiotic community composition among three wintering stages. The gut pathogenic community composition in the late stage was significantly different from that in early and middle stages (Supplementary Figure S6). There was little difference in saprotrophic diversity among three wintering stages (Figure 4A). In the late stage, hooded cranes had the highest relative abundance of plant saprotroph (Figure 4B), the lowest diversity and relative abundance of animal pathogens and endosymbionts relative to other stages (Figures 4C,D; Supplementary Figure S7).
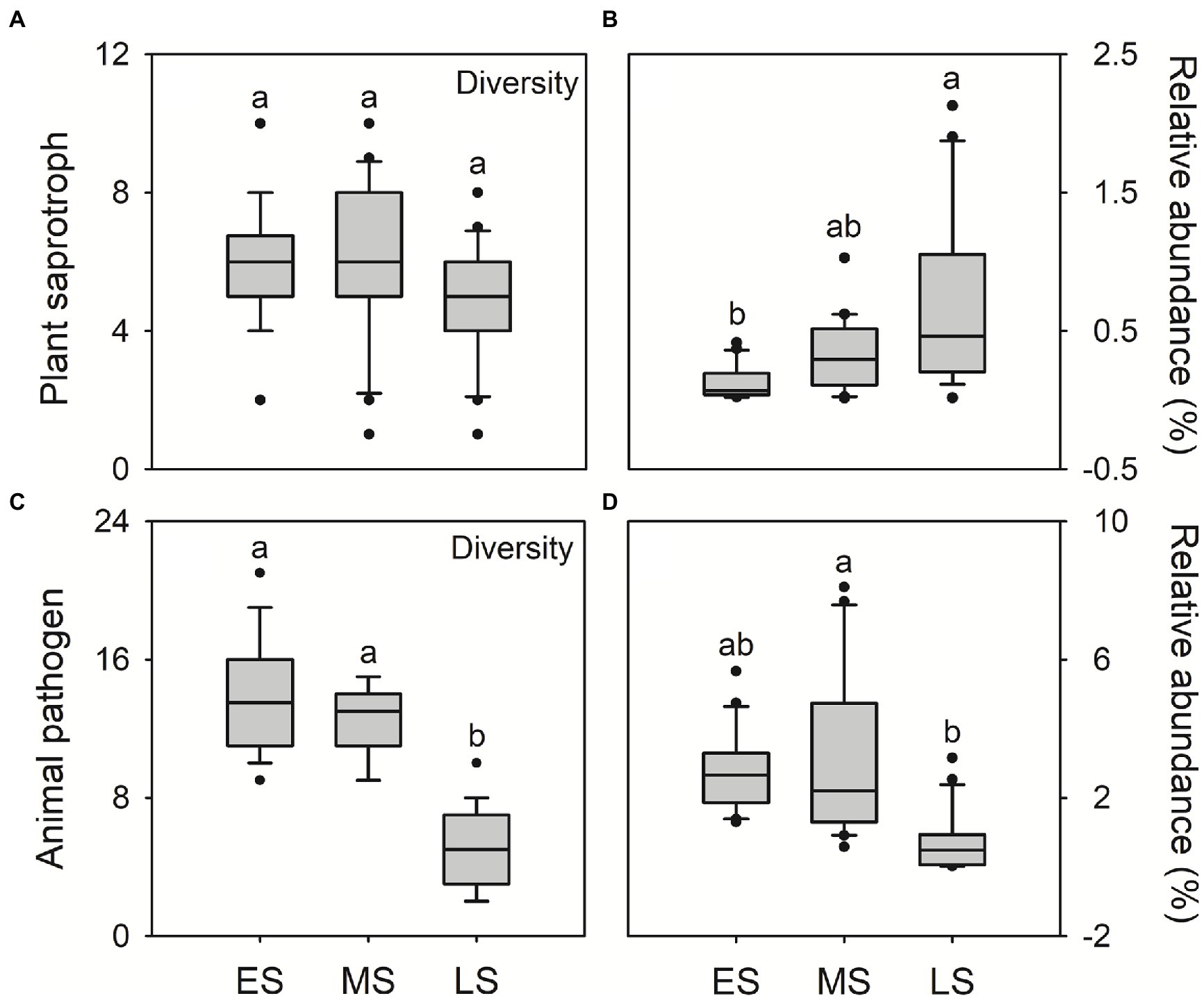
Figure 4. The plant saprotrophic diversity (i.e., saprotrophic OTU; A); the relative abundance of plant saprotroph (B); animal pathogenic diversity (i.e., pathogenic OTU; C); the relative abundance of animal pathogen (D). Letters represent significant differences from the Kruskal-Wallis test (p < 0.05). ES, early stage; MS, middle stage; LS, late stage. OTU, operational taxonomic units.
Co-occurrence network analysis was used to understand the interaction within intestinal fungal taxa of hooded cranes among three wintering stages. Generally, the majority of links in the intestinal network were related to Ascomycota (Supplementary Figure S8). The late stage had the highest clustering coefficient and average degree, the lowest average path length and modularity relative to early and middle stages (Supplementary Table S3). The network stability was assessed by natural connectivity, with the highest network stability in the late stage compared to early and middle stages (Supplementary Figure S9). The different roles of network nodes were identified by Zi-Pi plots (Supplementary Figure S10). There were 33, 35 and 20 keystone fungal OTUs in the early, middle and late stages, respectively (Supplementary Table S4).
Empirical studies have demonstrated that seasonal variation triggered dramatic shifts in animal gut microbial community (Maurice et al., 2015; Ren et al., 2017), which was consistent with this study. In addition, we found an increasing tread of deterministic process in gut fungal community assembly along wintering periods. Microbial community structure could be shaped by stochastic or deterministic processes (Yuan et al., 2019). The increasing relative importance of deterministic process might strengthen the role of environmental selection (Stegen et al., 2012). Gut filtering was an important selection factor to influence the animal microbial community structure (Zhou and Ning, 2017; Yang et al., 2021). The strong gut filtering would select specific microbial taxa and exclude certain taxa in the intestinal ecosystem, triggering the decrease of microbial diversity (Costello et al., 2012; Yan et al., 2016). Thus, the increasing effect of gut filtering might lead to the changes of fungal community composition and the decrease of fungal diversity of hooded cranes along wintering periods (Figures 1, 2).
The co-occurrence network showed the most complexity with the highest stability, average degree and the lowest average path length in late wintering stage (Supplementary Figure S9; Supplementary Table S3). The network with more stability might be associated with stronger resistance to environmental fluctuations and support greater multifunctionality (Deng et al., 2016; Yang et al., 2021). The higher average degree and lower average path length contributed to better organized pathways for metabolites and information exchange among gut fungal taxa (Zhou et al., 2010). The Ascomycota exhibited the major links of co-occurrence network and acted as keystone taxa in guts of hooded cranes (Supplementary Figure S8; Supplementary Table S4). We also found that the highest relative abundance of Ascomycota in the late wintering stage (Supplementary Figure S2A). Ascomycota were the main cellulolytic fungi (Zheng et al., 2020), and they could secrete massive cellulase and hemicellulase to decompose complex polysaccharides (Lichius et al., 2015; Linton, 2020; Sun et al., 2022) and improve the nutrient utilization rate for their hosts (Zhang et al., 2019).
In this study, the relative abundance of plant saprotroph was higher in the late wintering stage compared to early and middle stages (Figure 4B). Previous studies have shown that hooded cranes mainly forage Vallisneria spinulosa and Potamogeton malaianus at Shengjin lake in wintering periods (Wei et al., 2020; Xiang et al., 2021). Therefore, the higher relative abundance of plant saprotroph might be related to the rapid digestion of food for hooded cranes. Moreover, the Alternaria and Preussia were detected as indicator genera in the late stage (Table 1). The two enriched genera have been demonstrated to facilitate the digestion of food (Sugino, 2007; Sun et al., 2011; Liang et al., 2015). Overall, these results demonstrated that, in late wintering stage, hooded cranes might depend on their gut microbiota to enhance digestion and acquire more nutrients from food in preparation for long-term migration.
We also detected the potential pathogens in guts of hooded cranes. Hooded cranes might suffer from various pathogenic invasion to cause inflammation during wintering periods (Xiang et al., 2019). A total of 28 potentially pathogenic OTUs has been detected in the gut of hooded cranes (Supplementary Figure S4B), with higher diversity and relative abundance in early and middle stages than in the late stage (Figures 4C,D; Supplementary Figure S7). Diseases could interrupt appropriate intestinal function to reduce the co-nutrition interaction within gut fungal taxa, which might be a reason resulting in the intestinal network with lower stability in early and middle stages (Zhu et al., 2016; Supplementary Figure S9). In addition, the Cordyceps bassiana was detected as keystone species in the late stage (Supplementary Table S4). Related studies have shown that the Cordyceps bassiana showed anti-inflammatory potential and improved immunity for hosts (Wu et al., 2011; Yang et al., 2015). Thus, these results suggested that intestinal certain fungal taxa might increase hosts’ immunity to resist pathogens for hooded cranes in the late wintering stage.
In this study, hooded cranes suffered invasion from a variety of pathogens under harsh living environment (Supplementary Table S5). More attention should be paid to protect hooded cranes as they are endangered species. These pathogens in hooded cranes might cause a series of diseases not only for birds, but also other animals, even humans (Supplementary Table S5). Previous study has shown that wintering hooded cranes often foraged with poultry at Shengjin lake (Xiang et al., 2019). Therefore, the overlap of foraging niches could trigger pathogenic transmission from hooded cranes to poultry. Notably, poultry could in turn act as vectors to spread pathogens to local residents. Thus, more studies should be paid to clarify the underlying pathway of pathogenic transmission from wild migratory birds to poultry and human beings.
In summary, the increasing gut filtering might trigger the significant differences in fungal community composition of hooded cranes along wintering periods, with the strongest gut selection/filtering to retain probiotics (i.e., Ascomycota and plant saprotroph) and exclude certain pathogens in the late wintering stage. The results suggested that hooded cranes might regulate their gut fungal community to enhance digestion and immunity in preparation for long-term migration. This work contributed to a more complete picture of the ecological function of the gut microbiota in migratory birds. However, there were certain limitations in this study. Previous studies have demonstrated that sex and age were important factors to shape animal gut microbial communities (O’Toole and Jeffery, 2015; Org et al., 2016). We have not yet studied the effects of sex and age of hooded cranes on gut fungal communities. These limitations should be clarified in future studies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, SRP383975.
YW, ZC, and XX designed the experiments. YW, ZL, and JZ completed the field sampling. YW and XX performed the data analysis and prepared the figures. YW wrote the manuscript. XX and ZC contributed to the revision of manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (31801989), and Anhui University Scientific Research Foundation (S020118002/038).
We thank Miss Nazhong Zhang and Wei Wang from Anhui University for assistance in sample collection.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.991998/full#supplementary-material
Behary, J., Amorim, N., Jiang, X. T., Raposo, A., Gong, L., McGovern, E., et al. (2021). Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 12, 187–114. doi: 10.1038/s41467-020-20422-7
Bodawatta, K. H., Hird, S. M., Grond, K., Poulsen, M., and Jønsson, K. A. (2022). Avian gut microbiomes taking flight. Trends Microbiol. 30, 268–280. doi: 10.1016/j.tim.2021.07.003
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chi, L., Tu, P. C., Ru, H. Y., and Lu, K. (2021). Studies of xenobiotic-induced gut microbiota dysbiosis: from correlation to mechanisms. Gut Microbes 13:1921912. doi: 10.1080/19490976.2021.1921912
Costello, E. K., Stagaman, K., Dethlefsen, L., Bohannan, B. J. M., and Relman, D. A. (2012). The application of ecological theory toward an understanding of the human microbiome. Science 336, 1255–1262. doi: 10.1126/science.1224203
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. doi: 10.1038/nature12820
Deng, Y., Zhang, P., Qin, Y. J., Tu, Q. C., Yang, Y. F., He, Z. L., et al. (2016). Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation. Environ. Microbiol. 18, 205–218. doi: 10.1111/1462-2920.12981
Duca, F. A., Waise, T. M., Peppler, W. T., and Lam, T. K. T. (2021). The metabolic impact of small intestinal nutrient sensing. Nat. Commun. 12, 903–912. doi: 10.1038/s41467-021-21235-y
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Fox, A. D., Cao, L., Zhang, Y., Barter, M., Zhao, M. J., Meng, F. J., et al. (2011). Declines in the tuber-feeding waterbird guild at Shengjin Lake National Nature Reserve, China-a barometer of submerged macrophyte collapse. Aquat. Conserv. Mar. Freshwat. Ecosyst. 21, 82–91. doi: 10.1002/aqc.1154
Fu, R., Xiang, X. J., Dong, Y. Q., Cheng, L., and Zhou, L. Z. (2020). Comparing the intestinal bacterial communies of sympatric wintering hooded crane (Grus monacha) and domestic goose (Anser anser domesticus). Avian Res. 11, 1–9. doi: 10.1186/s40657-020-00195-9
Grond, K., Santo Domingo, J. W., Lanctot, R. B., Jumpponen, A., Bentzen, R. L., Boldenow, M. L., et al. (2019). Composition and drivers of gut microbial communities in arctic-breeding shorebirds. Front. Microbiol. 10:2258. doi: 10.3389/fmicb.2019.02258
Hebert, P. D. N., Stoeckle, M. Y., Zemlak, T. S., and Francis, C. M. (2004). Identification of birds through DNA barcodes. PLoS Biol. 2:e312. doi: 10.1371/journal.pbio.0020312
Li, D. T., Wang, P., Wang, P. P., Hu, X. S., and Chen, F. (2016). The gut microbiota: a treasure for human health. Biotechnol. Adv. 34, 1210–1224. doi: 10.1016/j.biotechadv.2016.08.003
Liang, X., Fu, Y. M., and Liu, H. (2015). Isolation and characterization of enzyme-producing bacteria of the silkworm larval gut in bioregenerative life support system. Acta Astronaut. 116, 247–253. doi: 10.1016/j.actaastro.2015.07.010
Lichius, A., Bidard, F., Buchholz, F., Le Crom, S., Martin, J., Schackwitz, W., et al. (2015). Genome sequencing of the Trichoderma reesei QM9136 mutant identifies a truncation of the transcriptional regulator XYR1 as the cause for its cellulase-negative phenotype. BMC Genomics 16, 326–320. doi: 10.1186/s12864-015-1526-0
Linton, S. M. (2020). Review: The structure and function of cellulase (endo-β-1, 4-glucanase) and hemicellulase (β-1, 3-glucanase and endo-β-1, 4-mannase) enzymes in invertebrates that consume materials ranging from microbes, algae to leaf litter. Comp. Biochem. Physiol. B: Biochem. Mol. Biol. 240:110354. doi: 10.1016/j.cbpb.2019.110354
Luan, L., Liang, C., Chen, L. J., Wang, H. T., Xu, Q. S., Jiang, Y. J., et al. (2020). Coupling bacterial community assembly to microbial metabolism across soil profiles. mSystems 5, e00298–e00220. doi: 10.1128/mSystems.00298-20
Maurice, C. F., Knowles, S. C. L., Ladau, J., Pollard, K. S., Fenton, A., Pedersen, A. B., et al. (2015). Marked seasonal variation in the wild mouse gut microbiota. ISME J. 9, 2423–2434. doi: 10.1038/ismej.2015.53
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
O’Toole, P. W., and Jeffery, I. B. (2015). Gut microbiota and aging. Science 350, 1214–1215. doi: 10.1126/science.aac8469
Org, E., Mehrabian, M., Parks, B. W., Shipkova, P., Liu, X. Q., Drake, T. A., et al. (2016). Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 7, 313–322. doi: 10.1080/19490976.2016.1203502
Pan, D., and Yu, Z. T. (2014). Intestinal microbiome of poultry and its interaction with host and diet. Gut Microbes 5, 108–119. doi: 10.4161/gmic.26945
Paul, S. S., Chatterjee, R. N., Raju, M. V. L. N., Prakash, B., Rao, S. V. R., Yadav, S. P., et al. (2021). Gut microbial composition differs extensively among Indian native chicken breeds originated in different geographical locations and a commercial broiler line, but breed-specific, as well as across-breed core microbiomes, are found. Microorganisms 9:391. doi: 10.3390/microorganisms9020391
Ren, T. T., Boutin, S., Humphries, M. M., Dantzer, B., Gorrell, J. C., Coltman, D. W., et al. (2017). Seasonal, spatial, and maternal effects on gut microbiome in wild red squirrels. Microbiome 5, 163–114. doi: 10.1186/s40168-017-0382-3
Rothschild, D., Weissbrod, O., Barkan, E., Kurilshikov, A., Korem, T., Zeevi, D., et al. (2018). Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215. doi: 10.1038/nature25973
Ryu, H., Grond, K., Verheijen, B., Elk, M., Buehler, D. M., and Domingo, J. W. S. (2014). Intestinal microbiota and species diversity of campylobacter and helicobacter spp. in migrating shorebirds in Delaware Bay. Appl. Environ. Microbiol. 80, 1838–1847. doi: 10.1128/AEM.03793-13
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60–R18. doi: 10.1186/gb-2011-12-6-r60
Song, S. J., Sanders, J. G., Delsuc, F., Metcalf, J., Amato, K., Taylor, M. W., et al. (2020). Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. MBio 11, e02901–e02919. doi: 10.1128/mBio.02901-19
Sonnenburg, J. L., and Bäckhed, F. (2016). Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64. doi: 10.1038/nature18846
Stegen, J. C., Lin, X. J., Konopka, A. E., and Fredrickson, J. K. (2012). Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 6, 1653–1664. doi: 10.1038/ismej.2012.22
Sturm-Ramirez, K. M., Hulse-Post, D. J., Govorkova, E. A., Humberd, J., Seiler, P., Puthavathana, P., et al. (2005). Are ducks contributing to the endemicity of highly pathogenic H5N1 influenza virus in Asia? J. Virol. 79, 11269–11279. doi: 10.1128/JVI.79.17.11269-11279.2005
Sugino, H. (2007). Comparative genomic analysis of the mouse and rat amylase multigene family. FEBS Lett. 581, 355–360. doi: 10.1016/j.febslet.2006.12.039
Sun, X., Guo, L. D., and Hyde, K. D. (2011). Community composition of endophytic fungi in Acer truncatum and their role in decomposition. Fungal Divers. 47, 85–95. doi: 10.1007/s13225-010-0086-5
Sun, Y., Liu, X., Sun, L., Men, M. Q., Wang, B., Deng, L. T., et al. (2022). Microecological insight to fungal structure and key fungal communities regulating nitrogen transformation based on spatial heterogeneity during cow manure composting by multi-angle and multi-aspect analyses. Waste Manag. 142, 132–142. doi: 10.1016/j.wasman.2022.02.013
Trosvik, P., and de Muinck, E. J. (2015). Ecology of bacteria in the human gastrointestinal tract-identification of keystone and foundation taxa. Microbiome 3, 1–12. doi: 10.1186/s40168-015-0107-4
Vujkovic-Cvijin, I., Sklar, J., Jiang, L. J., Natarajan, L., Knight, R., and Belkaid, Y. (2020). Host variables confound gut microbiota studies of human disease. Nature 587, 448–454. doi: 10.1038/s41586-020-2881-9
Waite, D. W., and Taylor, M. W. (2014). Characterizing the avian gut microbiota: membership, driving influences, and potential function. Front. Microbiol. 5, 223. doi: 10.3389/fmicb.2014.00223
Walter, J., Britton, R. A., and Roos, S. (2011). Host-microbial symbiosis in the vertebrate gastrointestinal tract and the lactobacillus reuteri paradigm. Proc. Natl. Acad. Sci. 108:4645–4652. doi: 10.1073/pnas.1000099107
Wei, Z. H., Zheng, M., Zhou, L. Z., and Xu, W. B. (2020). Flexible foraging response of wintering hooded cranes (Grus monacha) to food availability in the lakes of the Yangtze River floodplain, China. Animals 10:568. doi: 10.3390/ani10040568
Wu, G., Li, L., Sung, G. H., Kim, T. W., Byeon, S. E., Cho, J. Y., et al. (2011). Inhibition of 2, 4-dinitrofluorobenzene-induced atopic dermatitis by topical application of the butanol extract of Cordyceps bassiana in NC/Nga mice. J. Ethnopharmacol. 134, 504–509. doi: 10.1016/j.jep.2010.12.012
Xiang, X. J., Jin, L. L., Yang, Z. Q., Zhang, N. Z., and Zhang, F. (2021). Dramatic shifts in intestinal fungal community between wintering Hooded Crane and Domestic Goose. Avian Res. 12, 1–6. doi: 10.1186/s40657-020-00238-1
Xiang, X. J., Zhang, F. L., Fu, R., Yan, S. F., and Zhou, L. Z. (2019). Significant differences in bacterial and potentially pathogenic communities between sympatric hooded crane and greater white-fronted goose. Front. Microbiol. 10:163. doi: 10.3389/fmicb.2019.00163
Yan, Q. Y., Li, J. J., Yu, Y. H., Wang, J. J., He, Z. L., Van Nostrand, J. D., et al. (2016). Environmental filtering decreases with fish development for the assembly of gut microbiota. Environ. Microbiol. 18, 4739–4754. doi: 10.1111/1462-2920.13365
Yang, W. S., Ratan, Z. A., Kim, G., Lee, Y., Kim, M. Y., Kim, J. H., et al. (2015). 4-Isopropyl-2, 6-bis (1-phenylethyl) aniline 1, an analogue of KTH-13 isolated from Cordyceps bassiana, inhibits the NF-κB-mediated inflammatory response. Mediat. Inflamm. 2015:143025. doi: 10.1155/2015/143025
Yang, J., Yu, Z. S., Wang, B. B., and Ndayisenga, F. (2021). Gut region induces gastrointestinal microbiota community shift in Ujimqin sheep (Ovis aries): from a multi-domain perspective. Environ. Microbiol. 23, 7603–7616. doi: 10.1111/1462-2920.15782
Youngblut, N. D., Reischer, G. H., Walters, W., Schuster, A., Walzer, C., Stalder, G., et al. (2019). Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 10, 2200–2215. doi: 10.1038/s41467-019-10191-3
Yuan, H. Y., Mei, R., Liao, J. H., and Liu, W. T. (2019). Nexus of stochastic and deterministic processes on microbial community assembly in biological systems. Front. Microbiol. 10:1536. doi: 10.3389/fmicb.2019.01536
Zhang, M., Cai, G. Y., Zheng, E. Q., Zhang, G. G., Li, Y., Li, Z., et al. (2019). Transgenic pigs expressing β-xylanase in the parotid gland improve nutrient utilization. Transgenic Res. 28, 189–198. doi: 10.1007/s11248-019-00110-z
Zhao, N., Wang, S. P., Li, H. Y., Liu, S. L., Li, M., Luo, J., et al. (2018). Influence of novel highly pathogenic avian influenza A (H5N1) virus infection on migrating whooper swans fecal microbiota. Front. Cell. Infect. Microbiol. 8:46. doi: 10.3389/fcimb.2018.00046
Zheng, F. L., Cao, Y. L., Yang, R. F., Wang, L., Lv, X. X., Zhang, W. X., et al. (2020). Trichoderma reesei XYR1 activates cellulase gene expression via interaction with the Mediator subunit TrGAL11 to recruit RNA polymerase II. PLoS Genet. 16:e1008979. doi: 10.1371/journal.pgen.1008979
Zheng, M., Zhou, L. Z., Zhao, N. N., and Xu, W. B. (2015). Effects of variation in food resources on foraging habitat use by wintering Hooded Cranes (Grus monacha). Avian Res. 6, 1–10. doi: 10.1186/s40657-015-0020-3
Zhou, J. Z., Deng, Y., Luo, F., He, Z. L., Tu, Q. C., and Zhi, X. Y. (2010). Functional molecular ecological networks. mBio 1, e00169–e00110. doi: 10.1128/mBio.00169-10
Zhou, J. Z., and Ning, D. L. (2017). Stochastic community assembly: does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 81, e00002–e00017. doi: 10.1128/MMBR.00002-17
Keywords: hooded crane, gut fungal community, wintering stage, high-throughput sequencing, pathogen
Citation: Wu Y, Li Z, Zhao J, Chen Z and Xiang X (2022) Significant differences in intestinal fungal community of hooded cranes along the wintering periods. Front. Microbiol. 13:991998. doi: 10.3389/fmicb.2022.991998
Edited by:
Jinbo Xiong, Ningbo University, ChinaReviewed by:
Gang Liu, Anhui Medical University, ChinaCopyright © 2022 Wu, Li, Zhao, Chen and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhong Chen, Y2hlbnpob25nMjAyMjA3QDE2My5jb20=; Xingjia Xiang, eGp4aWFuZ0BhaHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.